

Abstract
Bispecific T-cell engager (BiTE) molecules are biologic T cell–directing immunotherapies. Blinatumomab is approved for treatment of B-cell malignancies, but BiTE molecule development in solid tumors has been more challenging. Here, we employed intravital imaging to characterize exposure and pharmacodynamic response of an anti-muCD3/anti-huEGFRvIII mouse surrogate BiTE molecule in epidermal growth factor receptor variant III (EGFRvIII)-positive breast tumors implanted within immunocompetent mice. Our study revealed heterogeneous temporal and spatial dynamics of BiTE molecule extravasation into solid tumors, highlighting physical barriers to BiTE molecule function. We also discovered that high, homogeneous EGFRvIII expression on cancer cells was necessary for a BiTE molecule to efficiently clear tumors. Additionally, we found that resident tumor-infiltrating lymphocytes (TILs) were sufficient for optimal tumor killing only at high BiTE molecule dosage, whereas inclusion of peripheral T-cell recruitment was synergistic at moderate to low dosages. We report that deletion of stimulatory conventional type I DCs (cDC1) diminished BiTE molecule–induced T-cell activation and tumor clearance, suggesting that in situ antigen-presenting cell (APC) engagements modulate the extent of BiTE molecule efficacy. In summary, our work identified multiple requirements for optimal BiTE molecule efficacy in solid tumors, providing insights that could be harnessed for solid cancer immunotherapy development.
Introduction
In the past decade, dramatic improvements for the treatment of cancers have come from immunotherapy, particularly in the field of T cell–targeting therapies, where immune checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR)-expressing T cells are currently approved for clinical use (1-3). Another promising strategy is the bispecific T-cell engager (4, 5), or BiTE, a recombinant bispecific protein that has two linked single-chain variable fragments (scFvs), one targeted to the CD3 co-receptor on T cells (e.g. CD3ε) and the other targeting a tumor-associated antigen (TAA) (6). BiTE molecules have been shown to redirect and stimulate both CD4+ and CD8+ T-cell populations to kill tumor cells (7), independent of intrinsic T-cell receptor (TCR) and MHC-restricted recognition, and this mechanism-of-action has been validated clinically, most notably via the approval of CD19-targeting blinatumomab (Blincyto®) for the treatment of B-cell acute lymphoblastic leukemia (5, 8, 9).
Looking beyond hematologic cancers to targeting solid tumor indications, clinical success has thus far been limited. For example, BiTE molecules specific for epithelial cell adhesion molecule (EpCAM) and carcinoembryonic antigen (CEA) show limited therapeutic responses and dose limiting toxicities in clinical trials targeting solid tumors (6, 10, 11). However, encouraging antitumor activity has been observed in Phase 1 clinical studies with the epidermal growth factor receptor variant III (EGFRvIII)-targeted BiTE molecule AMG 596 (12), the prostate-specific membrane antigen (PSMA)-targeted BiTE molecule AMG 212 (13), AMG 160 in metastatic castrate-resistant prostate cancer (mCRPC) (14), and the delta-like ligand 3 (DLL3)-targeted BiTE AMG 757 (15). Although the clinical data are emerging, there are still many unknowns as to what characteristics of the solid tumor microenvironment (TME) drive BiTE molecule responses.
One potential limitation of a BiTE molecule when targeting solid tumors is drug accessibility to the intended site of action. The molecular weights of BiTE molecules span a large range [∼54 kDa for first-generation molecules through ∼110 kDa for half-life extended (HLE) BiTE molecules], and solid tumor exposures could potentially be limiting (16), given the often narrow therapeutic index of this modality (4). Furthermore, the tumor vasculature is often heterogeneous, leading to additional physical barriers to drug extravasation and sufficient T-cell infiltration (17). There have been few reports that characterize BiTE molecule biodistribution to solid tumors (18), and it is anticipated that a combination of poor drug exposure within the often highly immunosuppressive TME remain as hurdles to BiTE efficacy (5). Ideally, real-time observation within tumors to define anatomical, cellular, and/or biochemical barriers to BiTE efficacy in the immunocompetent state could be beneficial for optimizing BiTE-based immunotherapy, including dissecting the potential role of ancillary immune components in the solid TME.
A second limitation of BiTE molecules is the selection of TAAs, which have minimal background tissue expression to avoid off-tumor effects in non-malignant cells and yet are of sufficiently high expression to allow high valency crosslinking of T cells. One putative TAA is epidermal growth factor receptor (EGFR) with activating mutations specific to cancers, including the extensively studied EGFRvIII (deletion of exons 2–7) that promotes cell proliferation, angiogenesis, and invasion in different model systems (19). The EGFRvIII mutation occurs most commonly in glioblastoma (GBM) with inconsistent expression in breast, head and neck, lung, and prostate cancers reported (20, 21). Despite early evidence of clinical activity with the EGFRvIII BiTE AMG 596 in patients with GBM, the relationship between EGFRvIII expression and BiTE molecule activity remain unclear (12), especially given its known heterogeneity and possible downregulation in tumors (22).
Monitoring intratumoral T-cell responses has become important to understand the tumor response to immune manipulation and address questions related to effective immunotherapies (23). Intravital imaging (IVM) in tumors has revealed previously unknown immune processes inside the TME, including the link between T-cell behavior and dysfunctional differentiation (24), as well as the interaction of stimulatory, conventional type I dendritic cells (cDC1) and cytotoxic T cells (CTLs) (25) via real-time analysis of an immunocompetent breast cancer mouse model. IVM has also uncovered unexpected modes of action of ICIs and CAR T cells (26). In the current study, we examined a mouse surrogate EGFRvIII-targeting BiTE molecule in a similarly adapted (syngeneic) breast cancer tumor system to visualize BiTE molecule diffusion into solid tumors, with the goal of understanding the tissue pharmacokinetics in relation to the pharmacodynamic exposure-immune response in a fully immunocompetent mouse model. We visualized tumor exposure to a fluorescently labeled BiTE molecule and found it to be characterized by multiple distinct diffusion profiles from heterogenous intratumoral vasculatures. By engineering variable target expression on tumor cells and by modulating tumor-infiltrating CTLs and DC populations, we discovered critical, and sometimes unexpected, requirements for maximum BiTE molecule efficacy that included: (i) high, uniform target expression; (ii) a dose-dependent peripheral lymphocyte recruitment sensitivity that could be overcome at a high dosage when resident T-cell populations proved sufficient; and (iii) a role for ancillary CD103+ DC populations that enhanced T-cell expansion and recruitment.
Materials and Methods
Study design
The main objective of this study was to visualize and characterize human(hu)EGFRvIII/murine(mu)CD3ε-muFc BiTE molecule tissue extravasation into EGFRvIII-expressing breast tumors and understand the key requirements for BiTE molecule efficacy in solid tumors. An immunocompetent mouse tumor model, as described below, was used in this study to better understand the complex interactions among the immune cells in both the peripheral immune system and the local TME of the solid tumor. Data for tumor intravital imaging was collected from 3 regions of interest (ROIs) of a tumor, as indicated below, and was representative of >5 tumors from different mice. In the orthotopic tumor growth experiments, mice were randomly assigned to groups based on the tumor size so that control and experimental groups had the same average size before the start of treatment. The number of mice used for each group is indicated in the figure or figure legends. Animal numbers were chosen to ensure adequate statistical power and were based on the consistency of tumor growth observed in preliminary studies.
Mouse strains
All mice were housed and bred at University of California, San Francisco (UCSF) and were maintained under specific pathogen-free conditions in accordance with the regulatory standards of the National Institutes of Health (NIH) and American Association of Laboratory Animal Care standards and were with the approval of UCSF Institution of Animal Care and Use Committee (IACUC approval no. AN170208–01B). C57BL/6J and Batf3−/− mice were purchased from The Jackson Laboratory. PyMT-ChOVA transgenic mice were generated as previously described (25). CD2-DsRed mice were obtained from Mark Coles (University of Oxford) (24, 27). XCR1DTR mice were a gift from Tsuneyasu Kaisho (Osaka University). All mice used in experiments were female age 8 to 20 weeks and bred onto a C57BL/6 background.
Cell lines
A PyMT-ChOVA cell line (PyMT-Control) was derived from PyMT-ChOVA spontaneous mammary tumors as previously described and verified by sequencing for the transgene cassettes (28). Cells tested negative for Mycoplasma on October 19, 2018, by PCR. Cell lines were used at less than 5 passages after thawing for the described experiments. To generate the PyMT-EGFRvIII cell line, a pLenti-humanEGFRvIII construct was generated by inserting the human EGFRvIII gene (NCBI Reference Sequence: NP_001333870.1) into the pLenti-puro vector (Addgene; #39481) and sequenced for accuracy. Lentivirus was produced in HEK293 cells (ATCC) using TransIT-Lenti Transfection Reagent (Mirus Bio, Madison, WI). 500 µl of lentivirus containing supernatant were collected from HEK293 T cells after 48 hours and were applied to 5 × 105 PyMT-ChOVA cells. Transduced cells were sorted for EGFRvIII-positive cells on day 2 after infection on sorting on a BD FACSAria III using a chimeric EGFRvIII human variable/murine IgG1 constant domain antibody generated as previously described (29). Following an additional week of culture, cells were sorted for a second time based on the expression EGFRvIII to establish PyMT-EGFRvIII-Hi (high expression of EGFRvIII) and PyMT-EGFR-Lo (low expression) cell lines. These two cell lines were stained for EGFRvIII and sorted into hi- and lo-expressing cells to maintain the proper EGFRvIII expression when needed. All the cell lines were cultured at 37 °C in 5% CO2 in DMEM (Invitrogen) with 10% FCS (Benchmark) and 1% penicillin–streptomycin–glutamine (Thermo Fisher Scientific).
BiTE molecule construction
The mouse surrogate BiTE molecule consisted of a human anti-huEGFRvIII scFv, anti-muCD3 (KE3) scFv linked via a SG4S linker, and a single-chain murine IgG1 Fc domain for half-life extension (HLE). The mouse surrogate CD3 engager moiety, KE3, was based on a rat anti-mouse CD3ε scFv derived from the KT3 monoclonal antibody sequence (30). KT3 engagement on CD3ε has been demonstrated by competition for T-cell binding with the monoclonal CD3ε antibody, 145–2C11 (31). An analogous human-CD3ε–targeted HLE BiTE construct was used as a control in human breast cancer cell co-culture assays that harbored an identical huEGFRvIII scFv, but with an anti-huCD3ε scFv sequence and single-chain human IgG1 Fc domain. The BiTE molecules were constructed by recombinant DNA technology, produced in stably transfected CHO cells, and purified by chromatography as described previously (32).
Characterization of CD3ε and EGFRvIII target binding affinity by surface plasmon resonance (SPR)
The binding kinetics and affinity of the mouse surrogate anti-EGFRvIII/anti-muCD3ε BiTE-Fc (muIgG1) to either EGFRvIII or muCD3ε target was characterized by surface plasmon resonance (SPR) using a Biacore™ T-200 biosensor (Cytiva). For characterization of TAA (EGFRvIII) binding, a standard streptavidin biosensor chip was functionalized as a capture surface with 600 Response Units (RU)of a biotinylated anti-6xHis tag (GenScript, #A00613) and used to load recombinant EGFRvIII-His tag protein (Sino Biologial, #29662-H08B) to a maximum density of 180 RU. A serial dilution (2000 – 0.64nM; 1:5 fold) of BiTE analyte was prepared in running buffer (HBS-P+, Cytiva), and binding analysis was conducted at a flow rate of 50 µL/min. with 60-sec. injections during association, and a 400-sec. interval of dissociation. The surface was regenerated between cycles using 10 mM glycine, pH = 1.5, with new EGFRvIII protein captured before each subsequent injection. The specificity of the SPR response was verified with controls where the BiTE analyte was injected over the same flow cell without previously captured EGFRvIII-His protein. The data were double referenced by subtraction of the SPR response from both reference flow cell lacking EGFRvIII protein, and the response was observed with running buffer alone across the EGFRvIII active flow cell. The resultant sensorgram data were fit to a Langmuir (1:1) binding model using Scrubber (BioLogic Software) to estimate the parameters kon, koff, and KD. A similar approach was used to characterize the BiTE-muCD3ε interaction. To supplement the full length CD3ε TCR co-receptor and enable in vitro binding analysis, a muCD3ε (1–100)-huFc peptide fusion construct, developed at Amgen, was used and contains the binding epitope of our anti-muCD3ε scFv. Briefly, biotinylated a monoclonal anti-huFc (Amgen) was immobilized on a streptavidin biosensor surface to a level of 300 RU and used as a capture surface for loading muCD3ε (1–100)-huFc to levels up to 120 RU. A standard BiTE analyte dilution series (1000 – 1.6 nM; 5-fold) was injected at a flow rate of 50 µL/min. for 75 sec. to characterize association and 300 sec. for monitoring the dissociation phase. The data were processed similarly to EGFRvIII binding kinetics data.
Pharmacokinetic study and non-compartmental analysis of serum PK in C57BL/6 mice
A single-dose pharmacokinetics study of anti-huEGFRvIII mouse surrogate BiTE-Fc (muIgG1) was conducted in naive female C57BL/6 mice. The BiTE molecule was administered by intravenous (IV) bolus injection at either 0.05 mg/kg or 0.5 mg/kg using a formulated dosing solution of 0.0125 mg/mL and 0.125 mg/mL, respectively. Each study arm included n = 6 animals that were split into two staggered subgroups (n = 3) for sparse sampling to obtain concentration-time profiles over 240 hours post-dose (time points: 0.083, 1, 4, 8, 24, 48, 96, 168, & 240 hours). At each time point, approximately 0.06 mL of whole blood was collected via submandibular or tail venipuncture puncture in SARSTEDT Microvette® serum separator tubes (Thermo Fisher Scientific) for processing serum. After whole blood was collected into the SARSTEDT tubes, the tubes were inverted several times and remained at room temperature for approximately 20 minutes or until fully clotted. Clot formation was verified by manual inversion. The specimens were centrifuged using a calibrated Eppendorf 5417R Centrifuge System (Brinkmann Instruments, Inc., Westbury, NY 11590). Blood specimens were centrifuged at 2–8°C at approximately 11,500 rpm for 15 minutes. To quantify the concentration of anti-EGFRvIII BiTE molecule, an electrochemiluminescent (ECL) immunoassay was developed for the Meso Scale Discovery Analyzer (MSD) QuickPlex platform. Briefly, a streptavidin functionalized MSD plate (Meso Scale Diagnostics, LLC) was treated with biotinylated-huEGFRvIII protein (R&D Systems, #9565-ER-050) at 2 µg/mL concentration to facilitate analyte capture. Serum samples were diluted up to 1:10 in Blotto solution (Thermo Fisher Scientific) prior to dispensation on the plate for room temperature incubation/capture (2 hours), along with a standard anti-EGFRvIII BiTE analyte curve (range 10,000 – 0.61 ng/mL) and quality control samples consisting of serum-spiked EGFRvIII BiTE analyte at variable nominal concentration to assess accuracy throughout the full concentration range of the prepared standard curve. Post-incubation, the MSD plates were washed with KPL buffer (LGC Clinical Diagnostics, Inc.), followed by addition of polyclonal anti-mouse IgG1 (Jackson ImmunoResearch, #115–005-205) in Blotto (1 µg/mL) that had been previously conjugated to a MSD SULFO-TAG™ (Meso Scale Diagnostics, LLC).. After incubation and multiple plate wash steps in KPL buffer, tripropylamine read buffer (Meso Scale Diagnostics, LLC) was added to enable detection of electrically stimulated light emission at 620 nm wavelength, where the instrument responses are proportional to the amount of anti-EGFRvIII BiTE molecule contained in a given serum sample. For quantitation, the ECL assay response versus anti-EGFRvIII BiTE molecule concentration relationship was regressed using a four-parameter logistic (Marquardt) regression model to estimate lower and upper curve plateaus, the Hill slope, and EC50 using a weighting factor of 1/Y2. Noncompartmental analysis (NCA) was performed on serum concentration-nominal time data using Phoenix WinNonlin® (Version 6.4.0.768, Pharsight Corporation). The PK parameters estimates for area under the curve (AUC), clearance (CL), volume of distribution at steady state (Vss), mean residence time (MRT), and terminal phase elimination half-life (t1/2,z) are reported.
Tumor model
2 × 105 PyMT-Control (Ctrl) cells, or PyMT- EGFRvIII-Hi, or PyMT-EGFRvIII-Lo tumor cells were suspended in PBS and mixed 1:1 with Matrigel GFR (Corning) for a final injection volume of 50 μL. For the mixed tumor experiment, PyMT-Ctrl and PyMT-EGFRvIII-Hi cells were mixed at ratio of 3:1 or 1:3 for a total number of 2 × 105. Cells were injected in the fat pad of C57BL/6J mice anesthetized with 3% isoflurane (Henry Schein). Tumors were then measured 3 times per week using electronic calipers. Tumor volume was calculated through the formula V = 0.5*(w2 * L). Mice were euthanized when tumors exceeded a volume of 1000 mm3. To block lymphocyte egress from lymph nodes (LNs), 50 µL FTY720 (Cayman Chemical Company, Ann Arbor, MI) at 1 mg/kg was injected intraperitoneally everyday beginning on day 21 post tumor injection and continued through the end of the experiment. 50 µL DMSO was injected as mock treatment in control groups. To deplete cDC1s in XCR1DTR mice, diphtheria toxin (DT) (List Biological Laboratories) at 500 ng/mouse was injected intraperitoneally every three days beginning on day 10 to day 25 post tumor injection. The same dosage of DT was injected into wild type C57BL6/J mice as the control groups.
Intravital imaging of mouse tumor and analysis
PyMT-EGFRvIII-Hi tumor-bearing mice were kept under anesthesia using 2% isofluorane, and mammary tumors were surgically exposed when palpable. Confocal imaging was performed using a Nikon A1R laser scanning confocal microscope with NIS-Elements software (Nikon Instruments, Melville, NY) and a 16X LWD water-dipping objective. Before surgery, 75 μg DyLight 649–labeled Griffonia Simplicifolia Lectin I isolectin B4 (Vector Laboratories) was injected by retro-orbital injection for visualization of blood vessels. During imaging, 20 μg AF488-labeled BiTE molecule was injected by retro-orbital injection as described in figure legends. Time-lapse videos were acquired every 90 seconds with a z-step of 3 μm. Images were taken at 100 µm deep inside the tumor and in the center areas with obvious blood vessels, where tumor cells were also abundant. Data were analyzed using Imaris (Bitplane), including drift correction, video generation, cell surface detection, and generation of cell speed and track displacement data.
Analysis of in vivo BiTE molecule dispersal and target binding
The fluorescence intensity from AF488-labeled HLE-BiTE was used to calculate the spatiotemporal kinetics of intratumoral BiTE diffusion. Using Distance Transformation on Imaris, the distance of each voxel from the nearest isolectin-labeled blood vessel was calculated and binned into 5 µm blocks. The average BiTE intensity of all voxels within each bin was then calculated and centered spatially at the midpoint of the bin (e.g., the average intensity of the bin from 10–15 µm would be centered at 12.5 µm). Next, we used the BiTE channel intensity prior to BiTE injection (usually a 15–20–minute time-lapse was obtained before BiTE injection) to subtract the background autofluorescence. Finally, to dissociate the absolute amount of dispersal from its spatiotemporal dynamics, the intensities were normalized to the minimum and maximum intensities observed at that field of view (Fig. 3B-G), as follows:
Figure 3. Intratumoral vasculature diversity results in heterogeneous BiTE diffusion in solid tumors.
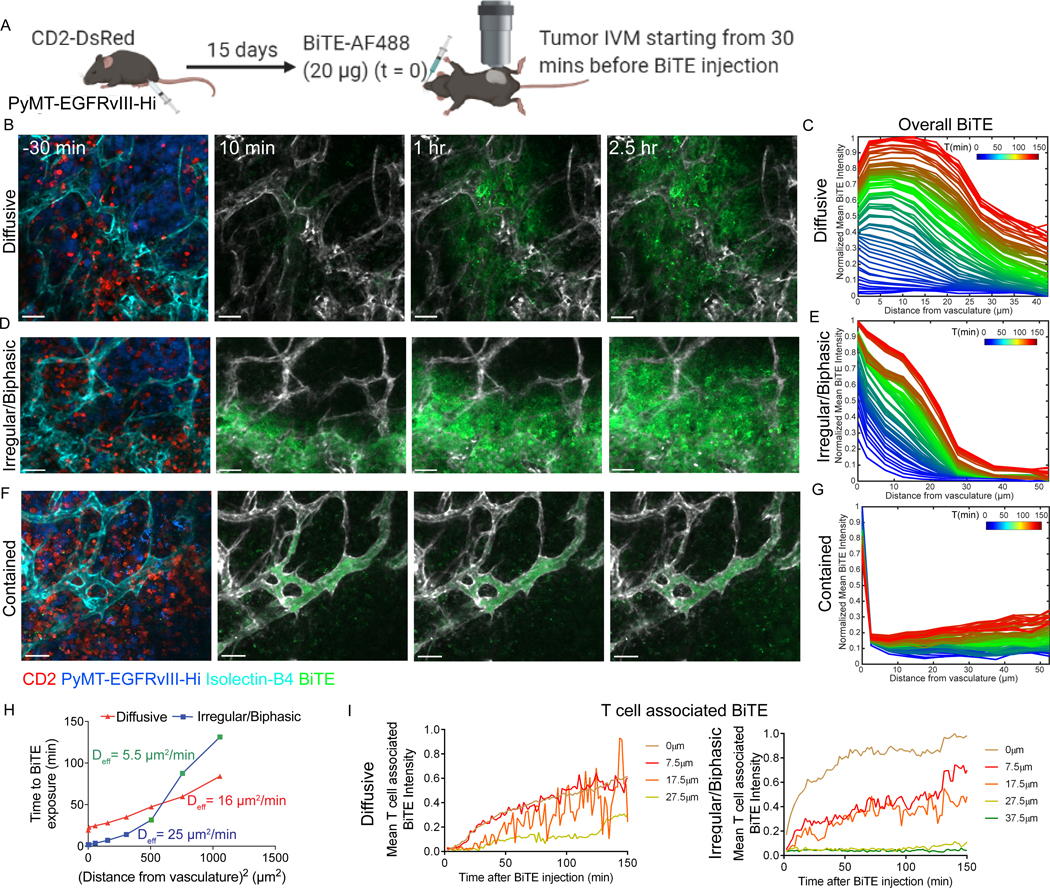
(A) Diagram showing protocol for intravital imaging of PyMT-EGFRvIII-Hi tumors. Representative images of AF488 fluorophore-labeled BiTE (BiTE-AF488, 20 μg injected at time=0) diffusion from vessels in a PyMT-EGFRvIII-Hi tumor. 3 diffusion patterns are shown: Diffusive (B), Irregular/Biphasic (D), and Contained (F). Scale bar = 50 μm. Also see Supplementary Video S4. (C,E,G) Corresponding quantifications of the BiTE accumulation at increasing distance from the vessels over time (color-coded). (H) The effective diffusion coefficient (Deff) for BiTE diffusion from vessels under conditions in panels B and D was calculated using the relationship R2= Deff x t and plotted (see methods). (I) Plots of means of BiTE intensity on T cells over time at different distances away from vessels (color coded) in the Diffusive and Irregular diffusion patterns shown in panels B and D. Data are representative of two independent experiments.
To quantify the essence of the patterns of BiTE dissemination, an effective diffusion coefficient (Deff) was calculated (Fig. 3H) using the relationship
where R is distance traversed by the molecule from its source in time t. Although this relationship is only approximate for complex mass transport scenarios, such as drug dispersal in a tumor, it offers a quick and reliable comparison of transport kinetics across fields of view. The time of entry (t) of the BiTE at a particular spatial location x-µm away from the nearest labeled vasculature was taken to be the first time point when the intensity threshold at that spatial bin reached a value >10. The theoretical Deff of AF488-labeled HLE-BiTE (MW ∼ 120kD) in the interstitial fluid at 37°C was calculated using Stokes-Einstein equation , where k = 1.38 × 10−23 J/K, T = 310K, η ∼ 7 × 10−3 kg·m−1·s−1, r ∼ 4 nm.
The frequency of the specific diffusion type was calculated using the volume of the ROI in which the specific type of diffusion appeared divided by the total volume of the tissue that we imaged from that tumor. For the contained type, the percentage was calculated using 15 ROIs from 5 mice using the same method. To calculate BiTE binding kinetics on target cells, the target cells were first delineated by surface rendering on Imaris, and the average BiTE fluorescence intensity on these surfaces was calculated. Using similar distance transformations as described above, the minimum distance of these target cells from the vasculature were also concurrently obtained. These were then used to plot traces of T cell–associated BiTE accumulation over time on individual cells proximal to the vasculature (Fig. 2C) or an aggregate of cells at different distances from the vasculature (Fig. 3I).
Figure 2. BiTE-mediated target-binding and tumor killing in breast tumors.

(A) Schematic of the experimental design. PyMT-EGFRvIII-Hi cells and CD2-DsRed mice were used. Panels with corresponding data are indicated. (B) Representative images showing BiTEs (20 μg) binding to T cells and tumor cells near vessels (< 10 μm). Blue outlines: tumor cells; red outlines: T cells. Scale bar = 15 μm. (C) Quantification of individual T cell– or tumor cell–associated BiTE intensity over time. Each colored line represents one individual cell. (D) Histogram and (E) quantification of BiTE-AF488 binding on CD8+ T cells or PyMT-EGFRvIII-Hi mcherry+ tumor cells at two dosages and different time points as indicated. NT: no treatment. (F) Representative flow plots and (G) quantification of live tumor cells out of all CD45– live cells or all live cells in the tumors at 72 hours with high or low dosage of BiTEs. Live cells were gated using Zombie Aqua fixable viability dye and then mcherry+ cells were gated as live tumor cells. N=3 per group (panels D-G). Data are representative of two independent experiments; ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05 as determined by one-way ANOVA and multiple comparison test.
Similarly, larger images obtained from PyMT-Ctrl or PyMT-EGFR-Hi or PyMT-EGFRvIII-Lo tumor tissue sections fixed by 2% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for overnight were used to quantify BiTE distribution and retention in the tumor microenvironment. Average voxel-by-voxel intensity and distance transformations were used, as described above. A sum of all BiTE intensity across same-sized fields of view were used as a surrogate to measure total amount of BiTE dispersed a given time after injection, as shown in Supplementary Fig. S4C. Analysis of Imaris-processed images was done on Matlab (Mathworks Inc.) using custom-Matlab scripts and either Matlab or GraphPad Prism was used to plot the analyzed data. Matlab scripts are available for share upon request.
Breast cancer samples, digestion, and coculture assays
Three breast tumor samples were acquired from the National Cancer Institute’s (NCI) Cooperative Human Tissue Network (CHTN) and from the Department of Surgery, Feinberg School of Medicine, Northwestern University. Specimens were anonymized and then collected and distributed by CHTN. Specimens are covered under collection/distribution of tissues under consent or waiver of consent. Samples were minced with surgical scissors and transferred to a GentleMACs C Tube (Miltenyi Biotec) with 100 μg/mL Liberase TL (Roche) and 200 μg/mL DNase I (Roche) at 3 mL per gram of tissue. C tubes were incubated in the GentleMACS octo dissociator (Miltenyi Biotec) with heaters, following the manufacturers dissociation protocol (Miltenyi Biotec Tumor Dissociation Kit). 10 mL of FACS buffer (PBS + 2% fetal calf serum (FCS)) was added to samples and filtered through a 100 μm filter and centrifuged at 350g for 5 minutes at 4 °C, and red blood cells were lysed with 175 mM ammonium chloride. CD45– live cells from the breast cancer tumor were then sorted on a FACSAria II (BD) and co-cultured with CD8+ T cells at a 1 (12500 cells) to 8 (1 × 105 cells) ratio for 72 hours. The huCD3ε/huEGFRvIII BiTE molecule (obtained from Amgen Inc.) wasadded at the indicated concentrations. CD8+ T cells were isolated from PBMCs of a healthy donor using EasySep™ Human CD8+ T-cell Isolation Kit (StemCell Technologies, Vancouver, Canada). T cells stimulated with Dynabeads™ Human T-Activator CD3/CD28 (ThermoFisher Scientific) were used as controls.
Mouse tumor and lymph node digestion
For tumor digests, tumors from mice were isolated, minced, and incubated in digestion buffer containing 100 U/mL collagenase type I (Roche), 500 U/mL collagenase type IV (Roche), and 200 μg/mL DNAse I (Roche) in RPMI-1640 (GIBCO) for 30 minutes on a shaker at 37 °C. For lymph node (LN) digests, tumor-draining LNs (the inguinal, axillary, and brachial) were dissected, pierced, and torn with sharp forceps, and incubated in the same digestion buffer as tumors for 15 minutes at 37 °C. Digestion mixtures were then pipetted repeatedly, followed by another 15-minute incubation. For both tumor and LNs, cells were washed with PBS plus 2% FCS and filtered through a 100 μm cell strainer before staining for flow cytometry. The number of BiTE molecules bound to T cells or tumor cells was calculated using quantitative fluorescence cytometry as described below.
Quantitative fluorescence cytometry
The number of BiTE molecules bound to T cells or tumor cells was calculated based on the degree of labeling (1.5 fluorophore per BiTE) using quantitative fluorescence cytometry of BiTE molecules. The Quantum™ Alexa Fluor 488 MESF kit (Bangs Laboratories, Inc) was used per the manufacturer’s instructions. The numbers of molecules of the fluorochrome were measured to be 9000 for T cells and 4500 for tumor cells, so the numbers of BiTEs bound to T cells and tumor cells were 6000 and 3000, respectively.
Flow cytometry antibodies
The following mouse antibodies were purchased from Biolegend and were used for surface staining: anti-CD45–Brilliant Violet (BV)510, –BV605, or –Alexa Fluor (AF)488 (clone 30-F11); anti-CD90.2–AF700 (clone 30-H12); anti-CD8a–PerCP/Cyanine5.5 (clone 53–6.7); anti-CD4–BV421 (clone RM4–5); anti-CD11b–BV785 or –BV510 (clone M1/70); anti-CD44–BV711 (clone IM7); anti-CD69–BV650 or –PE (clone H1.2F3); anti-CD279 (PD-1)–BV605 (clone 29F.1A12); anti–I-A/I-E–AF711 (clone M5/114.15.2), anti-CD11c–BV650 (clone N418); anti-CD19–BV785 (clone 6D5); anti-B220–BV785 (clone RA3–6B2); anti-Ly6G–BV785 (clone 1A8); anti-CD24–PE/Cy7 (clone M1/69); anti-CD103–APC (clone 2E7); anti-NK1.1–BV785 (clone PK136); anti-F4/80–AF488 (clone BM8); anti-Ly6C–AF700 (clone HK1.4); anti-CD62L–APC (clone MEL-14); and anti-CD25–APC (clone 3C7) Anti-IFNγ–APC (clone XMG1.2); anti-TNFα–PE (clone MP6-XT22), and anti-granzyme B–PE/Cyanine7 (clone QA16A02) were purchased from Biolegend; anti–Ki-67–PE-eFluor 610 (clone SolA15) was purchased from ThermoFisher Scientific These antibodies were used for intracellular staining. Fluorescence minus one control was used to identify positive populations.
The following human antibodies were used for surface staining: anti-CD45–APC/Cy7 (clone 30-F11), anti-CD25–APC (clone BC96), anti-CD45RO–AF700 (clone UCHL1), anti-CD69–PE (clone FN50), and anti-CD8a–BV605 (clone RPA-T8). All were purchased from Biolegend.
Flow cytometry
Cells from tumor digests or cell lines were stained with fixable Live/Dead Zombie NIR (Biolegend) in PBS per the manufacturer’s instructions, followed by staining with anti-Fc receptor (clone 2.4G2) (Biolegend) and antibodies to surface antigens in FACS buffer for 30 minutes on ice. Intracellular staining was subsequently performed using the eBioscience Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) when needed (all antibodies listed in the section above). For intracellular cytokine staining, cells were incubated in RPMI-1640 plus 10% FCS supplemented with 10 ng/mL PMA (Sigma-Aldrich), 0.5 μg/mL ionomycin (Thermo Fisher Scientific), and 10 μg/mL Brefeldin A (Cayman Chemical) for 4 hours before staining. For sorting, cells were stained with anti-Fc receptor (clone 2.4G2) and antibodies to surface antigens, followed by DAPI staining (Biolegend) to exclude dead cells. Flow cytometry was performed on BD Fortessa or LSR II cytometer. Cell sorting was performed using BD FACS Aria 2 or Aria Fushion. Analysis of flow cytometry data was done using FlowJo (Treestar) software.
Coupling assay and T cell-tumor cell in vitro co-cultures
Mouse CD8+ T cells from naïve C57BL/6 mice were isolated from LNs using the CD8 EasySep enrichment kit (STEMCELL Technologies) and were stained with Violet Proliferation Dye 450 (VPD; BD Biosciences). For the coupling assay, 1 × 105 PyMT-Control or PyMT-EGFRvIII cells were labeled with CellTracker Orange CMTMR Dye (Thermo Fisher Scientific) per the manufacturer’s instructions. Labeled CD8+ T cells and PyMT cells at a 1 to 1 ratio were co-cultured in in 96-well V bottom plate for one hour at 37 °C in 100 μL RPMI-1640 plus 10% FCS with 1% penicillin–streptomycin–glutamine (Thermo Fisher Scientific) with a gentle spin-down before the incubation. After incubation, 100 μL 4% paraformaldehyde (PFA) per well was added into each well for 10 minutes, and cells were washed with FACS buffer. T cell-tumor doublets were measured by flow cytometry as described in the Flow cytometry session. For T cell-tumor cell long-term co-cultures, cells were collected at the indicated time points, and flow cytometry was performed to determine T-cell activation states and tumor killing effect.
Calcium influx imaging
Mouse CD8+ T cells from naïve C57BL/6 mice were isolated as described previously and were stimulated in vitro with Dynabeads Mouse T-activator CD3/CD28 for T-Cell Expansion and Activation (Thermo Fisher Scientific) plus IL2 (50 IU, Roche) for 2 to 3 days in RPMI-1640 plus 10% FCS with 1% penicillin–streptomycin–glutamine (Thermo Fisher Scientific). Activated T cells were harvested and labeled with Fura-2 AM (Thermo Fisher Scientific) per manufacturer’s instructions. PyMT-EGFRvIII cells were labeled with CellTracker Orange CMTMR Dye (Thermo Fisher Scientific). T cells and PyMT-EGFRvIII cells were mixed at 1 to 1 ratio and incubated in 0.5% low-melting agarose in RPMI-1640 without Phenol-red (Gibco) (T-cell imaging medium) in Nunc Lab-Tek II Chambered Coverglass (Thermo Fisher Scientific) for 15 minutes before imaging. Time-lapse videos were acquired every 90 sec. using a widefield inverted Zeiss Axioscope 200M microscope equipped with a 20x long working distance lens, an Andor Clara CCD Camera, and 340 and 380 excitation filters paired with a Lambda XL light source (Sutter Instruments). Samples were kept at 37 °C using a heated stage top incubator with 5% CO2: 95 O2. BiTEs were added after 270 sec. of imaging.
Cell death imaging
Mouse CD8+ T cells from naïve C57BL/6 mice were stained with Violet Proliferation Dye 450 (VPD; BD Biosciences). PyMT-EGFRvIII cells were labeled with the Live/Dead Cell Imaging Kit (Thermo Fisher Scientific) per manufacturer’s instructions. T cells and tumor cells were co-cultured in T-cell imaging medium, as described in the previous section, plus the Live/Dead staining reagent and imaged as indicated in the section above.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software. Data are shown as mean ± S.D.. Specifically, an unpaired Student’s t-test or one-way ANOVA with multiple comparison test was used as indicated in the figure legends, with p values described in detail.
Data and material availability
All data are available in the main text or the supplementary materials. Matlab code for imaging analysis is available to share upon request.
Results
HuEGFRvIII/muCD3ε surrogate BiTE molecule and PyMT-EGFRvIII+ model
A previously developed anti-huEGFRvIII/huCD3 BiTE construct (AMG 596) was investigated in patients with glioblastoma in clinical trial NCT03296696 (12). In the current study, we constructed an analogous EGFRvIII-targeted, mouse surrogate molecule with a similar half-life extended (HLE-BiTE) design based on murine IgG1 Fc. For this, an anti-EGFRvIII scFv moiety was fused to the anti-murine CD3ε engager (KE3), along with a murine IgG1 Fc domain harboring a N297G mutation to minimize Fc effector function (huEGFRvIII/muCD3ε-muFc BiTE) and to impart favorable pharmacokinetics (PK) (Fig. 1A). The single-dose serum PK was characterized in C57BL/6 mice and had a terminal elimination phase half-life of ∼3.3 days (Supplementary Fig. S1A-B). The affinity (KD) to EGFRvIII measured by SPR was 5.28 nM, and the affinity against a murine CD3ε (residues 1–100)-Fc construct was 28.4 nM (Supplementary Fig. S1C).
Figure 1. Dose- and target-dependent huEGFRvIII/muCD3ε-muFc BiTE function in vitro.

(A) General schematic depicting the surrogate BiTE design, consisting of an anti-huEGFRvIII TAA scFv domain, anti-murine CD3ε scFv (murinized rat anti-mouse CD3ε engager, KE3), and half-life extension moiety murine IgG1 single-chain Fc domain harboring a N297G mutation to minimize Fc effector function. MW: molecular weight; kDa: kilodaltons. (B) Flow cytometry analysis of the coupling between mouse CD8+ T cells (violet proliferation dye (VPD)-labeled) and PyMT-Ctrl (control) or PyMT-EGFRvIII-Hi cells (CMTMR-labeled) co-cultured for 1 hour in the presence of BiTEs as indicated. (C) Calcium influx in mouse CD8+ T cells indicated by the ratio of Fura-2 signal, with comparison of excitation at 340 nm vs. 380 nm. Left: Representative images of T cells in contact with PyMT-EGFRvIII-Hi cells (close) and T cells that were further away (far) from cancer cells (Scale bar = 10µm). Right: Quantification of the change of the Fura-2 ratio in the two T-cell populations (close and far), along with time. Also see Supplementary Video S1. N=10–20 cells per condition. (D) Live imaging of active killing of PyMT-EGFRvIII-Hi tumor cells, labeled with a mix of live green dye and dead red dye, by VPD-labeled mouse CD8+ T cells in co-culture for 2 hours in the presence of two concentrations of BiTEs (Scale bar = 30µm). Quantification of the number of dead cells over time is shown. Also see Supplementary Video S2. (E) Flow cytometry analysis of mouse CD8+ T-cell proliferation (indicated by VPD dilution), activation (indicated by surface CD25 and CD69 expression), and tumor killing induced by BiTEs when T cells were co-cultured with PyMT-EGFRvIII-Hi cells for 48 hours in the presence of different concentrations of BiTEs as indicated. Measurement of tumor cell death was done by staining with Live/Dead Zombie dye. N=3–4 per group. Data are representative of two independent experiments; ****p<0.0001, **p<0.01, *p<0.05 as determined by one-way ANOVA and multiple comparison test.
To better understand the effect of intratumoral T-cell activity dependent on BiTE molecule diffusion into solid tumors in an immunocompetent mouse model, we adapted an extensively characterized mouse mammary tumor virus-polyoma middle T (MMTV-PyMT-ChOVA) (25) breast cancer model to an orthotopic model, with PyMT-ChOVA cells implanted into the fat pad (28). PyMT-ChOVA cells express fluorescent mCherry protein to aid visualization of tumors (25). Given that altered T-cell function has been shown to correlate with changes in dynamic behavior inside PyMT tumors (24, 33), this model provides an ideal imaging platform to study the relationship between BiTE molecule exposure within the TME and pharmacodynamic response, including target cell engagement and cell killing. Moreover, normalization of the tumor vasculature in this mouse model enhances drug delivery and delays tumor growth (34), supporting it as a valid mouse model to study drug diffusion properties. We overexpressed huEGFRvIII on a PyMT-ChOVA cell line and sorted the cells with high expression (EGFRvIII-Hi) to access BiTE diffusion and efficacy. To understand if target expression influenced BiTE function, for some studies we also sorted cells with low EGFRvIII expression (PyMT-EGFRvIII-Lo), which was roughly 6–7 times lower than its expression on EGFRvIII-Hi cells (Supplementary Fig. S2A). The use of PyMT tumors engineered to express EGFRvIII antigen was further validated as a relevant model of human disease by screening several primary breast cancer samples, where we detected variable expression of EGFRvIII on CD45– populations (Supplementary Fig. S2B-D), with the highest expression present in a triple-negative breast cancer patient, which was about 50% lower compared to the PyMT-EGFRvIII-Hi cell line, whereas the EGFRvIII expression on other breast cancer samples was similar to that of PyMT-EGFRvIII-Lo cells. Furthermore, adding the analogous huCD3ε/huEGFRvIII BiTE molecule in an in vitro co-culture assay consisting of the isolated CD45- populations from the triple-negative breast cancer sample in combination with allogenic human CD8+ T cells from a healthy donor induced T-cell activation similar to that observed with the surrogate mouse BiTE molecule (Supplementary Fig. S3), suggesting this to be a high-fidelity mouse surrogate molecule.
CD8+ T-cell activation and tumor killing by huEGFRvIII/muCD3ε BiTE in vitro
To further validate the efficacy of the huEGFRvIII/muCD3ε BiTE molecule, we assessed its ability to simultaneously engage both effector and target cells using a flow cytometry-based trans-coupling assay, where T cell (CD8+) – PyMT-EGFRvIII-Hi cell doublet formation was monitored as a function of BiTE molecule concentration. The BiTE was observed to efficiently mediate the formation of T cell-PyMT cell doublets in an EGFRvIII-dependent manner (Fig. 1B), yet in line with theoretical predictions, cell-cell coupling was observed with increasing BiTE molecule concentration to a maximum doublet frequency, followed by a decrease in coupling efficiency above the 100 nM concentration, consistent with the classic Hook effect anticipated for three-body binding equilibria (35). The huEGFRvIII/muCD3 BiTE molecule also induced a dose-dependent calcium influx, but only when T cells were in close contact with PyMT-EGFRvIII-Hi cells (Fig. 1C, Supplementary Video S1). Additionally, more T cells were observed in close proximity to tumor cells with BiTE molecule treatment (Supplementary Fig. S4A).
We next examined the tumor killing efficiency of the mouse surrogate BiTE molecule in vitro and observed PyMT-EGFRvIII-Hi tumor cell death within 2 hours at ≥1nM BiTE molecule concentration in cocultures with CD8+ T cells (Fig. 1D, Supplementary Video S2). Additional immunophenotyping conducted at 48 hours demonstrated dose-dependent expression of the activation markers, CD25 and CD69, expansion of T-cell numbers, and tumor killing (Fig. 1E). We validated a similar upregulation of CD25 and CD45RO of allogenic human T cells when mixed with a EGFRvIII-expressing human breast cancer sample and the human BiTE molecule (Supplementary Fig. S3). No effect was seen if T cells were co-cultured with PyMT-control cells (PyMT-Ctrl), demonstrating that BiTE molecule-TCR binding alone is not sufficient for T-cell activation, consistent with the intended mechanism of action for this modality. In total, we successfully validated an in vitro tumor-T cell system and demonstrated that huEGFRvIII/muCD3ε BiTE molecule-activated T cells could kill tumors expressing EGFRvIII in a dose-dependent manner, with the anticipated potency observed with primary human cell types exposed to the human-targeted BiTE molecule.
Characterization of huEGFRvIII/muCD3ε BiTE molecule in solid tumors
To extend our characterization of the BiTE molecule dose-response to the added complexity inherent in an immunocompetent mouse, we examined the effects of target engagement on T cells in vivo with intravital imaging (IVM) of PyMT-EGFRvIII-Hi tumor and combined multicolor flow analysis of disaggregated tumor harvests to characterize the TME (Fig. 2A). To this end, an AF488-labeled BiTE molecule (average degree of labeling/DoL ∼ 1.5:1) was administered at 20 µg/animal (∼0.8 mg/kg) retro-orbitally, where fluorescence indicated accumulation on T cells or tumor cells predominantly near the intratumoral vasculature (Fig. 2B-C). Surface puncta-like structures were visible at T cell-tumor cell interfaces by 2.5 hours post-dose, suggesting that the clusters of CD3-BiTE-EGFRvIII bridged cellular complexes (Fig. 2B). We also visualized real-time T-cell recruitment to target tumor cells in regions near the vasculature (Supplementary Fig. S4B, Supplementary Video S3).
To better understand the pharmacokinetics in tumor tissues and to dissect BiTE cellular partitioning within the TME, we quantified the fluorescent BiTE molecule bound to PyMT-EGFRvIII tumor cells or CD3+ tumor-infiltrating lymphocytes (TILs) over 72 hours. At a low BiTE dosage (2 µg), preferential accumulation on CD8+ T cells was observed (Fig. 2D-E), with up to ∼75% of CD8+ T cells harboring a bound BiTE molecule, but only ∼2% of isolated PyMT-EGFRvIII cells produced a detectable BiTE molecule signal. In contrast, at the higher 20 µg dose, both cell types were equivalently saturated. This phenomenon would not be anticipated for a monovalent binding event, based on the target affinity indicated by Kd measurement of EGFRvIII and CD3e. (Supplementary Fig. S1A), nor given that target expression (and cellularity) within the TME would intuitively favor the higher affinity, higher abundance EGFRvIII target. However, the preferential accumulation on T cells could be due to the better accessibility of the BiTE molecule to T cells in blood and peripheral lymphoid organs (Supplemental Fig. S5D), followed by the migration of the BiTE molecule-bound T cells to the tumor. This effect would be more obvious with a low BiTE dosage because BiTE T-cell binding in blood could be significant, contributing to a sufficient ‘sink’ effect. We also noted that maximum BiTE molecule exposure occurred within the tumor at ∼12 hours (tmax) following a single dose, inferred by the absence of a further increase in BiTE-AF488 intensity on target cells at subsequent times (Fig. 2E, Supplementary Fig. S5A-C).
Using bead-based calibration, we determined that at tmax (∼12 hours post 20 µg dose), CD8+ T cells and mcherry+ tumor cells bound to ∼6000 and ∼3000 BiTE-AF488 molecules per cell respectively (Fig. 2E; see methods). A dose dependence of CD8+ target cell coverage was also observed in T cell-rich lymph nodes (Supplementary Fig. S5D), although a more dramatic difference in T-cell coverage at 20 µg vs. 2 µg was observed, possibly indicating that abundant EGFRvIII expression in tumor tissue may stabilize, or otherwise enhance, binding or retention of the drug to T cells. These drug concentrations were efficacious based on the induction of extensive tumor killing 72 hours post injection, particularly by high-dose BiTE therapy (Fig. 2F-G).
Heterogeneous tissue extravasation of BiTE molecule within solid tumor
We next visualized the impact of the heterogeneous tumor vasculature on early BiTE molecule extravasation into the tumor space in PyMT-EGFRvIII-Hi tumors (Fig. 3A, Supplementary Video S4). Following injection of BiTE-AF488, we observed three distinct patterns of diffusion (Fig. 3B, 3D, 3F). By plotting normalized pixel intensities against the distance from vasculature over time, we described these as normally diffusive, irregular diffusive, and contained. (Fig. 3C, 3E, 3G). Diffusive behavior was observed in 39.2% of fields as a gradual and steady decrease in BiTE molecule intensity away from vessels (Fig. 3B-C). This was best modeled by a constant slope, when time of BiTE molecule entry was plotted against the square of the distance (Fig. 3H), consistent with minimal physical barriers and/or indicating that binding sites for BiTE molecules were homogeneous throughout the region of interest. In stark contrast, in 28.2% of fields, we also often classified BiTE molecule extravasation as irregular (Fig. 3D), which was characterized by a sharp slope of BiTE molecule signal gain at less than 25 μm from the vessel boundary, followed by a blunt increase at 30 to 50 μm away from the vasculatures (Fig. 3E). BiTE molecule extravasation kinetics in this regime were found to be biphasic (Fig. 3H), with the calculated Deff being almost 5-times higher when BiTE molecules were closer to the vessels than when they were away. The theoretical Deff of this AF488-labeled, HLE-BiTE molecule in the interstitial fluid is about 450 µm2/min, calculated using Stokes-Einstein equation for the diffusion coefficient of particles undergoing Brownian motion (see Methods), which is an order or two magnitudes higher than the Deff estimated from IVM, indicating binding site barriers and/or physical barriers to BiTE molecule diffusion. Finally, a third, tightly restricted BiTE-AF488 (‘Contained’, Fig. 3F-G) profile was also observed in 33% of fields, with insignificant signal for BiTE molecule above the limit of detection emanating beyond the vessel boundary, even at 2.5 hours. This BiTE molecule diffusion heterogeneity was observed both across separate tumor-bearing animals, and within a given tumor for an individual animal.
Such variation has predicted consequences of accumulation of BiTE molecules on CD3+ cells (Fig. 3I, Supplementary Fig. S6). Under the diffusive condition, BiTE molecule binding to T cells accumulated at a steady rate, regardless of where T cells were located (Fig. 3I). However, in the irregular/biphasic case, appreciable BiTE molecule binding to T cells was restricted to the regions of rapid “leaky” dissemination (<25 μm from vessels, Fig. 3D-E). Such pronounced heterogeneity in HLE-BiTE molecule tumor extravasation could limit efficacy in a clinical setting.
High and homogenous target expression maximized BiTE efficacy.
To characterize the significance of these findings, we varied the requisite target expression for BiTE molecule efficacy in our immunocompetent mouse model, using the Hi- and Lo-expressing lines, spanning the range of human tumors examined (Supplementary Fig. S2). The BiTE diffused farther away from the vasculature in PyMT-EGFRvIII-Hi tumors compared to low-expressing tumors at 4 hours post BiTE injection, with a higher binding to tumor cells. This difference was more pronounced when measured at 72 hours (Supplementary Fig. S7). A single 10 μg dosage failed to clear PyMT-EGFRvIII-Lo tumors (Fig. 4A-B) and did not significantly increase overall CD8+ T cells compared to PyMT-Ctrl tumors (Fig. 4C). In contrast, the same dose given to mice bearing PyMT-EGFRvIII-Hi tumors resulted in complete tumor clearance (Fig. 4A-B) and significant CD8+ T-cell expansion (Fig. 4C), suggesting that high target expression either enhanced intratumoral T-cell proliferation or recruited more T cells from lymphoid tissues, or both. Immunophenotyping demonstrated increased CD44 and IFNγ expression in EGFRvIII-Hi tumors but not in EGFRvIII-Lo tumors, as well as increased proliferative capability indicated by Ki67 (Fig. 4D-E).
Figure 4. BiTE efficacy in solid tumors requires high target expression.

(A) Measurement of tumor size of indicated groups of mice over time. PyMT tumor cells (control (Ctrl), EGFRvIII-Hi, and EGFRvIII-Lo) were orthotopically injected into fat pad of the C57BL/6 mice. 10 μg BiTEs were injected intravenously (i.v.) at day 15 post tumor inoculation when tumors were palpable. (B) Measurement of the tumor growth. Curves for individual mice shown for each group. (C) Flow cytometry analysis of CD8+ T cells in the indicated groups. T cells were harvested on day 19 post tumor injection. (D) Representative flow cytometry plots and (E) quantification of activation markers expressed by CD8+ T cells in the indicated tumor groups. N=10, 7, and 8 mice in Ctrl, EGFRvIII-Hi, and EGFRvIII-Lo groups, respectively. Data are representative of two independent experiments; ****p<0.0001, ***p<0.001, **p<0.01 as determined by one-way ANOVA and multiple comparison test.
Given that the expression of a specific tumor antigen is often variable in solid tumors, we mixed EGFRvIII-Hi tumor cells with control tumor cells at two different ratios of 3 to 1 or 1 to 3 to determine whether the BiTE molecule could lead to clearance of non-targeted tumors. Despite that a higher percentage of EGFRvIII-expressed cells resulted in smaller overall tumor size at every time point, full tumor regression was not observed in either condition. This indicated that, although effective against EGFRvIII+ tumors, BiTE molecule-induced killing of tumors was incomplete when not every cell had high expression (Supplementary Fig. S8).
Peripheral T-cell recruitment for BiTE efficacy is dose-dependent
We next sought to understand whether the BiTE molecule-induced tumor clearance required peripheral T cells to be recruited from lymphoid tissues. T-cell egress from lymph nodes was blocked by treatment with FTY720, an S1P agonist (36), and this was evidenced by a reduction in total and naïve (CD44–CD62L+) circulating blood CD8+ T cells compared to mock-treated animals (Fig. 5A-B). At a high BiTE molecule IV dosage (20 µg), FTY720 treatment did not have a significant effect and permitted complete clearance of PyMT-EGFRvIII-Hi tumors, similarly to controls. In contrast, low BiTE molecule dosage (2 µg) was sufficient for complete regression of PyMT-EGFRvIII-Hi tumors, but FTY720 treatment significantly decreased BiTE molecule antitumor activity (Fig. 5C). IFNγ and granzyme B production from CD8+ T cells was not differentially upregulated by high-dose BiTE under FTY720 treatment compared to control groups. However, it was lower in FTY720-treated groups with low-dose BiTE therapy (Fig. 5D-E). The data taken together suggest a significant contribution of cell recruitment from the lymphatics that regulates T-cell function in the TME with BiTE treatment, but that limitations produced by this can be overcome with a high BiTE dosage due to sufficient activity from resident TILs.
Figure 5. The sufficiency of local T cells for BiTE efficacy is dose-dependent.

(A) Diagram of experimental design. BiTEs (20 or 2 μg/mouse, i.v. single dose) and FTY720 (1 mg/kg, intraperitoneally, i.p., daily until harvest) were injected when EGFRvIII-Hi tumor size on average reached 40 mm2. (B) Percentage of total CD8+ T cells (left) and CD62L+CD44-CD8+ T cells (right) in blood detected by flow cytometry. (C) Measurement of tumor size for the indicated groups of mice over time. (D) Representative flow plots and (E) quantification of IFNγ and granzyme B secretion from tumoral CD8+ T cells for the indicated groups. T cells were harvested on day 27 post tumor injection. N=4, 5, 8, 5, and 5 mice per group from left to right (D, E). Data are representative of two independent experiments; ****p<0.0001, **p<0.01, *p<0.05 as determined by one-way ANOVA and multiple comparison test. ns: not significant; NT: no treatment.
Stimulatory DCs boosted local T cell pool to enhance BiTE efficacy
We extended our investigation of the mechanisms of BiTE molecule-mediated T cell activation in solid tumor to focus on the role of other immune cells potentially involved. Specifically, we focused on CD103+ intratumoral cDC1 because they can potently stimulate T cells to drive anti-tumor responses (37). Batf3 is the master transcriptional factor for cDC1 development (38), and we used Batf3 knockout mice (Batf3−/−) to understand if cDC1s were required for CD8-mediated tumor killing in response to BiTE treatment. In PyMT-EGFRvIII-Hi tumors in the absence of BiTE molecule, the loss of CD103+ DCs did not change the already-robust tumor outgrowth nor affect the CD4+ T-cell population but did result in reduced numbers of CD8+ T cells in tumors, with those present having similar expression of effector molecules (e.g., granzyme B and IFNγ; Supplementary Fig. S9A-C). In contrast to controls, in Batf3−/− mice, the BiTE molecule was unable to elicit clearance of PyMT-EGFRvIII-Hi tumors (Fig. 6A-B). As expected, CD103+ DCs were largely absent in the tumors of the Batf3−/− group, whereas other DC populations remained unchanged (Supplementary Fig. S9D, Fig. 6C). Concordant with failed tumor clearance, we also observed significantly lower IFNγ and granzyme B production in the global absence of CD103+ DCs in treated mice (Fig. 6D). We also found that DCs were critical in a genetic model in which DCs were present throughout development but were depleted just days prior to BITE treatment using XCR1DTR mice (Supplementary Fig. S10A-B) (39, 40). Taken together, the data suggest that an established ongoing endogenous CD8+ T-cell response, mediated by specific APCs, is necessary to permit optimal BiTE function.
Figure 6. Stimulatory DCs are required for BiTE efficacy in solid tumors.

(A) Measurement of PyMT-EGFRvIII-Hi tumor size for the indicated groups of mice over time. 10 μg BiTEs were injected intravenously (i.v.) at day 18 post tumor inoculation when tumors were palpable. (B) Measurement of tumor growth from each individual mouse for the indicated groups. Tumors were harvested on day 24 post tumor injection. (C) Flow cytometry analysis of DC populations. (D) Quantification of IFNγ and granzyme B secretion from tumoral CD8+ T cells for the indicated groups. Data are representative of two independent experiments; ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05 as determined by the one-way ANOVA and multiple comparison test. NT: no treatment.
Discussion
Here, we identified key requirements of perfusion, target expression, lymph node involvement, and a DC-driven endogenous response for promoting BiTE efficacy. We found these requirements using a pre-clinical solid tumor mouse model with endogenous immunocompetency, whose features closely mimicked human disease, including target expression and heterogeneity, and the model also allowed a clinical drug to be studied to determine pharmacological barriers to its efficacy in the context of a solid tumor.
Prior to this work, other studies have described the efficacy of various types of T-cell bispecific molecules in diverse solid tumor models (41), including glioblastoma (42), prostate (43), colorectal (44), breast (45), and lung (46) cancers. In those models, immuno-deficient mice were typically transplanted with human cancer cell lines or primary tumor tissue and supplemented with exogenous human T cells. Although these studies provide strong evidence of target-dependent T-cell activation, proliferation, and infiltration into the tumor and tumor regressions induced by BiTE immunotherapy, they are incapable of mimicking the complex interactions of immune cells in both the peripheral immune system and the TME of solid tumors or depicting BiTE molecule diffusion patterns into solid tumors.
The pharmacokinetics of immunotherapeutic reagents defining the exposure of biologic immunotherapies within solid tumors has remained an underappreciated area of study to rationalize efficacy, or lack thereof, in patients. Numerous studies have shown how vascular transport of many drugs into tissue are limited by poorly perfused tumor vessels due to vessel compression, hyperpermeability, abnormal morphology, and organization (47). Lessons learned from immune checkpoint therapy development include strategies to normalize vascularization in solid tumors, highlighted by promising improvements in anti- PD-1/PD-L1 efficacy in lung and kidney cancers (48). Prior to our study, the actual biodistribution of BiTE molecules within the heterogeneous TME was poorly defined and remains a significant barrier to our understanding of the tumor accessibility of half-life–extended, T-cell directing therapies. Our study suggests that some tumors may impose additional diffusion barriers distal to the vessels themselves. At present, we are unable to directly measure how significantly this limits overall tumor exposure but surmise that such a barrier could potentially complicate BiTE therapy.
Our results provide a first and direct picture of BiTE molecule extravasation in a solid tumor model through intravital imaging. We found that within a tumor, the vascular permeability to the BiTE molecule exhibited a high degree of variability, with several distinct diffusion topographies, ranging from facile, or widespread “leakage”, to vessels of low apparent permeability that limited tissue/target exposure to therapy. Such variable diffusion patterns suggest that BiTE molecule extravasation within solid tumors may be inconsistent in the clinical setting, and that vascularization is just one of the criteria determining tumor accessibility. Given enough time and a high dosage of BiTE therapy, we observed ample coverage of the tumor by the drug in our model. However, the tumor vasculature usually differs between mouse and human tumors (49, 50) and such differences affect the efficacy of anti-tumor drugs (51). Therefore, an even more restricted diffusion pattern could theoretically exist in the clinical setting that potentially limits drug exposure. The heterogeneous diffusion patterns of the BiTE molecule revealed in our study highlighted that in some cases, better delivery of the drug may be needed to improve BiTE efficacy in solid tumors. One important caveat to our analysis is that our observations were conducted after single dose BiTE treatment, whereas typical clinical cycles involve multiple doses over several weeks. Thus, it is possible that sufficient tumor exposures are achieved when dosing to steady state under the variety of tumor architectures probed here. The relationships between total vascular surface area, vessel size, and receptor expression and their effects on both active and passive (paracellular) BiTE molecule transport to the tumor interstitium warrant further study.
The abundance of TILs is a positive prognostic indicator for some tumor types and is also a potential predictor of the responsiveness to immune checkpoint inhibitors in cancer patients (52, 53). The number of TILs is determined by both T-cell expansion in situ, and the recruitment of peripheral T cells. BiTEs have been shown to increase TILs in many xenograft models (54, 55), as well as in our immunocompetent PyMT-EGFRvIII model presented here; however, the additional requirement for T-cell recruitment from the periphery, most notably from the lymphoid organs, for efficient activity leading to tumor clearance was not possible to assess prior to this analysis. One possible explanation for this requirement is that suboptimal BiTE dosages preferentially bind to T cells in the periphery and not the tumor and thus their trafficking becomes a requirement for them to carry the agent into the tumor and affect clearance. Our study demonstrated that a high TIL count may be prognostic of the potential for maximal BiTE efficacy, and that at lower tumor exposures, we clearly delineated the role of peripheral T-cell populations in maintaining anti-tumor responses. Our numerous observations also suggested not only variable TIL content within a given tumor region, but also unpredictable dynamic behaviors for TILs, ranging from active swarm-like phenotypes to T-cells apparently at rest prior to BiTE molecule dosing (Supplementary Video S3 vs. Video S4, respectively). The clinical relevance of baseline T-cell dynamic cellular phenotypes in relation to BiTE molecule responses in solid tumor is unknown and is an area in need of further study.
Additionally, high, homogenous EGFRvIII target abundance was determined to be a critical component for maximal efficacy. In this investigation, we mimicked the potential for heterogeneous target expression within the TME by grafting tumor cell lines with varying EGFRvIII expression, which in turn was found to impact BiTE molecule potency significantly. Our study indicates that low target expressors may require a higher BiTE molecule exposure to be completely cleared. Non-uniform target expression is a potential limiting factor for efficacy of any T-cell directing therapy, and the impact of target heterogeneity is likely context-dependent.
Among the abundant immunosuppressive myeloid cells residing in the TME, CD103+ cDC1 in mice (defined rather by expression of BDCA-3 in humans), demonstrate potent cross-presentation capability and the ability to stimulate CD8+ T cells to drive anti-tumor immune responses (37). The abundance of this type of stimulatory DC in solid tumors positively correlates with increased patient survival and predicts responsiveness to anti-PD-1 immunotherapy (56). Although BiTE molecules stimulate T cells regardless of MHC restriction, the demonstration that BiTE efficacy required DCs for optimal outcomes was not previously known. Consistent with previous findings about the role of Batf3+ DCs in effector T-cell trafficking (57), it is most likely that CD103+ DC deficiency diminished BiTE efficacy in solid tumors due to a deficiency in the number of available T cells to be acted upon by BiTE molecules. Although clinical data is currently lacking, we propose that the abundance of CD103+/BDCA-3+ DCs, a rare myeloid population in solid tumors, may be a key predictive factor for responsiveness to BiTE immunotherapy in human solid tumors.
Supplementary Material
Synopsis:
Maximizing bispecific T-cell engager (BiTE) efficacy in solid tumors is challenging. Here, key requirements for BiTE efficacy are identified, including tumor diffusion, target expression level, lymphoid tissue involvement, and DC-driven responses, highlighting possible strategies to improve this therapy.
Acknowledgements
We thank the Biological Imaging Development Center at the University of California San Francisco (UCSF) for help with microscopy data collection and instrumentation. We also thank the Parnassus Flow Cytometry Core for flow cytometry instrumentation, supported by grant RRID:SCR_018206 and DRC Center Grant NIH P30DK063720. These studies were funded by Amgen, Inc. and were supported by NIH grant R01AI052116 (M.F.K.) and by the Cancer Research Institute (award number CRI2940 to A.R.).
Footnotes
Competing interests
KPC is an employee of Amgen Inc. and owns stock in Amgen Inc.
References
- 1.Guedan S, et al. Emerging Cellular Therapies for Cancer. Annu Rev Immunol. 2019;37:145–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callahan MK, et al. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldman AD, et al. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vafa O, et al. Perspective: Designing T-Cell Engagers With Better Therapeutic Windows. Front Oncol. 2020;10:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goebeler ME, et al. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–434. [DOI] [PubMed] [Google Scholar]
- 6.Slaney CY, et al. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018;8(8):924–934. [DOI] [PubMed] [Google Scholar]
- 7.Mack M, et al. Biologic properties of a bispecific single-chain antibody directed against 17–1A (EpCAM) and CD3: tumor cell-dependent T cell stimulation and cytotoxic activity. J Immunol. 1997;158(8):3965–70. [PubMed] [Google Scholar]
- 8.Dao T, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33(10):1079–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagorsen D, et al. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res. 2011;317(9):1255–60. [DOI] [PubMed] [Google Scholar]
- 10.Kebenko M, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7(8):e1450710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moek WMF KL, von Einem JC, Verheul HM, Seufferlein T, de Groot DJ, Heinemann V, Kebenko M, Menke-van CW der Houven van Oordt, Ettrich TJ, Rasmussen E, Bogner P, Sable B, Stienen S, de Vries EGE. Phase I study of AMG 211/MEDI-565 administered as continuous intravenous infusion (cIV) for relapsed/refractory gastrointestinal (GI) adenocarcinoma. Annals of Oncology. 2018;29(8):VIII139–140. [Google Scholar]
- 12.Rosenthal MA, et al. Atim-49 (Ltbk-01). Amg 596, a Novel Anti-Egfrviii Bispecific T Cell Engager (Bite®) Molecule for the Treatment of Glioblastoma (Gbm): Planned Interim Analysis in Recurrent Gbm (Rgbm). Neuro-Oncology. 2019;21(Supplement_6):vi283-vi283. [Google Scholar]
- 13.Hummel HD, et al. Pasotuxizumab, a BiTE((R)) immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy. 2021;13(2):125–141. [DOI] [PubMed] [Google Scholar]
- 14.Tran LH B, Dorff T, Rettig M, Lolkema MP, Machiels J, Rottey S, Autio K, Greil R, Adra N, Lemech C, Minocha M, Cheng F, Kouros-Mehr H, Fizazi K. Results from a phase I study of AMG 160, a half-life extended (HLE), PSMA-targeted, bispecific T-cell engager (BiTE®) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). Annals of Oncology; 2020:S507–S549. [Google Scholar]
- 15.Taofeek Kunle Owonikoko HB, Stéphane Champiat, Paz-Ares Luis G., Ramaswamy Govindan, Boyer Michael J., Melissa Lynne Johnson, Hibiki Udagawa, Horst-Dieter Hummel, Ravi Salgia, Fiona Helen Blackhall, Boosman Rene J., Wei-Chu Victoria Lai, Afshin Dowlati, Vokes Everett E., Hann Christine L., Chiang Anne C., Marichu Endraca, Neelesh Soman, Marie-Anne Damiette Smit. Phase I study of AMG 757, a half-life extended bispecific T-cell engager (HLE BiTE immune therapy) targeting DLL3, in patients with small cell lung cancer (SCLC). Journal of Clinical Oncology. 2020;38(15)(TPS9080) [Google Scholar]
- 16.Conner KP, et al. The biodistribution of therapeutic proteins: Mechanism, implications for pharmacokinetics, and methods of evaluation. Pharmacol Ther. 2020;212:107574. [DOI] [PubMed] [Google Scholar]
- 17.Schaaf MB, et al. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9(2):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moek KL, et al. (89)Zr-labeled Bispecific T-cell Engager AMG 211 PET Shows AMG 211 Accumulation in CD3-rich Tissues and Clear, Heterogeneous Tumor Uptake. Clin Cancer Res. 2019;25(12):3517–3527. [DOI] [PubMed] [Google Scholar]
- 19.An Z, et al. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37(12):1561–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan HK, et al. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 2013;280(21):5350–70. [DOI] [PubMed] [Google Scholar]
- 21.Rutkowska A, et al. EGFR(vIII): An Oncogene with Ambiguous Role. J Oncol. 2019;2019:1092587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Platten M EGFRvIII vaccine in glioblastoma-InACT-IVe or not ReACTive enough? Neuro Oncol. 2017;19(11):1425–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yee C, et al. Modulating T-cell immunity to tumours: new strategies for monitoring T-cell responses. Nat Rev Cancer. 2002;2(6):409–19. [DOI] [PubMed] [Google Scholar]
- 24.Boldajipour B, et al. Tumor-infiltrating lymphocytes are dynamically desensitized to antigen but are maintained by homeostatic cytokine. JCI Insight. 2016;1(20):e89289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelhardt JJ, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 2012;21(3):402–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boulch M, et al. Tumor Immunosurveillance and Immunotherapies: A Fresh Look from Intravital Imaging. Trends Immunol. 2019;40(11):1022–1034. [DOI] [PubMed] [Google Scholar]
- 27.Veiga-Fernandes H, et al. Tyrosine kinase receptor RET is a key regulator of Peyer’s patch organogenesis. Nature. 2007;446(7135):547–51. [DOI] [PubMed] [Google Scholar]
- 28.Hu KH, et al. ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat Methods. 2020;17(8):833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamblett KJ, et al. AMG 595, an Anti-EGFRvIII Antibody-Drug Conjugate, Induces Potent Antitumor Activity against EGFRvIII-Expressing Glioblastoma. Mol Cancer Ther. 2015;14(7):1614–24. [DOI] [PubMed] [Google Scholar]
- 30.Tomonari K A rat antibody against a structure functionally related to the mouse T-cell receptor/T3 complex. Immunogenetics. 1988;28(6):455–8. [DOI] [PubMed] [Google Scholar]
- 31.Fernandes RA, et al. T cell receptors are structures capable of initiating signaling in the absence of large conformational rearrangements. J Biol Chem. 2012;287(16):13324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brauchle B, et al. Characterization of a Novel FLT3 BiTE Molecule for the Treatment of Acute Myeloid Leukemia. Mol Cancer Ther. 2020;19(9):1875–1888. [DOI] [PubMed] [Google Scholar]
- 33.You R, et al. Active surveillance characterizes human intratumoral T cell exhaustion. J Clin Invest. 2021;131(18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmittnaegel M, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017;9(385) [DOI] [PubMed] [Google Scholar]
- 35.Douglass EF Jr., et al. A comprehensive mathematical model for three-body binding equilibria. J Am Chem Soc. 2013;135(16):6092–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cyster JG, et al. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. [DOI] [PubMed] [Google Scholar]
- 37.Broz ML, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell. 2014;26(6):938. [DOI] [PubMed] [Google Scholar]
- 38.Edelson BT, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207(4):823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamazaki C, et al. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J Immunol. 2013;190(12):6071–82. [DOI] [PubMed] [Google Scholar]
- 40.Gurka S, et al. Mouse Conventional Dendritic Cells Can be Universally Classified Based on the Mutually Exclusive Expression of XCR1 and SIRPalpha. Front Immunol. 2015;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu S, et al. Recent advances of bispecific antibodies in solid tumors. J Hematol Oncol. 2017;10(1):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma P, et al. Anti-CD3 x EGFR bispecific antibody redirects cytokine-induced killer cells to glioblastoma in vitro and in vivo. Oncol Rep. 2015;34(5):2567–75. [DOI] [PubMed] [Google Scholar]
- 43.Friedrich M, et al. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther. 2012;11(12):2664–73. [DOI] [PubMed] [Google Scholar]
- 44.Lutterbuese R, et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A. 2010;107(28):12605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y, et al. Fully human HER2/cluster of differentiation 3 bispecific antibody triggers potent and specific cytotoxicity of T lymphocytes against breast cancer. Mol Med Rep. 2015;12(1):147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hipp S, et al. A Bispecific DLL3/CD3 IgG-like T-cell Antibody induces anti-tumor responses in Small Cell Lung Cancer. Clin Cancer Res. 2020; [DOI] [PubMed] [Google Scholar]
- 47.Stylianopoulos T, et al. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer. 2018;4(4):292–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munn LL, et al. Vascular regulation of antitumor immunity. Science. 2019;365(6453):544–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lauk S, et al. Comparative morphometric study of tumor vasculature in human squamous cell carcinomas and their xenotransplants in athymic nude mice. Cancer Res. 1989;49(16):4557–61. [PubMed] [Google Scholar]
- 50.Goveia J, et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell. 2020;37(1):21–36 e13. [DOI] [PubMed] [Google Scholar]
- 51.Dong Z, et al. Xenograft tumors vascularized with murine blood vessels may overestimate the effect of anti-tumor drugs: a pilot study. PLoS One. 2013;8(12):e84236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mariathasan S, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tauriello DVF, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. [DOI] [PubMed] [Google Scholar]
- 54.Giffin MJ, et al. AMG 757, a Half-Life Extended, DLL3-Targeted Bispecific T-Cell Engager, Shows High Potency and Sensitivity in Preclinical Models of Small-Cell Lung Cancer. Clin Cancer Res. 2021;27(5):1526–1537. [DOI] [PubMed] [Google Scholar]
- 55.Benonisson H, et al. CD3-Bispecific Antibody Therapy Turns Solid Tumors into Inflammatory Sites but Does Not Install Protective Memory. Mol Cancer Ther. 2019;18(2):312–322. [DOI] [PubMed] [Google Scholar]
- 56.Barry KC, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24(8):1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spranger S, et al. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31(5):711–723 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.