

Carcinogen-induced rat mammary tumors are shown to recapitulate many aspects of human breast cancer, including the immune environment. Tumor growth is associated with immune escape, which can be reversed by immunotherapy, highlighting the potential utility of this model.
Abstract
Animal models are critical for the preclinical validation of cancer immunotherapies. Unfortunately, mouse breast cancer models do not faithfully reproduce the molecular subtypes and immune environment of the human disease. In particular, there are no good murine models of estrogen receptor–positive (ER+) breast cancer, the predominant subtype in patients. Here, we show that Nitroso-N-methylurea–induced mammary tumors in outbred Sprague-Dawley rats recapitulate the heterogeneity for mutational profiles, ER expression, and immune evasive mechanisms observed in human breast cancer. We demonstrate the utility of this model for preclinical studies by dissecting mechanisms of response to immunotherapy using combination TGFBR inhibition and PD-L1 blockade. Short-term treatment of early-stage tumors induced durable responses. Gene expression profiling and spatial mapping classified tumors as inflammatory and noninflammatory, and identified IFNγ, T-cell receptor (TCR), and B-cell receptor (BCR) signaling, CD74/MHC II, and epithelium-interacting CD8+ T cells as markers of response, whereas the complement system, M2 macrophage phenotype, and translation in mitochondria were associated with resistance. We found that the expression of CD74 correlated with leukocyte fraction and TCR diversity in human breast cancer. We identified a subset of rat ER+ tumors marked by expression of antigen-processing genes that had an active immune environment and responded to treatment. A gene signature characteristic of these tumors predicted disease-free survival in patients with ER+ Luminal A breast cancer and overall survival in patients with metastatic breast cancer receiving anti–PD-L1 therapy. We demonstrate the usefulness of this preclinical model for immunotherapy and suggest examination to expand immunotherapy to a subset of patients with ER+ disease.
See related Spotlight by Roussos Torres, p. 672
Introduction
Breast tumors progress through defined stages starting with atypical ductal hyperplasia then ductal carcinoma in situ (DCIS), invasive ductal carcinoma (IDC), and culminating in metastatic disease (1). We previously reported that immunosuppression progressively increases during breast tumor progression and identified the DCIS–IDC transition as a key step of immune escape (2, 3). Multiple mechanisms contribute to establishing an immunosuppressive environment, including amplification and overexpression of CD274, which encodes PD-L1, in a subset of triple-negative breast cancer (TNBC) and an increased frequency of regulatory T cells and myeloid-derived suppressor cells (MDSC) in IDC, potentially due to increased TGFβ signaling (2, 3).
Immune checkpoint inhibitors (ICI) have shown long-lasting responses in many metastatic cancers. In breast cancer, however, the efficacy of single-agent ICIs has been limited (4). Combining ICIs with chemotherapies and application at earlier stages leads to better results, predominantly in TNBC. Lower intratumor heterogeneity and more coordinated antitumor immune responses have been observed in early-stage cancers (4).
Given the stark immunologic differences between DCIS and IDC (3), immune escape could drive the eventual progression of breast tumors. To test this hypothesis, we chose the Nitroso-N-methylurea (NMU)-induced outbred Sprague-Dawley (SD) rat model of breast cancer, which is the only preclinical immunocompetent model of DCIS and hormone-dependent ER+ mammary tumors (5). In this model, the histology of early-stage mammary tumors resembles human DCIS, progesterone and estrogen signaling drives high incidence of hormone-dependent mammary tumors, and the genetic variation of the outbred strain mimics interpatient variability (5). Mammary tumors are induced by intraperitoneal injection of NMU in virgin female rats at peripubertal age, because age, reproductive status, and estrous cycle impact tumor incidence (6).
Here, we have characterized the cellular composition and molecular profiles of NMU-induced mammary tumors in SD rats, showing their remarkable similarity to human breast cancers, and we have demonstrated that combined PD-L1 and TGFBR inhibition induces effective regression of these tumors and prevents immune escape. Response to immunotherapy varied according to hormone receptor status, which recapitulates clinical data. However, in contrast to clinical observations, a subset of ER+ tumors responded to immunotherapy in this model, enabling us to identify a gene signature that was predictive of response. This gene signature was found to be prognostic in patients with ER+ breast cancer and classified tumors into immunologically distinct subgroups.
Materials and Methods
Rat experiments and tissue harvesting
All animals were housed in Dana-Faber Cancer Institute (DFCI, Boston, MA) Longwood Center Association for Assessment and Accreditation of Laboratory Animal Care International–approved animal facility. All animal experiments were performed following protocol #15-050 approved by the DFCI Institutional Animal Care and Use Committee. Virgin SD (SD:Hsd) female rats ages 3 to 4 weeks were purchased from Envigo. To induce tumors, rats were injected 1–3 times with 50 mg/kg NMU (BOC Sciences, 684-93-5) intraperitoneally at ages 35, 49, or 53 days. For immunotherapy studies, we performed one injection of 35-day-old rats. Treatment with LY2157299 (TGFBR inhibitor), anti–rat PD-L1 or both was initiated when palpable tumors were detected. Rats were randomly assigned to treatment groups. Tumor size was tracked by weekly caliper measurements. Researchers measuring tumors were blinded as to the animals’ treatment groups. LY2157299 powder was provided by Eli Lilly and stored at −20°C. LY2157299 was dissolved in vehicle (1% carboxymethylcellulose, 0.5% sodium lauryl sulfate, 0.085% povidone) and stored at 4°C for up to 1 week. Oral gavage of vehicle or 75 mg/kg of LY2157299 was performed twice daily (8–16 hours between doses) for 10 consecutive days. Anti–rat PD-L1 (clone 368.A.4H1, mouse IgG1, kappa) was produced by the Freeman lab by immunization of cd274, PDCD1LG2-deficient mice with human PD-L1-Fc fusion protein using a previously reported protocol (7, 8). It contained <2 endotoxin units/mg protein. Clone 368.A.4H1 (RRID:AB_2910261; ref. 9) was shown to have high affinity for human and rat PD-L1 by flow cytometry of 300.19 cells transfected with human or rat PD-L1 and to block PD-1 binding. Anti–PD-L1 or IgG1 isotype control (BioXcell, BE0083) were given at 10 mg/kg weekly for 6 consecutive weeks by intraperitoneal injection. Tumors were harvested and processed for paraffin embedding or tissue digestion as described previously (3). Briefly, a slice of the largest cross-section was saved for histologic analysis, fixed in 10% formalin overnight, stored in 70% ethanol, then paraffin embedded and processed into histologic slides (3). The remaining tumor tissue was dissociated to a single-cell suspension for FACS by digestion in 2 mg/mL collagenase type IV (Worthington, LS004189) in DMEM/F12 (Thermo Fisher Scientific, MT10090CV) with constant stirring at 37°C for 1 to 2 hours.
Tumor growth rates
Tumor growth rates were defined as “diametrical growth rate” computed by linear regression of all collected diameter points over time. As rats were sacrificed at different times, this parameter stays relatively constant throughout the growth of the tumor. The top two thirds of samples were considered “growing,” whereas the bottom third was considered as “slow growing or stable” with a growth rate of ≤ 2 mm/week, allowing for errors in measurement of tumor size.
Slide staining and analysis
Multicolor immunofluorescence, immunohistochemistry (IHC), trichrome, and hematoxylin–eosin (H&E) analyses were performed as described previously (3). Briefly, after heat-induced antigen retrieval in sodium citrate (pH = 6) or TRIS-EDTA buffer (pH = 9), the samples were permeabilized with 0.5% TritonX-100, blocked with 100% goat serum (Thermo Fisher Scientific, 16210072) and stained with various antibodies at the indicated dilutions. Supplementary Table S1 includes information on antigen retrieval conditions, antibody identity, and antibody dilution. Stained slides were imaged using the Panoramic MIDI II digital slide scanner (3DHistech), the Yokogawa Spinning disk confocal, or the Nikon Eclipse Ti2 microscope and quantified using QuPath (https://qupath.github.io/). For other staining where no quantifications were shown, four or more tumors per group were stained and visually inspected to confirm protein expression of hits from RNA sequencing (RNA-seq) analysis.
Rat organoid culture and growth assays
Rat mammary tumor and autologous healthy kidney tissues were minced into 1 to 2 mm fragments then digested with 1 mg/mL collagenase/dispase (Sigma, 11097113001) for 30 minutes. Digestion was stopped by adding equal volume of 1% BSA in DMEM, then centrifuged at 1,500 rpm × 5 minutes. Pellets were further digested with Accutase (Sigma, A6964) for 30 minutes then collected by centrifugation at 1,500 rpm for 5 minutes. Pellets were then resuspended in organoid growth medium containing Y-27632, 5% matrigel, and growth factors including B27 (Thermo Fisher Scientific, 1750404), insulin (Sigma, 12643), cholera toxin (Sigma, C8052), FGF (Peprotech, AF-100-18B), EGF (Peprotech, AF-100-15), and IL6 (Peprotech, 200-06) The suspension was seeded onto 12-well plates precoated with matrigel. Culture media was replaced every 4 days. For fulvestrant experiments, organoids were plated at 5,000 to 10,000 single cells per well on top of a layer of matrigel in 96-well plates. On day 6, organoids were treated with fulvestrant (MedChem Express, HY-13636; 100 and 750 nmol/L) or DMSO vehicle control. After 72 hours of treatment, CellTiter-Glo 3D reagent (Promega, G9682) was added to each well and luminescence signal was detected after 30 minutes using a Synergy H1 microplate reader.
Generation of rat organoid-primed T cells
Rat T cells were isolated from spleen by EasySep Rat T-Cell Isolation Kit (Stemcell Technologies, 19641). The cells were cultured with rat complete T-cell medium (RTM): Serum-free medium (CellGernix, 20801-0100), 10% FBS (Glico, 16140071), and Rat IL2 (1,000 IU/mL; Peprotech, 212-12), Rat IL15 (10 ng/mL; Peprotech, 210-15) and Rat IL21 (10 ng/mL; Peprotech, 210-21). On day 10, T cells were stimulated with autologous tumor organoids without matrigel. This was repeated 7 days later. Stimulation was performed in RTM, generating organoid-primed T cells (opT). The T cell:Tumor cell ratios ranged from 1:1 to 10:1.
Three-dimensional killing assay
Rat mammary tumor or autologous kidney was digested into single cells as described above (see Rat organoid culture and growth assays) and plated onto matrigel-coated 96-well flat bottom plates. A total of 10,000 cells were plated per well in 100 μL of organoid medium containing 5% matrigel. Tumor cells were allowed to form three-dimensional (3D) structures for 4 days and 100 μL per well of autologous opT cells in RTM were gently added to the top of the organoid culture at a cell ratio of 1:1, with final media containing 50:50 ratio of organoid media and RTM. The 1:1 T cell:tumor cells ratio was estimated by counting tumor cell number in parallel wells cultured for 4 days. No additional factors were added to the coculture. Cells were incubated at 37°C for 1 to 4 days. Phase-contrast images were acquired using a 4× objective (Leica). Supernatants were harvested after 1 and 2 days for analysis of IFNγ production as described below.
OpT killing after PD-L1 antibody blockade and/or TGFβ inhibition
Day 8 organoid cultures were digested with collagenase/dispase keeping the 3D structure of the organoids intact. The organoids were resuspended in RTM and plated in 96-well flat bottom plates with autologous opT cells at a ratio of 1:1 in the presence of the isotype control (InVivoMab mouse IgG1, clone MOPC-21, from BioXcell), anti–PD-L1 (final 10 μg/mL; 368A.4H1), LY2157299 (final 1μmol/L), or the combination of anti–PD-L1 mAb plus LY2157299. Phase-contrast images were acquired using a 4× objective (Leica). Supernatants were harvested after 1 and 2 days for analysis of IFNγ production as described below.
IFNγ production
The supernatants from rat opT and tumor cocultures were collected and measured for IFNγ by ELISA as per the manufacturer's instructions: rat IFNγ ELISA kit (Mabtech, 3220-1H-6).
Flow cytometry and FACS
Single-cell suspensions of dissociated tumors were blocked in PBE (PBS with 0.5% BSA and 2 mmol/L EDTA) and stained at 4°C for 30 minutes with antibodies (Supplementary Table S1). Cells were analyzed and sorted using BD FACSAria II SORP UV (Becton Dickinson). FlowJo v9 (Becton Dickinson) was used for analysis and graphing.
RNA-seq
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, 74106) and measured by Agilent 2100 Bioanalyzer. RNA-seq libraries were prepared using Clontech Low Input mRNA Library (Clontech SMARTer) v4 kit (Clontech, 634892) using <10 ng of purified total RNA according to the manufacturer's protocol. Library concentrations were measured by Qubit Fluorometer and Kapa Biosystems library quantification kit (Kapa Biosystems, 07960298001), and library fragment size by Agilent TapeStation 2200. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75 bp reads, or Illumina NovaSeq6000 with paired-end 100 bp reads by the DFCI Molecular Biology Core Facilities. RNA-seq datasets were aligned to the rat reference genome (rn6) and annotated with the UCSC rn6 refSeq annotation file using STAR2.7.0f.
Whole-genome sequencing
EpCAM+ cells from tumors in the characterization cohort were isolated by FACS and DNA was purified (Qiagen, 80284) prior to being sent to the New York Genome Centre for whole-genome sequencing (WGS). DNA from the liver of an untreated rat served as a normal control. Libraries were prepared using the Illumina TruSeq PCR-free DNA HT sample preparation kit (Illumina, 20015963). A total of 1 μg of intact genomic DNA was sheared using the Covaris sonicator (Adaptive Focused Acoustics), followed by end-repair and bead-based size selection (450 bp). Adenines were added to the 3′ ends of fragments, followed by Illumina sequence adaptor ligation (Illumina, 20022370), PCR amplification and final library QC, where library fragment size was measured by Agilent BioAnalyzer (Agilent, DNF-474-0500), total DNA concentration by PicoGreen (Life Technologies, P7589) and yield and efficiency of adaptor ligation with a quantitative PCR assay (Roche, KK4873). These steps were performed on the Caliper SciClone NGSx workstation (PerkinElmer), a robotics system developed and validated for automated library preparation. 2×150 bp sequencing was performed on the Illumina HiSeqXTen instrument to a depth of 30×. WGS data were aligned to the rn6 genome using bwa-mem.
Patient cohorts
We utilized the following external datasets:
The Cancer Genome Atlas (10) from which clinical and gene expression data were retrieved from firebrowse (http://firebrowse.org/) and immune-related properties including immune composition and TCR diversity retrieved from ref. 11. All primary tumor samples with available RNA-seq data were used (N = 1,080), comprising of 410 Luminal A, 172 Luminal B, 64 Her2+, 136 basal-like, 25 normal-like tumors, and 273 uncharacterized tumors.
Pembrolizumab trial in ER+ metastatic breast cancer cohort (12, 13) features a clinical trial of 88 patients treated with eribulin with or without the addition of pembrolizumab. We assessed a cohort of 30 patients (N = 16 vehicle, N = 14 treatment) for which RNA-seq data, clinical information, and plasma proteomics data were available.
Survival analysis, differential gene expression analysis, single-sample gene set enrichment analysis (GSEA), and association studies were performed on both cohorts in R using methods described below (see Genomic and imaging data analyses).
Genomic and imaging data analyses
Code for downstream analysis and figure generation can be obtained at https://github.com/polyak-lab/RatDCISbookdown.
Differential gene expression analysis
Raw counts obtained from STAR/HTSeq were processed using the DESeq2 package (https://bioconductor.org/packages/release/bioc/html/DESeq2.html). Genes were filtered to retain only those with at least 60 counts across all samples. Samples used for downstream analysis passed the following criteria: library size of 4M+ reads, 11K+ genes with nonzero reads, library read SD of 500+, and clustering by PCA within the same cell type. This left 108 samples: 31 EpCAM+, 46 CD45+, and 31 double negative (DN) cases across 40 rats. Differentially expressed genes (DEGs) were identified with an adjusted P value of ≤0.05, absolute fold change ≥ 1.5, and base mean number of counts of 80, using a model that considered batch effects. Gene expression heat maps were generated using row-scaled variance-stabilized transformed gene counts, using differentially expressed genes (limited to the top 50). For treatment-based comparisons, significant DEGs for each treatment compared with the control were combined to construct heat maps. The “luminal-growing” signature refers to the 50 DEGs identified by comparing EpCAM+ profiles from growing versus noninflammatory stable HRhigh tumors.
GSEA
GSEA was performed using log2 fold changes of all genes using the hallmark compendium from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/collection_details.jsp) and Metacore Process Network through the HTSAnalyzeR2 package (https://github.com/CityUHK-CompBio/HTSanalyzeR2). The top 40 significant pathways with a FDR < 0.1 were used to create enrichment network diagrams. The width of lines between two gene sets indicates the number of genes in common, and the size of the node indicates the number of genes.
Inferring immune composition
Gene names were converted to corresponding mouse gene IDs and TPM counts were used in TIMER cistrome (http://timer.cistrome.org/), which provides immune cell abundance estimates from tools including CIBERSORT and TIMER.
Mutation and copy-number analysis
Consensus mutation calling of WGS data was performed using MuTect1 (https://github.com/broadinstitute/mutect) and Strelka (https://github.com/Illumina/strelka) by the New York Genome Centre. Gene annotation was performed using SnpEff (http://pcingola.github.io/SnpEff/) using the UCSC rn6 refGene annotation file. Tumor mutational burden was calculated as the number of coding mutations (missense, nonsense, nonstop, frameshift insertion/deletion, splice site) per MB of exome (43.2 MB). The corresponding amino acid in human homologs was obtained by aligning a peptide containing the mutation site up to 11 amino acids long to the respective human protein sequence, allowing up to two mismatches. The corresponding human site was considered a hotspot mutation if found within a collated list of hotspot mutations in human cancer (14). Mappability files for the rn6 genome were prepared using GEM3 for 75 bp kmers, and copy number was estimated using BICSeq (ref. 15; smoothness parameter λ = 2), using a control liver as a normal input. Gains and losses were called using log2(fold change) of ±0.3.
Pathway Mapper (https://github.com/iVis-at-Bilkent/pathway-mapper) was used to overlay mutational and copy-number frequencies on key cancer pathways.
Single-base substitution mutational signatures (SBSv2: https://cancer.sanger.ac.uk/signatures/signatures_v2/) were inferred using SigProfilerSingleSamples using the trinucleotide context for each single-nucleotide variant mutation (https://github.com/AlexandrovLab/SigProfilerSingleSample).
Following marking duplicates, detecting split reads and base recalibration using GATK (https://gatk.broadinstitute.org), mutations from RNA-seq data were inferred using GATK HaplotypeCaller and annotated using SnpEff.
Whole-slide image analysis
Computational image analysis was performed firstly by detecting and segmenting cells using Qupath (https://qupath.github.io/), using an estimated cell radius of 8.0 μm, area between 10 and 100 μm2, intensity threshold of 2.0. Extracted shape, intensity, and haralick features were used to train a random forest classifier to differentiate between CD8+, EpCAM+, SMA+, EpCAM+SMA+, and triple-negative cells. SMA+EpCAM+ cells were present in only a few tissue samples and could not be unambiguously classified into one group or the other by manual inspection. Classifications were reviewed by an expert observer and cell classifications and co-ordinates were exported for downstream analysis. Spatial pattern analysis was assessed using k-nearest neighbor analysis, which computes the average distance between a cell type x to its k = 3 nearest neighbors of class y. In addition, the Morisita–Horn index for spatial overlap between two cell types of interest (16) was used as a metric for global cooccurrence which accounts for population frequency and can be interpreted as a correlation coefficient. We divided the tissue region of interest into S squares of size 100 μm × 100 μm and evaluated the frequency of each cellular population in each square. The Morisita-Horn index for two cell types x and y are calculated as:
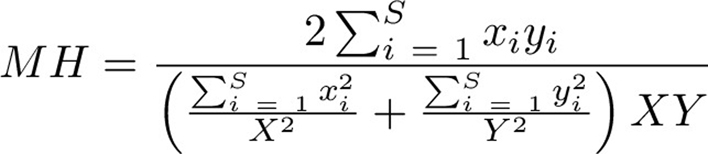 |
where X and Y are the total counts of the populations of interest, and xi and yi refers to the frequency of these cells in square Si.
BCR and TCR repertoire analysis
BCR and TCR repertoire was inferred from bulk RNA-seq data using TRUST4. For BCR repertoire, the reference VJC database was built using the reference genome (rn6 UCSC) and known IG sequences from IMGT (http://www.imgt.org/; ref. 17). Given the lack of references for rat TCR repertoire, the VJC database was built using the mouse reference genome (GRCm38 UCSC) and known TR sequences from IMGT. All clones with less than two reads or that had a CDR3 sequence with less than six amino acids were excluded from analysis. Clonality was computed using the Gini index, and diversity was computed using the Shannon index using the tcR package (https://github.com/imminfo/tcr).
Survival analysis
Vst-transformed values of single genes (ADAMST10, CD74) or gene signature scores for the luminal signature, OncotypeDX (18) and Mammaprint (19) signatures computed using single-sample GSEA (20) were used as inputs for survival analysis following normalization. Survival outcome was modeled using multivariate Cox proportional regression models accounting for PAM50 and stage where applicable. Overall and disease-free survival (DFS) was censored to 60 months. Outcome was reported as hazard ratio (HR) with 95% confidence intervals (CI). Model performance was assessed using log-rank P value. Outcome was plotted using the Kaplan–Meier method.
Statistical tests
Nonparametric Mann–Whitney Wilcoxon test (MWW) was used to compare differences in two distributions due to small sample size and possible deviation from normality, using a significance threshold of 0.05. P values were adjusted using the Benjamini–Hochberg method to correct for multiple hypothesis testing, and false discovery rate (FDR) <0.1 was used to designate significance.
Data and materials availability
WGS data have been deposited in SRA (SUB6887244) and RNA-seq data in NCBI Gene Expression Omnibus (GSE167102). All additional data associated with this study are available within the article and its Supplementary Data files or are available on request from the corresponding author.
Results
NMU-induced mammary tumors resemble human breast cancer
We first optimized NMU treatment by testing rats at varying ages (35–53 days) and after administration of a varying number of injections (1–3 injections). A total of 100% of rats developed mammary tumors after a single injection of NMU at ≤49 days of age; both increased number of NMU injections and younger age (35 days) increased tumor penetrance (more tumors per rat) and reduced latency (Supplementary Fig. S1A and S1B). No macroscopically visible tumors were observed in any other organs. We followed mammary tumor growth for 60 to 200 days after NMU injection, sacrificing animals at different timepoints to obtain tumors of different sizes (1–35 mm) to reflect different stages of progression (Supplementary Fig. S1C). Most tumors were well-differentiated solid adenocarcinomas (55/59) with a small subset of fibroadenomas (2/59) and mucinous carcinomas (2/59; Fig. 1A; Supplementary Fig. S1D; Supplementary Table S2). All tumors resembled intraductal lesions surrounded by collagen-rich stroma and a cell layer expressing p63 and smooth muscle actin (SMA) myoepithelial markers (Fig. 1B–D). We detected proliferative Ki67+ cells by immunofluorescence in both luminal and myoepithelial cells in all tumors (Fig. 1D), suggesting that these tumors may represent human adenomyoepitheliomas (21). All tumors expressed progesterone receptor (PR) and estrogen receptor (ER) based on immunofluorescence (Fig. 1E), consistent with the known ovarian hormone dependence of this model (5), which we confirmed by fulvestrant treatment of primary tumor organoid cultures (Supplementary Fig. S1E).
Figure 1.

Histology and molecular features of NMU-induced mammary tumors. Virgin SD female mice were injected intraperitoneally with NMU and tumors harvested at various times after NMU administration herein referred as the characterization cohort. A–E, Paraffin-embedded tumor sections were analyzed by H&E (A), trichrome (B), IHC analysis of p63 (C), immunofluorescence for Ki67 and SMA (D), and immunofluorescence for PR and ER (E). Shown are representative images of well-differentiated adenocarcinomas, which were 55 of 59 tumors in the cohort. Scale bar, 100 μm. F–I, WGS of EpCAM+ tumor epithelial cells harvested from 31 NMU-induced tumors from 23 unique rats. Liver from tumor-free animal was used as control. F, Tumor mutational burden and types of mutations. G, Mutations found in breast cancer–driver genes. H, Frequency of mutations in selected breast cancer–driver genes. I, Population frequency of genetic aberrations in key breast cancer pathways. J–L, FACS sorted EpCAM+ tumor epithelial cells from NMU-induced tumors harvested from 10 tumors from 8 unique rats were analyzed by RNA-seq. Red and blue indicates HRlow and HRhigh tumors, respectively. Green and orange marks small and large tumors, respectively. J, Heatmap of gene expression Z-scores depicting the RNA expression of hormone receptors and selected luminal transcription factors. K, Heatmap of row-normalized Z-scores of genes differentially expressed between large (>7 mm) and small (≤7 mm) tumors. L, Pathways enriched in large and small tumors. Red lines indicate pathways highlighted in text. Node size indicates the number of genes within the pathway, line width indicates the number of shared genes between two gene sets. All tests for significance used Mann–Whitney–Wilcoxon test using a threshold of P = 0.05 unless otherwise specified; error bars representative of SD; box-whisker plots indicate 0th, 25th, 50th, 75th, and 100th percentiles.
Molecular characteristics of NMU-induced tumors
NMU is an alkylating agent that causes AT:GC mutations (22). However, the specific mutations in NMU-induced mammary tumors have not been comprehensively characterized. Thus, we performed WGS on sorted EpCAM+ tumor epithelial cells (Supplementary Fig. S1F) and normal liver from one animal as germline control. Most coding mutations were missense, and tumor mutational burden ranged from 7.6/Mb to 19.5/Mb per tumor (Fig. 1F). This range is higher than most human breast cancers (∼0.1–10/Mb; Fig. 1F, red box) but comparable with human cancers with high (∼1–100/Mb) mutational burden such as melanoma (23). The mutational burden within coding regions did not correlate with the number of NMU doses nor with tumor size (Supplementary Fig. S1G). Most mutations were C>T and T>C transitions reflecting aging and alkylation, respectively. The most frequent COSMIC mutational signature was SBS5, which is a clock-like signature of an unknown etiology associated with tobacco smoking (24), but SBS11 (alkylation) and SBS32 (azathioprine) treatment signatures were also observed (Supplementary Fig. S1H). These results suggest that most mutations were acquired at the time of NMU administration.
We detected mutations in known breast cancer–driver genes (e.g., Pik3ca, Foxa1, and Tp53; Fig. 1G and H; Supplementary Table S3). Hras, Braf, Pik3ca, and Tp53 variants occurred at mutational hotspots commonly found in human tumors (Supplementary Fig. S1I and S1J; Supplementary Table S3) including HrasG12E, which is characteristic of this model (25). In addition, we assessed copy-number variations but found very few changes in regions harboring cancer driver genes (Supplementary Fig. S1K). We also explored whether these mutations converged on common breast cancer pathways. A total of 87% of tumors had genes in the p53 pathway impacted with most having an Atm mutation; this also impacted cell-cycle pathways, and mutations in Brca2 were observed in 10% of the cohort. In addition, 16% of tumors had a mutation in the PIK3 signaling pathway (Fig. 1I) and NOTCH signaling was affected in 74% of tumors (Supplementary Fig. S1L).
Next, we performed RNA-seq of CD45+ leukocyte and EpCAM+ epithelial cells to characterize gene expression profiles. We observed two distinct tumor profiles in the EpCAM+ cell population based on the expression of hormone receptors (Ar, Pgr, and Esr1) and luminal breast cancer–specific transcription factors (Foxa1 and Gata3), recapitulating the “luminal” hormone receptor high (HRhigh) and “basal” hormone receptor low (HRlow) phenotypes in human breast cancer (Fig. 1J). Hierarchical clustering of CD45+ leukocyte gene expression profiles segregated the tumors into two groups (small and large; Supplementary Fig. S1M) implying the need for substantial immune-related changes for tumors to grow beyond a certain size. This threshold of 7 mm was also the median size of measured tumors. Gene expression profiles of leukocytes from smaller tumors (<7 mm) were highly similar to each other and to the normal mammary gland, whereas leukocytes from each larger tumors (≥7 mm) had profiles different from any other tumor (Supplementary Fig. S1M). In contrast, neither hierarchical clustering nor principal component analysis (PCA) could separate the EpCAM+ cells based on tumor size (Supplementary Fig. S1N and S1O). HR status was also not associated with tumor size (χ2 test, P = 0.5).
DEG analysis and GSEA of the EpCAM+ cell population showed that small tumors were enriched for genes associated with proliferation, differentiation, protein folding, and transcription (Fig. 1K and L; Supplementary Table S4). Small tumors were also enriched for immune-response and inflammation-related gene sets including IL5 and IL2 signaling, inflammasome, and unfolded protein response. In large tumors, we found greater interactions with the microenvironment, evidenced by GSEA terms related to cell adhesion, matrix interactions, angiogenesis, and blood coagulation (Fig. 1L).
Similarity of NMU-induced mammary tumor and human breast cancer immune environment
We next investigated the immune environment of NMU-induced mammary tumors and found them to be immune-infiltrated with a mean proportion of CD45+ cells of 37.06% by flow cytometry (Supplementary Fig. S2A). We analyzed the cellular composition of the immune infiltrates using a flow cytometry marker panel (26) adapted for rat (Supplementary Fig. S2B and S2C), and found both innate and adaptive immune cells, including monocytes and T cells in the tumors (Fig. 2A). We also collected unaffected mammary glands, mammary gland–draining lymph nodes, spleen, and bone marrow from tumor-free (no NMU) and tumor-bearing animals. We found no significant differences between tumor-free and tumor-bearing animals in these tissues, except for a decrease of bone marrow T cells (MWW P = 0.006), and an increase in mammary gland monocytes (P = 0.03) in tumor-bearing animals (Fig. 2A). In addition, a decrease in B cells (P = 0.01) was observed in tumors compared with the mammary gland of tumor-free animals. We also assessed the activation status of cytotoxic immune cells [natural killer (NK) and CD8+ T cells] in the tumors by immunofluorescence. Most NK cells expressed granzyme B (Fig. 2B) and a fraction of CD8+ T cells expressed CD44 and/or Ki67, indicating activation (Fig. 2C). Overall, these results demonstrate the recruitment and activation of both innate and adaptive immune cells in NMU-induced mammary tumors.
Figure 2.

The immune microenvironment of NMU-induced mammary tumors. A, Leukocyte composition of the indicated tissues from tumor-bearing (TB) or tumor-free (TF) animals from the characterization cohort (see Fig. 1) determined by polychromatic flow cytometry. LN, mammary gland-draining lymph node. BM, bone marrow. MG, mammary gland. *, P < 0.05 on Mann–Whitney–Wilcoxon test. B, Immunofluorescence analysis for CD244 (NK cells) and granzyme b (GZMB). C, Immunofluorescence analysis for CD8 (cytotoxic T cells), CD44, and Ki67. Yellow arrows, Ki68+CD44+CD8+ cells. D, Immunofluorescence analysis of CD3 T-cell and SMA myoepithelial markers in normal, DCIS-like, and IDC-like regions. E, Immunofluorescence analysis of Ki67+CD8+ T cells (yellow arrow) and SMA+ myoepithelial cells in small and large tumors. Graph depicts frequency of Ki67+CD8+ T cells in small and large tumors. Error bars, SD. P value calculated using a Mann–Whitney Wilcoxon test. F, Immune cell type frequency predicted by CIBERSORT based on RNA-seq data of CD45+ cells from 10 NMU-induced tumors. P values calculated by Mann–Whitney Wilcoxon test and difference between means between small and large tumors shown. *, P < 0.05. G, Heat map of row-normalized Z-scores of DEGs in CD45+ cells from large and small tumors, defined as having an adjusted p value of ≤ 0.05 and absolute fold change ≥ 1.5. Red and blue indicates HRlow and HRhigh tumors, respectively. Green and orange marks large and small tumor, respectively. H, Enriched pathways in CD45+ cells from large and small tumors. Green and orange bars depict pathways enriched in stable and growing tumors, respectively. Node size indicates the number of genes within the pathway, line width indicates the number of shared genes between two gene sets. I, Gini index measure of BCR heterogeneity in tumor and normal mammary gland (MG) infiltrating B cells and the number of unique BCR clonotypes. Box and whisker plot, quartiles. P values were calculated using Mann–Whitney Wilcoxon test. J, Immunofluorescence analysis of NMU-induced tumor organoid culture stained for EpCAM (red) and SMA (green). K, Representative phase-contrast images of rat mammary tumor organoid or autologous normal kidney organoids incubated with or without opT for indicated times. L, INFγ levels in the medium of rat tumor organoids cocultured for 2 days with opT under mIgG1 mAb, PD-L1 mAb, LY, or PD-L1+LY treatment measured by ELISA. P value calculated using Mann–Whitney Wilcoxon test. Error bars, SD. B–E, J and K, Scale bar, 100 μm. All tests for significance used Mann–Whitney–Wilcoxon test using a threshold of P = 0.05 unless otherwise specified; error bars representative of SD; box-whisker plots indicate 0th, 25th, 50th, 75th, and 100th percentiles.
Given that changes in the composition and spatial localization of T cells are characteristic of human breast tumor progression (3, 27, 28), we analyzed the spatial topology of CD3+ T cells by immunofluorescence. Similar to normal human breast, normal rat mammary glands had very few T cells, and any present were commonly intraepithelial within the ducts (Fig. 2D). In the DCIS-like regions where an intact layer of SMA+ myoepithelial cells surround the tumor epithelium, T cells were mostly excluded from the ducts and localized in the stroma. In IDC-like regions where the myoepithelium was not continuous or was lacking, the T cells were intermixed with the malignant tumor cells (Fig. 2D). We also found that smaller tumors had a higher percentage of Ki67+CD8+ T cells and this decreased with increasing tumor size (Fig. 2E; Supplementary Fig. S2D).
We performed immune deconvolution using CIBERSORT (29) and DEG analysis to identify differences in CD45+ profiles between small and large tumors. We observed an inverse correlation between tumor size and the frequency of memory activated CD4+ T cells, suggesting an increasingly immunosuppressive environment as tumors grow (Fig. 2F). Higher expression of genes involved in adaptive immunity, including cytotoxic T-cell responses, Th1 lineage differentiation, B-cell response, and Nkfb1, a transcription factor for inflammation, was observed in small tumors (Fig. 2G and H; Supplementary Table S4), which is indicative of an active immune response. Furthermore, small tumors were enriched for transcription- and lymphocyte proliferation–related terms, implying clonal expansion. In contrast, larger tumors were enriched for chemoattractants that support monocyte infiltration/differentiation and lymphocyte activation down the Th2 lineage, which may contribute to the upregulation of complement that is associated with immunosuppressive environments (30). CD45+ cells in large tumors were enriched for cell adhesion–related terms, including interactions with the endothelium or matrix, suggesting interactions with the extracellular matrix (ECM).
To investigate adaptive immunity further, we first analyzed BCR and TCR clonotypes from RNA-seq data. Compared with the B cells of unaffected mammary glands from tumor-bearing animals, B cells from tumors displayed a significantly lower number of unique clonotypes but lower Gini index, indicating higher diversity (Fig. 2I). This suggests that there is greater richness of B cells in the mammary tumors as well as a more targeted antitumor response. However, we did not find an association between BCR Gini index and tumor size and TCR diversity differences were not significant (Supplementary Fig. S2E and S2F). Next, we assessed the ability of T cells to exert targeted antitumor responses using 3D organoid killing assays. Organoids grown from rat tumors retained both an EpCAM+ epithelial and SMA+ myoepithelial layer (Fig. 2J). We generated opTs by exposing spleen-derived T cells to autologous tumor cells. The opTs were subsequently cocultured with autologous tumor or normal kidney cells. After 4 days of coculture, we observed specific killing of the tumor cells and not of the autologous kidney cells (Fig. 2K). Finally, we assessed EpCAM+ cell RNA-seq data for immune checkpoint proteins and saw heterogeneous expression of inhibitory markers, including CD274 (PD-L1), the macrophage “don't eat me” signal CD47, and CD276 (B7-H3), which is involved in immune regulation and NFκB signaling (Supplementary Fig. S2G). Altogether, these results suggest that smaller tumors have a more active immune environment that becomes muted during tumor progression.
Immunotherapy limits NMU-induced mammary tumor growth
We hypothesized we could prevent tumor progression by targeting PD-L1 and TGFBR because in large tumors we detected a decrease in activated CD8+ T cells and an increase in ECM-related genes, including many known targets of TGFβ. We used an anti–PD-L1 (PD-L1 mAb; see Supplementary Fig. S3 for characterization of the mAb) and the small-molecule inhibitor of TGFβ receptor type I (Galunisertib/LY2157299, referred to hereafter as LY; ref. 31). We first tested these agents in opT and tumor cell cocultures and measured cytotoxic activity by ELISA for IFNγ in the supernatant (Fig. 2L). Although statistically underpowered (MWW P = 0.33), the combination treatment resulted in a higher effect size relative to the isotype control compared with either of the single treatments.
Subsequently, we tested the efficacy of these agents in preventing progression of NMU-induced mammary tumors. Following the emergence of palpable tumors induced by a single injection of NMU, animals were randomly assigned a treatment regimen of LY or vehicle twice daily for 10 days, PD-L1 or isotype control mAb weekly for 6 weeks, or the combination of PD-L1 mAb and LY (Fig. 3A). Tumor size was monitored weekly up to 100 days. We observed heterogeneous growth in all treatment arms, including spontaneously regressing tumors in vehicle-treated rats (Fig. 3B). Collection of tumors at a uniform timepoint after treatment was not feasible as we sought to determine the long-term effects of the treatment and collect sufficient tissue for molecular profiling, and thus we used the growth rate in mm/week rather than final size as a measurement of outcome. Overall, tumors treated with PD-L1 mAb and its combination with LY had significantly lower growth rates compared with the control (Fig. 3C; MWW, PD-L1 P = 0.03, LY P = 0.07, and PD-L1+LY P = 0.002). We divided tumors into “slow-growing/stable” and “growing” by comparing the bottom tercile with the rest of the samples. This threshold of (2 mm growth/week) appeared as the nadir in the bimodal distribution of growth rates (Fig. 3D). There was a significant increase in the proportion of stable tumors with combination LY and PDL1 mAb treatment (Fig. 3E, χ2P = 0.01), showing that early treatment for even a short duration has sustained long-term benefit.
Figure 3.

The effect of immunotherapy on tumor progression. A, Schematic experimental outline. Yellow and turquoise lines indicate treatment with LY2157299 (LY) and anti–PD-L1, respectively. Yellow lightning marks NMU injection. Red hexagon, experimental endpoint. B, Graphs depicting changes in diameter of tumors from the immunotherapy cohort. C, Individual tumor growth rates. Quartiles and range are shown. P value calculated on the basis of comparison of treated versus vehicle groups using a Mann–Whitney–Wilcoxon test. D, Histogram depicting growth rate distribution and the local minimum (red dashed line) between the two peaks used for growing/stable classification. E, Summary of tumor growth categories in each treatment group. P values were calculated using χ2 test. F, Representative images of H&E and trichrome staining. Scale bar, 100 μm. G, Graph depicting quantification of collagen content in stable and growing tumors and in the indicated treatment groups. Shown are quartiles and range. P values calculated by Mann–Whitney Wilcoxon test. H, Immunofluorescence analysis of SMA, CD8, and Ki67 in 10 stable and nine growing tumors. Scale bar, 100 μm. White arrows, Ki67+CD8+ cells in direct contact with malignant cells. Blue arrows, stroma-restricted Ki67+CD8+ cells. I, Graph illustrating quantification of immunofluorescence analysis of SMA, CD8, and Ki67 in stable and growing tumors. Shown are quartiles and range. P values calculated by Mann–Whitney–Wilcoxon test. Tumor cellular composition analysis by automated cell classification from WSI scans of immunofluorescence images stratified by treatment arm (J) or growth rate (K). Quartiles and range are shown. P values were calculated using the Mann–Whitney Wilcoxon test against vehicle. L, Nearest neighbor distance analysis for CD8+ cells to all other cell types from WSI. Quartiles and range are shown. P values were calculated using the Mann–Whitney–Wilcoxon test. All tests for significance used Mann–Whitney–Wilcoxon test using a threshold of P = 0.05 unless otherwise specified; error bars representative of SD; box-whisker plots indicate 0th, 25th, 50th, 75th, and 100th percentiles.
Tumor growth is associated with fibrosis and spatial topology of CD8+ T cells
To explore mechanisms of response and resistance, we first assessed tumor histology and found most tumors (n = 77/84) to be well-differentiated solid adenocarcinomas regardless of treatment (Supplementary Table S2; Supplementary Fig. S4A). However, irrespective of treatment, growing tumors had an “encapsulated” tissue architecture where large areas of tumor epithelium were encased by thick stromal cell layers suggestive of fibrosis, whereas stable tumors had significantly smaller and more dispersed epithelial patches surrounded by stroma (MWW, P = 9 × 10–5; Fig. 3F; Supplementary Fig. S4B). Trichrome staining revealed collagen-rich stroma, which was present in lower amounts in the combination treatment group compared with vehicle irrespective of growth rate (MWW, P = 0.03; Fig. 3G). However, stable tumors in general had more collagen than growing tumors (MWW, P = 0.045; Supplementary Fig. S4C). We also performed immunofluorescence for CD8, SMA, and Ki67 to assess whether differences in proliferation could explain differences in growth rate, but no significant differences were found in the relative fraction of Ki67+CD8− cancer cells or Ki67+CD8+ T cells (Fig. 3H and I; Supplementary Fig. S4D).
To quantitatively assess the topologic distribution of CD8+ T cells and their interaction with tumor cells, we performed whole-slide immunofluorescence (WSI) imaging for SMA, CD8, and EpCAM on the largest cross-section of these tumors (Supplementary Fig. S4E and S4F). Cells were segmented into individual nuclei and classified into five groups (DAPI+ unclassified cells, CD8+ T cells, and EpCAM+, SMA+, EpCAM+SMA+ tumor cells) using QuPath (32). In several tissue samples, an EpCAM+SMA+ cell population most likely represents a mixture of EpCAM+ or SMA+ tumor cells in close contact with one another. The frequencies of CD8+ T cells obtained by WSI correlated with frequencies obtained by flow cytometry, expression levels in CD45+ cell RNA-seq data, and manual quantitative scoring (Supplementary Fig. S4G). The proportion of CD8+ T cells did not vary according to treatment nor growth rate. However, LY-treated tumors had a lower fraction of EpCAM+ tumor (MMW P = 0.03) and DAPI+ unclassified cells (P = 0.05), and a higher fraction of SMA+ tumor cells compared to vehicle (P = 0.03; Fig. 3J and K). In addition, stable tumors had a higher proportion of EpCAM+ tumor cells compared to growing tumors (P = 0.06).
To assess the spatial localization of CD8+ T cells, we considered three different spatial metrics: nearest neighbor distances, an interacting fraction defined as the proportion of CD8+ T cells within a 15 μm distance of another cell, and the Morisita-Horn index for colocalization (16). The average nearest neighbor distance from CD8+ T cells to EpCAM+ cells was higher in growing tumors compared with stable tumors (MWW, P = 0.02); however, colocalization associations were not found with the other cell types (Fig. 3L). Similar results were obtained using the interacting fraction (P = 0.002) and Morisita–Horn index (P = 0.1) (Supplementary Fig. S4H and S4I). With respect to treatment, the spatial architecture in LY-treated samples differed the most from vehicle-treated ones, characterized by shorter distances between CD8+ T and SMA+ cells and higher interacting fractions, and low colocalization between CD8+ T and DAPI+ unclassified cells (Supplementary Fig. S4J and S4K). In the combination treatment, there was higher colocalization of CD8+ T and DAPI+ unclassified cells, evidenced by shorter nearest neighbor distances and higher interacting fractions (Supplementary Fig. S4J and S4K). The majority of these DAPI+ unclassified cells are likely to be CD45+CD8− leukocytes, suggesting that these direct interactions might be required for CD8+ T-cell activation and a coordinated immune response.
In summary, fibrosis and low EpCAM+–CD8+ T-cell interactions are associated with tumor growth.
Molecular profiles of cellular subsets
To investigate how immunotherapy affected molecular profiles, we collected CD45+, EpCAM+, and DN cells by FACS and performed RNA-seq. Although the relative abundance of these cells did not vary by treatment, growing tumors had a significantly lower proportion of CD45+ cells (MWW, P = 0.05) compared with stable tumors (Supplementary Fig. S5A). PCA showed that DN and EpCAM+ cells were more similar to each other than to CD45+ cells (Supplementary Fig. S5B). Key immune-related genes, including Cd3, Cd8, Cd4, showed CD45+ cell-specific expression, confirming sample purity (Supplementary Fig. S5C). The EpCAM+ and DN populations shared expression of epithelial-specific genes, including Krt8 and Epcam, but the EpCAM+ cells had higher expression of Esr1 and Pgr, consistent with their luminal features (Supplementary Fig. S5C). Genes specific to DN cells included myoepithelial (Tp63), fibroblast (Fap), and endothelial (Cdh5) cell markers, suggesting that this fraction was a mixture of these cell types. Both EpCAM+ and DN cells expressed Mki67, implying proliferation.
When comparing vehicle versus immunotherapy-treated samples (all treatments pooled), the biggest separation was observed in the CD45+ cell population, whereas EpCAM+ and DN cells overlapped regardless of treatment (Fig. 4A). DEG analysis between vehicle- and immunotherapy-treated tumors showed treatment-induced downregulation of inflammatory genes in the CD45+ fraction, including cytokines (Cxcl13) and complement factors (C1qc; Fig. 4B; Supplementary Table S3). Very few genes were consistently upregulated following immunotherapy except for B cell–related gene (Cd79b) and Spp1 (osteopontin). Polychromatic flow cytometry did not show significant differences in relative frequencies of specific immune cells across treatments or growth rates (Supplementary Fig. S5D and S5E).
Figure 4.

Tumor stromal characteristics of NMU-induced tumors in the immunotherapy-treated cohort. A, PCA plot of RNA-seq data of 34 CD45+, double 29 negative (DN), and 20 EpCAM+ cell samples from NMU-induced tumors harvested from immunotherapy treated rats. B, Heat map of row-normalized Z-scores of genes differentially expressed in CD45+ cells between vehicle (N = 12) and all immunotherapy-treated tumors (N = 22). Red and blue indicates HRlow and HRhigh tumors, respectively. Green and orange marks stable and growing tumors, respectively.C, Immune cell type frequency estimation based on RNA-seq of CD45+ cells using CIBERSORT. P values calculated by Mann–Whitney–Wilcoxon test and difference between means shown *, P < 0.05. D, Volcano plot of genes differentially expressed in CD45+ cells between growing and stable tumors. Red points have a (log2 fold change)>1.5 and adjusted P < 0.05, and named genes are known to be immune related. E, GSEA normalized enrichment scores of pathways enriched in CD45+ cells from growing and stable tumors (FDR < 0.1). Gini index and number of unique clonotypes describing BCR (F) and TCR (G) repertoire heterogeneity in growing versus stable tumors. P values calculated using Mann–Whitney–Wilcoxon test. Immunofluorescence analysis of SMA, CD163, and complement factor D (CFD) (H) and CD11b, CD3, and MHCII (I) in growing and stable tumors. Scale bar, 100 μm. All tests for significance used Mann–Whitney–Wilcoxon test using a threshold of P = 0.05 unless otherwise specified; error bars representative of SD; box-whisker plots indicate 0th, 25th, 50th, 75th, and 100th percentiles.
Analysis of the immunotherapy-treated DN fraction showed two major phenotypes: inflammatory with higher expression of Il6, Cxcl2, and Il13ra1, and metabolically active with activated replication and heat shock signaling (Supplementary Fig. S5F). The vehicle-treated DN fraction lay between these two extremes. Treatment-specific comparisons revealed minimal differences in the LY and PD-L1 mAb treated–tumors compared with the vehicle-treated ones, suggesting that stromal composition and activity are relatively similar (Supplementary Table S4). However, the combination treatment showed significant differences in ECM remodeling and collagen genes, including the downregulation of Mmp2, collagen, and fibrillin, consistent with the difference observed with trichrome staining (Supplementary Fig. S5G; Supplementary Table S3). The EpCAM+ cell population did not show major differences upon immunotherapy, however, HRhigh samples were transcriptionally more similar to each other compared with the HRlow samples (Supplementary Fig. S5H and S5I; Supplementary Table S3).
To assess whether cancer-driver gene mutations contributed to treatment response or growth rate, we searched for these mutations in our RNA-seq data (Supplementary Fig. S5J). We detected mutations in Foxa1, Tp53, Pten, Esr1, and Brca1, often in both EpCAM+ and DN fractions. Setd2 (7/20 vs. 1/9, Fisher test P = 0.4), Pms2 (10/20 vs. 1/9, P = 0.1), Casp3 (7/20 vs. 1/9 P = 0.4), and Hras (5/20 vs. 1/9 P = 0.7) were more frequently mutated in growing tumors, although the difference was not significant due to small sample size. Hence, tumor stromal and immune features may have a larger role in tumor progression.
Overall, we found that immunotherapies decreased tumor growth mainly by affecting CD45+ cells. However, both “growing” and “stable” responses were noted in all treatment arms, reflecting the heterogeneity in response often observed in patients.
Myeloid cells and a complement-rich immune environment are associated with tumor growth
To further explore changes in immune populations during tumor growth, we analyzed differences in CD45+ cells of growing and stable tumors from all treatment groups. CIBERSORT deconvolution of CD45+ cell RNA-seq data revealed higher activated CD4+ T-cell frequencies and lower frequencies of myeloid dendritic cells in stable tumors (Fig. 4C). Genes enriched in stable tumors include Il21r, a receptor involved in the differentiation of B, T, and NK cells (Fig. 4D). GSEA showed an enrichment of terms associated with BCR, TCR, IL2, and IFNγ signaling, and NK-cell cytotoxicity in stable tumors reflective of an antitumor immune response (Fig. 4E; Supplementary Table S4). Consistent with this, stable tumors had significantly higher BCR and TCR Gini index and higher numbers of unique BCR clonotypes, which may reflect antitumor immune response–associated clonal expansion (Fig. 4F and G).
In contrast, proportions of M2 macrophages, myeloid dendritic cells, and monocytes correlated positively with tumor growth rate (Fig. 4C). Growing tumors also had higher expression of Cd163 (33), an M2-polarized macrophage marker, complement-related genes (C1qa-c), and chemokines (e.g., Ccl12) commonly induced in macrophages (Fig. 4D; Supplementary Table S4). Immunofluorescence for CD163 and complement factor D (CFD, aka adipsin) showed a higher fraction of CD163+CFD+ macrophages in growing tumors (Fig. 4H). Furthermore, CD11b+CD3−MHCII− myeloid-derived suppressor cells (MDSC) were frequently found in growing tumors but only rarely in stable tumors (Fig. 4I). These results suggest that recruitment of MDSC and cells with a protumor M2-polarized macrophage phenotype in growing tumors helps to establish an immunosuppressive microenvironment (34), and that immunotherapies could inhibit tumor growth by reversing this effect.
Stable HRlow tumors are characterized by an inflammatory phenotype
Tumor immune microenvironments are modulated by both extrinsic and tumor-cell intrinsic factors that enable immune escape. GSEA demonstrated an upregulation of NFκB-mediated TNFα signaling in stable tumors across all cell populations (CD45+, DN, EpCAM+), and IFN signaling was upregulated in EpCAM+ and DN but not in CD45+ cells (Fig. 5A). The EpCAM+ fraction of stable tumors showed enrichment for IL6–JAK–STAT3, IFNγ, IFNα, and IL2–STAT5 signaling pathways, reflecting an inflamed immune environment (Fig. 5B). This inflammatory phenotype was largely driven by a few, mostly HRlow (4/5) stable (4/5) tumors with high expression of inflammatory cytokine receptors, including Il6r and Tnsfr1a. The only tumor in this group (P17T1) that was classified as “growing” had started regressing before the animal was sacrificed (Supplementary Fig. S6A).
Figure 5.

Molecular profiles of inflammatory tumor epithelial cells from immunotherapy-treated cohort of NMU-induced tumors. A, GSEA between growing versus stable tumors in CD45+, DN (double negative), and EpCAM+ cells using the MSigDB Hallmark compendium. Red and blue indicates stable and growing enriched, respectively, FDR < 0.05. B, Normalized single-sample gene set enrichment scores (ssGSEA NES) of significant pathways in EpCAM+ cells of each tumor. Inset, gene expression row z-scores of genes in the IL6–JAK–STAT3 signaling pathway. Hyperinflammatory tumors indicated in purple C, Immunofluorescence analysis of inflammatory and noninflammatory tumors for CD8, SMA, and CD74. Scale bar, 50 μm. D, Comparison of CD8+ T-cell frequency and intermixing with EpCAM+ cells in hyperinflammatory and noninflammatory tumors in WSI. Quartiles and range are shown. P values were calculated using Wilcoxon rank-sum test. E, Spearman correlation of CD74 expression with leukocyte infiltration and TCR diversity in TCGA breast cancer cohort (N = 1,054 complete observations). F, Forest plot showing HRs with 95% CIs from multivariable Cox regression analysis using CD74 gene expression z-score, PAM50 subtype and tumor stage in TCGA breast cancer cases. Log-rank test P value: 3 × 10−9. All tests for significance used Mann–Whitney–Wilcoxon test using a threshold of P = 0.05 unless otherwise specified; error bars representative of SD; box-whisker plots indicate 0th, 25th, 50th, 75th, and 100th percentiles.
Across these inflammatory samples we observed an upregulation of many genes with immune ontologies, including MHCII genes (RT1-Ba, RT1-DR, Cd74), neutrophil and macrophage stimulants (Csf3, Cxcl3), and regulators of TCR signaling (Lck) as well as RAS (Rac2) signaling (Supplementary Fig. S6B). By immunofluorescence, the antigen presentation protein CD74 (35) was highly expressed in inflammatory stable tumors in both tumor epithelial and SMA+ cells (Fig. 5C). These inflammatory samples also had a higher fraction of infiltrating CD8+ T cells (MWW, P = 0.006), and higher CD8+-EpCAM+ intermixing measured by the Morisita-Horn index (P = 0.04) from WSI (Fig. 5D).
To confirm the clinical relevance of our findings, we analyzed The Cancer Genome Atlas (TCGA) human breast cancer cohort (11) and found that CD74 expression correlated with leukocyte recruitment and a more diverse TCR clonotype (Fig. 5E). Moreover, CD74 expression was associated with longer overall survival after adjusting for patient PAM50 subtype and tumor stage [HR = 0.73 (95% CI, 0.55–1.00), log-rank P = 3 × 10–9] (Fig. 5F). This inflammatory, high MHCII phenotype has been observed in human cancers and is associated with basal subtypes, high CD4+ and CD8+ T-cell infiltration and longer progression-free survival (35, 36), which underscores the fidelity with which this rat model recapitulates human breast tumor biology and immunotherapy responses.
Adamst10 expression and fibrosis is associated with HRhigh growing tumors
Next, we explored differences between stable and growing tumors in the noninflammatory subgroup. As the noninflammatory subgroup was enriched for HRhigh tumors compared with the inflammatory subgroup (10/16 vs. 1/5, Χ2 test, P = 0.055), we focused specifically on comparing stable versus growing HRhigh tumors to avoid subtype-specific confounders. We derived a “luminal-growing” signature, which featured upregulated genes involved in immune regulation (Ikbke, Lmbr1L, Leng8); ECM remodeling and interaction with other cells (Col5a1, Cdc42bpg, Adamst10); RNA processing and epigenetic regulation (Pnisr, Setd7, Kmt5c; Fig 6A; Supplementary Table S4). In contrast, immune pathways enriched in stable tumors included leukocyte chemotaxis, IFN, IL, as well as BCR and TCR signaling (Supplementary Fig. S6C; Supplementary Table S4).
Figure 6.

Molecular profiles of HRhigh tumor epithelial cells. A, Heat map of row-normalized Z-scores of DEGs between HRhigh growing and stable tumors in the EpCAM+ fraction. B, Trichrome staining quantification comparing inflammatory with noninflammatory growing and noninflammatory stable tumors. Quartiles and range are shown. P values were calculated using Wilcoxon rank-sum test between growing and all other tumors. C, Immunofluorescence analysis of HRhigh tumors for SMA and ADAMTS10. Scale bar, 50 μm. D, Spearman correlation of ADAMTS10 expression with inferred stromal content in TCGA luminal breast cancer cases (N = 564 complete observations). E, Forest plot showing HRs with 95% CIs from multivariable Cox regression analysis using ADAMTS10 gene expression z-score, PAM50 subtype and tumor stage in TCGA luminal breast cancer cases HR = 1.37 (0.97–1.95), log-rank P value = 1 × 10−6. F, Kaplan–Meier plot of DFS of patients with TCGA Lum A breast cancer segregated by “Luminal-growing” signature expression. Dark, medium, and light blue curves represent terciles with high, medium, and low expression of the signature, respectively. HR, 95% CI, and log-rank P value from a Cox model using the signature score as a continuous variable and accounting for stage are shown. G, Forest plot of showing HR and 95% CIs modeling contributions of the luminal-growing signature, OncotypeDx, and Mammaprint to DFS in a Cox proportional hazards regression accounting for tumor stage in TCGA Lum A breast cancers. HR = 1.78 (1.16–2.72), log-rank P = 0.03. H, Spearman correlation of “Luminal-growing” gene signature with leukocyte infiltration and TCR diversity in TCGA Luminal breast cancer cases (N = 553 complete observations) I, Spearman correlation between Luminal-growing signature score and plasma MICA protein levels in ER+ metastatic breast cancer cohort (N = 19). J, Boxplot of luminal-growing signature score in PD-L1–positive and PD-L1–negative metastatic breast cancer samples. All tests for significance used Mann–Whitney–Wilcoxon test using a threshold of P = 0.05 unless otherwise specified; error bars representative of SD; box-whisker plots indicate 0th, 25th, 50th, 75th, and 100th percentiles.
As we observed greater encapsulation and lower collagen content in growing tumors (Fig. 6B), we explored whether differences in immune environments could be explained by fibrosis. ADAMST10, a protease predicted to cleave fibrillins (37), was highly expressed in HRhigh growing tumors, which we confirmed by immunofluorescence in both SMA+ myoepithelial and tumor epithelial cells (Fig. 6C). In contrast, in HRhigh stable tumors, ADAMTS10 expression was only detected in a fraction of SMA+ myoepithelial cells and was absent in the epithelium. In TCGA cohort, ADAMTS10 expression correlated with stromal content (ρ = 0.36, P = 7 × 10−9; Fig. 6D) and was associated with poor prognosis in ER+ tumors [HR = 1.37(0.97–1.95), P = 1 × 10−6; Fig. 6E), indicating the clinical relevance of our findings. The high expression of Adamts10, Mgp (Matrix Gla protein), and Spp1 (osteopontin) in luminal-growing tumors in combination with high levels of Plg (plasminogen) and Slpi (secretory leukocyte peptidase inhibitor) in luminal-stable samples highlight the importance of ECM remodeling and angiogenesis in tumor growth and immune responses.
Relatedness of immunotherapy-responsive HRhigh rat tumors to human LumA immune-hot breast cancer
To determine the human relevance of our “luminal-growing” gene signature, we computed signature scores in ER+ tumors in TCGA cohort using single-sample GSEA (Supplementary Table S4). This “luminal-growing” signature was associated with shorter DFS specifically in Luminal A samples [HR = 1.7 (1.15–2.52), P = 0.02] (Fig. 6F) and not in other subtypes (Supplementary Fig. S6D). It was also associated with poor DFS in a Cox proportional hazards model including Mammaprint (19) and OncotypeDx (18), indicating that the signature captures prognostic information orthogonal to these existing signatures [HR = 1.78 (1.16–2.72), P = 0.03] (Fig. 6G; Supplementary Table S4). In addition, the signature negatively correlated with inferred leukocyte fraction (ρ = −0.11, P = 0.025) and TCR diversity (ρ = −0.28, P = 7 × 10−9; Fig. 6H) indicating that tumors with high expression of the “luminal-growing” signature could be more immune cold. Consistent with this, TCGA Luminal A tumors enriched for the “luminal-growing” signature showed lower activity of antitumor immune pathways including leukocyte chemotaxis, phagosome in antigen presentation, Th cell differentiation, IFNγ signaling, TCR and BCR signaling, and NK-cell cytotoxicity (Supplementary Fig. S6E and S6F; Supplementary Table S4).
Finally, to specifically test if our “luminal-growing” signature was able to predict response to immunotherapy in patients with ER+ breast cancer, we applied it to RNA-seq data of primary tumors in an ER+ metastatic breast cancer cohort treated with eribulin with or without pembrolizumab (anti-PD-1; refs. 12, 13). Patients whose tumors had high expression of the “luminal-growing” signature showed a trend for longer overall survival only in the pembrolizumab-treated group (Supplementary Fig. S6G). The “luminal-growing” signature was also inversely correlated with plasma MHC class I chain-related gene A (MICA) protein levels (Fig. 6I) and was enriched in tumors lacking PD-L1 expression (Fig. 6J). MICA is a ligand for NKG2D present on T cells and NK cells (38). Tumor-cell shedding of MICA leading to higher plasma levels has been associated with lower NKG2D expression on intratumoral T cells promoting immune evasion (38, 39). These data imply that downregulation of NKG2D may be a mechanism of immune escape in a subset of luminal tumors and upregulation of PD-L1 may not confer additional advantage explaining their lack of response to pembrolizumab. However, due to the small size of this cohort, validation in larger studies is necessary.
Discussion
Carcinogen-induced rat models of breast cancer were first developed over 50 years ago, but lost popularity following the advent of genetically engineered mouse and transplantable models (5). However, rat models are regaining popularity as CRISPR–Cas9 enables genome editing of any organism, and the higher similarity of rat mammary tumors to human disease makes them especially useful as immunocompetent preclinical models of breast cancer. Here, we describe what we believe to be the first comprehensive genomic and immune characterization of the NMU-induced SD rat mammary tumor model, and we demonstrate its similarity to human breast cancer. Although this model holds great potential for preclinical testing of novel targeted and immunotherapies, it is important to note differences from human breast cancers, including the biphasic nature of these tumors, which potentially reflects human adenomyoepitheliomas; their mutational burden, which aligns more closely to human melanoma; and their limited invasiveness and metastatic spread.
However, gene expression patterns clearly show resemblance to a spectrum of luminal and TNBC. Moreover, the NMU-induced mutations and the evolution of these tumors in their endogenous microenvironment under immune selection mimics the immunoediting that occurs in human breast cancer, making this model useful for immunotherapy studies. A subset of the NMU-induced mammary tumors were luminal ER+, which is the most common form of human breast cancer but is not well represented by most murine models. ER+ mouse mammary tumors driven by PIK3CAH1047R (40) or deletion of STAT1 (41) do not reproduce the genetic and phenotypic heterogeneity of NMU-induced mammary tumors in outbred SD rats. Furthermore, the mutation and neoantigen repertoire of those mouse tumors are more limited, which impacts their immune environment and response to immunotherapies. Given that immunotherapy response and mechanisms of immune evasion are tightly associated with tumor-intrinsic subtype, intratumor heterogeneity, and interindividual differences, the ability to recapitulate these key aspects of the human disease makes this rat model particularly relevant.
As a proof of principle, we tested the response of NMU-induced mammary tumors to combined PD-L1 and TGFβ inhibition given that our gene expression data showed high activity of TGFβ-regulated processes, including ECM remodeling and MDSC accumulation in large tumors. Combined PD-L1 and TGFβ inhibition has also shown promise in urothelial cancer (42), and in mouse models of breast and colon cancer (43). We found durable responses after a single cycle of combination treatment in early tumors. Our data suggest that immune exclusion could be due to high expression of complement proteins leading to macrophage recruitment and M2 polarization. The complement system plays an important role in cell–cell interactions and has been implicated in protumor inflammation in human cancers (30). An inflammatory microenvironment has been reported to promote the recruitment of MDSCs via C5a (44) and M2-polarized macrophages via CCL2 (45), and to inhibit activation of CD8+ T cell via C3-mediated IL10 blocking of T cells (46). Consistent with this, we saw an enrichment of CFD+CD163+ macrophages and CD11b+CD3−MHCII− MDSCs in growing tumors, both features that have been associated with resistance to immunotherapy (47). These data support the targeting of complement, macrophages, and MDSCs to improve the efficacy of immune therapies; indeed, for macrophages there is already promising preclinical data along these lines (48). However, systemic inhibition of TGFβ is not a feasible approach due to cardiovascular and skin toxicities and induction of secondary tumors in animal models (49). Nonetheless, localized, cell type–specific targeting of TGFβ has been achieved along the integrin αVβ6–TGFβ axis (50), which was shown to sensitize PD-L1–resistant TNBC murine models to PD-L1 blockade (51).
Another key challenge for immunotherapies is the lack of effective predictive biomarkers to identify the patients most likely to respond. PD-L1 expression has been commonly used for patient enrollment but does not necessarily predict patient response to treatment (52). Here, we show that intratumoral spatial distribution of immune cells and ECM are associated with tumor growth. Although recruitment of CD8+ T cells was not significantly different between stable and growing tumors, the mean distance between these and cancer cells was significantly smaller in stable tumors. Intrinsic tumor characteristics might modulate the ECM and stromal cells resulting in immune exclusion. In line with this, resistance mechanisms include fibrosis, cancer-associated fibroblast-mediated upregulation of immune checkpoint proteins in T cells (53), and downregulation of MHC machinery. We found CD74, a protein involved in antigen processing, as an alternate biomarker for hyperinflammatory HRlow tumors that may benefit from immunotherapy. Furthermore, contrary to the current literature advising against immunotherapy for ER+ patients due to low HLA expression and T-cell recruitment (54), we demonstrated that some HRhigh tumors respond to immunotherapy. Characterization of the nonresponding HRhigh tumors revealed an immune-cold phenotype, higher expression of epigenetic regulators and ECM modulators including ADAMTS10. Inhibition of epigenetic enzymes (55) and microenvironmental reprogramming by antiangiogenic agents or angiotensin receptor blockers have been shown to enhance the efficacy of immunotherapies in preclinical models (56). Although further validation is warranted, the NMU-induced mammary tumor model provides an effective starting point to identify potential biomarkers, mechanisms of resistance, and novel therapeutic strategies that may synergize with immunotherapy, including endocrine therapies and CDK4/6 inhibitors, which are the current standard of care in ER+ breast cancer.
It is important to note that our observations were made in preinvasive tumors that are distinctive to this rat model. However, the predictive power of our gene signatures in advanced human breast tumors suggests that immune evasion is an early step of tumor evolution and that the underlying mechanisms are maintained in late-stage disease. Our observation that short-term treatment with immunotherapy, especially combined TGFβ and PD-L1 inhibition, of early-stage tumors results in durable responses underscores the importance of tumor stage for immunotherapy. Clinical trial results showing improved responses in neoadjuvant and adjuvant setting compared with metastatic disease also confirm this (57).
In summary, we characterized a carcinogen-induced model of breast cancer in rats that is amenable to preclinical testing and elucidated mechanisms of response and resistance to immunotherapy. Our results using this model warrant the evaluation of PD-1/PD-L1 immunotherapy in combination with TGFβ inhibition in early-stage breast cancer and identified a subset of ER+ patients that may benefit from immunotherapy. This is an important first step toward the larger goal of widening patient eligibility and patient response to immunotherapy, in particular by preventing progression of disease at an earlier stage.
Supplementary Material
Acknowledgments
We thank members of the Polyak lab for their critical reading of this article and useful discussions. We thank Zach Herbert and the staff of the DFCI Molecular Biology Core Facility for their dedication and technical expertise with genomic studies. We also thank the DFCI Animal Facility staff for their help with gavage of the rats. We thank Lakshmi Muthuswamy for her help with rat genome sequence analyses and Kyla E. Driscoll and Eli Lilly for providing LY2157299. This work was supported by the Claudia Adams Barr Program (C.R. Gil Del Alcazar), DFCI Helen Gurley Brown Presidential Initiative Award (A. Trinh), the Breast Cancer Research Foundation (K. Polyak, S.K. Muthuswamy), and NIH R35 CA197623 (K. Polyak), T32CA009172 (T.E. Keenan), and P50 CA168504 (G.J. Freeman).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Disclosures
A. Trinh reports grants from Helen Gurley Brown Foundation during the conduct of the study. M. Alečković reports personal fees from Shasqi Inc. outside the submitted work; and Shasqi Inc. as current employer. T.E. Keenan reports personal fees from Merck outside the submitted work. E.M. Van Allen reports personal fees from Tango Therapeutics, Genome Medical, Genomic Life, Monte Rosa Therapeutics, Manifold Bio, Illumina, Enara Bio, Janssen, and Foaley & Hoag; grants from Novartis and BMS outside the submitted work; in addition, E.M. Van Allen has a patent for Institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation pending. S.M. Tolaney reports grants and personal fees from AstraZeneca, Eli Lilly, Merck, Novartis, Nektar, Pfizer, Genentech/Roche, Gilead, BMS, Eisai, Sanofi, Odonate, Seattle Genetics; grants from Nanostring, Exelixis, Cyclacel; personal fees from Puma, Daiichi Sankyo, ARC Therapeutics, Zentalis, Athenex, OncoPep, Kyowa Kirin Pharmaceuticals, Samsung Bioepsis Inc., CytomX, Mersana Therapeutics, Certara, OncoSec Medical Inc, Ellipses Pharma, 4D Pharma, BeyondSpring Pharmaceuticals, OncXerna, Zymeworks, Blueprint Medicines, Reveal Genomics, and Chugai Pharma outside the submitted work. G.J. Freeman reports grants from NIH during the conduct of the study; personal fees from Roche, Bristol Myers Squibb, Xios, Origimed, Triursus, iTeos, NextPoint, IgM, Jubilant, Trillium, GV20, and Geode outside the submitted work; in addition, G.J. Freeman has a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Roche, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Merck MSD, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Bristol Myers Squibb, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Merck KGA, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Boehringer-Ingelheim, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from AstraZeneca, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Dako, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Leica, a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Mayo Clinic, and a patent for PD-L1/PD-1 pathway issued, licensed, and with royalties paid from Novartis; and equity in Nextpoint, Triursus, Xios, iTeos, IgM, GV20, and Geode. D.A. Dillon reports other support from Oncology Analytics and non-financial support from Canon, Inc outside the submitted work. S.K. Muthuswamy reports personal fees from KAHR Bio outside the submitted work; in addition, S.K. Muthuswamy has a patent for Method for culturing organoids pending and licensed to Stem Cell Technologies and a patent for Method for generation of organoid-primed T cells pending. K. Polyak reports grants from Breast Cancer Research Foundation and NCI during the conduct of the study; personal fees from Acrivon Therapeutics, Vividion Therapeutics, Scorpion Therapeutics, Aria Pharmaceuticals, AstraZeneca, and grants from Novartis outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
C.R. Gil Del Alcazar: Conceptualization, data curation, formal analysis, funding acquisition, methodology, writing–original draft. A. Trinh: Data curation, formal analysis, writing–original draft. M. Alečković: Methodology. E. Rojas Jimenez: Formal analysis, methodology. N.W. Harper: Formal analysis. M.U.J. Oliphant: Formal analysis, methodology. S. Xie: Methodology. E.D. Krop: Methodology. B. Lulseged: Methodology. K.C. Murphy: Methodology. T.E. Keenan: Formal analysis. E.M. Van Allen: Formal analysis. S.M. Tolaney: Supervision, funding acquisition. G.J. Freeman: Resources. D.A. Dillon: Methodology. S.K. Muthuswamy: Resources, supervision. K. Polyak: Conceptualization, resources, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. Punglia RS, Bifolck K, Golshan M, Lehman C, Collins L, Polyak K, et al. Epidemiology, biology, treatment, and prevention of ductal carcinoma in situ (DCIS). JNCI Cancer Spectr 2018;2:pky063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gil Del Alcazar CR, Alečković M, Polyak K. Immune escape during breast tumor progression. Cancer Immunol Res 2020;8:422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov 2017;7:1098–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. J Natl Compr Canc Netw 2020;18:479–89. [DOI] [PubMed] [Google Scholar]
- 5. Russo J, Russo IH. Atlas and histologic classification of tumors of the rat mammary gland. J Mammary Gland Biol Neoplasia 2000;5:187–200. [DOI] [PubMed] [Google Scholar]
- 6. Rivera ES, Andrade N, Martin G, Melito G, Cricco G, Mohamad N, et al. Induction of mammary tumors in rat by intraperitoneal injection of NMU: histopathology and estral cycle influence. Cancer Lett 1994;86:223–8. [DOI] [PubMed] [Google Scholar]
- 7. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res 2016;4:124–35. [DOI] [PubMed] [Google Scholar]
- 8. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol 2003;170:1257–66. [DOI] [PubMed] [Google Scholar]
- 9. Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, Freeman GJ. PD-L1 binds to B7–1 only In Cis on the same cell surface. Cancer Immunol Res 2018;6:921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tolaney SM, Barroso-Sousa R, Keenan T, Li T, Trippa L, Vaz-Luis I, et al. Effect of eribulin with or without pembrolizumab on progression-free survival for patients with hormone receptor-positive, ERBB2-negative metastatic breast cancer: a randomized clinical trial. JAMA Oncol 2020;6:1598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tolaney SM, Kalinsky K, Kaklamani V, D'Adamo DR, Aktan G, Tsai ML, et al. Eribulin plus pembrolizumab in patients with metastatic triple-negative breast cancer (ENHANCE 1): a phase 1b/2 study. Clin Cancer Res 2021;27:3061–8. [DOI] [PubMed] [Google Scholar]
- 14. Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 2018;8:174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xi R, Lee S, Xia Y, Kim TM, Park PJ. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res 2016;44:6274–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horn H. Measurement of “Overlap” in comparative ecological studies. Am Nat 1966;100:419–24. [Google Scholar]
- 17. Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, et al. IMGT(R), the international ImMunoGeneTics information system(R) 25 years on. Nucleic Acids Res 2015;43:D413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26. [DOI] [PubMed] [Google Scholar]
- 19. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530–6. [DOI] [PubMed] [Google Scholar]
- 20. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hayes MM. Adenomyoepithelioma of the breast: a review stressing its propensity for malignant transformation. J Clin Pathol 2011;64:477–84. [DOI] [PubMed] [Google Scholar]
- 22. Horsfall MJ, Gordon AJ, Burns PA, Zielenska M, van der Vliet GM, Glickman BW. Mutational specificity of alkylating agents and the influence of DNA repair. Environ Mol Mutagen 1990;15:107–22. [DOI] [PubMed] [Google Scholar]
- 23. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sukumar S, Notario V, Martin-Zanca D, Barbacid M. Induction of mammary carcinomas in rats by nitroso-methylurea involves malignant activation of H-ras-1 locus by single point mutations. Nature 1983;306:658–61. [DOI] [PubMed] [Google Scholar]
- 26. Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep 2017;19:203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim M, Chung YR, Kim HJ, Woo JW, Ahn S, Park SY. Immune microenvironment in ductal carcinoma in situ: a comparison with invasive carcinoma of the breast. Breast Cancer Res 2020;22:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Agahozo MC, Westenend PJ, van Bockstal MR, Hansum T, Giang J, Matlung SE, et al. Immune response and stromal changes in ductal carcinoma in situ of the breast are subtype dependent. Mod Pathol 2020;33:1773–82. [DOI] [PubMed] [Google Scholar]
- 29. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roumenina LT, Daugan MV, Petitprez F, Sautes-Fridman C, Fridman WH. Context-dependent roles of complement in cancer. Nat Rev Cancer 2019;19:698–715. [DOI] [PubMed] [Google Scholar]
- 31. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther 2015;9:4479–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fabriek BO, Dijkstra CD, van den Berg TK. The macrophage scavenger receptor CD163. Immunobiology 2005;210:153–60. [DOI] [PubMed] [Google Scholar]
- 34. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002;23:549–55. [DOI] [PubMed] [Google Scholar]
- 35. Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC-II expression by tumor cells in cancer. Clin Cancer Res 2019;25:2392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang ZQ, Milne K, Webb JR, Watson PH. CD74 and intratumoral immune response in breast cancer. Oncotarget 2017;8:12664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karoulias SZ, Taye N, Stanley S, Hubmacher D. The ADAMTS/fibrillin connection: insights into the biological functions of ADAMTS10 and ADAMTS17 and their respective sister proteases. Biomolecules 2020;10:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmiedel D, Mandelboim O. NKG2D ligands-critical targets for cancer immune escape and therapy. Front Immunol 2018;9:2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002;419:734–8. [DOI] [PubMed] [Google Scholar]
- 40. Stratikopoulos EE, Kiess N, Szabolcs M, Pegno S, Kakit C, Wu X, et al. Mouse ER+/PIK3CA(H1047R) breast cancers caused by exogenous estrogen are heterogeneously dependent on estrogen and undergo BIM-dependent apoptosis with BH3 and PI3K agents. Oncogene 2019;38:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res 2012;14:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med 2018;10:eaan5488. [DOI] [PubMed] [Google Scholar]
- 44. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol 2008;9:1225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 2015;160:700–14. [DOI] [PubMed] [Google Scholar]
- 46. Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov 2016;6:1022–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res 2017;5:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann N Y Acad Sci 2020. [DOI] [PubMed] [Google Scholar]
- 49. Colak S, Ten Dijke P. Targeting TGF-beta signaling in cancer. Trends Cancer 2017;3:56–71. [DOI] [PubMed] [Google Scholar]
- 50. Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, et al. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 2003;422:169–73. [DOI] [PubMed] [Google Scholar]
- 51. Bagati A, Kumar S, Jiang P, Pyrdol J, Zou AE, Godicelj A, et al. Integrin alphavbeta6-TGFbeta-SOX4 pathway drives immune evasion in triple-negative breast cancer. Cancer Cell 2021;39:54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Isaacs J, Anders C, McArthur H, Force J. Biomarkers of immune checkpoint blockade response in triple-negative breast cancer. Curr Treat Options Oncol 2021;22:38. [DOI] [PubMed] [Google Scholar]
- 53. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov 2020;10:1330–51. [DOI] [PubMed] [Google Scholar]
- 54. Lee HJ, Song IH, Park IA, Heo SH, Kim YA, Ahn JH, et al. Differential expression of major histocompatibility complex class I in subtypes of breast cancer is associated with estrogen receptor and interferon signaling. Oncotarget 2016;7:30119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mokhtari RB, Sambi M, Qorri B, Baluch N, Ashayeri N, Kumar S, et al. The next-generation of combination cancer immunotherapy: epigenetic immunomodulators transmogrify immune training to enhance immunotherapy. Cancers 2021;13:3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc Natl Acad Sci U S A 2019;116:10674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379:2108–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.