

Summary
Patient-derived xenografts (PDX) and organoids (PDO) have been shown to model clinical response to cancer therapy. However, it remains challenging to use these models to guide timely clinical decisions for cancer patients. Here we used droplet emulsion microfluidics with temperature control and dead-volume minimization to rapidly generate thousands of Micro-Organospheres (MOS) from low-volume patient tissues, which serve as an ideal patient-derived model for clinical precision oncology. A clinical study of recently diagnosed metastatic colorectal cancer (CRC) patients using a MOS-based precision oncology pipeline reliably assessed tumor drug response within 14 days, a timeline suitable for guiding treatment decisions in clinic. Furthermore, MOS capture original stromal cells and allow T cell penetration, providing a clinical assay for testing immuno-oncology (IO) therapies such as PD-1 blockade, bispecific antibodies, and T cell therapies on patient tumors.
Keywords: micro-organosphere, precision oncology, colorectal cancer, lung cancer, droplet microfluidics, immune-oncology, bispecific antibody, adoptive cell therapy
Graphical Abstract
Cancer patients often receive ineffective treatments while oncology drugs frequently fail in clinical trials because every patient is unique. Precision oncology hinges on selecting the right drug for the right patient. Micro-organospheres (MOS) capture each patient tumor with its microenvironment for rapid clinical drug selection and immunotherapy profiling.
Introduction
The success of precision oncology relies on models that capture the morphological, molecular, and functional characteristics of patient tumors to accurately predict drug response and resistance. The development of various patient-derived models of cancer (PDMC) has provided important tools in this effort. Drug sensitivity assays using PDMC have recapitulated antitumor response in the clinic, underscoring their potential for guiding personalized care (Barretina et al., 2012; Gao et al., 2015b; Lu et al., 2017; Vlachogiannis et al., 2018).
However, the promise of clinical translation of PDMC—using drug sensitivity assays on patient-derived tissue to drive clinical decision-making—remains largely unrealized, in part, due to technical limitations with each model. Patient-derived cell lines exhibit genetic and morphological changes over time that make them unsuitable for clinical screening (Lorio et al., 2016; Stein et al., 2004). Patient-derived xenografts (PDX) can be repeatedly passaged with retention of intra-tumoral clonal architecture and genetic diversity (Bruna et al., 2016), while serving as a model to study patient therapeutic response (Gao et al., 2015a; Hidalgo et al., 2014). However, PDX are expensive and slow to generate, limiting their utility for diagnostic drug screens and precision medicine. In comparison, patient-derived organoids (PDO) offer a lower cost and higher throughput model for clinical applications (Jenkins et al., 2018b; Neal et al., 2018; Yuki et al., 2020). With large-scale biobanks of breast (Sachs et al., 2018), colorectal (Sato et al., 2011; van de Wetering et al., 2015a), ovarian (Kopper et al., 2019), pancreatic (Driehuis et al., 2019b), brain (Jacob et al., 2020), kidney (Calandrini et al., 2020), head and neck (Driehuis et al., 2019a) and gastric cancers (Seidlitz et al., 2021; Yan et al., 2018), PDO have been shown to capture patient diversity. Additionally, broad-based drug screens have reproduced known associations between genetic mutations and sensitivity to targeted therapies (Gao et al., 2014; Skardal et al., 2015). Thus, PDO have been exploited as a potential functional precision medicine technology for guiding treatment decisions in the clinic (Ganesh et al., 2019; Ooft et al., 2019; Pauli et al., 2017; Tiriac et al., 2018; Vlachogiannis et al., 2018; Yao et al., 2020).
However, expanding sufficient numbers of PDO for drug screening remains too slow and inefficient for adoption into the clinic (2018; van de Wetering et al., 2015b). Since clinical treatment decisions are typically made within 14 days of diagnosis, the existing timeframe for PDO generation would result in unacceptable treatment delays. Thus, to develop a clinically useful diagnostic assay, it is necessary to both accelerate PDO generation and functional testing as well as develop automated procedures from a core biopsy.
Furthermore, given the growing clinical importance of immuno-oncology (IO), there is significant interest to reproduce physiological immune activity in organoid cultures. For example, peripheral blood lymphocyte and tumor organoid co-culture models have been used to test tumor-reactive T cells (Dijkstra et al., 2018). In addition to organoid culture, patient-derived organotypic spheroids and minced primary tissue fragments embedded in collagen gels in air-liquid interface cultures have been developed to study interactions between tumor and local immune cells (Jenkins et al., 2018a; Neal et al., 2018).
In the current study, we report the development of an automatic microfluidics droplet platform that can generate patient-derived Micro-Organospheres (MOS) in a high-throughput fashion to facilitate drug screening and personalized medicine treatment. A clinical study of eight metastatic CRC patients showed that MOS assay readout within 14 days (10 days on average) correlates with later clinical outcomes. Furthermore, MOS preserved stromal cells of the original tumor tissue and potency of immune cells, providing a powerful tool for testing IO therapy.
Results:
MOS Generation and Establishment
To establish a precision medicine pipeline that can be used to guide patient care, we developed droplet-based microfluidics technology to rapidly generate patient-derived models of cancer in a reliable manner (Figure 1A). The core principle involves adding suspended cells from primary tissue to a 3D-extracellular matrix (Matrigel) followed by mixing with a biphasic liquid (oil) to generate microfluidic-based droplet Micro-Organospheres (MOS). The generated MOS are demulsified to remove excess oil and then cultured as suspension droplets.
Figure 1.
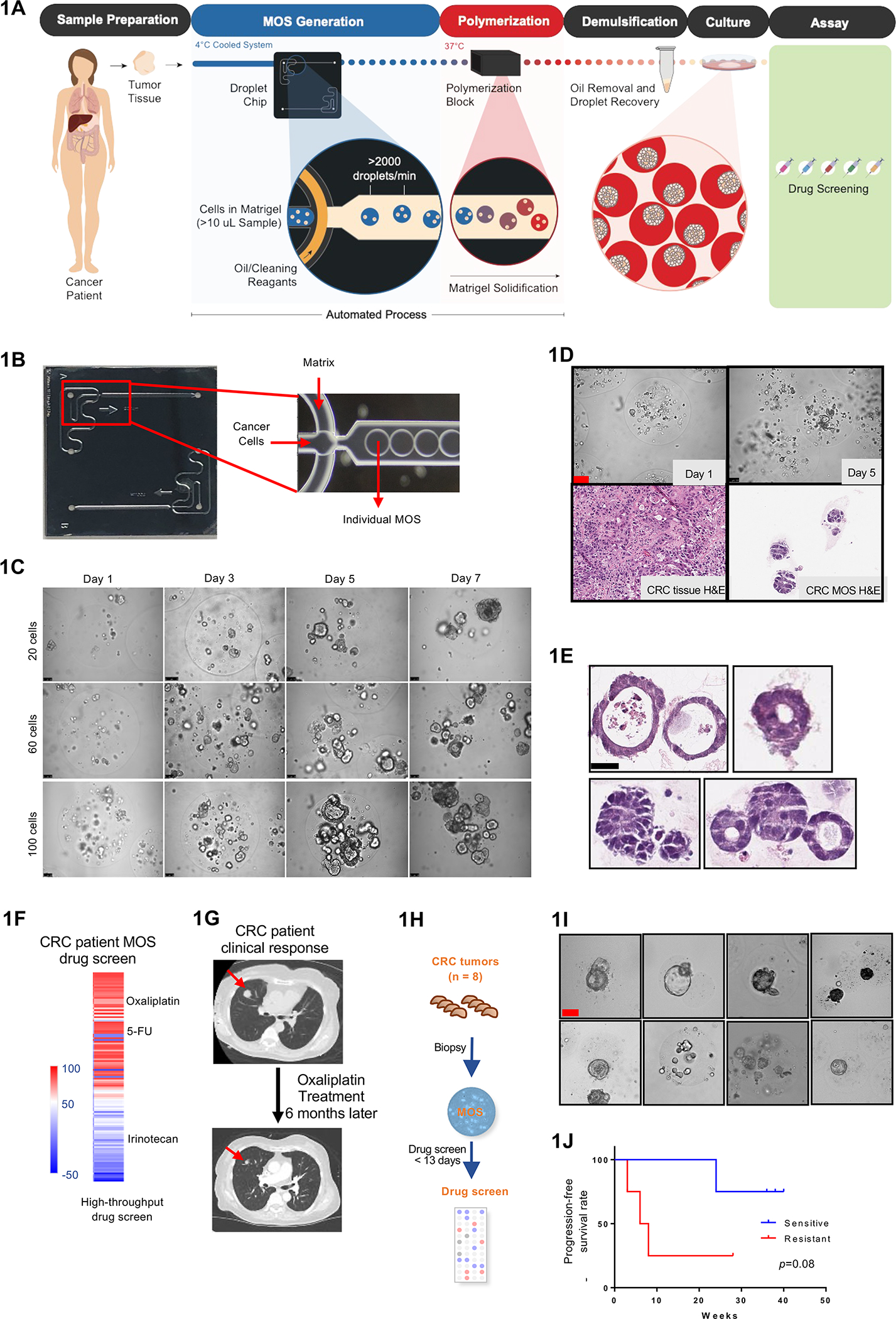
Establishing CRC MOS for drug screen and clinical validation. (A) Scheme of CRC MOS generation and drug screening. (B) Images of the microfluidic MOS chip. (C) Bright field microscope images of CRC MOS generated with different cell numbers per MOS. (D) Representative images of generated MOS from patient CRC tumor tissue and hematoxylin and eosin (H&E) staining of the primary CRC tumor tissue and derived MOS. (E) H&E staining of CRC MOS established from different patient tumor tissues. (F) Heat map of high throughput drug screen using CRC tumor-derived MOS indicates sensitivity to oxaliplatin and resistance to Irinotecan. (G) The same patient showed response to oxaliplatin after 6 months of treatment in clinic. (H) Schematic illustration of the clinical study design. MOS are established from CRC biopsy for drug testing. (I) Representative images of patient-derived MOS. (J) Survival outcomes from all eight CRC patients are correlated with MOS drug sensitivity. Scale bar: 100 um.
The basis of our pipeline is a benchtop machine for the generation of MOS (Figure 1B, S1A, Supplementary Table 1, Movie S1). Important design features of our device include reservoirs for loading both the oil and sample phases directly onto a custom microfluidic chip followed by positioning of the sample outlet on the backside of the chip for direct dispensing into a MOS recovery vessel. Attached pressure sources (e.g. Fluigent FlowEZ) are used to control the flow of oil and sample fluids into the custom microfluidic chip through tubing connected via a clamped manifold. The sample and oil meet at a ‘T’ junction (Figure 1B) where the sample is ‘pinched’ into droplets by the oil phase as it enters a collection channel. The system is compatible with temperature sensitive Matrigel. Both the 4°C sample and 37°C collection blocks are integrated into the device, which allows Matrigel to flow through microfluidic channels and then quickly solidify at higher temperatures. The channel and chamber heights were engineered to generate MOS that average 250 um - 450 um in diameter, as these dimensions provide a 3D environment that is well-suited for a variety of cell numbers and sizes. The device can generate MOS from as few as 15,000 cells from 18-gauge core biopsies, a sample size typically too small for reliable generation of conventional organoids for therapeutic profiling within the clinical time constraint.
As a proof of concept, we first used our device to generate MOS from CRC PDX cells. We monitored CRC MOS growth at different seeding densities (20–100 cells per droplet) and discovered that MOS establish tumorsphere-like structures (Figure 1C). The number and size of tumorspheres increased with the seeding density per droplet. MOS were then generated from clinical CRC biopsies (Figure 1D) and shown to have various morphologies (Figure 1E). The number of MOS are determined by the number of viable cells divided by the number of cells per droplet.
MOS Rapidly Assess Patient Drug Response in a Prospective Clinical Study
Since clinical treatment decisions are often made within 10–14 days of diagnosis, the ideal diagnostic assay should give results within 14 days and use minimal tissue (i.e., core biopsies) to predict clinical outcome. As a proof-of-concept study, we obtained a biopsy from a patient who presented with metastatic rectal cancer and established MOS (30 tumor cells per MOS) within 8 days of biopsy. We performed an in vitro high-throughput drug screen by treating the MOS with the Approved Oncology Set VI panel (provided by the NCI Developmental Therapeutics Program), which contained 119 different FDA-approved small molecule inhibitors at 1 uM concentrations, and then analyzed treatment responses. The MOS were sensitive to oxaliplatin (% killing >50%) and resistant to irinotecan (% killing < 50%) (Figure 1F). The entire process was performed within 11 days of biopsy acquisition. Consistent with the MOS drug response, the patient’s tumor still responded to oxaliplatin-based therapy 6 months later (Figure 1G).
We next designed and conducted a prospective clinical study wherein we obtained 18-gauge core biopsies from seven additional patients presenting with metastatic CRC, generated MOS, and performed drug testing (Figure 1H and 1I). Patient demographic information and mutation status from these two protocols are shown in Table 1. We generated MOS (30 tumor cells per MOS) and tested responses to oxaliplatin within 13 days (9.9 days on average) from time of biopsy for all eight biopsy samples with a success rate of 100% (8/8) (Table 2). Given the limited tissue volume, dosages such as 1uM and 10 uM were selected based on previous literature (Vlachogiannis et al. 2018, Ooft et al, 2019, Ganesh et al, 2019, Yao et al, 2020). We used the same cut-off as measured via Cell Titer Glo. Among the eight patients, four had oxaliplatin-sensitive MOS and four had oxaliplatin-resistant MOS (Figure 1J).
Table 1:
CRC Patient Demographics and clinical diagnosis
| ID | Age | Gender | Race | Histology | Grade | Primary Site | Metastatic Site | Molecular profiling |
|---|---|---|---|---|---|---|---|---|
| CRC-MOS-001 | 87 | F | Caucasian | Adenocarcinoma | Moderately differentiated | Rectal | Lung | MSS |
| CRC-MOS-002 | 68 | M | Caucasian | Adenocarcinoma | Moderately differentiated | Colon | Liver | MSS, TMB 1 Muts/Mb, KRAS (G12V), APC (L674fs), p53 (I254S) |
| CRC-MOS-003 | 71 | M | Caucasian | Adenocarcinoma | Moderately differentiated | Colon | Liver | MSS, TMB 3 Mut/Mb, KRAS (G12V), APC (Q1303), SMAD4 loss, TP53 (L257Q) |
| CRC-MOS-004 | 62 | F | Caucasian | Adenocarcinoma | Moderately differentiated | Colon | Liver | MSS, TMB 3 Mut/Mb, KRAS WT APC (R1450), ATM (R805), PIK3CA (E545K) |
| CRC-MOS-005 | 73 | M | Caucasian | Adenocarcinoma | Moderately differentiated | Colon | Liver | MSS, TMB 4 Mut/Mb, KRAS (G12D), APC (R876), TP53 (R158fs), PIK3CA (E545Q) |
| CRC-MOS-006 | 31 | F | Asian | Adenocarcinoma | Poorly differentiated | Rectal | Pelvis | MSS, TMB (4 Muts/Mb) KRAS (G12D), TP53 (R175H) |
| CRC-MOS-007 | 37 | M | Caucasian | Adenocarcinoma | Colon | Liver | MSS, TMB (1 Mut/Mb), KRAS (G12D), APC (R216), TP53 (G226E) | |
| CRC-MOS-008 | 68 | M | Caucasian | Adenocarcinoma | Moderately differentiated | Colon | Liver | MSS, TMB (6 Mut/Mb) KRAS WT, APC (Q1367), EGFR amplification TP53 (R248W) |
Table 2:
MOS Correlation to CRC Patient Outcome
| Patient ID | MOS Prediction | Clinical Outcome | Response time | Drug Screen |
|---|---|---|---|---|
| 001 | Sensitive | Response | 46 wks | 8 days |
| 002 | Resistance | Response | 28 wks | 9 days |
| 003 | Sensitive | Response | 38 wks | 12 days |
| 004 | Resistance | No response | 8 wks | 8 days |
| 005 | Sensitive | Response | 36 wks | 8 days |
| 006 | Resistance | No response | 3 wks | 10 days |
| 007 | Sensitive | No response | 24 wks | 11 days |
| 008 | Resistance | No response | 6 wks | 13 days |
All eight patients received oxaliplatin-based therapy per usual treatment guidelines. Patient outcomes were subsequently evaluated by CT scan before and after oxaliplatin treatment (Figure S1B–S1H), and we compared time-on-treatment to MOS oxaliplatin sensitivity. The four patients whose MOS were sensitive to oxaliplatin all responded clinically and stayed on treatment past 20 weeks (and three of four still remained on treatment close to 40 weeks), whereas three out of the four patients with resistant MOS did not respond to oxaliplatin treatment and were taken off treatment within 10 weeks (Figure 1J). The remaining patient (Case #1, ID #002) from the resistant MOS group on initial CT scan had a mixed response to therapy, but given clinical benefit, patient was continued on therapy. Subsequent CT scan showed response to therapy and patient continue to remain on treatment until 28 weeks when liver section was performed to remove the metastatic lesion (Figure S1B, Table 2).
This proof of concept clinical study suggests that MOS can be reliably generated from 18-gauge biopsies of metastatic CRC tumors and then be used to test sensitivity to frontline chemotherapy within 14 days. Our initial results demonstrate that this workflow largely correlate with patient outcomes, albeit larger trials are needed to further validate clinical applicability.
We further measured cell death in each MOS by imaging the Caspase 3/7 fluorescence signal and normalizing it by the cell surface area inside each MOS. Treatment of two available CRC MOS lines (20 cells per MOS) that are resistant to oxaliplatin showed that only the highest dosages induced significant cell killing with heterogeneity among different MOS (Figure S2A and S2B).
Tumor Stromal and Immune Cells in MOS
As the tumor microenvironment, in particular immune components, has been shown to affect cancer therapy, we sought to characterize the stromal components of patient-derived MOS. We focused on lung tumor due to its response to immunotherapy, but also characterized renal, breast, CRC and ovarian tumors to lesser degrees. We generated MOS at a density of 30 tumor cells per MOS in 70% Matrigel diluted in culture medium and concurrently established bulk organoids using the same density of cells for comparison. Representative pictures of MOS generated from each tumor type as well as H&E from each tumor tissue and MOS are shown in Figure 2A and Figures S2C and S2D. Formation and growth of MOS and bulk organoids at days 2, 5, and 7 were comparable (Figure S2E).
Figure 2.
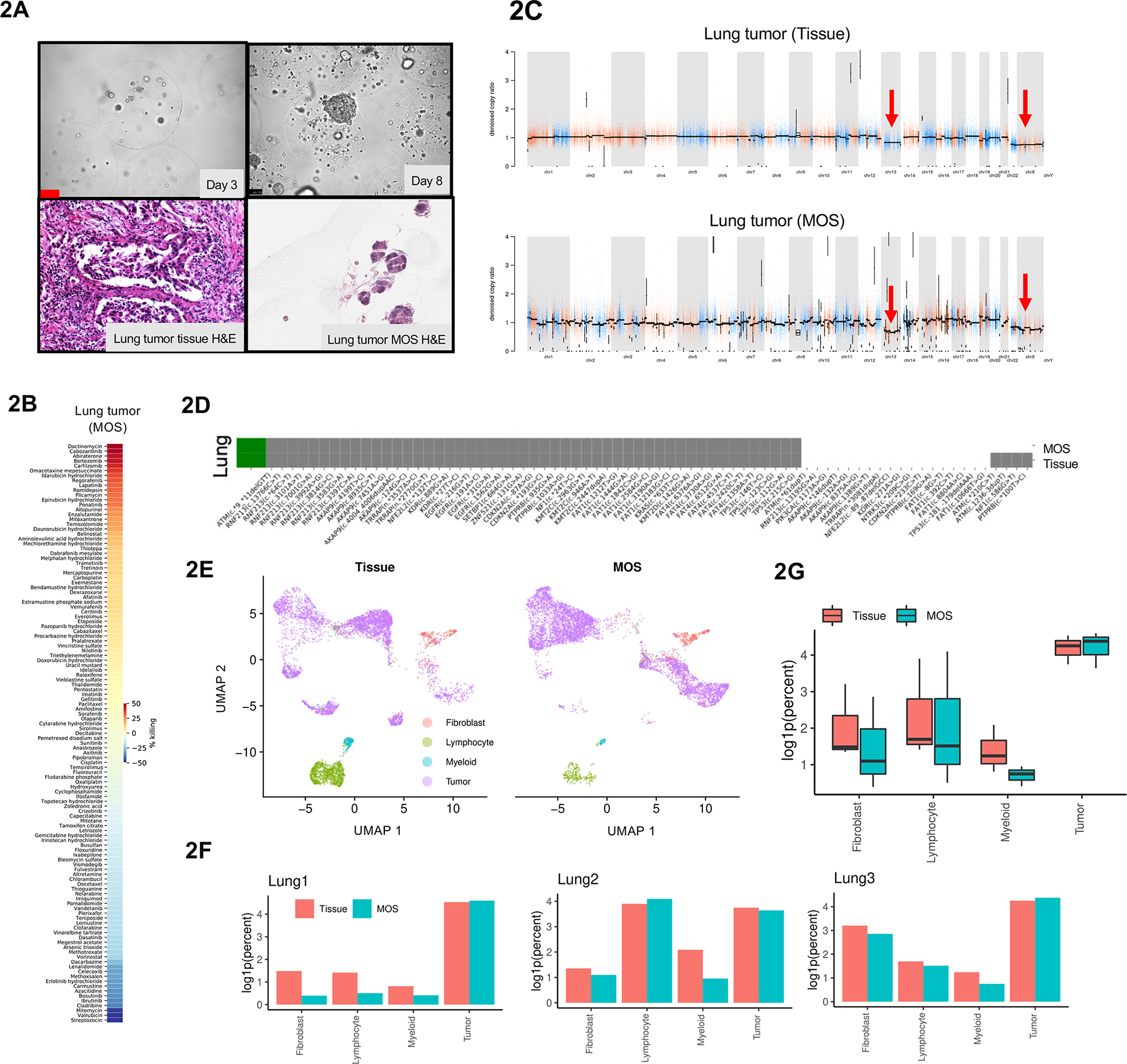
Genomic and transcriptomic characterization of MOS generated from patient lung tumor. (A) Representative images of generated MOS from patient lung tumor tissue and H&E staining of the primary lung tumor tissue and derived MOS. (B) High-throughput drug screen demonstrates feasibility of using lung MOS to identify other targets in cancer therapy. (C) Copy number variation (CNV) profiles with correlations of lung tumor tissue and derived MOS. (D) Driver mutations in commonly mutated genes for lung cancer is largely preserved in MOS compared to respective original tissues. Grey: driver mutations present. White: driver mutations absent. (E) UMAP of cells from primary lung tumor tissue or derived MOS labeled by cell types. (F) Comparison of log-transformed relative abundance of each cell type for lung tumor samples and derived MOS. (G) Relative abundance of cell types represented in either tissue (n=3) or MOS (n=3) samples. Abundances reported as log1p(percentage out of 1).
Overgrowth of fibroblasts is often a challenge for establishing organoids from clinical samples of certain cancer types. We compared the number of fibroblasts in MOS and bulk organoid cultures between days 7–9. Fewer fibroblasts were observed in MOS compared to bulk organoid cultures (Figure S2F), as confirmed by flow cytometry analysis of Vimentin expression (Figure S3A). We then performed rapid, high-throughput chemotherapeutic drug screening on MOS generated from lung, ovarian, and kidney cancer patients and measured sensitivities to commonly used agents in the treatment of these cancers (Figure 2B and S3B).
We compared whole exome sequencing of MOS to the matched original tumor specimen to determine if genomic alterations were maintained (Supplementary Table 2). First, we characterized copy number variations (CNVs). A similar pattern of amplifications and losses were seen in MOS and original tissue from lung cancer (Figure 2C) and other cancer types (Figure S3C). Second, we characterized somatic mutations in the genomes of matched MOS and original tumor samples. For each cancer type, mutation profiles for matched tissue specimen and MOS were highly correlated, while unmatched samples were not (Figure S3D). Variants were common between tissue specimens and matched MOS (Figure S3E). Driver mutations were largely consistent between tissue specimens and MOS among commonly affected genes for each cancer type (Figure 2D and S3F), with sensitivity (mutations detected in tumor tissue also detected in MOS) of 85% ± 0.007 and specificity (mutations absent in tumor tissue also absent in MOS) of 95% ± 0.003 (Figure S3G). These findings suggest that MOS largely capture the genomic profiles of patient tumors from which they are derived.
To compare the tumor and stromal cell types between tissue and derived MOS, we performed single cell RNA sequencing (Macosko et al., 2015) on six pairs of matched patient tumor specimens (three lung cancers, one kidney cancer, one ovarian cancer, and one CRC) and derived MOS (7–9 days). QC summary is shown in Figure S4A and S4B. Cells from all three lung tumor samples were clustered using UMAP reductions into four groups marked as tumor cells, cancer-associated fibroblasts, and either lymphoid or myeloid immune cells, which were concordant between tissue and MOS (Figure 2E) with comparable relative abundance levels (Figure 2F, Figure S4C). Similar single cell RNA-seq analyses were performed on the kidney cancer, ovarian cancer and CRC pairs (Figure S4D–S4H). The presence of major immune cell populations in CRC MOS was confirmed by flow cytometry analysis (Figure S5A). Pseudo-bulk analysis showed comparable overall gene expression levels in each of these cell populations between primary tissue and MOS (Figure 2G), with relatively few differentially expressed genes (Figure 3A, S5B and S5C). Analysis of each cell type in lung tumor pairs revealed that lymphoid cells had more differentially expressed genes than the other cell types (Figure 3B).
Figure 3.
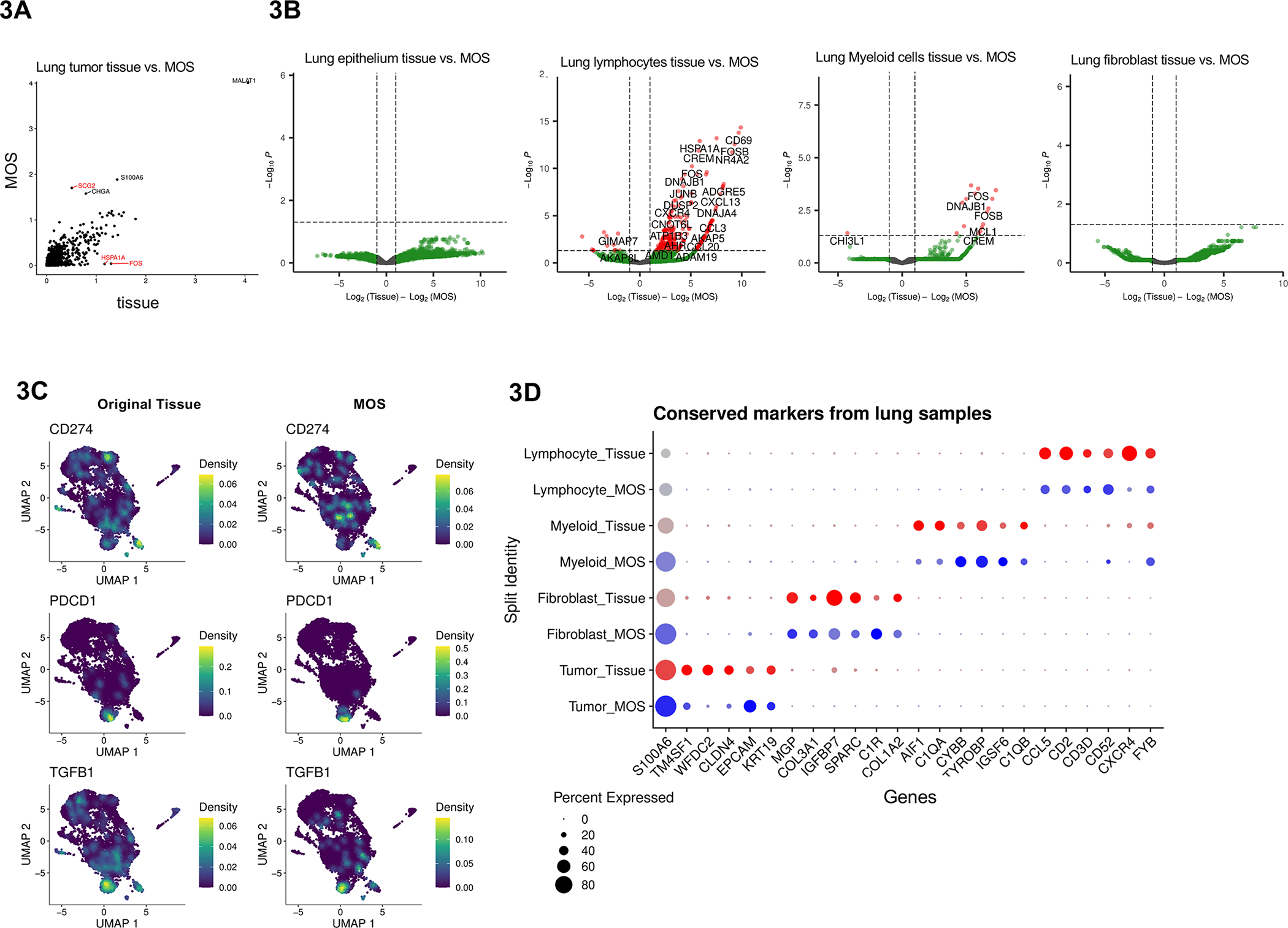
Differential gene analysis on lung tumor tissue vs. derived MOS. (A) Pseudo-bulk differential expression analysis comparing lung cancer primary tissue and derived MOS datasets (n=3). Genes with absolute log-fold change > 1.5 are labeled in red. (B) Volcano plots of differentially expressed genes from fibroblasts, lymphoid cells, myeloid cells, and tumor cells from lung tumor samples. (C) Expression of cancer-associated marker genes CD274 (PD-L1), PDCD1 (PD-1), and TGFB1 (TGF-beta) plotted as UMAPs. Cells are plotted on separate UMAPs depending on source: primary tissue (left) or MOS (right). (D) Top five identified conserved markers for each cell type and labeled by cell source.
Additionally, expression patterns of immunosuppressive markers were largely consistent between lung tumor tissue and MOS. Cell type-specific gene marker expression was visualized using UMAPs following automated cell type labeling with the SingleR package. CD274 (PD-L1) was primarily expressed in tumor and myeloid cell clusters, while PDCD1 (PD-1) and TGFB1 (TGF-β) had elevated expression in lymphoid cells (Figure 3C). The top five genes with the highest log-fold change enrichment in each cell type were visualized to confirm concordant expression for each cell type and sample preparation (Figure 3D). These conserved markers, including key cell type-specific markers EPCAM, PDGFRA, LYZ, and CD3E for tumor cells, fibroblasts, myeloid cells, and lymphoid cells, respectively, were largely consistent between tissue and derived MOS (Figure S5D).
Immune Cells Preserved in MOS Respond to Immune Therapy
We then examined whether the patient immune cells in MOS are functional and responsive to IO therapies. Addition of anti-CD3 and anti-CD28 antibodies to MOS medium increased CD4+, CD8+, and CD56+ (Natural Killer cell marker) populations and had modest effect on the CD11b+ (dendritic cell marker) populations (Figure S6A), suggesting that resident immune cells encapsulated in MOS are viable and responsive to immune stimulation.
Immune checkpoint inhibitors, specifically those targeting the programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) axis, have demonstrated promising activity in non-small cell lung cancer (NSCLC) (Han et al., 2020). However, there remains a crucial need for an in vitro assay to better guide IO treatment for patients with advanced NSCLC, as PD-L1 expression and tumor mutation burden do not completely predict patient response. MOS generated from NSCLC patient samples, at a density of 30 tumor cells per MOS, formed tumorsphere-like structures in the presence of tumor-resident immune cells. We then treated MOS (day 4) with anti-PD1 therapy nivolumab at 10 ug/ml and used Annexin V to evaluate cell apoptosis. Nivolumab induced death in the tumorspheres within MOS (Figure 4A, 4B, Movie S2). The Incucyte measurements also contained background signals outside tumorspheres from cell debris in the MOS microenvironment, giving rise to the rising curves in the control. In MOS (day 3) derived from a kidney cancer patient, nivolumab treatment alone did not enhance killing of tumorspheres inside MOS while a combination of nivolumab and T cell activator enhanced tumorsphere killing (Figure S6B–S6D).
Figure 4.
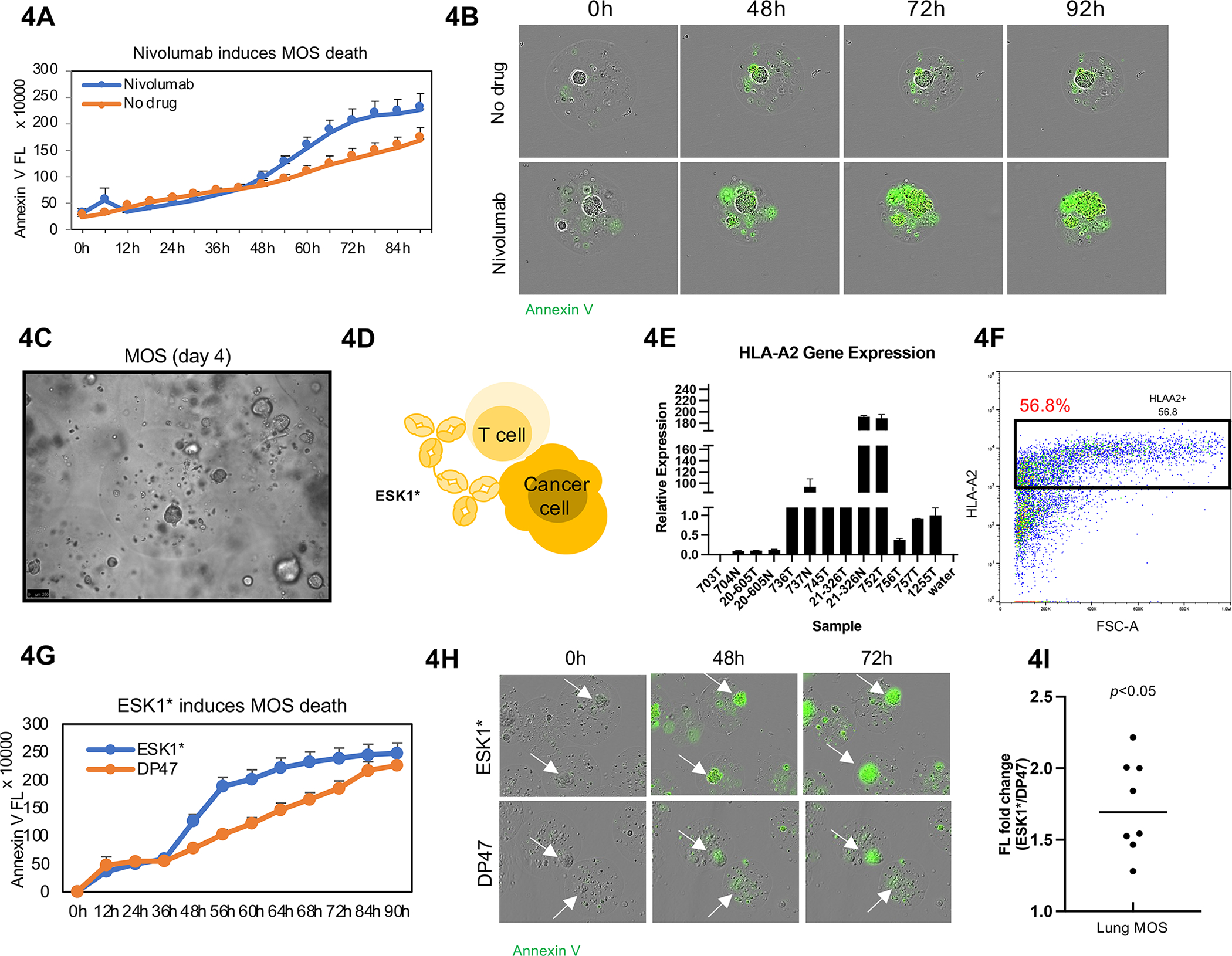
Immune cells preserved in MOS are responsive to immunotherapy. (A) Nivolumab induced significant cytotoxicity in tumorspheres within MOS. Incucyte images were taken every 2 hours for 4 days, and Annexin V Green dye was added to indicate apoptosis. (B) Representative images from Incucyte demonstrated Nivolumab induces cell apoptosis within MOS. (C) Established MOS (day 4) derived from lung tumor tissue. (D) Animation of how ESK1* TCB drug induces CTL-mediated killing in MOS. (E) HLA-A2 gene expression in lung tumor tissues. (F) HLA-A2 expression detected by flow cytometry in established MOS derived from lung tumor tissue. (G) ESK1* induced higher apoptosis signal (indicated by Annexin V signal) in MOS. (H) Representative images of apoptosis induced by ESK1* treatment. (I) ESK1* induced killing of lung cancer MOS in all eight lung cases (p<0.005).
Intracellular antigens presented on the cell surface by human leukocyte antigen (HLA) molecules have been targeted by T cell-based therapies. We tested whether a non-selective HLA-A*02/WT1 targeting antibody ESK1* (ESK-1 tumor binder, Roche proprietary CD3), a T-cell receptor mimic monoclonal antibody (mAb) that binds both human leukocyte antigen HLA-A2/WT1 and CD3, can induce cytotoxic T cell (CTL)-mediated killing in MOS derived from patient lung tumor (Figure 4C and 4D) (Dao et al., 2015). HLA-A2 genotype was validated by qRT-PCR (Figure 4E) and flow cytometry (Figure 4F). We compared ESK1* to negative control DP47, a non-tumor targeted T-Cell bispecific (CD3 arm only) antibody (TCB). ESK1* or DP47 was added into the MOS culture medium (without Y compound) on day 5. ESK1* induced apoptosis (indicated by Annexin V signal) in MOS (Figure 4G, Movie S3). Although DP47 was also capable of activating T cells via CD3 and causing cell death, ESK1* induced more killing in all eight lung cancer patient cases (Figure 4H and 4I).
We then treated CRC MOS (HLAA2+) with ESK1*. MOS was generated at density of 30 tumor cells per MOS. Higher dose of ESK1* (10 ug/ml) induced more tumorsphere death in MOS than lower dose of ESK1* (1 ug/ml) (Figure S6E and S6F). Quantification of Annexin V fluorescence signals from individual tumorspheres confirmed ESK1-mediated killing in MOS but not in organoids embedded in traditional Matrigel dome (Figure S6G–S6I).
To further understand how MOS respond to ESK1*, we performed 10X single cell RNA-seq on original lung tumor tissue cells at day 0, and MOS treated with ESK1*, negative TCB (DP47), as well as no added treatment at day 5 (Figure S6J). The clusters profiled from tissue sample and treated MOS were consistent (Figure S6K and S6L). The abundance of each cell type from MOS with DP47 or without treatment were similar to original tissue cells but decreased drastically in ESK1*-treated MOS (Figure S6M). Collectively, these results suggest that the MOS assay can rapidly assess the impact of IO therapy molecules such as PD-1 blockade and TCB on patient tumor and stromal cells.
A MOS Potency Assay for T-Cell Therapies
Adoptive T cell therapies (ACT), such as chimeric antigen receptor T-cell (CAR-T) therapy and tumor-infiltrating lymphocyte (TIL) therapy, have the potential to transform cancer treatment (June et al., 2018; Waldman et al., 2020). However, an area of unmet need is an assay to assess the potency of manufactured T cells against a patient’s tumor, which is required by regulatory authorities like the FDA for approving cell therapies (HHS and FDA, 2011). Interferon gamma release has been used to evaluate TILs against patient tumors, but at least four studies have shown that it does not correlate with clinical response (Besser et al., 2009; Dudley et al., 2010; Nguyen et al., 2019; Radvanyi et al., 2012). For ACT with TILs, the patient tumor model has to be established rapidly from a fraction of the biopsy (as the majority has to be used to extract and expand TILs), making it particularly challenging.
To explore whether MOS can be used as a potential potency assay, we first compared penetration of autologous patient-derived tumor infiltrating lymphocytes (TILs) into bulk Matrigel vs. MOS (20 cells per MOS). Most T cells stayed on the periphery of the bulk Matrigel gel; conversely, T cells readily infiltrated into MOS (due to its smaller size and larger surface-to-volume ratio) and adhered to tumor cells (Figure 5A and 5B). We also compared peripheral blood mononuclear cell (PBMC) penetration into Matrigel vs. MOS (20 cells per MOS) using the Incucyte live imaging system and confirmed that immune cells can easily infiltrate MOS (Figure S7A). We then used time-lapse fluorescence imaging to measure immune cytotoxicity against target tumor cells with TILs and PBMCs. For the TIL potency assay, we simultaneously grew MOS, generated at density of 30 tumor cells per MOS, and TILs from the same lung tumor tissue. Increased killing (indicated by Annexin V) was observed in MOS treated with autologous TILs (Figure 5C and 5D). This assay confirmed the potency of rapid expansion protocol (REP) TILs against matched lung tumor MOS, thus providing promising preliminary data as a TIL potency assay.
Figure 5.
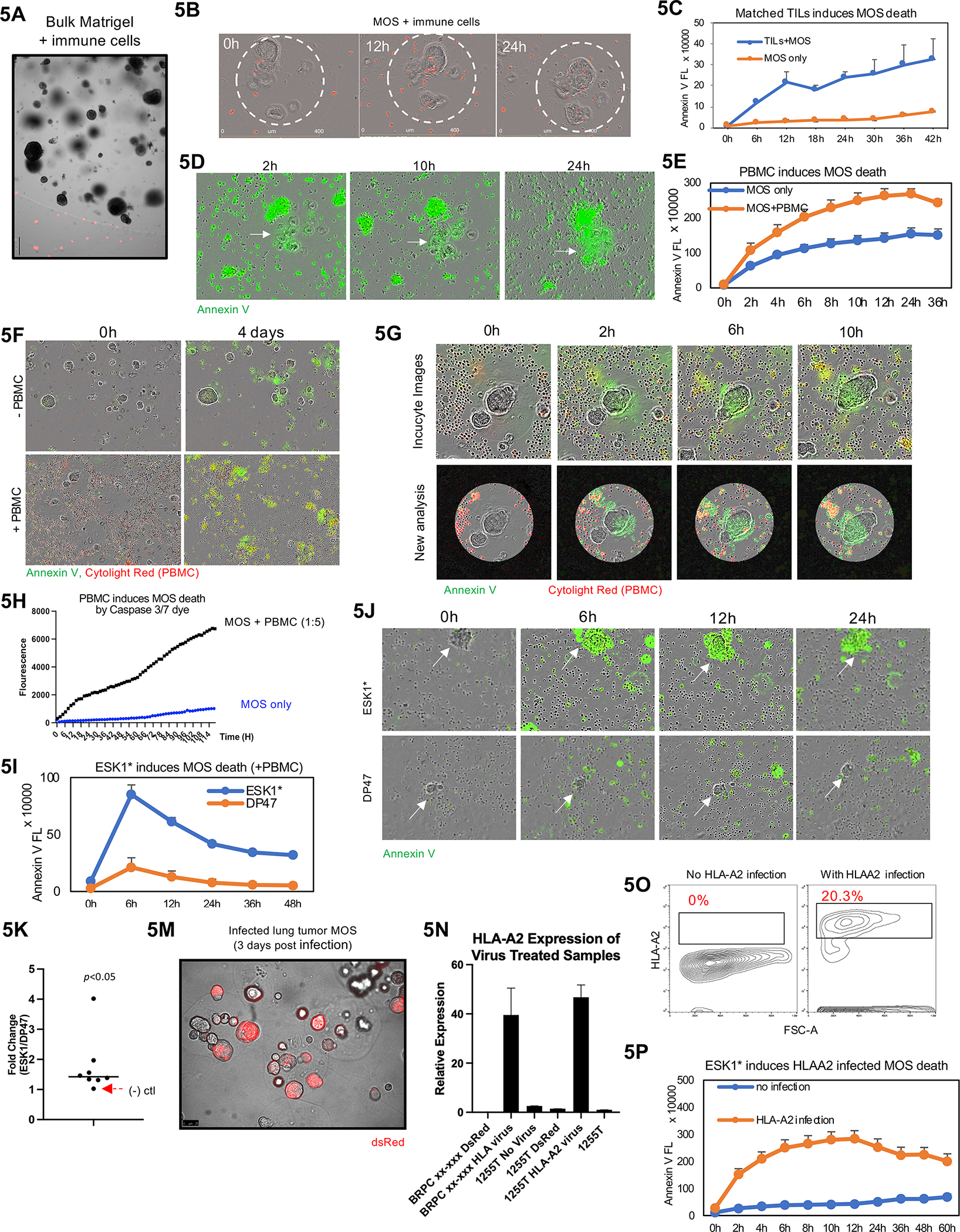
A MOS potency assay for T-Cell therapies. (A) TILs cannot penetrate traditional Matrigel. (B) TILs can penetrate MOS and adhere to tumor cells. Immune cells were stained with Cytolight Red dye before images taken using Incucyte. (C) Increased killing indicated by Annexin V was observed in MOS treated with autologous TILs. (D) Representative images of MOS killing by TILs in MOS (indicated by Annexin V dye). (E) Activated PBMCs induce MOS killing (indicated by Annexin V Green dye). (F) Representative images of MOS killing by PBMCs. (G) Representative images to illustrate an imaging analysis pipeline that identifies droplet area to minimize background noise from outside immune cells. (H) Quantification analysis suggest PBMCs induce MOS killing (indicated by Caspase 3/7 dye). (I) ESK1* enhanced PBMC-induced tumor cell killing compared to the DP47 (CD3 only TCB). (J) Representative images of induced death of ESK1* treated MOS combined with PBMCs. White arrows indicating lung cancer tumorspheres within MOS. Compared to ESK1*, the negative control TCB, DP47, did not induce significant apoptosis of tumorspheres within MOS. (K) Dotted plot suggests ESK1* induces PBMC-mediated lung tumor MOS death in seven patient cases (p<0.005). (M) Image of dsRed expressing vector infection on MOS derived from lung tumor (3 days post infection). Significant higher gene expression of HLA-A2 (N) and antigen expression (O) were observed in HLA-A2-infected MOS. (P) HLA-A2-infected MOS underwent higher cell death than matched uninfected MOS in the presence of ESK1* and activated PBMCs (as indicated by Annexin V dye).
We then assessed the potency of PBMCs against lung tumor MOS to prove the concept of using MOS as in vitro platform for cell therapy. We derived MOS from lung cancer patients and added allogeneic PBMCs from a different normal patient. Tumorspheres within MOS remained viable, appearing orange as labeled by Cytolight Rapid Red, after 96 hours of co-culture with PBMCs; however, when PBMCs were activated by anti-CD3 and anti-CD28 antibodies, the tumorspheres had increased cell death as shown by Annexin V staining (Figure S7B).
We characterized the response of lung cancer MOS (20 cells per MOS) to activated PBMC using Annexin V (early-stage cell surface apoptosis), Caspase 3/7 (enzyme-mediated cell apoptosis), and Cytotox (cell membrane integrity). PBMCs were pre-stained with live cell marker Cytolight Red dye. Both Annexin V and Caspase 3/7 can detect MOS apoptosis, while Caspase 3/7 had higher specificity (Figure 5E, 5F, S7C, S7D, Movie S4). An imaging analysis pipeline was developed to identify MOS area to mask out background noise from outside immune cells (Figure 5G), which confirmed PBMC-induced MOS apoptosis with less background signal from outside the MOS (Figure 5H). PBMCs also induced tumorsphere death in CRC MOS (20 cells per MOS), which was enhanced by cytokine activation (Figure S7E and S7F), and in kidney cancer MOS (20 cells per MOS), which was enhanced by higher effector:target cell ratio (Figure S7G and S7H).
We explored adjunctive therapies by first combining PD-1 blockade (nivolumab) with autologous TILs against matched lung tumor MOS. PD-1 blockade enhanced TIL-mediated killing inside MOS, which was abrogated by blocking MHC (Figure S7I). We then combined TCB with autologous TILs or allogeneic PBMCs to treat lung cancer MOS (20 cells per MOS) expressing HLA-A2. ESK1* enhanced both TIL- and PBMC-induced tumor cell death compared to DP47 (Figure 5I, 5J, S7J, S7K). Annexin V signals were higher in MOS treated with ESK1* vs. DP47 in all seven lung cancer samples (Figure 5K). As a negative control, ESK1* did not enhance killing of HLA-A2(−) lung cancer MOS, as indicated by the red arrow. Heterogeneity in drug response between MOS from the same patient was observed and quantified (Figure S7K and S7L).
Conventional bulk organoids require single cell dissociation for viral gene delivery before re-embedding into Matrigel. Due to their small size and large surface-to-volume ratio, MOS can be infected by directly adding lentiviruses into culture medium without dissociation. This provides a convenient way to edit MOS at passage 0. Lung cancer MOS (20 cells per MOS) from an HLA-A2(−) patient was infected with a lentiviral HLA-A2 expression vector for 3 days with dsRed as a control (Figure 5M and S7M). The infected MOS showed high expression of HLA-A2 (Figure 5N and 5O). We then performed combinatorial treatments with ESK1* and activated PBMCs on HLA-A2-infected MOS. HLA-A2-infected MOS underwent more cell death than matched uninfected MOS in the presence of ESK1* and activated PBMCs, thus validating that HLA-A2 expression level mediates the efficacy of ESK1*+PBMC treatment (Figure 5P, Movie S5).
Discussion:
Over the past two decades, genomic profiling has been the primary precision medicine strategy to guide patient care, but recent estimates suggest that <10% of cancer patients benefit from this approach (Marquart et al., 2018; Prasad, 2016). Issues using genomic profiling include the lack of actionable genomic alterations in a vast majority of cases (Meric-Bernstam et al., 2015), and even when they are present, a significant proportion of patients do not respond to indicated targeted therapy (Schwaederle et al., 2016). As we learn more about the staggering complexity of tumor biology, it has become increasingly clear that understanding factors such as oncogenic alterations (Blakely et al., 2017), tumor heterogeneity (de Bruin et al., 2014), epistatic interactions (Sulkowski et al., 2017), and adaptive cellular circuitry (Chandarlapaty et al., 2011; Manchado et al., 2016; Poulikakos et al., 2010) are needed to match the right therapy with the right patients. As the number of validated drug targets and FDA-approved cancer therapies against those targets continues to grow rapidly, the success and evolution of our fundamental approach to precision medicine have lagged.
The development of a PDO precision oncology platform to identify the most effective therapy for each patient could deliver on the promise of personalized medicine in oncology. The ability to identify non-responders would spare patients from unnecessary side effects of an ineffective treatment, and PDO from non-responders could then be funneled through a drug screening workflow to identify new potential treatment options. Indeed, functional assays have already shown that cancer cells harbor unpredictable vulnerabilities in the form of synthetic lethal interactions (Huang et al., 2020) and collateral sensitivities (Wang and Bernards, 2018), such as the homologous recombination defect seen in IDH1/2-mutant cancers that renders unexpected PARP inhibitor sensitivity (Sulkowski et al., 2017), and PDO drug screening could detect these treatment opportunities.
The main barrier preventing clinical implementation of PDO for precision oncology is the current methodology for generation. Despite their favorable comparison to PDXs, the process of deriving and expanding sufficient numbers of PDO for drug screening nevertheless remains too slow, too complicated, and too costly for wide adoption into the clinic (2018; van de Wetering et al., 2015b). Since clinical treatment decisions are often made within 14 days, the current timeframe for PDO generation would result in unacceptable treatment delays. Thus, to create a functional precision medicine platform that can reliably deliver results, new approaches are needed for establishing PDO more quickly and efficiently.
To circumvent these issues, we have developed patient-derived MOS, which utilize microfluidic-based technology to establish a model that is clinically relevant because it recapitulates the tumor microenvironment, can be made from minimal patient tissue, which is particularly relevant for biopsy samples which have few cells to begin with, and can reliably provide results within 7–14 days of obtaining a biopsy. Our prospective clinical study showed that MOS drug response correlated with sensitivity to standard-of-care first-line treatment in CRC. Further larger scale study is required for validation and, ideally, development of a streamlined process that can guide patients to effective treatments.
PDX and PDO typically do not recapitulate the original patient tumor stromal cells, especially the immune cells. In humanized PDX, the reconstituted immune cells often do not match the HLA of the patient tumor unless deliberate effort is made to find HLA-matched cord blood containing hematopoietic stem cells (HSC). Immune cells are usually lost when PDX are passaged repeatedly to expand the number of cells for functional testing. By performing the clinical assay quickly, MOS were able to retain most of the original tumor-resident immune cells and the immunosuppressive microenvironment; furthermore, we demonstrated that immunotherapies such as PD-1 blockade and bispecific antibodies can activate TILs to attack tumor cells within MOS.
The lack of an effective potency assay to evaluate whether expanded patient-derived TILs or engineered T cells (such as CAR-T) are still effective against targeted patient tumors has hindered the development of new immune cell therapies such as adoptive TIL therapy. Although a potency assay is required by the FDA for approval of cellular therapy products, recent studies have shown that interferon release assays do not correlate with clinical response (Besser et al., 2009; Dudley et al., 2010; Nguyen et al., 2019; Radvanyi et al., 2012). For example, in one protocol using unselected TIL, four out of thirteen patients with a treatment response had a negative interferon release assay, and there were nine nonresponding patients who had a positive interferon release assay (Dudley et al., 2010). However, to our knowledge, no ACT trials have managed to specifically address potency assays outside of IFN release. The small dimensions and high surface-to-volume ratio of MOS make them amenable to penetration by patient-derived immune cells and allow for high content imaging. Furthermore, the ability to generate thousands of MOS from a small fraction of tissue biopsy is critical for ACT, as most of the limited tissue must be used for TIL extraction and expansion. These unique features enable MOS to provide a clinically feasible T cell potency assay against the targeted patient tumors for such cell therapies.
Limitations of the study: In the CRC clinical study as only 8 patients were enrolled in this initial study, this proof of concept pilot study only provides initial data that MOS can potentially be used as a clinical tool. In order to further validate the clinical value of MOS in terms of reproducibility, sensitivity and specificity of drug response, MOS must next be validated in clinical trials to determine applicability. We are currently expanding our pilot trial of 8 patients to a trial of 250 patients to move from anecdotal case studies to a platform that will focus on reproducibility of results from individual patients, generating drug concentration curves for each patient to develop chemotherapy classifiers and further develop our device so that it can be generally used for this approach. This will then allow us to design trials in colorectal cancer (NCT05189171, MicroOrganoSphere Drug Screen Pilot Trial in Colorectal Cancer) where the primary objective is to determine the feasibility of generating sufficient MOS from a biopsy of a patient’s colorectal cancer liver metastasis to determine sensitivity to standard of care drug used in the treatment of colorectal cancer (oxaliplatin, irinotecan, 5-FU/Xeloda, Bevacizumab, Panitumumab or Cetuximab, Lonsurf, Regorafenib and Pembrolizumab or Nivolumab) in < 14 days and the secondary objective of this study is to assess the association between standard of care drug sensitivity in MOS to clinical outcome of patient treated with standard of care therapy from which the MOS was derived. Such trial design does not pose a significant risk to patients as only standard-of-care options are considered and more importantly, clinically meaningful improvement in overall survival, progression-free survival and response rate will be obtained to demonstrated that MOS are capable of correlating outcome and guiding precision oncology.
STAR Methods
RESCOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiling Shen (xiling.shen@terasaki.org)
Materials availability:
This study did not generate new unique reagents.
Data and code availability:
Single-cell RNA-seq data and DNA WES data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients’ age and gender of the subjects are provided in Table 1 and Supplementary Table 2. Biopsies were obtained through two Duke IRB approved protocols (Pro00089222 and Pro00100141 from patient with CRC liver metastasis. Patient enrolled under Pro00089222 received a standard of care biopsy and an additional research biopsy was obtained to generate MOS for further studies. Pro00100141 (A Pilot Study of a Precision Medicine Strategy for Patients with Colorectal Cancer Liver Metastasis, PI Jingquan Jia) was subsequently designed to obtain research biopsies from patients presenting with colorectal cancer liver metastasis to specifically generate MOS and perform drug screen. The primary objective of this study is to determine the feasibility of generating sufficient MOS from a biopsy of a patient’s colorectal cancer liver metastasis to determine sensitivity to oxaliplatin and irinotecan and the secondary objective of this study is to assess the association between oxaliplatin or irinotecan sensitivity in MOS to clinical outcome of patient treated with oxaliplatin or irinotecan-based therapy from which the MOS was derived from. Informed consent was obtained from all subjects.
METHODS DETAILS
Microfluidic chip fabrication and design:
Microfluidic chips were fabricated out of silicon wafers (Wafer Pro, Santa Clara, CA). Details of manufacturing microfluidic features in silicon can be found in published handbook (Rius G., et al, 2017). Briefly, designs were imprinted onto a 6” silicon wafer using standard photolithography techniques and features were etched using Deep Reactive Ion Etching (DRIE) in a clean room facility. Once cleaned, a borofloat glass cover slide (PG&O, Santa Ana, CA) was bonded to the silicon chip using anodic bonding. After bonding, the microfluidic channels were coated with Aquapel (Aquapel Glass, Cranberry Twp, PA) to create a hydrophobic surface. Following coating, channels were rinsed with 3 mL of Novec 7500 engineered fluid (3M, Saint Paul, MN) and then baked at 60°C for 20 min.
MOS generator assembly:
MOS generation took place inside a 1.7 cu. ft. miniature refrigerator to keep the temperature-sensitive gel from polymerizing during generation. Fluigent FlowEZ (Fluigent, La Kremlin-Bicetre, France) pressure sources were attached to the top of the refrigerator. Air tubing was connected to the reagent and sample reservoir PCaps (Fluigent, La Kremlin-Bicetre, France) through the top of the refrigerator via two drilled holes. Pumps were operated manually according to manufacturer’s recommendations. Chips were assembled inside a custom fabricated manifold that contained ports to connect the reagent and sample reservoirs to the chip. All components were placed inside the refrigerator. The door is kept closed when processing temperature sensitive material. MOS generation can be imaged by assembling the camera and lens components found in Supplementary Table 1 and placing the camera directly over the chip.
Patient specimens:
Tissue sections (approximately 1–2 cm3) of metastatic colorectal cancer, lung cancer, ovarian cancer, kidney cancer, breast cancer, and non-tumor tissue were obtained from surgically resected specimens provided by Duke BioRepository & Precision Pathology Center (BRPC) with the patients’ consent. The entire experimental protocol was conducted in compliance with institutional guidelines. Samples were confirmed as tumor or normal tissue via histopathological assessment.
Tumor tissue processing and MOS generation:
All tumor and non-tumor tissues were kept in transfer media and on ice after dissection. Ten percent of the tissue sample was frozen down in OCT immediately, and the remainder was minced before mixing with 10 ml of enzymatic solution. The enzymatic solution consisted of a collagenase-based digestion solution containing CaCl2 (3 mM), Collagenase (1 mg/ml) (Sigma Cat# 11088858001), DNase I (0.1 mg/ml) (STEMCell technology, Cat# 07900), Y-27632 (10 μM) (STEMCell technology Cat# 72302), and Primocin (100 ug/ml) (Fisher Scientific, Cat# NC9141851). Minced tissue samples were dissociated with gentle agitation in enzymatic solution for 30 minutes at 37°C before first cell quality check. If large cell clumps were observed, an additional 15–20 minutes of digestion was performed until the tissue was mostly dissociated into single cells. After digestion, cells were filtered through a 70 μM cell strainer, and yield and cell viability were determined by a Countess II cell counter using a previously described Trypan blue method. The initial cell number inserted into MOS depends on the intended application. Single cell per MOS is used for clonal diversity studies. 20 tumor cells per MOS is often used for testing chemotherapy, providing the best tradeoff between tumorsphere establishment speed and the number of MOS for testing different conditions. 30–50 tumor cells (and proportional number of immune cells from the same digested sample) per MOS is more appropriate for IO assays. The same cell density was seeded using traditional Matrigel methods as a comparison. Demulsified MOS were layered with tumor medium and seeded in 6-well low binding plates. Cells in Matrigel were loaded in 24-well plates and grown in the tumor medium. Medium was changed every 3 days.
H&E staining of original tumors and MOS:
Tissues and MOS were processed for paraffin sectioning as following described. Matrigel-embedded MOS were collected after spinning in a 15 mL tube at 100 g for 3 minutes. The supernatant was removed, and MOS were fixed in 2% paraformaldehyde (PFA) with 0.1% Glutaldehyde for 30 min at room temperature before washing in 1X PBS and embedding in Histogel. Fresh cancer tissue was embedded in paraffin after formalin fixation. After deparaffinization, 5-μm sections were stained with hematoxylin–eosin (H&E). MOS and primary tumor sections were evaluated for morphological characterization by a pathologist.
MOS and organoid imaging:
Images of MOS and organoids in bulk Matrigel were acquired using a Leica microscope (Leica, USA) at day 1, day 3, day 5, and day 7 after initial plating, and organoid surface area was quantified using ImageJ software (Wayne Rasband, NIHR, USA, http://imagej.nih.gov/ij). To calculate the average size (area) of the organoids, more than 40 tumorspheres in MOS or organoids in Matrigel for each tumor sample were manually quantified, and statistical analysis was performed using Prism 8.
Genomic and transcriptomic analysis:
DNA extraction and WES sequencing:
MOS developed on day 7 were harvested for DNA extraction. DNA was extracted using a Zymo Quick-DNA Microprep kit (Zymo Research #D2030) according to the manufacturer’s protocol. DNA was quantified using a NanoDrop. Tumor samples and matched tumor-derived MOS were analyzed using whole-exome sequencing (WES) by Novagene using an Illumina Novaseq 6000 sequencer.
WES mutation analysis on tumors and MOS:
A total of >0.4μg DNA per sample was used as input. The effective sequencing depth was above 50× (6G) per sample. Prior to alignment, adaptors were trimmed from raw sequencing data using TrimGalore. The resulting fastq files were then aligned to the human reference genome (hg38) using BWA. Duplicate BAM files from matched samples were then merged and filtered to remove duplicates and non-chromosomal reads. Sequence variants were then called using GATK’s HaplotypeCaller pipeline (version 4.2.0). Variants were then filtered based on quality by depth (QD < 2.0), mapping quality (MQ < 40.0), Fisher strand (FS > 60.0), strand odds ratio (SOR > 4.0), mapping quality rank sum (MQRankSum < −12.5), and read position rank sum (ReadPosRankSum < −8.0). Finally, variants were annotated using snpEff.
Disruptive variants (e.g., missense, stop-gained, disruptive inframe indels, 3/5’ UTR, splice acceptors, and splice donor variants) were selected for downstream analysis. Each unique mutation (classified as a specific position-base-alternate combination) was binarized for each sample according to presence-absence. The resulting binary vectors were used to calculate Jaccard similarity scores and generate Venn diagrams and the presence-absence table. Genes represented in the presence-absence table in Figure D were limited to the 25 most commonly mutated genes by cancer type according to The Cancer Genome Atlas (TCGA).
Drop-seq single cell assay and analysis:
Frozen PBMCs were thawed, and count and cell viability were measured by Countess II. For single cell RNA-seq, 200K cells were aliquoted, spun down, resuspended in 30 μl PBS+0.04%BSA+0.2U/ul RNase inhibitor, and counted using Countess II.
The scRNA Drop-seq libraries were generated using a Dolomite Nadia machine following the manufacturer’s protocol. Libraries were then pooled and sequenced using Illumina NovaSeq platform with the goal of reaching saturation or 20,000 unique reads per cell on average. Sequencing data were used as inputs to the Drop-seq pipeline published by the Broad Institute (https://github.com/broadinstitute/Drop-seq). Gene count matrices were produced using the first 4,000 cellular barcodes with the largest number of reads associated with each index.
10x Next GEM 3’ scRNA assay and analysis:
For single cell RNA-seq on ESK1* drug treated MOS and original tissue tumor cells, we first generated MOS from lung tumor tissue (case #805), ESK1* drug (1μg/ml) was added into culture medium on day5 MOS for 24 hours before cells were collected to perform scRNA seq library preparation. During library preparation, 200K cells were aliquoted, spun down, resuspended in 30 ul PBS+0.04%BSA+0.2U/μl RNase inhibitor, and counted using Countess II. GEM generation, post GEMRT cleanup, cDNA amplification, and library construction were performed following 10X Genomics Single Cell 3’ v3.1 chemistry. Quality was assessed using Agilent DNA tape screen assay. Libraries were then pooled and sequenced using Illumina NovaSeq platform with the goal of reaching saturation or 20,000 unique reads per cell on average. Sequencing data were used as inputs to the 10x Genomics Cell Ranger pipeline to demultiplex BCL files, generate FASTQs, and generate feature counts for each library.
Cell type annotation and clustering:
Gene-barcode matrices generated using the DigitalExpression script from the Broad Drop-seq pipeline were analyzed using Seurat 3 with the default parameters unless otherwise specified. Cells with > 2,500 genes detected were removed from the analysis. Counts were log-normalized, and the top 2,000 variable features were identified. Principal component analysis was performed using these variable genes, and the top 30 principal components were used for downstream analysis. UMAP dimensionality reduction was performed using the top 20 principal components identified using the Harmony package. Graph-based clustering was performed with resolution = 1. Cell types were inferred by using the HumanPrimaryCellAtlasData() function from the SingleR package. Labels were confirmed by identification of differentially expressed genes using the FindAllMarkers() function from Seurat and visualization of marker genes plotted as kernel density on UMAPs using the Nebulosa package. To perform differential expression analysis, cell type labels were grouped into four groups: tumor cells, fibroblasts, lymphoid cells, and myeloid cells.
Pseudo-bulk Differential Expression Analysis:
Three biological replicates from patients with lung cancer were used for pseudo-bulk differential expression analysis. Specifically, datasets generated from primary tissue were compared with datasets generated from MOS to determine changes in gene expression between the two platforms. Gene count values from cells with the same cell type label were aggregated into a single matrix. The model design formula included a term indicating which samples were produced from primary tissue or MOS. Significance testing was performed using the glmQLFit() function from the EdgeR package, and false discovery rate adjustment was performed for the p-values. Genes with an absolute log-fold change > 1 and adjusted p-value < 0.05 were considered significantly differentially expressed between the two conditions. Intersections in the differentially expressed gene list for each cell type were visualized as an UpSet plot using the UpSetR package. A volcano plot was generated for the pseudo-bulk results from each of the cell types using the EnhancedVolcano package and the same significance thresholds.
The remaining two samples were collected from patients with either kidney cancer (n=1) or ovarian cancer (n=1). For these samples, log-fold change in gene expression was compared for libraries produced from primary tissue or MOS, and no p-values were reported. Gene counts were averaged for each cell type using the AverageExpression() function from Seurat, and log-transformed values were plotted to compare the samples produced from primary tissue and MOS. Genes with a log-fold change > 1 were labeled in red, and genes with average log expression > 1.5 labeled in black to provide context for highly expressed genes in each cell type.
Identification of Conserved Gene Expression:
The FindConservedMarkers() function from Seurat was applied to cells of each cell type, and genes with conserved expression and log-fold change enrichment > 0.5 were identified. The top five markers with the highest log-fold change enrichment for each cell type were visualized using the DotPlot() function from Seurat. Important cancer gene expression markers CD274 (PD-L1), PDCD1 (PD-1), and TGFB1 (TGF-beta) were also specifically visualized to compare expression in the tumor cells, lymphoid cells, and fibroblasts, respectively. Expression of these markers was plotted as UMAPs using the Nebulosa package and labeled on the respective volcano plots.
Flow cytometry analysis:
MOS and bulk Matrigel established by Day 7–9 were dissociated into single cells using TrypLE treatment and incubated in 37 °C for 5 minutes. Dissociated cells were washed with PBS + 0.04% BSA and stained with either anti-human Vimentin antibody (CST Cat#, 1:100) combined with PE or anti-human EpCAM (Biolegend Cat#324205, 1:250), at room temperature for 20 minutes. Cells were washed once again with PBS + 0.04% BSA before staining with goat anti-mouse Alexa Fluro 488 secondary antibody (Invitrogen Cat# A32723) for 15 minutes at room temperature. Cells were washed once more with PBS+ 0.04% BSA before flow assay. Sytox blue dead cell stain (A34857) was added as 1:1000 dilution to gate out dead cells in the assays. All flow assays were performed using a Sony SH800 FACS sorter, and flow data were analyzed using FlowJo.
Drug High-Throughput Screening:
Automated liquid handling was provided by the Echo Acoustic Dispenser (Labcyte) for drug administration or Well mate (Thermo Fisher) for cell plating, and assays were performed using a Clarioscan plate reader (BMG Labtech). Immediately prior to cell plating, 384 well plates were stamped with 119 FDA-approved drug compounds at a final concentration of 1 μM. The compound library (Approved Oncology Set VI) was provided by the NCI Developmental Therapeutics Program (https://dtp.cancer.gov/). MOS were plated in these drug pre-coated plates at 100 MOS/well with each MOS containing 30 cells/droplet. Cell viabilities were assessed via CellTiter-Glo Luminescent Cell Viability Assay (Promega, USA) 72 hours after cell plating. Percent cytotoxicity was quantified using the following formula: 100*[1-(average CellTiterGlodrug/average CellTiterGlocontrol)].
HLAA2 lentivirus packaging and MOS infection:
The HLA-A2 (insert) was amplified from cDNA library prepared with RNA from NCI-H1755(ATTC, CRL-5892) using the flowing set of primers: sense primer: GGTCGCCACCATGGCCGTCATGGCTCCCCG; antisense primer: GGCCGCTTTACACTTTACAAGCTGTGAGAG. The linearized plasmid (recipient) was amplified from pLenti CMV GFP Puro plasmid (Addgene: 17748) using the following set of primer: sense primer: TTGTAAAGTGTAAAGCGGCCGCGTCGACAA; antisense primer: TGACGGCCATGGTGGCGACCGGTGGATCCT. The PCR products (both insert and vector) were purified using Gel DNA Recovery Kits (Zymo, D4007). The insert was then cloned into the vector by Gibson assembly (NEB, E2611S). Lentiviral particles were produced by co-transfection of HEK 293T cells using Lipofectamine 2000 transfection. Briefly, HEK293T cells were co-transfected with 10 μg of transgene plasmid, 10 μg of packaging plasmid pCMVR8.74 (Addgene: 22036) and 5 μg envelope plasmid pMD2.G(Addgene: 12259). After 12h, the transfection medium was changed, and recombinant lentiviruses were harvested at 24 and 48 hours. The supernatant containing the viral particles was then concentrated using the Lenti-X Concentrator kit (Takara, 631232). Concentrated lentiviral particles were then aliquoted and stored at −80 °C until needed. MOS infection with HLA-A2 expressing lentivirus: HLA-A2- and DsRed-expressing lentivirus was added into lung tumor MOS culture (MOI=5–6) when MOS was established. After 3 days of incubation, infection efficiency was evaluated by observing DsRed expression under the microscope. HLA-A2 gene expression and HLA-A2 antigen expression were evaluated using flow cytometry.
RNA extraction and qRT-PCR:
To quantify HLA-A2 gene expression in lung tumor samples, RNA was extracted using a Norgen single cell RNA purification kit (Norgen Biotek Cat# 51800). cDNA reverse transcription was performed using SuperScript IV Vilo MasterMix with ezDNase (Thermo Fisher Cat# 11756050). HLA-A2 gene was amplified using the following primers: Forward primer: TGA AGG CCC ACT CAC AGA CTC. Reverse primer: CCC ACG TCG CAG CCA TAC ATC
Immuno-Oncology potency assay
Human PBMC and patient tumor TILs expansion:
Human PBMC was purchased from STEMCell technology (Cat# 70025.1). Tumor TILs were generated from dissociated tumor tissue cells. Dissociated cells (0.5 × 106) were collected for the purpose of TIL expansion. Cells were resuspended in ImmunoCult™-XF T Cell Expansion Medium supplemented with 6000 IU/mL Recombinant Human IL-2 (Miltenyi Biotec Cat# 130–097-743). TILs were maintained for 1 week before splitting and the medium was changed to one with CD3/CD28/CD2 T cell activator (STEMCell technology, Cat# 10971) for further expansion.
ESK1* drug preparation:
ESK1* TCB and Negative TCB (DP47) were supplied by Roche. Drugs were aliquoted immediately after receiving to avoid multiple freeze-thaw. Drugs were used at 1 ug/ml or 10ug/ml in all potency assays.
IO assay and Incucyte live cell imaging:
MOS generated from primary tumor tissue were plated into 96-well plates with a density of 30–50 MOS per well supplied with culture medium without Y compound. Day 3 or day 4 MOS were treated with ESK1*, DP47 or Nivolumab for at least 3 days and imaged in Incucyte during the treatment. When performing immune cell potency assay, pre-activated PBMCs or matched TILs were stained with Cytolight Rapid red dye following manufacturer instructions. Briefly, Cytolight Rapid Red dye in one vial was diluted with 20 μl DMSO and further diluted 10-fold in PBS. PBMC or TILs were incubated at 37 °C with 5 μl diluted Cytolight Red dye (500X) in PBS for 25 minutes. After one wash with PBS, PBMC or TILs were counted and resuspended into wells containing MOS and culture medium at an effector:target ratio of 5:1 or 10:1. Annexin V green dye, Caspase 3/7 green dye, or Cytotox green dye was added into each well following manufacturer instruction. Plates were loaded into Incucyte S3 and images were taken every 2 hours for 4–5 days.
IO assay with immunotherapy and MHC block:
Lung tumor MOS were incubated with anti-MHC I/II antibodies (W6/32; Tu39, Cat# 361702, Biolegend) at a concentration of 20 ug/ml for 45 minutes at 37°C before seeding into a 96-well plate at a density of 30–50 MOS per well supplied with lung tumor culture medium without Y-27632. Non-MHC-blocked MOS were used as controls. Matched TILs were added to each well at a 5:1 effector:target ratio. Nivolumab was added to wells at a working concentration of 10 ug/ml. The CD2/CD3/CD28 T cell activator reagent was added at a working concentration of 25 ul/ml. Annexin V was added into each well following manufacturer instructions.
Incucyte imaging data analysis:
Raw images from phase wand green and red fluorescence channels were exported, and MOS were manually drawn using the “Labelme” image annotation software. The fluorescent images and labels were then fed into a Python script that binarized the images using a constant threshold, counting all pixels in the red image above the threshold as “red”, all pixels in the green image above the threshold as “green”, and all pixels that were above the threshold in both the red and green images as “yellow”. These pixels were then grouped according to which MOS (if any) they belonged to, and the script then exported a CSV file containing, for each well, for each time, for each MOS labeled in the associated image, the count of red, green, and yellow pixels contained within that MOS at that time.
QUANTIFICATION AND STATISTICAL ANALYSIS
T-tests were performed using Prism 8.0. p<0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE
S1: MOS generation. Related to Figure 1.
S2: Nivolumab induces lung tumor MOS apoptosis. Related to Figure 4. (A) Nivolumab (10ug/ml) treatment. Apoptosis indicated by Annexin V Green. (B) No treatment.
S3: TCB induces lung tumor MOS apoptosis. Related to Figure 4. (A) ESK1* (1ug/ml) treatment (apoptosis indicated by Annexin V Green). (B) DP47 treatment.
S4: Co-culture with activated PBMC (apoptosis indicated by Caspase3/7 Green). Related to Figure 5. (A) Without PBMC. (B) With PBMC (PBMC:target cells= 5:1).
S5: HLAA2 infected lung tumor MOS treated with ESK1*. Related to Figure 5. (A) Uninfected MOS treated with ESK1*. (B) HLA-A2-infected MOS with PBMC (PBMC:target cells= 5:1) treated with ESK1*.
Clinical biopsy derived MOS enable rapid drug testing in 14 days
MOS maintain patient tumor microenvironment
MOS capture patient tumor response to immunotherapy
MOS assay to test T cell potency against patient tumor for adoptive cell therapy
Acknowledgement:
Funding sources: NIH U01 CA217514, U01 CA214300, Duke Woo Center for Big Data and Precision Health
Footnotes
Declaration of interests
X.S., D.H., and H.C are co-founders of Xilis Inc. H.C. is an employee of Roche and on the advisory board of Cell Stem Cell. H.C.’s full disclosure is given at https://www.uu.nl/staff/JCClevers/. S.D. and Z.W. performed the majority of the study at Duke University and now are employees of Xilis Inc. Patents WO2020242594 and US 2021/0285054 are related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (2018). Organoids May Point to Best Therapy. Cancer discovery 8, 524–524. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, Kubi A, Shalmon B, Hardan I, Catane R, et al. (2009). Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother 32, 415–423. [DOI] [PubMed] [Google Scholar]
- Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, McGranahan N, Wilson GA, Birkbak NJ, Olivas VR, et al. (2017). Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 49, 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, Pogrebniak K, Sandoval J, Cassidy JW, and Tufegdzic-Vidakovic A (2016). A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 167, 260–274. e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandrini C, Schutgens F, Oka R, Margaritis T, Candelli T, Mathijsen L, Ammerlaan C, van Ineveld RL, Derakhshan S, and de Haan S (2020). An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nature communications 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, and Rosen N (2011). AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, Xiang J, Yan S, de Morais Guerreiro MD, Veomett N, et al. (2015). Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nature biotechnology 33, 1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, et al. (2014). Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, and Kelderman S (2018). Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598. e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E, Spelier S, Beltran Hernandez I, de Bree R, S MW, Clevers H, and Oliveira S (2019a). Patient-Derived Head and Neck Cancer Organoids Recapitulate EGFR Expression Levels of Respective Tissues and Are Responsive to EGFR-Targeted Photodynamic Therapy. J Clin Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E, van Hoeck A, Moore K, Kolders S, Francies HE, Gulersonmez MC, Stigter EC, Burgering B, Geurts V, and Gracanin A (2019b). Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proceedings of the National Academy of Sciences 116, 26580–26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, Phan GQ, Kammula US, Hughes MS, Citrin DE, et al. (2010). CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res 16, 6122–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh K, Wu C, O’Rourke KP, Szeglin BC, Zheng Y, Sauve CG, Adileh M, Wasserman I, Marco MR, Kim AS, et al. (2019). A rectal cancer organoid platform to study individual responses to chemoradiation. Nat Med 25, 1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, and Arora VK (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, and Zhang Y (2015a). High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature medicine 21, 1318–1325. [DOI] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al. (2015b). High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature medicine 21, 1318–1325. [DOI] [PubMed] [Google Scholar]
- Han Y, Liu D, and Li L (2020). PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res 10, 727–742. [PMC free article] [PubMed] [Google Scholar]
- HHS, and FDA (2011). https://www.fda.gov/files/vaccines,%20blood%20&%20biologics/published/Final-Guidance-for-Industry--Potency-Tests-for-Cellular-and-Gene-Therapy-Products.pdf. [Google Scholar]
- Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, and Mælandsmo GM (2014). Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer discovery 4, 998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Garraway LA, Ashworth A, and Weber B (2020). Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 19, 23–38. [DOI] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Salinas RD, Zhang DY, Nguyen PT, Schnoll JG, Wong SZH, Thokala R, Sheikh S, Saxena D, and Prokop S (2020). A patient-derived glioblastoma organoid model and biobank recapitulates inter-and intra-tumoral heterogeneity. Cell 180, 188–204. e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, and Miao D (2018a). Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer discovery 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. (2018b). Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC (2018). CAR T cell immunotherapy for human cancer. Science 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- Kopper O, de Witte CJ, Lõhmussaar K, Valle-Inclan JE, Hami N, Kester L, Balgobind AV, Korving J, Proost N, and Begthel H (2019). An organoid platform for ovarian cancer captures intra-and interpatient heterogeneity. Nature medicine 25, 838–849. [DOI] [PubMed] [Google Scholar]
- Lu M, Zessin AS, Glover W, and Hsu DS (2017). Activation of the mTOR Pathway by Oxaliplatin in the Treatment of Colorectal Cancer Liver Metastasis. PLoS One 12, e0169439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchado E, Weissmueller S, Morris J.P.t., Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT, Gainor JF, Corcoran RB, et al. (2016). A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart J, Chen EY, and Prasad V (2018). Estimation of the Percentage of US Patients With Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol 4, 1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, Routbort M, Piha-Paul SA, Janku F, Ueno N, et al. (2015). Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol 33, 2753–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou SH, Salahudeen AA, Smith AR, et al. (2018). Organoid Modeling of the Tumor Immune Microenvironment. Cell 175, 1972–1988 e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Saibil SD, Sotov V, Le MX, Khoja L, Ghazarian D, Bonilla L, Majeed H, Hogg D, Joshua AM, et al. (2019). Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol Immunother 68, 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, Schipper L, Hoes L, Vis DJ, van de Haar J, et al. (2019). Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Science translational medicine 11. [DOI] [PubMed] [Google Scholar]
- Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, Sailer V, Augello M, Puca L, Rosati R, et al. (2017). Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer discovery 7, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, and Rosen N (2010). RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad V (2016). Perspective: The precision-oncology illusion. Nature 537, S63. [DOI] [PubMed] [Google Scholar]
- Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, et al. (2012). Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res 18, 6758–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius G, Baldi A, Ziaie B, Atashbar MZ (2017) Introduction to Micro-/Nanofabrication. In: Bhushan B (eds) Springer Handbook of Nanotechnology. Springer Handbooks. Springer, Berlin, Heidelberg. [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, and Begthel H (2018). A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386. e310. [DOI] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van Den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, and Siersema PD (2011). Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- Schwaederle M, Zhao M, Lee JJ, Lazar V, Leyland-Jones B, Schilsky RL, Mendelsohn J, and Kurzrock R (2016). Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysis. JAMA Oncol 2, 1452–1459. [DOI] [PubMed] [Google Scholar]
- Seidlitz T, Koo B-K, and Stange DE (2021). Gastric organoids—an in vitro model system for the study of gastric development and road to personalized medicine. Cell Death & Differentiation 28, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skardal A, Devarasetty M, Rodman C, Atala A, and Soker S (2015). Liver-tumor hybrid organoids for modeling tumor growth and drug response in vitro. Annals of biomedical engineering 43, 2361–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein WD, Litman T, Fojo T, and Bates SE (2004). A Serial Analysis of Gene Expression (SAGE) database analysis of chemosensitivity: comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res 64, 2805–2816. [DOI] [PubMed] [Google Scholar]
- Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR, et al. (2017). 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang GH, et al. (2018). Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer discovery 8, 1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, and Kester L (2015a). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015b). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman AD, Fritz JM, and Lenardo MJ (2020). A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, and Bernards R (2018). Taking advantage of drug resistance, a new approach in the war on cancer. Front Med 12, 490–495. [DOI] [PubMed] [Google Scholar]
- Yan HH, Siu HC, Law S, Ho SL, Yue SS, Tsui WY, Chan D, Chan AS, Ma S, and Lam KO (2018). A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell stem cell 23, 882–897. e811. [DOI] [PubMed] [Google Scholar]
- Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, Xia F, Fu G, Deng Y, Pan M, et al. (2020). Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell stem cell 26, 17–26 e16. [DOI] [PubMed] [Google Scholar]
- Yuki K, Cheng N, Nakano M, and Kuo CJ (2020). Organoid Models of Tumor Immunology. Trends Immunol 41, 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
KEY RESOURCES TABLE
S1: MOS generation. Related to Figure 1.
S2: Nivolumab induces lung tumor MOS apoptosis. Related to Figure 4. (A) Nivolumab (10ug/ml) treatment. Apoptosis indicated by Annexin V Green. (B) No treatment.
S3: TCB induces lung tumor MOS apoptosis. Related to Figure 4. (A) ESK1* (1ug/ml) treatment (apoptosis indicated by Annexin V Green). (B) DP47 treatment.
S4: Co-culture with activated PBMC (apoptosis indicated by Caspase3/7 Green). Related to Figure 5. (A) Without PBMC. (B) With PBMC (PBMC:target cells= 5:1).
S5: HLAA2 infected lung tumor MOS treated with ESK1*. Related to Figure 5. (A) Uninfected MOS treated with ESK1*. (B) HLA-A2-infected MOS with PBMC (PBMC:target cells= 5:1) treated with ESK1*.
Data Availability Statement
Single-cell RNA-seq data and DNA WES data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.