


Abstract
Advances in transcriptomic and translatomic techniques enable in-depth studies of RNA activity profiles and RNA-based regulatory mechanisms. Ribosomal RNA (rRNA) sequences are highly abundant among cellular RNA, but if the target sequences do not include polyadenylation, these cannot be easily removed in library preparation, requiring their post-hoc removal with computational techniques to accelerate and improve downstream analyses. Here, we describe RiboDetector, a novel software based on a Bi-directional Long Short-Term Memory (BiLSTM) neural network, which rapidly and accurately identifies rRNA reads from transcriptomic, metagenomic, metatranscriptomic, noncoding RNA, and ribosome profiling sequence data. Compared with state-of-the-art approaches, RiboDetector produced at least six times fewer misclassifications on the benchmark datasets. Importantly, the few false positives of RiboDetector were not enriched in certain Gene Ontology (GO) terms, suggesting a low bias for downstream functional profiling. RiboDetector also demonstrated a remarkable generalizability for detecting novel rRNA sequences that are divergent from the training data with sequence identities of <90%. On a personal computer, RiboDetector processed 40M reads in less than 6 min, which was ∼50 times faster in GPU mode and ∼15 times in CPU mode than other methods. RiboDetector is available under a GPL v3.0 license at https://github.com/hzi-bifo/RiboDetector.
INTRODUCTION
rRNA is the predominant form of RNA in both prokaryotic and eukaryotic cells (1–6). The RNA content in a prokaryotic or an eukaryotic cell consists of 80–90% rRNA, 10–15% transfer RNA (tRNA) and 3–7% messenger RNA (mRNA) and regulatory ncRNA (1–6). RNA sequencing (RNAseq) of microbial communities and prokaryotic isolates is widely used for activity profiling in microbiology (7–9). Sequencing of functional non-coding RNAs (ncRNAseq) has also expanded our knowledge of the regulatory roles of various ncRNAs (10). Furthermore, sequencing of ribosome-protected mRNA fragments, called ribosome sequencing (Riboseq) or ribosome profiling, provides insight into the translatome (11). However, the lack of polyadenylation (polyA) tails in prokaryotic mRNAs, all ncRNAs, and ribosome-protected mRNA fragments in Riboseq complicates the enrichment of these non-rRNA sequences before sequencing using polyA tails. Currently, numerous rRNA depletion kits are available that can drastically remove rRNA sequences for model organisms (12). However, the efficacy for non-model organisms and unknown bacteria is limited, which results in highly abundant rRNA reads being retained in the sequencing data. For example, rRNA reads correspond to 78.44 ± 11.41% of RNAseq data after rRNA depletion with the MicrobEnrich and MicrobExpress kits in our previous oral metatranscriptome study (13). In other metatranscriptomic and bacterial transcriptomic studies, in which the RiboZero kit was used, the rRNA reads account for 22.05% ± 20.18% to 31.98% ± 10.07% of total reads (14–16).
The presence of abundant rRNA can introduce substantial bias to transcriptome data. A number of protein coding genes contain some rRNA-like sequence segments (17–20), so their expression levels can be strongly overestimated unless the rRNA reads are removed. Furthermore, rRNA removal can greatly reduce the data size for downstream analysis and accelerate the entire workflow. To facilitate the removal of rRNA sequences from large-scale sequencing data, a number of methods have been developed, including Meta-RNA (21), rRNASelector (22) and rRNAfilter in the RNA-QC-chain pipeline (23) (named RQC_rRNAfilter below), which use hidden Markov models (HMMs) trained on curated rRNA sequences; RiboPicker (24) and SortMeRNA (25), which are based on sequence alignment; and rRNAFilter (26), which applies an expectation-maximization algorithm to discriminate rRNA reads and non-rRNA reads based on their k-mer profiles. Some short-read aligners such as BWA (27) can also be used to remove rRNA reads. Most of these methods are either based on alignment algorithms that search for sequence homologies or based on a first-order HMM, which makes probabilistic predictions based on the assumption that the current state only depends on the state one step before. Moreover, RNAseq datasets are usually large: one sample can comprise 10G bases and one dataset may have 100 such samples, and the existing methods take hours to process one such sample. Thus, those methods may require weeks to remove rRNA from an entire dataset.
Deep neural networks, particularly recurrent neural networks (RNNs), are able to capture the sequence patterns in a long-range context (28–30). Therefore, we proposed a novel method named RiboDetector, which is based on a sophisticated RNN architecture, the Long Short-Term Memory (LSTM) (31) network, and detects rRNA sequences from large sequencing datasets rapidly and accurately with a very low level of bias for downstream functional analyses.
MATERIALS AND METHODS
Data collection and RiboDetector training
We collected all the rRNA sequences from the SILVA database, version 138.1 (32), and coding sequences (CDSs) from the Orthologous MAtrix (OMA) database (August 2020 version) (33). SILVA v138.1 contains curated sequences for 510 508 small subunit (SSU) and 95 286 large subunit (LSU) rRNAs. The August 2020 version of OMA includes 5 979 441 prokaryotic and 9 522 432 eukaryotic non-redundant protein coding DNA sequences. To further reduce the number of sequences but keep the diversity of the remaining sequences, we performed sequence clustering using MMseqs2 v12.113e3 (34) at a sequence identity cutoff of 0.7 and a coverage of 0.7 with ‘easy-cluster –min-seq-id 0.7 -c 0.7 –cov-mode 1’, which resulted in 6 935 571 representative sequences from both prokaryotes and eukaryotes. The resulting CDSs were mapped against the SILVA sequences using minimap2 v2.17-r941 (35) with the CIGAR (Concise Idiosyncratic Gapped Alignment Report) output option ‘-c –secondary = no’ to detect possible rRNA sequences in the CDS collection. Thus, 354 CDSs, which shared 98% similarity and 90% coverage with any rRNA sequences in the SILVA database, were removed, as they were probably rRNA sequences erroneously included in the OMA database. The rest of the CDS collection, consisting of 6 935 217 sequences, comprises the OMA_id07_cov07 dataset. Next, ∼300 000 rRNA sequences and 300 000 CDSs were randomly selected from SILVA and OMA_id07_cov07 as training data for training the RiboDetector models. A validation dataset was also generated for selecting the best model. It consists of 1 million paired-end rRNA reads and 1 million paired-end CDS reads simulated from sequences that are not included in the training data.
To determine the optimal hyperparameter settings, such as use of a one-directional or bi-directional LSTM, one or two LSTM layers, and different numbers of hidden units (64, 128, 256), we compared models trained from the first 30 epochs with different hyperparameter settings. For training, one-hot encoded full-length rRNA and CDSs were provided as input. The classification models were trained with a batch size of 256, a maximum sequence length of 100 (varying from 70 to 100), and a sliding window step size of 25 for rRNA sequences and 30 for CDSs. We chose 100 as the maximum sequence length because most of the current next-generation sequencing short reads are around 100 bp long. The trained models can be also used for the classification of reads longer than 100 bp and shorter than 70 bp. All the models using 256 hidden units and models based on two layers of BiLSTM with 128 hidden units were not trained successfully, because of an ‘out of CUDA (Compute Unified Device Architecture)’ memory issue. The architecture using one BiLSTM layer with 128 hidden units showed the best performance in terms of Matthews correlation coefficients (MCCs, 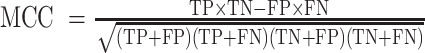 ) (Supplementary Figure S1A) for the validation dataset.
) (Supplementary Figure S1A) for the validation dataset.
We then trained the RiboDetector models with the selected optimal hyperparameters (one input layer, one BiLSTM layer with 128 hidden units, and one fully connected linear layer with two outputs representing two classes) on the same training dataset with 150 epochs and an initial learning rate of 0.001 (Supplementary Figure S1B). The softmax values of the outputs from the linear layer representing the predicted probabilities of the input sequences being rRNA and non-rRNA were computed to calculate the cross-entropy loss. The implementation uses PyTorch v1.6 and Python v3.8. Parameter optimization was performed using the stochastic gradient descent method Adaptive Moment Estimation (Adam). The learning rate decayed by a factor of 0.5 every 10 epochs. After 150 epochs of training, the best model was selected from these 150 models (last model of each epoch) based on the MCC on the validation dataset. The reads used in the tSNE visualization were generated from Firmicutes mRNA, Tuf genes, virus sequences obtained from the European Nucleotide Archive (ENA) database, and rRNA sequences downloaded from the Human Oral Microbiome Database (HOMD) (36).
To further improve the runtime performance of RiboDetector in CPU mode, we used the Open Neural Network Exchange (ONNX) technique to accelerate RiboDetector on a CPU. When taking full advantage of multiprocessing, it was only about three times slower in CPU mode with 40 CPU cores than in GPU mode with one NVIDIA Tesla V100 GPU and 40 CPU cores. For low-memory computers, we provided a ‘–chunk_size’ parameter that can substantially reduce the memory use but does not affect much of the runtime. Additionally, there is also an ‘–ensure’ option (with option values of ‘none’, ‘rrna’, ‘norrna’ or ‘both’) for paired-end reads. Specifically, the option ‘rrna’ will output rRNAs with high confidence; in other words, the read pair is considered as rRNA only when both ends are predicted as rRNAs, whereas ‘norrna’, conversely, outputs non-rRNAs with high confidence. For the option ‘both’, the discordantly predicted read pair will be discarded. The option ‘none’, which is the default option, averages the output probabilities of both ends and chooses the class with the higher average probability.
In the benchmark, we considered rRNA reads as positives and non-rRNA reads, including mRNA, regulatory ncRNA, and viral reads as negatives. Therefore, correctly classified rRNA reads were considered to be true positives (TPs), whereas incorrectly classified rRNA reads were considered to be false negatives (FNs). Further, correctly classified non-rRNA reads were true negatives (TNs) while misclassified non-rRNA reads were false positives (FPs). The false positive rate (FPR) was formulated as 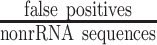 ; the false negative rate (FNR) was defined as
; the false negative rate (FNR) was defined as 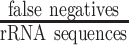 . The misclassifications include FPs as well as FNs, and the misclassification rate was calculated as
. The misclassifications include FPs as well as FNs, and the misclassification rate was calculated as 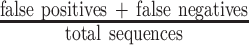 . The true positive rate (TPR) was calculated as
. The true positive rate (TPR) was calculated as 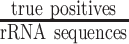 .
.
Generation of benchmark datasets
To evaluate the performance of RiboDetector and other rRNA detection methods, we created eight benchmark datasets with sequences that were not used in the training and validation datasets described above. The datasets were simulated using ART_Illumina v2.3.7 (37) with the parameter settings ‘-p -l 100 -ss HS25 -m 150 -s 10’. The benchmark datasets are:
SILVA_rRNA to assess the false negative rate of rRNA detection methods: 20M paired-end reads simulated from 50 474 SSU and LSU rRNA sequences from the SILVA database; these sequences are distinct from the sequences used for training and validation. It includes 18 269 416 rRNA reads generated from 46 107 bacterial rRNA sequences, 556 229 rRNA reads generated from 1401 archaeal rRNA sequences, and 1 174 355 rRNA reads generated from 2966 eukaryotic rRNA sequences;
OMA_CDS to assess the false positive rate of rRNA detection methods on prokaryotic and eukaryotic mRNAs: 20M paired-end reads simulated from 500 000 CDSs from the OMA_id07_cov07 dataset excluding the sequences used for training and validation. It contains 7 634 440 reads from 1639 bacterial species, 650 320 reads from 153 archaeal species, and 11 715 240 from 480 eukaryotic species;
ENA_virus to evaluate the false positive rate of rRNA detection methods on virus sequences: 27 206 792 paired-end reads simulated from 13 848 viral gene sequences downloaded from the ENA database;
Amplicon_16S to evaluate the false negative rate of rRNA detection methods on real 16S rRNA gene sequencing data: 7 917 920 real paired-end amplicon sequencing reads targeting the V1–V2 region of 16s rRNA genes from an oral microbiome study (38);
Human_ncRNA to evaluate the false positive rate of rRNA detection methods on regulatory ncRNAs: 6 330 381 paired-end reads simulated from 106 880 human non-coding RNA sequences;
MetaT to evaluate the false negative rate and false positive rate of rRNA detection methods on metatranscriptome data: 9 165 829 paired-end oral metatranscriptome reads consisting of 4 735 326 prokaryotic mRNA reads from 50 species, 2 474 450 human mRNA reads, 73 100 viral mRNA reads and 1 882 953 rRNA reads. The four sequence components were simulated separately and then combined into one dataset. To simulate prokaryotic mRNA reads, we used the abundance of the 50 most abundant species in an oral microbiome study (38) as the multiplication factor
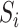
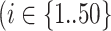 ). As the expression levels of genes for an organism follow the log-normal distribution (39–41)
). As the expression levels of genes for an organism follow the log-normal distribution (39–41) 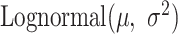 , we determined the mean
, we determined the mean 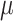 and standard deviation
and standard deviation 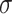 of the distribution (Supplementary Figure S1C) on the basis of a previous oral bacterial transcriptome dataset (15). We then generated the expression levels, represented as the fragments per kilobase per million reads (FPKM), of all genes (e.g. N genes) for each species which was denoted as
of the distribution (Supplementary Figure S1C) on the basis of a previous oral bacterial transcriptome dataset (15). We then generated the expression levels, represented as the fragments per kilobase per million reads (FPKM), of all genes (e.g. N genes) for each species which was denoted as 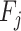 (
(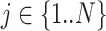 ) following the log-normal distribution
) following the log-normal distribution 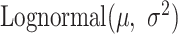 . The coverage of gene
. The coverage of gene 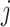 of species
of species 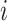 was calculated as
was calculated as 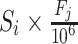 . Prokaryotic mRNA reads were simulated from the genes of these 50 species in OMA with the computed coverage. Similarly, we then simulated transcriptome reads of the human host for which the FPKMs of the genes also followed the distribution
. Prokaryotic mRNA reads were simulated from the genes of these 50 species in OMA with the computed coverage. Similarly, we then simulated transcriptome reads of the human host for which the FPKMs of the genes also followed the distribution 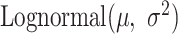 with a coverage of
with a coverage of 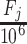 , and the reads of the top 10 most active viruses from a previous oral microbiome dataset (42) with a coverage of 50. Finally, we added 1 882 953 rRNA reads simulated from 10 000 rRNA sequences from the 50 prokaryotic species extracted from the SILVA database with a coverage of 50. The proportion of human mRNA reads to all mRNA reads was set to 0.34 corresponding to the human mRNA reads fraction in an oral metatranscriptome study in periodontitis (42). The proportion of microbial rRNA reads to all reads was set to 0.21 according to the average rRNA reads proportion in a previous study (14). We did not set this to the proportion of rRNA reads in the above mentioned oral metatranscriptome study, as a rarely used and inefficient rRNA depletion protocol (MicrobEnrich and MicrobExpress kits) was applied, resulting in about 90% of the generated reads being rRNA reads. We set the viral mRNA reads proportion to all mRNA reads to 0.01, which is ten times the fraction found in the above referenced oral metatranscriptome study, to allow us to benchmark on viral data;
, and the reads of the top 10 most active viruses from a previous oral microbiome dataset (42) with a coverage of 50. Finally, we added 1 882 953 rRNA reads simulated from 10 000 rRNA sequences from the 50 prokaryotic species extracted from the SILVA database with a coverage of 50. The proportion of human mRNA reads to all mRNA reads was set to 0.34 corresponding to the human mRNA reads fraction in an oral metatranscriptome study in periodontitis (42). The proportion of microbial rRNA reads to all reads was set to 0.21 according to the average rRNA reads proportion in a previous study (14). We did not set this to the proportion of rRNA reads in the above mentioned oral metatranscriptome study, as a rarely used and inefficient rRNA depletion protocol (MicrobEnrich and MicrobExpress kits) was applied, resulting in about 90% of the generated reads being rRNA reads. We set the viral mRNA reads proportion to all mRNA reads to 0.01, which is ten times the fraction found in the above referenced oral metatranscriptome study, to allow us to benchmark on viral data;The OMA_SILVA dataset to estimate false positive rate of rRNA detection methods on mRNAs sharing sequence similarity to rRNAs in Figure 1C contains 1 027 675 paired-end reads simulated from OMA_id07_cov07 CDSs which share similarity (identity ≥ 70%) to rRNA genes. Sequences with identity ≥98% and query coverage ≥90% to rRNAs and sequences used for training and validation were excluded;
The HOMD_FP dataset in Figure 1C has 100 558 paired-end reads simulated from HOMD CDSs from human oral microbes, which share similarity (identity ≥ 70%) to high-FPR (FPR ≥ 0.5) sequences of BWA, RiboDetector, and SortMeRNA in the OMA_CDS dataset above; again, sequences with identity ≥98% to and query coverage ≥90% of rRNAs were excluded. This dataset was used to estimate false positive rate of rRNA detection methods on mRNAs sharing sequence similarity to high FPR sequences.
Figure 1.

Performance of RiboDetector on different datasets and hardware. (A) Visualization of the output of the BiLSTM hidden layer for different sequence types. (B) Comparison of the misclassification rates (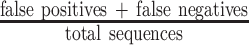 ), runtime, maximum memory use, and output size of RiboDetector and other methods. The colored dots show different datasets and the red diamond indicates the mean. The metrics are specified on the top of each plot and the four plots share the same x-axis. (C) Benchmarking on the microbiome dataset consisting of prokaryotic mRNA reads, human mRNA reads, virus mRNA reads, and rRNA reads. Note: to show all the differences in different ranges, three intervals with breaks are shown on the y-axis. (D) The performance of all methods on the microbiome dataset demonstrated by TPR (
), runtime, maximum memory use, and output size of RiboDetector and other methods. The colored dots show different datasets and the red diamond indicates the mean. The metrics are specified on the top of each plot and the four plots share the same x-axis. (C) Benchmarking on the microbiome dataset consisting of prokaryotic mRNA reads, human mRNA reads, virus mRNA reads, and rRNA reads. Note: to show all the differences in different ranges, three intervals with breaks are shown on the y-axis. (D) The performance of all methods on the microbiome dataset demonstrated by TPR (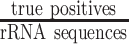 ) and FPR (
) and FPR (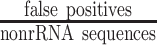 ) or ROC curve. Only the ROC curve of RiboDetector is shown because the other methods do not report values that can be used as confidence or probability for all input sequences. The ROC curve is based on 20 000 randomly selected reads from the whole metatranscriptome dataset, while the TPR-FPR calculation was performed using all reads, therefore the curve does not intersect with the TPR-FPR values for RiboDetector. Logarithmic scale was applied in the x-axis. The black dots on the curve with values beside indicate the cutoffs of the probabilities generating the ROC curve. The y-axis starts from 0.6 as the curve below 0.6 is a vertical line and not informative. (E) The FPR of RiboDetector, BWA and SortMeRNA on two additional benchmark datasets. The details of the datasets used in A–C can be found in the Material and Methods.
) or ROC curve. Only the ROC curve of RiboDetector is shown because the other methods do not report values that can be used as confidence or probability for all input sequences. The ROC curve is based on 20 000 randomly selected reads from the whole metatranscriptome dataset, while the TPR-FPR calculation was performed using all reads, therefore the curve does not intersect with the TPR-FPR values for RiboDetector. Logarithmic scale was applied in the x-axis. The black dots on the curve with values beside indicate the cutoffs of the probabilities generating the ROC curve. The y-axis starts from 0.6 as the curve below 0.6 is a vertical line and not informative. (E) The FPR of RiboDetector, BWA and SortMeRNA on two additional benchmark datasets. The details of the datasets used in A–C can be found in the Material and Methods.
We evaluated RiboDetector alongside other rRNA detection methods including BWA v0.7.17-r1188, RiboPicker v0.4.3, rRNAFilter v1.1, SortMeRNA v4.2.0 and RQC_rRNAfilter v1.0. We also attempted to include Meta-RNA v3 and rRNASelector in the benchmarking. However, Meta-RNA did not finish generating the final output for the SILVA_rRNA dataset after running for 5 days with 40 CPU cores, whereas rRNAselector is not maintained anymore and the download link is unavailable. For BWA, we used the BWA-MEM algorithm with the default parameter settings. The training rRNA sequences of RiboDetector were used as rRNA reference sequences for mapping. The unmapped reads were extracted by SAMtools (43) with ‘-f 12 -F 256’ and BEDTools bamtofastq (44). The mapped reads were retrieved using SAMtools with ‘-F 12 -F 256’, and read counting of the genes was carried out using SAMtools idxstats. RiboDetector with ‘–ensure norrna’ was used for the datasets SILVA_rRNA and Amplicon_16S; for the rest of the datasets, ‘–ensure rrna’ was used. We also evaluated RiboDetector with the default setting ‘–ensure none’ for all benchmark datasets. The number of misclassifications was 513 ± 359, which was much lower than that of other methods (Supplementary Table S1). Minimap2 was used to determine the similarity, query coverage, and sequence divergence between sequences in the benchmark datasets and the training sequences or reference sequences. The runtime, memory (maximum unique set size), and output size were determined with the Snakemake (45) benchmark functionality on a computer with a NVIDIA Tesla V100 GPU, Intel Xeon processor (2.20GHz) virtualized to 80 cores (40 cores were used in the benchmark) and 500GB of main memory. Gene ontology (GO) terms for each CDS were extracted from the OMA database. The enrichment analysis was performed using Fisher's exact test based on the contingency table of FPs or TNs assigned or not assigned to a given GO term. Benjamini–Hochberg correction was applied to the Fisher's exact test p-values to control false discovery rate (FDR) and account for multiple comparisons.
RESULTS
rRNA reads may be mapped to protein coding genes by short read alignment
To demonstrate that activity profiling without rRNA removal introduces considerable bias towards certain functional groups, we mapped rRNA reads from the SILVA_rRNA dataset against CDSs in the OMA database with BWA (Material and Methods). Over 83.37% of the rRNA reads were mapped to CDSs. Genes belonging to 27 GO terms had over 1% of the rRNA reads mapped (Supplementary Figure S2A). Base coverage analysis showed that the reads were most likely to be mapped onto certain regions of these protein-coding genes, indicating that these genes have one or several short regions that share sequence similarity to rRNA sequences (Supplementary Figure S2B). The results suggest that functional groups such as membrane, translation, ATP-binding, and DNA-binding proteins can be strongly overestimated, if rRNA reads are not removed before activity profiling. We also calculated the fraction of rRNA reads mapped to CDSs after filtering by the mapping quality score (MAPQ) and using a longer exact match seed length in BWA. The MAPQ quantifies the probability that a read is misplaced because of ambiguity, poor base quality, and/or bad alignment. A larger MAPQ represents a higher certainty that the alignment is correct and unambiguous. A longer seed length setting in BWA will generate a more stringent and unambiguous alignment. This analysis showed that 56.26% of the rRNA reads were mapped uniquely to specific CDSs with MAPQ ≥5, 47.15% with MAPQ ≥ 10 and 41.59% with MAPQ ≥20. In comparison to the default seed length of 19, with a seed length of 29, the fraction of rRNA reads being mapped to CDSs was reduced to 25.49%. However, a longer seed length will decrease the alignment rate for mRNA reads to the reference sequences in real data analysis.
Highly accurate discrimination of rRNA reads and non-rRNA reads with neural networks
We developed a discriminative neural network model based on a BiLSTM that detects rRNA reads from the non-rRNA background with high accuracy (Supplementary Figure S1B, Materials and Methods). Visualization of the last step's outputs from the BiLSTM layer for 8000 randomly selected short reads (including four types of sequence, each containing 2000 reads; details regarding the data are given in Material and Methods) using tSNE demonstrated that the features captured by the model clearly discriminated rRNA sequences from the non-rRNA sequences (Figure 1A).
We then evaluated the performance of RiboDetector alongside BWA, RiboPicker, rRNAFilter, SortMeRNA and RQC_rRNAfilter on five benchmark datasets derived from SILVA rRNA sequences, 16S rRNA gene amplicon sequences, OMA CDSs, viral sequences, and ncRNA sequences (Materials and Methods). On average, RiboDetector had the fewest misclassifications, namely 165 ± 114 per 1 million (M) reads, compared with 8956 ± 7219, 1488 ± 1302, 373 257 ± 509 856, 948 ± 1438, and 12 800 ± 28 544 for BWA, RiboPicker, rRNAFilter, SortMeRNA and RQC_rRNAfilter, respectively (Figure 1B, Supplementary Table S1). The number of misclassifications of RiboDetector was six times lower than the method ranked second (i.e. SortMeRNA). SortMeRNA is the most commonly used method for rRNA read detection at present. RQC_rRNAfilter made the fewest FP predictions, but had the highest FNR (6.39%) for the SILVA_rRNA dataset. For the OMA_CDS dataset which was simulated from 500 000 CDSs, RQC_rRNAfilter and RiboDetector had only 24 and 35 genes, respectively, with a FPR ≥0.5 in classifying the corresponding reads, while it was 549 for BWA, 114 for RiboPicker, 579 for rRNAFilter, and 153 for SortMeRNA (Supplementary Figure S3). For each method, the FPR of a gene was computed as the number of FP reads produced by the method from the gene divided by the total number of reads from the gene. Nineteen out of 35 high-FPR (≥0.5) genes of RiboDetector were also high-FPR genes of all other methods, except for rRNAFilter, which had no overlap with all the other methods (Supplementary Figure S3). Remarkably, following RQC_rRNAfilter, RiboDetector also had a very low level of FPs on the human ncRNA dataset (Figure 1B, Supplementary Table S1). Specifically, RQC_rRNAfilter falsely predicted 504 out of 6 330 381 human ncRNA reads as rRNA reads and RiboDetector misclassified 2021 human ncRNA reads, whereas BWA falsely predicted 113 816; RiboPicker, 11 085; rRNAFilter, 5 815 406; and SortMeRNA, 21 898 reads.
Next-generation sequencing datasets are usually large, containing over 10M reads per sample. Therefore, in addition to predictive quality, the runtime is another important factor of software performance. We evaluated the runtimes and memory uses of all methods with the benchmark functionality of Snakemake (Material and Methods). RiboDetector required 0.43 ± 0.05 minutes per million reads, which was 6.9 times faster than BWA (the second fastest method) and 33.2 times faster than SortMeRNA on average (Figure 1B, Supplementary Table S1). RiboDetector also had the smallest output size (420.21 ± 232.83 MB per million reads). In terms of memory usage, the high-memory mode of RiboDetector used slightly more memory (1955.50 ± 463.05 MB) per million reads than BWA (1674.22 ± 1420.67 MB). However, in low-memory mode with a chunk size of 64, the memory use of RiboDetector was reduced to 485.55 MB per million reads (a quarter of the high-memory mode) while increasing the runtime by only 1.5 times (0.59 minutes per million reads for the SILVA_rRNA dataset).
To assess the performance of different methods on microbiome data, we benchmarked these methods on a metatranscriptome dataset consisting of oral microbial mRNA, human mRNA, viral mRNA, and rRNA reads (Materials and Methods). RiboDetector made the fewest misclassifications (70 misclassifications per million reads), followed by SortMeRNA (269), RiboPicker (344), rRNAFilter (5142), RQC_rRNAfilter (6111) and BWA (16839) (Figure 1C, Supplementary Table S2). RiboDetector had low levels of misclassification for all sequence types, whereas RQC_rRNAfilter and rRNAFilter made few misclassifications for bacterial and human mRNA reads, but had very high misclassification levels for rRNA and viral mRNA reads. The receiver operating characteristic (ROC) curve of the RiboDetector predictions on this metatranscriptome dataset (area under the curve: 0.9999942) also shows that it performs well on microbiome data (Figure 1D). SortMeRNA and RiboPicker were performant as well on this dataset in terms of their TPR and FPR.
Based on all benchmark datasets that we evaluated above, RiboDetector produced 6, 9, 69, 78 and 2088 times less misclassifications than SortMeRNA, RiboPicker, BWA, RQC_rRNAfilter and rRNAFilter, respectively. Since SortMeRNA ranked second in the benchmarks and is the most used method for rRNA removal, we investigated whether its performance could be further improved when using the training rRNA sequences of RiboDetector as reference database. However, in this setting SortMeRNA produced 1883 misclassifications per 1 million reads on the metatranscriptome dataset, 7 times more than with the original SortMeRNA reference database. The result suggests that its original database was highly optimized for SortMeRNA. RiboDetector performs better than SortMeRNA is not because of the training sequences.
Test on CDSs similar to FPs and rRNAs
To compare the performance of different methods on non-rRNA sequences sharing similarity to rRNA sequences, we used two additional non-rRNA datasets named OMA_SILVA and HOMD_FP for this evaluation (Material and Methods). RiboPicker, rRNAFilter, and RQC_rRNAfilter, which had runtimes of >30 min per 1M reads in the Snakemake runtime benchmark (Figure 1B), were not included in this evaluation and following analyses. The HOMD database contains a collection of well-annotated human oral microbial genomes, CDSs and rRNA genes, the CDSs of which can be used as negative samples for evaluation. The FPRs of RiboDetector were 1.1% and 1.5% for OMA_SILVA and HOMD_FP, respectively (Figure 1E). BWA had a FPR of 17.4% for OMA_SILVA and 32.0% for HOMD_FP, whereas SortMeRNA had a FPR of 10.1% for OMA_SILVA and 4.3% for HOMD_FP. Thus, the FPRs of RiboDetector were about 15–20 times lower than those of BWA and 3–10 times lower than those of SortMeRNA for these two datasets. The results suggest that RiboDetector performs well even for the CDSs sharing similarity to rRNA sequences.
Generalizability of different methods for novel rRNA detection
It is important for rRNA detection methods to be able to identify novel rRNA sequences that are not in the public databases. This is particularly critical in microbiome datasets, in which numerous novel and unknown microbes are likely to be present. We analyzed the probabilities of the correct class assignment (termed confidence) to given rRNA or non-rRNA sequences predicted by RiboDetector. In general, the confidence was not smaller for sequences that were more divergent from the training data (Figure 2A). To compare the generalizability of RiboDetector with that of BWA and SortMeRNA, we grouped the reads from the SILVA_rRNA dataset into different divergence bins on the basis of the least divergence from the training dataset or reference database sequences and summarized their FPRs (Figure 2B). The divergence was determined by mapping the rRNA sequences used in SILVA_rRNA to the training or reference rRNA sequences with minimap2 (Material and Methods). BWA and SortMeRNA showed a clear increase in their FNR, along with an increase in the divergence between the benchmark data and sequences in their reference databases or training datasets (Figure 2B). The FNR of BWA was close to 20% when the divergence was 20%, and SortMeRNA was close to 15%, whereas the FNR of RiboDetector remained at a very low level across all the divergence bins. There are no sequences in the SILVA_rRNA benchmark dataset that have a sequence divergence from the training dataset and reference database of more than 20%. To demonstrate the performance of RiboDetector on more divergent (≥20%) rRNA sequences from training and reference sequences, we identified 144 such rRNA sequences from the SILVA database. We then analyzed the FNRs of different methods on 38,083 reads simulated from these 144 rRNA sequences (Supplementary Figure S4). The FNRs for these more divergent sequences were indeed higher than those for the sequences in the SILVA_rRNA dataset for all methods. However, the FNRs of RiboDetector are still about two times smaller than those of the other methods, rising from 12.8% to 23.0% with increasing sequence divergence. These results show that identifying novel sequences that are not in databases is a big challenge for currently widely used methods, whereas RiboDetector demonstrated a substantial generalizability for divergent rRNA sequences, which should facilitate the accurate identification of novel rRNA sequences. Still, for rRNA sequences with a divergence ≥20% from training and reference sequences, all the methods showed a high FNR (≥10%). In practice, this case may be very rare though, as the sequences in the training data and reference database well represent rRNA sequence diversity.
Figure 2.

Evaluation of the generalizability of the prediction model. (A) The predicted probabilities of the correct label for rRNA in the benchmark dataset SILVA_rRNA and non-rRNA in the dataset OMA_CDS, and the divergence of the sequence from the training dataset. The x-axis shows the divergence of each testing sequence from its most closely related sequence in the training dataset. The probability in the y-axis shows the mean confidence (i.e. predicted probabilities) of all reads of a gene. The probabilities were calculated as the softmax values of outputs from the final linear layer, which represent the predicted probabilities for the correct class. (B) The false negative rate (FNR) of RiboDetector, BWA and SortMeRNA on rRNA sequences from SILVA_rRNA dataset with different ranges of divergence from the closest rRNA genes used in the reference database. Since the rRNA sequences are relatively conserved, there were no sequences with divergence >20% from the training and reference rRNA sequences in the SILVA_rRNA dataset. The three panels share the same y-axis.
Analysis of the FPs of BWA, RiboDetector and SortMeRNA
To elucidate why some non-rRNA reads from the OMA_CDS dataset were classified as rRNA by different methods, we analyzed the alignments for the FPs of BWA and SortMeRNA. As RiboDetector does not generate alignments, to show whether the FPs of RiboDetector are similar to rRNAs, we mapped its FPs to the training rRNA sequences with BWA and analyzed the alignments. Around 30% of its FP reads could be mapped. In this analysis, the alignment match reflects the length of the aligned region and the edit distance represents the total bases of mismatches, insertions and deletions (indels) within the alignment. The mapped FPs of RiboDetector had a much longer alignment match length than those of BWA and SortMeRNA, as well as fewer mismatches and indels within the alignment (Supplementary Figure S5A). For BWA, 84.5% of the alignments had an alignment match length between 19 (the default seed length) and 21 nucleotides to the reference, and the remaining read regions were not aligned to rRNA (Supplementary Figure S5A). The default seed length of BWA is 19, thus allowing for one mismatch in 20 nucleotides, which corresponds to 95% identity or 5% divergence on average. Therefore, the number of FPs could be reduced by increasing the seed length of BWA. However, our generalizability analysis showed that the FNR of BWA started to increase for tested rRNA sequences with a divergence of over 5% from the reference rRNA sequences. Together, these points indicate that a larger seed length will result in a low FPR but a higher FNR. To prove this, we ran BWA on the OMA_CDS and SILVA_rRNA datasets with a larger seed length, namely 29 (one mismatch in 30 nucleotides, corresponding to ∼97% identity). As expected, the number of FPs for the OMA_CDS dataset decreased from 283 517 to 17 021, whereas the number of FNs on SILVA_rRNA increased from 58 736 to 85 610. With SortMeRNA, the FPs tended to have a larger alignment match with the reference rRNA sequences, but more mismatches and indels within the alignment than BWA (Supplementary Figure S5A). Only 16.68% of the mapped FPs of RiboDetector had an aligned length shorter than half of the read (50 nucleotides), but this value rose to 98.15% for BWA and 56.23% for SortMeRNA. Moreover, 95.60% of the mapped FPs of RiboDetector had an edit distance (mismatches and indels, not including clipping) smaller than five; this was 99.48% for BWA and 57.96% for SortMeRNA. This result suggests that the FPs of RiboDetector that could be mapped to rRNA are very similar to rRNA sequences, whereas the FPs of BWA and SortMeRNA are less similar to rRNAs overall, but contain either short exact matches to rRNA or long alignment matches with numerous mismatches and indels. We also analyzed the predicted probabilities of FPs that could be mapped to rRNAs, FPs that could not be mapped to rRNAs, and the TPs of RiboDetector. Remarkably, the unmapped FPs tended to have smaller probabilities than the mapped FPs and the TPs (Supplementary Figure S5B). Overall, the FPs have intermediate probabilities and all TPs have very high probabilities close to one (Supplementary Figure S5B). This suggests that RiboDetector generates FPs by predicting non-rRNA reads as rRNA reads with low confidence.
rRNA removal methods must ensure not to introduce bias towards certain functional groups by FP predictions. That is, the FP sequences being removed should not be significantly enriched in certain functional groups. Therefore, we performed a GO term enrichment analysis with Fisher's exact test based on the hypergeometric distribution of the number of FP and TN reads from the OMA_CDS dataset for each GO term (Material and methods). The FPs of RiboDetector were enriched in three GO terms with a FDR of ≤0.01 and an odds ratio of 10 (Figure 3A, Supplementary Table S3), but none of them had FPRs higher than 5%. In comparison, BWA’s FPs were significantly enriched in 39 GO terms; all of these had FPRs over 5% and 22 had FPRs higher than 15% (Figure 3B, Supplementary Table S3). The FPRs can be up to 40.5% for certain GO terms. The FPs of SortMeRNA were enriched in 62 GO terms, 18 of which had FPRs higher than 5% (Figure 3C, Supplementary Table S3). Its FPRs were up to 22.5% for certain GO terms. Overall, analysis suggests that the FPs of RiboDetector were enriched in fewer functional groups than other methods, which will enable a more accurate functional analysis of the data.
Figure 3.

GO term enrichment analysis for FPs of RiboDetector (A), BWA (B) and SortMeRNA (C) with a false discovery rate (FDR) of ≤0.01 and an odds ratio of ≥10. For BWA, only GO terms with a FPR of ≥15% are shown; GO terms with a FPR of ≥5% are shown for SortMeRNA. BP: biological process, CC/MF: cellular component/molecular function. The three panels share the same x-axis.
Benchmarking computational resource requirements of RiboDetector
To systematically evaluate the computational performance of RiboDetector, we ran the software on different computers with limited CPU, GPU, and memory resources. First, we compared RiboDetector with other methods on a personal workstation computer with a consumer-grade GPU NVIDIA RTX 2080 Ti (Figure 4A). Compared to the consumer-grade GPU 2080 Ti, V100 has more CUDA cores, and it is an advanced data center GPU (https://www.nvidia.com/en-us/data-center/v100/). Interestingly, the runtime on the 2080 Ti computer was shorter than that on one with a V100 (17.6 s versus 22.8 s for 1 million paired-end reads), possibly because RiboDetector can take advantage of the high input/output speed of the solid-state drive of the workstation. Since the memory on the workstation is limited, we used the low memory mode of RiboDetector and it used only 4.5GB memory for 20M paired-end reads. To make RiboDetector suitable for most use cases, including on computer without GPU, we optimized RiboDetector for CPUs using ONNX (Material and Methods). It was then tested on different computational servers with a V100 GPU, a T4 GPU, or only CPUs with different memory settings by changing the chunk size (Figure 4B). In CPU mode, RiboDetector was only 3.3 times slower than in the GPU mode but still 5.6 times faster than the second-fastest method (BWA) and 12.3 times faster than SortMeRNA on the same dataset (Figure 4).
Figure 4.

The computational resource requirement benchmarks of RiboDetector on computers with limited resources. (A) Comparison of the resource consumption and runtime of different methods on a personal computer with eight CPUs and a 2080 Ti GPU. One million paired rRNA reads from the SILVA_rRNA dataset were used for the evaluation. RiboDetector had zero false misclassifications. (B) Performance of RiboDetector and other methods on different computers. V1001: 40 cores, no memory limit; V1002: 40 cores, chunk size 256 (∼20GB memory limit); T4: 40 cores, no memory limit; CPU: 40 core Intel Xeon processor (2.20 GHz), no GPU, no memory limit; 2080 Ti: eight cores, chunk size 64 (∼5GB memory limit).
A metatranscriptome data processing workflow usually consists of read quality control, rRNA reads removal, host reads removal, and reference gene mapping. To demonstrate that rRNA removal is the most time-consuming step in processing with the most widely used method (SortMeRNA), we ran the entire data processing workflow on the oral metatranscriptome dataset that was used in the previous benchmark with 40 CPU cores. We performed read quality control with Fastp (46), then removed rRNA reads with SortMeRNA, filtered out human reads by mapping the non-rRNA reads against the human reference with BWA, and finally mapped the cleaned reads against HOMD reference CDSs with BWA. The read quality control required 1 min, rRNA removal with SortMeRNA took 109 min, human reads removal took 4 min, and read alignment took 2 min of computing time. In comparison, with RiboDetector, rRNA removal required only 4 min in GPU mode and 15 min in CPU mode.
DISCUSSION
Here, we describe RiboDetector, a deep learning-based method leveraging GPU acceleration and a BiLSTM model to capture patterns from a long-range context for rapid and accurate rRNA sequence detection. Removal of rRNA reads is an essential step in prokaryotic transcriptome, metatranscriptome, ncRNAseq, and Riboseq data analysis. In rRNA detection, RiboDetector was very accurate with low levels of both false positives and negatives. Moreover, RiboDetector also demonstrated a notable generalizability for detecting rRNA sequences divergent from the training data over other methods. While alignment-based methods generated more false positives with partial matches to rRNAs, whereas the HMM-based method was substantially slower in terms of runtime and produced numerous false negatives. RiboDetector showed a false negative rate over 15% on the rRNA sequences with divergence >25% from training dataset, but this is about two times lower than those of other methods. In practice, this case will be very rare, as the sequences in the training dataset well represent rRNA sequence diversity and we identified only 144 rRNA sequences in the whole SILVA database with divergence ≥20% from the training dataset. The LSTM is capable of memorizing many previous steps in a sequence, which, together with the highly efficient implementation provided by RiboDetector based on the Pytorch package, allows for a more accurate and rapid prediction compared with alignment- and HMM-based methods.
The misclassification of reads by rRNA removal methods can introduce bias to the downstream analysis. More specifically, the abundance of a given non-rRNA gene will be underestimated if the reads originating from it are misclassified as rRNA reads and thus removed. On the other hand, the abundance of a non-rRNA gene will be overestimated if false negatives of a rRNA removal method can be mapped to this gene. If the genes with a biased abundance estimation are enriched in certain functional groups, the entire analysis of these functional groups will be biased. On the CDS benchmark dataset, the few false positives of RiboDetector introduced a very low level of functional bias compared with other methods. CDSs sharing partial sequence similarity to rRNAs are enriched in certain functional groups, which may cause the enrichment of false positives for alignment-based approaches in numerous functional groups. Additionally, many false positives of RiboDetector did not share sequence similarity to rRNA at all, which may also explain why these reads were not enriched in many functional groups.
The sample sizes in microbiome or ncRNA studies are generally large, with each sample consisting of tens of millions of reads. Processing such datasets is excessively time-consuming and computationally intensive. rRNA read removal can be the most time-consuming step in the entire data processing pipeline. RiboDetector is able to process tens of millions of reads in a few minutes and it is over 30 times faster than the most widely used method (SortMeRNA), yet achieves a misclassification rate six times smaller. RiboDetector does not need a large reference database, unlike other methods, and the size of the model file is around 1.5 MB.
Currently, RiboDetector is intended for rRNA short read detection from sequence data. As it demonstrated a remarkable performance in nucleotide sequence classification, we will develop methods with a similar architecture for rRNA gene annotation from genome assembly data or other sequence recognition problems in the future.
DATA AVAILABILITY
RiboDetector is available under a GPL v3.0 license at https://github.com/hzi-bifo/RiboDetector. A Docker image is available at https://hub.docker.com/r/dawnmy/ribodetector. The benchmark datasets used in this study are available via the link https://zenodo.org/record/5547691.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Megan Foster for editing the text and Dr. Ehsaneddin Asgari for helpful suggestions regarding the description of the deep learning architecture.
Contributor Information
Zhi-Luo Deng, Department for Computational Biology of Infection Research, Helmholtz Center for Infection Research, Braunschweig, Germany; Braunschweig Integrated Centre of Systems Biology (BRICS), Technische Universität Braunschweig, Braunschweig, Germany.
Philipp C Münch, Department for Computational Biology of Infection Research, Helmholtz Center for Infection Research, Braunschweig, Germany; Braunschweig Integrated Centre of Systems Biology (BRICS), Technische Universität Braunschweig, Braunschweig, Germany.
René Mreches, Department for Computational Biology of Infection Research, Helmholtz Center for Infection Research, Braunschweig, Germany; Braunschweig Integrated Centre of Systems Biology (BRICS), Technische Universität Braunschweig, Braunschweig, Germany.
Alice C McHardy, Department for Computational Biology of Infection Research, Helmholtz Center for Infection Research, Braunschweig, Germany; Braunschweig Integrated Centre of Systems Biology (BRICS), Technische Universität Braunschweig, Braunschweig, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2155 ‘RESIST’ [39087428]. Funding for open access charge: Helmholtz Centre for Infection Research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lodish H., Berk A., Lawrence Zipursky S., Matsudaira P., Baltimore D., Darnell J.. Processing of rRNA and tRNA. 2000; W. H. Freeman. [Google Scholar]
- 2. Karpinets T.V., Greenwood D.J., Sams C.E., Ammons J.T.. RNA:protein ratio of the unicellular organism as a characteristic of phosphorous and nitrogen stoichiometry and of the cellular requirement of ribosomes for protein synthesis. BMC Biol. 2006; 4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenow C., Saxena R.M., Durst M., Gingeras T.R.. Prokaryotic RNA preparation methods useful for high density array analysis: comparison of two approaches. Nucleic Acids Res. 2001; 29:E112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scott M., Gunderson C.W., Mateescu E.M., Zhang Z., Hwa T.. Interdependence of cell growth and gene expression: origins and consequences. Science. 2010; 330:1099–1102. [DOI] [PubMed] [Google Scholar]
- 5. Cooper G.M. RNA Processing and Turnover. 2000; Sinauer Associates. [Google Scholar]
- 6. Palazzo A.F., Lee E.S.. Non-coding RNA: what is functional and what is junk. Front. Genet. 2015; 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Croucher N.J., Thomson N.R.. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010; 13:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Filiatrault M.J. Progress in prokaryotic transcriptomics. Curr. Opin. Microbiol. 2011; 14:579–586. [DOI] [PubMed] [Google Scholar]
- 9. Bashiardes S., Zilberman-Schapira G., Elinav E.. Use of metatranscriptomics in microbiome research. Bioinform. Biol. Insights. 2016; 10:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arrigoni A., Ranzani V., Rossetti G., Panzeri I., Abrignani S., Bonnal R.J.P., Pagani M.. Lanzuolo C., Bodega B.. Analysis RNA-seq and noncoding RNA. Polycomb Group Proteins: Methods and Protocols. 2016; NY: Springer; 125–135. [DOI] [PubMed] [Google Scholar]
- 11. Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014; 15:205–213. [DOI] [PubMed] [Google Scholar]
- 12. Herbert Z.T., Kershner J.P., Butty V.L., Thimmapuram J., Choudhari S., Alekseyev Y.O., Fan J., Podnar J.W., Wilcox E., Gipson J.et al.. Cross-site comparison of ribosomal depletion kits for illumina RNAseq library construction. BMC Genomics. 2018; 19:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szafrański S.P., Deng Z.-L., Tomasch J., Jarek M., Bhuju S., Meisinger C., Kühnisch J., Sztajer H., Wagner-Döbler I.. Functional biomarkers for chronic periodontitis and insights into the roles of prevotella nigrescens and fusobacterium nucleatum; a metatranscriptome analysis. NPJ Biofilms Microbiomes. 2015; 1:15017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reck M., Tomasch J., Deng Z., Jarek M., Husemann P., Wagner-Döbler I.COMBACTE Consortium . Stool metatranscriptomics: a technical guideline for mRNA stabilisation and isolation. BMC Genomics. 2015; 16:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deng Z.-L., Sztajer H., Jarek M., Bhuju S., Wagner-Döbler I.. Worlds apart - Transcriptome Profiles of key oral microbes in the periodontal pocket compared to single laboratory culture reflect synergistic interactions. Front. Microbiol. 2018; 9:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng Z.-L., Gottschick C., Bhuju S., Masur C., Abels C., Wagner-Döbler I.. Metatranscriptome analysis of the vaginal microbiota reveals potential mechanisms for protection against metronidazole in bacterial vaginosis. Msphere. 2018; 3:e00262-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mauro V.P., Edelman G.M.. rRNA-like sequences occur in diverse primary transcripts: implications for the control of gene expression. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kermekchiev M., Ivanova L.. Ribin, a protein encoded by a message complementary to rRNA, modulates ribosomal transcription and cell proliferation. Mol. Cell. Biol. 2001; 21:8255–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Root-Bernstein R., Root-Bernstein M.. The ribosome as a missing link in prebiotic evolution III: over-representation of tRNA- and rRNA-Like sequences and plieofunctionality of ribosome-related molecules argues for the evolution of primitive genomes from ribosomal RNA modules. Int. J. Mol. Sci. 2019; 20:e00262-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elitzur S.B., Cohen-Kupiec R., Yacobi D., Fine L., Apt B., Diament A., Tuller T.. Prokaryotic rRNA-mRNA interactions are involved in all translation steps and shape bacterial transcripts. RNA Biol. 2021; 18:684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang Y., Gilna P., Li W.. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics. 2009; 25:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J.-H., Yi H., Chun J.. rRNASelector: a computer program for selecting ribosomal RNA encoding sequences from metagenomic and metatranscriptomic shotgun libraries. J. Microbiol. 2011; 49:689–691. [DOI] [PubMed] [Google Scholar]
- 23. Zhou Q., Su X., Jing G., Chen S., Ning K.. RNA-QC-chain: comprehensive and fast quality control for RNA-Seq data. BMC Genomics. 2018; 19:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmieder R., Lim Y.W., Edwards R.. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics. 2012; 28:433–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kopylova E., Noé L., Touzet H.. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012; 28:3211–3217. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y., Hu H., Li X.. rRNAFilter: a fast approach for ribosomal RNA read removal without a reference database. J. Comput. Biol. 2017; 24:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 28. Singh J., Hanson J., Paliwal K., Zhou Y.. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 2019; 10:5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang L., Liu Y., Zhong X., Liu H., Lu C., Li C., Zhang H.. DMfold: a novel method to predict RNA secondary structure with pseudoknots based on deep learning and improved base pair maximization principle. Front. Genet. 2019; 10:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mao K., Wang J., Xiao Y.. Prediction of RNA secondary structure with pseudoknots using coupled deep neural networks. Biophys. Rep. 2020; 6:146–154. [Google Scholar]
- 31. Hochreiter S., Schmidhuber J.. Long short-term memory. Neural Comput. 1997; 9:1735–1780. [DOI] [PubMed] [Google Scholar]
- 32. Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O.. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013; 41:D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altenhoff A.M., Glover N.M., Train C.-M., Kaleb K., Warwick Vesztrocy A., Dylus D., de Farias T.M., Zile K., Stevenson C., Long J.et al.. The OMA orthology database in 2018: retrieving evolutionary relationships among all domains of life through richer web and programmatic interfaces. Nucleic Acids Res. 2018; 46:D477–D485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steinegger M., Söding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 35. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen T., Yu W.-H., Izard J., Baranova O.V., Lakshmanan A., Dewhirst F.E.. The human oral microbiome database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database. 2010; 2010:baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang W., Li L., Myers J.R., Marth G.T.. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Conrads G., Wendt L.K., Hetrodt F., Deng Z.-L., Pieper D., Abdelbary M.M.H., Barg A., Wagner-Döbler I., Apel C.. Deep sequencing of biofilm microbiomes on dental composite materials. J. Oral Microbiol. 2019; 11:1617013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu H.-M., Yang D., Liu Z.-F., Hu S.-Z., Yan S.-H., He X.-W.. Density distribution of gene expression profiles and evaluation of using maximal information coefficient to identify differentially expressed genes. PLoS One. 2019; 14:e0219551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang J., Huang M., Torre E., Dueck H., Shaffer S., Murray J., Raj A., Li M., Zhang N.R.. Gene expression distribution deconvolution in single-cell RNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E6437–E6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bengtsson M., Ståhlberg A., Rorsman P., Kubista M.. Gene expression profiling in single cells from the pancreatic islets of langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005; 15:1388–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deng Z.-L., Szafrański S.P., Jarek M., Bhuju S., Wagner-Döbler I.. Dysbiosis in chronic periodontitis: key microbial players and interactions with the human host. Sci. Rep. 2017; 7:3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000 Genome Project Data Processing Subgroup . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Köster J., Rahmann S.. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 46. Chen S., Zhou Y., Chen Y., Gu J.. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RiboDetector is available under a GPL v3.0 license at https://github.com/hzi-bifo/RiboDetector. A Docker image is available at https://hub.docker.com/r/dawnmy/ribodetector. The benchmark datasets used in this study are available via the link https://zenodo.org/record/5547691.