

Abstract
Background:
Mast cells (MCs) are pleiotropic cells that accumulate in the esophagus of patients with eosinophilic esophagitis (EoE) and are thought to contribute to disease pathogenesis, yet their properties and functions in this organ are largely unknown.
Objective:
We aimed to perform a comprehensive molecular and spatial characterization of esophageal MCs in EoE.
Methods:
Esophageal biopsies obtained from patients with active EoE, patients with EoE in histologic remission, and individuals with histologically normal esophageal biopsies and no history of esophageal disease (i.e., control individuals) were subject to scRNA-sequencing, flow cytometry, and immunofluorescence analyses.
Results:
We probed 39,562 single esophageal cells by single-cell RNA sequencing; approximately 5% of these cells were MCs. We identified dynamic MC expansion across disease states. During homeostasis, TPSAB1highAREGhigh resident MCs were mainly detected in the lamina propria and exhibited a quiescent phenotype. In patients with active EoE, resident MCs assumed an activated phenotype, and two additional pro-inflammatory MC populations emerged in the intraepithelial compartment, each linked to a proliferating MKI67high cluster. One pro-inflammatory activated MC population, marked as KIThighIL1RL1highFCER1Alow, was not detected in disease remission (termed transient MC), whereas the other population, marked as CMA1highCTSGhigh, was detected in disease remission where it maintained an activated state (termed persistent MC). MCs were prominent producers of esophageal IL-13 mRNA and protein, a key therapeutic target in EoE.
Conclusions:
Esophageal MCs comprise heterogeneous populations with transcriptional signatures associated with distinct spatial compartmentalization and EoE disease status. In active EoE, they assume a pro-inflammatory state and locally proliferate, and remain activated and poised to re-initiate inflammation even during disease remission.
Keywords: Eosinophilic esophagitis, Mast cells, Single-cell RNA sequencing, IL-13, Proliferation
Graphical Abstract
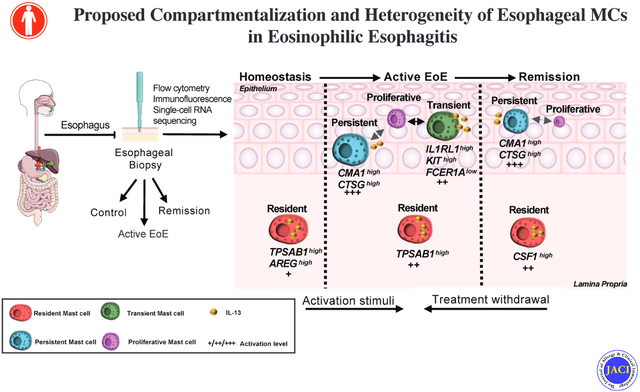
Capsule summary:
We have performed a comprehensive examination of esophageal MCs across EoE diagnostic states assessing their expansion and activation profile by transcriptional and protein analytic methods, providing insight into their role in disease pathogenesis.
INTRODUCTION
Eosinophilic esophagitis (EoE) is a chronic, allergic, type 2 (T2) inflammatory disease driven by food antigens1, 2. Histologic evaluation of eosinophil accumulation currently serves as the gold-standard diagnostic criteria for diagnosis and monitoring of disease activity. Active disease is defined as the presence of greater than or equal to 15 eosinophils per high-power field (HPF) in biopsy samples of the esophageal mucosa. The disease is clinically managed through dietary restrictions, anti-inflammatory treatment (e.g., topical glucocorticoids, proton pump inhibitors, biological treatment), and esophageal dilatation3, 4. However, there is currently no cure nor FDA-approved treatment for EoE, and the disease typically follows a chronic, relapsing course5.
Genome-wide transcriptional profiling of the esophagus in patients with active EoE has revealed abundant expression of mast cell (MC)-associated genes, including those encoding tryptase (TPSAB1), carboxypeptidase A3 (CPA3), and c-KIT (KIT), implying MC accumulation6. Indeed, there is an accumulation of esophageal MCs in the epithelial layer but not in the lamina propria in patients with EoE7. Additionally, in patients with active EoE, MCs are degranulated, and their levels correlate with cardinal symptoms, such as dysphagia8, 9. Furthermore, in patients with EoE in remission (defined by reduced esophageal eosinophil levels), increased MC concentrations are associated with persistent endoscopic and histologic abnormalities10. Additionally, increased esophageal CPA3 and chymase (CMA1) expression are associated with a steroid-refractory endotype of EoE11, and MC reduction is associated with clinical response to emerging biological therapies, including IL-5, IL-13, and Siglec-8 blockade12–14. Thus, tissue MC expansion and transcriptional activity are common in EoE and may be a key pathogenic contributor.
MCs are tissue-resident immune effector cells that exhibit context-dependent expression of pro-inflammatory mediators (e.g., proteases, cytokines) and receptors (e.g., activating, inhibiting, and growth factor receptors) and can demonstrate variable responses to pharmacologic inhibitors15. In homeostasis, MCs reside within barrier tissues, including the skin, airways, and gastrointestinal tract16, 17. Tissue mastocytosis is a common feature of T2 inflammation that is thought to arise from the recruitment of rare, bone marrow-derived circulating MC progenitors8, 18, 19. However, SCF can promote local proliferation of differentiated tissue mast cells20, 21. Overall, the mechanisms directing MC hyperplasia in these settings are not well understood.
The two canonical subsets of human MCs are recognized on the basis of their protease expression. MCs containing tryptase, chymase, and CPA3, termed MCTC, are commonly found in the skin and the intestinal submucosa. In contrast, MCs that express tryptase but not chymase, termed MCT, are commonly found at mucosal surfaces23–24. Nuanced phenotyping of murine MCs has suggested that MC heterogeneity results from their plasticity and interaction with the microenvironment25–27. The transcriptomic profile of murine MCs isolated from multiple organs, including the trachea, esophagus, tongue, skin, and peritoneum, demonstrated wide transcriptional heterogeneity across the different tissues28. Moreover, studies using single-cell RNA sequencing (scRNA-seq) demonstrate multiple, distinct MC subsets in the human intestine29, lung30, and nasal polyp31.
Herein, we conduct a comprehensive examination of esophageal MCs in EoE using scRNA-seq, flow cytometry, and immunofluorescence on esophageal biopsies obtained from patients with active EoE (active EoE), patients with EoE in histologic remission (remission), and individuals with histologically normal esophageal biopsies and no history of esophageal disease (i.e., control individuals). Through these approaches, we assess MC composition and transcriptional heterogeneity across the full spectrum of EoE and evaluate the potential contribution of MCs to both disease pathophysiology and relapse. We found that MCs in control individuals were present at relatively low levels (termed resident MCs). The esophageal MC pool expanded in individuals with active EoE due to the accumulation of two additional MC populations, each associated with local proliferation and activation. One of the EoE-specific MC populations waned with disease remission (termed transient MCs), whereas the other MC population persisted in individuals with EoE in remission (termed persistent MCs). We demonstrate that esophageal MCs further express disease-associated-transcriptional signatures, with the persistent population exhibiting an increased percentage of activation-associated genes even during disease remission. Notably, histologic and scRNA-seq analyses demonstrated that MCs are a prominent source for the key EoE driver cytokine IL-13 in all disease states. Collectively, the data presented herein show that the esophageal MC pool comprises dynamic populations that assume a pro-inflammatory phenotype in patients with active EoE and that specific MC populations (resident, persistent) remain transcriptionally activated and poised to re-initiate inflammation even during remission.
METHODS
Human subjects
Written informed consent was obtained before a patient’s enrollment in the study, and all human studies were approved by the CCHMC Institutional Review Board (IRB #2008–0090). Control patients were defined as having no history of EoE diagnosis, having 0 eosinophils per 40X microscopic high-power field (HPF) of esophageal biopsy samples, and having no evidence of esophagitis within esophageal biopsies. Patients with active EoE had clinician-diagnosed EoE defined by a histologically active disease with ≥ 15 eosinophils in distal esophageal biopsies. Patients with inactive EoE (i.e., remission) had clinician-diagnosed EoE in the past and were defined by a histologically inactive disease with a peak eosinophil count of < 15 eosinophils per HPF in distal esophageal biopsies. An independent cohort of 10 patients was enrolled for the scRNA-seq analyses (control, n= 2; active EoE, n = 5; EoE remission, n = 3), an independent cohort of 13 patients was enrolled for the flow cytometry to assess mast cell levels (control, n = 3; active EoE, n = 6; EoE remission, n = 4), and an independent cohort of 20 patient biopsies was used for immunofluorescence of proteins (control, n = 6; active EoE, n = 7; EoE remission, n = 7) (Supplementary Table 1). A previously reported cohort of patients with active EoE (n = 147) analyzed by the EoE diagnostic panel11, 32 and an independent cohort with active EoE (n =10) analyzed by bulk RNAseq6 were also analyzed in this study.
Enzymatic digestion of esophageal biopsies to produce a single-cell suspension
A single biopsy from the distal esophagus was collected into RPMI-1640 medium supplemented with 10% FBS, kept on ice, and transported to the research laboratory within 30 minutes for processing. The biopsy was then transferred into EDTA buffer (5 mM EDTA, 25 mM HEPES, 10% FBS in HBSS) for 15 minutes at 37°C, washed once with 1X phosphate-buffered saline (PBS) (pH 7.4), minced, and then subjected to collagenase A (2.4 mg/mL, Roche) digestion for 30 minutes at 37°C. The resulting suspension was diluted with ice-cold PBS, passed through a 19-gauge needle, filtered through a 70-μm cell strainer, and washed with ice-cold PBS.
Flow cytometry staining
A single-cell suspension, generated by enzymatic digestions, was pelleted in PBS before viability staining (Invitrogen L34957, 1:1000) for 10 minutes; cells were washed with 2% FBS in PBS for antibody staining. Cells were incubated with Fc Block (BD Biosciences 564219, 1:50) for 15 minutes before adding labeled antibodies. Cells were stained for CD45 (BV711, Biolegend 103147,1:50), FcεR1α (FITC, Biolegend 334608, 1:50), and c-KIT (PE, Biolegend 313204, 1:50) for 30 minutes at 4°C. Samples were then resuspended in PBS + 2% FBS and analyzed on a Fortessa cytometer in the Cincinnati Children’s Hospital Medical Center (CCHMC) Research Flow Cytometry Core (RFCC). Single-color compensation was calculated, and data were analyzed in FlowJo (Version 10.6.1).
Immunofluorescence staining
Distal esophageal biopsies were fixed with 10% formalin and embedded in paraffin (FFPE). FFPE samples were sectioned and de-paraffinized using xylene and then subjected to graded ethanol washes. Heat-induced epitope retrieval in R-UNIVERSAL Epitope Recovery Buffer (EMS, 62719–20) was performed. Slides were blocked in PBS with 10% donkey serum for 1 hour followed by overnight incubation at 4°C in the following primary antibodies: mouse anti-human tryptase (Biolegend, 369402), rabbit anti-human KI-67 (Invitrogen, PA-19462), or rat anti-human IL-13 (Biolegend, 501902). Slides were then washed, incubated for 1.5 hours at room temperature with Hoechst 33342 nuclear staining (Invitrogen, H3570) and the following secondary antibodies: donkey anti-mouse Alexa Fluor 647 (Jackson, 715605151), donkey anti-rat Alexa Fluor 488 (Invitrogen, A21208), or donkey-anti-rabbit Alexa Fluor 488 (Jackson, 711545152). Then slides were washed and mounted with ProLong™ gold antifade reagent (Invitrogen, P10144). Images were obtained using the NIKON A1 inverted LUNV confocal microscope. Quantifying confocal sections were carried out with Imaris and Elements software.
Single-cell RNA sequencing analysis
Single-cell suspensions were prepared from biopsies as described above. Bulk population cells were directly subjected to the 10X mass genomics chip (10X Genomics, Inc.), targeting 10,000 simultaneously captured live events for next-generation sequencing. Each cell was uniquely barcoded during the cDNA library generation, subsequently sequenced on an Illumina HiSeq 2500 at CCHMC’s DNA sequencing core, allocating a total read of ~320M (2 lanes/flow cell). Sparse data matrices, provided by 10X genomics, were used as input into Seurat analyzed in R studio (R version 3.6.1) as previously described33, 34. For analysis of all sequenced esophageal samples, cells were filtered on the basis of unique feature counts of over 200 or less than 4,800, with less than 20% mitochondrial counts. In addition, genes that were expressed in fewer than three cells were excluded. In sum, 39,562 cells and 20,208 genes passed these filter criteria. A gene’s relative expression was calculated by log normalization and centering. Principal component analysis (PCA) was performed with the list of the top 2000 variable genes. Data were subjected to Uniform Manifold Approximation and Projection (UMAP) and shared nearest neighbor (SNN) modularity optimization-based clustering, identifying 21 distinct clusters using 12 dimensions and 0.8 resolution parameters. Top marker genes with high specificity were used to classify cell markers into seven known cell types: epithelium, fibroblast, endothelium, T cells, B cells, myeloid cells, and MCs. The MC population, a total of 1,897 cells, was subject to subsetting and re-PCA and SNN modularity optimization-based clustering, which identified six distinct clusters using 4 dimensions and 0.3 resolution parameters. They were classified and merged into 4 distinct populations on the basis of their differential presence: resident, transient, persistent, and proliferative populations.
Non-epithelial clusters identified as strongly expressed marker genes associated with esophageal epithelium were considered contaminated and discarded. PCA and SNN modularity optimization-based clustering steps were performed before the beginning of the analysis.
Gene enrichments were calculated using the FindAllMarkers function in Seurat. The Wilcoxon test was implemented in Seurat with the default parameter threshold set of 0.25 unless mentioned otherwise to identify cluster-defined genes.
MST pseudotime analysis
A minimum spanning tree (MST) map was created in R by first extracting the cell data and cluster metadata from the Seurat object. Using the dyno package35, as previously described at https://github.com/dynverse/dyno, the cell data were competently converted to a wrapper object containing the cell expression, counts, and cluster metadata and compatible with the dyno package. Then, an MST trajectory was inferred and plotted using the dyno package. This trajectory was then added back to the original Seurat object for further visualization by grouping the cells onto their nearest milestone and adding these data to the Seurat object.
Gene ontology (GO) analysis
GO analysis was performed using the ToppGene website (https://toppgene.cchmc.org/)
Statistical analyses
Statistical significance was determined for normal distribution using one-way ANOVA with Tukey’s multiple comparisons post-test or a Student’s t-test (unpaired, two-tailed). For non-normal distribution, Mann-Whitney test or Kruskal-Wallis’s test with Dunn’s multiple comparison post-test were used. Correlation analysis was performed using Spearman’s rank correlation coefficient, followed by Bonferroni-Dunn’s adjustment. Statistical analyses were performed using GraphPad Prism (version 8.0.1, GraphPad Software Incorporated. La Jolla, CA. USA)
RESULTS
Esophageal MCs increase in human active EoE, persist during remission, and strongly associate with the EoE cytokine milieu
Flow cytometry analysis of a cohort of human esophageal biopsies (Supplementary Table 1), including control (n = 3), active EoE (n = 6), and remission (n = 4), identified FcεR1αhighc-KIThigh MCs in the tissue (Figure 1A). MCs were a highly prevalent white blood cell during active EoE, accounting for 22 ± 7.5% of total white blood cells in patients with active EoE compared to 4.2 ± 1.8% in control individuals (Figure 1B). Notably, although a trend of decreased MC levels was observed in remission biopsies compared to active EoE, MCs remained elevated in disease remission compared to control, accounting for 11.7 ± 4.1% of total white blood cells (Figure 1B). This finding contrasts with that of eosinophils which were identified as FcεR1αintc-KITint FSChighSSChigh 36 (Figure 1A); eosinophils increased to 18.7 ± 11.1% of total white blood cells in active EoE compared to 0% in control esophageal tissue but were nearly fully depleted in remission biopsies (Figure 1A–B). Eosinophil levels were confirmed by histologic quantification of the tissue sections and accounted for 0 eosinophils/HPF in control, 46 ± 14 eosinophils/HPF in active EoE, and 1 ± 2 eosinophils/HPF in remission (Supplementary Figure S1).
Figure 1: MC levels by EoE disease state and association with EoE cytokine milieu.
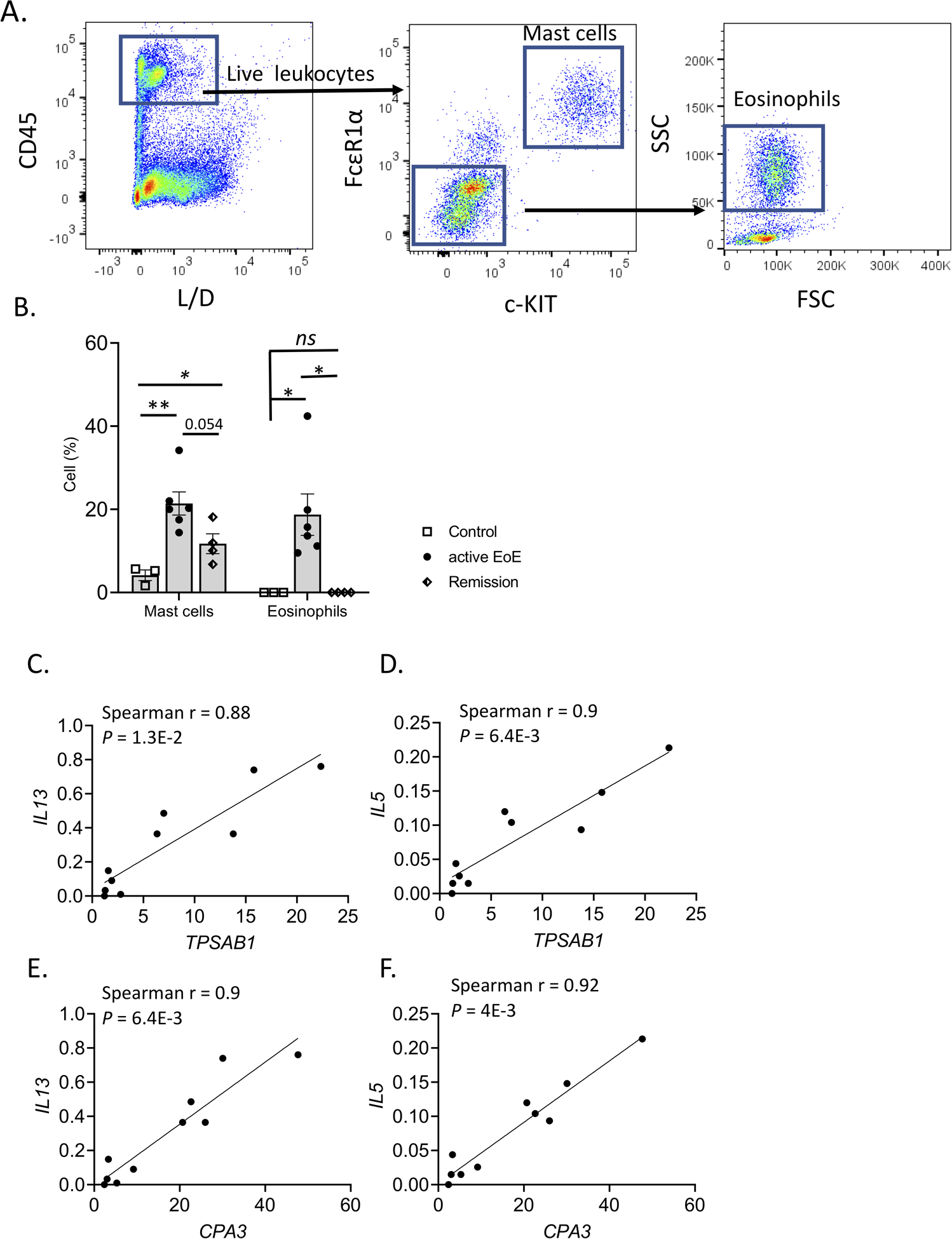
A–B A cohort of esophageal tissue (control, n = 3; EoE, n = 6; EoE remission, n = 4) was processed to obtain a single-cell suspension. Cells were stained and analyzed via flow cytometry to identify mast cells (MCs) (CD45highc-KIThighFcεRIα high); (A) the gating strategy is shown, and the L/D X-axis label represents staining for live/dead cells. (B) The percentage of mast cells and eosinophils out of the total white blood cell population (CD45high) is graphed. Individual markers represent distinct individuals, and bars represent mean ± SEM, which was analyzed using a t-test for mean comparison between two groups; **, P < 0.01 *, P < 0.05; ns, not significant. C–F Spearman correlation of mast cell markers (TPSAB1 and CPA3) with key gene expression levels (IL13 and IL5) and comparison to other immunocyte markers in active EoE. Individual markers represent distinct individuals. Data are derived from bulk RNA sequencing of patients with active EoE (n = 10), as reported6, 92. P-value Bonferroni-Dunn’s correction was calculated for each correlation.
To determine the relationship between MC concentration and EoE-related inflammatory mediators, we examined bulk RNA sequencing data from the esophageal tissue of patients with active EoE (n = 10)6, 93. In active disease, the MC marker genes TPSAB1 and CPA38 had a high, positive correlation with the EoE-associated cytokines IL13 (Spearman r = 0.88, P = 1.3E-2; and Spearman r = 0.90, P = 6.4E-3, respectively), IL5 (Spearman r = 0.90, P = 6.4E-3 and Spearman r = 0.92, P = 4.0E-3, respectively). (Figure 1C–F)37. These data suggested the potential importance of MCs as a source of several major components of the inflammatory milieu in active EoE.
ScRNA-seq: esophageal MCs are poised to generate pro-inflammatory mediators
To further assess the potential contribution of MCs to EoE pathobiology relative to other cell populations, we conducted scRNA-seq on 39,562 single cells from esophageal biopsies (n = 5 patients with active EoE; n = 3 patients with EoE in remission, and n = 2 non-EoE controls) (Supplementary Table 1). Seven major cell lineages were identified on the basis of hallmark gene expression (shown in parentheses): epithelial cells (KRT13 and KRT5), T cells (CD3E and CD7), endothelial cells (SPARC and VWF), myeloid cells (HLA-DRB and HLA-DRA), fibroblasts (DCN and COL1A1), B cells (MS4A1 and CD79A), and MCs (TPASB1, CPA3, GATA2, and MITF) (Figure 2A–B and Supplementary Table S2). Although eosinophils were present in histologic samples (Supplementary Figure S2A), no cluster was detected expressing the eosinophil marker transcript CLC. This finding is consistent with prior scRNA-seq studies of respiratory tissue38, 39 that failed to detect eosinophils, likely due to their high expression of RNases, such as eosinophil cationic protein (RNASE3) and eosinophil-derived neurotoxin (RNASE2)40. MCs were a prominent cell type in the esophagus, comprising 7.3 ± 2.4% of total recovered cells during active disease (Supplementary Figure S2B). In active EoE patients, in addition to the enriched expression of the hallmark proteases TPSAB1, CPA3, CTSG, and CMA1, the MC cluster exhibited enrichment of the cathepsin family proteases CTSD and CTSW; however, the expression of these genes was not exclusively restricted to MCs (Figure 2C and Supplementary Table S2). MCs were also the most prevalent population that expressed genes associated with histamine biosynthesis (HDC), prostaglandin biosynthesis (HPGDS, PTGS1, and PTGS2), and leukotriene biosynthesis (LTC4S, ALOX5AP, and ALOX5) (Figure 2C and Supplementary Table S2). MCs together with T cells significantly expressed the type 2 cytokine–encoding genes IL5 and IL13 and colony-stimulating factor 1 (CSF1, aka macrophage-colony stimulating factor 1) (Figure 2C and Supplementary Table S2). MCs were also the only cells that significantly expressed the IL-6 cytokine family member leukemia inhibitory factor (LIF) (Figure 2C and Supplementary Table S2). MCs further expressed mRNA encoding diverse cytokines, chemokines, and growth factors, including CCL2, CCL4, TGFB1, AREG (encoding Amphiregulin), and VEGFA, although expression of these genes was not restricted to MCs (Figure 2C and Supplementary Table S2). MCs were also the main esophageal population expressing the receptors for IL-33 (IL1RL1) and c-KIT (KIT), expressed the highest level of the aryl hydrocarbon receptor (AHR), and were the only esophageal cell population that expressed all three subunits of the high-affinity IgE receptor FcεR1 (i.e., FCER1A, FCER1G, and MS4A2) (Figure 2C).
Figure 2: esophageal MC identification and characterization by scRNA-seq.
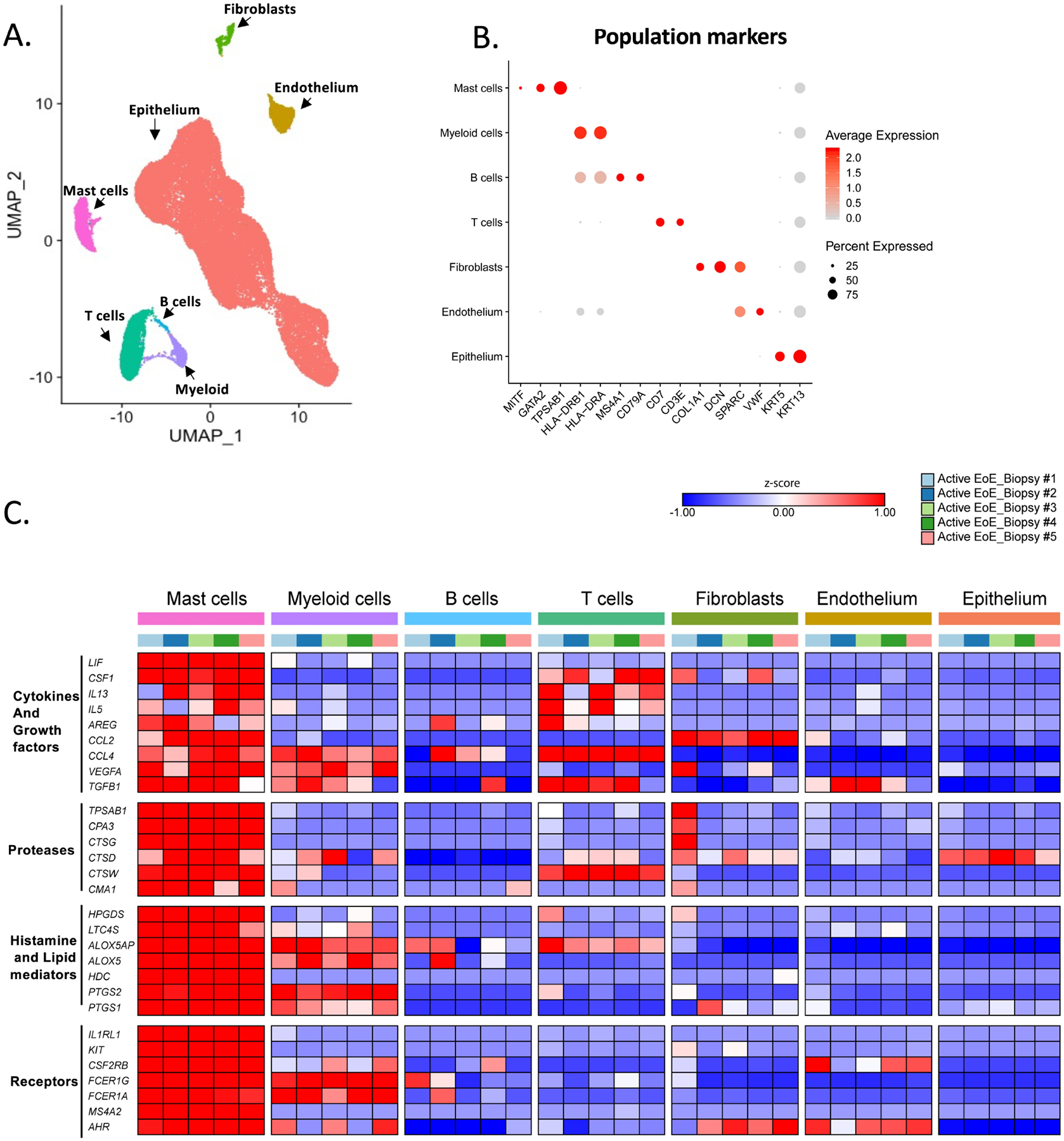
A–B Examination of esophageal cell populations on the basis of gene expression. (A) Results were analyzed using an experimental workflow that led to a uniform manifold approximation and projection (UMAP) plot for dimension reduction displaying 39,562 single cells. The plot is colored by cell types derived from the shared nearest neighbor (SNN) clustering and enriched marker genes. Red, epithelium; yellow, endothelium; green, fibroblasts; magenta, mast cells (MCs); turquoise, T cells; light blue, B cells; and purple, myeloid cells. (B) A dot plot of marker genes for indicated cell types is shown. C Heatmap of MC–enriched genes for proteases, histamine and lipid mediator related, cytokines and growth factors and receptors in the 5 active EoE donors. The map’s color for each gene is proportional to the average per-cell gene expression within the given patient in the esophageal cell populations. MCs were isolated from esophageal biopsies derived from patients with active EoE (n = 5) or remission EoE (n = 3) and non-EoE controls (n = 2).
scRNA-seq: MCs are engaged in multiple cellular axes of the esophageal ecosystem
We next directly evaluated MCs within the context of the cellular signaling axes thought to promote inflammation in EoE. We identified cellular expression of key type 2 inflammation–associated cytokines and the corresponding receptors within the scRNA-seq dataset, identifying potential interactions between MCs and the other detected esophageal populations, including myeloid cells, fibroblasts, endothelial cells, and epithelial cells (Supplementary Figure 3A–C). The MC survival factor KITLG was expressed by structural cells, including fibroblasts, endothelial, and epithelial cells; the latter cell demonstrated increased expression in active EoE (Figure 3A–B and Supplementary Figure 3A–C)41, 42. In addition, MC expression of IL1RL1 indicated a possible networking with cells expressing IL33, a cytokine that can promote human MC activation, survival, and adhesion43. Interestingly, although IL33 was mainly detected in endothelial cells (Figure 3C–D), during active EoE, epithelial cells exhibited increased IL33 expression. MC’s IL13 expression was predominantly observed in active EoE (Figure 3E–F), supporting the correlation between TPSAB1 and IL13 observed in bulk sequencing of EoE biopsies (Figure 1C and Figure 1E).
Figure 3: EoE-associated MCs axes.
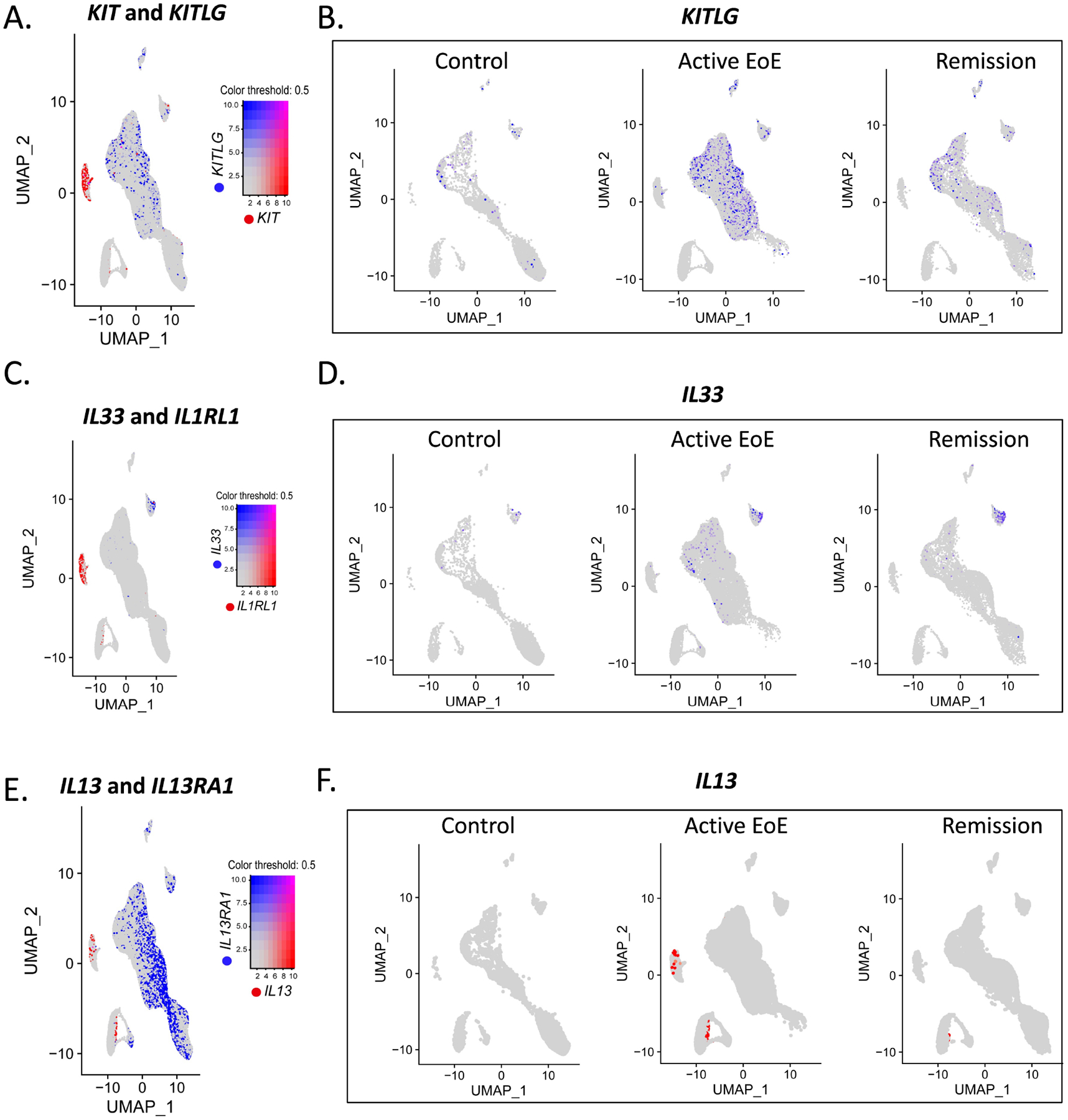
A–F Examination of mast cells (MC) potential interaction with other esophageal cell populations in cellular networking. Feature plot analyses of EoE hallmarked genes of the scRNA-seq of the ten biopsies obtained from patients with active EoE (n = 5), remission EoE (n = 3), and non-EoE controls (n = 2). Feature plots of co-expression of the key type 2 cytokines and the compatible receptors in MCs (red) and other esophageal cell population(s) (blue) (A, C, E) and feature plots split by diagnosis (B, D, F) are presented.
In addition, in control biopsies, MCs expressed AREG, indicating a potential communication axis with EGFR+ epithelial cells (Supplementary Figure 3A), an interaction that was previously demonstrated as essential for normal epithelial differentiation44, 45.
In active EoE, MCs expressed several additional inflammatory mediators, including IL5, potentially allowing them to interact with eosinophils, and CSF1, potentially allowing them to interact with CSF1R-expressing myeloid cells, an essential interaction for myeloid differentiation and expansion46, 47 (Supplementary Figure 3B). MCs also expressed TGFB1, suggesting interaction with TGFBR2-expressing fibroblasts, which has the potential to promote fibroblast proliferation and collagen production48 and contribute to the Mendelian-inherited form of EoE caused by TGFBR2 mutations (Supplementary Figure 3B)49. MCs also expressed elevated LIF, allowing possible interactions with endothelial cells that express LIFR, a receptor that regulates angiogenesis50–52. Finally, MCs expressed VEGFA, congruent with a previous report which demonstrated that activated MCs secrete VEGFA53, suggesting an additional axis of communication with FLT1+ endothelial cells, an interaction previously shown to promote endothelial differentiation54 (Supplementary Figure 3B). Interestingly, many potential interactions established in active disease persisted in remission biopsies (Supplementary Figure 3C).
We also evaluated the cellular expression of transcripts encoding lipid mediators and histamine biosynthetic enzymes along with the corresponding receptors. MCs expressed the leukotriene-associated genes LTC4S, ALOX5AP, and ALOX5, indicating potential interactions with CYSLTR1+ endothelial cells and CYSLTR1+ CYSLTR2+ fibroblasts, all of which could be observed across disease states. These interactions have been shown to promote inflammatory signals and proliferation in endothelial cells and fibroblasts55, 56. MCs expressed the histamine biosynthesis–associated gene HDC, indicating the potential to interact with esophageal HRH1+ endothelial cells across disease states. In contrast, myeloid cell expression of HRH2 was only observed in active EoE. MC expression of the prostaglandin biosynthesis–associated genes HPGDS, PTGS1, and PTGS2 indicated the potential to associate with PTGDR2+ fibroblasts. This interaction has been shown to promote fibroblast activation and is associated with fibrosis57 (Supplementary Figure 3D–F).
ScRNA-seq: increased MC heterogeneity during EoE, including an actively proliferating MC population
After re-clustering 1,897 single-cell MC transcriptomes, scRNA-seq analysis identified six MC clusters (Supplementary Figure S4A). Three of them were present in biopsies from all groups, termed resident MC 1–3 (Supplementary Figure S4B–D). To overcome the limitation of MCs numbers in control biopsies and as pooling resident MC 1–3 populations mostly maintained the expression levels of hallmark proteases, lipids, cytokines, and receptors compared to the other populations across the disease and control samples (Supplementary Figure S4E), these populations were pooled for subsequent downstream analysis and termed “resident MCs” (Figure 4A). The revised clustering was conserved across multiple donors (Supplementary Figure S5A–B). The resident population accounted for 100 ± 0% of control MCs, 42 ± 27% of active EoE MCs, and 83 ± 22% of remission MCs (Figure 4B). Further analysis identified three additional MC populations in patients with active EoE. The first was designated transient MCs, as it was only detected in active disease, accounting for 19 ± 22% of esophageal MCs (Figure 4B). The second EoE-associated population was designated persistent MCs, as it was observed in both active EoE (accounting for 37 ± 24% of total MCs in active EoE) and in remission (accounting for 17 ± 21% of the total MCs during remission) (Figure 4B). The third EoE-associated MC cluster was designated as proliferative MCs on the basis of enriched expression of proliferation-associated genes, including MKI67, TOP2A, PCNA, and KIAA0101 (aka PCLAF, PCNA clamp-associated factor58) (Figure 4C and Supplementary Table S3). GO analysis corroborated enriched expression of genes involved in the cell cycle (corrected p-value = 3.8E-70) and cell division (corrected p-value = 1.9E-56) within the proliferative MC cluster (Supplementary Table S4).
Figure 4: Esophageal MC populations across the diagnostic spectrum.
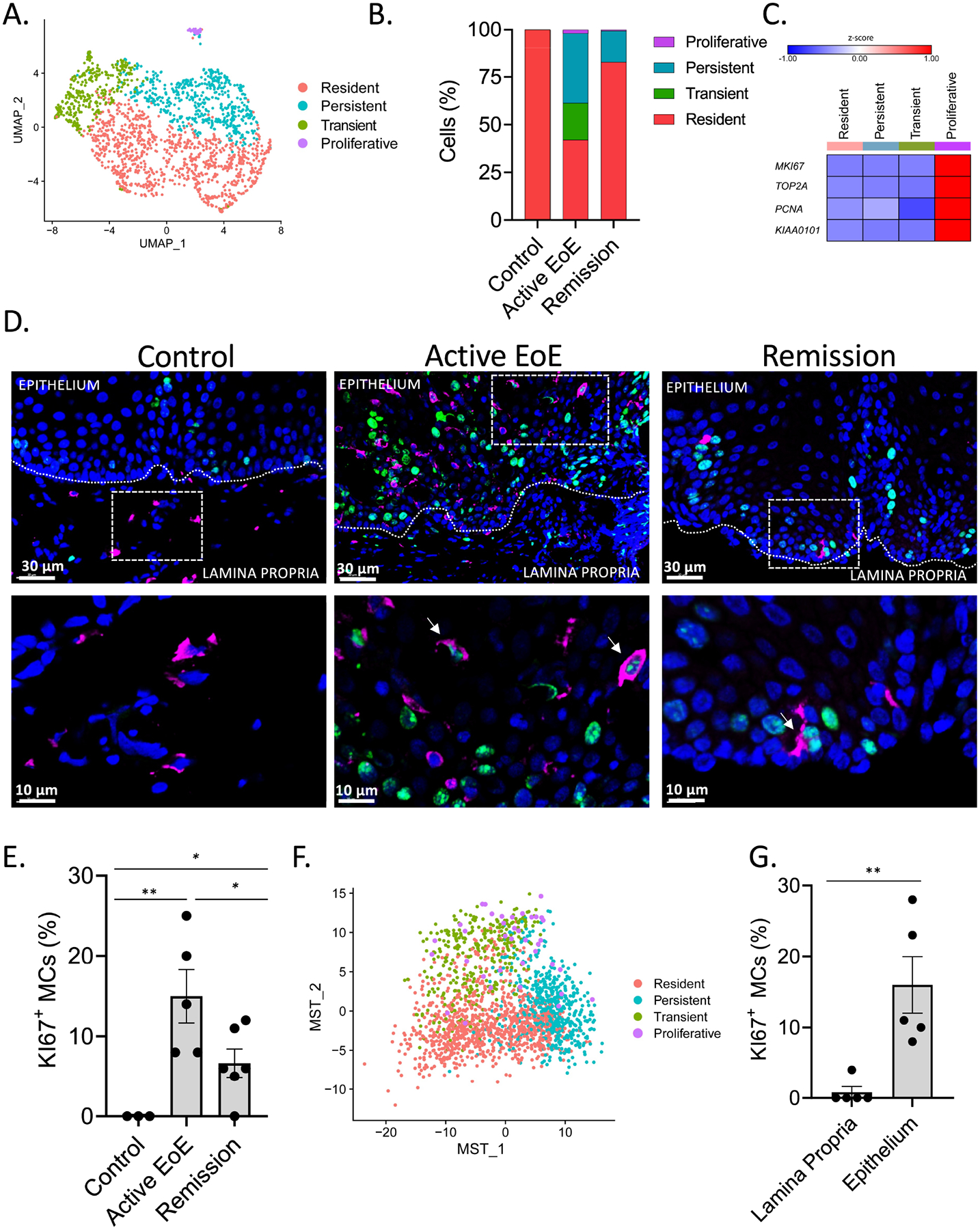
A–C, F Examination of esophageal mast cell (MC) populations on the basis of gene expression. Esophageal biopsies were subjected to scRNA-seq. (A) Results were analyzed using an experimental workflow leading to a UMAP plot of 1,897 single mast cells (n = 10 samples) that were subsetted and reclustered from the total esophageal single cells (39,562 cells) colored by SNN clustering. Red, resident; green, transient; turquoise, persistent; and magenta, proliferative. (B) The stacked bar graph shows a given mast cell subpopulation as a percentage of the total mast cells in each sample. (C) Heatmap of proliferation marker genes; (F) MST plot of pseudotime analysis reclustered and organized mast cell populations on the basis of genetic similarity, colored by the original SNN clusters. A, F Individual markers represent individual cells. D–E, G A separate cohort of esophageal tissue (active EoE, n = 5; remission, n = 6; control, n = 3) was subject to immunofluorescent staining for tryptase (magenta), as a marker of MCs, and KI67 (green), as a marker of proliferation. Representative pictures (D) and quantification of the percentage of KI67+ MC in the entire esophageal biopsy of all patients (E) and specifically in the epithelial and lamina propria in esophageal biopsies of patients with active EoE (G) are being shown in magnifications of 20X (D, upper row) and 100X (D lower row, magnified area of 20X indicated by dashed line boxes). D, double-positive cells for tryptase and KI67 depicted by the arrows E, G Individual markers represent distinct individuals, and bars represent mean ± SEM, which was analyzed using the t-test for comparison between each two groups. *, P < 0.05, **, P < 0.01.
Immunofluorescence analysis of biopsies from an independent patient cohort (control, n = 6; active EoE, n = 7; remission, n = 7) (Supplementary Table 1) revealed that MCs in control biopsies were located mainly in the lamina propria and the level of this population did not significantly change in active EoE or remission (Supplementary Figure S6A, Supplementary Figure 6C–D, and Supplementary Figure 6F). As control samples were only found to contain resident MCs present by scRNA-seq analysis (Figure 4B), we concluded that resident MCs are likely localized in the lamina propria. MC numbers were significantly increased in the epithelium layer, but not in the lamina propria, in active EoE compared with control biopsies (Supplementary Figure S6A–B), consistent with previous reports7. Biopsies obtained from patients with EoE in remission exhibited variability in the intraepithelial MC levels (Supplementary Figure S6D–F), consistent with a recent report10.
The proliferative cluster was predominantly observed in active EoE biopsies, accounting for 2.0 ± 2.7% of total MCs (Figure 4B). Immunofluorescent staining of an orthogonal patient cohort (control, n = 3; active EoE, n = 5; remission, n = 6) (Supplementary Table 1) confirmed the presence of KI67+ MCs within the esophagus during active and remission (Figure 4D–E).
The MC clusters were further analyzed through pseudotime using independent Minimum Spanning Tree (MST) trajectory analysis59, 60 (Figure 4F). The proliferative population was distributed between both transient and persistent MCs through this approach, suggesting that the increased MC proliferation observed histologically during EoE (Figure 4D–E) could be attributed to these populations (Figure 4F). Indeed, quantification of KI67+ MCs in each compartment in biopsies obtained from patients with active EoE, in which all populations were present, demonstrated that proliferation occurs mainly within the epithelium (Figure 4G). The MST trajectory analysis demonstrated that the proliferating cells were observed in the persistent population during remission; however, the number of cells was too low to be defined accordingly (n = 2) (Supplementary Figure S7).
Marker gene analysis identified disease state-dependent MC transcriptional signatures
To further examine the transcriptional differences in esophageal MCs across disease states, we evaluated differential gene expression across both the entire MC population as a whole and within each subpopulation. This analysis demonstrated that MCs obtained from control biopsies were composed of resident MCs which, compared with MC’s that were obtained from active EoE and remission biopsies, exhibited increased expression of TPSAB1, ALOX5, and AREG; the latter encodes for a protein that maintains epithelial homeostasis44, 45 (Figure 5A and Supplementary Table S5). MCs from active EoE were enriched in the expression of the protease-encoding transcripts CPA3 and CTSG; the lipid mediator biosynthetic enzyme-encoding transcripts ALOX5AP and PTGS2; the cytokine, chemokine, and growth factor-encoding transcripts CCL2, VEGFA, IL5, and IL13 and the receptor-encoding transcripts IL1RL1, FCER1A, AHR, CSF2RB, MS4A2, and KIT, compared with MCs derived from control and remission biopsies (Figure 5A and Supplementary Table S5). Next, we were interested in evaluating if the higher level of mRNA transcripts in MCs obtained from active EoE patients also translated to an increased protein level. As a proof of principle, we evaluated FcεR1A protein levels. We found that MCs from active EoE patients exhibited higher FcεR1A levels compared with MCs from control and remission (Supplementary Figure S8), consistent with FCER1A mRNA levels. Subset analysis (Figure 5B) indicated that resident MCs were most enriched for expression of IL5, transient MCs were enriched for expression of the receptor-encoding genes IL1RL1 and KIT and expressed the lowest level of FCER1A, and persistent MCs were enriched for expression of CTSG and CMA1, suggesting an MCTC phenotype (Supplementary Table 6). In biopsies obtained from patients with EoE in remission, MCs as a whole exhibited the highest enrichment of the protease-encoding transcripts CTSD, CTSW, and CMA1; lipid mediator–associated genes LTC4S, HPGDS, HDC, and PTGS1; the cytokine and growth factor–encoding transcripts LIF, TGFB1, and CSF1; and the signaling adaptor–encoding transcript FCER1G compared with that of MCs identified in control and active EoE biopsies (Figure 5A and Supplementary Table S5). In remission, subset analysis indicated that the resident MC population was most enriched for CSF1. The persistent population exhibited enrichment of the protease-encoding transcripts CMA1 and CTSG (Figure 5B and Supplementary Table S6).
Figure 5: Characterization of the diversity of esophageal MC populations.
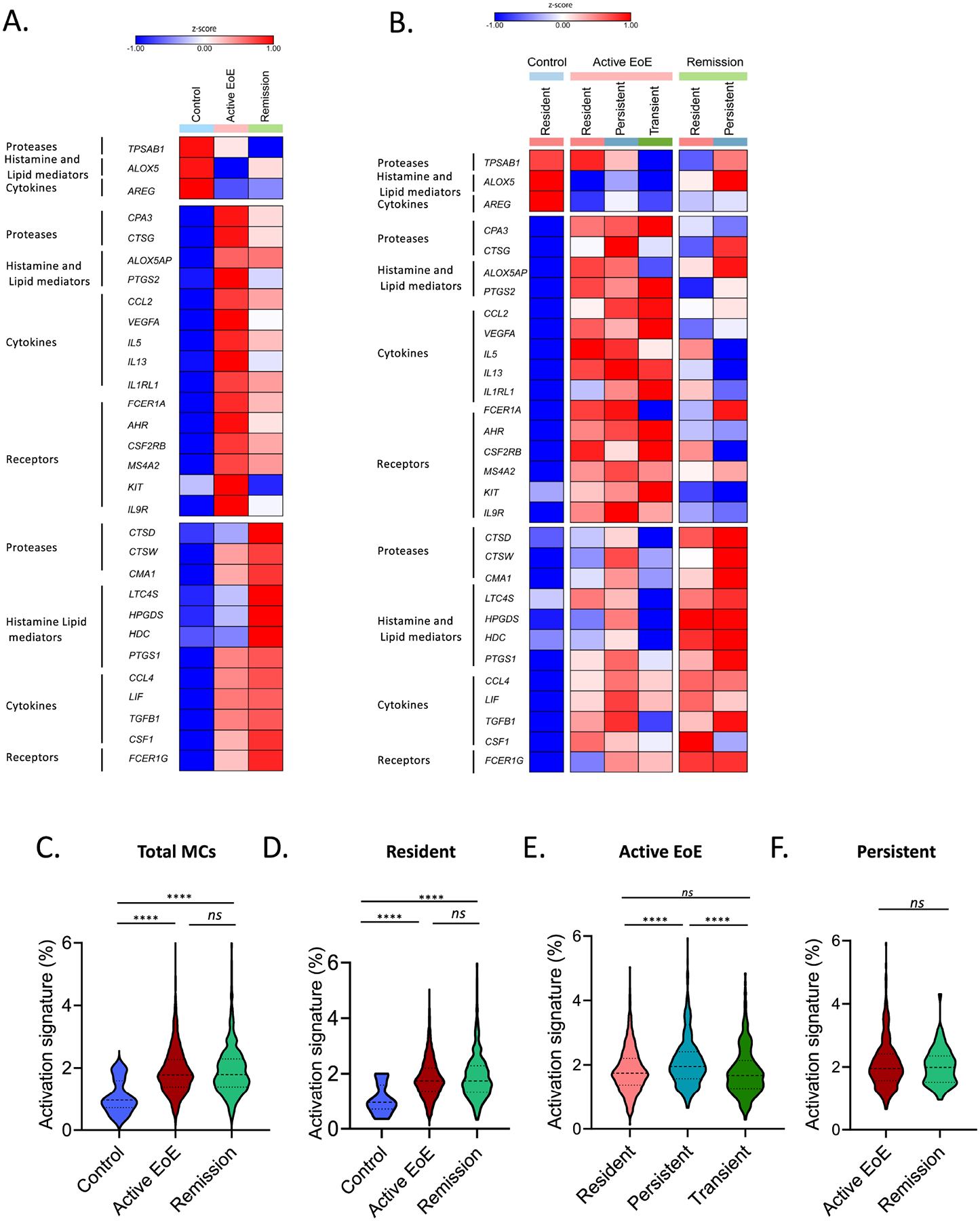
A–F Examination of esophageal mast cell (MC) populations on the basis of gene expression. Heatmaps of enriched marker gene expression in the entire MC population (A) and divided by MC subpopulations (B) as a function of the disease state. The map’s color for each gene is proportional to the average per-cell gene expression within the given MC population. Violin plots indicate the percentage of genes out of the total gene number included in the activation signature expressed by the MC population as a whole (C), the resident population in each diagnostic state (D), all MC populations/cluster present in active EoE (E), and the persistent population in EoE compared to remission (F). Bars represent mean ± SEM, which were analyzed using one-way ANOVA with Tukey post-test; ns = not significant, ****, P ≤ 0.0001.
MC activation signatures indicate transcriptional activation during active disease and remission
The transcriptional signature of MCs across disease activity states (Figure 5C) suggested that all MC populations, including the resident population, upregulated inflammatory genes during active EoE and remission, consistent with a heightened activation state. We thus examined the expression of MC activation genes of the total MCs transcriptome across the different disease states (Supplementary Table S8)61. MCs from active EoE and remission biopsies were more activated than those derived from control biopsies (Figure 5D). Notably, despite differences in transcripts encoding pro-inflammatory mediators observed between active EoE and remission (Figure 5A), no difference was found in total MC activation signature expression between active EoE and remission (Figure 5C). Subset analysis indicated that the resident MC population exhibited an increased activation signature in both active EoE and remission compared with non-EoE controls (Figure 5D). Specific analysis of active EoE, the only disease state in which all three major populations could be observed, indicated that the persistent population exhibited the highest overall activation signature expression (Figure 5E). No difference in activation signature expression was observed in persistent MCs between active EoE and remission (Figure 5F). Relative to the other MC populations, persistent MCs exhibited increased expression of 307 unique genes and decreased expression of only 17 genes. GO analysis of biological processes of the 307 elevated genes found enrichment in genes associated with an activation process (corrected p-value = 2.7E-25) and immune effector process (corrected p-value = 1.2E-22) (Supplementary Table S3 and Supplementary Table S7). Thus, the resident and persistent populations maintained the increased activation signature despite disease remission.
MCs were a prominent source of esophageal IL-13
IL13 was enriched in MCs compared with other esophageal cells in active EoE biopsies (Figure 2C) and undetectable in control biopsies (Figure 5A and Supplementary Figure S9A) and correlated with MC protease expression during active EoE in bulk RNAseq analysis (Figure 1C and Figure 1E). Using immunofluorescence, we evaluated IL-13 protein content in MCs from control (n = 6) and active EoE (n = 7) (Supplementary Table 1). In control biopsies, although resident MCs did not express detectable IL13 mRNA (Figure 5A and Supplementary Figure S9A), they contained a readily detectable intracellular IL-13 (Figure 6A, top row). In active EoE, resident MCs upregulated their IL13 transcripts (Figure 5B) and IL-13+ MC numbers in the lamina propria were unchanged with 214 ± 117 IL-13+ MCs/mm2 lamina propria in control samples and 263 ± 143 IL-13+ MCs/mm2 lamina propria in active EoE (Figure 6B). During active EoE, although most MCs within the epithelium lacked intracellular IL-13 (e.g., 20 ± 9% were IL-13+), the concentration of IL-13+ intraepithelial MC numbers increased significantly to 40 ± 18 MCs/mm2 epithelium in active EoE compared with 12.5 ± 5 MCs/mm2 epithelium in controls, consistent with the enrichment of IL13 observed in the transient and persistent intraepithelial populations during active EoE (Figure 6A–B and Figure 5A–B). The high degree of activation observed in all MCs in active EoE (Figure 5E) coupled with the accumulation of IL13+ MCs in the intraepithelial compartment, and MC’s ability to secrete stored intracellular cytokines upon activation62, 63 suggests that the activated intraepithelial MCs may actively secrete IL-13 during disease. This possibility is supported by the positive correlation of TPSAB1, a reliable marker of MC tissue burden8, with canonical IL-13 induced epithelial genes including CAPN14, CCL26, CDH26 and POSTN (n = 147 patients with active EoE in an independent cohort)11, 32 (Supplementary Figure S9B–E).
Figure 6: Examination of MC IL-13 protein expression.
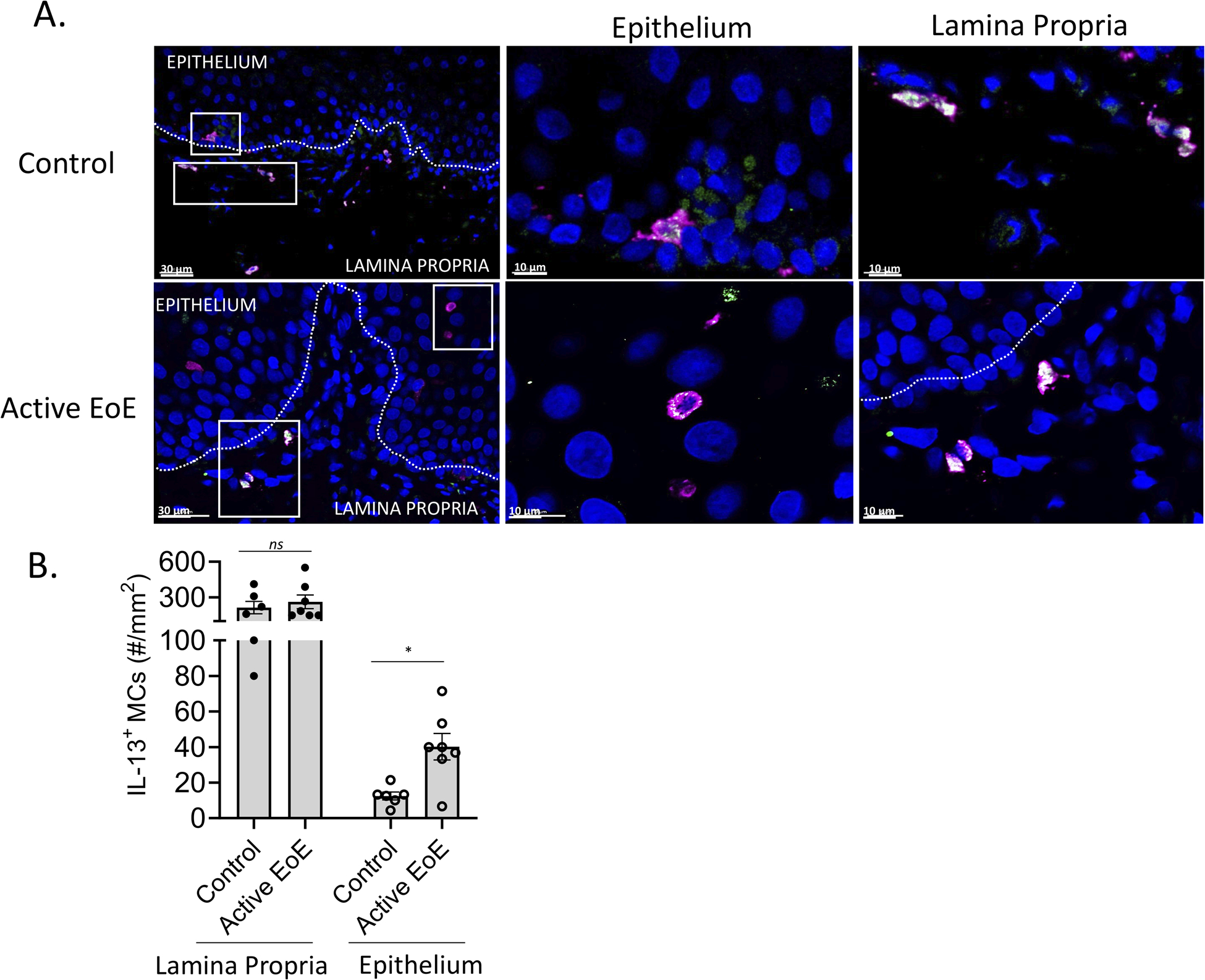
A–B A cohort of esophageal tissue (control, n = 6; active EoE, n = 7) was subject to immunofluorescent staining of tryptase (magenta) as a marker of mast cells (MCs) and IL-13 (green). (A) Representative pictures from control and active EoE are shown in magnification of 20X (left panels) and 40X (epithelium middle panel and lamina propria right panel). Dashed line boxes represent the magnified area in epithelium and lamina propria, respectively. Double positive staining display as a combination of magenta and green, generating white staining. (B) Quantification of IL-13+ MC numbers per mm2 in the lamina propria and epithelial layers. B Bars represent mean ± SEM, which were analyzed using the t-test; *, P < 0.05, ns = not significant.
DISCUSSION
MCs have well-described roles in type 2 diseases associated with IgE-mediated degranulation, such as urticaria and anaphylaxis64. However, therapeutic targeting of IgE was largely unsuccessful in EoE65–67. Herein, we demonstrate that MCs infiltrate the inflamed esophagus in EoE, exist in at least three distinct populations, proliferate locally, remain activated in disease remission, and generate IL-13, a primary mediator and therapeutic target in EoE68, 69. In addition, the MC markers TPSAB1 and CPA3 are highly associated with type 2 cytokine–induced genes in the esophageal epithelium. These data highlight the importance of human MC effector function beyond IgE-mediated degranulation and suggest that control of tissue MC expansion will be required for complete remission of EoE.
We have found that MCs are present in the healthy esophagus and become highly prevalent cell type detected in active EoE. Furthermore, we find that specific MC subpopulations can remain elevated despite disease remission. We have established that MCs are present mainly in the esophageal lamina propria during homeostatic conditions, characterized as TPSAB1highAREGhigh, expressing a low level of MC activation–associated genes but can be readily activated as they store inflammatory proteins such as IL-13. In active EoE, MCs expand and infiltrate into the esophageal epithelium and proliferate locally. They are characterized by the accumulation of KIThighIL1RL1highFCER1Alow transient and CMA1highCTSGhigh persistent EoE-specific MCs populations, each of which is associated with the MKI67high proliferative cluster. In remission, the transient population is abolished, consistent with the decreased level of the intraepithelial MCs; however, the CMA1highCTSGhigh persistent population, in some patients, remains detectable in the intraepithelial compartment. Both CSF1high resident and CMA1highCTSGhigh persistent populations remain transcriptionally activated even in disease remission. Supplementary Figure 10 summarizes these primary findings.
MC accumulation is a central feature of EoE7, 8 and is linked to multiple disease pathologies10. Thus, understanding their tissue accumulation is essential. Until now, evidence has demonstrated that MCs in the intestinal mucosa arise from bone marrow–derived progenitors that constitutively home to the intestine during T cell–mediated inflammation70–72. We report that in biopsies obtained from patients with EoE, MCs exhibit increased local proliferation in the esophagus associated with the increased proliferation of the transient and the persistent MC populations. Increased local proliferation during active EoE is aligned with a recent paper that reported local MC proliferation in another type 2 inflammatory disease, chronic rhinosinusitis with nasal polyposis31. Therefore, MC expansion by proliferation is likely a common hallmark feature of type 2 diseases. Several factors upregulated in the esophagus in active EoE are candidates for inducing local MC proliferation. This includes stem cell factor (SCF), which is a prominent candidate as human and rodent MCs cultured with SCF undergo extensive proliferation ex vivo and in vivo, respectively22, 20. Our data demonstrate the SCF-encoding gene, KITLG, is expressed mainly by fibroblasts and present in all EoE disease states although it is upregulated in epithelial cells in active disease (Figure 3A–B and Supplementary Figure 3A–C). In addition, the receptor KIT is upregulated in active EoE, especially by the transient population that is linked to MC proliferation (Figure 2C, Figure 3A, Figure 5A–B, and Supplementary Figures 3A–C). Another potential regulator of MC proliferation is IL-9, which also promotes MC maturation and activation12, 73, 74. IL9R is enriched in MCs during active EoE, and remission compared with MCs obtained from control biopsies (Figure 5A–B and Supplementary Table S5). IL9 is absent from the scRNA-seq dataset, perhaps due to the technological limitations (e.g., the absence of eosinophil gene signatures) or because it is produced by relatively rare cell populations (e.g., ILC2 and TH9 cells)73, 75. Understanding the mechanisms that promote the local proliferation of MCs and whether they are specific to the esophagus or common to other type 2 diseases remains largely unexplored.
In homeostasis, MCs localize around the vasculature and nerves, and their wide range of receptors led to the notion that they serve as tissue sentinels. As MCs are pre-armed with an arsenal of proteases and inflammatory proteins, they have the potential to be one of the first responders to local stimuli, promoting acute local inflammation15. In control biopsies, MCs reside in the lamina propria and are characterized by the expression of proteases, such as chymase, which is known to increase epithelial permeability in the intestine7, 76. During homeostasis, MCs contain a large amount of IL-13 protein (Figure 6A–B).This cytokine has potential to promote key inflammatory processes in EoE, such as activating the epithelium to secrete CCL26, inducing tissue remodeling, and impairing epithelial differentiation and barrier function37. Although MCs are known to generate IL-13 in response to various stimuli77, 78, Th2 lymphocytes are thought to be the primary source of IL-13 in EoE. Th2 lymphocytes are not present in the esophagus under homeostatic conditions but are instead present in the tissue only during active EoE and remission83. In addition, evidence is accumulating that Th2 lymphocytes may be clonal and home to the esophagus via GPR15 during active EoE84, 85. Moreover, in active disease, we found that MC express abundant IL-13 protein and there was a high correlation between MC marker genes and expression of IL-13–elicited genes in bulk RNA-seq (Figure 6A–B and Supplementary Figure 9B–E), indicating that MCs also contribute to IL-13 generation in disease. Indeed, MCs are an essential source of IL-13 in other T2 inflammatory diseases, such as asthma78 and nasal polyposis31. Other potential sources for esophageal IL-13 during active EoE are eosinophils, basophils and ILC279, 80, 86, 87. While eosinophils, similar to Th2, are present only during active disease, basophils and ILC2 are present in the esophagus also under homeostatic conditions36, 81, 82. However, only basophils, similar to MCs, can store preformed IL-13 as ILC2 require T2 stimuli to generate and secrete IL-1381, 82, 86, 87.
EoE exhibits defined histopathology, including local type 2 inflammation, angiogenesis, epithelial barrier disruption, epithelial hyperplasia, and fibrosis88. During active disease, MCs expand in the epithelial compartment7 (Supplementary Figure S6A–C) and exhibit an enriched transcriptional activation signature in both the lamina propria and epithelial compartments (Figure 5). Our data suggest that their expansion allows them to interact with a wide range of esophageal cell populations, including epithelial cells (e.g., via IL-13–induced CCL26), eosinophils (e.g., via IL-5–induced eosinophil activation and survival), other myeloid cells (e.g., via CSF-induced dendritic cell activation), endothelial cells (e.g., via LIF and leukotriene-induced angiogenesis), and fibroblasts (e.g., via TGFβ1-induced proliferation) (Supplementary Figure 3)48, 52, 56, 89–91. These findings provide rationale for therapeutic targeting of MCs in EoE; this is a timely finding as MC-depleting and/or MC-inhibiting drugs, such as anti-KIT (CDX-0159) and anti–Siglec-8 (lirentelimab), respectively, are in clinical testing14, 92.
A recent study demonstrated that patients with EoE in histologic remission (i.e., less than 15 eosinophils/HPF) exhibited variability in histologic and clinical remission positively associated with MC levels10. In patients who exhibited persistent endoscopic and histologic abnormalities, MC numbers were elevated. Conversely, in patients showing complete histologic and clinical remission (i.e., no symptoms nor endoscopic abnormalities), MC numbers were decreased10. Our findings add increased resolution about the dynamic level and function of MCs in EoE by identifying a persistent population of activated MCs during histologic remission (defined by eosinophil levels) (Figure 4A–B, Figure 5B, and Figure 5E–F). The presented findings emphasize that MC resident and persistent subpopulations are activated in remission and poised to promote inflammation and that increased MC transcript expression is associated with increased IL-13 stimulation (Figures 5A–D and Figure 5F). Thus, we suggest that assessing MC burden may be a needed component in defining EoE remission.
Chymase and cathepsin G are classically associated with submucosal MCTC22–24. In control biopsies, resident MCs were primarily located in the lamina propria compartment and exhibited an MCTC phenotype7. Our study identifies the CMA1highCTSGhigh persistent population located in the intraepithelial compartment in active disease and remission (Figure 5B, Supplementary Figure S6, and Supplementary Table S3). This finding is consistent with a previous study that demonstrated that during active EoE, MCTC are also found in the intraepithelial compartment7. As the differences between mRNA and protein levels may exist due to storage of proteins in MC granules94, further study will be required to determine whether the persistent intraepithelial MCs exhibit a MCTC phenotype.
Our data demonstrate increased variation in the proportion of resident MC subpopulation between donors (Supplementary Figure 4D). These finding may result from differences in disease severity that influence the proportion of EoE-associated MC populations. Alternatively, variation in the size of the lamina propria in each biopsy may contribute. As resident MCs are localized in the lamina propria, the proportion of the resident MC is likely dependent upon the size of the lamina propria in each biopsy. To overcome this limitation, computational analysis verified that MCs subpopulations do not derive from donor variation but are rather dependent upon disease status. In addition, we combined resident subpopulations 1–3 to enable focus on diagnostic-dependent differences (Figure 4 and Supplementary Figure 5). To evaluate the possibility of the existence of multiple subpopulations in the resident MC population, a larger cohort will be examined in the future.
In conclusion, we have identified three populations of esophageal MCs that exhibit heterogenous properties defined by their (1) selective accumulation at different stages of disease and health; (2) expression of inflammatory genes; (3) level of transcriptional activation; (4) association with local proliferation; and (5) spatial compartmentalization. We demonstrate that MCs constitute a prominent source of key EoE mediators (e.g., IL-13) even under homeostatic conditions. In particular, during homeostasis, TPSAB1highAREGhigh resident MCs are mainly present in the lamina propria and exhibit a quiescent phenotype. In patients with active EoE, resident MCs assume an activated phenotype, and two additional pro-inflammatory MC populations emerge in the intraepithelial compartment, each linked to a proliferating MKI67high cluster. The transient MC population, marked as KIThighIL1RL1highFCER1Alow, is not detected in disease remission, whereas the persistent MC population is marked by CMA1highCTSGhigh expression and persists even in disease remission. The finding that MC subpopulations remain present and primed for activation even in patients without esophageal eosinophilia provides a paradigm shift in defining disease remission as the mere absence of eosinophils and suggests that eosinophil levels may not be a sufficient marker of disease activity. Our data substantiate the importance of considering the presence of MCs, especially the persistent CMA1highCTSGhigh MC population identified herein. Thus, restraining different MC populations and their activity should be a therapeutic consideration for EoE.
Supplementary Material
Key messages:
Esophageal MCs are a dynamic population that can increase their numbers, heterogeneity, activation, and compartmentalization in active EoE and remain activated in remission.
Esophageal MCs expand by in situ proliferation.
Esophageal MCs constitute a prominent cellular source of inflammatory mediators (e.g., IL-13) even under homeostatic conditions.
Acknowledgement
Single-cell RNA sequencing data was acquired by the Gene expression core by Shawn Smith and Kelly Rangel. All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Core at Cincinnati Children’s Hospital Medical Center. All immunofluorescence data were acquired using equipment maintained by the Imaging Core at Cincinnati Children’s Hospital Medical Center. The authors would like to thank Shawna Hottinger for editorial support and the Cincinnati Center for Eosinophilic Disorders team and Michael Eby for their assistance.
Grant support
This study was supported by the NIH R01 AI124355 and R01 AI045898-21.
Competing Interests:
M.E.R. is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celldex Therapeutics, Celgene, Astra Zeneca, Adare/Ellodi Pharma, GlaxoSmith Kline, Regeneron/Sanofi, Revolo Biotherapeutics, and Guidepoint and has an equity interest in the first five listed, and royalties from reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust) and UpToDate. M.E.R. is an inventor of patents owned by Cincinnati Children’s Hospital. T.S. has received research support from JSPS Overseas Research Fellowships and is a co-inventor of patents owned by Cincinnati Children’s Hospital Medical Center. V.A.M is a consultant for Takeda and Allakos.
All other authors declare that they have no competing interests.
Abbreviations:
- EoE
Eosinophilic Esophagitis
- MC
Mast cell
- scRNA-seq
Single-cell RNA sequencing
- T2
Type 2
- HPF
High Power Field
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Richter JE. Endoscopic Treatment of Eosinophilic Esophagitis. Gastrointest Endosc Clin N Am 2018; 28:97–110. [DOI] [PubMed] [Google Scholar]
- 2.Schoepfer AM, Straumann A, Safroneeva E. Pharmacologic Treatment of Eosinophilic Esophagitis: An Update. Gastrointest Endosc Clin N Am 2018; 28:77–88. [DOI] [PubMed] [Google Scholar]
- 3.Schoepfer AM, Gonsalves N, Bussmann C, Conus S, Simon HU, Straumann A, et al. Esophageal dilation in eosinophilic esophagitis: effectiveness, safety, and impact on the underlying inflammation. Am J Gastroenterol 2010; 105:1062–70. [DOI] [PubMed] [Google Scholar]
- 4.Gonsalves NP, Aceves SS. Diagnosis and treatment of eosinophilic esophagitis. J Allergy Clin Immunol 2020; 145:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuta GT, Katzka DA. Eosinophilic Esophagitis. N Engl J Med 2015; 373:1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherrill JD, Kiran KC, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014; 15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-beta1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol 2010; 126:1198–204 e4. [DOI] [PubMed] [Google Scholar]
- 8.Abonia JP, Blanchard C, Butz BB, Rainey HF, Collins MH, Stringer K, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol 2010; 126:140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin LJ, Franciosi JP, Collins MH, Abonia JP, Lee JJ, Hommel KA, et al. Pediatric Eosinophilic Esophagitis Symptom Scores (PEESS v2.0) identify histologic and molecular correlates of the key clinical features of disease. J Allergy Clin Immunol 2015; 135:1519–28 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolton SM, Kagalwalla AF, Arva NC, Wang MY, Amsden K, Melin-Aldana H, et al. Mast Cell Infiltration Is Associated With Persistent Symptoms and Endoscopic Abnormalities Despite Resolution of Eosinophilia in Pediatric Eosinophilic Esophagitis. Am J Gastroenterol 2020; 115:224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shoda T, Wen T, Aceves SS, Abonia JP, Atkins D, Bonis PA, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross-sectional study. Lancet Gastroenterol Hepatol 2018; 3:477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otani IM, Anilkumar AA, Newbury RO, Bhagat M, Beppu LY, Dohil R, et al. Anti-IL-5 therapy reduces mast cell and IL-9 cell numbers in pediatric patients with eosinophilic esophagitis. J Allergy Clin Immunol 2013; 131:1576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothenberg ME, Wen T, Greenberg A, Alpan O, Enav B, Hirano I, et al. Intravenous anti-IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J Allergy Clin Immunol 2015; 135:500–7. [DOI] [PubMed] [Google Scholar]
- 14.Dellon ES, Peterson KA, Murray JA, Falk GW, Gonsalves N, Chehade M, et al. Anti-Siglec-8 Antibody for Eosinophilic Gastritis and Duodenitis. N Engl J Med 2020; 383:1624–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol 2014; 14:478–94. [DOI] [PubMed] [Google Scholar]
- 16.Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol 2010; 40:1843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metcalfe DD, Boyce JA. Mast cell biology in evolution. J Allergy Clin Immunol 2006; 117:1227–9. [DOI] [PubMed] [Google Scholar]
- 18.Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med 2011; 183:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawakami T, Ando T, Kimura M, Wilson BS, Kawakami Y. Mast cells in atopic dermatitis. Curr Opin Immunol 2009; 21:666–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai M, Shih LS, Newlands GF, Takeishi T, Langley KE, Zsebo KM, et al. The rat c-kit ligand, stem cell factor, induces the development of connective tissue-type and mucosal mast cells in vivo. Analysis by anatomical distribution, histochemistry, and protease phenotype. J Exp Med 1991; 174:125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kambe N, Miyachi Y. A possible mechanism of mast cell proliferation in mastocytosis. J Dermatol 2002; 29:1–9. [DOI] [PubMed] [Google Scholar]
- 22.Miller JS, Schwartz LB. Human mast cell proteases and mast cell heterogeneity. Curr Opin Immunol 1989; 1:637–42. [DOI] [PubMed] [Google Scholar]
- 23.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev 1997; 77:1033–79. [DOI] [PubMed] [Google Scholar]
- 24.Pejler G, Abrink M, Ringvall M, Wernersson S. Mast cell proteases. Adv Immunol 2007; 95:167–255. [DOI] [PubMed] [Google Scholar]
- 25.Kitamura Y Heterogeneity of mast cells and phenotypic change between subpopulations. Annu Rev Immunol 1989; 7:59–76. [DOI] [PubMed] [Google Scholar]
- 26.Friend DS, Ghildyal N, Gurish MF, Hunt J, Hu X, Austen KF, et al. Reversible expression of tryptases and chymases in the jejunal mast cells of mice infected with Trichinella spiralis. J Immunol 1998; 160:5537–45. [PubMed] [Google Scholar]
- 27.Austen KF, Boyce JA. Mast cell lineage development and phenotypic regulation. Leuk Res 2001; 25:511–8. [DOI] [PubMed] [Google Scholar]
- 28.Dwyer DF, Barrett NA, Austen KF, Immunological Genome Project C. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol 2016; 17:878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and Intercellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019; 178:714–30 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020; 587:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Sci Immunol 2021; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wen T, Stucke EM, Grotjan TM, Kemme KA, Abonia JP, Putnam PE, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology 2013; 145:1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018; 36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell 2019; 177:1888–902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol 2019; 37:547–54. [DOI] [PubMed] [Google Scholar]
- 36.Wen T, Kuhl J, Putnam P, Mukkada V, Farrell M, Kaul A, et al. A flow cytometry-based diagnosis of eosinophilic esophagitis. J Allergy Clin Immunol 2017; 140:1736–9 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis BP, Rothenberg ME. Mechanisms of Disease of Eosinophilic Esophagitis. Annu Rev Pathol 2016; 11:365–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 2018; 560:649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med 2019; 25:1153–63. [DOI] [PubMed] [Google Scholar]
- 40.Acharya KR, Ackerman SJ. Eosinophil granule proteins: form and function. J Biol Chem 2014; 289:17406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iemura A, Tsai M, Ando A, Wershil BK, Galli SJ. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am J Pathol 1994; 144:321–8. [PMC free article] [PubMed] [Google Scholar]
- 42.Galli SJ, Tsai M, Wershil BK, Tam SY, Costa JJ. Regulation of mouse and human mast cell development, survival and function by stem cell factor, the ligand for the c-kit receptor. Int Arch Allergy Immunol 1995; 107:51–3. [DOI] [PubMed] [Google Scholar]
- 43.Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest 2007; 87:971–8. [DOI] [PubMed] [Google Scholar]
- 44.Jones JT, Akita RW, Sliwkowski MX. Binding specificities and affinities of egf domains for ErbB receptors. FEBS Lett 1999; 447:227–31. [DOI] [PubMed] [Google Scholar]
- 45.Macdonald-Obermann JL, Pike LJ. Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J Biol Chem 2014; 289:26178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacDonald KP, Rowe V, Bofinger HM, Thomas R, Sasmono T, Hume DA, et al. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J Immunol 2005; 175:1399–405. [DOI] [PubMed] [Google Scholar]
- 47.Jones CV, Ricardo SD. Macrophages and CSF-1: implications for development and beyond. Organogenesis 2013; 9:249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark RA, McCoy GA, Folkvord JM, McPherson JM. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: a fibronectin matrix-dependent event. J Cell Physiol 1997; 170:69–80. [DOI] [PubMed] [Google Scholar]
- 49.Sherrill JD, Rothenberg ME. Genetic and epigenetic underpinnings of eosinophilic esophagitis. Gastroenterol Clin North Am 2014; 43:269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen CW, Okada M, Proto JD, Gao X, Sekiya N, Beckman SA, et al. Human pericytes for ischemic heart repair. Stem Cells 2013; 31:305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicola NA, Babon JJ. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev 2015; 26:533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kubota Y, Hirashima M, Kishi K, Stewart CL, Suda T. Leukemia inhibitory factor regulates microvessel density by modulating oxygen-dependent VEGF expression in mice. J Clin Invest 2008; 118:2393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boesiger J, Tsai M, Maurer M, Yamaguchi M, Brown LF, Claffey KP, et al. Mast cells can secrete vascular permeability factor/vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of fc epsilon receptor I expression. J Exp Med 1998; 188:1135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature 2005; 438:937–45. [DOI] [PubMed] [Google Scholar]
- 55.Holgate ST, Peters-Golden M, Panettieri RA, Henderson WR, Jr. Roles of cysteinyl leukotrienes in airway inflammation, smooth muscle function, and remodeling. J Allergy Clin Immunol 2003; 111:S18–34; discussion S-6. [DOI] [PubMed] [Google Scholar]
- 56.Duah E, Adapala RK, Al-Azzam N, Kondeti V, Gombedza F, Thodeti CK, et al. Cysteinyl leukotrienes regulate endothelial cell inflammatory and proliferative signals through CysLT(2) and CysLT(1) receptors. Sci Rep 2013; 3:3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stratton R, Shiwen X. Role of prostaglandins in fibroblast activation and fibrosis. J Cell Commun Signal 2010; 4:75–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emanuele MJ, Ciccia A, Elia AE, Elledge SJ. Proliferating cell nuclear antigen (PCNA)-associated KIAA0101/PAF15 protein is a cell cycle-regulated anaphase-promoting complex/cyclosome substrate. Proc Natl Acad Sci U S A 2011; 108:9845–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei J, Zhou T, Zhang X, Tian T. SCOUT: A new algorithm for the inference of pseudo-time trajectory using single-cell data. Comput Biol Chem 2019; 80:111–20. [DOI] [PubMed] [Google Scholar]
- 60.Herring CA, Chen B, McKinley ET, Lau KS. Single-Cell Computational Strategies for Lineage Reconstruction in Tissue Systems. Cell Mol Gastroenterol Hepatol 2018; 5:539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cildir G, Toubia J, Yip KH, Zhou M, Pant H, Hissaria P, et al. Genome-wide Analyses of Chromatin State in Human Mast Cells Reveal Molecular Drivers and Mediators of Allergic and Inflammatory Diseases. Immunity 2019; 51:949–65 e6. [DOI] [PubMed] [Google Scholar]
- 62.Gordon JR, Galli SJ. Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J Exp Med 1991; 174:103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bischoff SC, Lorentz A, Schwengberg S, Weier G, Raab R, Manns MP. Mast cells are an important cellular source of tumour necrosis factor alpha in human intestinal tissue. Gut 1999; 44:643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galli SJ. The Mast Cell-IgE Paradox: From Homeostasis to Anaphylaxis. Am J Pathol 2016; 186:212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clayton F, Fang JC, Gleich GJ, Lucendo AJ, Olalla JM, Vinson LA, et al. Eosinophilic esophagitis in adults is associated with IgG4 and not mediated by IgE. Gastroenterology 2014; 147:602–9. [DOI] [PubMed] [Google Scholar]
- 66.Foroughi S, Foster B, Kim N, Bernardino LB, Scott LM, Hamilton RG, et al. Anti-IgE treatment of eosinophil-associated gastrointestinal disorders. J Allergy Clin Immunol 2007; 120:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loizou D, Enav B, Komlodi-Pasztor E, Hider P, Kim-Chang J, Noonan L, et al. A pilot study of omalizumab in eosinophilic esophagitis. PLoS One 2015; 10:e0113483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirano I, Collins MH, Assouline-Dayan Y, Evans L, Gupta S, Schoepfer AM, et al. RPC4046, a Monoclonal Antibody Against IL13, Reduces Histologic and Endoscopic Activity in Patients With Eosinophilic Esophagitis. Gastroenterology 2019; 156:592–603 e10. [DOI] [PubMed] [Google Scholar]
- 69.Gann PH, Deaton RJ, McMahon N, Collins MH, Dellon ES, Hirano I, et al. An anti-IL-13 antibody reverses epithelial-mesenchymal transition biomarkers in eosinophilic esophagitis: Phase 2 trial results. J Allergy Clin Immunol 2020; 146:367–76 e3. [DOI] [PubMed] [Google Scholar]
- 70.Crapper RM, Schrader JW. Frequency of mast cell precursors in normal tissues determined by an in vitro assay: antigen induces parallel increases in the frequency of P cell precursors and mast cells. J Immunol 1983; 131:923–8. [PubMed] [Google Scholar]
- 71.Friend DS, Ghildyal N, Austen KF, Gurish MF, Matsumoto R, Stevens RL. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J Cell Biol 1996; 135:279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xing W, Austen KF, Gurish MF, Jones TG. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc Natl Acad Sci U S A 2011; 108:14210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rojas-Zuleta WG, Sanchez E. IL-9: Function, Sources, and Detection. Methods Mol Biol 2017; 1585:21–35. [DOI] [PubMed] [Google Scholar]
- 74.Matsuzawa S, Sakashita K, Kinoshita T, Ito S, Yamashita T, Koike K. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J Immunol 2003; 170:3461–7. [DOI] [PubMed] [Google Scholar]
- 75.Gounni AS, Nutku E, Koussih L, Aris F, Louahed J, Levitt RC, et al. IL-9 expression by human eosinophils: regulation by IL-1beta and TNF-alpha. J Allergy Clin Immunol 2000; 106:460–6. [DOI] [PubMed] [Google Scholar]
- 76.Groschwitz KR, Ahrens R, Osterfeld H, Gurish MF, Han X, Abrink M, et al. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc Natl Acad Sci U S A 2009; 106:22381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol 2007; 179:2051–4. [DOI] [PubMed] [Google Scholar]
- 78.Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev 2018; 282:121–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmid-Grendelmeier P, Altznauer F, Fischer B, Bizer C, Straumann A, Menz G, et al. Eosinophils express functional IL-13 in eosinophilic inflammatory diseases. J Immunol 2002; 169:1021–7. [DOI] [PubMed] [Google Scholar]
- 80.Jacobsen EA, Helmers RA, Lee JJ, Lee NA. The expanding role(s) of eosinophils in health and disease. Blood 2012; 120:3882–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doherty TA. At the bench: understanding group 2 innate lymphoid cells in disease. J Leukoc Biol 2015; 97:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Doherty TA, Baum R, Newbury RO, Yang T, Dohil R, Aquino M, et al. Group 2 innate lymphocytes (ILC2) are enriched in active eosinophilic esophagitis. J Allergy Clin Immunol 2015; 136:792–4 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wen T, Aronow BJ, Rochman Y, Rochman M, Kc K, Dexheimer PJ, et al. Single-cell RNA sequencing identifies inflammatory tissue T cells in eosinophilic esophagitis. J Clin Invest 2019; 129:2014–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morgan DM, Ruiter B, Smith NP, Tu AA, Monian B, Stone BE, et al. Clonally expanded, GPR15-expressing pathogenic effector TH2 cells are associated with eosinophilic esophagitis. Sci Immunol 2021; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morgenstern NB, Rochman M, Rothenberg ME. Zooming in on T cell clones: Are we heading to personalized treatment of allergy? Sci Immunol 2021; 6. [DOI] [PubMed] [Google Scholar]
- 86.Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol 2013; 14:536–42. [DOI] [PubMed] [Google Scholar]
- 87.Li H, Sim TC, Alam R. IL-13 released by and localized in human basophils. J Immunol 1996; 156:4833–8. [PubMed] [Google Scholar]
- 88.Collins MH. Histopathology of eosinophilic esophagitis. Dig Dis 2014; 32:68–73. [DOI] [PubMed] [Google Scholar]
- 89.Sewell WA, Scurr LL, Orphanides H, Kinder S, Ludowyke RI. Induction of interleukin-4 and interleukin-5 expression in mast cells is inhibited by glucocorticoids. Clin Diagn Lab Immunol 1998; 5:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol 2006; 24:147–74. [DOI] [PubMed] [Google Scholar]
- 91.Bieber T The pro- and anti-inflammatory properties of human antigen-presenting cells expressing the high affinity receptor for IgE (Fc epsilon RI). Immunobiology 2007; 212:499–503. [DOI] [PubMed] [Google Scholar]
- 92.Metz M, Maurer M. Use of biologics in chronic spontaneous urticaria - beyond omalizumab therapy? Allergol Select 2021; 5:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wheeler JC, Vanoni S, Zeng C, Waggoner L, Yang Y, Wu D, et al. 17beta-Estradiol protects the esophageal epithelium from IL-13-induced barrier dysfunction and remodeling. J Allergy Clin Immunol 2019; 143:2131–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Siddhuraj P, Clausson CM, Sanden C, Alyamani M, Kadivar M, Marsal J, et al. Lung Mast Cells Have a High Constitutive Expression of Carboxypeptidase A3 mRNA That Is Independent from Granule-Stored CPA3. Cells 2021; 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.