

Abstract
Articular cartilage (AC) is essential for body movement but is highly susceptible to degenerative diseases and has poor self-repair capacity. To improve current subpar regenerative treatments, developmental mechanisms of AC should be clarified and, specifically, how its postnatal multizone organization is acquired. Primary cilia are cell surface organelles crucial for mammalian tissue morphogenesis. Although their importance for chondrocyte function is appreciated, their specific roles in postnatal AC morphogenesis remain unclear. To explore these mechanisms, we used a murine conditional loss-of-function approach (Ift88-flox) targeting joint-lineage progenitors (Gdf5Cre) and monitored postnatal knee AC development. Joint formation and growth up to juvenile stages were largely unaffected. However, mature AC (aged 2 months) exhibited disorganized extracellular matrix, decreased aggrecan and collagen II due to reduced gene expression (not increased catabolism), and marked reduction of AC modulus by 30%–50%. In addition, and unexpectedly, we discovered that tidemark patterning was severely disrupted, as was hedgehog signaling, and exhibited specificity based on regional load-bearing functions of AC. Interestingly, Prg4 expression was markedly increased in highly loaded sites in mutants. Together, our data provide evidence that primary cilia orchestrate postnatal AC morphogenesis including tidemark topography, zonal matrix composition, and ambulation load responses.
Keywords: PRIMARY CILIA, Ift88, ARTICULAR CARTILAGE DEVELOPMENT, HEDGEHOG, TIDEMARK, Prg4
Introduction
Body movement requires uniquely shaped synovial joints to maximize range and type of motion based on anatomical location and load-bearing needs.(1) Much has been learned about the structure and function of adult joint tissues—articular cartilage (AC), meniscus, and intrajoint ligaments—including the biomechanical and lubricating roles each tissue plays in sustaining motion and resilience.(2) Although it is clear that the majority of morphogenesis and terminal organization of mature AC occur during postnatal life in coordination with growth of the skeleton,(3–6) the underlying cellular mechanisms remain poorly understood. A full understanding of this morphogenetic process would be critical to inform on how joint developmental defects/diseases are established and, as importantly, how defective and damaged joint tissues could be repaired and regenerated.
In adults, the fully mature AC displays a stereotypic zonal organization of cells and extracellular matrix (ECM) and contains a top non-mineralized portion and a bottom mineralized portion. The nonmineralized cartilage includes: (i) a superficial zone of flat cells juxtaposed with the synovial space and parallel to the surface, producing ECM components such as lubricin/Prg4; (ii) an intermediate zone of round chondrocytes amidst aggrecan and randomly aligned collagen II fibrils; and (iii) a thick, deep zone of polarized chondrocytes oriented into columns and containing abundant ECM rich in aggrecan and collagen II fibrils aligned perpendicular to the surface. The mineralized cartilage comprises a bottom zone with large hypertrophic-like chondrocytes that conjoins AC to the underlying subchondral bone.(7) The tidemark represents the mineralization front demarcating the boundary between nonmineralized and mineralized cartilage. Although little is known of tidemark developmental origin, significant evidence exists that it advances toward the cartilage surface with age(8–12) or with alterations in mechanical load,(13–15) reducing nonmineralized cartilage thickness. In addition, the transitioning of aquatic axolotl salamanders to terrestrial life results in the formation of a tidemark,(16,17) suggesting that ambulation influences its initiation and positioning. Despite these observations, no cellular mechanisms on tidemark patterning or disease-state repositioning have been clarified.
Postnatal AC morphogenesis into zones is complex because it needs to eventually establish a highly structured, anisotropic tissue critical for joint mechanical function.(7,18,19) Recent studies by our group and others have led to a better understanding of basic tenets of postnatal morphogenesis of AC in mouse knee joints.(4–6) At birth, nascent AC is disorganized and contains a pool of highly proliferative progenitors that give rise to all the cells of adult AC. By 2–3 weeks of age, AC is matrix-rich, but still lacks a clear zonal organization. By 8 weeks, the stereotypical zonal organization is established. Importantly, we found that cell proliferation decreases rapidly soon after birth, whereas the tissue continues to thicken through matrix production/accumulation and increases in average cell size.(4) Thus, there are three stages to postnatal AC development that have become clear: proliferation (neonatal), growth (adolescent), and maturation (adult). Lineage tracing in our recent study strongly suggests that chondrocytes establish zonal organization and vertical columns during the maturation phase,(4) but the mechanisms by which this occurs have not been elucidated.
Primary cilia are mechano-transducing and morphogen-transducing cell surface organelles present on nearly every cell in mammals and are crucial for tissue morphogenesis. Cytoskeletal regulation, cell polarity, and a number of signaling pathways (eg, Hedgehog, Wnt, and transforming growth factor β [TGFβ]) are governed, at least in part, by the primary cilium.(20,21) The chondrocyte primary cilium is an important regulator of cartilage function(22–24) and lengthening has been correlated with human osteoarthritis (OA).(25) In mice, conditional loss of primary cilium function in chondrocytes (including in the growth plate) results in AC deficits that often lead to early onset OA.(26–31) It is thus clear that primary cilia are important to AC health, but the specific mechanisms by which they function during postnatal morphogenesis remain unclearly defined. In addition, the concurrent ablation of cilia in growth plate carried out in previous studies could have had secondary and severe influences on AC, thus requiring new studies selectively interrogating primary cilia function in AC. To identify mechanisms of postnatal AC development in mice and how cilia may function in this context, we developed a joint-specific (Gdf5Cre) disruption of primary cilia (Ift88-flox) and delineated AC development up to 8 weeks of age.
Materials and Methods
Animals
All studies involving mice were reviewed by our institutional IACUC at The Children’s Hospital of Philadelphia. All animals were handled, treated, and cared for according to the approved protocols in conventional group housing and provided standard diet (Lab Diet 5001; LabDiet, St. Louis, MO, USA). IFT88-flox (Stock no. 022409), Rosa-TdTomato (Stock no. 007914), and Gli1-LacZ (Stock no. 008211) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Gdf5Cre mice (kindly provided by Dr. D. Kingsley [Dept. of Developmental Biology, Stanford University School of Medicine]) were described previously(32) and line B was used in the present study. Wild-type, sex-matched littermate animals (either Ift88F/F or Ift88F/+ depending on litter genotyping) were used for controls. Males and females were used equally. Cage activity was not monitored and is a limitation of the study. In approximately one-third of Gdf5Cre animals, Cre recombines randomly early in embryonic development resulting in a mosaic recombination throughout the adult animal. These animals were not used in the study, were monitored by genotyping, and were neonatal lethal consistent with global loss of IFT88 function.(33)
Atomic force microscopy–based nanoindentation
Atomic force microscopy (AFM) nanoindentation was applied to freshly dissected femoral condyle and tibial plateau cartilage from mice at 8 weeks of age (n = 5 animals), following the established procedure.(34,35) Immediately after euthanasia, cartilage was dissected free of tendon and ligament tissues and glued onto AFM sample discs by a cyanoacrylate adhesive gel (Loctite 409; Henkel Corp., Rocky Hill, CT, USA). Throughout the procedure, tissues were immersed in PBS with protease inhibitors (Pierce 88266; Fisher Scientific, Rockford, IL, USA) to minimize postmortem degradation. Nanoindentation was performed on the surfaces of cartilage on a Dimension Icon AFM (Bruker Nano, Santa Barbara, CA, USA) using a colloidal AFM probe. The probe was custom-made by attaching a polystyrene microsphere (radius [R] ≈ 12.5 μm; PolySciences, Warrington, PA, USA) to a tipless cantilever (nominal force constant [k] ≈ 5.4 N/m, cantilever C, HQ:NSC35/tipless/Cr–Au; NanoAndMore, Watsonville, CA, USA) using liquid epoxy glue (M-bond 610; SPI Supplies, West Chester, PA, USA). For each sample, at least 10 random, different indentation locations were tested on each selected region of interest up to ~1μN force at 10 μm/s AFM z-piezo displacement rate. The effective indentation modulus (Eind) was calculated by fitting the entire loading portion of each indentation force delpth (F–D) curve to the Hertz model, where R is the tip radius and ν is the Poisson’s ratio (0.1 for cartilage).(36)
β-galactosidase staining
To visualize β-galactosidase (β-gal, LacZ) in Gli1-LacZ animals, β-gal staining was performed by wholemount (n = 4). Following euthanasia, joints were disarticulated and cleaned to expose the articular cartilage and fixed for 1 hour at 4°C in 1% paraformaldehyde (PFA). Tissues were washed three times for 5 minutes in LacZ wash buffer (2mM MgCl2, 0.01% Na Deoxycholate, and 0.02% NP-40 in 1× PBS) followed by staining at 37°C with rocking in the dark for either 3 hours (3-week-old samples) or 16 hours (8-week-old samples). To stain, X-gal (Millipore, Billerica, MA, USA; 71077-3) was freshly diluted 1:40 in LacZ base staining solution (5mM K3Fe(CN)6, 5mM K4Fe(CN)6 in LacZ wash buffer). All staining was performed with samples from Gli1--LacZ–negative animals to control for background staining. Following staining, samples were fixed in 4% PFA and imaged by wholemount using a Nikon dissecting microscope (Nikon, Tokyo, Japan) equipped with camera and SPOT imaging software (Diagnostic Instruments, Sterling Heights, MI, USA). For sectioning, samples were decalcified in 20% EDTA and processed for paraffin embedding. Six-micrometer-thick (6-μm-thick) sections were counterstained with eosin.
Tissue processing
Tissues processed for frozen sectioning were injected with 4% PFA and fixed for 2 days at 4°C, followed by decalcification in 20% EDTA for 6–10 days (depending on age), washes in 1× PBS, equilibration in 30% sucrose for 16–36 hours (depending on age), and embedding in optimal cutting temperature (OCT) compound. Sections were either taken at 18 μm for normal slide collection or at 10 μm on Kawamoto’s cryofilm(37) to preserve tissue morphology. Tissues processed for paraffin sectioning (basic histology and immunofluorescence) were injected with 10% neutral buffered formalin (NBF) and fixed for 2 days at 4°C, followed by decalcification in 20% EDTA for 6–10 days (depending on age), washes in 1× PBS and water, and dehydration in 70% ethanol overnight before automated processing into paraffin overnight. Tissues processed for RNAscope (Advanced Cell Diagnostics, Inc., San Jose, CA, USA) were decalcified using Morse’s solution as described(38) and processed into paraffin overnight. All sections were cut at 6-μm-thick and slides were prepared with mutant and littermate control sections on a single slide for each probe (n = 5 per time point).
Histology/immunofluorescence and imaging
Safranin O staining was performed using standard protocols and imaged using a Nikon Eclipse Ci-L upright microscope with attached DS-Fi3 camera (Nikon). Immunofluorescence staining was carried out on frozen or paraffin sections depending on antibody optimization. For staining on frozen sections, 1× PBS + 0.1% Tween was used for all washes and incubations. Sections were blocked with 5% donkey serum and incubated overnight at 4°C with primary antibody and for 2 hours at room temperature with a Cy3-conjugated secondary antibody (1:500; Jackson Immunoresearch, West Grove, PA, USA). For staining on paraffin sections, antigen retrieval was performed as indicated in Table 1. For non-amplified staining, sections were blocked with 5% donkey serum (and Mouse-on-Mouse block as necessary; Vector Laboratories, Inc., Burlingame, CA, USA) and incubated overnight at 4C with primary antibody and for 2 hours at room temperature with a Cy3- or AF488-conjugated secondary antibody (1:500; Jackson Immunoresearch). For amplified staining, sections were treated with 3.6% H2O2 (made from 30% stock in water) and permeabilized with 1× PBS + 0.1% Triton for 20 minutes. 1× PBS + 0.1% Tween was used for all following washes and incubations. Sections were blocked with 5% donkey serum (and Mouse-on-Mouse block as necessary; Vector Laboratories). Sections were incubated at room temperature for 90 minutes with primary antibody, for 1 hour with biotin-conjugated secondary antibodies (1:500; Jackson Immunoresearch), for 1 hour with Vectastain Elite ABC amplification kit (Vector Laboratories) and developed for 10 minutes with fluorescein-conjugated or Cy3-conjugated Tyramide amplification system (Perkin Elmer, Shelton, CT, USA). Amplified staining for Aggrecan was performed without detergent (1× PBS only). All sections were counterstained with DAPI (1 mg/mL stock per manufacturer guidelines; Sigma-Aldrich, St. Louis, MO, USA; D9542) diluted 1:10,000 in wash buffer for 10 minutes and coverslipped using Prolong Gold (Invitrogen, Waltham, MA, USA; P36930). A Leica DMi8CEL Advanced inverted SP8 confocal was used to collect fluorescent and DIC images (Leica Camera, Wetzlar, Germany).
Table 1.
Primary Antibody Information and Immunofluorescence Protocols
| Antibody | Manufacturer | Catalog # | Protocol | Amplified | Antigen retrieval | Dilution |
|---|---|---|---|---|---|---|
|
| ||||||
| MMP13 | Abcam (Cambridge, UK) | Ab39012 | Frozen | No | - | 1:100 |
| Perlecan | Life Technologies (Carlsbad, CA, USA) | MA1-06821 | Paraffin | No | (ii) | 1:50 |
| Col2a1 | MilliporeSigma (Burington, MA, USA) | MAB8887 | Paraffin | No | (ii) | 1:50 |
| ColX | MilliporeSigma | 234196 | Paraffin | No | (ii) | 1:50 |
| Acetylated tubulin | MilliporeSigma | T7451 | Paraffin | No | (ii) | 1:200 |
| Sox9 | Novus Biologicals (Littleton, CO, USA) | AF3075 | Paraffin | Yes | (ii) | 1:2000 |
| Aggrecan | Dev Studies Hyb Bank (Iowa City, IA, USA) | 12/21/1-C-6 | Paraffin | Yes | (ii) + (iii) | 1:25 |
| pSmad2 | Cell Signaling (Danvers, MA, USA) | 3108S | Paraffin | Yes | (ii) | 1:1500 |
| pSmad159 | Cell Signaling | 13820 | Paraffin | Yes | (i) | 1:800 |
Antigen retrieval methods
1 mg/mL trypsin (Sigma-Aldrich; T7168) for 20 minutes at 37°C.
10 μg/mL Proteinase K for 20 minutes at 37°C.
10mM dithiothreitol (DTT) for 2 hours at 37°C (reduction) followed by 40mM iodoacetamide for 2 hours at room temperature (alkylation) followed by 5 mg/mL Hyaluronidase (in 10% serum) for 1 hour at 37°C.
RNAscope
RNAscope in situ hybridization (n = 5 per time point) was carried out using RNAscope2.5 HD Detection reagent-RED (Advanced Cell Diagnostics) to visualize the spatiotemporal expression of genes listed in Table 2. Briefly, sections were pretreated with a custom reagent and the probe was hybridized for 2 hours at 40°C in a custom oven. Signal was amplified with multiple reagents as per manufacturer’s protocols and final signal was detected and visualized by reaction with Fast Red substrate for 10–20 minutes (dependent on age) at room temperature. Companion sections were hybridized with positive (Advanced Cell Diagnostics; Cat. No. 313911) or negative control probes (Advanced Cell Diagnostics; Cat. No. 310043) to assure signaling specificity (data not shown). Sections imaged with brightfield were counterstained with hematoxylin, dried, and sealed with Permount. Sections imaged with confocal microscopy were counterstained with Hoescht and coverslipped with Prolong Gold. A Nikon Eclipse Ci-L upright microscope with attached DS-Fi3 camera was used to capture brightfield images. A Leica DMi8CEL Advanced inverted SP8 confocal was used to collect fluorescent and differential interference contrast (DIC) images.
Table 2.
RNAscope Probes
| Gene name | Probe name | Catalog number |
|---|---|---|
|
| ||
| Adamts4 | Mm-Adamts4 | 497161 |
| Adamts5 | Mm-Adamts5 | 427621 |
| Col2a1 | Mm-Col2a1 | 407221 |
| Acan | Mm-Acan | 439101 |
| Prg4 | Mm-Prg4 | 437661 |
| Gli1 | Mm-Gli1 | 311001 |
| Ptch1 | Mm-Ptch1 | 402811 |
| Ihh | Mm-Ihh | 413091 |
Quantification from tissue sections
All quantification was carried out using ImageJ software (National Institutes of Health [NIH], Bethesda, MD, USA; https://imagej.nih.gov/ij/) with FIJI plugins. All sections were chosen from the central region of the medial knee that includes that concentrated load-bearing region of the tibial plateau. For cilia incidence, acetylated tubulin immunofluorescence DAPI signal was quantified manually using the cell counter plugin in FIJI. For AC thickness measurements (n = 5 controls and four mutants at 3 weeks; n = 6 controls and 7 mutants at 8 weeks). Safranin O-stained sections were used. For each section (three to six per animal), the thickest region of AC was identified, and eight lines were drawn manually and measured. An average and standard deviation (SD) was calculated from all lines in all sections per region of interest. Percent Sox9-positive (n = 5 at 3 weeks, n = 6 at 8 weeks), pSmad2-positive (n = 7 at 8 weeks), and pSmad159-positive (n = 7 at 8 weeks) cells was calculated from immunofluorescent-stained slides counterstained with DAPI and additionally imaged with DIC to outline nonmineralized and mineralized cartilage zones (two sections per animal per stain). The total number of cells in each region was calculated by thresholding the DAPI signal in ImageJ and analyzing particles to count each nucleus. The same thresholding technique was applied to count Sox9, pSmad2, and pSmad159-positive cells and divided by the total (DAPI) number of cells per region. Cell density at 3 weeks and 8 weeks was calculated from DAPI staining and DIC imaging (from Sox9, pSmad2 and pSmad159-stained sections). AC composition at 8 weeks was calculated from DIC imaging (from Sox9, pSmad2 and pSmad159-stained sections). To quantify Collagen II immunofluorescence, total AC area for was manually outlined on each section (two sections per animal) and the mean gray value was measured separately for femoral condyle and tibial plateau. RNAscope for Prg4 was quantified by thresholding the signal from brightfield images manually and applying the same threshold value to all images. The thresholded area was quantified over total AC area (drawn manually) in all sections (one section per animal, n = 5). Gli1-LacZ in tissue sections at 8 weeks was quantified manually using the cell counter plugin in FIJI (n = 6). The approximate location of the tidemark in control animals was drawn, as described in the Results section, using knowledge from DIC images of previous quantification, and applied to all control and mutant tissue sections. The femoral condyle tidemark was drawn four cell layers from the surface and the tibial plateau tidemark was drawn seven cell layers from the surface.
Statistical methods
Sample sizes were justified based on prior experimentation using the described techniques. A relative standard deviation of 15%–20% is found in cartilage biomechanics for mice. Based on a 30% estimated difference between groups, a statistical power (1 – β) or 80% at a significance level of α = 5% is obtained with a sample size of n = 6 biological repeats per group. For histologic assessments, we also aimed for n = 6 for most measurements but used n = 4 for quantification analyses of 3-week samples where no visual differences were detected for multiple stains and n = 5 for RNAscope where the procedure limits the number of slides per experiment. Data were analyzed using Prism 9 (Graphpad Software, Inc., San Diego, CA, USA). For all analyses, two groups were compared to each other (control versus mutant) and we used unpaired t tests with Welch correction where p < 0.05 was considered statistically significant. All data are shown as box plots with interquartile range and minimum and maximum.
Results
Gdf5Cre is joint specific, and conditional deletion of Ift88 results in loss of primary cilia
To target primary cilia specifically in synovial joints, we used Gdf5Cre in combination with an Ift88 conditional allele. Gdf5 is a unique marker for the embryonic mesenchymal progenitor cells of synovial joints called the interzone. Gdf5Cre faithfully marks these cells in the embryo and lineage-traces to all of the mature tissues of synovial joints(4,39) (Fig. 1Aa–c) but not growth plate chondrocytes (Fig. 1Ad). Thus, we combined Gdf5Cre with an Ift88 conditional allele whose product (intraflagellar transport protein 88, IFT88) is important in anterograde transport in cilia and maintenance of cilium structure. We visualized primary cilia using an antibody against acetylated tubulin. They were evident as expected in growth plate chondrocytes in control and mutant animals (Fig. 1B). They were also present and clear in control AC (Fig. 1Ca,b) but much reduced in the AC of mutant animals (Fig. 1Cc,d). The incidence of primary cilia was quantified in control and mutant AC (Fig. 1D), confirming that our model was reliable and the loss of IFT88 resulted in a significant reduction of primary cilia specifically in articular chondrocytes.
Fig. 1.
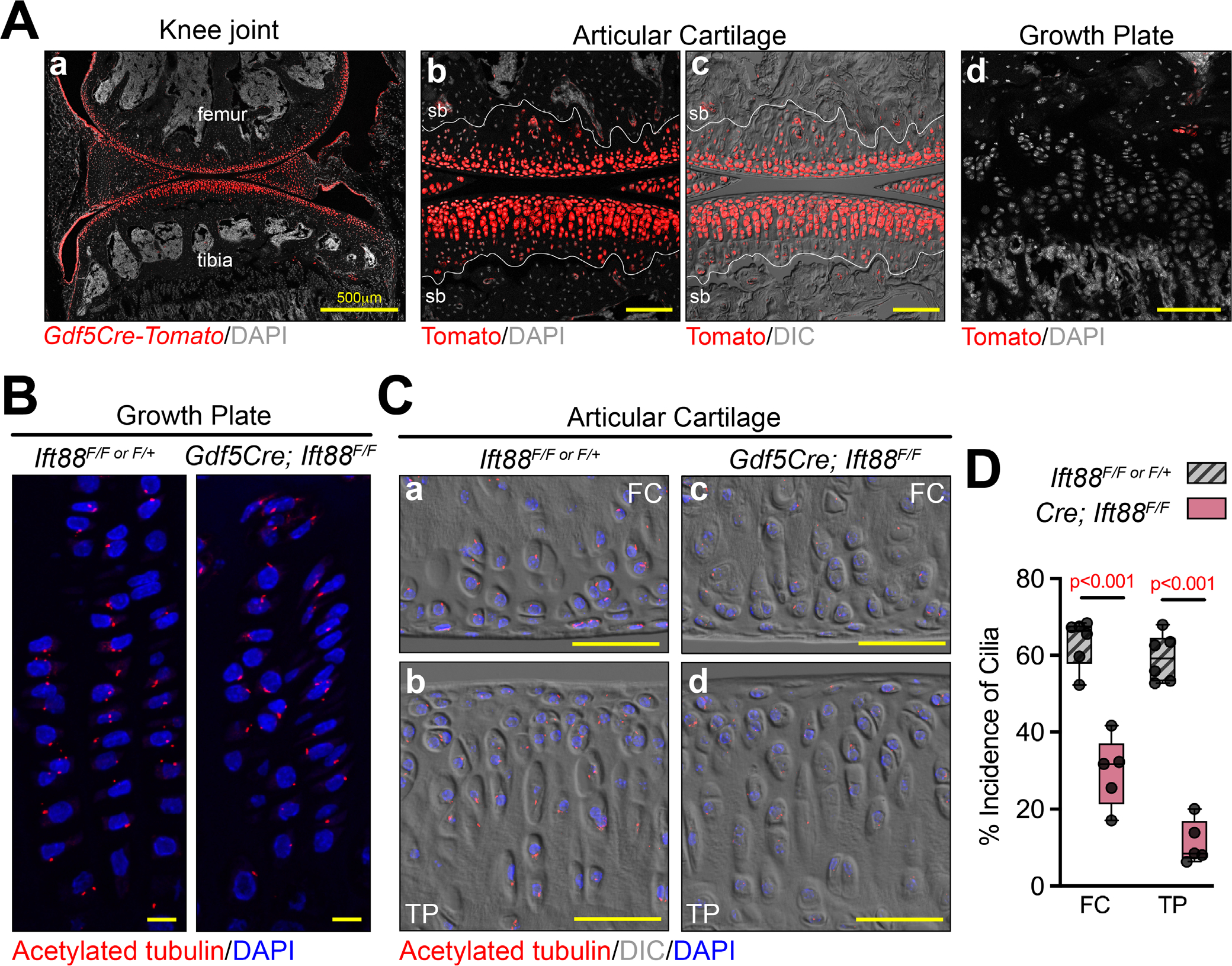
Gdf5Cre is joint specific and conditional deletion of Ift88 results in loss of primary cilia. (A) Gdf5Cre/+; Rosa-tdTomato lineage cells are present in adult joints (Aa) and all zones of AC as shown with DIC imaging to visualize the tidemark (Ab,c) but are absent in growth plate (Ad) (n = 3). (B,C) Primary cilia are visualized by immunofluorescence staining for acetylated tubulin. They are present in control and mutant growth plate chondrocytes (B) and in control AC (Ca,b), and visibly reduced in mutant AC (Cc,d). (D) The incidence of cilia in AC is quantified as a percentage of AC nuclei (n = 5). Statistical analysis was performed by unpaired t tests with Welch correction. Scale bar = 100 μm (Ab–d), 10 μm (B), 50 μm (C). All images displayed with FC on top and TP on bottom unless otherwise noted. FC = femoral condyle; sb = subchondral bone; TP = tibial plateau.
Joint-specific deletion of Ift88 leads to disrupted AC tidemark patterning and reduced AC stiffness
Next, we carried out histological analysis and performed mechanical testing to assess the consequences of primary cilia loss on postnatal AC morphogenesis. For all images provided, the femoral condyle is on top and the tibial plateau is on the bottom, unless otherwise noted. We used Safranin-O staining to visualize basic histology and proteoglycan content in the ECM (Fig. 2A). At birth, nascent AC of mutants was indistinguishable from that in controls (Fig. 2Aa,b, brackets). Up to 3 weeks, immature AC developed similarly in all animals (Fig. 2Ac,d) and higher magnification images revealed no discernible differences in cell morphology or patterning (Fig. 2Ae–h). At 8 weeks, however, we noted irregular accumulation of interterritorial ECM (arrows) and varying chondrocyte size (asterisks) in the femoral condyle (Fig. 2Ai–l) and a consistent reduction of Safranin-O in the tibial plateau (Fig. 2i,j,m,n). Thus, Ift88 deletion primarily affected the maturation phase of postnatal AC morphogenesis. Growth plates were unaffected at all stages consistent with no recombination from Gdf5Cre (Fig. S1). To assess possible functional consequences of these morphologic changes in mature AC, we used AFM nanoindentation to measure tissue stiffness. We measured two regions on the medial tibial plateau (the edge covered by meniscus and the center load-bearing region) and one region on the femoral condyle. In all regions of interest, the indentation modulus (ie, resistance to deformation) was significantly decreased in mutants (Fig. 2B). Thus, Ift88 deletion in AC results in impaired load-bearing function during tissue maturation.
Fig. 2.
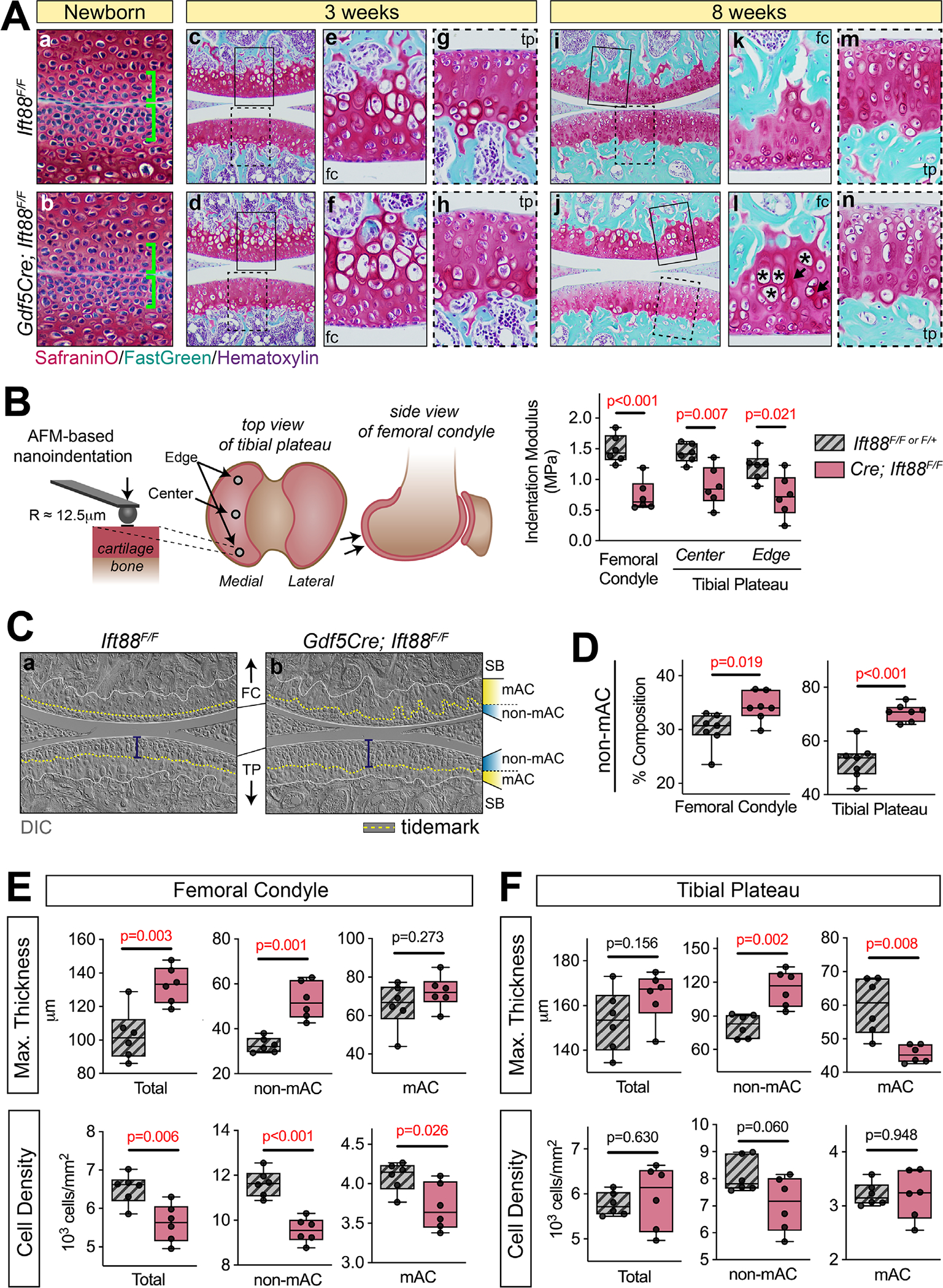
Joint-specific deletion of Ift88 leads to disrupted AC tidemark patterning and reduced AC stiffness. (A) Safranin-O staining at birth through 8 weeks. Nascent AC at birth is unchanged in mutant joints (Aa,b, brackets). AC is also unchanged at 3 weeks. Low magnification (Ac,d) and high magnification images of femoral condyle (Ae,f) or tibial plateau (Ag,h) are comparable. At 8 weeks, control AC (Ai,k,m) displays strong safranin O staining throughout the femoral condyle (Ak) and tibial plateau (Am). In mutant AC (Aj,l,n), the femoral condyle displays aberrant ECM deposition (Al, arrows) and increased cell size (Al, asterisks), while the tibial plateau displays reduced Safranin-O staining (An). (B) Biomechanical function was measured by AFM-snanoindentation from three regions. Schematic shows approximate location of the two regions of tibial plateau AC. Indentation modulus was significantly decreased in all locations. (C) DIC imaging of the tidemark (dashed yellow line) reveals ordered patterning in control AC (Ca). In mutant AC, the tidemark of the femoral condyle is disordered and its distance from the surface is increased in the tibial plateau (Cb, bracket). (D) Nonmineralized (non-mAC) composition is shown as a percent area from multiple tissue sections and is significantly increased in mutant AC. (E,F) Measurements of maximum AC thickness and cell density. In the femoral condyle (E), maximum AC thickness of total cartilage and non-mAC is significantly increased. Cell density is significantly decreased throughout mutant AC. In the tibial plateau (F), maximum AC thickness of total cartilage is unchanged, however non-mAC is significantly increased and mAC is significantly decreased. Cell density is unchanged. Statistical analyses were performed by unpaired t tests with Welch correction. Scale bar = 100 μm. All images displayed with FC on top and TP on bottom unless otherwise noted. AC = articular cartilage; FC = femoral condyle; SB = subchondral bone; mAC = mineralized AC; non-mAC = nonmineralized AC; TP = tibial plateau.
Next, we utilized a combination of Safranin-O–stained sections and sections imaged with DIC to visualize the tidemark and quantify AC composition of mineralized (mAC) and nonmineralized regions (non-mAC), maximum AC thickness, and cell density. These analyses revealed several notable changes in mutants. First, in healthy AC the tidemark (Fig. 2Ca, dashed yellow line) closely followed the contour of the femoral condyle surface and was two to three cell layers below the surface. Instead, it was relatively flat in the central load-bearing region of the tibial plateau and was located about five to six cell layers from the surface (Fig. 2Ca, bracket). In mutants, the tidemark was more difficult to visualize, but followed an irregular path in the femoral condyle and was flat, but noticeably further from the surface, in the tibial plateau (Fig. 2Cb, bracket). Quantifying AC composition as a percent area (Fig. S2A), we found that nonmineralized articular cartilage constituted approximately 31% of the femoral condyle and 51% of the tibial plateau AC in control animals. In mutants, this fraction increased to 36% and 70% in the femoral condyle and tibial plateau, respectively (Fig. 2D) and was accompanied by respective decreases in mineralized AC (Fig. S2B). We measured maximum AC thickness and cell density to further understand changes in zonal composition. In the femoral condyle, we found that the transition of mutant AC to subchondral bone tended to be more irregular and significantly increased maximal non-mAC thickness (Fig. 2E, top). Cell density was in general decreased in femoral condyle AC (Fig. 2E, bottom). In the tibial plateau, although there was no overall change in AC thickness, the maximal thickness of non-mAC increased whereas that of mAC decreased (Fig. 2F, top). Cell density remained unchanged in the tibial plateau (Fig. 2F, bottom). Cell density and AC thickness were both unchanged at 3 weeks (Fig. S2C).
Primary cilia regulate chondrocyte fate and matrix deposition into mineralized and nonmineralized zones
Next, we performed immunofluorescence staining to examine several key ECM and chondrocyte-specific markers. Collagen II is a major structural component of cartilage ECM, responsible for tissue tensile strength.(40) In control animals, it was uniformly dispersed in AC at 3 and 8 weeks (Fig. 3Aa,b). In mutant animals, we noted no changes at 3 weeks (Fig. 3Ac,B, top) but a reduction at 8 weeks (Fig. 3Ad,B, bottom). Aggrecan is the major proteoglycan in cartilage ECM that yields compressive load-bearing and energy dissipation properties. In healthy tissue, it was uniformly deposited over the entire thickness of developing AC at 3 weeks (Fig. 3Ca) and was enriched in nonmineralized cartilage at 8 weeks (Fig. 3Cb). A clear boundary (ie, tidemark) could be visualized where the bottom aggrecan-low mineralized cartilage met the top aggrecan-rich nonmineralized cartilage (Fig. 3Cb, arrows). In Ift88 mutants, aggrecan deposition was comparable to controls in immature cartilage at 3 weeks (Fig. 3Cc), but at 8 weeks the mutant tissue displayed notable disorganization, lack of tidemark, and variable aggrecan deposition (high, arrows; low, arrowheads) (Fig. 3Cd). Sox9 is considered a master transcriptional regulator of chondrocyte fate and can directly regulate the expression of aggrecan and collagen II.(41) In control immature AC, Sox9 was present uniformly in articular chondrocytes (Fig. 3Da) consistent with the uniform distribution of collagen II and aggrecan at this 3-week stage. In mature AC, Sox9-positive cells were largely restricted to the nonmineralized cartilage above the tidemark (Fig. 3Db, dashed line), as visualized by DIC imaging (Fig. S3a–c), and positive cell distribution was consistent with previous reports of Sox9 function in AC.(42,43) Mutant tissues at 3 weeks showed comparable Sox9 localization to controls (Fig. 3Dc,E, top). At 8 weeks Sox9-positive chondrocytes continued to be broadly present in nonmineralized cartilage (Fig. 3Dd and Fig. S3d–f), but positive cells could also be seen beneath the tidemark in femoral condyle (Fig. 3D, arrows; Fig. 3E, bottom). Taken together, these data show that Ift88 mutants display alterations in the patterning of nonmineralized chondrocytes and in the distribution of ECM components in mature AC, but not in immature AC.
Fig. 3.
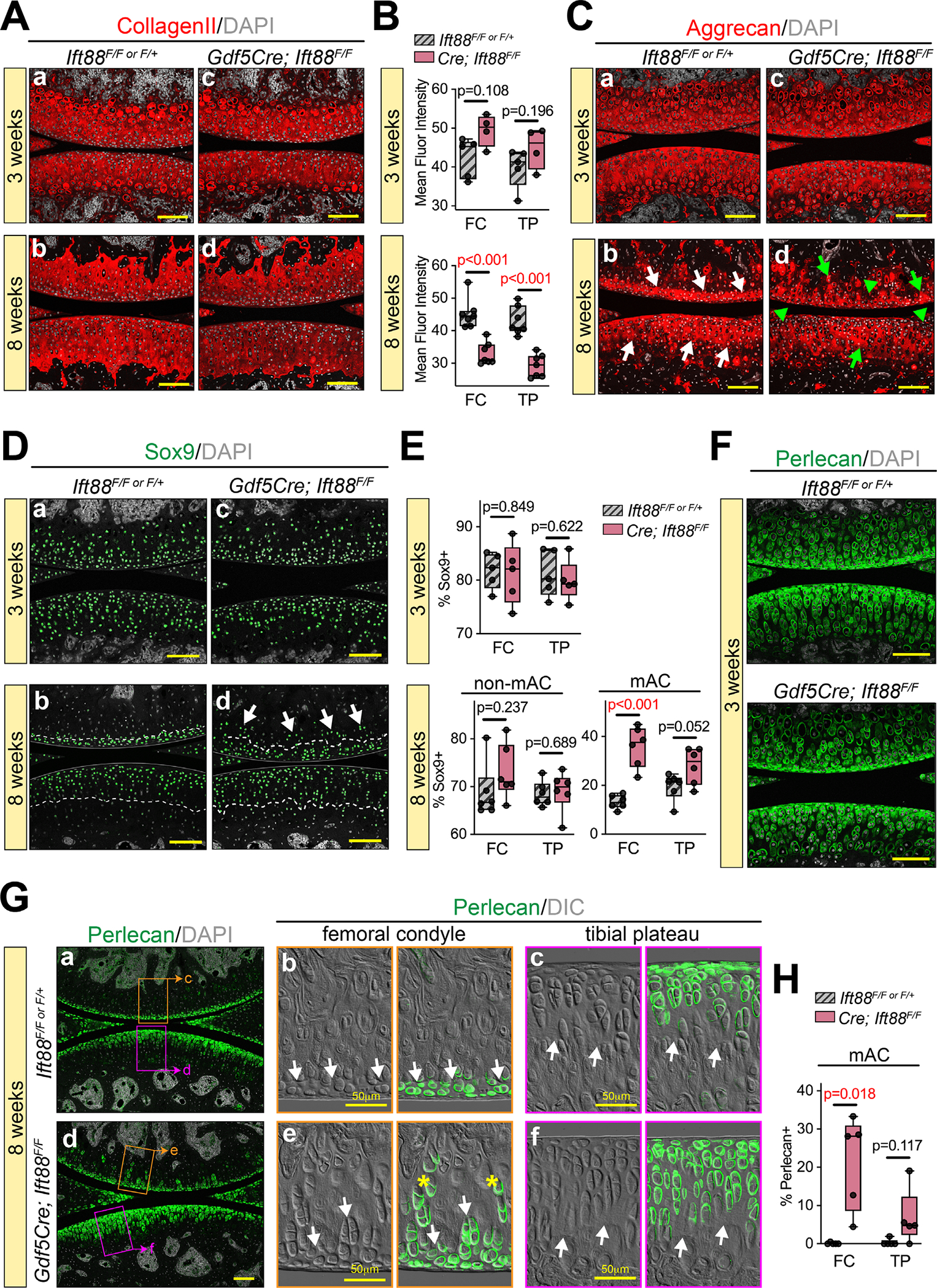
Primary cilia regulate chondrocyte fate and matrix deposition in mineralized and nonmineralized zones. (A,B) Collagen II immunostaining is broadly distributed in AC and comparable at 3 weeks (Aa–c) and reduced at 8 weeks in mutants (Ad–f). The mean fluorescence intensity is quantified at 3 weeks (top) and 8 weeks (bottom) in B. (C) Aggrecan immunostaining is broadly distributed and comparable at 3 weeks (Ca,c). It is enriched in nonmineralized 8-week AC of controls (Cb, white arrows point to approximate tidemark) and is disorganized in 8-week AC of mutants (Cd), with irregular areas of high staining (green arrows) and low staining (green arrowheads). (D,E) Sox9 is broadly distributed and comparable in AC at 3 weeks (Da,c). It is also broadly distributed in nonmineralized AC of controls (Db) and mutants (Dd) at 8 weeks (dashed line represents the tidemark), but mutant femoral condyle AC also displays ectopic Sox9-positive cells beyond the tidemark (Dd, white arrows). The percentage of Sox9-positive cells is quantified of all cells at 3 weeks (top) and in zones at 8 weeks (bottom) in E. (F–H) Perlecan immunostaining detects the pericellular chondrocyte matrix. At 3 weeks (F), Perlecan is broadly distributed in AC and comparable in controls and mutants. At 8 weeks (G), Perlecan is enriched nearest the surface in both controls (Ga) and mutants (Gd). High magnification with DIC shows most perlecan staining in nonmineralized AC above the tidemark (Gb–c, e–f, white arrows). Ectopic perlecan-positive cells are present below the mutant femoral condyle tidemark (e, asterisks), quantified in H. Statistical analyses were performed by unpaired t tests with Welch correction. Scale bar = 100 μm unless otherwise noted. All images displayed with FC on top and TP on bottom unless otherwise noted. FC = femoral condyle; TP = tibial plateau; mAC = mineralized articular cartilage; non-mAC = nonmineralized articular cartilage.
Pericellular matrix (PCM) creates a thin microenvironment surrounding chondrocytes within the broader ECM and is considered a crucial transducer of biomechanical signals.(44) The PCM is rich in certain matrix macromolecules including collagen VI and perlecan; together with the enclosed chondrocytes, the PCM is referred to as a chondron.(45–47) To analyze the PCM, we performed immunofluorescence staining for perlecan. In immature AC at 3 weeks, perlecan was broadly distributed in both control and mutant tissue (Fig. 3F). In mature AC at 8 weeks, perlecan was localized to chondrocytes nearest to the surface in controls (Fig. 3Ga) and high magnification images overlaid with DIC imaging revealed restriction to the nonmineralized cartilage above the tidemark (Fig. 3Gb,c, arrows). Perlecan deposition was significantly disrupted in mutant AC at 8 weeks (Fig. 3Gd). In the femoral condyle, chondrocytes with a perlecan-rich PCM could be visualized far below the tidemark (Fig. 3Ge, asterisks; Fig. 3H), but in tibial plateau AC there was an expansion of perlecan-positive chondrocytes extending deeper into the concentrated load-bearing region above the tidemark (Fig. 3Gf). Together, these data provide strong evidence that deletion of Ift88 in AC disrupts tidemark patterning and chondrocyte fate/microenvironment during postnatal maturation.
Ift88 mutants do not show indications of AC matrix degradation or precocious hypertrophy
Previous in vitro studies have shown that primary cilia, and intraflagellar transport (IFT) proteins, regulate ECM proteases in chondrocytes.(48) To test whether this applied to our in vivo model, we performed immunofluorescence staining for MMP13 (collagenase) and in situ hybridization for Adamts4 and Adamts5 (aggrecanases) at 8 weeks. Matrix metalloproteinase 13 (MMP13) was restricted to the base of AC at the transition to subchondral bone in controls (Fig. 4Aa) and was unchanged in mutants (Fig. 4Ab). We used RNAscope (FastRed detection) to perform in situ hybridization and took advantage of FastRed fluorescence for imaging low-expressing genes. In AC, we found no expression of either Adamts4 or Adamts5 in controls (Fig. 4Ba,Ca) or mutants (Fig. 4Bb,Cb). To ensure these data were reliable, we compared to other tissues in and around the joint. Adamts4 (Fig. S4A) was expressed in primary spongiosa (PS, arrowheads) beneath the growth plate and in periosteum (PO, arrows), and Adamts5 (Fig. S4B) was highly expressed by inner synovial fibroblasts in both controls and mutants. These data indicate that the changes observed in collagen II and aggrecan deposition in mutant AC (Fig. 2A–C) were likely not caused by matrix degradation. Though protease gene expression and enzymatic activity does not always correspond with each other, the lack of detectable protease gene expression supports our conclusion. Collagen X is a marker of hypertrophic and mineralizing chondrocytes and often accompanies precocious matrix degradation. We found collagen X similarly localized in control and mutant cells juxtaposed to the developing secondary ossification center at 3 weeks (Fig. 4Da, b) and 8 weeks (Fig. 4Dc,d), consistent with a lack of precocious hypertrophy closer to the AC surface.
Fig. 4.
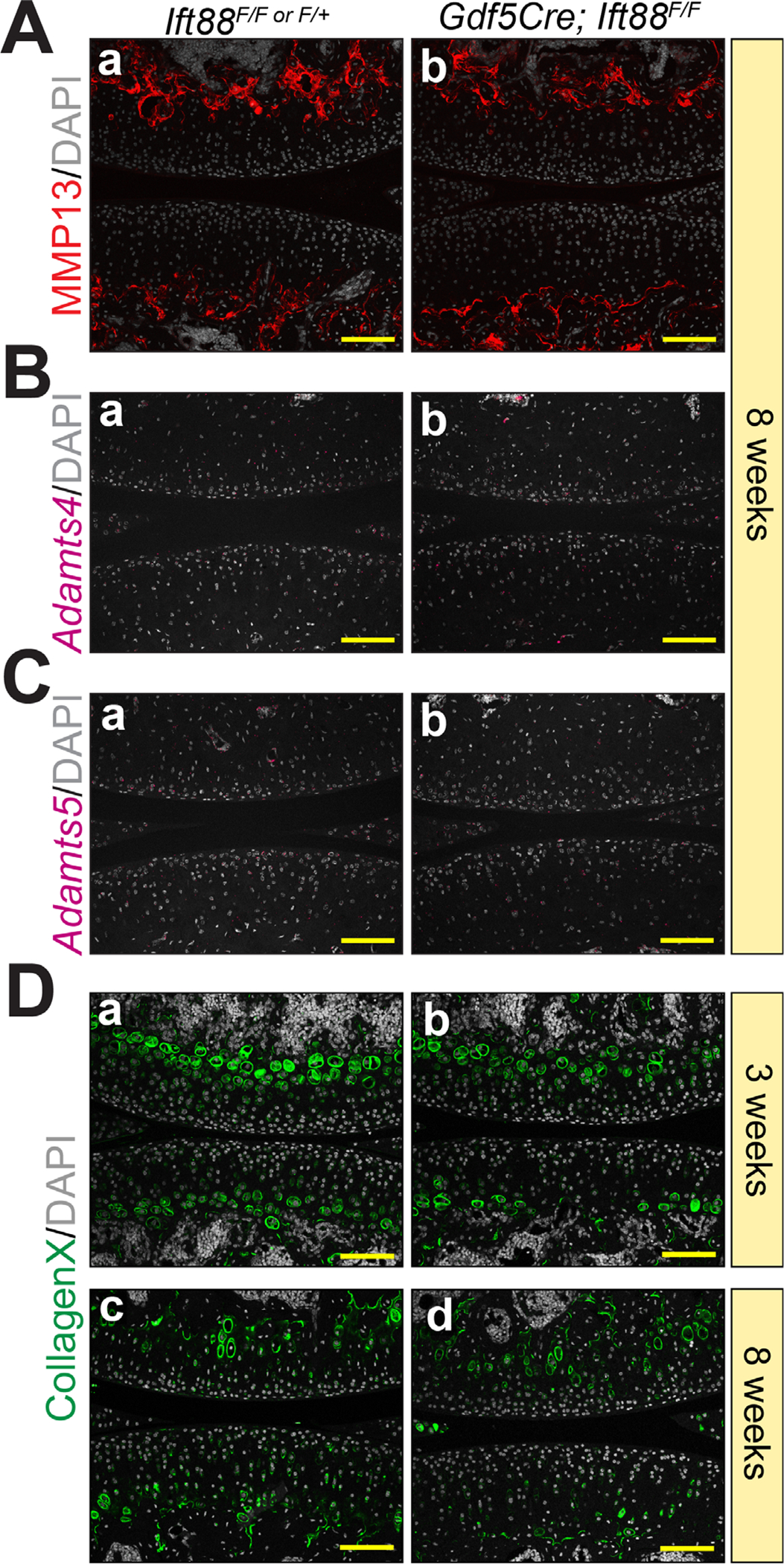
Ift88 mutants do not show indications of AC matrix degradation or precocious hypertrophy. (A) Immunofluorescence for MMP13 is restricted to the subchondral bone at 8 weeks and is unchanged in controls (Aa) and mutants (Ab). (B,C) Adamts4 and Adamts5 are not detectable by RNAscope in control (Ba, Ca) or mutant (Bb, Cb) AC at 8 weeks. (D) Immunofluorescence for CollagenX is restricted to chondrocytes near the subchondral bone and is comparable in controls and mutants at 3 weeks (Da,b) and 8 weeks (Dc,d). Scale bar = 100 μm. All images displayed with FC on top and TP on bottom. FC = femoral condyle; TP = tibial plateau.
Altered ECM in mature mutant AC correlates with abnormal gene expression
To assess transcriptional changes of Col2a1, Acan, and Prg4, we utilized RNAscope (FastRed detection) and imaged these high-expressing genes with brightfield. At 3 weeks, Col2a1 and Acan were broadly expressed in immature AC of both controls (Fig. 5Aa,Ba) and mutants (Fig. 5Ab,Bb), consistent with our findings from immunofluorescent staining at this stage described in Fig. 3A–C. In mature control AC at 8 weeks, Col2a1 (Fig. 5Ac,d) and Acan (Fig. 5Bc,d) transcripts were restricted to layers of AC corresponding with the domain of nonmineralized cartilage, two to three layers deep in the femoral condyle and five to six layers deep in the tibial plateau. In mutant AC, both Col2a1 (Fig. 5Ae,f) and Acan (Fig. 5Be,f) expression appeared much more disorganized, particularly in the femoral condyle (arrows), consistent with deviant Sox9-positive cells outside of the nonmineralized cartilage (Fig. 3D,E). High-magnification images (Fig. 5Af, Bf) highlighted the clear disorganization of gene expression in all mutant cartilage, consistent with dysregulated tidemark patterning and ECM deposition (Figs. 2 and 3) and reduced expression per cell of Col2a1 and Acan in the concentrated load-bearing region of the tibial plateau compared to this region in controls. Prg4 (lubricin) is important for protecting AC from OA.(49–53) Prg4 was highly expressed and uniformly distributed in cell layers closest to the AC surface in control animals at 3 weeks (Fig. 5Ca) and at 8 weeks (Fig. 5Cc–e), extending slightly farther in the tibial plateau (Fig. 5Ce, bracket). In mutant femoral condyle cartilage, we found a significant reduction of Prg4 at both 3 weeks (Fig. 5Cb) and at 8 weeks (Fig. 5Cd,f; Fig. 5D top). In mutant tibial plateau, there was no change in Prg4 at 3 weeks (Fig. 5Cb) and a striking increase in the concentrated load-bearing region at 8 weeks (Fig. 5Cd,f, bracket; Fig. 5D, bottom). These data suggest that reduction/disorganization of collagen II and aggrecan deposition in mature cartilage were due to changes at the transcriptional level and that, surprisingly, Prg4 expression was altered in both immature and mature cartilage of mutants.
Fig. 5.
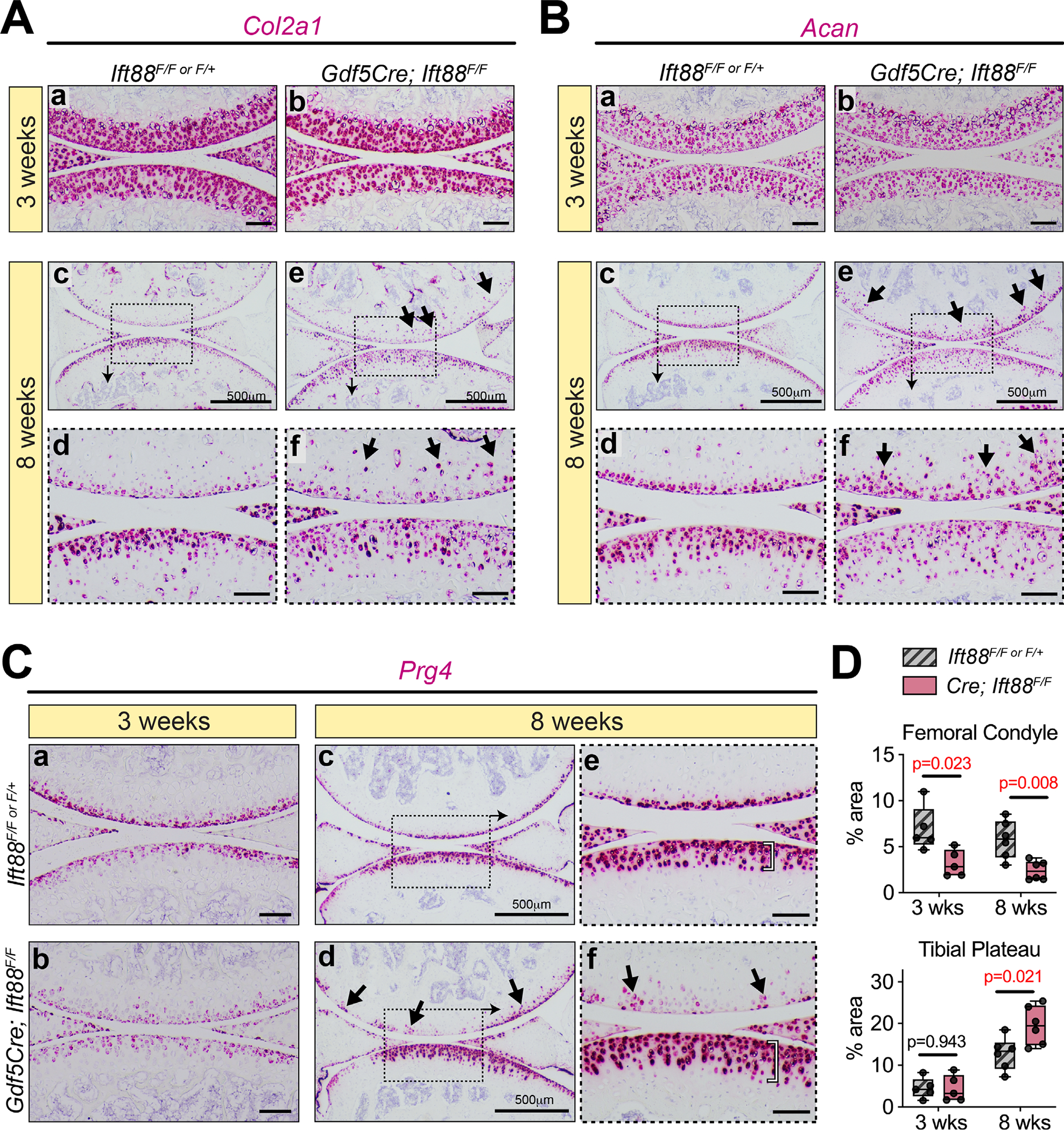
Altered ECM in mature mutant AC correlates with abnormal gene expression. (A,B) RNAscope for Col2a1 (A) and Acan (B) is comparable in controls and mutants at 3 weeks (Aa,b, Ba,b). In control AC at 8 weeks, low magnification images (Ac, Bc) and high magnification images (Ad, Bd) show organization and high levels of expression closest to the surface. In mutant AC, low magnification images (Ae, Be) and high magnification images (Af, Bf) show generally disorganized expression that is displayed deeper into the tissue than in controls (arrows) and decreased particularly in the tibial plateau. (C,D) RNAscope for Prg4 is shown with quantification in D. In the femoral condyle, Prg4 is decreased at 3 weeks (Ca,b, top). At 8 weeks, low magnification images (Cc,d, top) and high magnification images (Ce,f, top) show generally decreased but patchy expression (arrows). In the tibial plateau, Prg4 expression is unchanged at 3 weeks (Ca,b, bottom). At 8 weeks, low-magnification images (Cc,d, bottom) and high-magnification images (Ce,f, bottom) show a significant increase of Prg4 that extends further from the surface (bracket). Quantification of Prg4 in D is shown as a percentage area of AC per section after staining area was measured by thresholding the red signal in ImageJ. Statistical analyses performed by unpaired t tests with Welch correction. M (with arrow) denotes regions of mAC in images. Scale bar = 100 μm. All images displayed with FC on top and TP on bottom unless otherwise noted. FC = femoral condyle; TP = tibial plateau; mAC = mineralized articular cartilage; non-mAC = nonmineralized articular cartilage.
Primary cilia impart load/location-dependent functions for TGFβ and bone morphogenetic protein signaling
Primary cilia are considered a signaling nexus that exert regulatory influences on most major signaling pathways.(54) TGFβ and bone morphogenetic protein (BMP) regulate chondrocyte fate and ECM anabolism by distinct mechanisms via downstream SMAD-dependent mechanisms.(55) Although TGFβ signaling largely maintains nonmineralized chondrocyte function and prevents hypertrophy,(56–64) BMP signaling can influence both nonmineralized and mineralized functions dependent on context.(65–70) To assess the involvement of these pathways, we used immunofluorescence for pSmad2 (TGFβ) or pSmad159 (BMP). In immature AC at 3 weeks, pSmad2-positive cells were broadly distributed in all layers (Fig. 6Aa), whereas pSmad159-positive cells were largely present in the deepest layers of cartilage (Fig. 6Ba). These patterns were unchanged in mutant cartilage (Fig. 6Ab,Bb). In mature control AC at 8 weeks, pSmad2 was abundantly present (Fig. 6Ca) and localized to non-mineralized AC above the tidemark (Fig. 6Cb,c, dashed line). In mutant femoral condyle AC, pSmad2 was similarly localized to non-mineralized cartilage despite tidemark topographical irregularities (Fig. 6Cd,e,D). However, in mutant tibial plateau AC, areas of pSmad2-negative cells could be visualized (Fig. 6Cf, arrow) and were quantifiably reduced (Fig. 6D). We performed parallel experiments for pSmad159 and found similar localization in nonmineralized cartilage in control animals (Fig. 6Ea–c) and in mutant femoral condyle (Fig. 6Ed,e), but a significant, quantifiable and specific loss only in mutant tibial plateau (Fig. 6Ef,F). These data suggest that disruption of IFT88 leads to alterations in BMP signaling and, to a lesser degree, TGFβ signaling in the concentrated load-bearing region of the tibial plateau simultaneously with ECM compositional alterations to collagenII and aggrecan described in Fig. 3 and Fig. 5.
Fig. 6.
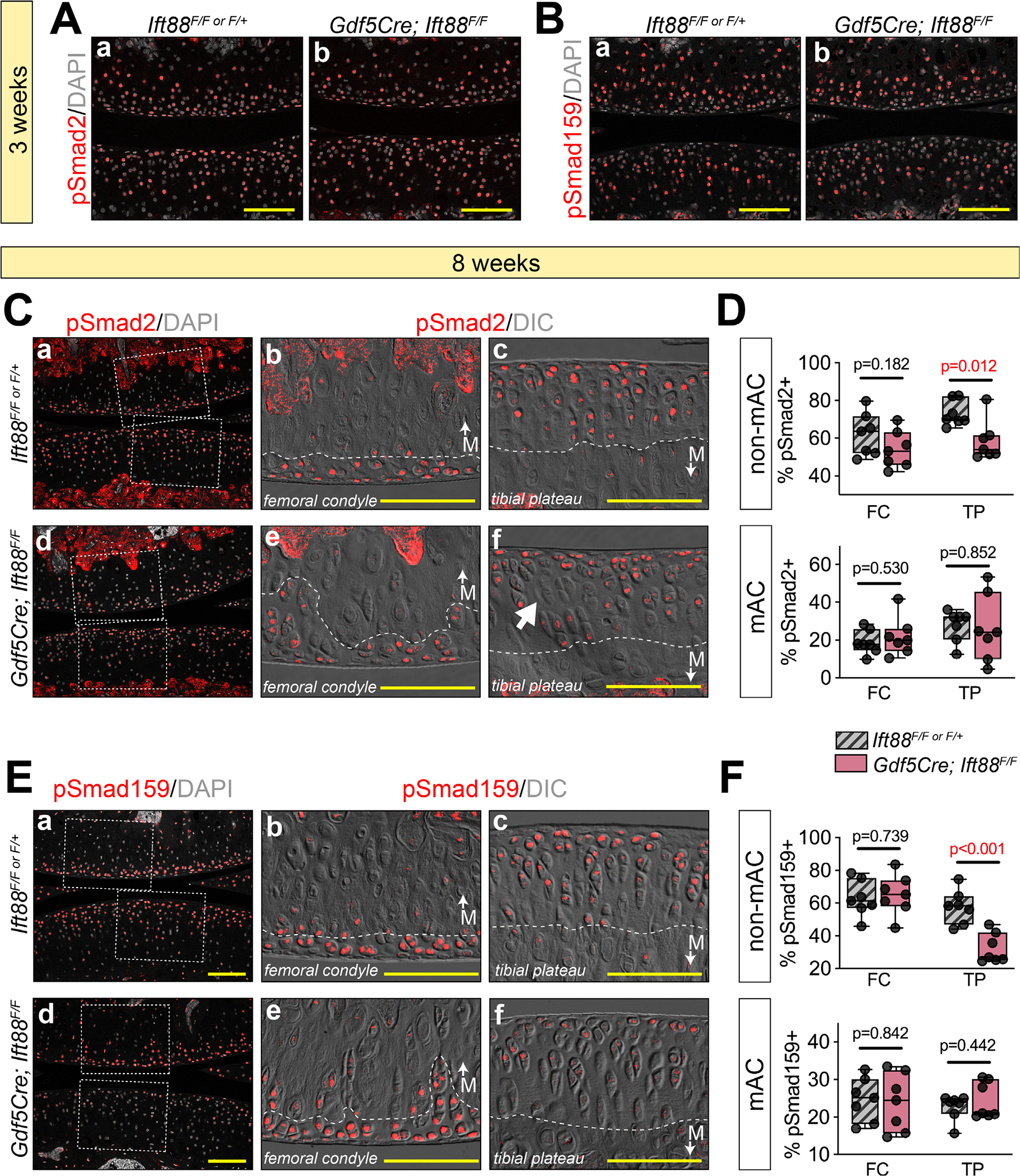
Primary cilia impart load/location-dependent functions for TGFβ and BMP signaling. (A,B) Immunofluorescence for pSmad2 (TGFβ) and pSmad159 (BMP) at 3 weeks. pSmad2-positive chondrocytes are distributed throughout AC in both controls (Aa) and mutants (Ab). pSmad159-positive chondrocytes at 3 weeks are localized in middle and bottom layers of AC in both controls (Ba) and mutants (Bb). (C–F) Immunofluorescence and quantification for pSmad2 (TGFβ) and pSmad159 (BMP) at 8 weeks. DIC imaging is used to visualize the tidemark (dashed white line). (C) Low magnification (Ca,d) and high magnification of femoral condyle (Cb,e) and tibial plateau (Cc,f) shows that pSmad2 is mostly localized to non-mAC (above the tidemark). Positive cells are unchanged in the femoral condyle (Ce) and displays abnormal negative regions in the tibial plateau (Cf, arrow). Quantification (D) shows a significant reduction in the non-mAC of the tibial plateau. (E) Low magnification (Ea,d) and high magnification of femoral condyle (Eb,e) and tibial plateau (Ec,f) shows that pSmad159 is mostly localized to non-mAC (above the tidemark). Positive cells are unchanged in the femoral condyle (Ee) and displays a significant reduction in the tibial plateau (Ef). Quantification confirms this in F. Statistical analyses were performed by unpaired t tests with Welch correction. M (with arrow) denotes regions of mAC in images. Scale bar = 100 μm. All images displayed with FC on top and TP on bottom unless otherwise noted. FC = femoral condyle; TP = tibial plateau; mAC = mineralized articular cartilage; non-mAC = nonmineralized articular cartilage.
Patterning of Hedgehog-responsive cells is coupled to tidemark patterning
Hedgehog signaling is a regulator of chondrocyte function and is dependent on the primary cilium for proper transduction through processing of glioma-associated oncogene (Gli) transcriptional effectors (Gli1, Gli2, and Gli3) for pathway activation or repression.(21) In the absence of hedgehog ligands (Sonic, Desert, or Indian), the transmembrane receptor Patched1 (Ptch1) inhibits the transmembrane protein Smoothened (Smo) to sequester Gli factors in the cytoplasm. When hedgehog ligand is present, it binds to Ptch1 and, in turn, the inhibition on Smo is relieved and Gli proteins are processed into active forms translocating to the nucleus for transcriptional activation. Thus, loss of primary cilia generally results in loss of hedgehog signaling. During embryonic development, hedgehog signaling is tightly linked to tissue patterning through established gradients of ligand distribution. A notable example of this is neural tube formation where cells establish specific identities dependent on their exposure, and response, to hedgehog ligands(71) and finetuned by a number of co-activators/antagonists.(72–74) Zonal chondrocyte patterning of the growth plate is also dependent on hedgehog exposure in a signaling loop that requires Indian Hedgehog (Ihh)-expressing prehypertrophic chondrocytes and Parathyroid related protein (Pthrp)-expressing reserve zone chondrocytes.(75,76) Hedgehog signaling patterns have not been well established in AC.
We sought to elucidate a possible role for hedgehog signaling in our AC model. To visualize active signaling, we used the Gli1--LacZ reporter mouse in combination with whole mount staining and imaging. Background staining was monitored using Gli1--LacZ–negative tissues (Fig. S5). We obtained several key findings. First, hedgehog signaling was highly active in immature AC of control animals at 3 weeks (Fig. 7Aa,Ba), and tissue sections revealed differences in Gli1-LacZ patterning of the femoral condyle and concentrated load-bearing region of the tibial plateau, localized to the first three to four cells layers of the femoral condyle (Fig. 7Ac) and to the bottom three to four cells layers in the tibial plateau (Fig. 7Bc). Gli1-LacZ persisted in mature AC at 8 weeks (Fig. 7Ca,Da) with similar cell layer patterning of immature cartilage and near the approximate location of the tidemark (Fig. 7Cc,Dc, dashed line). In immature cartilage of mutants, Gli1--LacZ was strongly downregulated in both the femoral condyle (Fig. 7Ab,d) and the tibial plateau (Fig. 7Bb,d). In mature mutant femoral condyle AC, Gli1-LacZ repression was maintained from 3 weeks (Fig. 7Cb), but exhibited randomly distributed positive cells (Fig. 7Cd). In the concentrated load-bearing region of mutant AC tibial plateau, Gli1-LacZ was strongly upregulated (Fig. 7Db, arrows) including at the surface (Fig. 7Dd), a pattern not seen in controls. The zonal properties of Gli1-LacZ in mature AC was quantified by approximating the tidemark location of control tissues based on knowledge from DIC imaging (Fig. 7Cc,d,Dc,d, dashed line). Staining with eosin and brightfield imaging to visualize LacZ did not allow actual tidemark visualization. Quantification confirmed a significant reduction of Gli1--LacZ in nonmineralized femoral condyle AC of mutants and a sharp increase in the tibial plateau (Fig. 7E, left). Interestingly, Gli1-LacZ was increased in mineralized AC of both regions, suggesting extension beyond the normal tidemark location (Fig. 7E, right). Taken together, the data show significant hedgehog signal disruption before a structural AC phenotype is apparent, largely consistent with known roles for primary cilia in hedgehog signaling. However, our data also shows hyperactive Gli1-LacZ activity in the absence of primary cilia but in conjunction with high mechanical load during ambulation, suggesting load-dependent alterations.
Fig. 7.
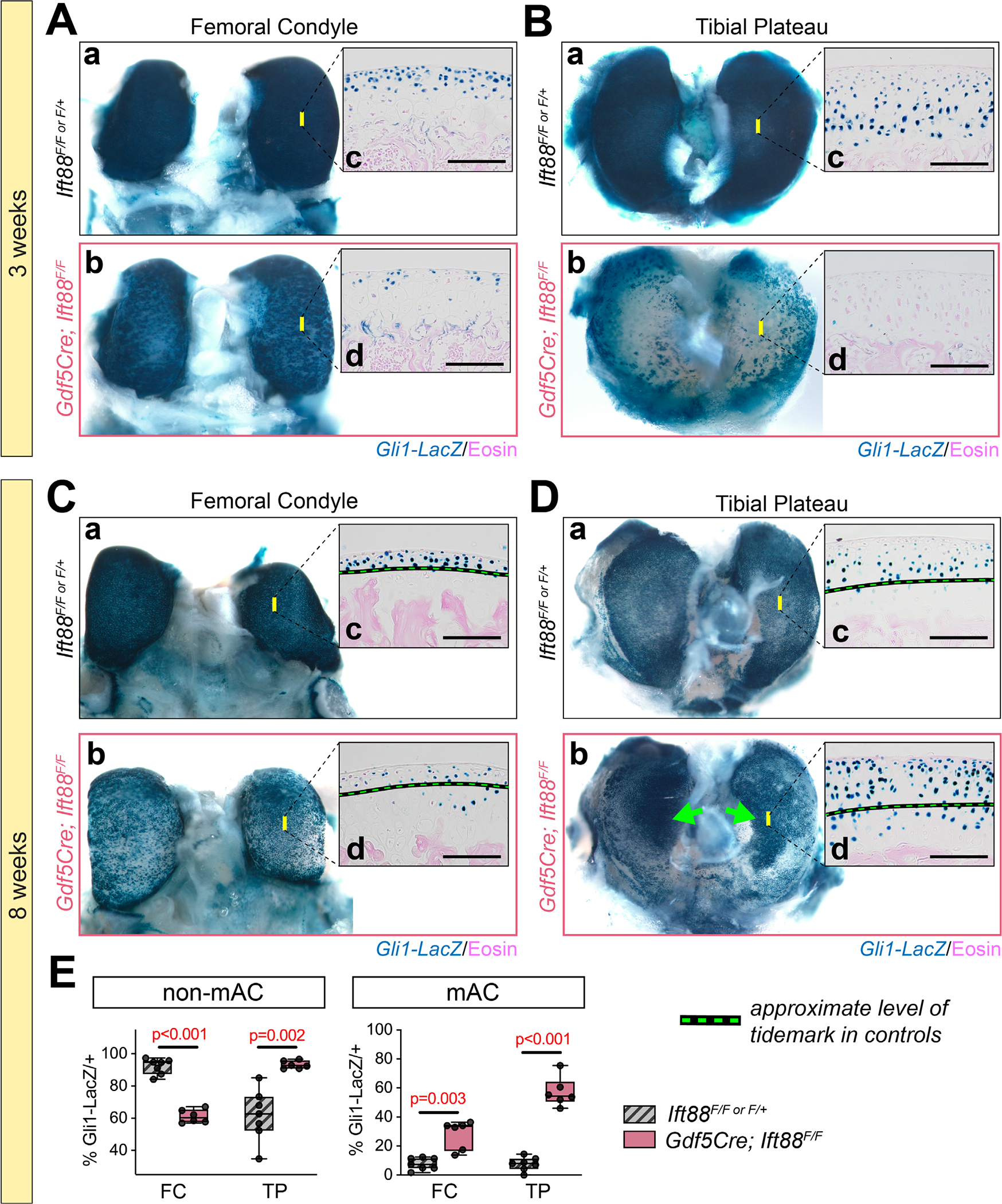
Primary cilia impart age-dependent and anatomic location-dependent functions for Hedgehog signaling. Gli1-LacZ (active hedgehog signaling) was developed by whole mount β-gal staining. Yellow bars on whole mount images indicate the approximate location of tissue section images. (A,B) At 3 weeks, Gli1-LacZ staining is strong in control femoral condyle (Aa) and tibial plateau (Ba). Staining is reduced in mutant femoral condyle (Ab) and tibial plateau (Bb). Tissue sections at 3 weeks from controls reveal Gli1-LacZ-expressing cells localized near the surface in the femoral condyle (Ac) and three to four cell layers from the surface in the tibial plateau (Bc). In mutant tissue sections, Gli1-LacZ is patchy in the femoral condyle (Ab) and nearly absent in the tibial plateau (Bd). (C–E) Dashed line denotes approximate location of the tidemark in control tissues. At 8 weeks, Gli1-LacZ staining is strong in control femoral condyle (Ca) and tibial plateau (Da). Staining is reduced in mutant femoral condyle (Cb) and increased in the central region of the tibial plateau (Db, arrows). Tissue sections at 8 weeks from controls reveal Gli1-LacZ–expressing cells localized near the surface in the femoral condyle (Cc) and three to four cell layers from the surface in the tibial plateau (Dc). In mutant tissue sections, Gli1-LacZ is patchy in the femoral condyle (Cb) and dramatically increased in the tibial plateau up to the surface (Dd). The percent of Gli1-LacZ postitive cells was quantified by zone using an approximation of location that is known in control tissues. Mutants display decreased positive cells in non-mAC of the femoral condyle and increases in all other regions. Statistical analyses performed by unpaired t-tests with Welch correction. M (with arrow) denotes regions of mAC in images. Scale bar = 100 μm. FC = femoral condyle; TP = tibial plateau; mAC = mineralized articular cartilage; non-mAC = nonmineralized articular cartilage.
Using RNAscope, we assessed two major hedgehog response genes (Gli1 and Ptch1) as well as Ihh in immature and mature AC and compared these patterns to the response when Ift88 was disrupted. Once again, we used confocal microscopy to image the FastRed detection and overlaid these results with DIC to visualize cell morphology, the tidemark (Fig. 8, dashed blue line), and the subchondral bone (Fig. 8, sb, solid white line). First, we established patterning in immature AC at 3 weeks. In control AC, Gli1 and Ptch1 were expressed in overlapping domains surrounding the future tidemark located three to four cell layers below the surface in femoral condyle (Fig. 8Aa,b) and four to six cell layers in tibial plateau (Fig. 8Ba,b). Importantly, these data were consistent with those from our Gli-LacZ studies (Fig. 7A,B). In both regions only cells juxtaposed to the underlying subchondral bone expressed Ihh (Fig. 8Ac,Bc). Two things were particularly notable about these patterns in immature AC: (i) the location of the future tidemark correlated strongly with Gli1 and Ptch1 expression; and (ii) despite Ihh-expressing cells being located always nearest the subchondral bone, the responding cells were closer to the surface in condylar cartilage than in tibial plateau. In mutant AC at this stage, Gli1 was significantly reduced in both femoral condyle and tibial plateau AC (Fig. 8Ad,Bd), consistent with Gli1-LacZ studies (Fig. 7C,D). Ptch1 expression generally followed the same profile, but with some notable groups of positive cells in the femoral condyle (Fig. 8Ae, arrows; Fig. 8Be). Importantly, Ihh expression was unchanged compared to controls (Fig. 8Af,Bf). Thus, downregulation of signaling in this context is due to lack of response rather than lack of ligand expression.
Fig. 8.
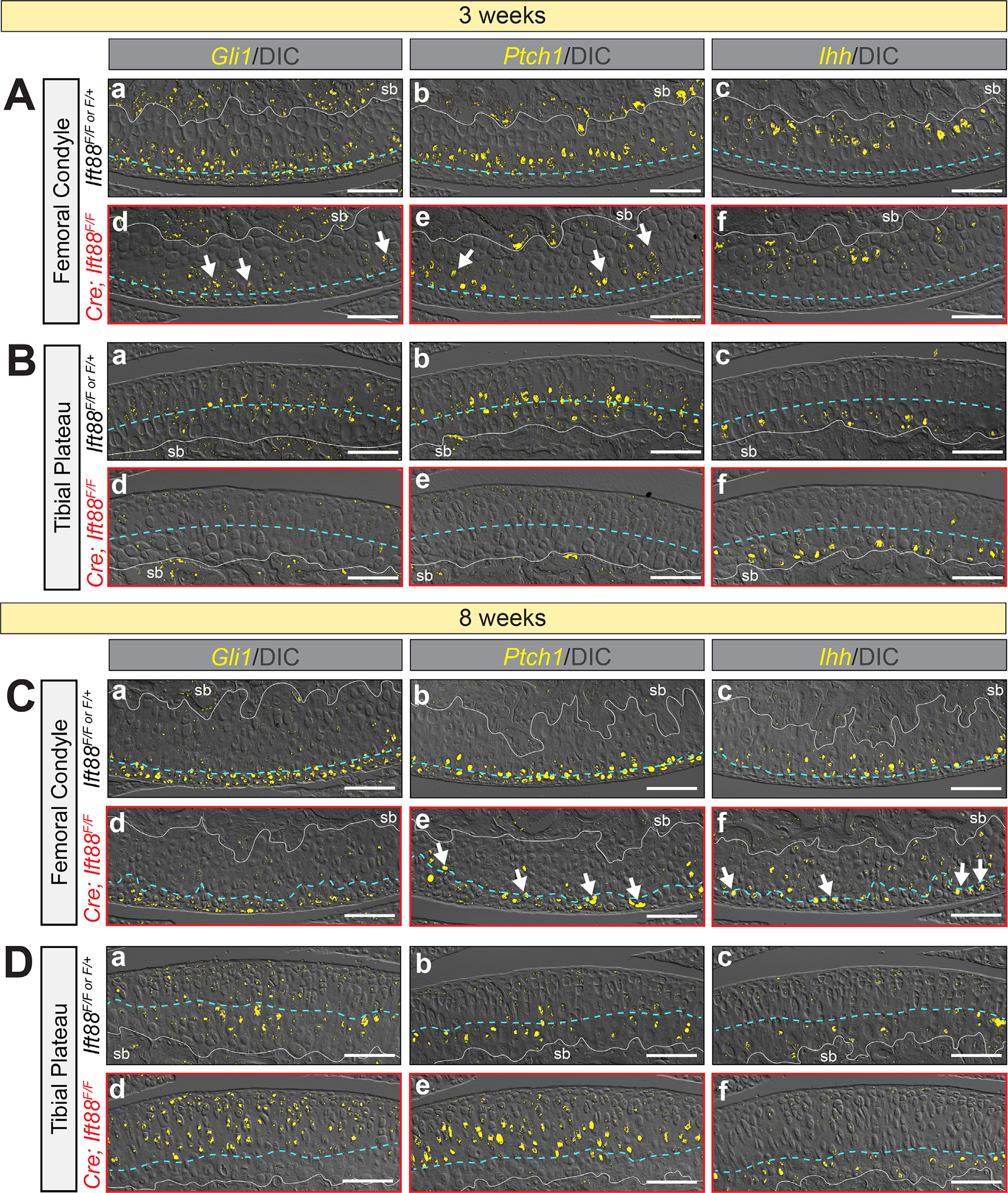
Patterning of hedgehog responsive cells correlates with tidemark patterning. RNAscope detected by FastRed is visualized by confocal microscopy and overlaid with DIC imaging to visualize cell morphology and tidemark patterning. The blue dashed line in (A,B) represents the approximate level of the future tidemark in control AC and in (C,D) it follows the tidemark visualized by DIC. (A) Femoral condyle AC at 3 weeks. Control AC expresses Gli1 in multiple cell layers near the AC surface (Aa), Ptch1 in one to two cell layers around the presumptive tidemark (Ab) and Ihh in chondrocytes adjacent to the subchondral bone (Ac). Mutant AC displays decreased but patchy Gli1 (Ad, arrows) and Ptch1 (Ae, arrows) expression and normal Ihh in chondrocytes adjacent to the subchondral bone (Af). (B) Tibial plateau AC at 3 weeks. Control AC expresses Gli1 (Ba) and Ptch1 (Bb) in similar layers surrounding the presumptive tidemark and Ihh in chondrocytes adjacent to the subchondral bone (Bc). Mutant AC displays decreased Gli1 (Bd) and Ptch1 (Be) expression and normal Ihh in chondrocytes adjacent to the subchondral bone (Bf). (C) Femoral condyle AC at 8 weeks. Control AC expresses Gli1 in multiple cell layers near the AC surface (Ca), Ptch1 in one to two cell layers around the tidemark (Cb) and Ihh in chondrocytes just beneath the tidemark (Cc). Mutant AC displays decreased expression of Gli1 (Cd), decreased and patchy expression of Ptch1 that follows the irregular tidemark (Ce, arrows) and ectopic Ihh in nonmineralized chondrocytes (Cf, arrows). (D) Tibial plateau AC at 8 weeks. Control AC expresses Gli1 (Da) and Ptch1 (Db) in similar layers surrounding the tidemark and Ihh in chondrocytes just beneath the tidemark (Dc). Mutant AC displays increased and expanded expression of Gli1 (Dd) and Ptch1 (De) but normal Ihh just beneath the tidemark (Df). Scale bar = 100 μm.
We performed identical analyses in mature AC at 8 weeks of age. Femoral condyle AC of controls displayed Gli1 expression in nonmineralized chondrocytes including surface zone cells (Fig. 8Ca), Ptch1 expression in cells at the level of the tidemark (Fig. 8Cb), and Ihh expression in mineralized chondrocytes beneath the tidemark (Fig. 8Cc). Tibial plateau AC displayed overlapping Gli1 and Ptch1 in a zone two to three cell layers thick surrounding the tidemark (Fig. 8Da,b) and Ihh in mineralized chondrocytes beneath the tidemark (Fig. 8Dc). Femoral condyle cartilage of mutants displayed generally reduced Gli1 above the tidemark (Fig. 8Cd), consistent with Gli1-LacZ studies. Interestingly, although Ptch1 was also significantly reduced compared to control cartilage, the random distribution of persistent Ptch1-expressing chondrocytes correlated with the irregularly shaped tidemark (Fig. 8Ce, arrows). Ihh also persisted but was ectopically expressing in some nonmineralized chondrocytes (Fig. 8Cf, arrows). In tibial plateau cartilage, Gli1 and Ptch1 expression was highly increased in the concentrated load-bearing region (Fig. 8Dd,e) consistent with Gli1-LacZ, but, unlike the condylar cartilage, Ihh remained exclusively expressed by nonmineralized chondrocytes beneath the tidemark (Fig. 8Df). Taken together, these results provide evidence that tidemark patterning is closely tied to hedgehog ligand gradients and local ambulation load, and that primary cilia integrate cellular responses to appropriately pattern the location of the tidemark during postnatal development.
Discussion
Our study of joint-specific loss of primary cilia function provides new and probing insights into AC zonal patterning and morphogenesis during postnatal growth and development. We find that the loss of IFT88 function (and primary cilia) has little consequence on embryonic joint formation, in line with previous in vivo studies.(26,28–31) However, we have now uncovered the that primary cilia exert major roles in AC structural and functional maturation between adolescence (3 weeks) and adulthood (8 weeks). During this postnatal period loss of primary cilia alters chondrocyte behavior and fate in both mineralized or nonmineralized cartilage, with consequent disruption of ECM component production and overall organization. However, we observed no clear evidence of cartilage matrix destruction in mutants, a result consistent with recent work by Coveney and colleagues,(27) who conditionally ablated Ift88 in adult articular cartilage and growth plate using AggrecanCreER mice. Interestingly, that study showed that Ift88 ablation at 6 or 8 weeks of age resulted in articular tissue thinning, but no such effect was noted when ablation was induced at 4 weeks.(27) We also did not observe tissue thinning in adults when Ift88 is ablated starting in embryonic joint progenitors. Similarly, we observed no obvious phenotype in adult synovium despite its cells being targeted by Gdf5Cre. Together, the data lead to the notion that conditional IFT88 deficiency in embryonic joint progenitors and early postnatal life may at least in part be compensated to maintain certain articular tissue traits such as overall thickness and synovial fibroblast function. However, other important traits cannot be compensated for, and mutants do display articular tissue defects as we clearly document in the present study.
One of our most important and surprising findings is that genetic deletion of Ift88 alters tidemark patterning and topography in a manner that mirrors alterations in hedgehog signaling. These analyses allow us to establish, for the first time, the interrelationships between spatial patterning of Ihh and cellular responsiveness in developing and mature AC. Our transcriptional analysis shows that even before the tidemark emerges, hedgehog-responding cells topographically align with the future location of the tidemark eventually seen in mature AC. This is further supported by our joint-specific loss of primary cilia model that disrupts cellular responsiveness to hedgehog signaling and alters tidemark topography. Cells that chance to avoid such loss and continue to be hedgehog-responsive—such as those in femoral condyle—remain located where the tidemark is patterned (Fig. 9, femoral condyle). The disruptions to hedgehog signaling lead to irregular zonal patterning, particularly in much of the femoral condyle where hedgehog signaling remains low throughout postnatal growth and likely contributes to ectopic Sox9-positive and Perlecan-positive cells that become mislocated in the mineralized zone (Fig. 9, non-mACs in mineralized zone). In the tibial plateau, the tidemark is relatively well patterned closer to the subchondral bone likely due to a switch from low hedgehog response to high hedgehog response in the timeframe where the tidemark is formed (Fig. 9, tibial plateau). In future work, the mechanisms linking hedgehog signaling to tidemark patterning should be tested directly. Is the tidemark repositioning dependent on a changing hedgehog response? What is the response in tissue integrity? Tidemark duplication and advancement toward the cartilage surface are highly associated with OA disease progression, but it is not understood whether this movement enhances disease progression or whether it is correlative.(8–12) In addition, overactivation of hedgehog signaling in mice leads to early onset of OA,(77) possibly through inhibition of ectonucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1), a suppressor of pathological calcification.(78) In sum, future studies delineating how the tidemark forms during postnatal development can inform why it is altered in disease, offering additional mechanistic insight into disease progression.
Fig. 9.
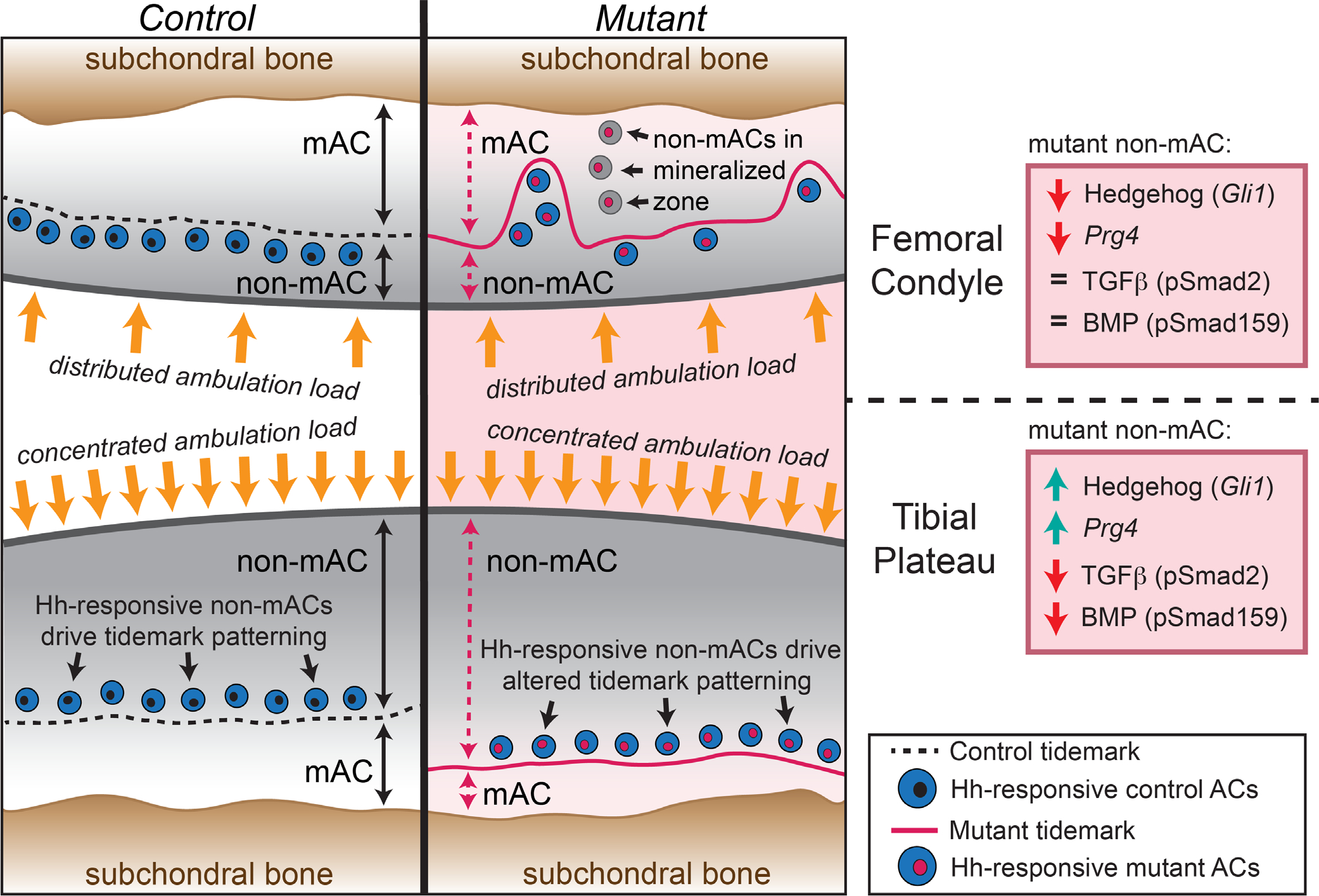
Summary schematic of findings. Patterning of control AC (left) and joint-specific (Gdf5Cre) loss of primary cilia (Ift88-flox) mutant AC (right). In the femoral condyle (top), the tidemark is located three to four cell layers from the surface in both controls and mutants but includes additional irregular patterning in mutants. In the tibial plateau (bottom), the tidemark is located five to six cell layers from the surface in controls and seven to eight cell layers from the surface in mutants. Alterations to mutant nonmineralized AC is shown in the boxed panels on the far right. Hh = hedgehog; mAC = mineralized articular cartilage; non-mAC, nonmineralized articular cartilage.
On a broad level, our study underscores the differential topographical character of AC in normal joints and the differential phenotype due to the loss of primary cilia during postnatal growth. Each joint must be uniquely fitted to the load-bearing and movement requirements at each anatomic location, and our study provides evidence that tidemark patterning is involved in this evolution and adaptation. Specifically, we highlight differential tidemark patterning in opposing AC in the same joint during normal tissue postnatal growth that bear unique ambulatory loads (Fig. 9, control). In the femoral condyle, ambulatory load is dispersed throughout cartilage as the entire surface slides along the underlying meniscus and tibial plateau with joint flexion and body movement. In the tibial plateau instead, ambulatory load is highly concentrated within a central location that is not covered by the meniscus. Tissue and tidemark patterning reflects such differing load-bearing requirements. In the femoral condyle, nonmineralized cartilage makes up ~30% of the tissue in control mice, with the tidemark located two to three cell layers beneath the surface. In the tibial plateau, nonmineralized cartilage makes up a significantly larger percentage of the tissue (~52%), with the tidemark located five to six cell layers beneath the surface. It is likely that the differing load-bearing topography in femur and tibia is also at play in response to the loss of primary cilia in articular cartilage and contributes to differential tissue responses. In this context, we have identified three major distinctions in the response of femoral condyle and tibial plateau to loss of primary cilia (highlighted in Fig. 9): (i) cellular responsiveness to hedgehog; (ii) cellular responsiveness to BMP and TGFβ; and (iii) expression of Prg4.
Our study shows that joint-specific loss of primary cilia leads to decreased hedgehog responsiveness in the femoral condyle and increased responsiveness in the tibial plateau in mature tissues. Most tissues follow the dogma that loss of the primary cilia leads to loss of hedgehog signaling. However, in the musculoskeletal system, Gli3 repressor plays a large role in tissue function and is also regulated by the primary cilium. Several studies have provided evidence that the loss of primary cilia, in addition to preventing Gli1 and Gli2 processing to active forms, affects the processing of Gli3 to its repressor form, resulting in abrogation of its repression on Gli1.(26,33,79,80) Our data are consistent with these studies and, in particular, with a recent study in skeletal muscle suggesting that Gli1 overactivation also has a mechanical load component.(80) However, in the femoral condyle, hedgehog signaling in mutants is decreased compared to controls with only patchy expression where ambulation load is distributed across the tissue. Of note, the loss of ciliation in the femoral condyle is not as strong as in the tibial plateau and the remaining hedgehog response could be the result of some ciliated cells still able to respond. In future work testing hedgehog signaling more directly, it will be interesting to see if these differential loading responses of femur and tibia persist over time.
We explored TGFβ and BMP signaling in order to gain mechanistic insights into changes in ECM production in mature AC. Our data indicate that both signaling pathways remain unchanged at 3 weeks in both mutant femoral condyle and tibial plateau, consistent with no apparent changes in Col2a1 and Acan gene expression. However, at 8 weeks, BMP or TGFβ responsiveness is significantly decreased in the load-bearing region of the tibial plateau. Knowledge of the role(s) for BMP signaling in postnatal AC is limited, but two studies of conditional ablation of either Bmpr1a or Bmp2 in embryonic joints (Gdf5Cre) reported no embryonic phenotype and a progressive loss of ECM during postnatal stages, without hypertrophy or loss of Sox9.(32,81) These findings are strikingly similar to those we report here. Hedgehog signaling is also a known negative regulator of BMP during embryonic somitogenesis by enhancing the expression of BMP antagonists Noggin and Gremlin.(82,83) Perhaps, a similar mechanism is at play here. The loss of TGFβ responsiveness is less clear but might include negative regulation by pathways not assessed in this study or an altered adaptation to mechanical load without primary cilia. In the femoral condyle at 8 weeks, we detected no changes to BMP or TGFβ signaling in nonmineralized cartilage, although the tissue was disorganized in correlation with a disorganized tidemark. Changes to Col2a1 or Acan gene expression were also not as prominent in the femoral condyle. It is notable that tissue stiffness (by AFM nanoindentation) was equally decreased in femoral condyle and tibial plateau. Thus, despite a lack of signaling changes, the ECM is still not normal. Notable decreases in cell density in the femoral condyle could contribute to this altered ECM organization; however, we believe that irregular patterning of nonmineralized and mineralized zones also plays a significant role. The additional decrease of TGFβ and BMP signaling in mutant tibial plateau might explain the more significant loss of collagen II compared to mutant femoral condyle (p = 0.03), with more clearly visible reductions in Safranin-O and aggrecan immunostaining. To test this theory directly, future work could use fluorescence-guided AFM nanomechanical mapping(84) to delineate local micromechanical properties that could differentially contribute to ECM composition and properties in femoral condyle versus tibial plateau.
Prg4 (lubricin) is an important regulator of AC homeostasis(85) and prevents tissue and matrix degradation that would lead to OA.(49,53) Prg4 can be regulated by multiple pathways including TGFβ,(49) Creb5,(86) Yap/Taz,(87) and mechanically activated Ca2+ signaling via Cox2.(88,89) The primary cilia also function to transduce mechano-osmotic signals via the mechanically activated cation channels such asTrpv4,(90–92) Polycystins1 and 2,(93) and Piezo1/2(94) that are present through the cell surface, but also on the primary cilium.(95–98) The sensitivity of Prg4 expression to mechanical load may explain the differential expression of Prg4 in femoral condyle versus tibial plateau. Consistently decreased expression in the femoral condyle suggests that full transcriptional activation of Prg4 requires the function of the primary cilium where ambulation load is dispersed throughout the tissue. However, in the tibial plateau, where ambulation load is concentrated to the tissue center, Prg4 is initially unchanged at 3 weeks and then increases markedly by 8 weeks, possibly due to the presence of cation channels residing outside the primary cilia that respond to this more concentrated load. The presence of Trvp4 channels on the primary cilium could also be important for the broader irregular ECM in our Ift88 mutants due to known functions for Trpv4 in ECM remodeling (reviewed by Ji and McCullough(99)), an active area of research in cartilage.
Although our mouse study is limited in its direct translation to human cartilage development and disease, previous work has shown that primary cilia are lengthened during human osteoarthritis.(25) Whether this accelerates disease progression or is a failed tissue response to subvert disease progression is not understood due, in large part, to a lack of understanding of how primary cilia function in articular cartilage postnatal development and growth. Better understanding of these facets of AC biology could be exploited to create more effective strategies for cartilage regeneration and repair. Hence, our study is a significant step ahead in providing important and novel insights into zonal and tidemark patterning, but also highlights that there are many remaining questions left to resolve regarding articular cartilage development.
Supplementary Material
Acknowledgments
This study was supported by NIH grants NIH/NIAMS F32AR074227 (to DR), R01AR062908 (to MP), R01AR074490 (to LH), and P30AR069619 (to Penn Center for Musculoskeletal Disorders). We thank Drs. Rosa Serra, Robert Mauck, Veronique Lefebvre, and Nathaniel Dyment for meaningful discussion and guidance throughout the project.
Authors’ roles: DR and MP designed and managed the study. DR, KH, CC, and EK performed animal experiments, data analyses, imaging, and quantifications. BH and LH performed AFM-nanoindentation and analyzed data. DR wrote the manuscript with contributions and input from all authors.
Footnotes
Conflict of Interests
All authors state that they have no conflicts of interest.
Additional Supporting Information may be found in the online version of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Mow V, Sugalski M. Physiology of Synovial Joints and Articular Cartilage. 3rd ed. Boston, MA: Butterwoth Heinemann Pubs; 2001. [Google Scholar]
- 2.Longobardi L, Li T, Tagliafierro L, et al. Synovial joints: from development to homeostasis. Curr Osteoporos Rep. 2015;13(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Decker RS. Articular cartilage and joint development from embryogenesis to adulthood. Semin Cell Dev Biol. 2017;62:50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Decker RS, Um H-B, Dyment NA, et al. Cell origin, volume and arrangement are drivers of articular cartilage formation, morphogenesis and response to injury in mouse limbs. Dev Biol. 2017;426(1): 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozhemyakina E, Zhang M, Ionescu A, et al. Identification of a Prg4-expressing articular cartilage progenitor cell population in mice. Arthritis Rheumatol. 2015;67(5):1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Newton PT, Bouderlique T, et al. Superficial cells are self-renewing chondrocyte progenitors, which form the articular cartilage in juvenile mice. FASEB J. 2017;31(3):1067–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunziker EB, Kapfinger E, Geiss J. The structural architecture of adult mammalian articular cartilage evolves by a synchronized process of tissue resorption and neoformation during postnatal development. Osteoarthritis Cartilage. 2007;15(4):403–413. [DOI] [PubMed] [Google Scholar]
- 8.Johnson L Joint remodeling as the basis for osteoarthritis. J Am Vet Med Assoc. 1962;141:1237–1241. [Google Scholar]
- 9.Lane LB, Bullough PG. Age-related changes in the thickness of the calcified zone and the number of tidemarks in adult human articular cartilage. J Bone Joint Surg Br. 1980;62(3):372–375. [DOI] [PubMed] [Google Scholar]
- 10.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oegema TR, Carpenter RJ, Hofmeister F, Thompson RC. The interaction of the zone of calcified cartilage and subchondral bone in osteoarthritis. Microsc Res Tech. 1997;37(4):324–332. [DOI] [PubMed] [Google Scholar]
- 12.Radin EL, Burr DB, Caterson B, Fyhrie D, Brown TD, Boyd RD. Mechanical determinants of osteoarthrosis. Semin Arthritis Rheum. 1991;21(3 Suppl 2):12–21. [DOI] [PubMed] [Google Scholar]
- 13.Nomura M, Sakitani N, Iwasawa H, et al. Thinning of articular cartilage after joint unloading or immobilization. An experimental investigation of the pathogenesis in mice. Osteoarthritis Cartilage. 2017; 25(5):727–736. [DOI] [PubMed] [Google Scholar]
- 14.O’Connor KM. Unweighting accelerates tidemark advancement in articular cartilage at the knee joint of rats. J Bone Miner Res. 1997; 12(4):580–589. [DOI] [PubMed] [Google Scholar]
- 15.Tomiya M, Fujikawa K, Ichimura S, Kikuchi T, Yoshihara Y, Nemoto K. Skeletal unloading induces a full-thickness patellar cartilage defect with increase of urinary collagen II CTx degradation marker in growing rats. Bone. 2009;44(2):295–305. [DOI] [PubMed] [Google Scholar]
- 16.Cosden-Decker RS, Bickett MM, Lattermann C, MacLeod JN. Structural and functional analysis of intra-articular interzone tissue in axolotl salamanders. Osteoarthritis Cartilage. 2012;20(11):1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rux DR, Decker RS, Koyama E, Pacifici M. Joints in the appendicular skeleton: developmental mechanisms and evolutionary influences. Curr Top Dev Biol. 2019;133:119–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gannon AR, Nagel T, Bell AP, Avery NC, Kelly DJ. Postnatal changes to the mechanical properties of articular cartilage are driven by the evolution of its collagen network. Eur Cell Mater. 2015;29:105–121; discussion 121–123. [DOI] [PubMed] [Google Scholar]
- 19.Julkunen P, Harjula T, Iivarinen J, et al. Biomechanical, biochemical and structural correlations in immature and mature rabbit articular cartilage. Osteoarthritis Cartilage. 2009;17(12):1628–1638. [DOI] [PubMed] [Google Scholar]
- 20.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol. 2018;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haycraft CJ, Zhang Q, Song B, et al. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134(2): 307–316. [DOI] [PubMed] [Google Scholar]
- 23.Serra R Role of Intraflagellar transport and primary cilia in skeletal development. Anat Rec. 2008;291(9):1049–1061. [DOI] [PubMed] [Google Scholar]
- 24.Yuan X, Yang S. Primary cilia and Intraflagellar transport proteins in bone and cartilage. J Dent Res. 2016;95(12):1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGlashan SR, Cluett EC, Jensen CG, Poole CA. Primary cilia in osteoarthritic chondrocytes: from chondrons to clusters. Dev Dyn. 2008; 237(8):2013–2020. [DOI] [PubMed] [Google Scholar]
- 26.Chang C-F, Ramaswamy G, Serra R. Depletion of primary cilia in articular chondrocytes results in reduced Gli3 repressor to activator ratio, increased Hedgehog signaling, and symptoms of early osteoarthritis. Osteoarthritis Cartilage. 2012;20(2):152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coveney CR, Zhu L, Miotla-Zarebska J, et al. The ciliary protein IFT88 controls post-natal cartilage thickness and influences development of osteoarthritis. Arthritis Rheumatol. 2022;74(1):49–59. [DOI] [PubMed] [Google Scholar]
- 28.Irianto J, Ramaswamy G, Serra R, Knight MM. Depletion of chondrocyte primary cilia reduces the compressive modulus of articular cartilage. J Biomech. 2014;47(2):579–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheffield ID, McGee MA, Glenn SJ, et al. Osteoarthritis-like changes in Bardet-Biedl syndrome mutant ciliopathy mice (Bbs1M390R/M390R): evidence for a role of primary cilia in cartilage homeostasis and regulation of inflammation. Front Physiol. 2018;9:708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song B, Haycraft CJ, Seo H, Yoder BK, Serra R. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev Biol. 2007;305(1):202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan X, Yang S. Deletion of IFT80 impairs epiphyseal and articular cartilage formation due to disruption of chondrocyte differentiation. PLoS One. 2015;10(6):e0130618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rountree RB, Schoor M, Chen H, et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004;2(11):e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–87. [DOI] [PubMed] [Google Scholar]
- 34.Doyran B, Tong W, Li Q, et al. Nanoindentation modulus of murine cartilage: a sensitive indicator of the initiation and progression of post-traumatic osteoarthritis. Osteoarthritis Cartilage. 2017;25(1): 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han B, Li Q, Wang C, et al. Decorin regulates the Aggrecan network integrity and biomechanical functions of cartilage extracellular matrix. ACS Nano. 2019;13(10):11320–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buschmann MD, Kim YJ, Wong M, Frank E, Hunziker EB, Grodzinsky AJ. Stimulation of aggrecan synthesis in cartilage explants by cyclic loading is localized to regions of high interstitial fluid flow. Arch Biochem Biophys. 1999;366(1):1–7. [DOI] [PubMed] [Google Scholar]
- 37.Kawamoto T, Kawamoto K. Preparation of thin frozen sections from nonfixed and undecalcified hard tissues using Kawamoto’s film method. Methods Mol Biol. 2021;2230:259–281. [DOI] [PubMed] [Google Scholar]
- 38.de Charleroy C, Haseeb A, Lefebvre V. Preparation of adult mouse skeletal tissue sections for RNA in situ hybridization. Methods Mol Biol. 2021;2245:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koyama E, Shibukawa Y, Nagayama M, et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev Biol. 2008;316(1): 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mow V, Ratcliffe A. Structure and Function of Articular Cartilage and Meniscus. 2nd ed. Philadelphia, PA: Lippincott-Raven; 1997. [Google Scholar]
- 41.Lefebvre V, Angelozzi M, Haseeb A. SOX9 in cartilage development and disease. Curr Opin Cell Biol. 2019;61:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haseeb A, Kc R, Angelozzi M, de Charleroy C, et al. SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc Natl Acad Sci U S A. 2021;118(8):e2019152118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henry SP, Liang S, Akdemir KC, de Crombrugghe B. The postnatal role of Sox9 in cartilage. J Bone Miner Res. 2012;27(12):2511–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guilak F, Alexopoulos LG, Upton ML, et al. The pericellular matrix as a transducer of biomechanical and biochemical signals in articular cartilage. Ann N Y Acad Sci. 2006;1068:498–512. [DOI] [PubMed] [Google Scholar]
- 45.Poole CA, Flint MH, Beaumont BW. Chondrons in cartilage: ultrastructural analysis of the pericellular microenvironment in adult human articular cartilages. J Orthop Res. 1987;5(4):509–522. [DOI] [PubMed] [Google Scholar]
- 46.Kvist AJ, Nyström A, Hultenby K, Sasaki T, Talts JF, Aspberg A. The major basement membrane components localize to the chondrocyte pericellular matrix: a cartilage basement membrane equivalent? Matrix Biol. 2008;27(1):22–33. [DOI] [PubMed] [Google Scholar]
- 47.Wilusz RE, Defrate LE, Guilak F. A biomechanical role for perlecan in the pericellular matrix of articular cartilage. Matrix Biol. 2012;31(6): 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coveney CR, Collins I, Mc Fie M, Chanalaris A, Yamamoto K, Wann AKT. Cilia protein IFT88 regulates extracellular protease activity by optimizing LRP-1-mediated endocytosis. FASEB J. 2018;32(12): 6771–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chavez RD, Sohn P, Serra R. Prg4 prevents osteoarthritis induced by dominant-negative interference of TGF-ß signaling in mice. PLoS One. 2019;14(1):e0210601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coles JM, Zhang L, Blum JJ, et al. Loss of cartilage structure, stiffness, and frictional properties in mice lacking PRG4. Arthritis Rheum. 2010; 62(6):1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marcelino J, Carpten JD, Suwairi WM, et al. CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Nat Genet. 1999;23(3):319–322. [DOI] [PubMed] [Google Scholar]
- 52.Rhee DK, Marcelino J, Baker M, et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J Clin Invest. 2005;115(3):622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruan MZ, Erez A, Guse K, et al. Proteoglycan 4 expression protects against the development of osteoarthritis. Sci Transl Med. 2013;5 (176):176ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol. 2019;15(4):199–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thielen NGM, van der Kraan PM, van Caam APM. TGFβ/BMP signaling pathway in cartilage homeostasis. Cell. 2019;8(9):E969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen CG, Thuillier D, Chin EN, Alliston T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheumatol. 2012;64(10):3278–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferguson CM, Schwarz EM, Reynolds PR, Puzas JE, Rosier RN, O’Keefe RJ. Smad2 and 3 mediate transforming growth factorbeta1-induced inhibition of chondrocyte maturation. Endocrinology. 2000;141(12):4728–4735. [DOI] [PubMed] [Google Scholar]
- 58.Ionescu AM, Schwarz EM, Zuscik MJ, et al. ATF-2 cooperates with Smad3 to mediate TGF-beta effects on chondrocyte maturation. Exp Cell Res. 2003;288(1):198–207. [DOI] [PubMed] [Google Scholar]
- 59.Kang JS, Alliston T, Delston R, Derynck R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005;24(14):2543–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim K-O, Sampson ER, Maynard RD, et al. Ski inhibits TGF-β/phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J Cell Biochem. 2012;113(6):2156–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leboy P, Grasso-Knight G, D’Angelo M, et al. Smad-Runx interactions during chondrocyte maturation. J Bone Joint Surg Am. 2001;83-A Suppl 1(Pt 1):S15–S22. [PubMed] [Google Scholar]
- 62.Li T-F, Gao L, Sheu T-J, et al. Aberrant hypertrophy in Smad3-deficient murine chondrocytes is rescued by restoring transforming growth factor beta-activated kinase 1/activating transcription factor 2 signaling: a potential clinical implication for osteoarthritis. Arthritis Rheumatol. 2010;62(8):2359–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seo H-S, Serra R Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev Biol. 2007;310(2):304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang X, Chen L, Xu X, Li C, Huang C, Deng CX. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153(1): 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bae J-S, Gutierrez S, Narla R, et al. Reconstitution of Runx2/Cbfa1-null cells identifies a requirement for BMP2 signaling through a Runx2 functional domain during osteoblast differentiation. J Cell Biochem. 2007;100(2):434–449. [DOI] [PubMed] [Google Scholar]
- 66.Fujii M, Takeda K, Imamura T, et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol Biol Cell. 1999;10(11):3801–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horiki M, Imamura T, Okamoto M, et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J Cell Biol. 2004;165(3):433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Javed A, Bae J-S, Afzal F, et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J Biol Chem. 2008; 283(13):8412–8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Javed A, Afzal F, Bae J-S, et al. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs. 2009;189(1–4): 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phimphilai M, Zhao Z, Boules H, Roca H, Franceschi RT. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J Bone Miner Res. 2006;21(4):637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ribes V, Briscoe J. Establishing and interpreting graded Sonic Hedgehog signaling during vertebrate neural tube patterning: the role of negative feedback. Cold Spring Harb Perspect Biol. 2009;1(2): a002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21(10):1244–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allen BL, Song JY, Izzi L, et al. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20(6):775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holtz AM, Peterson KA, Nishi Y, et al. Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development. 2013;140(16):3423–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kronenberg HM. PTHrP and skeletal development. Ann N Y Acad Sci. 2006;1068:1–13. [DOI] [PubMed] [Google Scholar]
- 76.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rockel JS, Yu C, Whetstone H, et al. Hedgehog inhibits β-catenin activity in synovial joint development and osteoarthritis. J Clin Invest. 2016;126(5):1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jin Y, Cong Q, Gvozdenovic-Jeremic J, et al. Enpp1 inhibits ectopic joint calcification and maintains articular chondrocytes by repressing hedgehog signaling. Development. 2018;145(18):dev164830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005; 1(4):e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kopinke D, Roberson EC, Reiter JF. Ciliary hedgehog signaling restricts injury-induced Adipogenesis. Cell. 2017;170(2):340–351.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gamer LW, Pregizer S, Gamer J, et al. The role of Bmp2 in the maturation and maintenance of the murine knee joint. J Bone Miner Res. 2018;33(9):1708–1717. [DOI] [PubMed] [Google Scholar]
- 82.Murtaugh LC, Chyung JH, Lassar AB. Sonic hedgehog promotes somitic chondrogenesis by altering the cellular response to BMP signaling. Genes Dev. 1999;13(2):225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stafford DA, Brunet LJ, Khokha MK, Economides AN, Harland RM. Cooperative activity of noggin and gremlin 1 in axial skeleton development. Development. 2011;138(5):1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chery DR, Han B, Li Q, et al. Early changes in cartilage pericellular matrix micromechanobiology portend the onset of post-traumatic osteoarthritis. Acta Biomater. 2020;111:267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Das N, Schmidt TA, Krawetz RJ, Dufour A. Proteoglycan 4: from mere lubricant to regulator of tissue homeostasis and inflammation: does proteoglycan 4 have the ability to buffer the inflammatory response? Bioessays. 2019;41(1):e1800166. [DOI] [PubMed] [Google Scholar]
- 86.Zhang C-H, Gao Y, Jadhav U, et al. Creb5 establishes the competence for Prg4 expression in articular cartilage. Commun Biol. 2021;4(1):332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Delve E, Co V, Regmi SC, Parreno J, Schmidt TA, Kandel RA. YAP/TAZ regulates the expression of proteoglycan 4 and tenascin C in superficial-zone chondrocytes. Eur Cell Mater. 2020;39:48–64. [DOI] [PubMed] [Google Scholar]
- 88.Abusara Z, Krawetz R, Steele B, DuVall M, Schmidt T, Herzog W. Muscular loading of joints triggers cellular secretion of PRG4 into the joint fluid. J Biomech. 2013;46(7):1225–1230. [DOI] [PubMed] [Google Scholar]
- 89.Ogawa H, Kozhemyakina E, Hung H-H, Grodzinsky AJ, Lassar AB. Mechanical motion promotes expression of Prg4 in articular cartilage via multiple CREB-dependent, fluid flow shear stress-induced signaling pathways. Genes Dev. 2014;28(2):127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2003;100-(Suppl 2):14531–14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A. 2004;101(1):396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase c-dependent and -independent pathways. J Biolog Chem. 2003;278(29):27129–27137. [DOI] [PubMed] [Google Scholar]
- 93.Gees M, Colsoul B, Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol. 2010;2(10):a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coste B, Mathur J, Schmidt M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330(6000):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee KL, Guevarra MD, Nguyen AM, Chua MC, Wang Y, Jacobs CR. The primary cilium functions as a mechanical and calcium signaling nexus. Cilia. 2015;4(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qin H, Burnette DT, Bae Y-K, Forscher P, Barr MM, Rosenbaum JL. Intraflagellar transport is required for the vectorial movement of TRPV channels in the ciliary membrane. Curr Biol. 2005;15(18):1695–1699. [DOI] [PubMed] [Google Scholar]
- 97.Phan MN, Leddy HA, Votta BJ, et al. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheumatol. 2009;60(10):3028–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thompson CL, McFie M, Chapple JP, Beales P, Knight MM. Polycystin-2 is required for chondrocyte Mechanotransduction and traffics to the primary cilium in response to mechanical stimulation. Int J Mol Sci. 2021;22(9):4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ji C, McCulloch CA. TRPV4 integrates matrix mechanosensing with Ca2+ signaling to regulate extracellular matrix remodeling. FEBS J. 2021;288(20):5867–5887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.