

Abstract
Background:
Mineralocorticoid receptor (MR) antagonists are recommended for patients with resistant hypertension even when circulating aldosterone levels are not high. Although aldosterone activates MR to increase epithelial sodium channel (ENaC) activity, glucocorticoids also activate MR, but are metabolized by 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2). 11βHSD2 is expressed at increasing levels from DCT through CD. Here, we hypothesized that MR maintains ENaC activity in the DCT2 and early connecting tubule in the absence of aldosterone.
Methods:
We studied aldosterone synthase knockout (AS−/−) mice, which were backcrossed onto the same C57Bl6/J strain as kidney–specific MR knockout (KS-MR−/−). KS-MR−/− mice were used to compare MR expression and ENaC localization and cleavage with AS−/− mice.
Results:
MR was highly expressed along DCT2 through the CCD, whereas no 11βHSD2 expression was observed along DCT2. MR signal and apical ENaC localization were clearly reduced along both DCT2 and CCD in KS-MR−/− mice, but were fully preserved along DCT2 and were partially reduced along CCD in AS−/− mice. Apical ENaC localization and ENaC currents were fully preserved along DCT2 in AS−/− mice and were not increased along CCD after low salt (LS). AS−/− mice exhibited transient Na+-wasting under LS diet, but administration of the MR antagonist eplerenone to AS−/− mice led to hyperkalemia and decreased body weight with higher Na+ excretion, mimicking the phenotype of MR−/− mice.
Conclusions:
Our results provide evidence that MR is activated in the absence of aldosterone along DCT2 and partially CCD, suggesting glucocorticoid-binding to MR preserves sodium homeostasis along DCT2 in AS−/− mice.
Graphical Abstract

Introduction
Aldosterone is a steroid hormone secreted by the adrenal cortex in response to extracellular fluid volume depletion or hyperkalemia. It is essential for terrestrial life, in large part because it activates sodium reabsorption along the distal nephron through effects on the epithelial sodium channel, ENaC. As a steroid hormone, aldosterone binds to mineralocorticoid receptors (MR) in the cytoplasm of cells, leading them to traffic to the nucleus where they bind to genes and modulate transcription;1 MR are highly expressed along the aldosterone-sensitive distal nephron (ASDN).
Mineralocorticoid receptor antagonists (MRA) are widely used to treat cardiovascular, kidney, and liver disease, but indications for their use have recently broadened. The PATHWAY trial showed that MRA are the most effective add-on treatment in resistant hypertension,2 owing to their ability to reduce thoracic volume, emphasizing the importance of their natriuretic capacity.3 Yet, although MRA effectively treat resistant hypertension, the blood pressure response is only weakly correlated with plasma aldosterone concentration,4 and many patients with low or normal aldosterone still respond;5 thus, although screening patients with resistant hypertension for primary aldosteronism is important, treatment with MRAs is often recommended even when aldosterone concentrations are not elevated.6
This observation raises the possibility that some MR are activated by hormones other than aldosterone. A strong candidate is the glucocorticoid hormone cortisol (in rodents, corticosterone). Glucocorticoid hormones bind to glucocorticoid receptors (GR), which typically have only minor effects on salt transport.7,8 As the affinity of MR for glucocorticoid hormones is similar to that of mineralocorticoid hormones, most MR are typically activated by glucocorticoids, as glucocorticoids typically circulate at 100–1000-fold higher levels than mineralocorticoid hormones. Canonical aldosterone-target cells therefore express 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2), an enzyme that metabolizes glucocorticoid hormones, and protects MR from binding to them.9
The ASDN comprises a portion of the distal convoluted tubule (DCT2), the connecting tubule (CNT), and the collecting duct (CD). ENaC is expressed throughout this segment, but Korbmacher and colleagues showed that activity along the DCT2/CNT is independent on aldosterone, whereas it is aldosterone-dependent along the CD.10–12 This differential activation profile correlates inversely with 11βHSD2, which is minimally expressed along the DCT2 but increases along the CNT and CD,10,13–15 suggesting that aldosterone-independent MR activity may be mediated by glucocorticoid binding to MR. Here, we tested whether MR activity in the absence of aldosterone maintains ENaC activity in the kidney, and whether this is responsible for phenotypic differences between mice lacking aldosterone and mice lacking MR. Finally, we tested whether aldosterone-independent effects of MRA might be responsible for natriuresis when aldosterone levels are not elevated. The results suggest a unique signaling mechanism in which two hormones converge on a single receptor to achieve salt balance despite wide variation in salt consumption.
Concise Methods
All data and supporting materials have been provided with the published article
(Additional methods are in supplemental content)
Animals
Animal studies were approved by the Oregon Health and Science University Institutional Animal Care and Use Committee (protocol IP00286) and New York Medical College independent animal use committee. AS wild-type (AS+/+) and AS-deficient (AS−/−) mice were generously provided from Dr. J.M. Luther at Vanderbilt University.
Animal diets and measurement of food consumption
For baseline analysis, animals were maintained on standard chow for these studies (5L0D diet: 1.18% K+, 0.4% Na+, and 0.67% Cl−). For Na+ deprivation studies, normal salt (0.8% K+ and 0.49% NaCl) and Na+-deficient diet (0.8% K+ and 0.03% Na+) were prepared by adding NaCl and KCl to a Na+ deficient diet (TestDiet AIN-93G Semi-Purified Diet, 57W5 with no added sodium chloride (0.36% K+, 0.03% Na+)). A gel diet was prepared from 37.9% powder diet, 60.6% water, and 1.5% agar. Food intake was determined by tracking the dry weight of food present in cages of individually housed mice. Eplerenone (#2397, Tocris Bioscience, Bristol, UK) was mixed into the gel diet at a dose of 100 mg/kg of body weight, assuming a daily food intake of 5g).
Antibodies
Antibody sources, species, dilutions, and validation references are provided in Supplemental Table S1. The α-ENaC antibody was generously provided by Johannes Loffing.
Patch-Clamp Experiments along DCT2 and CCD
Patch-Clamp was performed as described.12 Mice were perfused with 2 mL L-15 medium (Life Technology, Grand Island, NY) containing type 2 collagenase (250 unit/mL). The renal cortex was separated and cut into small pieces for additional incubation in collagenase-containing L-15 media for 30–50 min at 37°C. The isolated tubules were placed on a small cover glass coated with poly-lysine and the cover glass was placed on a chamber mounted on an inverted microscope. ENaC currents were measured at DCT2, defined as the last 100 μm of the DCT before CNT attachment, and CCD. Data were analyzed using the pClamp software system 9.0 (Axon).
Results
MR expression is high along DCT2, CNT and CCD
RNA sequencing data from Lee, et al. showed that Nr3c2 (encoding MR) is expressed along distal nephron, and its expression along CNT/CCD is much higher than along DCT (Figure S2),16 but MR expression levels along DCT2, in which ENaC current is higher than in CCD,10–12 have not been as clear. To address this, immunofluorescence was performed on kidneys from control and kidney-specific KS-MR−/− mice. In control mice, nuclear MR expression was relatively low along DCT1 (Parvalbumin-positive and NCC-positive tubules), but was increased toward DCT2 (Parvalbumin-negative and NCC-positive tubules) (Figures S3A and S3B). Along CNT (weakly aquaporin −2-positive tubules) and CCD (strongly aquaporin-2-positive tubules), nuclear MR expression was higher in principal cells (PCs) than in intercalated cells (aquaporin-2-negative cells) (Figures S3C and S3D), but signal decreased toward OMCD (aquaporin-2-positive tubules in outer medulla) (Figures S3E and S3F). MR expression was weakly detected along cortical thick ascending limb (NKCC2-positive tubules), and even less along medullary thick ascending limb (NKCC2-positive tubules in outer medullar) (Figures S3G–J). Nuclear MR signal intensity along all segments was clearly reduced in KS-MR−/− mice (Figures 1A and S3). These data suggest MR expression is high along DCT2 and in PCs of CNT/CCD.
Figure 1. MR regulates apical ENaC localization along DCT2 and CCD.
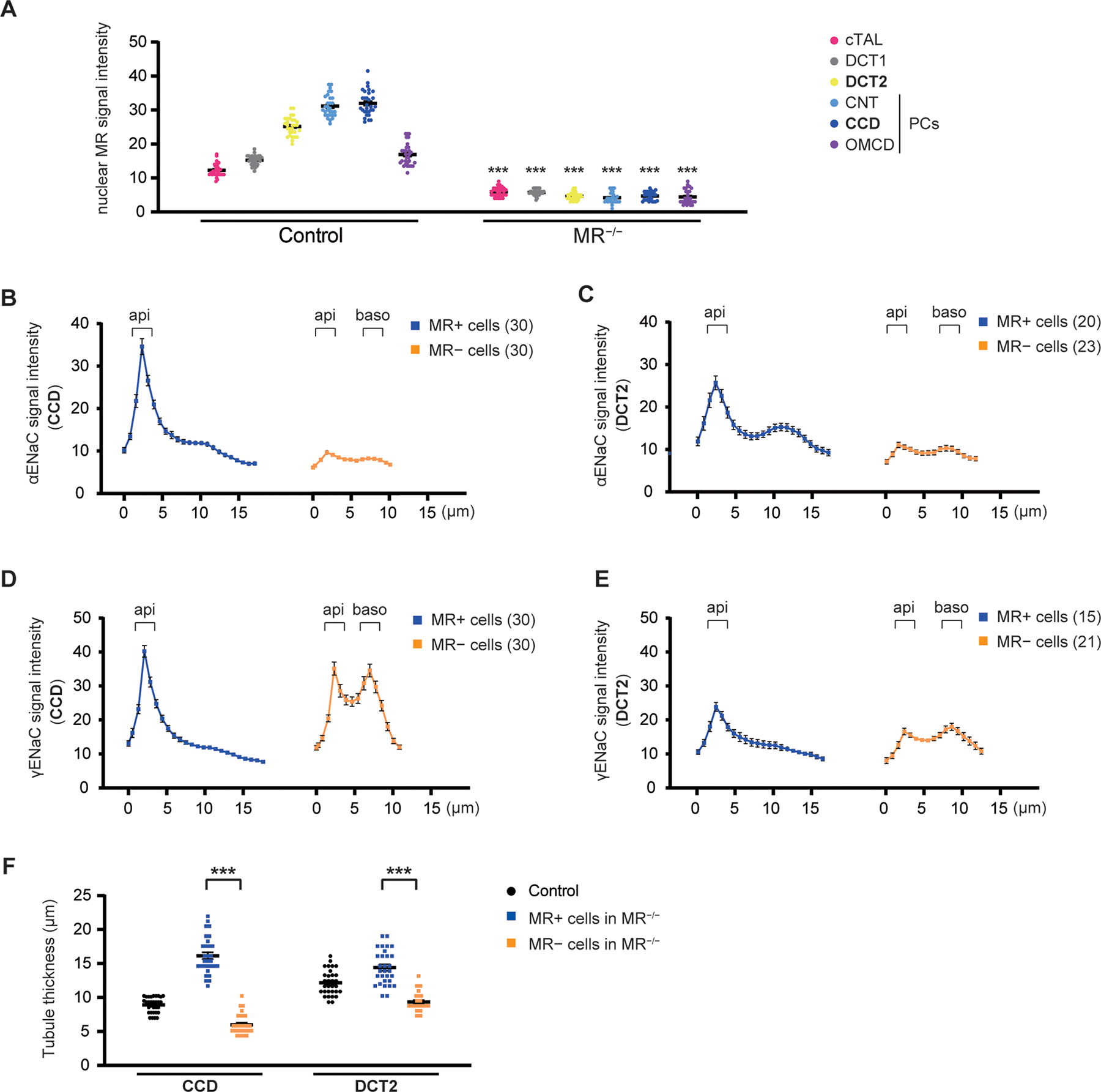
A, Nuclear MR signal intensity along the distal nephron. Analysis of raw data from Figure S3. MR is highly expressed along DCT2, CNT, and CCD in control mice (left). Nuclear MR signal intensities along these segments were nearly absent along the majority of cells in KS-MR−/− mice (right), although a few cells retained nuclear MR (Figure S6). B–E, Quantitative image analysis of α- and γ-ENaC signal intensity in MR-positive cells and MR-negative cells of KS-MR−/− mice (from Figure S7 1B–1E), measured from the lumen to the basolateral side at 0.78 μm-increment resolution. Along CCD and DCT2, MR-positive cells had strong α- and γ-ENaC apical signal (dark blue line), and both α- and γ-ENaC were exclusively cytoplasmic in MR-negative cells with extremely weak α-ENaC signal (orange line). F, Tubule thickness along CCD and DCT2 in control and KS-MR−/− mice. MR-positive cells had increased thickness compared with MR-negative cells in these segments. Data represent individual values and mean ± SEM. Statistical differences were examined by two-tailed unpaired t-tests (A and F). ***, P < 0.001.
MR regulates apical ENaC localization along DCT2 and CCD
Apical localization of α- and γ-ENaC is critical for ENaC activity. To determine their localization along DCT2, immunofluorescence was performed on serial sections in control mice on a normal salt diet. αENaC signal was mainly apical while γ-ENaC was cytoplasmic along CCD (Figures S4A–C). In contrast, α- and γ-ENaC were clearly apical along DCT2 (NCC-positive and strongly MR-positive tubules) (Figure S4D–F). Mean aldosterone levels in control mice were 318.4 pg/mL (Figure S5A), suggesting high aldosterone levels are not required for apical γ-ENaC localization along DCT2 in contrast to CCD. We previously showed that cleavage and apical localization of α- and γ-ENaC were severely reduced in KS-MR−/− mice, leading to high aldosterone with salt-wasting.17 Nuclear MR expression in KS-MR−/− mice was not detectable along the majority of distal tubules (Figure S6), but was still detected along a few tubules (Figures S6A and S6B). Thus, to investigate whether MR regulates apical ENaC localization along DCT2 and CCD, immunofluorescence staining was performed using MR-positive cells and MR-negative cells in KS-MR−/− mice. Note that, in the KS-MR−/− mice, plasma aldosterone concentrations were much higher in these mice than in controls (Figure S5). Along CCD, MR-positive cells had strong α- and γ-ENaC apical signal, but both α- and γ-ENaC were exclusively cytoplasmic in MR-negative cells with extremely weak αENaC signal (Figures 1B and D and Figure S7, 1B & 1D). Similar results were observed along DCT2, but α- and γ-ENaC signal intensities were weaker compared with CCD (Figures 1C & 1E and Figure S7 1C & 1E). Compared with control mice showing cytoplasmic γ-ENaC localization along CCD (Figure S4C), aldosterone was much higher in KS-MR−/− mice (owing to salt-wasting, 2576 pg/mL (mean), Figure S5A), suggesting that high aldosterone is required for apical γ-ENaC localization along CCD. Furthermore, MR-positive cells had increased thickness compared with MR-negative cells (Figure 1F). Together, these data suggest that MR regulates ENaC expression and apical localization along both DCT2 and CCD.
Aldosterone-independent MR activation regulates apical ENaC localization along DCT2 and partially CCD
To clarify whether MR is activated independently of aldosterone binding, we used AS+/+ and AS−/− mice, backcrossed onto the C57Bl6/J strain.18 Consistent with a previous study, plasma aldosterone levels were almost undetectable and plasma corticosterone levels trended lower, in AS−/− mice (Figures S5A and S5B). Compared with AS+/+ mice, nuclear MR signal intensity was partially reduced along CCD (Figure 2A and S7, Panel 2A) but was preserved along DCT2 (Figures 2B and S7, Panel 2B). Consistent with preserved MR activation, apical αENaC signal was only partially reduced along CCD (Figures 2C and S7, Panel 2C) and fully preserved along DCT2 in AS−/− mice (Figures 2D and S7, Panel 2D). Along CCD, γ-ENaC was not exclusively apical, but partially localized to the cytoplasm in both AS+/+ and AS−/− mice (Figures 2E and S7, Panel 2E). Similarly, apical γ-ENaC localization was preserved along DCT2 in AS−/− mice (Figures 2F and S7, Panel 2F). This suggests aldosterone-independent MR activation regulates ENaC activity along DCT2, and partially CCD. Western blotting showed that cleaved α- and γENaC abundances were reduced in AS−/− mice (Figures S8A–D), but the reductions were less than in KS MR−/− mice (Figures S9A–D). Compared with AS+/+ mice, AS−/− mice had significantly higher plasma [K+] (Figure S10A). NCC phosphorylation decreases as plasma [K+] rises;19,20 consistent with higher plasma [K+], abundance of phosphorylated NCC (pNCC) and the ratio of pNCC-T53 to total NCC were also lower in AS−/− mice (Figures S8E–H). Plasma [Na+], plasma [Cl−], TCO2, blood urea nitrogen (BUN), hemoglobin, urine volume, urinary Na+ excretion, urinary K+ excretion, and body weight were similar to those of AS+/+ mice (Figures S10B–J), suggesting AS−/− mice have a milder phenotype than KS-MR−/− animals.17,21 These results suggest aldosterone-independent activation of MR in AS−/− mice activates α- and γENaC along DCT2 and partially along CCD, preventing salt-wasting. Interestingly, although MR and its canonical regulated genes Nr3c3, Scnn1a, and Tsc22d3 were lower in both AS−/− and KS-MR−/− mice, Sgk1 was lower only in the KS-MR−/− mice (Figure S11).
Figure 2. Nuclear MR expression and apical ENaC localization are regulated along DCT2 and partially CCD in an aldosterone-independent manner.
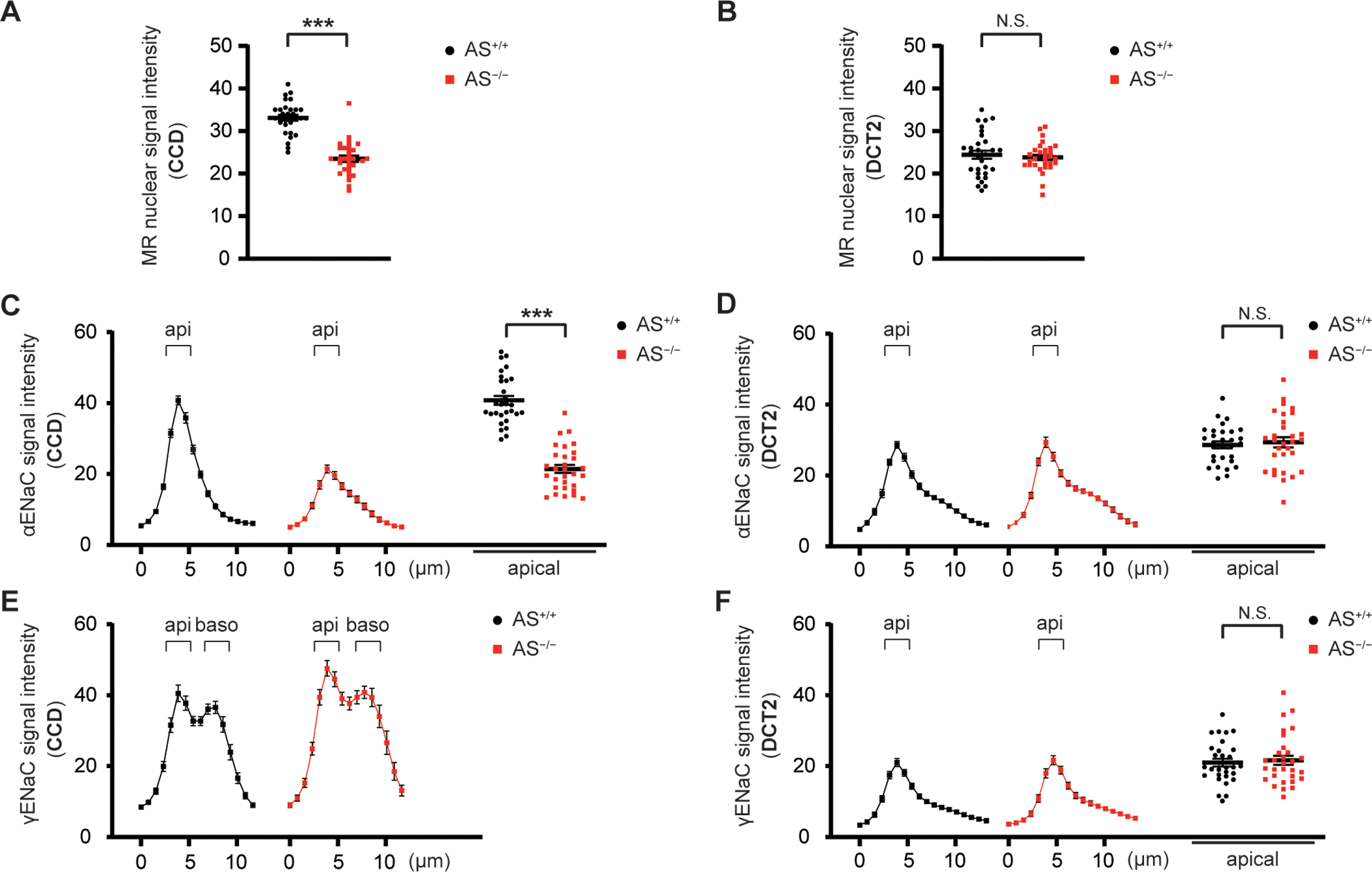
A and B, Nuclear MR signal intensity in CCD and DCT of AS+/+ and AS−/− mice. Analysis of raw data from Figure S7 2A–2B. MR signal intensity was partially reduced along CCD but was preserved along DCT2 in AS−/− mice. C–F, Quantitative image analysis of α- and γ-ENaC signal intensity in CCD and DCT of AS+/+ and AS−/− mice (from Figure S7 2B–2E), which was measured from the lumen to the basolateral side at 0.78 μm-increment resolution. Differences in apical signal intensity were analyzed only when apical localization could be discriminated (C, D, and F). α-ENaC signal was only partially reduced along CCD and fully preserved along DCT2 in AS−/− mice. Apical γ-ENaC localization was preserved along DCT2 in AS−/− mice, and γ-ENaC was localized to cytoplasmic in both AS+/+ and AS−/− mice. Data represent individual values and mean ± SEM. Statistical differences were examined by two-tailed unpaired t-tests (A–D and F). ***, P < 0.001; N.S., P > 0.05.
Dietary salt restriction leads to transient Na+-wasting in AS−/− mice
We next tested the hypothesis that a low salt diet exacerbates the phenotype of AS−/− mice, due to a lack of increased aldosterone-independent MR activation along CNT/CCD under this condition. Mice were treated with a low salt (LS) diet for 5 days to determine Na+ and K+ balance. There was significant weight loss on Day 1 in AS−/− mice compared with AS+/+ mice, but weight loss was similar between AS−/− mice and AS+/+ mice on Day 5 (Figure 3A). AS−/− mice had transiently elevated Na+ excretion and negative Na+ balance (Na+ intake – urinary Na+ excretion) on Day 1 compared with AS+/+ mice (Figures 3B, 3C, S12A and S12B). Although the interaction between genotype and time was statistically significant for urinary K+ excretion, analysis of K+ balance (K+ intake – urinary K+ excretion) indicated that AS−/− mice could maintain adequate K+ excretion on the LS diet (Figures 3D). Plasma [K+] tended higher on normal salt (NS) diet and was significantly higher on LS diet in AS−/− mice. Plasma [K+] rose significantly on LS diet in AS−/− but not in WT (Figure 3E). Consistent with the differences in plasma [K+], pNCC abundances (and the ratio of pNCC/NCC) were significantly lower in AS−/− mice than in AS+/+ mice when they were consuming a low salt diet (Figure S13). This is similar to the results of KS-MR−/− mice showing consumption of an Na+-deplete diet further increases plasma K+ and reduces pNCC abundance.17 LS had no effect on plasma [Na+] levels in AS−/− or AS+/+ mice (Figure 3F). AS−/− mice had significantly higher BUN levels on LS than on NS, whereas AS+/+ mice did not (Figure S12C). Plasma [Cl2−] and TCO2 tended toward being higher and lower respectively in both genotypes, whereas hemoglobin was higher in both genotypes compared with their baseline (Figures S12D–F). AS−/− mice thus displayed mild salt-wasting on a LS diet.
Figure 3. Dietary salt restriction leads to transient Na+-wasting in AS−/− mice.

AS+/+ and AS−/− mice were acclimated to normal salt diet (NS: 0.49% NaCl and 0.8% K+) then treated with low salt diet (LS: 0.03% Na+ and 0.8% K+) for 5 days. A, Body weight was not significantly changed in AS−/− mice fed LS diet compared with AS+/+ mice. B and C, Na+ excretion was higher on LS Day 1 in AS−/− mice but was similar to AS+/+ mice on LS Day 5. D, K+ balance (K+ intake – urinary K+ excretion) was similar between AS−/− mice and AS+/+ mice. E, Plasma potassium trended higher on NS diet and was significantly higher on LS diet in AS−/− mice compared with AS+/+ mice. F, Plasma sodium levels did not change in AS−/− or AS+/+ mice. Data represent individual values and mean ± SEM. Statistical differences were examined by two-tailed unpaired t-tests (C and D), two-way repeated measures ANOVA (A and B), or two-way ANOVA, followed by post hoc Tukey tests (E and F). **, P < 0.01; ***, P < 0.001; N.S., P > 0.05.
Na+ depletion does not stimulate apical ENaC localization or activity along DCT2 and CCD in AS−/− mice
To determine whether consumption of LS diet differentially affected apical ENaC localization along DCT2 and CCD, immunofluorescence was performed on serial kidney sections from AS+/+ mice and AS−/− mice. In AS+/+ mice fed LS diet with high plasma aldosterone levels (752.4 pg/mL (mean), Figure S5A), apical α-ENaC expression was higher along CCD than at baseline (Figures S14 and S15A and S15B), and γ-ENaC was mainly localized to the apical membrane along CCD (Figures S15C and S15D). In contrast, apical α-ENaC expression was not higher, and γ-ENaC remained localized to the cytoplasm along CCD in AS−/− mice fed LS diet (Figures S15A–D). However, apical α- and γ-ENaC localization was not changed along DCT2 in both AS+/+ mice and AS−/− mice fed LS diet (Figures S15E–H). Western blotting showed cleaved αENaC expression and the ratio of cleaved to uncleaved γ-ENaC was significantly higher in AS+/+ mice fed LS diet compared with NS diet, but not in AS−/− mice (Figures 4A–D). We previously showed that ENaC currents were higher along DCT2 than CCD, but ENaC currents along CCD were increased after LS diet (Figure 4E, left, redrawn from Wu et al.12). In the current study, patch-clamping showed ENaC currents were preserved along DCT2 in AS−/− mice, and were not higher along CCD in AS−/− mice after LS diet (Figure 4E, right). These results indicate that during Na+-depletion, the aldosterone-independent MR activation mechanism cannot activate ENaC.
Figure 4. Na+ depletion does not stimulate ENaC cleavage and its activity in AS−/− mice.

A–D, Western blot of ENaC when mice were switched from normal salt diet (NS: 0.49% NaCl and 0.8% K+) to low salt diet (LS: 0.03% Na+ and 0.8% K+), and quantification of results in A. Cleaved and total αENaC expression, and the ratio of cleaved to uncleaved γENaC, were all significantly higher in AS+/+ mice fed LS diet compared with NS diet, but not in AS−/− mice. White arrow head indicates non-specific band. E, ENaC currents measured at −60 mV along DCT2 and CCD in AS+/+ and AS−/− mice. Right, ENaC currents were preserved along DCT2 in AS−/− mice, and were not higher along CCD in AS−/− mice after LS diet. Left, ENaC currents were increased along CCD in WT mice fed LS diet. The bar graphs are redrawn from Wu et al.12 and are included for comparison with the normal response. Data represent individual values and mean ± SEM. Statistical differences were examined by one-way ANOVA (E) or two-way ANOVA, followed by post hoc Tukey tests (B–D). *, P < 0.01; **, P < 0.01; ***, P < 0.001; N.S., P > 0.05.
MR antagonist eplerenone exacerbates the phenotype of AS−/− mice fed LS diet
Consistent with a previous report,14 NCC and 11βHSD2 did not co-localize in either AS+/+ mice or AS−/− mice (Figure S16), suggesting that along DCT2 MR is activated by a steroid other than aldosterone. To determine whether aldosterone-independent MR activation plays an important role during Na+ deprivation, we treated AS−/− mice with the MR antagonist eplerenone. We first optimized eplerenone administration in C57Bl/6J mice. Eplerenone mildly increased urinary Na+ excretion and Na+/K+ ratio in C57Bl/6J mice on a normal salt diet (Figure S18), and to a greater degree on a LS diet (Figure S19). In AS−/− mice on a LS diet, urinary Na+ excretion and urinary Na+/K+ ratio were higher in mice treated with eplerenone, and plasma [Na+] was lower (Figures 5A–C). These mice also displayed lower body weight, negative Na+ balance, and higher BUN (Figure 5F and S19C–E), consistent with volume contraction as a result of enhanced Na+-wasting. Urinary K+ excretion was lower and plasma [K+] was higher (Figure 5E and 5F), consistent with impaired K+ secretion along CNT/CCD. Similar to KS-MR−/− mice,17 plasma [Cl−] and TCO2 trended to higher or lower respectively in the AS−/− mice treated that received eplerenone (Figures S19F and S19G). These data suggest that eplerenone treatment of AS−/− mice on a LS diet leads to a phenotype mimicking the phenotype of KS-MR−/− mice. Finally, to investigate whether eplerenone inhibits α- and γENaC cleavage, C57Bl/6J mice and AS−/− mice were treated with short-term eplerenone. Consistent with this, AS−/− mice treated with eplerenone for 8 hours had lower cleaved α- and γ-ENaC abundances, although the effect of LS diet was blunted in the AS−/− mice (Figures S20 and S21). Together, these data suggest that on a LS diet, eplerenone prevents aldosterone-independent activation of ENaC via MR in AS−/− mice, leading to salt-wasting.
Figure 5. Effect of eplerenone on urine Na+ excretion and Na+/K+ ratio in AS−/− mice fed LS diet.

AS−/− mice assigned to control group (AS−/−, LS) fed LS diet and eplerenone group (AS−/−, LS+Epl) treated with 100 mg/kg/day of eplerenone, as shown in detail in Figure S17A. A and B, Urine Na+ excretion (A) and urine Na+/K+ ratio (B), was significantly increased after eplerenone treatment. C and D, Plasma sodium was decreased and BUN was increased in eplerenone-treated AS−/− mice. E and F, Urine K+ excretion was significantly decreased and plasma potassium was increased after eplerenone treatment. Data represent individual values and mean ± SEM. Statistical differences were examined by two-tailed unpaired t-tests (C, D, and F) or two-way repeated measures ANOVA (A, B, and E). *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, P > 0.05.
Discussion
MRAs reduce cardiovascular mortality in patients with heart failure22,23 and chronic kidney disease,24,25 and are the most effective add-on treatment for resistant hypertension.3 It suggested that most of MRA’s beneficial effects result from the blockade of aldosterone actions on the MR, but several large studies show that blood pressure lowering efficacy does not correlate with pre-treatment aldosterone levels.3,5 Coupled with the knowledge that glucocorticoid hormones can also activate MR, this suggests that aldosterone-independent mineralocorticoid receptor activation contributes importantly to sodium homeostasis and human health.
The canonical renal target for aldosterone is the ASDN, where the effects derive predominantly from activating ENaC. Yet, ENaC activity along the DCT2 and early CNT is largely independent of circulating aldosterone,11 even though it is reduced in mice lacking MR.10,12 One possible mechanism for aldosterone-independent MR activation is through Rac1, which causes paradoxical activation in salt-sensitive models.26 Yet, the C57/BL6 mice we tested here are relatively salt-resistant, making this an unlikely explanation.
Here, we tested the hypothesis that MR are stimulated by hormones other than aldosterone by comparing the phenotypes of AS−/− and KS-MR−/− mice. AS−/− mice have had somewhat variable phenotypes, when evaluated in different laboratories,11,18,27–31 so we examined the phenotype on the same background that we used to assess the effects of MR deletion. The results show that AS−/− mice maintain normal sodium balance when they consume a control diet and adapt to salt restriction without progressive weight loss or hyperkalemia. In contrast, mice lacking MR display hyponatremia and salt-wasting even when consuming a control diet, and show progressive weight loss and hyperkalemia when dietary salt is restricted.17,21 The data also show that both ENaC and NCC are more severely reduced in the KS-MR−/− mice than in AS−/− mice; this likely reflects direct (ENaC) and indirect (NCC) effects, as we and others have shown that both ENaC and NCC must be reduced for the full salt-wasting phenotype.17,21,32 Together with results showing that amiloride-sensitive sodium currents along the DCT2/CNT are similar in control and AS−/− mice (Figure 4 and10), the results suggest an essential homeostatic role for non-aldosterone MR activation in the kidney. If this is true, then mice lacking aldosterone should still respond to MRA with natriuresis.
The results of experiments using eplerenone, a highly selective mineralocorticoid antagonist, support this hypothesis. in AS−/− mice, eplerenone caused natriuresis and increased the ratio of urinary sodium to potassium. As might be expected if a portion of MR are active in the absence of aldosterone, blocking MR in mice on a low salt intake led to substantial hyperkalemia, with evidence of extracellular fluid volume depletion, and a rise in BUN. In this way, pharmacological blockade of MR in AS−/− mice made them resemble KS-MR−/− mice.17,21
The current results confirm that MR are expressed all along the distal nephron, and that their abundance rises in more distal segments.15,16,33 Surprisingly, the work also confirms that MR are localized to nuclei along all these segments, even in the absence of aldosterone.15 Loffing and colleagues showed that nuclear MR localization in the absence of aldosterone depended on glucocorticoid hormones; they speculated that corticosterone degradation by 11βHSD2 was incomplete even in the CCD. In support of this, we found differences between nuclear MR intensity along the CCD of control versus AS−/− mice. Nevertheless, the current data indicate that factors other than nuclear trafficking of MR are required for aldosterone to stimulate ENaC, as ENaC activity along the CCD is highly dependent on aldosterone concentration (see Figure 4 and10–12). In contrast, no difference in nuclear trafficking between control and AS−/− mice was noted along the DCT2/CNT, which exhibits aldosterone-independent ENaC activity.
These data also clarify the nature of MR effects on ENaC. αENaC abundance is markedly reduced along both DCT2/CNT and CCD in cells lacking MR, an expected finding as MR regulate αENaC transcription;34 additionally, clear MR trophic effects are evident in the greater thickness of cells that express MR (note that all cells are exposed to the same extracellular environment, Figure 1F). Deletion of aldosterone itself, in contrast, causes a more modest decrease in αENaC along the CCD and has no apparent effect along the DCT2/CNT. The absence of aldosterone effect along DCT2/CNT is compatible with the absence of physiological effect in this segment. In contrast, along the CCD, ENaC currents are essentially absent in AS−/− mice, and yet apical staining persists. Our data are consistent with findings from neonatal AS−/− mice suggesting that corticosterone-MR signaling may be enough to support basal ENaC expression but not to support ENaC adaptation.35
These observations have substantial implications for the use of MRA clinically. The dissociation between sites of 11βHSD2 and MR expression has long been confusing. The present work suggests that there is sufficient corticosterone within cells all along the nephron to maintain MR predominantly in nuclei, but for MR along the DCT2/CNT this is sufficient to activate ENaC constitutively. One possibility is that corticosterone permits formation of an activating transcriptional complex in DCT2/CNT but not in CCD due to segment-specific differences in transcriptional regulators. Structural effects that depend on whether aldosterone or corticosterone is bound to MR might determine MR-mediated transcriptional activation along CCD.
MRAs inhibit ENaC along all segments of the ASDN. Yet, this inhibition may occur even when aldosterone concentrations are not elevated, because MRA also block glucocorticoid-induced MR stimulation. It is not surprising that direct ENaC inhibition with amiloride will have similar efficacy,3,4 as the stimulation in both situations involves ENaC. Reviews of resistant hypertension commonly invoke ‘excessive aldosterone’ as an explanation for the beneficial effects of these drugs;36 yet in many cases, it may be blockade of cortisol binding to MR, rather than aldosterone binding to MR, that is responsible for the salutary effects. It has long been recognized that MR are expressed in tissues, such as the heart and vasculature, which express little or no 11βHSD2. In these tissues, cortisol (or corticosterone) are believed to be the predominant ligands.37 The current work suggests that a similar phenomenon exists along the ASDN, where both aldosterone-dependent (regulatory) and aldosterone-independent (homeostatic) MR activation occurs.
Perspectives
MRAs are commonly used to reduce blood pressure and treat cardiovascular and kidney disease. In the kidney, aldosterone is typically assumed to be the agonist for MRs, which act along the ASDN to stimulate sodium current through ENaC. Yet, ENaC activity along a substantial portion of this segment is maintained even when aldosterone is absent. Here, we show that deleting aldosterone and deleting MR have very different effects on ENaC and sodium excretion. Sodium transport is highly dependent on both aldosterone and MR along CCD, with decreased α-ENaC abundance, decreased γ-ENaC apical localization, and decreased ENaC activity when either the hormone or the receptor is absent. In contrast, α-ENaC abundance and ENaC activity are preserved along the DCT2 when aldosterone is absent, but reduced when MRs are deleted. As might be expected based on these differences, blocking MR causes both natriuresis and potassium retention in mice lacking aldosterone. The results indicate that, like in other tissues, substantial MR effects are typically mediated by hormones other than aldosterone. This likely explains the potent blood pressure lowering effects of MR, even when circulating aldosterone levels are not elevated.
Supplementary Material
Figure 6. Aldosterone-independent MR activation regulates ENaC activity along DCT2 and partially CCD, contributing to sodium reabsorption.

MR is highly expressed along both DCT2 and CCD, but 11βHSD2 is not expressed along DCT2. Left, In a state of aldosterone (Aldo) excess, α- and γ-ENaC is localized to apical along DCT2 and CCD. Right, In KS-MR−/− mice, apical α- and γ-ENaC localization are severely reduced along both DCT2 and CCD. Middle, In AS−/− mice, nuclear MR expression and apical ENaC localization is preserved along DCT2 and is not fully reduced along CCD highly expressing 11βHSD2. This aldosterone-independent MR signaling along DCT2 and CCD, possibly regulated by corticosterone or cortisone (Cort), can explain mild their phenotype, and prevents from continuous salt wasting under LS diet.
Novelty and Relevance.
What is New? - drugs that block mineralocorticoid receptors are believed to act by blocking aldosterone. Yet, the current work shows that these drugs increase sodium excretion and decrease potassium excretion even when aldosterone is absent, indicating that many kidney mineralocorticoid receptors are normally bound by hormones other than aldosterone.
What Is Relevant? - Drugs that block mineralocorticoid receptors are first-line treatments for resistant hypertension. Yet, large studies indicate that high circulating aldosterone levels don’t correlate with their effects.
Clinical/Pathophysiological Implications - The current work supports using mineralocorticoid receptor antagonists, even when aldosterone is not elevated.
Sources of Funding
Y.M. received a postdoctoral award from the Uehara Foundation; J.A.M. is funded by NIDDK DK098141. DHE is funded by NIDDK DK054983 and Department of Veterans Affairs I01BX002228. DE and OS were funded by a Fondation LeDucq: Transatlantic Network of Excellence 17CVD05.
Non Standard Abbreviations
- 11βHSD2
11β-hydroxysteroid dehydrogenase
- ENaC
epithelial sodium channel
- AS
aldosterone synthase
- ASDN
aldosterone-sensitive distal nephron
- CCD
cortical collecting duct
- CNT
connecting tubule
- DCT
distal convoluted tubule
- GR
glucocorticoid receptor
- KS
kidney specific
- MR
mineralocorticoid receptor
- MRA
mineralocorticoid receptor antagonist
- NCC
thiazide-sensitive sodium chloride cotransporter
- NKCC2
bumetanide-sensitive sodium potassium chloride cotransporter
- OMCD
outer medullary collecting duct
- PC
principal cell
- pNCC
phosphorylated (activated) NCC
Footnotes
Disclosures
The authors have no conflicts of interest to declare.
References
- 1.Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev. 2015;95:297–340. doi: 10.1152/physrev.00011.2014 [DOI] [PubMed] [Google Scholar]
- 2.Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, Ford I, Cruickshank JK, Caulfield MJ, Salsbury J, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068. doi: 10.1016/S0140-6736(15)00257-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams B, MacDonald TM, Morant SV, Webb DJ, Sever P, McInnes GT, Ford I, Cruickshank JK, Caulfield MJ, Padmanabhan S, et al. Endocrine and haemodynamic changes in resistant hypertension, and blood pressure responses to spironolactone or amiloride: the PATHWAY-2 mechanisms substudies. Lancet Diabetes Endocrinol. 2018;6:464–475. doi: 10.1016/S2213-8587(18)30071-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams B, MacDonald TM, Morant SV, Brown MJ. PATHWAY-2: spironolactone for resistant hypertension - Authors’ reply. Lancet. 2016;387:1373–1374. doi: 10.1016/S0140-6736(16)00698-X [DOI] [PubMed] [Google Scholar]
- 5.Parthasarathy HK, Alhashmi K, McMahon AD, Struthers AD, McInnes GT, Ford I, Connell JM, MacDonald TM. Does the ratio of serum aldosterone to plasma renin activity predict the efficacy of diuretics in hypertension? Results of RENALDO. J Hypertens. 2010;28:170–177. doi: 10.1097/HJH.0b013e328332b79b [DOI] [PubMed] [Google Scholar]
- 6.Carey RM, Calhoun DA, Bakris GL, Brook RD, Daugherty SL, Dennison-Himmelfarb CR, Egan BM, Flack JM, Gidding SS, Judd E, et al. Resistant Hypertension: Detection, Evaluation, and Management: A Scientific Statement From the American Heart Association. Hypertension. 2018;72:e53–e90. doi: 10.1161/HYP.0000000000000084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivy JR, Jones NK, Costello HM, Mansley MK, Peltz TS, Flatman PW, Bailey MA. Glucocorticoid receptor activation stimulates the sodium-chloride cotransporter and influences the diurnal rhythm of its phosphorylation. American journal of physiology. 2019;317:F1536–F1548. doi: 10.1152/ajprenal.00372.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canonica J, Frateschi S, Boscardin E, Ebering A, Sergi C, Jager Y, Peyrollaz T, Merillat AM, Maillard M, Klusonova P, et al. Lack of Renal Tubular Glucocorticoid Receptor Decreases the Thiazide-Sensitive Na(+)/Cl(−) Cotransporter NCC and Transiently Affects Sodium Handling. Front Physiol. 2019;10:989. doi: 10.3389/fphys.2019.00989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, Horisberger JD, Rossier BC. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol. 2005;16:878–891. doi: 10.1681/ASN.2004121110 [DOI] [PubMed] [Google Scholar]
- 10.Nesterov V, Bertog M, Canonica J, Hummler E, Coleman R, Welling PA, Korbmacher C. Critical role of the mineralocorticoid receptor in aldosterone-dependent and aldosterone-independent regulation of ENaC in the distal nephron. American journal of physiology. 2021;321:F257–F268. doi: 10.1152/ajprenal.00139.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. American journal of physiology. 2012;303:F1289–1299. doi: 10.1152/ajprenal.00247.2012 [DOI] [PubMed] [Google Scholar]
- 12.Wu P, Gao ZX, Zhang DD, Duan XP, Terker AS, Lin DH, Ellison DH, Wang WH. Effect of Angiotensin II on ENaC in the Distal Convoluted Tubule and in the Cortical Collecting Duct of Mineralocorticoid Receptor Deficient Mice. J Am Heart Assoc. 2020;9:e014996. doi: 10.1161/JAHA.119.014996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bostanjoglo M, Reeves WB, Reilly RF, Velazquez H, Robertson N, Litwack G, Morsing P, Dorup J, Bachmann S, Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol. 1998;9:1347–1358. [DOI] [PubMed] [Google Scholar]
- 14.Hunter RW, Ivy JR, Flatman PW, Kenyon CJ, Craigie E, Mullins LJ, Bailey MA, Mullins JJ. Hypertrophy in the Distal Convoluted Tubule of an 11beta-Hydroxysteroid Dehydrogenase Type 2 Knockout Model. J Am Soc Nephrol. 2015;26:1537–1548. doi: 10.1681/ASN.2013060634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. American journal of physiology Renal physiology. 2010;299:F1473–1485. doi: 10.1152/ajprenal.00437.2010 [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Chou CL, Knepper MA. A Comprehensive Map of mRNAs and Their Isoforms across All 14 Renal Tubule Segments of Mouse. J Am Soc Nephrol. 2021. doi: 10.1681/ASN.2020101406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL, Ellison DH. Direct and Indirect Mineralocorticoid Effects Determine Distal Salt Transport. J Am Soc Nephrol. 2016;27:2436–2445. doi: 10.1681/ASN.2015070815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luther JM, Luo P, Wang Z, Cohen SE, Kim HS, Fogo AB, Brown NJ. Aldosterone deficiency and mineralocorticoid receptor antagonism prevent angiotensin II-induced cardiac, renal, and vascular injury. Kidney international. 2012;82:643–651. doi: 10.1038/ki.2012.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney international. 2016;89:127–134. doi: 10.1038/ki.2015.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell metabolism. 2015;21:39–50. doi: 10.1016/j.cmet.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canonica J, Sergi C, Maillard M, Klusonova P, Odermatt A, Koesters R, Loffing-Cueni D, Loffing J, Rossier B, Frateschi S, et al. Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch. 2016;468:895–908. doi: 10.1007/s00424-015-1785-2 [DOI] [PubMed] [Google Scholar]
- 22.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 23.Pitt B, Williams G, Remme W, Martinez F, Lopez-Sendon J, Zannad F, Neaton J, Roniker B, Hurley S, Burns D, et al. The EPHESUS trial: eplerenone in patients with heart failure due to systolic dysfunction complicating acute myocardial infarction. Eplerenone Post-AMI Heart Failure Efficacy and Survival Study. Cardiovasc Drugs Ther. 2001;15:79–87. [DOI] [PubMed] [Google Scholar]
- 24.Bakris GL, Agarwal R, Filippatos G, Group F-DS. Finerenone and Chronic Kidney Disease Outcomes in Type 2 Diabetes. Reply. N Engl J Med. 2021;384:e42. doi: 10.1056/NEJMc2036175 [DOI] [PubMed] [Google Scholar]
- 25.Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, Kolkhof P, Nowack C, Schloemer P, Joseph A, et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N Engl J Med. 2020;383:2219–2229. doi: 10.1056/NEJMoa2025845 [DOI] [PubMed] [Google Scholar]
- 26.Ayuzawa N, Fujita T. The Mineralocorticoid Receptor in Salt-Sensitive Hypertension and Renal Injury. J Am Soc Nephrol. 2021;32:279–289. doi: 10.1681/ASN.2020071041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makhanova N, Lee G, Takahashi N, Sequeira Lopez ML, Gomez RA, Kim HS, Smithies O. Kidney function in mice lacking aldosterone. American journal of physiology. 2006;290:F61–69. doi: 10.1152/ajprenal.00257.2005 [DOI] [PubMed] [Google Scholar]
- 28.Makhanova N, Sequeira-Lopez ML, Gomez RA, Kim HS, Smithies O. Disturbed homeostasis in sodium-restricted mice heterozygous and homozygous for aldosterone synthase gene disruption. Hypertension. 2006;48:1151–1159. doi: 10.1161/01.HYP.0000249902.09036.e7 [DOI] [PubMed] [Google Scholar]
- 29.Lea WB, Kwak ES, Luther JM, Fowler SM, Wang Z, Ma J, Fogo AB, Brown NJ. Aldosterone antagonism or synthase inhibition reduces end-organ damage induced by treatment with angiotensin and high salt. Kidney international. 2009;75:936–944. doi: 10.1038/ki.2009.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luther JM, Wang Z, Ma J, Makhanova N, Kim HS, Brown NJ. Endogenous aldosterone contributes to acute angiotensin II-stimulated plasminogen activator inhibitor-1 and preproendothelin-1 expression in heart but not aorta. Endocrinology. 2009;150:2229–2236. doi: 10.1210/en.2008-1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, Makhanova N, Korbmacher C, Wagner CA, Loffing J. Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol. 2015;26:425–438. doi: 10.1681/ASN.2013111156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perrier R, Boscardin E, Malsure S, Sergi C, Maillard MP, Loffing J, Loffing-Cueni D, Sorensen MV, Koesters R, Rossier BC, et al. Severe Salt-Losing Syndrome and Hyperkalemia Induced by Adult Nephron-Specific Knockout of the Epithelial Sodium Channel alpha-Subunit. J Am Soc Nephrol. 2015. doi: 10.1681/ASN.2015020154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ransick A, Lindstrom NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, Kim AD, Black HG, Kim J, McMahon AP. Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev Cell. 2019;51:399–413 e397. doi: 10.1016/j.devcel.2019.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stokes JB, Sigmund RD. Regulation of rENaC mRNA by dietary NaCl and steroids: organ, tissue, and steroid heterogeneity. The American journal of physiology. 1998. Jun;274:C1699–1707. [DOI] [PubMed] [Google Scholar]
- 35.Martinerie L, Viengchareun S, Meduri G, Kim HS, Luther JM, Lombes M. Aldosterone postnatally, but not at birth, is required for optimal induction of renal mineralocorticoid receptor expression and sodium reabsorption. Endocrinology. 2011;152:2483–2491. doi: 10.1210/en.2010-1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acelajado MC, Hughes ZH, Oparil S, Calhoun DA. Treatment of Resistant and Refractory Hypertension. Circ Res. 2019;124:1061–1070. doi: 10.1161/CIRCRESAHA.118.312156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young MJ, Clyne CD. Mineralocorticoid receptor actions in cardiovascular development and disease. Essays Biochem. 2021;65:901–911. doi: 10.1042/EBC20210006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.