

Abstract
CCR5 KO kidney transplant (KTx) recipients are extraordinarily high alloantibody producers and develop pathology that mimics human antibody-mediated rejection (AMR). C57BL/6 and CCR5 KO mice (H-2b) were transplanted with A/J kidneys (H-2a); select cohorts received adoptive cell therapy (ACT) with alloprimed CXCR5+CD8+ T cells (or control cells) on day 5 after KTx. ACT efficacy was evaluated by measuring posttransplant alloantibody, pathology, and allograft survival. Recipients were assessed for quantity of CXCR5+CD8+ T cells and CD8-mediated cytotoxicity to alloprimed IgG+ B cells. Alloantibody titer in CCR5 KO recipients was four-fold higher than in C57BL/6 recipients. The proportion of alloprimed CXCR5+CD8+ T cells 7 days after KTx in peripheral blood, lymph node, and spleen was substantially lower in CCR5 KO compared to C57BL/6 recipients. In vivo cytotoxicity towards alloprimed IgG+ B cells was also reduced six-fold in CCR5 KO recipients. ACT with alloprimed CXCR5+CD8+ T cells (but not alloprimed CXCR5−CD8+ or third-party primed CXCR5+CD8+ T cells) substantially reduced alloantibody titer, ameliorated AMR pathology, and prolonged allograft survival. These results indicate that a deficiency in quantity and function of alloprimed CXCR5+CD8+ T cells contributes to high alloantibody and AMR in CCR5 KO recipient mice, which can be rescued with ACT.
Introduction
The lack of definitive approaches to suppress the development of posttransplant alloantibody or to treat antibody-mediated rejection (AMR) is a key challenge following solid organ and cell transplantation [reviewed in (1)]. Clinical data support a pathogenic role for alloantibodies in both acute and chronic rejection following transplant (2, 3). Donor-specific alloantibody (DSA) develops de novo in 13–27% of kidney transplant recipients (4–9) and has been reported to occur in up to 40% of solid organ transplants (10). Such antibodies can be deleterious to kidney allografts (4, 11–14), with a 5–7% incidence of AMR in first-time kidney transplant recipients (3, 15). Over the long-term, recipients of kidney, pancreas, heart, lung, or liver who develop posttransplant DSA experience worse allograft survival than those without DSA [as reviewed in (16)]. Thus, there is an urgent need to investigate novel therapeutic approaches to reduce acute and chronic antibody-mediated graft injury and loss.
The importance of CD4+ T/B cell interactions for driving antibody production is well recognized (17, 18), yet despite the use of T cell depletion induction therapy and maintenance immunosuppressive agents that target CD4+ T cells, humoral alloimmunity readily develops in transplant recipients. Current therapies to treat AMR include removal of deleterious alloantibodies (e.g. by plasmapheresis, immunoadsorption), targeting IgG+ cells (e.g. intravenous immunoglobulin), cellular depletion (e.g. rituximab, alemtuzumab), complement inhibition, co-stimulation blockade (belatacept), proteasome inhibition, or a combination of strategies [reviewed in (3)]. These therapies carry risk associated with global non-specific immunosuppression, and in the case of co-stimulation blockade, an unexpected increased rate of acute rejection (19). Thus, current therapies can produce unpredictable or detrimental consequences. A recent expert consensus of immunotherapies to treat AMR concluded that there is a “low level of evidence for current strategies,” “no conclusive evidence to support any specific therapy,” and that research to identify new therapeutic targets is needed (20).
Our group spearheaded the discovery of a novel subset of antibody-suppressing CD8+ T cells (CD8+ TAb-supp cells) and their capacity to downregulate posttransplant alloantibody (21–24). We first reported a pivotal role of CD8+ T cells in inhibiting the magnitude of alloantibody produced after hepatocyte transplant in mice (21). CD8-mediated suppression of posttransplant alloantibody occurs after cell [hepatocyte and islet] and skin transplant across the same full MHC-disparate strain combination (C57BL/6 H-2b anti-FVB/N H-2q; Supplemental Figure 1). Our published data demonstrate that CD8+ TAb-supp cells require IFN-γ (21), as well as FasL and perforin (22), to inhibit antibody production. The function of CD8+ TAb-supp cells is associated with in vitro and in vivo killing of alloprimed (self-IgG+) B cell targets (22, 23). Antibody-suppressor CD8+ T cells express CXCR5 (23), a chemokine receptor important for homing to the germinal center (GC) where B cell maturation occurs (25–27). CD8-deficient recipient mice are high alloantibody producers and have increased number of antibody-producing B cells compared to wild type (WT) mice (22).
CCR5 KO KTx recipients are known to be extraordinarily high alloantibody producers and uniformly develop severe AMR (that mimics human AMR pathology (28–30)) and allograft failure (median survival time, MST=17 days) (28, 29). We hypothesized that the high alloantibody production reported in CCR5 KO KTx recipient mice could be associated with a deficit or dysfunction of antibody-suppressor CXCR5+CD8+ T cells, which could be mitigated by adoptive cell therapy (ACT). The current study investigated posttransplant alloantibody production, allograft pathology, allograft survival, and the proportion of CXCR5+CD8+ T cells and their cytotoxic effector function against alloprimed B cells in CCR5 KO and C57BL/6 KTx recipient mice. We report first evidence for the efficacy of ACT with antibody-suppressor alloprimed CXCR5+CD8+ T cells on alloantibody production, AMR pathology, and allograft survival after KTx, a vascularized solid organ transplant.
Materials and Methods
Experimental animals.
CCR5 KO, C57BL/6 (wild-type, WT), or CD8 KO (all H-2b) as well as A/J (H-2a) and FVB/N (H-2q) mouse strains (male and female at 8–20 weeks of age; Jackson Labs) were used in this study. All experiments were performed in compliance with the IACUC guidelines of The Ohio State University (Protocol 2019A00000124) and Nationwide Children’s Hospital (Protocol AR17–00045).
Kidney isolation, transplantation, and allograft monitoring.
Murine kidney transplantation with ureteral reconstruction was performed as previously described (31, 32). Briefly, the donor left kidney is isolated by ligating and dividing the adrenal and testicular vessels. The left ureter is dissected free from the renal hilus to the bladder. The aorta and inferior vena cava (IVC) are mobilized at their junction with the left renal artery and vein. After ligating the aorta above the renal artery, the graft is perfused in situ with 0.5 ml of cold, heparinized Ringer’s lactate solution. The kidney and its arterial and venous vascular supply along with the ureter attached to a small elliptical bladder patch are removed en bloc in preparation for transplantation. In the recipient mouse, vascular perfusion of the kidney graft is re-established by end-to-side anastomosis to the recipient aorta and inferior vena cava, respectively, using continuous 11–0 nylon sutures. Ureteral reconstruction is accomplished by anastomosis of the donor bladder patch to the recipient bladder using interrupted 10–0 monofilament nylon sutures.
To assess kidney transplant survival, some recipients underwent concurrent bilateral native nephrectomy (Figures 1B and 7). Graft function and survival were followed daily by examination of animal health and biweekly monitoring of serial serum creatinine levels by ELISA (Abcam, Cambridge, United Kingdom). Serum creatinine averages 21.2±0.9 μmol/L in naïve C57BL/6 mice (33) and 27±7 μmol/L in C57BL/6 kidney transplant recipient mice (with concurrent bilateral nephrectomy) (29). Allograft rejection was defined by a serum creatinine ≥100 μmol/L consistent with published work (29). Recipients with signs of poor health associated with uremia were euthanized.
Figure 1. CCR5 KO recipients produce a robust humoral alloimmune response following kidney transplant.
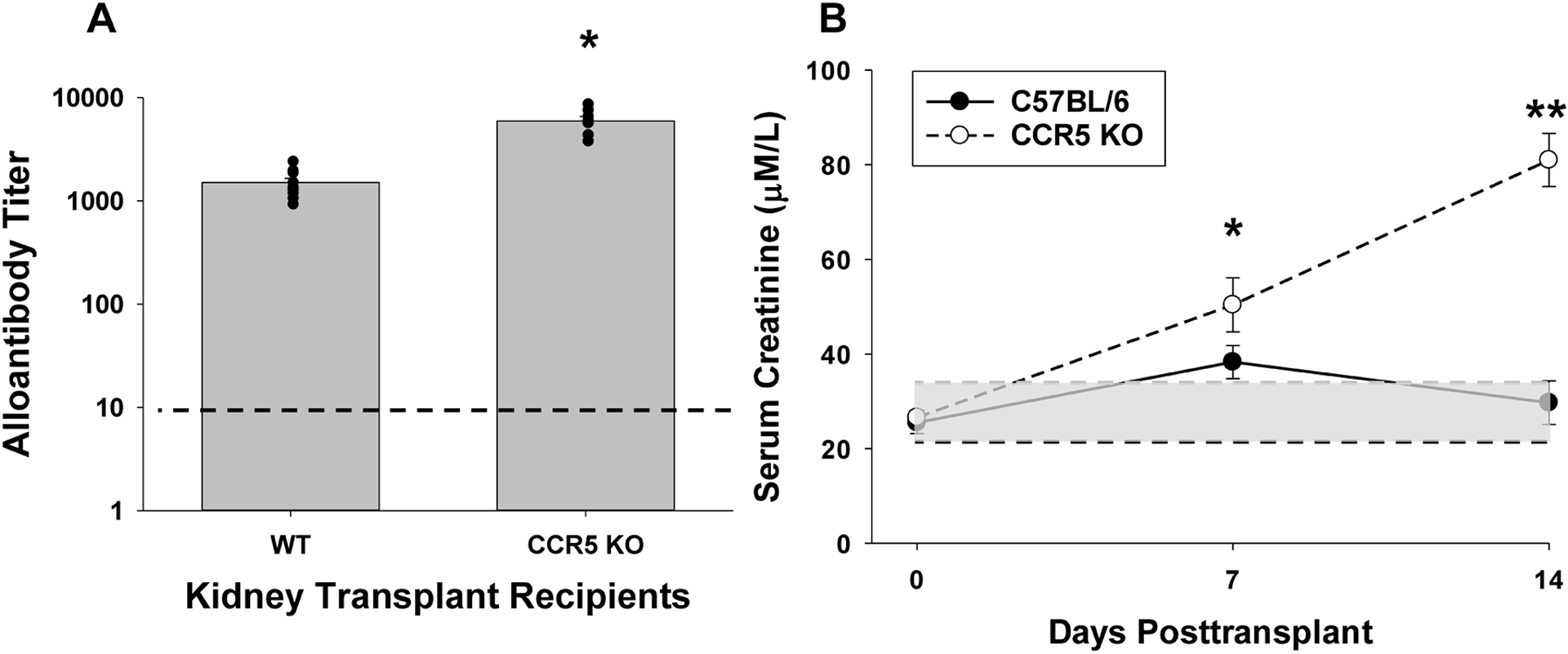
C57BL/6 (wild-type, WT; n=10) and CCR5 KO (n=7) mice (both H-2b) were transplanted with allogeneic (A/J; H-2a) kidneys. A) On day 14, serum was harvested and analyzed for alloantibody titer. CCR5 KO recipients produce significantly more alloantibody compared to WT recipients (6,000±630 vs. 1,500±150, respectively; *p<0.0001). Dashed line represents naïve control sera. B) Kidney allograft survival was assessed in a cohort of WT (n=6) and CCR5 KO (n=5) recipients that underwent concurrent bilateral nephrectomy. Recipient mice were evaluated for serum creatinine (SCr) before and serially following transplant. Posttransplant SCr progressively increased and was significantly higher in CCR5 KO recipients (day 7: 50.4±5.7 μM/L and day 14: 81.0±5.6 μM/L) compared to WT recipients (day 7: 38.3±3.5 μM/L, *p=0.047; day 14: 29.7±4.6 μM/L, **p<0.0001). No difference in SCr was observed prior to transplant between CCR5 KO and WT mice (WT: 25.5±2.4 μM/L, CCR5 KO: 26.6±1.4 μM/L, p=ns). Gray shaded area indicates normal range of SCr for C57BL/6 recipients (21–35 μM/L). Dark dotted line indicates mean serum creatinine in naïve C57BL/6 mice.
Figure 7. Adoptive cell therapy with alloprimed CXCR5+CD8+ T cells significantly enhances allograft survival in CCR5 KO kidney transplant recipients.
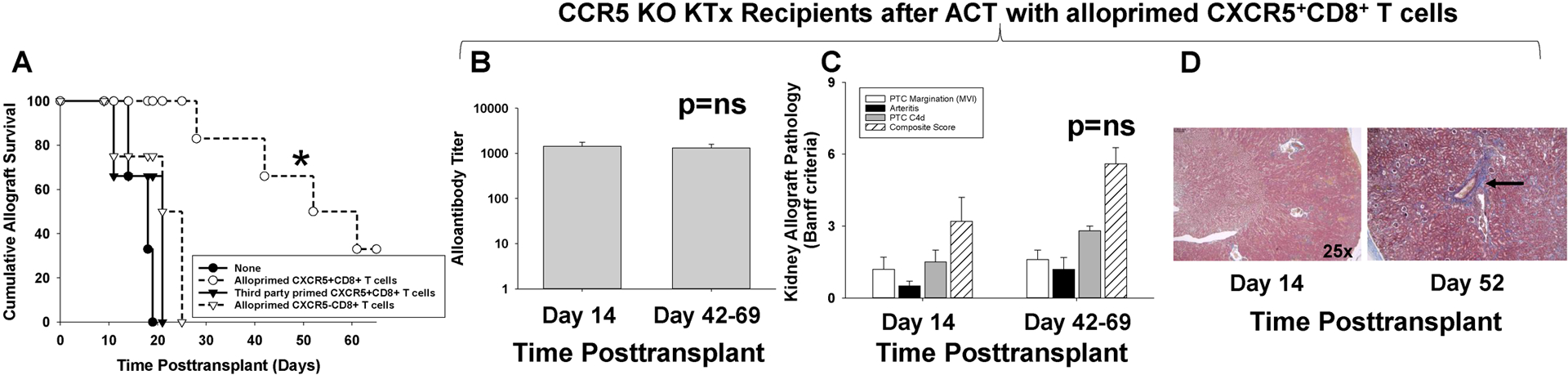
CCR5 KO (H-2b) mice were transplanted with allogeneic (A/J, H-2a) kidneys and underwent concurrent bilateral native nephrectomy. A cohort of CCR5 KO recipients received adoptive cell therapy (ACT) with 2×106 alloprimed CXCR5+CD8+ T cells (day 5 posttransplant). A) Recipients were monitored for serum creatinine (SCr) to determine allograft survival (SCr≥100 μmol/L indicates graft loss). Kidney allograft survival was significantly prolonged in recipients that received ACT with CXCR5+CD8+ T cells (MST= 52 days, n=6) as compared to CCR5 KO recipient without ACT (MST= 15 days, n=6; *p=0.0007). ACT with 2×106 third party-primed CXCR5+CD8+ T cells (MST=21, n=3) or 2×106 alloprimed CXCR5−CD8+ T cells (MST=25, n=4) into CCR5 KO kidney transplant recipients did not prolong allograft survival (p=ns for both). B-D) CCR5 KO recipients that received ACT with alloprimed CXCR5+CD8+ T cells were evaluated for alloantibody and pathology at day 14 and at time of late allograft loss. B) In CCR5 KO recipients that received ACT with alloprimed CXCR5+CD8+ T cells, alloantibody titer on day 14 (1,400±330, n=5) was similar to alloantibody titer at the time of late allograft loss (day 42–69; 1,300±260, p=ns). C) Composite AMR scores were not significantly different between kidney allografts analyzed on day 14 (3.2±1.0) or at the time of late allograft loss (5.6±1.5; p=ns). D) Trichrome staining demonstrated mild fibrosis (1.0±0.0) at the time of late allograft loss compared to day 14 kidney allograft specimens that had no evidence of fibrosis (0±0; p<0.0001). Images are representative of the sample groups. Black arrow indicates area of perivascular fibrosis.
Both CCR5 KO and C57BL/6 mice in these studies originated from Jackson labs and underwent kidney transplantation using the same surgical technique. Use of mouse strains from the same vendor source and application of the same surgical technique minimizes any differences in experimental outcomes that could be attributed these factors.
Splenocyte and lymph node cell isolation.
Splenocytes were isolated by mechanical disruption of the spleen followed by red blood cell lysis, as previously described (34). Lymph node cells were isolated from enlarged mesenteric lymph nodes (mLN) in a similar fashion. Mesenteric lymph nodes are noted to increase in size after kidney transplant, are readily accessible and are known to drain the peritoneal cavity (35); lymph nodes were not detected around the transplant kidney or in the peri-aortic regions.
Preparation of primed CXCR5+CD8+ T cells.
Lysate was prepared from allogeneic (A/J) or third-party (FVB/N) kidney tissue (5 freeze thaw cycles). Debris was removed by centrifugation. Lysate was administered to C57BL/6 mice by intraperitoneal injection (2 mg, i.p.) and primed CXCR5+CD8+ T cells were isolated from splenocytes of lysate-stimulated recipients on day 7.
CXCR5+CD8+ T cell isolation.
Isolation of CD8+ T cells from A/J lysate-primed (or third-party FVB/N-primed) hosts was performed using negative selection magnetic beads as per the manufacturer’s recommendations (StemCell Technologies, Vancouver, Canada; purity routinely >95%). Primed CD8+ T cells were sorted into CXCR5+CD8+ or CXCR5−CD8+ T cells by FACS Aria III flow cytometer (Becton Dickinson, Franklin Lakes, NJ) using anti-CXCR5 monoclonal antibody (clone 2G8; Becton Dickinson).
B cell isolation.
B cells (B220) and primed IgG+ B cells were purified from splenocytes using anti-mouse B220 (Miltenyi Biotech, Auburn, CA; purity routinely >95%) or anti-IgG magnetic beads (Miltenyi Biotech, Auburn, CA; purity routinely >95%) following the manufacturer’s instructions.
In vivo allocytotoxicity assay.
Detection of in vivo cytotoxic clearance of alloprimed IgG+ B cells was performed as previously described (22). Control target B220+ B cells were isolated from naïve wild-type C57BL/6 mice and were stained with 0.2μM carboxyfluorescein diacetate succinimidyl ester (CFSElow; Molecular Probes, Eugene, OR). Alloprimed IgG+ target B cells were isolated from high alloantibody producing CD8 KO mice primed with A/J kidney lysate and stained with 2.0μM CFSE (CFSEhigh). On day 7 posttransplant, kidney allograft recipient mice and control naïve mice received 10×106 CFSElow naïve B220+ B cells and 10×106 CFSEhigh alloprimed IgG+ B cells by tail vein injection. Eighteen hours following adoptive transfer, B cells were retrieved from the spleen and analyzed by flow cytometry (CFSE gating). Percent allospecific cytotoxicity was determined using a published formula that calculates the change in relative proportions of CFSEhigh alloprimed B cells compared to CFSElow control B cell targets (36).
In vitro cytotoxicity assay.
In vitro cytotoxicity was measured using a LIVE/DEAD cell-mediated cytotoxicity kit (Invitrogen, Eugene, OR) and performed according to the manufacturer’s instructions and as previously described (37). In brief, alloprimed CXCR5+CD8+ T cells were isolated from C57BL/6 or CCR5 KO mice 7 days after in vivo stimulation with allogeneic lysate. Target alloprimed IgG+ B cells (isolated from CD8 KO mice 7 days after stimulation with allogeneic lysate) were stained with CFSEhigh. CXCR5+CD8+ T cells and IgG+ B cells were co-cultured at a 10:1 ratio for 4 hours. Propidium iodide (PI) was added to the co-cultures to assess cell death, and PI uptake in CFSE+ B cells was immediately analyzed by flow cytometry, as previously described (22). In vitro cytotoxicity was determined by calculating the percentage of PI-positive B cells.
Donor-reactive alloantibody titer.
Alloantibody titer from recipient sera was quantitated using published methods (29). Briefly, serum was serially diluted and incubated with allogeneic target splenocytes. Splenocytes were then stained with FITC-conjugated goat anti-mouse IgG Fc (Organon Teknika, Durham, NC). The mean fluorescence intensity (MFI) was measured for each sample and the dilution that returned the MFI observed when splenocytes were incubated with a 1:4 dilution of naïve serum was divided by two and recorded as the titer.
Flow cytometric CD8+ T cell analysis and IFN-γ staining.
Peripheral blood mononuclear cells, mesenteric lymph node cells, and splenocytes were isolated from A/J kidney transplant recipients on days 0, 7, and 14 posttransplant and incubated for 4 hours with Leukocyte Activation Cocktail (PMA, ionomycin, and Brefeldin A; Becton Dickinson). Lymphocytes were then stained for CD8 (clone 53–6.7), CXCR5 (clone 614641), and CD44 (clone IM7), and subsequently stained and analyzed for expression of intracellular IFN-γ (clone XMG1.2). Intracellular staining was performed using the FIX&PERM cell permeabilization kit (Thermo Fisher). Flow cytometric analysis was performed by gating on single cell, lymphocyte populations of CD8+ T cells. Fluorescence minus one (FMO) was utilized as a negative control to set the positive/negative boundary for protein expression (38).
Flow cytometric B cell analysis.
Splenocytes were isolated from A/J kidney transplant recipients on day 14 posttransplant. Splenocytes were then stained for B220 (clone RA3–6B2), GL-7 (clone GL-7), and Fas (clone Jo2). Flow cytometric analysis was performed by gating on single cell, lymphocyte populations of B cells. Fluorescence minus one (FMO) was utilized as a negative control to set the positive/negative boundary for protein expression.
Histochemistry.
Renal allografts were excised and sectioned into halves. One half section was fixed in 10% buffered formalin (6 hours), rehydrated in 20% sucrose (18 hours), and embedded in paraffin. For histological analysis, 5μm sections were mounted on glass slides and stained with H&E. Images were captured and analyzed with an Aperio Scanscope XT (Leica, Wetzlar, Germany).
Immunofluorescence.
The other half section of renal allografts was immediately snap frozen with liquid nitrogen and OCT medium (Tissue-Tek, Torrance, CA). Frozen sections were cut at 3μm and analyzed for C4d deposition by FITC-conjugated polyclonal anti-mouse C4d antibody (39). Stained sections were analyzed by Olympus BX-43 microscope (confocal microscopy).
Pathological analysis.
Renal allografts were evaluated for AMR pathology, including microvascular inflammation/peritubular capillary (PTC) margination, arteritis, and PTC C4d deposition (29, 30). Following H&E and immunofluorescent C4d staining, pathological analysis of the samples was performed and scored in accordance to internationally accepted Banff criteria (grades 0–3 for each category) (40, 41). Individual scores were added together for a composite AMR score (PTC margination, arteritis, and PTC C4d deposition). Some renal allografts were analyzed for degree of fibrosis by Mason’s trichrome staining performed according to the manufacturer’s instructions (Abcam) and scored as previously described (42).
Statistical analysis.
Student’s t-tests were used to test differences in continuous outcomes between two experimental groups. When more than two experimental groups were assessed, continuous outcomes measured at one time point were compared using general linear models including experimental groups as an independent variable. Continuous outcomes measured at multiple time points were compared between relevant groups using general linear models including experimental groups, time, and their interaction as independent variables. As the measurements were not conducted on the same mice over time, measurements were assumed to be independent, and no additional correlation was considered. Assumptions of normally distributed residuals were assessed graphically using q-q plots and were not considered to be violated for any of the analyses. Graft survival between experimental groups was compared using a Kaplan-Meier survival curve and log-rank test. All analyses were conducted using SAS version 9.4 (SAS Institute, Inc., Cary, NC). Results are summarized as estimated mean ± standard error. Hypothesis testing was conducted at a 5% type I error rate (alpha=0.05) and p<0.05 was considered statistically significant.
Results
Proportion of CXCR5+CD8+ T cells is significantly reduced, and transplant renal function is worse in high alloantibody producing CCR5 KO recipients compared to low alloantibody producing C57BL/6 kidney transplant recipients.
CCR5 KO (H-2b) and C57BL/6 (H-2b) WT mice underwent complete MHC mismatch kidney transplant with allogeneic A/J (H-2a) donor kidneys. CCR5 KO recipients developed significantly higher alloantibody titer (4-fold) than WT recipients on day 14 posttransplant (6,000±630 vs. 1,500±150, respectively, p<0.0001; Figure 1A). The kinetics of alloantibody production are similar in CCR5 KO and WT recipients with alloantibody first detected on day 5 (29), increased titer on day 7 (WT= 580±60; CCR5 KO = 870±130; p=0.046), and peak titer on day 14 posttransplant.
Further, in cohorts of WT and CCR5 KO kidney transplant recipients that underwent concurrent bilateral nephrectomy to assess renal allograft survival, we noted that renal function was significantly worse on day 7 posttransplant and progressively deteriorated in the high alloantibody producing CCR5 KO recipients compared to stable and near normal renal function in the WT recipients. Posttransplant serum creatinine was significantly higher in CCR5 KO recipients (day 7: 50.4±5.7 μM/L and day 14: 81.0±5.6 μM/L) compared to WT recipients (day 7: 38.3±3.5 μM/L, p=0.047; day 14: 29.7±4.6 μM/L, p<0.0001; Figure 1B). These data are consistent with the reported severe AMR pathology and allograft loss observed in untreated high alloantibody producing CCR5 KO (but not WT) recipients. (28, 29).
There was no difference in the proportion of CXCR5+CD8+ T cells detected in peripheral blood lymph node, or spleen in WT (peripheral blood: 3.4±0.5%, lymph node: 9.3±1.4%, spleen: 2.8±0.5%) and CCR5 KO mice (peripheral blood: 3.3±0.2%, lymph node: 8.0±2.3%, spleen: 1.6±0.2%, p=ns for all) at baseline (naïve mice; Figure 2A,B). The proportion of CXCR5+CD8+ T cells significantly increased in WT recipients by day 7 posttransplant, in the peripheral blood (6.5-fold; 22.4±3.5%, p<0.0001), lymph node (1.7-fold; 16.3±3.3%, p=0.04), and spleen (4.0-fold; 11.4±1.7%, p<0.0001). Likewise, a substantial percentage of activated CXCR5+CD44+IFN-γ+CD8+ T cells was detected by day 7 posttransplant, in the peripheral blood (19.0±3.8%, p<0.0001) lymph node (3.1±0.5%, p=0.002), and spleen (7.3±0.9%, p<0.0001) compared to the baseline control in naïve WT mice (peripheral blood: 0.2±0.1%, lymph node: 0.4±0.2%, spleen: 0.1±0.1%; Figure 2C). In WT recipients, the proportion of both CXCR5+CD8+ T cells and CXCR5+CD44+IFN-γ+CD8+ T cells declined back to baseline by day 14 posttransplant (day 0 vs. day 14, p=ns).
Figure 2. The peak proportion of CXCR5+CD8+ T cells (and activated CXCR5+CD44+IFN-γ+CD8+ T cells) after kidney transplant is severely reduced in CCR5 KO compared to WT recipients.
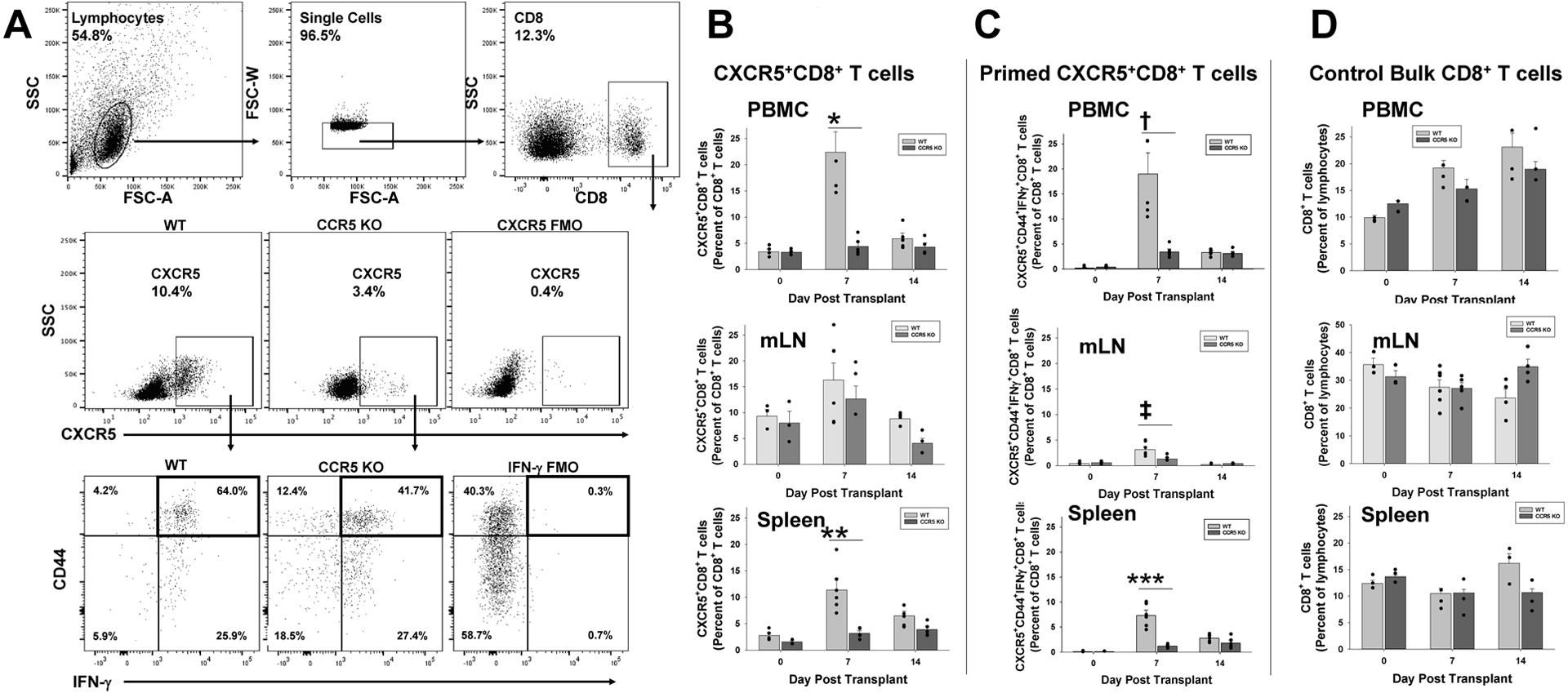
CCR5 KO and C57BL/6 (wild-type, WT; both H-2b) mice were transplanted with A/J (H-2a) kidney. Peripheral blood, mesenteric lymph node (mLN), and splenocytes were retrieved from cohorts of mice prior to transplant and on days 7 and 14 posttransplant. CD8+ T cells were analyzed by flow cytometry. A) Flow cytometric gating was on lymphocytes, single cells, CD8+ T cells, and CXCR5. Fluorescent minus one was used as a negative control. Representative flow panels of CXCR5+CD8+ and CXCR5+CD44+IFN-γ+CD8+ T cells from splenocytes of WT and CCR5 KO mice are shown. B) On day 7, CCR5 KO recipients compared to WT recipients have a significantly lower proportion of CXCR5+CD8+ T cells in the peripheral blood (4.4±0.8% vs. 22.4±3.5%; *p<0.0001) and spleen (3.2±0.4% vs. 11.4±1.7%; n=5 for both strains; **p<0.0001); however, no difference was detected in mLN (12.6±2.5% vs. 16.3±3.3%; p=ns). C) On day 7, CCR5 KO recipients compared to WT recipients have a significantly lower proportion of activated CXCR5+CD44+IFN-γ+CD8+ T cells in the peripheral blood (3.4±0.5% vs.19.0±3.8%; †p<0.0001), mLN (1.3±0.3% vs. 3.2±0.5%; ‡p<0.01) and spleen (1.2±0.1% vs. 7.3±0.9%; n=5 for both strains; ***p<0.0001). D) No significant difference in percentages of peripheral blood, mLN, or splenic CD8+ T cells in WT and CCR5 KO recipients was observed at any time point (p=ns).
In contrast, CCR5 KO KTx recipients did not develop a significant increase in total CXCR5+CD8+ T cells on day 7 (or 14) posttransplant (day 7, peripheral blood: 4.4±0.8%, lymph node: 12.6±2.5%, spleen: 3.2±0.4%) compared to baseline (peripheral blood: 3.3±0.2%, lymph node: 8.0±2.3%, spleen: 1.6±0.2%, p=ns; Figure 2A,B). Consequently, the proportion of CXCR5+CD8+ T cells on day 7 posttransplant was significantly less in CCR5 KO recipients compared to WT recipients (p<0.0001 for peripheral blood and spleen, but not lymph node). Similarly, the development of a significant population of activated CXCR5+CD44+IFN-γ+CD8+ T cells was not observed on day 7 (and 14) posttransplant in CCR5 KO recipient mice (day 7, peripheral blood: 3.4±0.5%, lymph node: 1.3±0.3%, spleen: 1.2±0.1%) when compared to WT recipients (Figure 2C). On day 7 posttransplant, the proportion of activated CXCR5+CD44+IFN-γ+CD8+ T cell subset was significantly less in CCR5 KO compared to WT recipients (3-fold less in peripheral blood, 2-fold less in lymph node, and 6-fold less in spleen, p<0.01 for all comparisons; Figure 2A,B). The observed differences in CXCR5+CD8+ T cell subsets in CCR5 KO and WT could not be attributed to differences in total CD8+ T cells (Figure 2C).
In contrast to the disparate proportions of CXCR5+CD8+ and CXCR5+CD44+IFN-γ+ CD8+ T cell subsets in CCR5 KO compared to WT KTx recipients, the proportions of alloprimed CXCR5−CD8+ (CXCR5−CD44+IFN-γ+CD8+) T cells are not significantly different between CCR5 KO and WT recipients (pretransplant or on day 7 or day 14 posttransplant; Supplementary Figure 2).
Alloprimed CXCR5+CD8+ T cell-mediated B cell cytotoxicity is significantly impaired in CCR5 KO (compared to C57BL/6) kidney transplant recipients.
To assay for CD8-mediated in vivo cytotoxicity to alloprimed B cell targets, CCR5 KO and WT kidney transplant recipients were adoptively transferred with CFSEhi labeled alloprimed (IgG+) B cells (and control CFSElo B220+ control naïve B cells) on day 7 posttransplant, as previously described (22). CCR5 KO recipient mice mediated significantly lower in vivo cytotoxicity to alloprimed B cells (6-fold; 12.9±4.0%, p<0.0001) compared to WT recipients (85.7±5.3%, Figure 3A,B). As CCR5 KO mice had relatively lower quantity of CXCR5+CD8+ T cells (Figure 2), the difference in magnitude of in vivo cytotoxic effector function observed in CCR5 KO compared to WT recipient mice could be associated with a lower effector to target cell ratio. To further investigate this possibility, 2×106 alloprimed CXCR5+CD8+ T cells (retrieved from alloprimed WT mice) were adoptively transferred into CCR5 KO recipients on day 5 posttransplant. CCR5 KO recipients were analyzed for in vivo cytotoxicity on day 2 following CD8 subset transfer (day 7 posttransplant). Adoptive transfer of alloprimed CXCR5+CD8+ T cells from WT mice into CCR5 KO recipient mice significantly enhanced in vivo cytotoxicity of alloprimed IgG+ B cells in CCR5 KO recipients (39.2±7.7%; p=0.0004) compared to untreated CCR5 KO recipients. In contrast, ACT of third party-primed CXCR5+CD8+ T cells into CCR5 KO recipients did not enhance in vivo cytotoxicity of alloprimed IgG+ B cells (12.7±7.7%; p=ns). Thus, in vivo cytotoxic clearance of alloprimed IgG+ B cells is mediated by allospecific CXCR5+CD8+ T cells.
Figure 3. CXCR5+CD8+ T cells from CCR5 KO (compared to WT) kidney transplant recipients mediate impaired in vitro and in vivo cytotoxicity to alloprimed B cell targets.
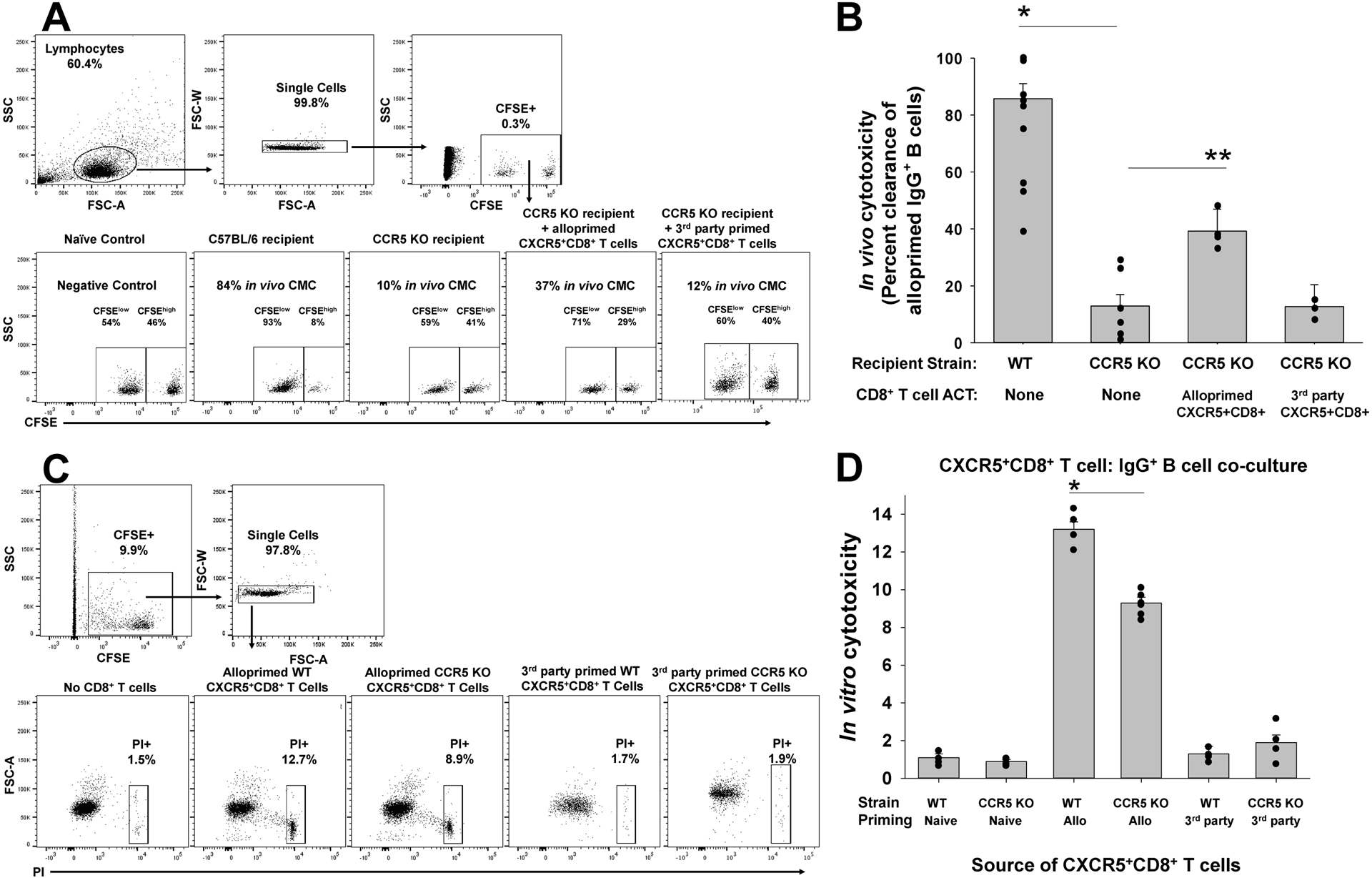
A) C57BL/6 (WT) and CCR5 KO mice (both H-2b) were transplanted with A/J (H-2a) kidney. On day 7, CFSEhi-labeled alloprimed IgG+ B cells were adoptively transferred (along with control, CFSElo naïve B220+ B cells) into transplant recipients to test for in vivo cytotoxic clearance of IgG+ B cells. A) Representative flow plots show gating on lymphocytes, single cells, and CFSElo naïve B cells and CFSEhi alloprimed IgG+ B cells that were used to determine vivo cytotoxicity. B) In vivo cytotoxicity to IgG+ B cells was significantly reduced in CCR5 KO recipients compared to WT recipients (12.9±4.0% vs. 85.7±5.3%, n=7 and n=10, respectively; *p<0.0001). Adoptive cell therapy (ACT) with alloprimed CXCR5+CD8+ T cells from WT mice significantly increased in vivo cytotoxicity to alloprimed B cell targets in CCR5 KO recipients (39.2±7.7%; n=4; **p=0.0004) compared to untreated CCR5 KO recipients. ACT with third party-primed CXCR5+CD8+ T cells was not associated with an increase in in vivo cytotoxicity to alloprimed IgG+ B cells (12.7±7.7%, n=4; p=ns) compared to CCR5 KO recipients without ACT. C) Next, we performed in vitro cytotoxicity using CXCR5+CD8+ T cells and alloprimed CFSE-labeled B cells in co-cultures. Propidium iodide (PI) was used to assess viability of B cells after 4 hours in co-culture. Representative flow plots show gating on CFSE+IgG+ B cell targets, single cells, and propidium iodide (PI). D) WT and CCR5 KO mice were alloprimed with A/J kidney lysate (2mg). On day 7, CXCR5+CD8+ T cells retrieved from the spleen were flow-sorted for in vitro cytotoxicity assays. Alloprimed CXCR5+CD8+ T cells from CCR5 KO mice mediated significantly less in vitro cytotoxicity to IgG+ B cells compared to those from WT mice (9.3±0.3 vs.13.2±0.4%, both groups n=6; *p=0.0001). Third-party primed WT or CCR5 KO CXCR5+CD8+ T cells did not mediate significant cytotoxicity to IgG+ B cells in vitro (1.3±0.4% and 1.9±0.4%, respectively; n=4 and p=ns for both).
In order to compare CXCR5+CD8+ T cell cytotoxic effector function from CCR5 KO or WT mice with equivalent effector to target cell ratios, we next tested in vitro cytotoxicity in CD8/B cell co-cultures. CXCR5+CD8+ T cells were purified from alloprimed WT and CCR5 KO mice and flow-sorted. Alloprimed CXCR5+CD8+ T cells were co-cultured with alloprimed self-IgG+ B cells for 4 hours. CXCR5+CD8+ T cells from alloprimed CCR5 KO mice (9.3±0.3%) mediated less cytotoxicity toward self-IgG+ B cell targets compared to CXCR5+CD8+ T cells from alloprimed WT mice (13.2±0.4%; p=0.0001; Figure 3C,D). Third-party primed WT or CCR5 KO CXCR5+CD8+ T cells did not mediate significant cytotoxicity to IgG+ B cells in vitro (1.3±0.4% and 1.9±0.4%, respectively; p=ns for both). Collectively, these data suggest that there is a relative deficiency in quantity and an impairment of cytotoxic effector function of allospecific, antibody-suppressor CXCR5+CD8+ T cells in CCR5 KO recipients compared to C57BL/6 recipients.
Adoptive Cell Therapy with CXCR5+CD8+ T cells into CCR5 KO mice significantly reduces posttransplant alloantibody after kidney transplant.
CCR5 KO mice were transplanted with A/J kidneys. On posttransplant day 5, cohorts of recipients underwent ACT with 0.5×106, 1×106, or 2×106 flow-sorted alloprimed CXCR5+CD8+ T cells. On day 14 posttransplant, recipient serum was collected and evaluated for alloantibody titer. Compared to untreated controls (titer= 6,000±630), ACT of 2×106 CXCR5+CD8+ T cells significantly inhibited the production of alloantibody in CCR5 KO recipients (4-fold reduction; 1,300±240, p<0.0001; Figure 4). Adoptive cell therapy with 1×106 alloprimed CXCR5+CD8+ T cells also inhibited alloantibody production (2-fold reduction; 2,800±1,100 p=0.0015), but ACT of 0.5×106 alloprimed CXCR5+CD8+ T cells did not (7,300±700, p=ns). The efficacy of ACT with CXCR5+CD8+ T cells for suppression of alloantibody was dose-dependent, since ACT with 2×106 or 1×106 cells reduced alloantibody titer to a greater extent than ACT with 0.5 × 106 cells (p<0.0002 for both). The reduction in alloantibody titer by ACT with 2×106 cells compared to 1×106 CXCR5+CD8+ T cells was not significantly different (p=0.07). Adoptive cell therapy with 2×106 third-party (FVB/N, H-2q) primed CXCR5+CD8+ T cells or alloprimed CXCR5−CD8+ T cells did not inhibit alloantibody production following A/J KTx (5,100±370 and 5,100±470 respectively) when compared to CCR5 KO recipients without ACT (6,000±630, p=ns for both; Figure 4). Alloantibody titer in CCR5 KO recipients that received ACT with third-party primed CXCR5+CD8+ T cells or alloprimed CXCR5−CD8+ T cells was substantially higher than in CCR5 KO KTx recipients that received an equivalent number of alloprimed CXCR5+CD8+ T cells (p<0.0001 for both comparisons). Thus, suppression of posttransplant alloantibody production by ACT with CXCR5+CD8+ T cells, in the setting of a vascularized solid organ transplant model, is dose-dependent and alloantigen specific.
Figure 4. Adoptive cell therapy with alloprimed CXCR5+CD8+ T cells into CCR5 KO kidney transplant recipients significantly inhibits alloantibody production.
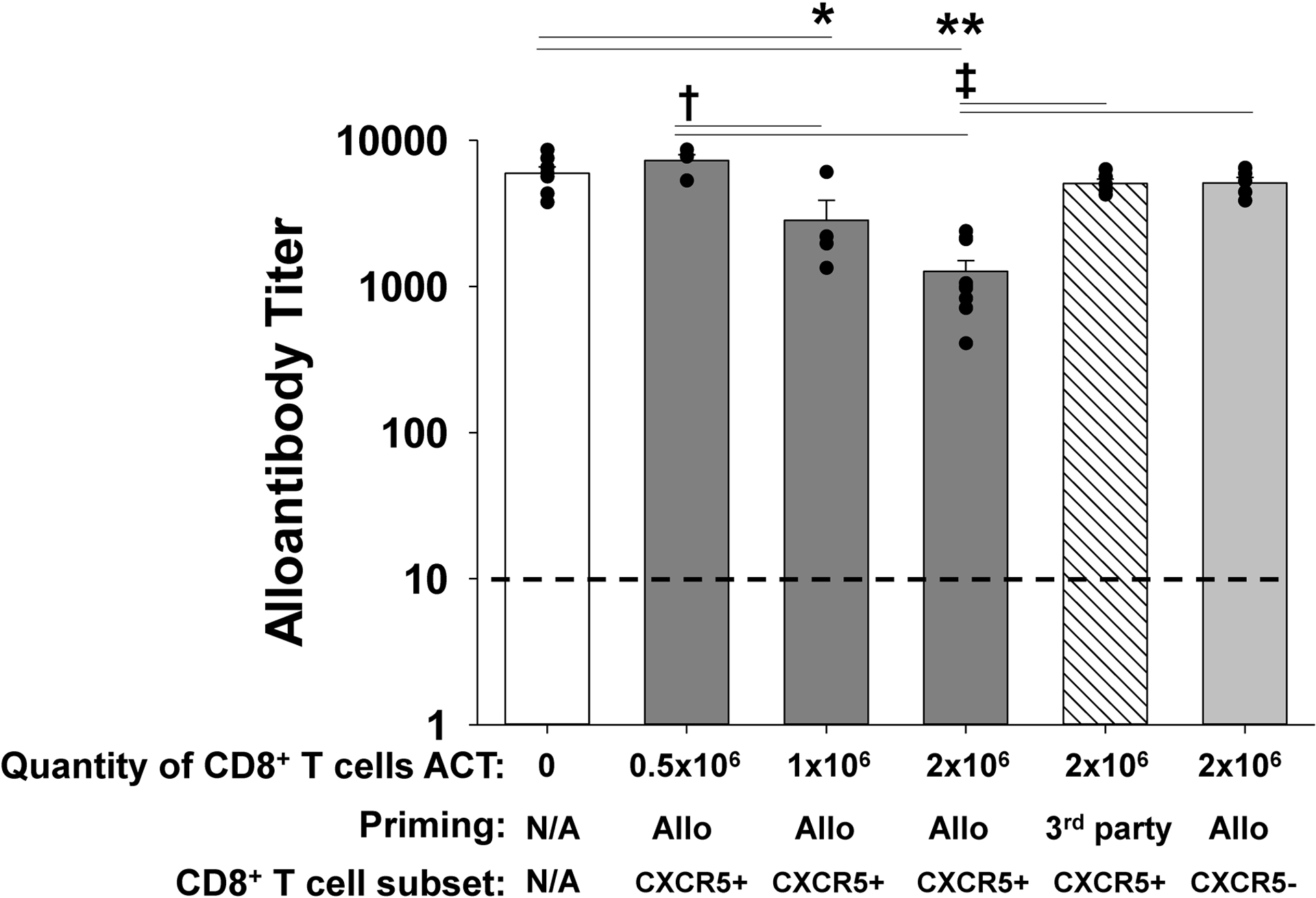
CCR5 KO (H-2b) mice were transplanted with an allogeneic kidney (A/J, H-2a). On day 5 posttransplant, a cohort of CCR5 KO recipients received adoptive cell therapy (ACT) with 0.5×106, 1×106, or 2×106 flow-sorted alloprimed CXCR5+CD8+ T cells (retrieved from A/J allolysate primed C57BL/6, H-2b, mice). Day 14 posttransplant serum from kidney transplant recipients was analyzed for alloantibody titer. Alloantibody production was significantly reduced following ACT with 1×106 CXCR5+CD8+ T cells (titer = 2,800±1,100, n=4; *p=0.0015) and ACT with 2×106 cells (titer = 1,300±240, n=9; **p<0.0001) compared to CCR5 KO recipients without ACT (titer= 6,000±630, n=7). Both ACT of 1×106 and 2×106 alloprimed CXCR5+CD8+ T cells inhibited alloantibody titer more than ACT with 0.5×106 alloprimed cells (†p<0.0002 for both). No significant reduction was observed with ACT with 0.5×106 alloprimed CXCR5+CD8+ T cells (titer = 7,300±700, n=4), 2×106 3rd party primed (FVB/N, H-2q) CXCR5+CD8+ T cells (titer = 5,100±370, n=5), or with 2×106 alloprimed CXCR5−CD8+ T cells (titer= 5,100±470, n=5). Recipient mice that received ACT with either 2×106 3rd party primed CXCR5+CD8+ T cells or alloprimed CXCR5−CD8+ T cells had significantly higher alloantibody titers compared to recipients that received 2×106 alloprimed CXCR5+CD8+ T cells (‡p<0.0001 for both). Dashed line represents naïve control sera.
Adoptive Cell Therapy with CXCR5+CD8+ T cells into CCR5 KO mice significantly reduces the proportion of splenic germinal center B cells after kidney transplant.
Based on the difference in alloantibody titer between CCR5 KO and WT KTx recipients (Figure 1), we expected the quantity of germinal center (GC) B cells to be higher in CCR5 KO recipients compared to WT recipients. The proportion of GC B cells was assessed on day 14 after KTx. WT recipients had a relatively low proportion (0.9±0.1%) of GC B cells compared to CCR5 KO recipients (7.3±0.9%; p<0.0001). ACT with 2×106 alloprimed CXCR5+CD8+ T cells into CCR5 KO recipients on day 5 after KTx resulted in a ~2-fold reduction in the proportion of splenic GC B cells (4.9±0.4%; p=0.002) compared to CCR5 KO recipients without ACT (Figure 5B). The difference in GC B cells could not be attributed to a difference in the percentage of B cells within the splenocyte population (Figure 5C). Similarly, we found that splenic plasma cells (CD138+B220−IgG−) on day 14 after KTx are significantly higher in CCR5 KO recipients compared to WT recipients. Following ACT, plasma cells are also significantly reduced in CCR5 KO recipients (Supplemental Figure 3). These data suggest that ACT of CXCR5+CD8+ T cells partially restores an intrinsic CXCR5+CD8+ T cell subset deficit in CCR5 KO mice that corresponds with reduced proportion of GC B cells (and plasma cells) and lower posttransplant alloantibody titer.
Figure 5. Adoptive cell therapy with alloprimed CXCR5+CD8+ T cells into CCR5 KO kidney transplant recipients significantly reduces the proportion of splenic germinal center B cells.
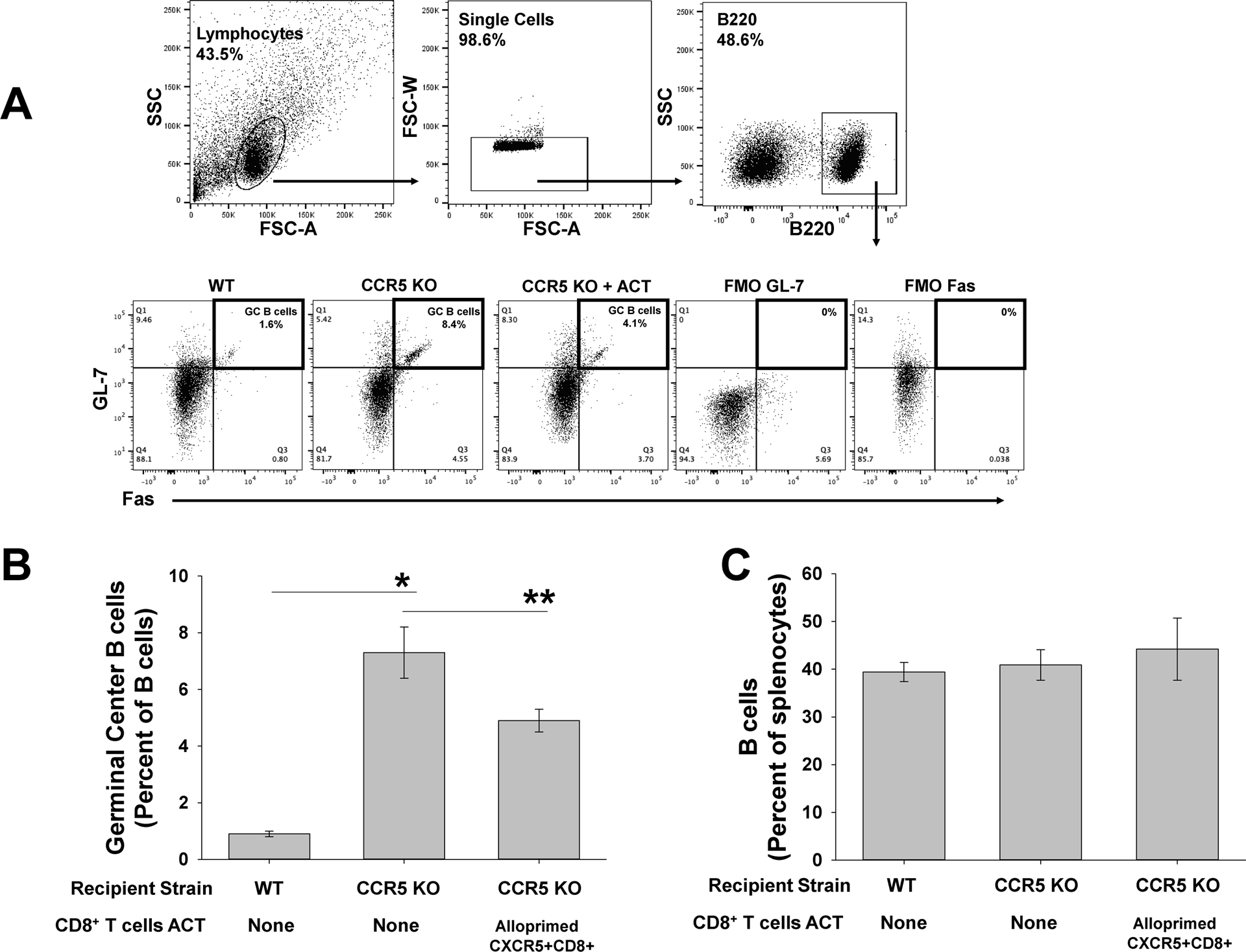
C57BL/6 (WT) and CCR5 KO mice (both H-2b) were transplanted with A/J (H-2a) kidney. On posttransplant day 5, a cohort of CCR5 KO recipients underwent ACT with 2×106 CXCR5+CD8+ T cells. Splenocytes were retrieved for analysis of germinal center (GC) B cells (B220+Fas+GL-7+) on day 14 posttransplant. A) Representative flow cytometric gating on lymphocytes, single cells, and B220+ cells are shown. Fluorescent minus one was used as a negative control. B) The proportion of GC B cells (GL-7+Fas+B220+) was significantly higher in CCR5 KO recipients (7.3±0.9%, n=5) compared to WT recipients (0.9±0.1%, n=5; *p<0.0001). Following ACT with 2.0×106 alloprimed CXCR5+CD8+ T cells, the proportion of splenic GC B cells was significantly reduced in CCR5 KO recipients (4.9±0.4%, n=6; **p=0.002). C) No significant difference in overall quantity of splenic B cells was observed between WT and CCR5 KO recipients (p=ns).
Adoptive Cell Therapy with CXCR5+CD8+ T cells ameliorates AMR pathology following kidney transplant in CCR5 KO mice.
The pathology of AMR in CCR5 KO KTx recipients mimics the pathologic features of AMR in humans (28–30). To compare AMR pathology in untreated CCR5 KO mice and those that received ACT with alloprimed CXCR5+CD8+ T cells, we developed a scoring system modeled on Banff criteria including peritubular capillary margination (PTC), arteritis, and C4d deposition (42, 43). Each of these pathologic features was assessed and scored as an individual parameter for blinded specimens. A composite score of all three parameters was also calculated. Kidney allografts from untreated high alloantibody producing CCR5 KO recipients had severe AMR pathology as reflected in a significantly higher composite AMR score on day 14 posttransplant compared to low alloantibody producing WT KTx recipients (7.6±0.3 vs. 2.4±0.2, respectively; p<0.0001; Figure 6). Adoptive cell therapy with 2×106 alloprimed CXCR5+CD8+ T cells into CCR5 KO KTx recipients significantly improved the day 14 pathology of transplanted kidneys, as reflected by lower composite AMR scores compared to untreated CCR5 KO recipients (3.2±1.0 vs. 7.6±0.3, p<0.0001). In fact, two kidney allografts from ACT-treated mice had histology and gross appearance similar to healthy non-transplanted kidneys of naïve A/J mice. Individual parameter scores were also significantly lower in treated compared to untreated CCR5 KO KTx recipients. Kidney allografts from CCR5 KO recipients that received ACT with 2×106 alloprimed CXCR5+CD8+ T cells exhibited significantly lower PTC margination (1.2±0.5; p=0.0005), arteritis (0.5±0.2; p<0.0001), and PTC C4d deposition (1.5±0.5; p=0.04) than untreated CCR5 KO recipients (2.7±0.2, 2.4±0.3, and 2.4±0.3, respectively). Adoptive cell therapy with 0.5×106 and 1.0×106 alloprimed CXCR5+CD8+ T cells, 2.0×106 3rd party primed CXCR5+CD8+ T cells, or 2.0×106 alloprimed CXCR5−CD8+ T cells did not significantly improve individual or composite AMR pathology scores.
Figure 6. Adoptive cell therapy with alloprimed CXCR5+CD8+ T cells into CCR5 KO kidney transplant recipients ameliorate antibody-mediated rejection pathology.
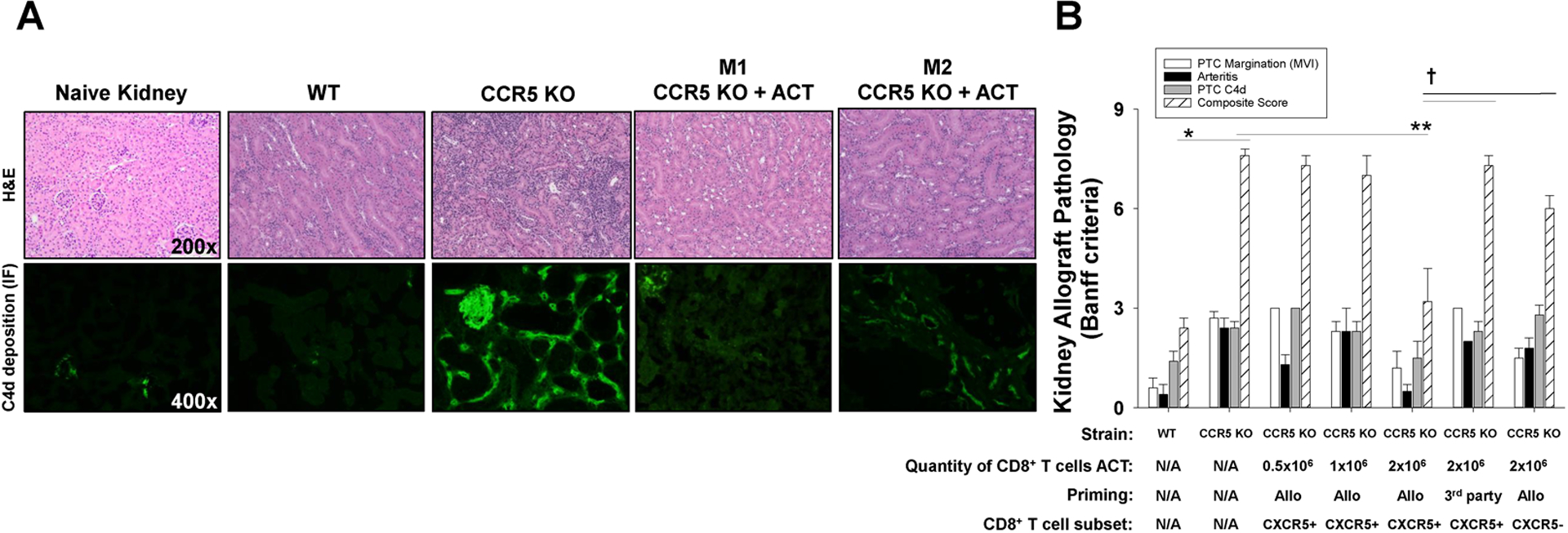
C57BL/6 and CCR5 KO (both H-2b) mice were transplanted with allogeneic (A/J, H-2a) kidneys. A cohort of CCR5 KO recipients received adoptive cell therapy (ACT) with alloprimed CXCR5+CD8+ T cells (0.5×106 to 2×106 cells; day 5 posttransplant) or 3rd party (FVB/N, H-2q) primed CXCR5+CD8+ T cells (2×106). On day 14 posttransplant, allografts were removed for histological analysis. A) Histology and C4d immunofluorescence of tissue samples from each group were analyzed (representative data shown). Kidney pathology in CCR5 KO recipients that received ACT had significantly less inflammation and C4d deposition compared to CCR5 KO recipients without ACT. There was some variability in the amount of inflammation in CCR5 KO mice that received ACT ranging from minimal PTC inflammation (or C4d deposition) observed in mouse 1 (M1: CCR5 KO + ACT) to reduced inflammation and C4d deposition as observed in mouse 2 (M2: CCR5 KO + ACT). B) Samples were scored according to Banff criteria. The individual histologic scores for PTC margination, arteritis, and PTC C4d deposition, as well as histologic composite score are shown for experimental groups. Composite AMR score in CCR5 KO recipients (7.6±0.4, n=7) was significantly higher than in C57BL/6 recipients (2.4±0.5, n=5; *p<0.0001). Composite AMR scores in CCR5 KO recipients that received ACT with 2.0×106 alloprimed CXCR5+CD8+ T cells (3.2±2.6, n=6) were significantly lower than in CCR5 KO recipients without ACT (7.6±0.4, n=7; **p<0.0001) indicating substantial amelioration of AMR pathology. In contrast, composite AMR scores in CCR5 KO recipients transferred with 0.5×106 (7.3±0.6, n=3) or 1.0×106 (7.0±1.0, n=3) alloprimed CXCR5+CD8+ T cells, 2.0×106 3rd party primed CXCR5+CD8+ T cells (7.25±0.5, n=4), or 2.0×106 alloprimed CXCR5−CD8+ T cells (6.0±0.4, n=4) were similar to AMR score in CCR5 KO recipients without ACT (p=ns for all comparisons) and significantly greater than AMR score in CCR5 KO recipients after ACT with 2.0×106 alloprimed CXCR5+CD8+ T cells (p<0.002 for all comparisons).
Adoptive Cell Therapy of CXCR5+CD8+ T cells enhances kidney allograft survival in CCR5 KO mice.
Given the significantly reduced alloantibody titer and improved AMR pathology following ACT with alloprimed CXCR5+CD8+ T cells, we next investigated the efficacy of ACT with alloprimed CXCR5+CD8+ T cells upon allograft survival. Assessment of KTx allograft survival necessitated modification of the model to include concurrent bilateral native nephrectomy at the time of KTx, followed by serial monitoring of recipient serum creatinine and general health. For reference, KTx allograft rejection and graft loss is reported to occur in all untreated CCR5 KO recipients by 2–3 weeks posttransplant (28).
Next, we tested the efficacy of ACT on kidney allograft survival in CCR5 KO recipients. On day 5 posttransplant, CCR5 KO kidney transplant recipients that underwent concurrent bilateral nephrectomy received ACT with 2×106 alloprimed CXCR5+CD8+ T cells. All control untreated CCR5 KO KTx recipients lost kidney transplant function by day 19 posttransplant (MST=15 days; Figure 7A). CCR5 KO recipients that received ACT with alloprimed CXCR5+CD8+ T cells experienced prolonged allograft survival (MST=52 days, p=0.0007). In contrast, ACT with 2×106 alloprimed CXCR5−CD8+ T cells did not enhance allograft survival (MST=25, p=ns). Furthermore, CCR5 KO recipients that received ACT with 2×106 third party-primed CXCR5+CD8+ T cells did not experience prolonged allograft survival (MST= 21, p=ns).
In CCR5 KO recipients that received ACT with 2×106 alloprimed CXCR5+CD8+ T cells, alloantibody levels remained at the reduced level noted on day 14 through later time points when allograft loss occurred (range day 28 to 69 posttransplant) (day 14 titer: 1300±710 vs later time points titer: 1300±580, p=ns; Figure 7B). The composite AMR scores for kidney allografts from CCR5 KO recipients that received ACT of CXCR5+CD8+ T cells were similar on day 14 (3.2±1.0) and at later time points when allograft loss occurred (5.6±1.5; p=ns; Figure 7C).
Prior studies have shown that KTx graft loss in CCR5 KO mice is AMR mediated and B cell dependent since in the absence of B cells, allograft loss does not occur (29). Thus, any contribution of T cell mediated rejection to pathologic changes in allografts from CCR5 KO recipients is superimposed upon AMR pathology. When we analyzed for histologic changes associated with T cell mediated rejection (arteritis, tubulitis, and interstitial inflammation) we did not detect any differences in allograft pathology between day 14 (2.8±1.0) and at the time of late allograft loss (3.2±0.5, p=ns; not shown). However, while kidney pathology at day 14 post transplant had no evidence of fibrosis, pathology at later time points had mild fibrosis (Figure 7D).
Discussion
CCR5 KO mice that undergo kidney transplantation serve as a rigorous animal model to study antibody-mediated rejection. They produce high quantities of alloantibody, develop AMR, and reject kidney transplants within 21 days posttransplant (28, 29). A similar exaggerated humoral alloimmune response and severe AMR observed in CCR5 KO heart allograft recipients has been attributed to a potential deficiency of CD4+ T regulatory cells (44), akin to CCR5-dependent regulation of effector immune responses in models of graft vs. host disease and Leishmania infection persistence (45, 46). Interestingly, Nozaki et al. also noted that CCR5 KO heart allograft recipients had a reduced population of IFN-γ+CD8+ T cells and an increased population of IL-4-producing CD4+ T cells compared to WT recipients (44). Some evidence that alloantibody production in CCR5 KO mice could be CD8-regulated arises from reports that CD8+ T cell depletion results in even higher (4-fold higher) alloantibody production following kidney transplant (28).
Our discovery of a novel IFN-γ-dependent antibody-suppressor CD8+ T cell subset in a hepatocyte transplant model prompted us to investigate the presence and function of antibody-suppressor CXCR5+CD8+ T cells in CCR5 KO mice after KTx, a vascularized solid organ transplant model that reproduces the pathology of human AMR. There is a substantial difference in magnitude of posttransplant alloantibody produced after KTx compared to after hepatocyte transplant in WT recipients (more than 10-fold). Alloantibody titer in CD8 KO hepatocyte transplant recipients on day 14 posttransplant is approximately 1,000 (23), while CCR5 KO kidney transplant recipients have alloantibody titer over 5,000 (28, 29). CD8-depleted WT kidney transplant recipients have enhanced alloantibody titers and severe AMR pathology, comparable to CCR5 KO recipients (data not shown). CD8-depletion in CCR5 KO KTx recipients results in even higher alloantibody titer (over 10,000) (28). Collectively, these data support CD8-mediated regulation of alloantibody production in multiple mouse strains and after multiple types of allografts (hepatocyte, islet, skin and kidney transplant). The efficacy of ACT to significantly suppress alloantibody titer, ameliorate severe AMR, and prolong allograft survival in CCR5 KO KTx mice that generate a robust humoral alloimmune response indicates the powerful antibody suppressor function of alloprimed CXCR5+CD8+ T cells.
In recent years, CXCR5+CD8+ T cells have garnered significant attention. This CD8+ T cell subset has been reported to impact biologic outcomes (infection, autoimmunity, malignancy) in various animal models of disease and in humans (reviewed in (47–49)). Subsets of CXCR5+CD8+ T cells have been reported to enhance antibody production (50–59), while others like the antibody-suppressor CXCR5+CD8+ T cells in our studies and Qa1-restricted CXCR5+CD8+ T cells reported by others inhibit alloantibody (60) and auto-antibody (61, 62) production (reviewed in (63)). Phenotypic, functional and mechanistic features of the antibody-suppressor CXCR5+CD8+ T cells in our studies that distinguish them from other subsets reported in the literature include the absence of PD-1 expression [(23, 63)], antigen specific cytotoxic effector function targeting antibody-producing B cells (22, 23), and regulatory functions that are not associated with FoxP3, IL-10, ICOS/ICOSL, and CD103 (63).
Our prior work indicates that alloprimed CD8+ T cell-mediated suppression of alloantibody production occurs, in part, by the killing of alloprimed self-IgG1+ B cells (MHC I-restricted alloantigen presentation), which is detected both in vitro and in vivo (22). In the current study, we found that compared to low alloantibody producing WT KTx mice, the high alloantibody producing CCR5 KO KTx mice have a relative deficit in quantity of CXCR5+CD8+ T cells (and activated CXCR5+CD44+IFN-γ+CD8+ T cells) in response to alloantigen stimulus (~5-fold reduction on day 7 posttransplant) that is accompanied by impaired alloprimed IgG+ B cell directed cytotoxic effector function and enhanced proportion of GC B cells and plasma cells. In the current study, as well as in previous reports, CCR5 KO transplant recipients produce detectable alloantibody as early as day 5–7 posttransplant (44). Thus, the reduction of alloantibody titer on day 14 in CCR5 KO KTx recipients that receive ACT with antibody-suppressor CXCR5+CD8+ T cells on day 5 posttransplant implies the efficacy of ACT for interrupting ongoing alloantibody production. Our data also demonstrates that the efficacy of alloprimed CXCR5+CD8+ T cell ACT for rescue of AMR and prolongation of KTx survival in CCR5 KO mice is dose-dependent and allospecific. Evidence that endogenous CD8+ T cells regulate alloantibody production following KTx, even in CCR5 KO recipients, is supported by the significant increase in alloantibody titer when CD8+ T cells are depleted (28). Thus, in future studies, it will be of interest to see if expansion of endogenous CXCR5+CD8+ T cells in CCR5 KO recipients can be optimized to significantly reduce alloantibody and preserve allograft function. There are no FDA-approved therapies for treatment of AMR and commonly adopted treatment strategies are based on “low level” evidence (20). Current approaches for treatment of AMR are associated with variable results and expose transplant recipients to the risk of complications associated with global immunosuppression. Thus, development of new therapies, including cellular immunotherapies, is an attractive avenue to pursue, especially if allospecific humoral immunity can be targeted with preservation of protective host immune responses.
We have pursued studies in humans to determine whether or not CXCR5+CD8+ T cells are detected in peripheral blood. Interestingly, we found in a prospective observational study that first-time KTx recipients who develop de novo DSA have significantly reduced quantity of peripheral blood CXCR5+CD8+ T cells (and activated CXCR5+IFN-γ+CD8+ T cells) compared to recipients that remain DSA-free at one year posttransplant, despite transplantation under cover of the same induction and maintenance immunosuppressive regimen and achievement of equivalent target drug levels (64). These findings raise the possibility that a homolog of murine antibody-suppressor CXCR5+CD8+ T cells exists in humans. Studies are ongoing to investigate the clinical utility of monitoring this cell subset as a biomarker to risk-stratify patients for development of de novo DSA. Further studies are warranted to investigate the potential for developing novel CXCR5+CD8+ T cell-based cellular therapies for the prevention or treatment of AMR after transplant.
Supplementary Material
Acknowledgments/Funding
This work was supported by a National Institutes of Health R01 grants AI139913 and AI083456 (to GLB), T32 AI106704-07 and F32 AI161844 (to JLH), CA016058, UL1TR002733, the OSU Division of Transplant Surgery, and the OSU College of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors would like to acknowledge the role of Tai Yi, M.D., Microsurgery Center Director in the Breuer laboratory, in facilitating team communication and scheduling of the mouse kidney transplant procedures.
Nonstandard Abbreviations:
- ACT
adoptive cellular therapy
- AMR
antibody-mediated rejection
- CD8+ TAb-supp cells
antibody-suppressing CD8+ T cells
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- MST
median survival time
- PTC
peritubular capillary
- WT
wild-type
Footnotes
Disclosure
The authors have no disclosures.
References
- 1.Schinstock CA, Mannon RB, Budde K, Chong AS, Haas M, Knechtle S et al. Recommended Treatment for Antibody-mediated Rejection After Kidney Transplantation: The 2019 Expert Consensus From the Transplantion Society Working Group. Transplantation 2020;104(5):911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKenna RM, Takemoto SK, Terasaki PI. Anti-HLA antibodies after solid organ transplantation. Transplantation 2000;69(3):319–326. [DOI] [PubMed] [Google Scholar]
- 3.Puttarajappa C, Shapiro R, Tan HP. Antibody-mediated rejection in kidney transplantation: a review. Journal of transplantation 2012;2012:193724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiebe C, Gibson IW, Blydt-Hansen TD, Pochinco D, Birk PE, Ho J et al. Rates and determinants of progression to graft failure in kidney allograft recipients with de novo donor-specific antibody. Am J Transplant 2015;15(11):2921–2930. [DOI] [PubMed] [Google Scholar]
- 5.Wiebe C, Pochinco D, Blydt-Hansen TD, Ho J, Birk PE, Karpinski M et al. Class II HLA epitope matching-A strategy to minimize de novo donor-specific antibody development and improve outcomes. Am J Transplant 2013;13(12):3114–3122. [DOI] [PubMed] [Google Scholar]
- 6.Lefaucheur C, Viglietti D, Bentlejewski C, Duong van Huyen JP, Vernerey D, Aubert O et al. IgG Donor-Specific Anti-Human HLA Antibody Subclasses and Kidney Allograft Antibody-Mediated Injury. Journal of the American Society of Nephrology : JASN 2016;27(1):293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devos JM, Gaber AO, Teeter LD, Graviss EA, Patel SJ, Land GA et al. Intermediate-term graft loss after renal transplantation is associated with both donor-specific antibody and acute rejection. Transplantation 2014;97(5):534–540. [DOI] [PubMed] [Google Scholar]
- 8.Cooper JE, Gralla J, Cagle L, Goldberg R, Chan L, Wiseman AC. Inferior kidney allograft outcomes in patients with de novo donor-specific antibodies are due to acute rejection episodes. Transplantation 2011;91(10):1103–1109. [DOI] [PubMed] [Google Scholar]
- 9.Chong AS. New insights into the development of B cell responses: Implications for solid organ transplantation. Human immunology 2019;80(6):378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valenzuela NM, Hickey MJ, Reed EF. Antibody Subclass Repertoire and Graft Outcome Following Solid Organ Transplantation. Frontiers in immunology 2016;7:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valenzuela NM, Reed EF. Antibodies in transplantation: the effects of HLA and non-HLA antibody binding and mechanisms of injury. Methods in molecular biology 2013;1034:41–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calp-Inal S, Ajaimy M, Melamed ML, Savchik C, Masiakos P, Colovai A et al. The prevalence and clinical significance of C1q-binding donor-specific anti-HLA antibodies early and late after kidney transplantation. Kidney international 2015. [DOI] [PubMed] [Google Scholar]
- 13.Hara S Banff 2013 update: Pearls and pitfalls in transplant renal pathology. Nephrology (Carlton) 2015;20 Suppl 2:2–8. [DOI] [PubMed] [Google Scholar]
- 14.Schmitz R, Fitch ZW, Schroder PM, Choi AY, Jackson AM, Knechtle SJ et al. B cells in transplant tolerance and rejection: friends or foes? Transpl Int 2020;33(1):30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nature reviews 2005;5(10):807–817. [DOI] [PubMed] [Google Scholar]
- 16.Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. The Journal of clinical investigation 2017;127(7):2492–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanchard D, Gaillard C, Hermann P, Banchereau J. Role of CD40 antigen and interleukin-2 in T cell-dependent human B lymphocyte growth. European journal of immunology 1994;24(2):330–335. [DOI] [PubMed] [Google Scholar]
- 18.Steele DJ, Laufer TM, Smiley ST, Ando Y, Grusby MJ, Glimcher LH et al. Two levels of help for B cell alloantibody production. The Journal of experimental medicine 1996;183(2):699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vincenti F, Charpentier B, Vanrenterghem Y, Rostaing L, Bresnahan B, Darji P et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant 2010;10(3):535–546. [DOI] [PubMed] [Google Scholar]
- 20.Wan SS, Ying TD, Wyburn K, Roberts DM, Wyld M, Chadban SJ. The Treatment of Antibody-Mediated Rejection in Kidney Transplantation: An Updated Systematic Review and Meta-Analysis. Transplantation 2018;102(4):557–568. [DOI] [PubMed] [Google Scholar]
- 21.Zimmerer JM, Pham TA, Sanders VM, Bumgardner GL. CD8+ T cells negatively regulate IL-4-dependent, IgG1-dominant posttransplant alloantibody production. J Immunol 2010;185(12):7285–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmerer JM, Pham TA, Wright CL, Tobin KJ, Sanghavi PB, Elzein SM et al. Alloprimed CD8(+) T Cells Regulate Alloantibody and Eliminate Alloprimed B Cells Through Perforin- and FasL-Dependent Mechanisms. Am J Transplant 2014;14(2):295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmerer JM, Ringwald BA, Elzein SM, Avila CL, Warren RT, Abdel-Rasoul M et al. Antibody-suppressor CD8+ T Cells Require CXCR5. Transplantation 2019;103(9):1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avila CL, Zimmerer JM, Elzein SM, Pham TA, Abdel-Rasoul M, Bumgardner GL. mTOR Inhibition Suppresses Posttransplant Alloantibody Production Through Direct Inhibition of Alloprimed B Cells and Sparing of CD8+ Antibody-Suppressing T cells. Transplantation 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol 2007;179(8):5099–5108. [DOI] [PubMed] [Google Scholar]
- 26.Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. The Journal of experimental medicine 2000;192(11):1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. The Journal of experimental medicine 2000;192(11):1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohei N, Tanaka T, Tanabe K, Masumori N, Dvorina N, Valujskikh A et al. Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney international 2016;89(6):1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bickerstaff A, Nozaki T, Wang JJ, Pelletier R, Hadley G, Nadasdy G et al. Acute humoral rejection of renal allografts in CCR5(−/−) recipients. Am J Transplant 2008;8(3):557–566. [DOI] [PubMed] [Google Scholar]
- 30.Abe T, Ishii D, Gorbacheva V, Kohei N, Tsuda H, Tanaka T et al. Anti-huCD20 antibody therapy for antibody-mediated rejection of renal allografts in a mouse model. Am J Transplant 2015;15(5):1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Schlachta C, Duff J, Stiller C, Grant D, Zhong R. Improved techniques for kidney transplantation in mice. Microsurgery 1995;16(2):103–109. [DOI] [PubMed] [Google Scholar]
- 32.Lerret NM, Li T, Wang JJ, Kang HK, Wang S, Wang X et al. Recipient Myd88 Deficiency Promotes Spontaneous Resolution of Kidney Allograft Rejection. Journal of the American Society of Nephrology : JASN 2015;26(11):2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fountain KJ, Kloss A, Garibyan I, Blitshteyn B, Brezzani A, Kyostio-Moore S et al. Analysis of creatinine in mouse and rat serum by ion exchange high performance liquid chromatography for in vivo studies of renal function. J Chromatogr B Analyt Technol Biomed Life Sci 2007;846(1–2):245–251. [DOI] [PubMed] [Google Scholar]
- 34.Lunsford KE, Horne PH, Koester MA, Eiring AM, Walker JP, Dziema HL et al. Activation and maturation of alloreactive CD4-independent, CD8 cytolytic T cells. Am J Transplant 2006;6(10):2268–2281. Epub 2006 Aug 2264. [DOI] [PubMed] [Google Scholar]
- 35.Parungo CP, Soybel DI, Colson YL, Kim SW, Ohnishi S, DeGrand AM et al. Lymphatic drainage of the peritoneal space: a pattern dependent on bowel lymphatics. Annals of surgical oncology 2007;14(2):286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horne PH, Koester MA, Jayashankar K, Lunsford KE, Dziema HL, Bumgardner GL. Disparate primary and secondary allospecific CD8+ T cell cytolytic effector function in the presence or absence of host CD4+ T cells. J Immunol 2007;179(1):80–88. [DOI] [PubMed] [Google Scholar]
- 37.Zimmerer JM, Ringwald BA, Elzein SM, Avila CL, Warren RT, Abdel-Rasoul M, and Bumgardner GL Antibody-suppressor CD8+ T cells require CXCR5. Transplantation 2019:Epub (March 1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tung JW, Heydari K, Tirouvanziam R, Sahaf B, Parks DR, Herzenberg LA et al. Modern flow cytometry: a practical approach. Clinics in laboratory medicine 2007;27(3):453–468, v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murata K, Fox-Talbot K, Qian Z, Takahashi K, Stahl GL, Baldwin WM 3rd et al. Synergistic deposition of C4d by complement-activating and non-activating antibodies in cardiac transplants. Am J Transplant 2007;7(11):2605–2614. [DOI] [PubMed] [Google Scholar]
- 40.Haas M The Revised (2013) Banff Classification for Antibody-Mediated Rejection of Renal Allografts: Update, Difficulties, and Future Considerations. Am J Transplant 2016;16(5):1352–1357. [DOI] [PubMed] [Google Scholar]
- 41.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant 2008;8(4):753–760. [DOI] [PubMed] [Google Scholar]
- 42.Roufosse C, Simmonds N, Clahsen-van Groningen M, Haas M, Henriksen KJ, Horsfield C et al. A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation 2018;102(11):1795–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant 2018;18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nozaki T, Amano H, Bickerstaff A, Orosz CG, Novick AC, Tanabe K et al. Antibody-mediated rejection of cardiac allografts in CCR5-deficient recipients. J Immunol 2007;179(8):5238–5245. [DOI] [PubMed] [Google Scholar]
- 45.Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood 2005;106(9):3300–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. The Journal of experimental medicine 2006;203(11):2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fousteri G, Kuka M. The elusive identity of CXCR5(+) CD8 T cells in viral infection and autoimmunity: Cytotoxic, regulatory, or helper cells? Molecular immunology 2020;119:101–105. [DOI] [PubMed] [Google Scholar]
- 48.Valentine KM, Hoyer KK. CXCR5+ CD8 T Cells: Protective or Pathogenic? Frontiers in immunology 2019;10:1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu D, Ye L. A Portrait of CXCR5. Trends in immunology 2018;39(12):965–979. [DOI] [PubMed] [Google Scholar]
- 50.Ye L, Li Y, Tang H, Liu W, Chen Y, Dai T et al. CD8+CXCR5+T cells infiltrating hepatocellular carcinomas are activated and predictive of a better prognosis. Aging (Albany NY) 2019;11(20):8879–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA et al. The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nature immunology 2017;18(8):931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Yu M, Zheng Y, Fu G, Xin G, Zhu W et al. CXCR5(+)PD-1(+) follicular helper CD8 T cells control B cell tolerance. Nature communications 2019;10(1):4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quigley MF, Gonzalez VD, Granath A, Andersson J, Sandberg JK. CXCR5+ CCR7- CD8 T cells are early effector memory cells that infiltrate tonsil B cell follicles. European journal of immunology 2007;37(12):3352–3362. [DOI] [PubMed] [Google Scholar]
- 54.Le KS, Ame-Thomas P, Tarte K, Gondois-Rey F, Granjeaud S, Orlanducci F et al. CXCR5 and ICOS expression identifies a CD8 T-cell subset with TFH features in Hodgkin lymphomas. Blood Adv 2018;2(15):1889–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao L, Jia L, Bai L, He L, Yang B, Wu C et al. Phenotypic and functional characteristics of IL-21-expressing CD8(+) T cells in human nasal polyps. Scientific reports 2016;6:30362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen J, Luo X, Wu Q, Huang J, Xiao G, Wang L et al. A Subset of CXCR5(+)CD8(+) T Cells in the Germinal Centers From Human Tonsils and Lymph Nodes Help B Cells Produce Immunoglobulins. Frontiers in immunology 2018;9:2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xing J, Zhang C, Yang X, Wang S, Wang Z, Li X et al. CXCR5(+)CD8(+) T cells infiltrate the colorectal tumors and nearby lymph nodes, and are associated with enhanced IgG response in B cells. Experimental cell research 2017;356(1):57–63. [DOI] [PubMed] [Google Scholar]
- 58.Valentine KM, Davini D, Lawrence TJ, Mullins GN, Manansala M, Al-Kuhlani M et al. CD8 Follicular T Cells Promote B Cell Antibody Class Switch in Autoimmune Disease. J Immunol 2018;201(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang H, Li L, Han J, Sun Z, Rong Y, Jin Y. CXCR5(+) CD8(+) T Cells Indirectly Offer B Cell Help and Are Inversely Correlated with Viral Load in Chronic Hepatitis B Infection. DNA and cell biology 2017;36(4):321–327. [DOI] [PubMed] [Google Scholar]
- 60.Choi JY, Eskandari SK, Cai S, Sulkaj I, Assaker JP, Allos H et al. Regulatory CD8 T cells that recognize Qa-1 expressed by CD4 T-helper cells inhibit rejection of heart allografts. Proceedings of the National Academy of Sciences of the United States of America 2020;117(11):6042–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leavenworth JW, Tang X, Kim HJ, Wang X, Cantor H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. The Journal of clinical investigation 2013;123(3):1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature 2010;467(7313):328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elzein SM, Zimmerer JM, Han JL, Ringwald BA, Bumgardner GL. CXCR5+CD8+ T cells: A Review of Their Antibody Regulatory Functions and Clinical Correlations. J Immunol:In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zimmerer JM, Basinger MW, Ringwald BA, Abdel-Rasoul M, Pelletier RP, Rajab A et al. Inverse Association Between the Quantity of Human Peripheral Blood CXCR5+IFN-gamma+CD8+ T Cells With De Novo DSA Production in the First Year After Kidney Transplant. Transplantation 2020;104(11):2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.