

Abstract
Background:
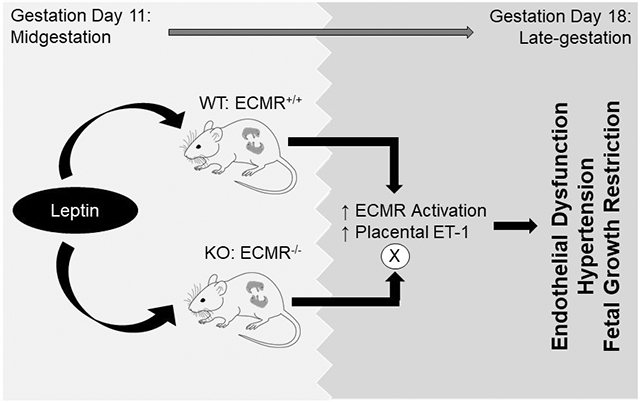
Preeclampsia (PE) patients demonstrate increases in placental leptin production in midgestation, and an associated increase in late gestation plasma leptin levels. The consequences of mid-late gestation increases in leptin production in pregnancy is unknown. Our previous work indicates that leptin infusion induces endothelial dysfunction in nonpregnant female mice via leptin-mediated aldosterone production and endothelial mineralocorticoid receptor (ECMR) activation, which is ablated by ECMR deletion. Therefore, we hypothesized that leptin infusion in mid-gestation of pregnancy induces endothelial dysfunction and hypertension, hallmarks of clinical PE, which are prevented by ECMR deletion.
Methods:
Leptin was infused via miniosmotic pump (0.9mg/kg/day) into timed-pregnant ECMR-intact (WT) and littermate-mice with ECMR deletion (KO) on gestation day (GD)11-18.
Results:
Leptin infusion decreased fetal weight and placental efficiency in WT mice compared to WT+vehicle. Radiotelemetry recording demonstrated that blood pressure (BP) increased in leptin-infused WT mice during infusion. Leptin infusion reduced endothelial-dependent relaxation responses to acetylcholine (ACh) in both resistance (2nd order mesenteric) and conduit (aorta) vessels in WT pregnant mice. Leptin infusion increased placental endothelin-1 production evidenced by increased prepro-endothelin-1 and endothelin converting enzyme-1 expressions in WT mice. Adrenal aldosterone synthase (CYP11B2) and angiotensin II type 1 receptor b (AT1Rb) expression increased with leptin infusion in pregnant WT mice. KO pregnant mice demonstrated protection from leptin-induced reductions in pup weight, placental efficiency, increased BP and endothelial dysfunction.
Conclusions:
Collectively, these data indicate that leptin infusion in midgestation induces endothelial dysfunction, hypertension and fetal growth restriction in pregnant mice which is ablated by ECMR deletion.
Keywords: Preeclampsia, leptin, hypertension, endothelial function, endothelial mineralocorticoid receptor, endothelin-1, aldosterone
Graphical Abstract
Introduction
Preeclampsia (PE) is a hypertensive disorder of pregnancy affecting around 5% of pregnancies in the US 1. PE prevalence 2 and severity 3 is increasing in the US and is diagnosed with late gestation (>20 weeks) hypertension presenting alongside a clinical marker of placental dysfunction. PE pregnancies increase the likelihood of adverse pregnancy events, notably fetal growth restriction (FGR) and also increase the risk of cardiovascular diseases for both mother and offspring post-pregnancy 4-7. Therefore, PE is currently recognized as a significant contributor to overall cardiovascular disease burden 8. PE is characterized by a pro-hypertensive milieu including endothelial dysfunction 9, 10, immune dysfunction 11-13 and altered renin-angiotensin aldosterone system (RAAS) activation 14, 15. Importantly, the placenta is a focal point of PE pregnancies and PE placentas demonstrate dysregulated secretions of hormones in mid-late gestation including increased secretion of antiangiogenic peptides (sFlt-1), decreased placental growth factor (PLGF), increased proinflammatory immune cytokines 16, 17, and also, increased secretion of leptin 18, 19.
Leptin is a hormone traditionally associated with obesity, and the primary source of leptin in nonpregnancy is the adipose tissue. However, the placenta is a potent producer of leptin in pregnant women and is believed to be responsible for the steady increase in plasma leptin levels observed in pregnancy across the gestational period 20-23. Although pregnancy increases placental leptin production, clinical studies indicate that placental leptin production and plasma leptin levels increase in PE patients following midgestation in association with the presentation of hypertension 18, 19. This leads to a dissociation of maternal adiposity with leptin levels in PE patients 24, 25. High leptin levels in late gestation of PE patients is associated with reductions in fetal birth weight and increased PE severity 26-33, however, the mechanisms via which leptin promotes late gestation PE pathology are unknown.
Endothelial dysfunction is a key characteristic of PE pregnancies in both PE patients 34 and rodent PE models 35. Emerging clinical data indicates that reductions in endothelial-dependent vascular relaxation responses predicts PE severity 35-37. Our laboratory previously demonstrated that increases in leptin receptor activation in nonpregnant female mice promotes endothelial dysfunction and hypertension 38, 39 and further, that leptin-mediated endothelial dysfunction in female mice is ablated by deletion of the endothelial mineralocorticoid receptor (ECMR) 40. Furthermore, we showed that high progesterone levels in pregnancy increases ECMR expression in females, indicating that high leptin levels and high ECMR expression in late gestation of PE pregnancies may serve to potentiate endothelial dysfunction 40. Therefore, we hypothesized in the current study that leptin infusion in midgestation induces endothelial dysfunction and hypertension of pregnancy, which are ablated by ECMR deletion.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
All procedures and protocols were approved by the Augusta University Institutional Animal Care and Use Committee (IACUC protocol #2011-0108) and are compliant with guidelines set forth by the NIH. All animals were housed in an American Association of Laboratory Animal Care-approved animal care facility at Augusta University. Animals were housed at ambient temperature with 12:12 hour light-dark cycles with food and water ad libitum. Mice with intact endothelial mineralocorticoid receptor (WT) or endothelial-specific mineralocorticoid receptor (ECMR) deletion (KO), originally provided by Dr. Iris Jaffe 41 (Tufts University), were bred and housed at Augusta University. KO mice (C57BL6 background) were generated by flanking loxP sites on MR exons in mice and breeding with vascular endothelial (VE)-cadherin Cre recombinase transgene (Cre+) mice to achieve an endothelial cell-specific MR deletion. Breeding across generations continued with Cre+ heterozygotes bred with Cre- homozygote mice, both with homozygote MR flox/flox, which generates an even distribution of KO mice and WT littermates. Female mice were bred with male littermates at 10-14 weeks of age and gestation day (GD)1 was determined via the detection of a vaginal plug. Mice were confirmed pregnant at GD11 and either saline sham surgery (vehicle) or miniosmotic pump (Alzet, Cupertino, CA) containing leptin (0.9mg/kg/kg, Prospec Ness-Ziona, Israel) was implanted in the subcutaneous area posterior to the subscapular region. This time-point was chosen for GD11 as a midgestation point in a mouse’s gestation period (~19.5 gestation days). Pregnant females were sacrificed at GD18 under isofluorane anesthesia and tissues and plasmas were harvested along with the collection of pup and placental weights ex-utero.
Statistics
Statistical analysis was performed in Graphpad Prism® software (La Jolla, CA). All bar data were expressed as mean±S.E.M. In non-repeated variables, effects of KO, leptin or their interaction were measured by two-way ANOVA and differences among means measured by Tukey’s multiple comparison test or student’s unpaired two-tailed t-test. Analysis of blood pressure and vascular concentration response curves utilized two-way ANOVA. P value of <0.05 was considered significant.
Additional Methods details can be found in the Supplemental Data File.
Results
Leptin infusion reduced fetal growth and placental efficiency without significantly affecting maternal weight
Compared to their vehicle counterparts, leptin infusion significantly reduced pup weight by GD 18 in WT mice without affecting pup weight in KO mice (Figure 1A). In addition, ECMR deficiency increased pup weight in pregnant mice. Neither leptin treatment nor ECMR deficiency affected litter size (Figure 1B). Placental efficiency, assessed as pup weight as a ratio to placental weight, decreased with leptin infusion in WT mice, but not KO mice (Figure 1C). However, ECMR deficiency significantly increased placental efficiency in pregnant mice. Furthermore, leptin infusion did not significantly affect placental weight (Figure 1D), resorptions (Figure 1E) or maternal body weight (Supplemental Table S2). Neither leptin infusion nor ECMR deficiency affected heart or kidney weights expressed as a ratio to body weight (Supplemental Table S2). Leptin infusion increased plasma leptin levels in WT and KO pregnant mice (Supplemental Table S2).
Figure 1. Leptin infusion induces fetal growth restriction in WT, but not KO, pregnant mice.
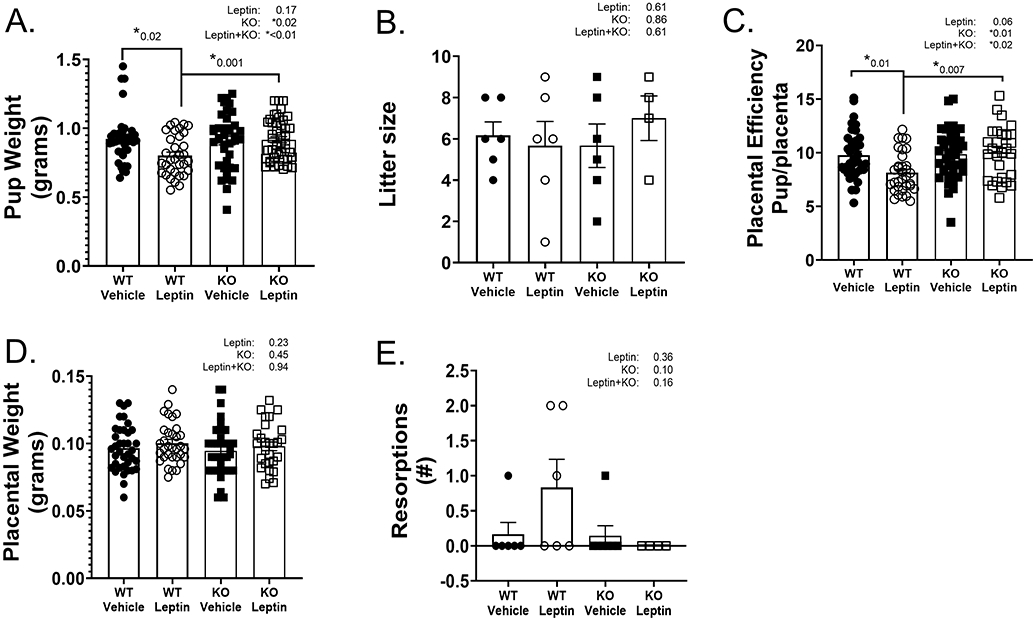
GD18 pup weights (A), average litter size by fetal number (B), placental efficiency (ratio of pup/placenta weight) (C), placental weights (D), reabsorbed pups (resorptions) per litter (E) in WT and KO pregnant mice with and without leptin infusion. 2-Way ANOVA with Tukey’s posthoc test for multiple comparisons. N=6 WT+Vehicle and WT+Leptin, N=7 KO+Vehicle, N=4 KO+Leptin. *P<0.05.
Leptin infusion increased blood pressure in late gestation of pregnant mice
Radiotelemetry recording of blood pressure and heart rate was performed prior to mating as well as across the pregnancy of WT pregnant mice with vehicle or leptin infusion (Figure 2A-D). WT pregnant mice demonstrated similar MAP, SBP, DBP and HR at prepregnancy baseline and from GD8-11. Leptin infusion increased MAP, SBP and DBP, but not heart rate, in WT pregnant mice (Figure 2A-D).
Figure 2. Leptin infusion increases blood pressure in WT pregnant mice.
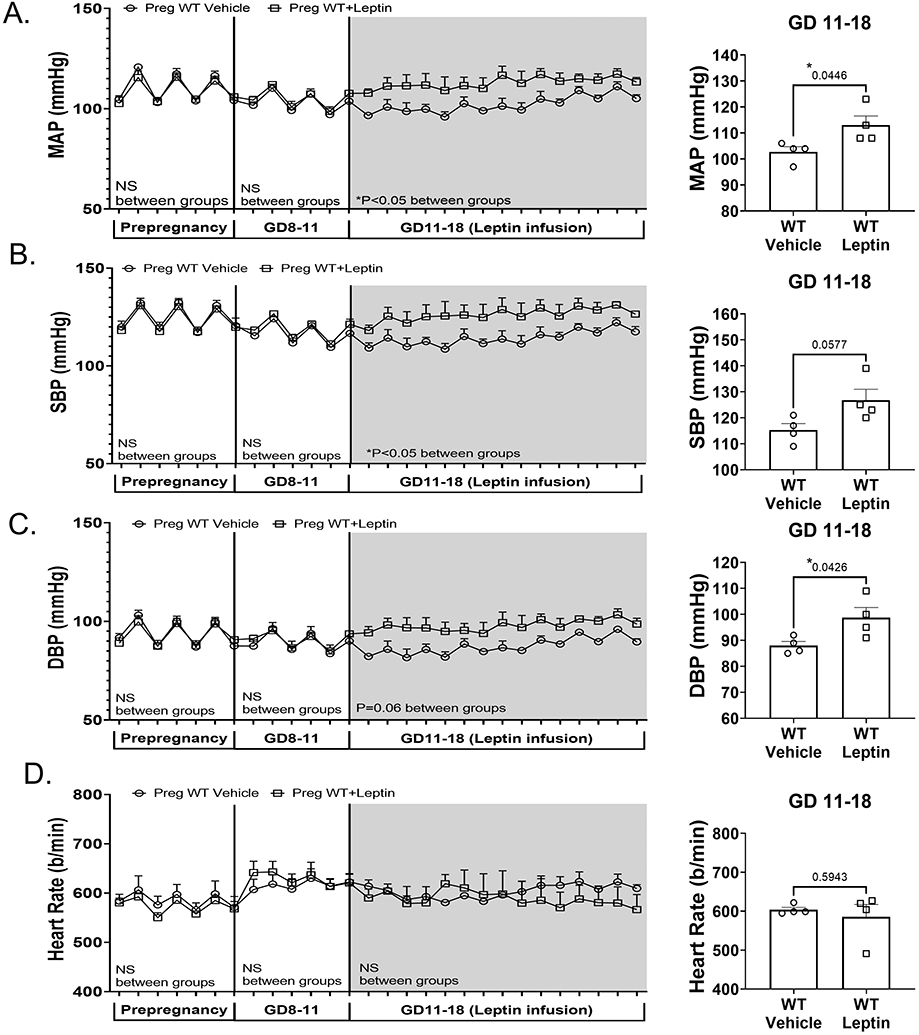
Radiotelemetry recording and average blood pressures at GD11-18 of blood pressure prior to mating (prepregnancy), during GD 8-11 and following vehicle or leptin miniosmotic pump implantation in WT mice (GD11-18). Mean arterial pressure (MAP) (A), systolic blood pressure (SBP) (B), diastolic blood pressure (DBP) (C) and heart rate (D) are depicted across all time points and the summary data of average recording from GD11-18. 2-way ANOVA with repeated measures depicted below each phase of recording and results of student’s t-test in the average pressures at GD11-18. N=4 WT+vehicle, N=4 WT+leptin, *P<0.05.
Leptin infusion decreased endothelial function in resistance and conduit vessels in WT, pregnant mice
Leptin infusion significantly reduced endothelial-dependent relaxation responses to acetylcholine (ACh) in 2nd order mesenteric arteries (resistance arteries) and aortas in WT mice (Figure 3A,B). Preincubation of mesenteric arteries (Figure 3A, B) with LNAME ablated leptin-induced differences in ACh-mediated relaxation in WT mice. Leptin infusion did not reduce endothelial-independent relaxation responses to sodium nitroprusside (SNP) (Figure 3C,D) nor increase contraction to phenylephrine (Phe) (Figure 3E,F), KCl (Supplemental Figure S1A,B) or endothelin-1 (ET-1) (Supplemental Figure S1C) in WT pregnant mice. Ex vivo incubation with leptin did not alter endothelial relaxation to ACh in aortas of female mice, indicating that leptin has no direct effect on endothelial relaxation in female mice (Supplemental Figure S2).
Figure 3. Leptin infusion reduces endothelial function in WT pregnant mice.
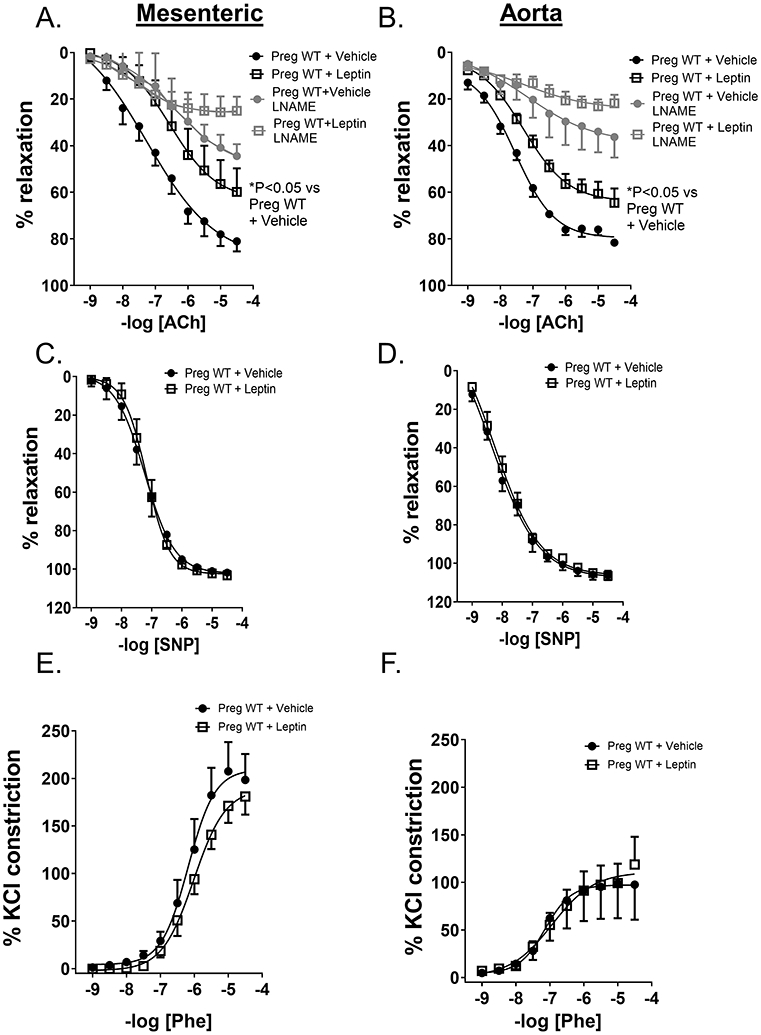
Vascular relaxation responses in 2nd order mesenteric arteries (Mesenteric) and thoracic aorta (aorta) in WT+vehicle and WT+leptin pregnant mice (GD 18). Leptin infusion decreased endothelial-dependent relaxation responses to acetylcholine (ACh) in both mesenteric arteries (A) and aorta (B) with and without LNAME preincubation. Leptin infusion did not reduce endothelial-independent relaxation responses to sodium nitroprusside (SNP) in either mesenteric arteries (C) or aorta (D) nor increase phenylephrine (Phe)-induced contraction in mesenteric arteries (E) or aorta (F). N=5 for both groups mesenteric arteries, N=5 WT+vehicle aorta, N=6 WT+leptin aorta. 2-way ANOVA with repeated measures.*P<0.05,
Endothelial MR deletion prevents leptin-induced increases in BP in pregnant mice
Vehicle- and leptin-infused mice with endothelial-specific MR knockout (KO) demonstrated similar MAP, SBP and DBP and HR both prior to pregnancy (prepregnancy) and at GD8-11 (Fig 4A-D). However, in contrast to WT pregnant mice, leptin infusion did not increase MAP, SBP, or DBP in pregnant mice with ECMR deletion, indicating that ECMR deletion prevents leptin-induced late gestation blood pressure increases in pregnant mice.
Figure 4. Endothelial MR deletion prevented leptin-mediated increases in BP in pregnant mice.
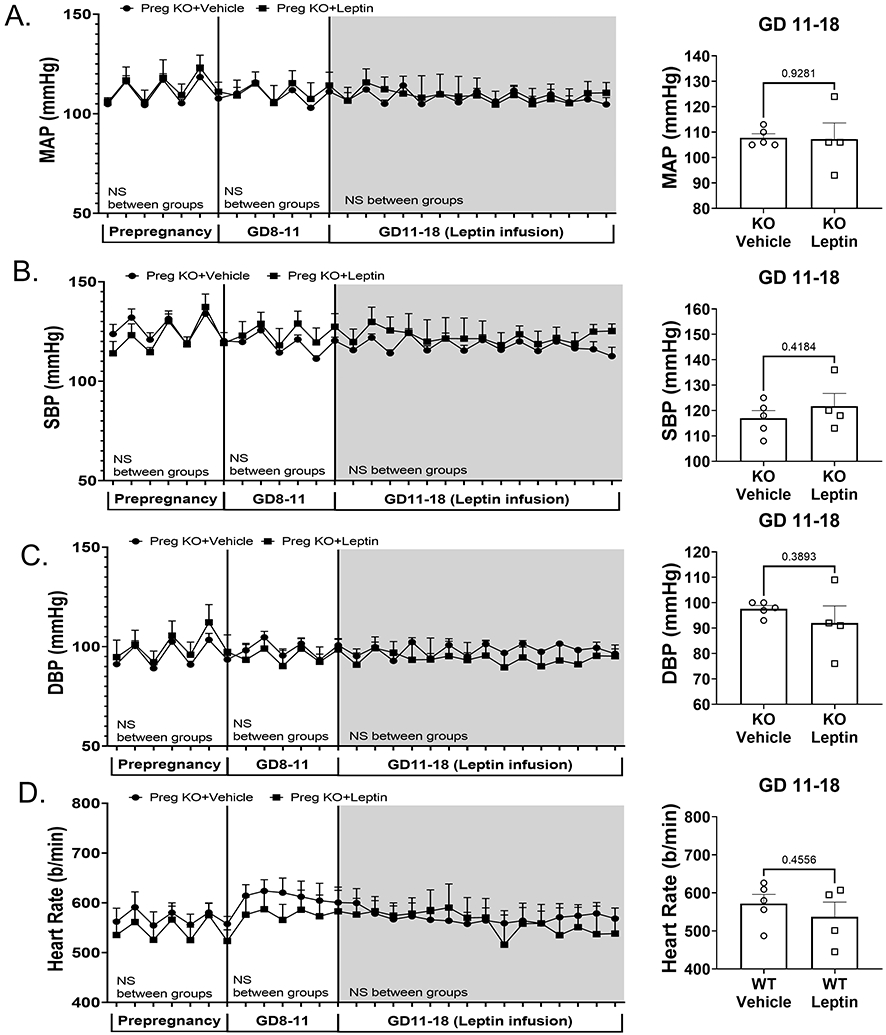
Radiotelemetry recording and average blood pressures at GD11-18 of blood pressure prior to mating (prepregnancy), during GD 8-11 and following vehicle or leptin miniosmotic pump implantation in KO mice (GD11-18). Mean arterial pressure (MAP) (A), systolic blood pressure (SBP) (B), diastolic blood pressure (DBP) (C) and heart rate (D) are depicted across all time points and the summary data of average recording from GD11-18. 2-way ANOVA with repeated measures depicted below each phase of recording and results of student’s t-test in the average pressures at GD11-18. N=5 KO+vehicle, N=4 KO+leptin, *P<0.05.
Endothelial MR deletion prevents leptin-induced endothelial dysfunction in pregnant mice
In contrast to WT pregnant mice, leptin infusion did not reduce ACh-mediated relaxation in aortas of KO pregnant mice and LNAME ablated ACh-mediated relaxation equally in vehicle- and leptin-infused KO pregnant mice (Figure 5A). Leptin infusion did not decrease SNP-mediated relaxation (Figure 5B), contraction to Phe (Figure 5C) nor KCl (Figure 5D) in KO pregnant mice. Supplemental Table S3 demonstrates ECMR deletion significantly increased maximal relaxation (Rmax) of thoracic aorta to ACh as well as relaxation to SNP and reduced maximal constriction (Emax) of Phe. In addition, pregnant leptin-infused KO mice demonstrated significantly higher maximal relaxation of thoracic aorta to ACh and lower maximal constriction to Phe compared to leptin-infused WT pregnant mice. No significant differences in vascular half maximum (EC50) were observed across all groups for any concentration response curve.
Figure 5. Endothelial MR deletion prevented leptin-mediated reduction in endothelial relaxation in pregnant mice.
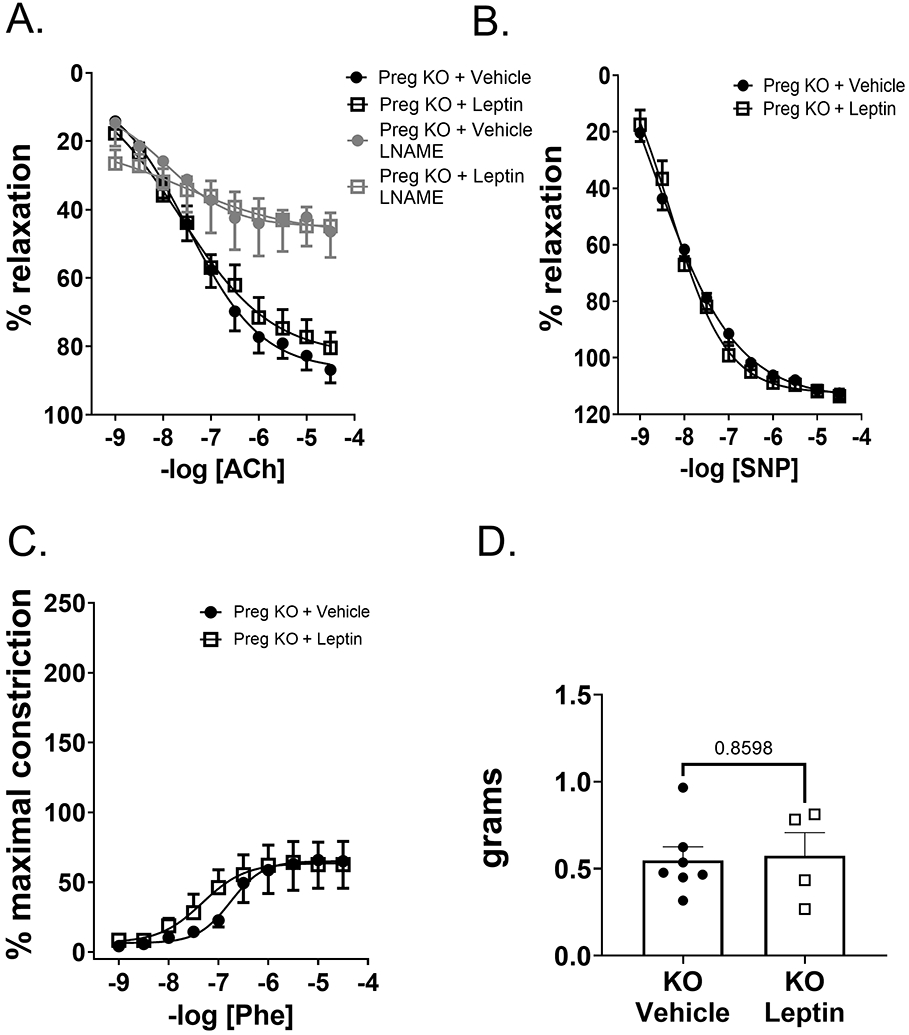
Vascular relaxation responses in thoracic aorta of KO+vehicle and KO+leptin pregnant mice (GD 18). Leptin infusion did not reduce endothelial-dependent relaxation responses to ACh in KO pregnant mice in the absence of or presence of LNAME preincubation (A). Leptin did not reduce SNP-mediated endothelial-independent relaxation responses in KO pregnant mice (B). Leptin infusion also did not increase vascular constriction responses to phenylephrine (Phe) (C) or KCl (D) in KO pregnant mice. N=6 KO+vehicle, N=4 KO+leptin. 2-way ANOVA with repeated measures.*P<0.05.
Leptin increases placental endothelin-1 production in pregnant mice
Previous studies indicate that increased endothelin-1 (ET-1) production is a crucial mediator of endothelial dysfunction in rodent PE models 42. Placental prepro-endothelin 1 (PPET-1) (Figure 6A) as well as endothelin converting enzyme-1 (ECE-1) (Figure 6B) mRNA expressions increased with leptin infusion in pregnant WT mice, but not KO mice. However, markers of placental dysfunction placental growth factor (PLGF) and soluble FMS-like tyrosine kinase-1 (sFLT-1) (Supplemental Figure S3A,B) did not increase with leptin infusion in WT mice, but PLGF expression increased with leptin infusion in pregnant KO mice. Leptin infusion did not increase plasma levels of proinflammatory cytokines as depicted in Supplemental Table S4. However, ECMR deficiency significantly decreased concentrations of IL-12, IL-23, IL-1a, IL-1b, IL-17a and GM-CSF, indicating a reduced inflammatory phenotype in KO pregnant mice.
Figure 6. Leptin increases placental endothelin-1 production and increases adrenal CYP11B2 and AT1Rb expression in pregnant mice.
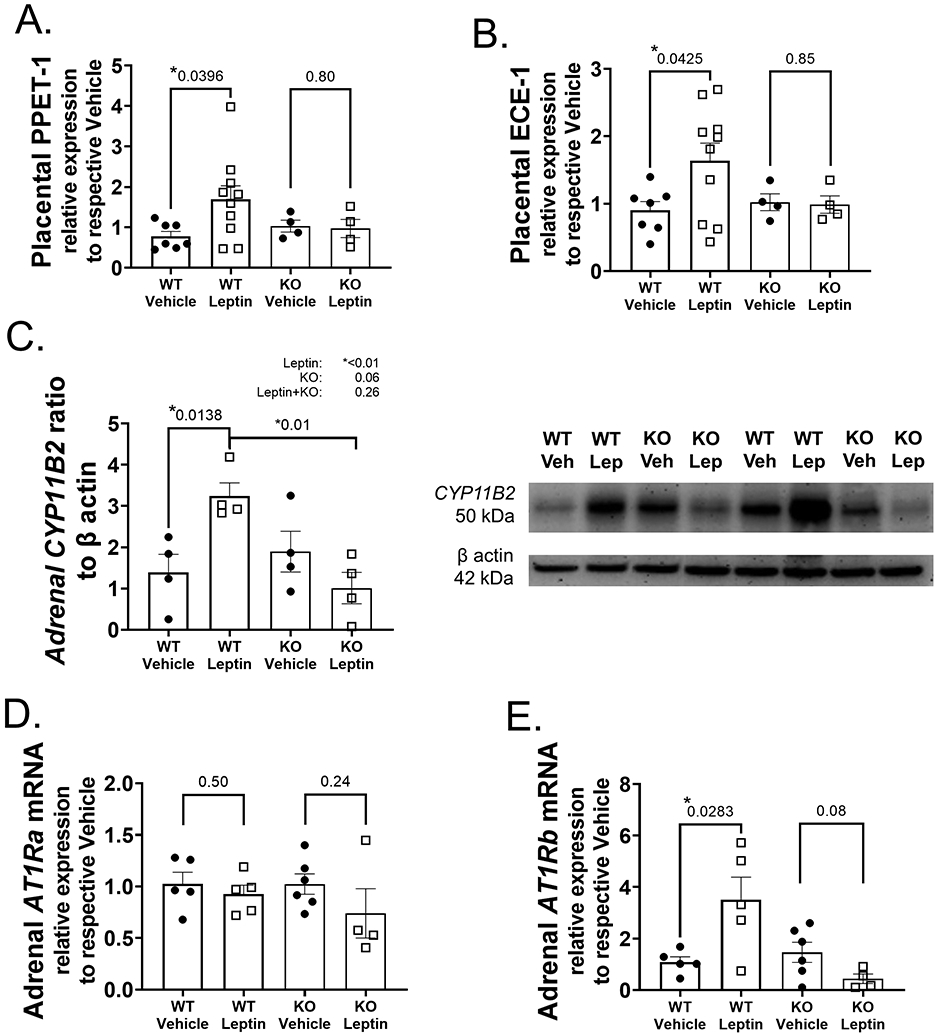
Placental mRNA expression of prepro-endothelin-1 (PPET-1) (A) and endothelin converting enzyme-1 (ECE-1) increased with leptin infusion in WT pregnant mice (B). Adrenal protein expression of CYP11B2 increased in WT mice infused with leptin (C). Adrenal angiotensin II type 1 receptor a (AT1Ra) mRNA expression did not increase with leptin infusion in WT pregnant mice (D) in contrast to the leptin-induced increase in AT1Rb observed in leptin-infused WT pregnant mice (E). Placental N values: N=7 WT+Vehicle, N=9 WT+Leptin, N=4 KO+Vehicle, N=4 KO+Leptin. Protein expression data N=4 for all groups. Adrenal N values: N=5 WT+Vehicle, N=4 WT+Leptin, N=5 KO+Vehicle, N=4 KO+Leptin. Student’s unpaired t-test for paired comparisons to respective Vehicle, 2-way ANOVA with Tukey’s posthoc test for multiple comparisons for protein expression. *P<0.05.
Leptin increases adrenal CYP11B2 and AT1Rb expression in WT pregnant mice
Our previous work demonstrates that leptin increases aldosterone synthase expression in nonpregnant female mice 39. Leptin infusion significantly increased adrenal CYP11B2 (aldosterone synthase gene) protein expression in WT pregnant mice, but not KO mice (Figure 6C). Leptin did not increase adrenal angiotensin II type 1 receptor a (AT1Ra) mRNA expression in WT pregnant mice (Figure 6D), however, did increase AT1Rb mRNA expression (Figure 6E). Leptin infusion failed to increase adrenal AT1Ra or AT1Rb mRNA expressions in KO pregnant mice.
Discussion
Leptin is a reproductively-tied hormone, evidenced in that leptin is required for fertility in both humans and in mice, and further, in that leptin levels steadily increase in a healthy pregnancy20-23. Some speculations indicate that leptin is a nutrient-sensor for healthy reproduction or a mechanism for angiogenesis and growth hormone stimulation, however, the role of leptin in pregnancy is incompletely understood at present43. Compelling and consistent clinical data report that midgestation rises in placental leptin production, and subsequently increases in plasma leptin levels, are associated with PE incidence and severity 26-33, 44, however, the physiological consequences of this increase in leptin in PE pathogenesis is largely unknown. The novel results of the current study indicate that mid-late gestation leptin infusion induces endothelial dysfunction, hypertension and fetal growth restriction (FGR) in murine pregnancy, which is characteristic of the pathogenesis of late gestation PE in human patients. This report introduces an important potential model isolating the effects of leptin in late gestation to promote endothelial impairment and indicates that high leptin levels of PE patients promotes adverse cardiovascular and fetal growth consequences of PE.
In previous studies, our laboratory showed that leptin infusion in female nonpregnant mice induces reductions in acetylcholine-mediated, endothelial-dependent vascular relaxation 38, 39. Endothelial dysfunction, and in particular reductions in NO bioavailability, is a hallmark of human PE patients 42. Endothelial dysfunction is a predictor of PE severity and indeed it has been proposed that vascular dysfunction is a crucial early event of PE pregnancies contributing to placental ischemia 45. Our study demonstrates for the first time that leptin infusion in midgestation in pregnant mice induces endothelial dysfunction in the resistance and conduit vasculature in a preclinical mouse model. Furthermore, we show that inhibition of nitric oxide synthase (NOS) with LNAME ablates differences in ACh-mediated relaxation responses between WT pregnant mice with and without leptin infusion. These data suggest that decreases in NO-mediated relaxation may play a role in leptin-induced endothelial-dysfunction, however, further studies investigating the degree of phosphorylation of eNOS and other mechanisms of vasodilation known to mediate relaxation in females, such as endothelial-derived hyperpolarizing factor (EDHF)46, are warranted to confirm this conclusion. No differences were observed between WT and WT+leptin pregnant mice in vascular responses to SNP or Phe responses and only the resistance vasculature demonstrated an increase in KCl-mediated constriction with leptin infusion in WT pregnant mice. These data indicate that smooth muscle function, was not significantly altered by leptin infusion in WT pregnant mice. Rather, these data indicate that leptin infusion leads to a decrease in endothelial relaxation capacity in late- gestation pregnancy.
We report novel data that leptin infusion in WT pregnant mice induces an increase in adrenal CYP11B2 expression in association with an increase in AT1Rb, but not AT1Ra, mRNA expression, indicating an increase in ligand for endothelial mineralocorticoid receptors. Our published works show that nonpregnant female mice increase CYP11B2 expression in response to adrenal leptin receptor activation, which is a Ca2+-dependent mechanism 39. A previous study by our laboratory also reported sodium restricted diet decreased vascular relaxation to acetylcholine female mice in association with increased adrenal AT1Rb and CYP11B2 adrenal expression levels in females compared to males 47. These data indicate that CYP11B2 and AT1Rb adrenal activity may be a female-specific relationship that further increases in pregnant females with high leptin levels. KO mice demonstrated a lack of increase in CYP11B2 and AT1Rb expression in response to leptin, which indicates that endothelial MR activation may serve as a feedback role to increase CYP11B2 and AT1Rb expression in response to stimuli. Indeed, studies demonstrate that MR inhibition (eplerenone) increases CYP11B2 activity in female mice48 and that endothelial MR deletion reduces CYP11B2 activity in female KO mice with diet-induced obesity46. These data indicate that endothelial MR activation is a crucial mediator of CYP11B2 expression and activation, which may play a role in the protection of pregnant KO mice from leptin-induced endothelial dysfunction, hypertension and FGR. Whether this increase in CYP11B2 expression is mediated by AT1Rb-mediated adrenal signaling warrants further investigation.
Endothlin-1 (ET-1) is a potent vasoconstrictive peptide whose levels significantly increase in PE patients 42, 49 and animal models of PE50, 51. Previous studies demonstrate an association of leptin and ET-1 in the pathogenesis of FGR and adverse pregnancy 52, 53, however, a causative relationship is unreported to-date. Leptin increased both placental PPET-1 and ECE-1 in the placentas of WT mice, indicating that leptin plays a role in PE-associated increases in ET-1 production. Arteries of leptin-infused WT mice did not contract moreso to maximal ET-1 dose than sham WT mice, indicating that vascular ET-1 sensitivity did not increase with leptin infusion in pregnant WT mice. Therefore, future studies are warranted to determine if ET-1 receptor blockade may prevent endothelial dysfunction in pregnant leptin-infused mice.
Endothelial MR deletion protected pregnant mice from leptin-induced endothelial dysfunction. Previous studies by our laboratory demonstrated that high progesterone levels, in particular those of pregnancy, induces increased ECMR expression and is the rationale for our previously observed sex-difference favoring higher ECMR expression in female humans and mice 40. Ablation of ECMR has shown to improve endothelial function in several female mouse models of endothelial dysfunction both in our laboratory 40, 54 and in others’ 46. In the current study, we surmise that improvement of endothelial function by ECMR deletion in leptin-infused pregnant mice protected these mice from hypertension and resulted in improved placental blood flow, preventing FGR and improving placental efficiency. The mechanism whereby leptin promotes placental efficiency is most likely linked to endothelial dysfunction, which is a hallmark of PE, particularly severe PE 34, 55, 56. These data indicate that the primary cardiovascular risk and risk to placental function, which reduces efficiency, that leptin poses in late gestation of PE is to injure the vascular endothelium.
PE is currently believed to be initiated by early gestation placental ischemia and failure of the spiral arteries to remodel into distensible vessels. In the current study we mimic the placental-derived surge in leptin production of PE patients and we observed that this hormone promotes endothelial dysfunction, hypertension and FGR as assessed by reduced pup weight and placental efficiency. We show that blood pressure begins to rise in leptin-infused pregnant mice immediately following leptin infusion, however, as endothelial function is a terminal measurement we are unable to determine whether the development of endothelial dysfunction predisposed these mice to increases in blood pressure or vice versa. Future studies are warranted to elucidate the timeline of pathology of hypertension in response to leptin in pregnant mice. Interestingly, this model also provides some insight into whether midgestation leptin propagates placental dysfunction. Markers of placental dysfunction PLGF and sFLT-1 as well as proinflammatory cytokine levels neither decreased nor increased in response to leptin infusion in WT pregnant mice. These data indicate that placental dysfunction resulting in increases in proinflammatory cytokines, sFLT-1 and decreases in PLGF may precede increases in placental leptin secretion in PE patients. Whether these factors may play a role in promoting leptin production in the placenta is a compelling notion that remains to be investigated.
The model presented in this report provides a valuable preclinical tool in that the cardiovascular consequences of hyperleptinemia are mimicked only in late gestation, without leptin “priming” in early gestation. Leptin is a reproductive hormone that induces fetal growth and trophoblast invasion in early pregnancy, however, high leptin levels in early gestation are also associated with adverse pregnancy outcomes, including PE and FGR which is most clinically apparent in the growing prevalence of obesity in pregnancy. Rising rates of obesity in premenopausal women predispose to PE, FGR, gestational diabetes, among other complications. These patients whom go on to develop PE maintain high leptin levels throughout their pregnancy which likely contribute to their cardiovascular and fetal risks, however, the assessment of adipose leptin regulation in pregnancy as well as whether early gestation leptin infusion regulates ET-1 and CYP11B2 remains unexplored. Leptin itself is a placental preeclampsia-associated hormone, in that several reports indicate that placental leptin production increases in PE patients in midgestation and that the placental trophoblasts are a source of the increased leptin production. Trophoblast shedding and release of these cells into the circulation is a phenomenon increased by PE in pregnancy57, further studies are warranted to determine if trophoblastic shedding increases placental leptin secretion from these cells.
An important notion in our current study is that our female leptin-infused mice demonstrated endothelial dysfunction, hypertension and FGR in the absence of a significant change in body weight or in litter size. A recent report in rats demonstrated that leptin infusion in Sprague Dawley rats failed to result in an increase in blood pressure, however did result in adverse fetal outcomes and reduced NO bioavailability 58. The rats in this particular study lost a significant amount of weight and body fat in response to the leptin infusion, a phenotype that we do not observe in the mouse model, therefore, the metabolic effects of leptin are a consideration that must be monitored in all studies of leptin infusion of pregnancy and offer an intriguing hypothesis of the variable effects of leptin dependent of- and independent of- its hypothalamic appetite suppression effects.
Perspectives
Increases in placental leptin production, and subsequent plasma leptin levels, in PE patients indicates an increased risk for adverse PE outcomes for both mother and fetus. Leptin infusion in midgestation induces an endothelial dysfunction phenotype in pregnant WT mice and further results in hypertension and fetal growth restriction. Genetic deletion of ECMR ablates leptin-induced endothelial dysfunction, hypertension and fetal growth restriction in pregnant mice. In association, leptin infusion increases adrenal CYP11B2 protein and adrenal AT1Rb mRNA expression in pregnant mice. These data indicate that increases in plasma leptin levels in late-gestation PE potentiate PE pathology via increases in aldosterone production and ECMR activation, promoting endothelial dysfunction and placental dysfunction and resulting in hypertension and fetal growth restriction. Further studies are needed to better understand the evolutionary role of the leptin and ECMR relationship in pregnancy to determine if therapeutics targeted at these pathways may be advantageous to PE women presenting with endothelial dysfunction.
Supplementary Material
Pathophysiological Novelty and Relevance.
What is New?
Leptin infusion in mid-late gestation pregnancy induces a model of clinical PE characteristics including endothelial dysfunction, hypertension and fetal growth restriction
Endothelial MR deletion protects pregnant mice from endothelial dysfunction, hypertension and fetal growth restriction induced by leptin infusion
Leptin infusion increases adrenal aldosterone synthase and placental ET-1 production in pregnant mice, which is prevented by endothelial MR deletion
What is Relevant?
Preclinical data in this report indicates that midgestation rises in plasma leptin levels in PE patients contributes to cardiovascular and fetal consequences on PE in late gestation
Placental ET-1 production and adrenal aldosterone may contribute to late-gestation pathology in PE patients with high leptin levels
Clinical/Pathophysiological Implications
The data in this report indicates that therapeutics targeting downstream, mechanisms of leptin secretion in pregnancy (such as eplerenone) may be a promising target to improve cardiovascular and fetal outcomes in PE patients presenting with elevated leptin levels.
Acknowledgements
The authors would like to acknowledge Drs Valentyna Fesenkova and Rafal Pacholczyk in the Immune Monitoring Core (Georgia Cancer Center at the Medical College of Georgia, Augusta University, Augusta, GA) for their assistance with the LEGENDplex cytokine assay. The authors would also like to thank Drs Elise and Celso Gomez-Sanchez at the University of Mississippi Medical Center in Jackson, MS for providing the antibodies for the detection of mouse CYP11B2 protein.
Funding Sources
Support for this work was provided by NIH 1R01HL130301-01, 1R01HL155265-01 and AHA 19EIA34760167 to E.JBdC and 4 R00 HL146948-03 and AHA CDA858380 to JLF.
Abbreviations
- PE
Preeclampsia
- FGR
Fetal Growth Restriction
- RAAS
Renin Angiotensin Aldosterone System
- sFlt-1
Soluble FMS-like tyrosine kinase-1
- PLGF
Placental growth factor
- ECMR
Endothelial mineralocorticoid receptor
- WT
Wild-type (ECMR intact littermate)
- KO
Transgenic (ECMR deletion)
- VE
Vascular endothelial
- GD
Gestation day
- ANOVA
Analysis of variance
- MAP
Mean arterial pressure
- SBP
Systolic blood pressure
- DBP
Diastolic blood pressure
- HR
Heart rate
- Ach
Acetylcholine
- LNAME
L-NG-Nitro arginine methyl ester
- SNP
Sodium nitroprusside
- Phe
Phenylephrine
- KCl
Potassium chloride
- ET-1
Endothelin-1
- Rmax
Maximal relaxation
- Emax
Maximal constriction
- EC50
Half-maximal relaxation
- PPET-1
Pre-proendothelin-1
- ECE-1
Endothelin converting enzyme-1
- IL-
Interleukin
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- AT1Ra
Angiotensin II type I receptor-a
- AT1Rb
Angiotensin II type I receptor-b
- NO
Nitric Oxide
- NOS
Nitric Oxide synthase
- EDHF
Endothelial-derived hyperpolarizing factor
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest
References
- 1.Minhas AS, Ogunwole SM, Vaught AJ, Wu P, Mamas MA, Gulati M, Zhao D, Hays AG, Michos ED. Racial disparities in cardiovascular complications with pregnancy-induced hypertension in the united states. Hypertension. 2021:HYPERTENSIONAHA12117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fingar KR, Mabry-Hernandez I, Ngo-Metzger Q, Wolff T, Steiner CA, Elixhauser A. Delivery hospitalizations involving preeclampsia and eclampsia, 2005-2014: Statistical brief #222. Healthcare cost and utilization project (hcup) statistical briefs. Rockville (MD); 2006. [PubMed] [Google Scholar]
- 3.Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the united states, 1980-2010: Age-period-cohort analysis. BMJ. 2013;347:f6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell MJ. A historical overview of preeclampsia-eclampsia. J Obstet Gynecol Neonatal Nurs. 2010;39:510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutcheon JA, Lisonkova S, Joseph KS. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol. 2011;25:391–403 [DOI] [PubMed] [Google Scholar]
- 6.Amaral LM, Wallace K, Owens M, LaMarca B. Pathophysiology and current clinical management of preeclampsia. Curr Hypertens Rep. 2017;19:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okoth K, Chandan JS, Marshall T, Thangaratinam S, Thomas GN, Nirantharakumar K, Adderley NJ. Association between the reproductive health of young women and cardiovascular disease in later life: Umbrella review. BMJ. 2020;371:m3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogel B, Acevedo M, Appelman Y, Bairey Merz CN, Chieffo A, Figtree GA, Guerrero M, Kunadian V, Lam CSP, Maas A, Mihailidou AS, Olszanecka A, Poole JE, Saldarriaga C, Saw J, Zuhlke L, Mehran R. The lancet women and cardiovascular disease commission: Reducing the global burden by 2030. Lancet. 2021. doi: 10.1016/S0140-6736(21)00684-X [DOI] [PubMed] [Google Scholar]
- 9.Qu H, Khalil RA. Vascular mechanisms and molecular targets in hypertensive pregnancy and preeclampsia. Am J Physiol Heart Circ Physiol. 2020;319:H661–H681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goulopoulou S, Davidge ST. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol Med. 2015;21:88–97 [DOI] [PubMed] [Google Scholar]
- 11.Cornelius DC, Cottrell J, Amaral LM, LaMarca B. Inflammatory mediators: A causal link to hypertension during preeclampsia. Br J Pharmacol. 2019;176:1914–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW Jr., Wallace K, LaMarca B. The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond). 2016;130:409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaMarca B, Cornelius DC, Harmon AC, Amaral LM, Cunningham MW, Faulkner JL, Wallace K. Identifying immune mechanisms mediating the hypertension during preeclampsia. Am J Physiol Regul Integr Comp Physiol. 2016;311:R1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell N, LaMarca B, Cunningham MW Jr. The role of agonistic autoantibodies to the angiotensin ii type 1 receptor (at1-aa) in pathophysiology of preeclampsia. Curr Pharm Biotechnol. 2018;19:781–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gathiram P, Moodley J. The role of the renin-angiotensin-aldosterone system in preeclampsia: A review. Curr Hypertens Rep. 2020;22:89. [DOI] [PubMed] [Google Scholar]
- 16.Ibrahim HS, Omar E, Froemming GR, Singh HJ. Leptin increases blood pressure and markers of endothelial activation during pregnancy in rats. Biomed Res Int. 2013;2013:298401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palei AC, Spradley FT, Granger JP. Chronic hyperleptinemia results in the development of hypertension in pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2015;308:R855–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mise H, Sagawa N, Matsumoto T, Yura S, Nanno H, Itoh H, Mori T, Masuzaki H, Hosoda K, Ogawa Y, Nakao K. Augmented placental production of leptin in preeclampsia: Possible involvement of placental hypoxia. J Clin Endocrinol Metab. 1998;83:3225–3229 [DOI] [PubMed] [Google Scholar]
- 19.Park MJ, Lee DH, Joo BS, Lee YJ, Joo JK, An BS, Kim SC, Lee KS. Leptin, leptin receptors and hypoxia-induced factor-1alpha expression in the placental bed of patients with and without preeclampsia during pregnancy. Mol Med Rep. 2018;17:5292–5299 [DOI] [PubMed] [Google Scholar]
- 20.Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T, Nakao K. Nonadipose tissue production of leptin: Leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3:1029–1033 [DOI] [PubMed] [Google Scholar]
- 21.Hoggard N, Haggarty P, Thomas L, Lea RG. Leptin expression in placental and fetal tissues: Does leptin have a functional role? Biochem Soc Trans. 2001;29:57–63 [DOI] [PubMed] [Google Scholar]
- 22.Bajoria R, Sooranna SR, Ward BS, Chatterjee R. Prospective function of placental leptin at maternal-fetal interface. Placenta. 2002;23:103–115 [DOI] [PubMed] [Google Scholar]
- 23.Henson MC, Castracane VD. Leptin in pregnancy: An update. Biol Reprod. 2006;74:218–229 [DOI] [PubMed] [Google Scholar]
- 24.Williams MA, Havel PJ, Schwartz MW, Leisenring WM, King IB, Zingheim RW, Zebelman AM, Luthy DA. Pre-eclampsia disrupts the normal relationship between serum leptin concentrations and adiposity in pregnant women. Paediatr Perinat Epidemiol. 1999;13:190–204 [DOI] [PubMed] [Google Scholar]
- 25.Song Y, Gao J, Qu Y, Wang S, Wang X, Liu J. Serum levels of leptin, adiponectin and resistin in relation to clinical characteristics in normal pregnancy and preeclampsia. Clin Chim Acta. 2016;458:133–137 [DOI] [PubMed] [Google Scholar]
- 26.Hao S, You J, Chen L, Zhao H, Huang Y, Zheng L, Tian L, Maric I, Liu X, Li T, Bianco YK, Winn VD, Aghaeepour N, Gaudilliere B, Angst MS, Zhou X, Li YM, Mo L, Wong RJ, Shaw GM, Stevenson DK, Cohen HJ, McElhinney DB, Sylvester KG, Ling XB. Changes in pregnancy-related serum biomarkers early in gestation are associated with later development of preeclampsia. PLoS One. 2020;15:e0230000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daskalakis G, Bellos I, Nikolakea M, Pergialiotis V, Papapanagiotou A, Loutradis D. The role of serum adipokine levels in preeclampsia: A systematic review. Metabolism. 2020;106:154172. [DOI] [PubMed] [Google Scholar]
- 28.Bhattacharya S, Campbell DM, Liston WA, Bhattacharya S. Effect of body mass index on pregnancy outcomes in nulliparous women delivering singleton babies. BMC Public Health. 2007;7:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bodnar LM, Ness RB, Markovic N, Roberts JM. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann Epidemiol. 2005;15:475–482 [DOI] [PubMed] [Google Scholar]
- 30.Duckitt K, Harrington D. Risk factors for pre-eclampsia at antenatal booking: Systematic review of controlled studies. BMJ. 2005;330:565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mbah AK, Kornosky JL, Kristensen S, August EM, Alio AP, Marty PJ, Belogolovkin V, Bruder K, Salihu HM. Super-obesity and risk for early and late pre-eclampsia. BJOG. 2010;117:997–1004 [DOI] [PubMed] [Google Scholar]
- 32.Tracy TA, Miller GL. Obstetric problems of the massively obese. Obstet Gynecol. 1969;33:204–208 [PubMed] [Google Scholar]
- 33.Singh HJ, Abu Bakar A, Che Romli A, Nila A. Raised leptin concentrations in feto-placental tissues from women with preeclampsia. Hypertens Pregnancy. 2005;24:191–199 [DOI] [PubMed] [Google Scholar]
- 34.Mannaerts D, Faes E, Cornette J, Gyselaers W, Spaanderman M, Goovaerts I, Stoop T, Roelant E, Jacquemyn Y, Van Craenenbroeck EM. Low-flow mediated constriction as a marker of endothelial function in healthy pregnancy and preeclampsia: A pilot study. Pregnancy Hypertens. 2019;17:75–81 [DOI] [PubMed] [Google Scholar]
- 35.George EM, Granger JP. Linking placental ischemia and hypertension in preeclampsia: Role of endothelin 1. Hypertension. 2012;60:507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilbert JS, Nijland MJ, Knoblich P. Placental ischemia and cardiovascular dysfunction in preeclampsia and beyond: Making the connections. Expert Rev Cardiovasc Ther. 2008;6:1367–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of hypertension during preeclampsia linking placental ischemia with endothelial dysfunction. Hypertension. 2001;38:718–722 [DOI] [PubMed] [Google Scholar]
- 38.Huby AC, Otvos L Jr., Belin de Chantemele EJ. Leptin induces hypertension and endothelial dysfunction via aldosterone-dependent mechanisms in obese female mice. Hypertension. 2016;67:1020–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, Belin de Chantemele EJ. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. 2015;132:2134–2145 [DOI] [PubMed] [Google Scholar]
- 40.Faulkner JL, Kennard S, Huby AC, Antonova G, Lu Q, Jaffe IZ, Patel VS, Fulton DJR, Belin de Chantemele EJ. Progesterone predisposes females to obesity-associated leptin-mediated endothelial dysfunction via upregulating endothelial mr (mineralocorticoid receptor) expression. Hypertension. 2019;74:678–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, Jaffe IZ. Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension. 2015;66:988–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bakrania BA, Spradley FT, Drummond HA, LaMarca B, Ryan MJ, Granger JP. Preeclampsia: Linking placental ischemia with maternal endothelial and vascular dysfunction. Compr Physiol. 2020;11:1315–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor BD, Ness RB, Olsen J, Hougaard DM, Skogstrand K, Roberts JM, Haggerty CL. Serum leptin measured in early pregnancy is higher in women with preeclampsia compared with normotensive pregnant women. Hypertension. 2015;65:594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Q, Hao S, You J, Yao X, Li Z, Schilling J, Thyparambil S, Liao WL, Zhou X, Mo L, Ladella S, Davies-Balch SR, Zhao H, Fan D, Whitin JC, Cohen HJ, McElhinney DB, Wong RJ, Shaw GM, Stevenson DK, Sylvester KG, Ling XB. Early-pregnancy prediction of risk for pre-eclampsia using maternal blood leptin/ceramide ratio: Discovery and confirmation. BMJ Open. 2021;11:e050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pereira MM, Torrado J, Sosa C, Zocalo Y, Bia D. Role of arterial impairment in preeclampsia: Should the paradigm shift? Am J Physiol Heart Circ Physiol. 2021;320:H2011–H2030 [DOI] [PubMed] [Google Scholar]
- 46.Davel AP, Lu Q, Moss ME, Rao S, Anwar IJ, DuPont JJ, Jaffe IZ. Sex-specific mechanisms of resistance vessel endothelial dysfunction induced by cardiometabolic risk factors. J Am Heart Assoc. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faulkner JL, Harwood D, Kennard S, Antonova G, Clere N, Belin de Chantemele EJ. Dietary sodium restriction sex specifically impairs endothelial function via mineralocorticoid receptor-dependent reduction in no bioavailability in balb/c mice. Am J Physiol Heart Circ Physiol. 2021;320:H211–H220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faulkner JL, Harwood D, Bender L, Shrestha L, Brands MW, Morwitzer MJ, Kennard S, Antonova G, Belin de Chantemele EJ. Lack of suppression of aldosterone production leads to salt-sensitive hypertension in female but not male balb/c mice. Hypertension. 2018;72:1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Granger JP, Spradley FT, Bakrania BA. The endothelin system: A critical player in the pathophysiology of preeclampsia. Curr Hypertens Rep. 2018;20:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gohar EY, Pollock DM. Sex-specific contributions of endothelin to hypertension. Curr Hypertens Rep. 2018;20:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LaMarca BD, Alexander BT, Gilbert JS, Ryan MJ, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension in response to placental ischemia during pregnancy: A central role for endothelin? Gend Med. 2008;5 Suppl A:S133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arslan M, Yazici G, Erdem A, Erdem M, Arslan EO, Himmetoglu O. Endothelin 1 and leptin in the pathophysiology of intrauterine growth restriction. Int J Gynaecol Obstet. 2004;84:120–126 [DOI] [PubMed] [Google Scholar]
- 53.Nezar MA, el-Baky AM, Soliman OA, Abdel-Hady HA, Hammad AM, Al-Haggar MS. Endothelin-1 and leptin as markers of intrauterine growth restriction. Indian J Pediatr. 2009;76:485–488 [DOI] [PubMed] [Google Scholar]
- 54.Faulkner JL, Lluch E, Kennard S, Antonova G, Jaffe IZ, Belin de Chantemele EJ. Selective deletion of endothelial mineralocorticoid receptor protects from vascular dysfunction in sodium-restricted female mice. Biol Sex Differ. 2020;11:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meeme A, Buga GA, Mammen M, Namugowa A. Endothelial dysfunction and arterial stiffness in pre-eclampsia demonstrated by the endopat method. Cardiovasc J Afr. 2017;28:23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weissgerber TL, Milic NM, Milin-Lazovic JS, Garovic VD. Impaired flow-mediated dilation before, during, and after preeclampsia: A systematic review and meta-analysis. Hypertension. 2016;67:415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buurma AJ, Penning ME, Prins F, Schutte JM, Bruijn JA, Wilhelmus S, Rajakumar A, Bloemenkamp KW, Karumanchi SA, Baelde HJ. Preeclampsia is associated with the presence of transcriptionally active placental fragments in the maternal lung. Hypertension. 2013;62:608–613 [DOI] [PubMed] [Google Scholar]
- 58.Palei AC, Martin HL, Wilson BA, Anderson CD, Granger JP, Spradley FT. Impact of hyperleptinemia during placental ischemia-induced hypertension in pregnant rats. Am J Physiol Heart Circ Physiol. 2021;320:H1949–H1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.