

Summary
Background
Progressive fibrosing interstitial lung disease (ILD) is characterised by parenchymal scar formation, leading to high morbidity and mortality. The ability to predict this phenotype remains elusive. We conducted a proteomic analysis to identify novel plasma biomarkers of progressive fibrosing ILD and developed a proteomic signature to predict this phenotype.
Methods
Relative plasma concentrations for 368 biomarkers were determined with use of a semi-quantitative, targeted proteomic platform in patients with connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, or unclassifiable ILD who provided research blood draws at the University of California (discovery cohort) and the University of Texas (validation cohort). Univariable logistic regression was used to identify individual biomarkers associated with 1-year ILD progression, defined as death, lung transplant, or 10% or greater relative forced vital capacity (FVC) decline. A proteomic signature of progressive fibrosing ILD was then derived with use of machine learning in the University of California cohort and validated in the University of Texas cohort.
Findings
The discovery cohort comprised 385 patients (mean age 63·6 years, 59% female) and the validation cohort comprised 204 patients (mean age 60·7 years, 61% female). 31 biomarkers were associated with progressive fibrosing ILD in the discovery cohort, with 17 maintaining an association in the validation cohort. Validated biomarkers showed a consistent association with progressive fibrosing ILD irrespective of ILD clinical diagnosis. A proteomic signature comprising 12 biomarkers was derived by machine learning and validated in the University of Texas cohort, in which it had a sensitivity of 0·90 and corresponding negative predictive value of 0·91, suggesting that approximately 10% of patients with a low-risk proteomic signature would experience ILD progression in the year after blood draw. Those with a low-risk proteomic signature experienced an FVC change of +85·7 mL (95% CI 6·9 to 164·4) and those with a high-risk signature experienced an FVC change of −227·1 mL (−286·7 to −167·5). A theoretical clinical trial restricted to patients with a high-risk proteomic signature would require 80% fewer patients than one designed without regard to proteomic signature.
Interpretation
17 plasma biomarkers of progressive fibrosing ILD were identified and showed consistent associations across ILD subtypes. A proteomic signature of progressive fibrosing ILD could enrich clinical trial cohorts and avoid the need for antecedent progression when defining progressive fibrosing ILD for clinical trial enrolment.
Funding
National Heart Lung and Blood Institute.
Introduction
Progressive fibrosing interstitial lung disease (ILD) is a devastating condition characterised by parenchymal scar formation, leading to deteriorating lung function and early death.1 Whereas almost all patients with idiopathic pulmonary fibrosis (IPF) progress, a variable proportion of patients with other common ILDs, including connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, and unclassifiable ILD, develop progressive fibrosing ILD.2 In 2021, criteria for identifying progressive fibrosing ILD were proposed as part of therapeutic clinical trials in this population.3,4 Although these criteria effectively identify those experiencing ILD progression,5 they do not allow patients who are at risk to be identified before progression occurs.
Several blood-based biomarkers have been linked to differential progression in patients with IPF and other fibrosing ILDs.6 In a 2020 study, we showed that plasma concentration of CXCL13, a chemokine responsible for B-lymphocyte activation, was consistently associated with increased progression risk across a diverse group of ILDs, including connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, and unclassifiable ILD.7 Although inflammation is a prominent feature of connective tissue disease-associated ILD and chronic hypersensitivity pneumonitis, inflammatory signalling also appears to have a prominent role in fibrotic-predominant ILDs.8,9 As such, cytokines, interleukins, and other immune mediators might serve as useful biomarkers of progressive fibrosing ILD, irrespective of ILD clinical diagnosis.
We conducted a multicentre, targeted proteomic investigation of inflammation-related proteins to identify and validate novel biomarkers of progressive fibrosing ILD in patients with fibrotic connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, and unclassifiable ILD. We hypothesised that the association between plasma biomarkers and progressive fibrosing ILD phenotype would be robust to ILD clinical diagnosis. We then derived and validated a proteomic signature to predict progressive fibrosing ILD in patients who would qualify for a theoretical ILD clinical trial and estimated sample sizes for clinical trials designed with and without regard to proteomic signature classification.
Methods
Study design and participants
For this multicentre cohort analysis, we identified patients with fibrotic ILD due to connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, or unclassifiable ILD, who provided a research blood draw at the University of California Davis (UC Davis; May, 2016, to August, 2019), University of California San Francisco (UCSF; March, 2006, to October, 2018) and University of Texas Southwestern Medical Center (UTSW; February, 2007, to August, 2019). Patients from UC Davis and UCSF comprised the discovery cohort and those from UTSW comprised the validation cohort. No other eligibility criteria were applied. Longitudinal pulmonary function testing and vital status were used to classify patients as having non-progressive ILD or progressive fibrosing ILD. Patients who were alive with less than 10% relative decline in forced vital capacity (FVC) at 12 months after blood draw were considered to have non-progressive ILD. Patients who died of any cause, underwent lung transplant, or experienced 10% or greater relative FVC decline within 12 months of blood draw were deemed to have progressive fibrosing ILD. All patients included in the analysis had a verified progression status at 12 months (plus or minus 3 months).
This study was approved by the institutional review boards at UC Davis (protocol 875917), UCSF (protocol 10-01592), and UTSW (protocols 092017-007 and 082010-127). All participants provided written informed consent as part of providing research blood draws.
Procedures
Peripheral blood was collected from consenting patients at each centre and plasma was isolated according to centre-specific protocols. After isolation, plasma was aliquoted and stored at −80°C without thawing. Frozen plasma aliquots were shipped in a single batch to the Olink US headquarters in Boston, MA, for processing. Samples were randomised at the time of plating according to centre, sex, ILD diagnosis, and progression status to mitigate batch effects.
The Explore Inflammation panel from Olink (Uppsala, Sweden) was used to quantify 368 inflammation-related proteins for each patient. Multiplex proximity extension assay panels were used to quantify each protein, as previously described.10 A proximity extension assay uses a dual-recognition immunoassay, in which two matched antibodies labelled with unique DNA oligonucleotides simultaneously bind to the target protein in solution. This brings the two antibodies into proximity, allowing hybridisation and serving as a template for a DNA polymerase-dependent extension step. The double-stranded DNA is unique to a specific antigen and amplifies using P5 and P7 Illumina adaptors along with sample indexing, which is quantitatively proportional to the plasma concentration of the target protein. Amplified targets are then quantified with next generation sequencing using an Illumina (San Diego, CA, USA) NovaSeq 6000. Semi-quantitative protein estimates were normalised across batches to mitigate systematic error and log2-transformed to ensure normality, which was also confirmed visually using histograms. Analytes with more than 10% missing data were excluded from the analysis. No imputation for missing data was performed.
Biomarkers of progressive fibrosing ILD
With an estimated progressive fibrosing ILD prevalence of around 0·4 in the discovery cohort, we estimated that 385 patients would be needed to detect biomarkers with an odds ratio (OR) greater than 1·8 (effect size >0·6), assuming a β of 0·8 and two-sided α of 0·00014, which was adjusted for multiple testing across 368 biomarkers using the Bonferroni procedure. The association between continuously modelled, log2-transformed biomarkers and categorical ILD progression at 12 months was assessed using univariable logistic regression, with discovery cohort results presented graphically with a volcano plot. Biomarkers with a false discovery rate q value of less than 0·05 in the UC Davis and UCSF discovery cohort were advanced for testing in the UTSW validation cohort. The association of biomarkers with progressive fibrosing ILD was considered statistically significant in the validation cohort if the p value was less than 0·05. 12-month progression-free survival was displayed for top biomarkers using the Kaplan-Meier estimator. Linearity in the relationship between validated biomarkers and progressive fibrosing ILD was confirmed using a goodness-of-fit test. We tested our hypothesis that validated biomarkers of progressive fibrosing ILD were robust to ILD clinical diagnosis by fitting a logistic regression model with a biomarker-by-diagnosis interaction term and performing a global heterogeneity test that determined whether biomarker progressive fibrosing ILD association varied by ILD subtype. An interaction term Wald p value of less than 0·05 was considered statistically significant. A similar approach was used when assessing associations of biomarkers with progressive fibrosing ILD after cohort stratification according to high-resolution CT morphology, gender, age, and physiology (ILD-GAP stage11). A sensitivity analysis was then performed to assess biomarker association with individual outcomes (>10% FVC decline, and death or transplant modelled together). Canonical pathways and interaction networks enriched with proteins associated with progressive fibrosing ILD at a false discovery rate p value of less than 0·05 were identified by Ingenuity Pathway Analysis software (version Q4 2021; Ingenuity Systems, Redwood City, CA, USA) using a one-tailed Fisher’s exact test.
Proteomic signature of progressive fibrosing ILD
Least absolute shrinkage and selection operator (LASSO) logistic regression with ten-fold cross-validation was performed to identify a variable set predictive of progressive fibrosing ILD, with patients from UC Davis and UCSF serving as the derivation cohort. The analysis was independent of our primary analysis and was restricted to patients who would qualify for a hypothetical ILD clinical trial—namely, those with percent predicted FVC of more than 45% and diffusing capacity for carbon monoxide of 30–80%.4 Point estimates for variables selected by LASSO were used to generate a progressive fibrosing ILD score for each patient. An exploratory analysis was performed to identify a threshold for score dichotomisation that prioritised sensitivity. Those with a score greater than the threshold were classified as having a high-risk proteomic signature and those with a score less than the threshold were classified as having a low-risk signature. The progressive fibrosing ILD proteomic signature was then applied to patients from UTSW, which served as an independent validation cohort, with test performance characteristics reported. Mixed-effects regression was then performed to estimate the 1-year change in FVC for patients stratified by proteomic signature classification. The mixed-effects model included an autoregressive correlation structure and a random intercept term. Centre and baseline ILD-GAP stage were modelled as fixed-effects terms. Time was modelled in 4-month intervals for longitudinal plotting. No lung function data were imputed.
Clinical trial enrichment
The sample size needed to conduct a theoretical ILD therapeutic clinical trial with 1:1 randomisation and using mean change in FVC as the primary endpoint was determined using a Student’s t test. Therapeutic efficacy was defined as a 50% reduction in FVC decline between treatment groups, in accordance with observed treatment effect for previous antifibrotic clinical trials in IPF and progressive fibrosing ILD.4,12
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
The discovery cohort comprised 385 patients (mean age 63·6 years) and the validation cohort comprised 204 patients (mean age 60·7 years; table 1). The majority of patients in both cohorts were female and of White ethnicity. Most patients underwent blood draw in the same year as being diagnosed with ILD in the discovery cohort, whereas longer intervals were observed in the validation cohort. Chronic hypersensitivity pneumonitis was the most common ILD diagnosis in the discovery cohort, whereas connective tissue disease-associated ILD was the most common in the validation cohort. Among those with connective tissue disease-associated ILD, there were similar proportions of patients with ILD due to systemic sclerosis, rheumatoid arthritis, and idiopathic inflammatory myopathy between cohorts. Each cohort showed moderate reductions in percent predicted FVC and diffusing capacity for carbon monoxide, with diffusing capacity for carbon monoxide being significantly lower in the validation cohort. Differences in high-resolution CT patterns were observed between cohorts, with non-specific interstitial pneumonia predominating in the validation cohort and patterns other than non-specific interstitial pneumonia and usual interstitial pneumonia predominating in the discovery cohort. Outcomes were similar between cohorts, with 164 (43%) patients in the discovery cohort and 92 (45%) in the validation cohort developing progressive fibrosing ILD. Among outcomes, categorical decline in FVC was the most common, followed by death and lung transplant. A similar proportion of patients experienced progressive fibrosing ILD irrespective of the interval between ILD diagnosis and blood draw (appendix p 2).
Table 1:
Baseline characteristics and outcomes for the discovery and validation cohorts
| Discovery cohort (n=385) | Validation cohort (n=204) | p value | |
|---|---|---|---|
| Age, years | 63·6 (12·7) | 60·7 (11·6) | 0·0085 |
| Sex | |||
| Female | 228 (59%) | 124 (61%) | .. |
| Male | 157 (41%) | 80 (39%) | 0·71 |
| Race or ethnicity | |||
| White | 273 (71%) | 151 (74%) | 0·42 |
| Black | 15 (4%) | 26 (13%) | <0·0001 |
| Hispanic | 55 (14%) | 21 (10%) | 0·17 |
| Asian | 32 (8%) | 6 (3%) | 0·012 |
| Mixed or other | 10 (3%) | 0 | 0·02 |
| Years from diagnosis to blood draw | <0·001 | ||
| <1 | 292 (76%) | 44 (22%) | .. |
| 1 | 33 (9%) | 56 (27%) | .. |
| 2 | 19 (5%) | 42 (21%) | .. |
| 3 | 8 (2%) | 27 (13%) | .. |
| >3 | 33 (9%) | 35 (17%) | .. |
| Ever smoker | 259 (67%) | 114 (56%) | 0·0063 |
| ILD classification | |||
| CTD-ILD | 148 (38%) | 97 (48%) | 0·033 |
| Systemic sclerosis ILD | 42 (11%) | 30 (15%) | 0·67 |
| Rheumatoid arthritis ILD | 38 (10%) | 21 (10%) | 0·47 |
| Idiopathic inflammatory myopathy ILD | 42 (11%) | 26 (13%) | 0·79 |
| Other CTD-ILD | 26 (9%) | 20 (10%) | 0·55 |
| Chronic hypersensitivity pneumonitis | 185 (48%) | 57 (28%) | <0·0001 |
| Unclassifiable ILD | 52 (14%) | 50 (25%) | 0·0008 |
| Pulmonary function | |||
| FVC, % predicted | 64·4 (18·4) | 62·5 (19·2) | 0·23 |
| DLCO, % predicted | 46·7 (17·3) | 40·7 (17·7) | 0·0002 |
| High-resolution CT pattern | |||
| Definite or probable UIP | 75 (19%) | 60 (29%) | 0·0063 |
| Non-specific interstitial pneumonia | 111 (29%) | 101 (50%) | <0·0001 |
| Other pattern | 199 (52%) | 43 (21%) | <0·0001 |
| Outcomes | |||
| ILD progression | 164 (43%) | 92 (45%) | 0·56 |
| Death | 59 (15%) | 32 (16%) | 0·91 |
| Lung transplant | 6 (2%) | 12 (6%) | 0·0037 |
| ≥10% FVC decline | 99 (26%) | 48 (24%) | 0·56 |
| Months to progression | 6·7 (3·5–9·5) | 7·0 (4·0–9·3) | 0·69 |
Data are mean (SD), n (%), or median (IQR). ILD=interstitial lung disease. CTD-ILD=connective tissue disease-associated ILD. FVC=forced vital capacity. DLCO=diffusion capacity of the lung for carbon monoxide. UIP=usual interstitial pneumonia.
Of 368 biomarkers assessed, 43 were excluded due to more than 10% missing data and 325 were included in the final analysis. 31 biomarkers were associated with progressive fibrosing ILD in the discovery cohort (figure 1, appendix pp 3–12), with KRT19 (OR 2·10 per one-unit increase, 95% CI 1·69–2·66), ITGB6 (3·17, 2·20–4·70), and PLAUR (3·78, 2·34–6·29) showing the strongest association with progressive fibrosing ILD based on FDR p value. Of these 31 biomarkers, 17 maintained progressive fibrosing ILD association in the validation cohort (table 2), with the strongest associations observed for ITGB6 (3·04, 1·88–5·15), KRT19 (1·83, 1·39–2·48), and IL17C (1·70, 1·31–2·24), based on p value. After combining discovery and validation cohorts, the strongest progressive fibrosing ILD associations were observed for PLAUR, ITGB6, SPON1, HGF, PRSS8, and KRT19 (appendix p 13), which persisted after adjustment for centre, ILD diagnosis, and baseline ILD-GAP stage.13 Differential progression-free survival was observed after stratifying the combined cohort by biomarker concentration quartiles, with median survival of less than 12 months for those with the highest concentration (quartile 4) of each biomarker (figure 2). When assessing the association between validated biomarkers and individual progressive fibrosing ILD outcomes, all biomarkers showed a stronger association with death or lung transplant than with 10% or greater relative FVC decline (appendix p 14).
Figure 1: Volcano plot showing discovery cohort results.
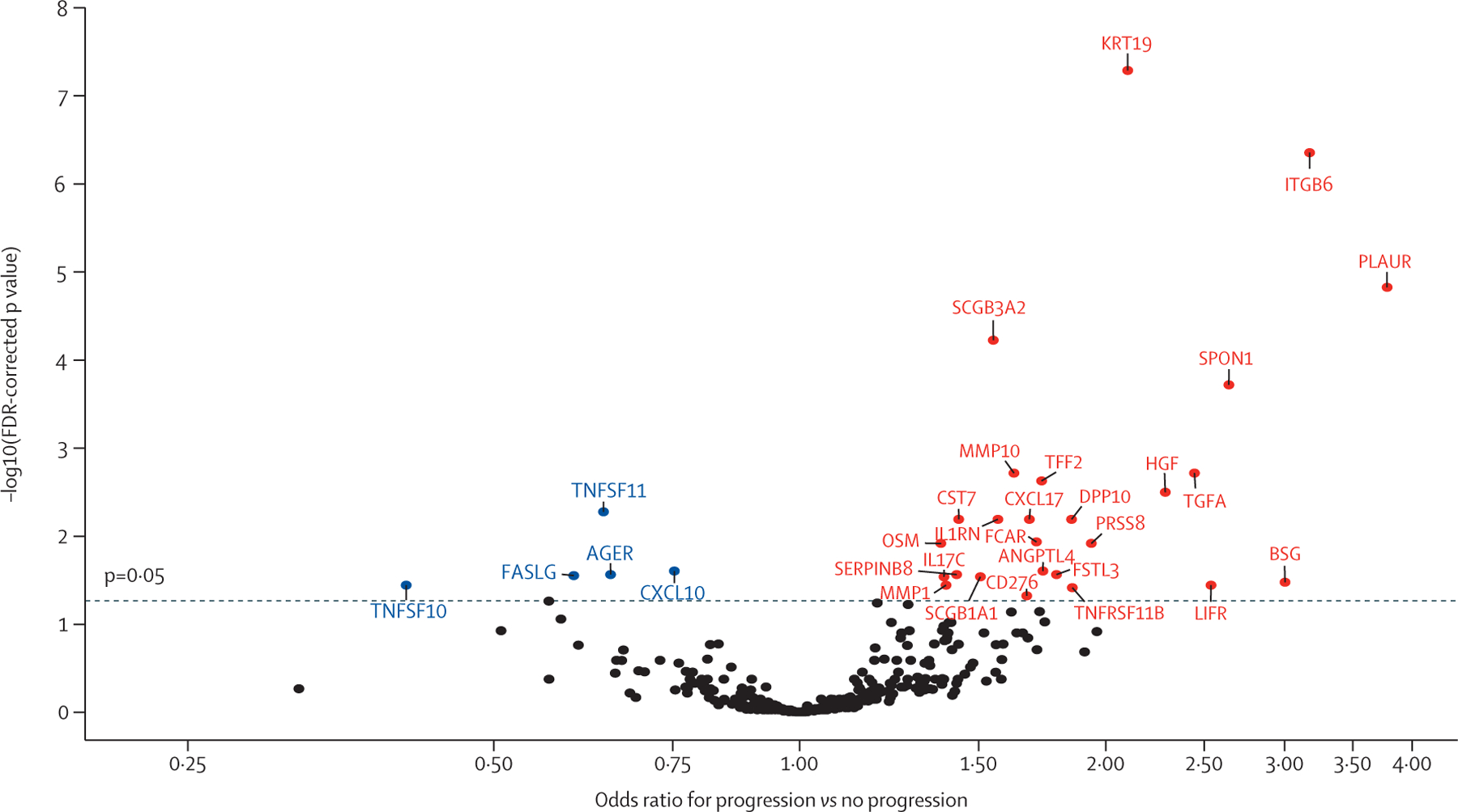
The dashed line corresponds to the FDR-corrected p value threshold. Progressive fibrosing interstitial lung disease risk decreases with each unit-change increase in the biomarkers shown in blue and increases with each unit-change in biomarkers shown in red. FDR=false discovery rate.
Table 2:
Associations of biomarkers advanced from discovery analysis with progressive fibrosing interstitial lung disease in the validation cohort
| OR (95% CI) | p value | |
|---|---|---|
| AGER | 0·51 (0·35–0·73) | 0·0003 |
| ANGPTL4 | 1·87 (1·20–2·98) | 0·0064 |
| BSG | 0·96 (0·41–2·22) | 0·92 |
| CD276 | 1·39 (0·89–2·19) | 0·16 |
| CST7 | 1·18 (0·95–1·48) | 0·15 |
| CXCL10 | 0·98 (0·78–1·23) | 0·88 |
| CXCL17 | 1·68 (1·22–2·36) | 0·0020 |
| DPP10 | 1·70 (1·10–2·68) | 0·018 |
| FASLG | 0·63 (0·41–0·97) | 0·038 |
| FCAR | 1·63 (1·11–2·43) | 0·014 |
| FSTL3 | 1·49 (0·90–2·52) | 0·13 |
| HGF | 2·09 (1·29–3·48) | 0·0033 |
| IL17C | 1·70 (1·31–2·24) | <0·0001 |
| IL1RN | 1·18 (0·93–1·49) | 0·18 |
| ITGB6 | 3·04 (1·88–5·15) | <0·0001 |
| KRT19 | 1·83 (1·39–2·48) | <0·0001 |
| LIFR | 1·49 (0·72–3·16) | 0·29 |
| MMP1 | 1·07 (0·84–1·36) | 0·59 |
| MMP10 | 1·65 (1·19–2·31) | 0·0031 |
| OSM | 1.19 (0·96–1·48) | 0·11 |
| PLAUR | 2·59 (1·42–4·89) | 0·0025 |
| PRSS8 | 2·52 (1·50–4·39) | 0·0007 |
| SCGB1A1 | 1·33 (0·94–1·91) | 0·11 |
| SCGB3A2 | 1·58 (1·25–2·04) | 0·0002 |
| SERPINB8 | 1·05 (0·87–1·28) | 0·59 |
| SPON1 | 2·14 (1·24–3·80) | 0·0074 |
| TFF2 | 1·24 (0·90–1·70) | 0·19 |
| TGFA | 1·65 (1·14–2·42) | 0·0089 |
| TNFRSF11B | 2·20 (1·31–3·79) | 0·0034 |
| TNFSF10 | 0·83 (0·43–1·57) | 0·57 |
| TNFSF11 | 0·86 (0·66–1·10) | 0·24 |
ORs are per unit change in log2-transformed biomarker concentration. p values are based on univariable logistic regression. OR=odds ratio.
Figure 2: 1-year progression-free survival in the combined cohort for the top biomarkers associated with progressive fibrosing interstitial lung disease after stratification by plasma biomarker concentration quartile.
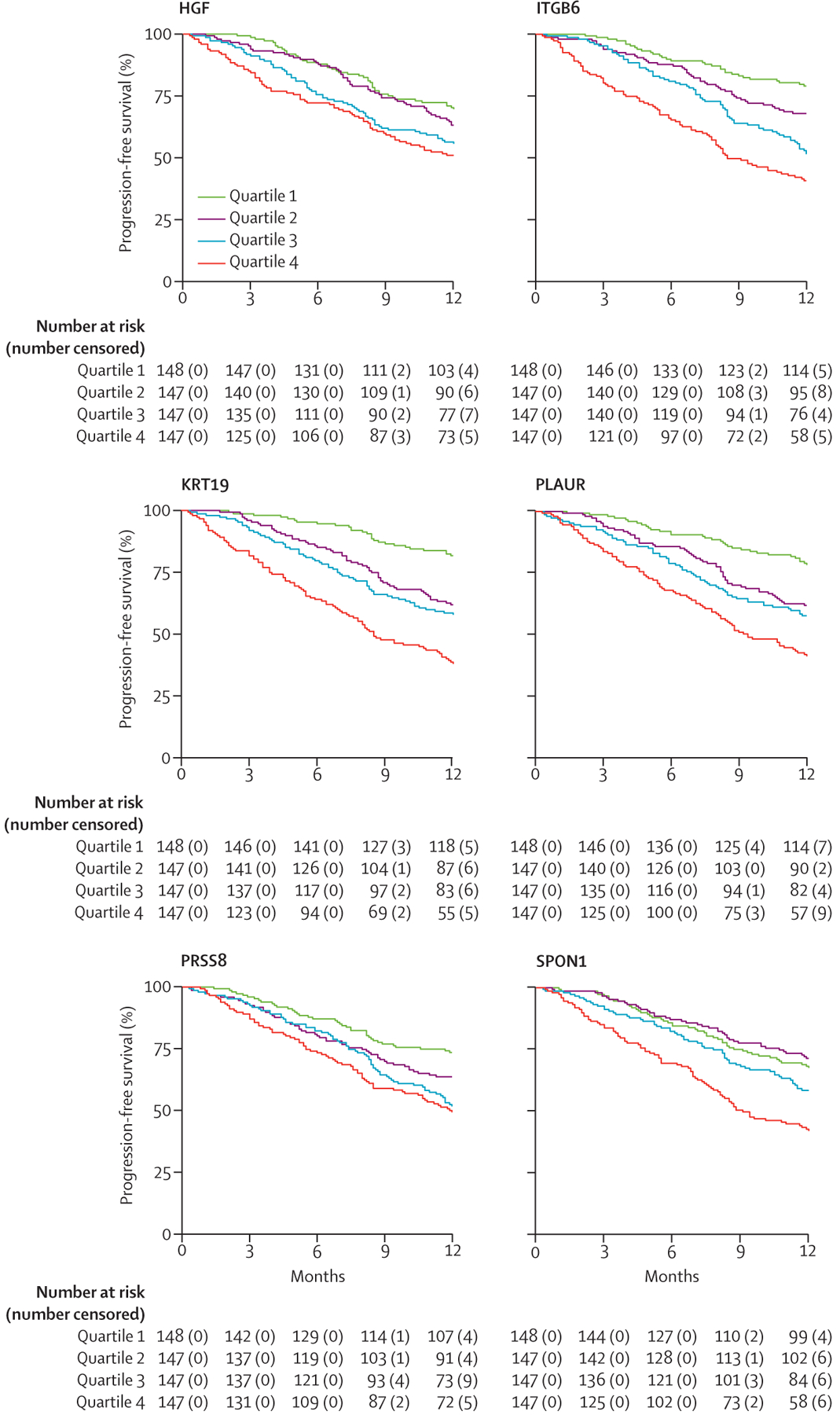
Logrank p<0·05 for all presented biomarkers.
After stratification of the combined cohort by ILD clinical diagnosis, heterogeneity in the association with progressive fibrosing ILD was observed for PRSS8 and a suggestion of heterogeneity was observed for FASLG (although this was not statistically significant; table 3), but it was not observed for any other validated biomarkers. When assessing subgroups stratified by morphological pattern on high-resolution CT, heterogeneity in the association with progressive fibrosing ILD was observed for ITGB6 and a suggestion of heterogeneity was observed for FASLG, PRSS8, and SPON1 (appendix p 15). When assessing ILD-GAP stage subgroups, significant heterogeneity in the association with progressive fibrosing ILD was observed for AGER, ITGB6, and SCGB3A2, with a suggestive association observed for KRT19 and SPON1 (appendix p 16). Linear regression showed mostly positive associations between biomarker concentration and ILD-GAP stage (appendix pp 17–19), suggesting that plasma concentration for some biomarkers might track with disease severity.
Table 3:
ILD subtype-stratified associations between validated biomarkers and progressive fibrosing ILD phenotype in the overall cohort (n=589)
| OR (95% CI) | Heterogeneity p value* | |||
|---|---|---|---|---|
| Chronic hypersensitivity pneumonitis (n=242) | Connective tissue disease-associated ILD (n=245) | Unclassifiable ILD (n=102) | ||
| AGER | 0·59 (0·41–0·84) | 0·56 (0·40–0·78) | 0·75 (0·45–1·24) | 0·63 |
| ANGPTL4 | 1·55 (1·02–2·38) | 1·95 (1·29–2·95) | 1·82 (0·92–3·60) | 0·75 |
| CXCL17 | 1·34 (0·95–1·89) | 1·71 (1·21–2·40) | 1·78 (1·09–2·91) | 0·52 |
| DPP10 | 2·24 (1·40–3·57) | 1·47 (0·99–2·19) | 1·66 (0·93–2·94) | 0·4 |
| FASLG | 0·32 (0·26–0·65) | 0·84 (0·58–1·22) | 0·57 (0·29–1·13) | 0·06 |
| FCAR | 2·24 (1·49–3·38) | 1·62 (1·12–2·33) | 1·45 (0·83–2·52) | 0·36 |
| HGF | 2·58 (1·51–4·40) | 2·40 (1·46–3·93) | 1·51 (0·80–2·83) | 0·40 |
| IL17C | 1·37 (1·08–1·73) | 1·41 (1·11–1·81) | 1·62 (1·06–2·47) | 0·79 |
| ITGB6 | 2·71 (1·76–4·19) | 3·16 (1·83–5·46) | 3·43 (1·66–7·11) | 0·83 |
| KRT19 | 1·92 (1·48–2·50) | 2·02 (1·50–2·71) | 2·03 (1·29–3·19) | 0·96 |
| MMP10 | 1·85 (1·36–2·52) | 1·52 (1·10–2·08) | 1·28 (0·86–1·91) | 0·34 |
| PLAUR | 2·50 (1·34–4·66) | 5·10 (2·73–9·51) | 2·11 (0·94–4·74) | 0·15 |
| PRSS8 | 1·20 (0·76–1·88) | 3·15 (1·87–5·34) | 2·37 (1·05–5·35) | 0·02 |
| SCGB3A2 | 1·54 (1·23–1·92) | 1·71 (1·35–2·16) | 1·47 (1·07–2·02) | 0·72 |
| SPON1 | 2·89 (1·69–4·94) | 2·96 (1·70–5·13) | 1·62 (0·84–3·13) | 0·32 |
| TGFA | 2·34 (1·44–3·80) | 1·76 (1·18–2·61) | 1·48 (0·90–2·43) | 0·42 |
| TNFRSF11B | 2·02 (1·19–3·45) | 1·53 (0·95–2·56) | 4·33 (1·76–10·67) | 0·13 |
ORs are per unit change in log2-transformed biomarker concentration. OR=odds ratio. ILD=interstitial lung disease.
Test of whether progressive fibrosing ILD biomarker association varies by ILD subtype.
Pathways associated with progressive fibrosing ILD (appendix p 20) were primarily involved in immunity and host response (IL-17 signalling, pattern recognition receptors for bacteria and viruses, and granulocyte adhesion and diapedesis) and fibrogenesis (hepatic cholestasis, cardiac hypertrophy signalling, HMGB1 signalling, hepatic fibrosis pathway, and regulation of the epithelial mesenchymal transition by growth factors pathway). Network analysis (appendix p 20) identified IL12B as a primary hub, with smaller hubs identified at pro-inflammatory cytokines and chemokines.
After applying clinical trial exclusion criteria according to baseline lung function,4 270 patients remained in the derivation cohort and 119 patients in the validation cohort. 12 biomarkers were selected by LASSO for proteomic signature development in the derivation cohort: AGER, CST7, CXCL10, DPP10, FASLG, ITGB6, KRT19, MEPE, PLAUR, PNPT1, TNFSF11, and WFIKKN2. Based on final model point estimates, a proteomic risk score was determined by the following equation using log2-transformed biomarker values: proteomic risk score = 0·053 + (0·884 × AGER) + (1·285 × CST7) + (0·715 × CXCL10) + (1·657 × DPP10) + (0·745 × FASLG) + (1·443 × ITGB6) + (1·451 × KRT19) + (0·578 × MEPE) + (2·070 × PLAUR) + (0·709 × PNPT1) + (0·902 × TNFSF11) + (0·490 × WFIKKN2).
The proteomic risk score resulted in an area under the curve of 0·79 in the derivation cohort and 0·73 in the validation cohort. After exploratory analysis of potential proteomic score thresholds (appendix p 21), those with a score of less than 0·3 were classified as having a low-risk proteomic signature and those above this threshold classified as having a high-risk signature. This resulted in a sensitivity of 0·85 and a specificity of 0·58, with a positive predictive value of 0·54 and a negative predictive value of 0·54 and 0·87 (appendix p 22). When applied to the validation cohort, the proteomic signature performed similarly, with a sensitivity of 0·90 and a specificity of 0·53, with a positive predictive value of 0·50 and a negative predictive value of 0·91 (appendix p 22). The high negative predictive value across both cohorts suggests that approximately 10% of patients with a low-risk proteomic signature would experience ILD progression in the year after blood draw.
Patients with a high-risk proteomic signature had almost a seven-times higher risk of progressive fibrosing ILD compared with those with a low-risk signature (OR 6·73, 95% CI 4·00–11·33). When modelling individual outcomes, a high-risk proteomic signature was associated with an almost six-times higher risk of experiencing 10% or greater relative FVC decline (5·77, 3·31–10·05) and an eight-times higher risk of death or transplant (8·0, 2·39–26·8). Those with a high-risk proteomic signature showed substantially larger 1-year change in FVC compared with those with a low-risk proteomic signature (figure 3A–C). This observation persisted after stratification by high-resolution CT pattern (figure 3D–F).
Figure 3: Longitudinal plots comparing 1-year change in FVC between patients with high-risk and low-risk proteomic signature.
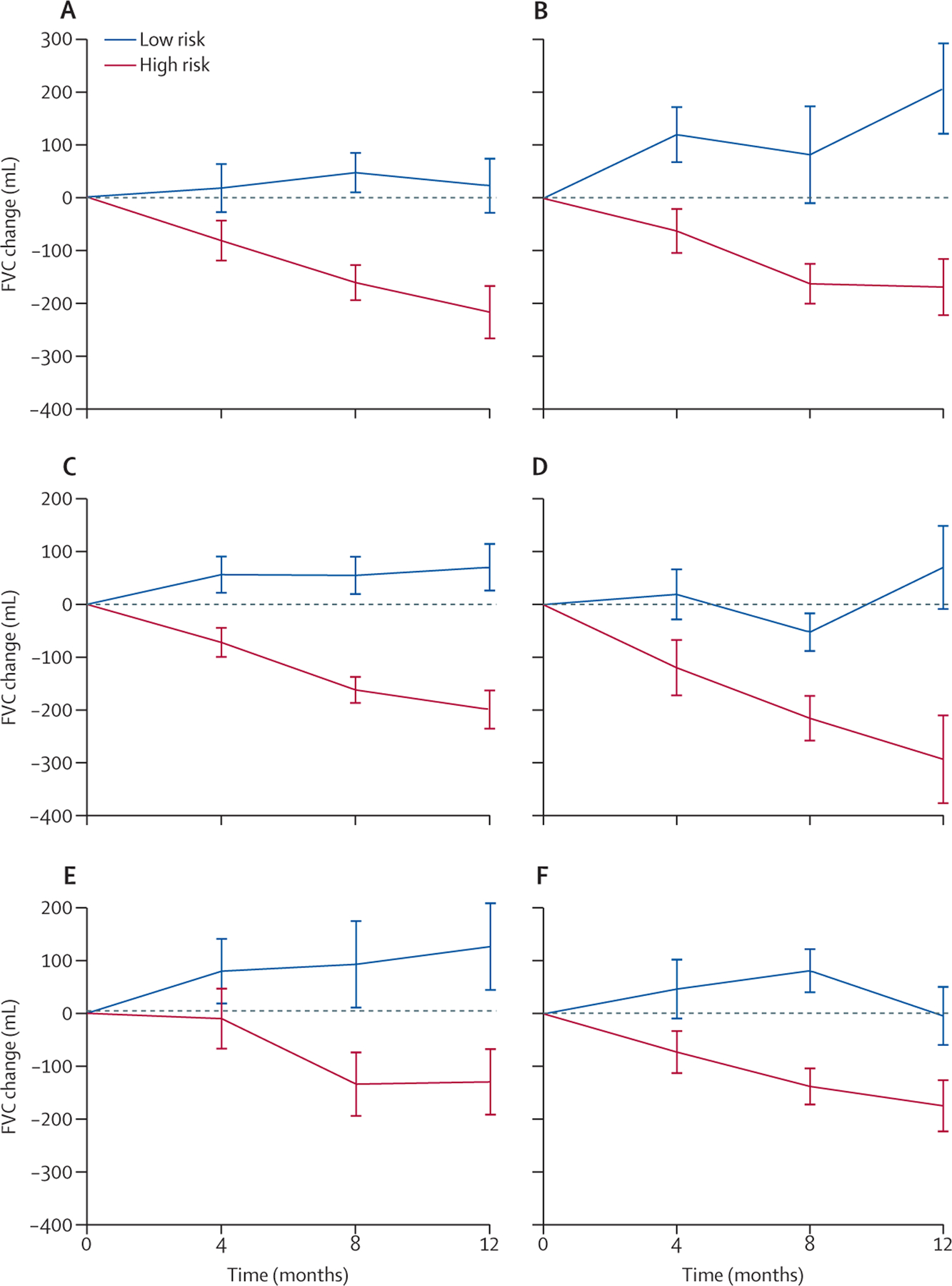
Plots show 1-year change in FVC in the derivation cohort (A), validation cohort (B), and the combined cohort (C). After stratification of the combined cohort by proteomic signature classification and high-resolution CT pattern, FVC trajectories were compared between high-risk and low-risk proteomic signature groups with definite or probable usual interstitial pneumonia (D), non-specific interstitial pneumonia (E), and other high-resolution CT patterns (F). FVC=forced vital capacity.
The overall cohort experienced an FVC change of −99·6 mL (95% CI −150·2 to −49·8). Patients with a low-risk proteomic signature experienced an FVC change of +85·7 mL (6·9 to 164·4) and those with a high-risk signature experienced an FVC change of −227·1 mL (−286·7 to −167·5). A theoretical randomised controlled trial with 1:1 randomisation designed without regard to proteomic signature would require 676 patients to detect a 50% reduction in FVC decline at 90% power, assuming a standard deviation of 200 mL and two-tailed α of 0·05. A similar trial restricted to patients with a high-risk proteomic signature would require 142 patients, assuming the same parameters (appendix p 23).
Discussion
In this investigation, we identified and validated 17 circulating plasma biomarkers of progressive fibrosing ILD, most of which were novel, in a combined cohort of patients with chronic hypersensitivity pneumonitis, connective tissue disease-associated ILD, and unclassifiable ILD. Biomarker association with progressive fibrosing ILD phenotype was robust to ILD clinical diagnosis and high-resolution CT morphology for most analytes assessed. Heterogeneity in progressive fibrosing ILD association was detected after stratification by GAP stage, suggesting that biomarker concentration might track with disease severity. We then derived a proteomic signature of progressive fibrosing ILD, which showed good performance when tested in an independent validation cohort. Although a number of circulating protein biomarkers have been linked to progressive fibrosing ILD,6 and proteomic investigations have identified novel biomarkers that discriminate IPF from healthy controls,14 to our knowledge, this study is the first to use proteomic technology to identify novel biomarkers of progressive fibrosing ILD in patients with non-IPF forms of ILD.
Although a targeted proteomic platform of inflammatory biomarkers was used for this analysis, the majority of validated biomarkers of progressive fibrosing ILD are of epithelial and mesenchymal cell origin.15 ITGB6 had the strongest and most consistent progressive fibrosing ILD association; it is the β6 subunit for the integrin αvβ6, an activator of TGF-β that is long known to be a critical mediator of fibrogenesis in IPF.16 In addition to the role it plays in fibrotic remodelling, αvβ6 integrin expression in lung tissue has been associated with reduced IPF survival and appears to track with disease activity.17 Although a trial assessing systemic αvβ6 blockade was stopped due to safety concerns (NCT1371305), two early-stage inhalation trials testing small molecule inhibitors of αvβ6 integrin in patients with IPF are ongoing.18,19 Our findings suggest that αvβ6 activity could contribute to fibrogenesis across a diverse set of ILDs and support investigation of αvβ6 blockade in patients with non-IPF ILD.
Another progressive fibrosing ILD-associated biomarker primarily expressed in epithelial cells was MMP10. The matrix metalloproteinase family has long been implicated in the pathogenesis of IPF, because these proteins can promote epithelial-to-mesenchymal transition, recruit profibrotic mediators, and promote abnormal wound healing.20 MMP7 is perhaps the most commonly measured matrix metalloproteinase in ILD, because this protein is elevated in the lungs of patients with IPF and has been linked to differential outcomes in IPF and other ILDs.6,7,20 Blockade of MMP9 was shown to ameliorate pulmonary fibrosis in an animal model, suggesting it could be a viable target in the treatment of IPF and other progressive fibrosing ILDs.21 MMP10, also called stromelysin 2, is expressed by endothelial cells, fibroblasts, and macrophages, and is known to be increased in the lung tissue of patients with IPF.20 A study from 2015 reported that serum MMP10 concentration predicted near-term progression in IPF,22 and our results extend these findings to non-IPF ILDs. MMP1 was also associated with progressive fibrosing ILD in our discovery cohort; however, this association was not maintained in our validation cohort.
Other biomarkers predominantly expressed in epithelial cells include AGER, CXCL17, DPP10, KRT19, PRSS8, and SCGB3A2. Decreasing AGER concentration was associated with progressive fibrosing ILD in our study, which is consistent with a previous study showing similar results in IPF.23 By preventing formation of advanced glycation end products, the AGER ligand aminoguanidine protected against bleomycin induced fibrosis in mouse models,24 which supports interaction between AGER and advanced glycation end products as a potential therapeutic target in IPF and progressive fibrosing ILD. Differential expression of CXCL17,25 KRT19,26 PRSS8,27 and SCGB3A228 has also been shown in patients with ILD compared with healthy controls, but our findings represent a novel association with progressive fibrosing ILD.
Progressive fibrosing ILD biomarkers predominantly expressed in mesenchymal cells included ANGPTL4, HGF, SPON1, and TNFRSF11B. Two of these, ANGPTL429 and SPON1,14 have been linked to systemic sclerosis ILD progression, whereas HGF has been shown to predict worsening liver fibrosis,30 suggesting shared pathobiology underpinning fibrogenesis across organ systems. Osteoprotegerin (TNFRSF11B) inhibits bone matrix turnover by acting as a decoy receptor protein for RANKL. As a decoy receptor for TRAIL, it can also prevent resolution of acute pulmonary inflammation. A 2020 study linked osteoprotegerin expression to TGF-β activity and showed plasma osteoprotegerin concentration to predict disease progression in IPF.31 In an ex-vivo study, administration of a RANKL mutant bypassed osteoprotegerin while still interacting with RANK to stimulate degradation of extracellular matrix and was proposed as a potential therapeutic target for pulmonary fibrosis.32
The remaining biomarkers identified are predominantly expressed in immune cells (FASLG and FCAR) or widely expressed (IL17C, PLAUR, and TGFA). FASLG has been previously implicated in IPF progression,33 whereas TGFA is upregulated in patients with ILD compared with healthy controls34 and PLAUR35 has been linked to systemic sclerosis ILD progression. IL17C and FCAR have been linked to several inflammatory conditions but, to our knowledge, our study is the first to link plasma concentration of these biomarkers to ILD outcome.
Besides biomarker discovery and validation, our analysis yielded a semi-quantitative proteomic signature of progressive fibrosing ILD, with high potential for clinical implementation if it were to be prospectively validated using a quantitative platform. Criteria for identifying those with a progressive fibrosing ILD phenotype have been proposed as part of therapeutic clinical trials.3,4 Although these criteria effectively identify those experiencing ILD progression,5 they do not allow patients at risk to be identified before irreversible progression occurs. Our findings suggest that our proteomic signature would be a reliable screening tool for progressive fibrosing ILD based on the high negative predictive value, because less than 10% of patients with a low-risk proteomic signature experienced ILD progression in the year after blood draw. Validation of this signature could justify a conservative approach in the majority of such patients. Conversely, approximately 50% of those with a high-risk proteomic signature are likely to experience ILD progression in the year after blood draw. Although the low specificity and positive predictive value suggest that this would be a poor confirmatory tool for progressive fibrosing ILD, the signature did effectively identify a group for whom the rate of FVC decline was higher than that observed for placebo-treated patients in progressive fibrosing ILD clinical trials.3,4 Whereas those trials required objective evidence of antecedent ILD progression, our proteomic signature does not, supporting its potential use to enrich clinical trial cohorts. Furthermore, we found that a clinical trial restricted to those with a high-risk proteomic signature could require roughly 80% fewer patients than a trial designed for all patients with non-IPF ILD.
Although we found that biomarker progressive fibrosing ILD association was robust to clinical diagnosis, heterogeneity in this association was observed for several biomarkers when stratifying the cohort by radiological pattern and GAP stage. These findings suggest that some biomarkers could reflect biological processes inherent to usual interstitial pneumonia, non-specific interstitial pneumonia, and other patterns, whereas others might better reflect advanced disease than early-stage, biologically active disease. Together, these observations support biomarker investigation in patients with early ILD and in appropriately powered cohorts to assess morphological subtypes.
Our study has several limitations. First, the retrospective nature of our design did not allow for standardisation of ILD diagnosis or management across centres, nor protocolised follow-up. However, we showed that progressive fibrosing ILD biomarker association was robust to ILD clinical diagnosis and our focus on near-term outcomes is likely to have resulted in less variability in biomarker association than studies focused on long-term survival. Second, although our intent was to identify biomarkers of progressive fibrosing ILD that manifested in the year after blood draw, the relationship between these biomarkers and long-term survival remains unclear. However, our analysis of individual outcomes suggests a strong association between these biomarkers and survival. Categorical decline in FVC is also a strong predictor of subsequent mortality in ILD, further suggesting that biomarkers identified in this analysis are likely to predict long-term survival. Finally, we did not account for ILD therapy in this analysis. Immunosuppressant therapy is regularly used to treat non-IPF forms of ILD; however, data supporting this approach is generally weak.2 Nintedanib was approved for the treatment of progressive fibrosing ILD in 2020, but all patients included in this analysis were recruited before approval of this therapy for progressive fibrosing ILD in those with non-IPF forms of ILD.4 Finally, because proteomic data are semi-quantitative, prospective validation and calibration of our proteomic signature using a quantitative platform is required before clinical implementation is feasible.
Although the progressive fibrosing ILD construct has led to successful testing of anti-fibrotic therapy in this patient population, the ability to predict progressive fibrosing ILD before progression remains elusive. Numerous plasma biomarkers have been linked to differential outcomes in IPF and other fibrotic ILDs, but few have resulted in clinical implementation. Here, we identified plasma biomarkers of near-term ILD progression and derived a proteomic signature that reliably predicted this endpoint. This proteomic signature has high potential to inform clinical decision making in the future, including identifying patients at high risk and enriching clinical trial cohorts. These findings support ongoing proteomic investigation in progressive fibrosing ILD and prospective validation of these and other relevant biomarkers.
Supplementary Material
Research in context.
Evidence before this study
On July 5, 2021, we searched PubMed for studies published in English up to June 30, 2021, using a matrix of keywords that included “biomarker,” “progression,” “progressive,” “interstitial lung disease,” “hypersensitivity pneumonitis”, and “pulmonary fibrosis,” which yielded 285 unique results. We excluded review articles, studies that did not assess outcomes, and those focused solely on idiopathic pulmonary fibrosis or sarcoidosis, resulting in 25 remaining studies. Most were either single-centre studies or had a small sample size and only two assessed pooled interstitial lung disease (ILD) cohorts. The first study showed CXCL13, CA-125, MMP7, YKL-40, SP-D, and VCAM-1 to be variable predictors of progressive ILD in a multicentre cohort of patients with connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, and unclassifiable ILD, and the second study showed serum KL-6 to predict progressive ILD in a similar cohort that also included patients with idiopathic pulmonary fibrosis. The search also returned the published protocol for an ongoing prospective cohort study (INJUSTIS, NCT03670576) that will recruit a heterogeneous group of 250 patients with ILD to identify prespecified biomarkers that predict progressive ILD. No studies were identified that used proteomics to identify biomarkers of ILD outcome.
Added value of this study
To our knowledge, this is the first study to use proteomics to identify novel plasma biomarkers of progressive fibrosing ILD in those with forms of ILD other than idiopathic pulmonary fibrosis, including connective tissue disease-associated ILD, chronic hypersensitivity pneumonitis, and unclassifiable ILD. We observed that associations of validated biomarkers with progressive fibrosing ILD were robust to ILD clinical diagnosis and that a proteomic signature comprising 12 biomarkers showed high sensitivity for discriminating progressive from non-progressive ILD. When applied in an independent validation cohort, less than 10% of patients with a low-risk proteomic signature experienced ILD progression in the year after blood draw, suggesting that such a tool could effectively screen for near-term progressive fibrosing ILD if validated using a quantitative platform.
Implications of all the available evidence
We and others have shown the high potential for plasma and serum biomarkers to inform progression risk in patients with ILD. Our collective findings indicate that shared pathobiology underpins progressive fibrosing ILD, irrespective of ILD subtype. Despite these advances, no biomarkers have been widely adopted for clinical use, in part due to modest test performance for individual biomarkers for predicting ILD progression. The proteomic signature presented here overcame this limitation by capturing the aggregated risk explanation provided by multiple biomarkers. Prospective validation of these findings using a quantitative protein platform could allow for clinical trial enrichment, thereby reducing the sample size needed to detect therapeutic efficacy and obviating the need for antecedent ILD progression when defining the progressive fibrosing ILD phenotype.
Acknowledgments
We thank all the patients that contributed blood samples for this research. This study was funded by the National Heart, Lung, and Blood Institute (grant number T32HL007013 to WSB and JVP; K23HL148498 to CAN; R01HL093096 to CKG; R01HL139897 to PJW; K23HL138190 and R03HL157895 to JMO). In addition, CAN was funded by the National Center for Advancing Translational Sciences (UL1TR001105), PJW was funded by the Nina Ireland Program for Lung Health, and CKG was funded by a Boehringer Ingelheim Investigator Initiated Grant.
Declaration of interests
CAN reports personal fees from Boehringer Ingelheim, outside of the submitted work. CKG reports a grant from Boehringer Ingelheim and previous advisory board service for Pliant Therapeutics, both unrelated to the submitted work. PJW reports grants from Sanofi, grants and personal fees from Boehringer Ingelheim and Roche/Genentech, and personal fees from Gossamer Bio, Blade Therapeutics, and Pliant, unrelated to the submitted work. JMO reports an unrelated grant from Boehringer Ingelheim and personal fees from Genentech, United Therapeutics, Gatehouse Bio, and AmMax Bio, unrelated to the submitted work. All other authors declare no competing interests.
Footnotes
Data sharing
Data will be made freely available without restriction through BioLINCC (keyword: pulmonary fibrosis) within 1 year of publication (https://biolincc.nhlbi.nih.gov).
References
- 1.Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev 2019; 28: 180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med 2020; 383: 958–68. [DOI] [PubMed] [Google Scholar]
- 3.Behr J, Prasse A, Kreuter M, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 2021; 9: 476–86. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019; 381: 1718–27. [DOI] [PubMed] [Google Scholar]
- 5.Maher TM, Brown KK, Kreuter M, et al. Effects of nintedanib by inclusion criteria for progression of interstitial lung disease. Eur Respir J 2021; 2004587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowman WS, Echt GA, Oldham JM. Biomarkers in progressive fibrosing interstitial lung disease: optimizing diagnosis, prognosis, and treatment response. Front Med (Lausanne) 2021; 8: 680997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alqalyoobi S, Adegunsoye A, Linderholm A, et al. Circulating plasma biomarkers of progressive interstitial lung disease. Am J Respir Crit Care Med 2020; 201: 250–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune mechanisms in pulmonary fibrosis. Am J Respir Cell Mol Biol 2016; 55: 309–22. [DOI] [PubMed] [Google Scholar]
- 9.Heukels P, Moor CC, von der Thusen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med 2019; 147: 79–91. [DOI] [PubMed] [Google Scholar]
- 10.Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 2014; 9: e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest 2014; 145: 723–28. [DOI] [PubMed] [Google Scholar]
- 12.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–82. [DOI] [PubMed] [Google Scholar]
- 13.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156: 684–91. [DOI] [PubMed] [Google Scholar]
- 14.Todd JL, Neely ML, Overton R, et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir Res 2019; 20: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumark N, Cosme C Jr, Rose KA, Kaminski N. The idiopathic pulmonary fibrosis cell atlas. Am J Physiol Lung Cell Mol Physiol 2020; 319: L887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatler AL, Jenkins G. TGF-β activation and lung fibrosis. Proc Am Thorac Soc 2012; 9: 130–36. [DOI] [PubMed] [Google Scholar]
- 17.Saini G, Porte J, Weinreb PH, et al. αvβ6 integrin may be a potential prognostic biomarker in interstitial lung disease. Eur Respir J 2015; 46: 486–94. [DOI] [PubMed] [Google Scholar]
- 18.Maher TM, Simpson JK, Porter JC, et al. A positron emission tomography imaging study to confirm target engagement in the lungs of patients with idiopathic pulmonary fibrosis following a single dose of a novel inhaled αvβ6 integrin inhibitor. Respir Res 2020; 21: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.John AE, Graves RH, Pun KT, et al. Translational pharmacology of an inhaled small molecule αvβ6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat Commun 2020; 11: 4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015; 53: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espindola MS, Habiel DM, Coelho AL, et al. Differential responses to targeting matrix metalloproteinase 9 in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2021; 203: 458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sokai A, Handa T, Tanizawa K, et al. Matrix metalloproteinase-10: a novel biomarker for idiopathic pulmonary fibrosis. Respir Res 2015; 16: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manichaikul A, Sun L, Borczuk AC, et al. Plasma soluble receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Ann Am Thorac Soc 2017; 14: 628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Wang T, Wang X, et al. Blockade of advanced glycation end product formation attenuates bleomycin-induced pulmonary fibrosis in rats. Respir Res 2009; 10: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burkhardt AM, Tai KP, Flores-Guiterrez JP, et al. CXCL17 is a mucosal chemokine elevated in idiopathic pulmonary fibrosis that exhibits broad antimicrobial activity. J Immunol 2012; 188: 6399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobashi N, Fujita J, Ohtsuki Y, et al. Elevated serum and BAL cytokeratin 19 fragment in pulmonary fibrosis and acute interstitial pneumonia. Eur Respir J 1999; 14: 574–78. [DOI] [PubMed] [Google Scholar]
- 27.Raghu G, Richeldi L, Jagerschmidt A, et al. Idiopathic pulmonary fibrosis: prospective, case-controlled study of natural history and circulating biomarkers. Chest 2018; 154: 1359–70. [DOI] [PubMed] [Google Scholar]
- 28.Cai Y, Kimura S. Secretoglobin 3A2 Exhibits anti-fibrotic activity in bleomycin-induced pulmonary fibrosis model mice. PLoS One 2015; 10: e0142497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito M, Mitani A, Ishimori T, et al. Active mTOR in lung epithelium promotes epithelial-mesenchymal transition and enhances lung fibrosis. Am J Respir Cell Mol Biol 2020; 62: 699–708. [DOI] [PubMed] [Google Scholar]
- 30.Bellan M, Castello LM, Pirisi M. Candidate biomarkers of liver fibrosis: a concise, pathophysiology-oriented review. J Clin Transl Hepatol 2018; 6: 317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Habibie H, Putri KSS, Boorsma CE, et al. Osteoprotegerin is elevated in pulmonary fibrosis and associates with IPF progression. bioRxiv 2020; published online Dec 3. 10.1101/2020.12.02.408062 (preprint). [DOI] [Google Scholar]
- 32.Wang Y, Michiels T, Setroikromo R, van Merkerk R, Cool RH, Quax WJ. Creation of RANKL mutants with low affinity for decoy receptor OPG and their potential anti-fibrosis activity. FEBS J 2019; 286: 3582–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallach-Dayan SB, Petukhov D, Ahdut-HaCohen R, Richter-Dayan M, Breuer R. sFasL—the key to a riddle: immune responses in aging lung and disease. Int J Mol Sci 2021; 22: 2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasam RK, Reddy GB, Jegga AG, Madala SK. Dysregulation of mesenchymal cell survival pathways in severe fibrotic lung disease: the effect of nintedanib therapy. Front Pharmacol 2019; 10: 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butt S, Jeppesen JL, Iversen LV, et al. Association of soluble urokinase plasminogen activator receptor levels with fibrotic and vascular manifestations in systemic sclerosis. PLoS One 2021; 16: e0247256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.