


Abstract
The spread of drug-resistant bacteria represents one of the most significant medical problems of our time. Bacterial fitness loss associated with drug resistance can be counteracted by acquisition of secondary mutations, thereby enhancing the virulence of such bacteria. Antibiotic rifampicin (Rif) targets cellular RNA polymerase (RNAP). It is potent broad spectrum drug used for treatment of bacterial infections. We have investigated the compensatory mechanism of the secondary mutations alleviating Rif resistance (Rifr) on biochemical, structural and fitness indices. We find that substitutions in RNAP genes compensating for the growth defect caused by βQ513P and βT563P Rifr mutations significantly enhanced bacterial relative growth rate. By assaying RNAP purified from these strains, we show that compensatory mutations directly stimulated basal transcriptional machinery (2–9-fold) significantly improving promoter clearance step of the transcription pathway as well as elongation rate. Molecular modeling suggests that compensatory mutations affect transcript retention, substrate loading, and nucleotidyl transfer catalysis. Strikingly, one of the identified compensatory substitutions represents mutation conferring rifampicin resistance on its own. This finding reveals an evolutionary process that creates more virulent species by simultaneously improving the fitness and augmenting bacterial drug resistance.
Graphical Abstract
Graphical Abstract.
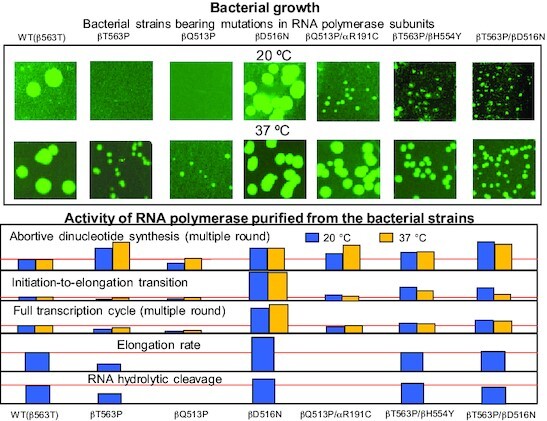
Growth rates at 20 °C and 37 °C for the E. coli strains studied in this work (top) and the activity of RNA polymerase purified from these strains in various in vitro assays at the same temperatures (bottom).
INTRODUCTION
The spread of antibiotic resistance among bacterial pathogens is occurring at an alarming rate over the recent years (1,2) as large pharmaceutical companies have virtually withdrawn from drug development (3). Resistance is associated with longer treatment time and poorer outcomes. Antibiotic rifampicin (Rif) is a potent broad spectrum antibacterial agent widely used in the clinic. It is the first-line drug in anti-tuberculosis chemotherapy. Studies of laboratory-generated rifampicin resistant (Rifr) mutants or clinical isolates of Escherichia coli (4–11),Mycobacterium tuberculosis (12–16), Staphylococcus aureus (17), Salmonella enterica (18–20), Enterococcus faecium (21) and Pseudomonas aeruginosa (22) suggest that acquisition of Rif resistance impacts bacterial fitness as measured by in vitro growth rates. Rifr bacteria that suffer reduced fitness can further evolve to acquire secondary, compensatory mutations that eliminate the cost of resistance (11–22). Since these additional mutations can render the organisms equally or even more fit than wild type bacteria and therefore, more virulent, a better understanding of the underlying compensatory mechanisms and their relevance to drug-resistant epidemiology is warranted.
The Rif group of antibiotics target bacterial RNA polymerase, RNAP (23–26), which is a vital cellular enzyme executing transcription, the first step of gene expression (Figure 1). Transcription (for review see (27)) starts with promoter recognition by RNAP followed by the melting of the DNA double helix to expose a single strand segment that is used as a template for RNA synthesis. Short RNA transcripts produced at the starting stage of RNA synthesis in the transcription initiation complex (TIC) are easily released by the active center in cycles of abortive initiation. Initiation-to-elongation transition (promoter escape, or promoter clearance) is manifested by relinquishing the promoter recognition contacts by RNAP and dramatic stabilization of transcription complex. Resulting ternary elongation complex (TEC) is highly processive, but eventually disintegrates in response to termination signal encoded in DNA at the end of a transcription unit. During transcription RNA products remain transiently base-paired to the template DNA forming RNA:DNA hybrid about 9–10 bp-long (28). Advancement of the RNAP along DNA upon RNA synthesis is occasionally interrupted by enzyme's back-translocation (28,29), which releases 3′ RNA segment from the hybrid, thus creating catalytically inactive state. Such transcription complex can be re-activated through hydrolytic removal of the disengaged RNA segment (30–32), which is performed by the same active center that normally executes RNA synthesis (33).
Figure 1.

Structural and genetic context of rifampicin resistance determining region (RRDR) of bacterial RNA polymerase and location of the rifampicin resistant and compensatory mutations selected in this study. (A) Top, location of the selected mutations in RNAP subunits. Bottom, sequence alignment of RRDR from various organisms. Positions of known Rifr mutations along with those identified in this work (black font) and corresponding compensatory substitutions (red font) are indicated. (B) Locations of the selected mutations in RNAP TEC (panel I). Panels II-VI represent enlarged images of I showing the location of particular mutations. Primary Rifr mutations and corresponding compensatory mutations have the same color coding. Green sphere represents catalytic Mg2+ ion of the active center. Structure of E. coli RNAP TEC is from Supporting Information of (54).
The Rif agents—notably rifampicin, rifapentine, and rifabutin function by binding to and inhibiting RNAP (34,35). These drugs insert into a site on RNAP adjacent to the active center (Figure 1B and Figure 8B) and prevent synthesis of RNA products >2–3 nt in length by plugging the transcript exit channel. Superior performance of Rif is due to extremely high affinity to RNAP (2–4 nM dissociation constant). The primary mechanism of rifampicin resistance is due to mutations (4–22) along a 186-bp hotspot RRDR (rifampicin resistance determining region) within the RNAP β subunit encoded in rpoB gene (Figure 1A). Rif resistance is attributable to mutations of the residues within, or immediately adjacent to the drug binding site on RNAP, i.e. changes that directly decrease binding of the inhibitors (35). Since these residues participating in Rif binding are also involved in retention of nascent RNA transcript (Figure 1B, panels II–IV) the mutations conferring resistance to the drug affect the normal pathway of RNA synthesis resulting in multiple catalytic defects and therefore, reduced fitness (4–22).
Figure 8.

Location of some Rifr compensatory mutations in Mtb (12) and E. coli (present work) (A) Sequence alignments of the regions of β′, β and α subunits of various organisms bearing compensatory mutations. Compensatory mutations are indicated. Mutations found in the present work are circled. Identical residues in the sequences are highlighted. (B) Structural context of the compensatory mutations. Location of the primary Rifr and compensatory mutations (both in space-fil representation) in the RNAP elongation complex is shown. Residues numbering is as in E. coli. Active center residues (pink) coordinating catalytic Mg2+ ion (green sphere) are indicated along with emerging RNA transcript (red). Position of Rif binding pocket is marked. Note, that all indicated residues are identical in E. coli and Mtb. E. coli RNAP structure is from (54).
In recent years, a number of mutations in various bacteria that are associated with primary mutations leading to Rif resistance have been detected within RNAP coding genes, rpoA-C (11–22). Since these secondary substitutions alleviated fitness cost of the drug resistance, they have been named ‘compensatory’ mutations. Studies on the molecular mechanism of the compensatory mutations are at the initial stage (36). Two principal scenarios for the compensatory action can be envisioned. Thus, these substitutions can either influence RNAP catalytic function, or act indirectly (e.g. through engaging transcription factors that suppress the defect of primary mutation as they bind to RNAP).
In the present research to explore compensatory mechanism we have conducted a systematic study to relate the fitness of a wild type (WT) ancestral, Rif resistant and corresponding evolved bacterial strain with compensatory mutation to transcriptional activity of RNAP purified from these strains. For these studies we’ve chosen E. coli, well characterized microorganism. Since the RNA polymerase residues involved in Rif binding as well as the identified loci for compensatory mutations are identical in many bacteria (Figures 1A and 8A), the results and conclusions of our study are relevant to other pathogenic species including M. tuberculosis (MTB).
We demonstrate that tested compensatory mutations directly affect RNA synthesis by increasing the RNAP performance at several steps of transcription cycle including first phosphodiester bond formation, promoter clearance, elongation, and RNA hydrolytic cleavage, thereby exerting pleiotropic stimulatory action on catalysis.
This approach aided by molecular modeling revealed: (i) how the change in RNAP basal transcription activity caused by the mutations affects bacterial growth, (ii) new interaction networks in transcribing complex that modulate RNA synthesis and (iii) the details of the transcription mechanism on the atomic level. Our multidisciplinary study represents a milestone on the way to detailed understanding the compensatory evolution and its impact on epidemiology of bacterial pathogens, which is necessary for development relevant measures to mitigate such infections.
MATERIALS AND METHODS
All chemicals were from Sigma-Aldrich (St Louis, MO, USA). Ultra-pure ribonucleoside-5′-triphosphates were from Pharmacia (Uppsala, Sweden). [α-32P]CTP was from MP Biomedicals (Santa Ana, CA, USA). Radioactive products of enzymatic reactions were resolved by electrophoresis in 20% polyacrylamide gel in the presence of 7 M urea and quantified by phosphoimagery using a Molecular Dynamics device GE Healthcare (Chicago, IL, USA). Molecular modeling was performed using WebLab ViewerLight 4.0, Molecular Simulations Inc (San Diego, CA, USA). Oligonucleotides were from Integrated DNA Technologies (Coralville, IA, USA). RL721 E. coli strain containing a His-6 tag fused to the C terminus of RNAP β′ subunit (37) was a gift of R. Landick.
Selection and characterization of Rifr mutants
Rifr mutants were selected by plating ca. 1010 CFU of E. coli RL721 strain on the LB agar medium containing Rif (25 μg/ml) and Kan (50 μg/ml). After overnight incubation at 37°C the material from the grown colonies was spread on the fresh agar plates with the same concentration of Rif and Kan to obtain single colonies. After three more passages the cells from the colonies were grown in liquid LB medium, mixed with glycerol to final glycerol concentration 30% and kept at −80°C. The mutations conferring Rif resistance were mapped as described below.
Compensatory evolution of Rifr strains
For these experiments we have chosen βQ513P and βT563P mutants, displaying the slowest relative growth rate at 20°C. The grown bacterial cells (∼1010 CFU) were plated on agar medium containing Kan (50 μg/ml). After 72 h incubation single colonies were spotted and the material from these colonies was transferred to agar plates with Kan and grown at 20°C to obtain single colonies, which were re-plated 3 times onto the same medium. The cells from final passage were transferred to liquid LB medium, grown to the density 0.2–0.3 au (at 600 nm), supplemented with glycerol to final concentration 30% and stored at −80°C. The compensatory mutations were mapped as described below.
Determination of the bacterial strains growth rate
The suspension of the cells from selected bacterial strains in PBS were adjusted to the same turbidity measured at 600 nm. Aliquots of the cells were transferred to the wells of microtiter plate filled with LB medium containing Kan (50 μg/ml). The plates were kept at either 20°C or 37°C. The time course for bacterial grows was monitored by measuring absorption at 600 nm using a plate reader. OD600 curves were blanked and the growth rates were determined by using the absorption values from log-transformed growth curves. For the assessment of the bacterial growth in solid medium cell suspensions were plated on agar LB medium containing Kan (50 μg/ml) and incubated at either 20°C or 37°C. The colonies’ size was measured using Biorad ChemiDoc XRS Imaging System. Twenty colonies of each strain were chosen to determine the average size. In both approaches, the growth experiments were repeated three times using material from different colonies of the same bacterial strain.
PCR and sequence analysis
The Rifr mutations in rpoB were mapped by PCR analysis using primers GTAGAGCGTGCGGTGAAAGAGCGTCTGTCT (sense), GAAGACCTGGTAACTTGCCGTAGCAAAGGC (antisense) flanking the region 1467–1903 of rpoB gene containing RRDR. Genomic DNA was harvested from each strain of interest, and the indicated above gene's fragment was amplified by PCR using QIAGEN Taq DNA polymerase in conjunction with the above primers. Compensatory mutations were located using the whole genome sequencing.
Purification of RNAP
RNAP was purified by combination of the protocols described in (38,39). Briefly, the purification steps included cell lysis, precipitation of RNAP/DNA complexes with polymin P, extraction of the enzyme from the pallet by sodium chloride buffered solution, affinity chromatography on Ni-nitrylotriaceto agarose (NTA) and heparin-agarose, followed by size-exclusion chromatography on Superpose 6. The resulting material was collected, concentrated to 1 mg/ml (measured by absorption at 280 nm and using extinction coefficient 0.693 au/mg/ml (38)) and stored at −80°C in the 10 mM Tris–HCl buffer containing 0.1 M KCl and 50% glycerol.
Abortive initiation assay
In multiple-round version the reaction mixture (9 μl) containing 0.5 pmol T7A2 promoter (40), 2 pmol RNAP, 5 pmol purified σ subunit in transcription buffer, TB (20 mM Tris–HCl pH 8.0, 0.1 M NaCl, 10 mM MgCl2) was incubated 5 min at 37°C. The reaction was initiated by addition of 1 μl of the mixture of 2 mM GTP, 20 μM [α-32P]CTP (specific activity 30 Ci/mmol) and after 30 min incubation at 20°C, or at 37°C, quenched by addition of 10 μl of 10 M urea with 0.05% BPB dye. Aliquots of the reaction mixture (1 μl) were applied on PEI cellulose sheet (Sigma-Aldrich) and the reaction products separated by TLC using 0.2 M KH2PO4 as developing solvent. The products were quantified by phosphoimagery. The reaction mixture for single-round assay was essentially the same but the final concentration of [α-32P]CTP was 3 nM (specific activity 3000 Ci/mmol). The reaction time was 4 min at 20°C and 1 min at 37°C.
Promoter clearance assay
The composition of the reaction mixture was the same as in the previous assay, but the reaction was started by addition of 1 μl of the mixture containing 2 mM ATP, UTP, GTP, and 10 μM [α-32P]CTP (specific activity 30 Ci/mmol). Incubation time was 30 min at 20°C and 10 min at 37°C. The reaction was stopped as descried above and the reaction products analyzed by electrophoresis in 20% polyacrylamide gel containing 7 M urea. The products were visualized and quantified by phosphoimagery.
Full transcription cycle assay
The reaction mixture (9 μl) containing 2 pmol RNAP, 0.05 pmol pTZ plasmid with cloned T7A1 promoter (41) and 5 pmol purified σ subunit in transcription buffer was incubated for 5 min at 37°C and supplemented with 1 μl of 0.25 mM ATP, UTP, GTP, and [α-32P]CTP (specific activity 30 Ci/mmol). After incubation for 2 h at 20°C or 40 min at 37°C the reaction was quenched by addition of 10 M urea and the aliquots (5 μl) applied on Whatman 3MM paper discs and washed by 5% trichloroacetic acid (3 × 100 ml, 20 min each wash). The discs were rinsed by ethanol, air-dried and the remaining radioactive material quantified by phosphoimagery.
Elongation assay
Ternary elongation complexes (TEC) were assembled from RNAP and synthetic oligonucleotides as described elsewhere (42). DNA template strand: 5′ ACCAGCAGGCCGATTGGGATGGGTATTCGCCGTGTACCTCTCCTAGCCCCCAAGTATCCTATAGG 3′ DNA non-template strand: 5′CCTATAGGATACTTGGGGGCTAGGAGAGGTACACGGCGAATACCCATCCCAATCGGCCTGCTGGT 3′; RNA: 5′ GGAGAGGUA 3′. Briefly, 1 pmol of DNA template strand was mixed with equimolar amount of RNA and RNAP holoenzyme in 10 μl of TB. After 10 min incubation at room temperature 1 pmol of DNA non-template strand was added and the incubation continued at 37°C for another 10 min. The mixture was supplemented with 0.5 μl of [α-32P]CTP (specific activity 3000 Ci/mmol, 1 mCi/ml) and kept for 2 min at room temperature. The labeled TECs were immobilized on NTA beads and washed with TB (3 × 1 ml) to remove non-incorporated labeled CTP. A portion of beads suspension (9 μl) was mixed with 1 μl of 4 NTPs (final concentration 1 μM) and after 30 s incubation at ambient temperature, the reaction was stopped by addition of equal volume of 10 M urea. The reaction products were analyzed by denaturing gel electrophoresis and phosphoimaging as described above.
Molecular modeling of the effect of T563P substitution
To model the mutation we inserted proline residue into β563 locus by aligning the position of the corresponding atoms of the backbone of the proline and threonine residue followed by deletion of the atoms constituting Thr side chain. This substitution eliminated an H-bond between hydrogen of the amido group connecting βE562 and βT563 residues and a carbonyl oxygen of the peptide bond between βV561 and βE562 residues. In addition, the substitution created a steric hindrance between γ carbon of the proline side chain and the carbonyl oxygen of βV561. This clash can be resolved in three principal ways by: (i) 38° rotation around the bound connecting the Cα and carbonyl carbon of βE562 residue, so that ψ backbone dihedral angle changes from 6° to 44°; (ii) 16° rotation around carboxamide bond between βV561 and βE562 resulting in ω backbone dihedral angle changes from −173° to −157° and (iii) 17° rotation around the bond between nitrogen and Cα of βE562 resulting in φ backbone dihedral angle change from −50° to −67°. In all cases, the immediate effect of the rotation was distancing of the βP564 side chain from the phosptate group of the RNA residue at −3 position by about 0.9 Ǻ in the first and the third cases and by 3.7 Ǻ in the second case.
RESULTS
Rifr mutations affect E. coli growth rate
We have generated collection of spontaneous Rifr mutants by plating the bacteria on selective agar medium. Sequencing the genomic DNA from the grown colonies revealed the presence of 13 various non-synonymous substitutions in the region of rpoB gene coding for 512–574 (amino acid numbering) protein segment of RNAP β subunit, which are a subset of previously reported (4–11). The identity of mutations and the frequency of their occurrence are presented in Supplementary Table S1. The ancestral strain used for mutant selection (RL-721) included an additional change conferring kanamycin resistance along with His6 tag genetically fused to the C-terminus of RNAP β′ subunit to facilitate the enzyme purification.
As suggested by our previous data (10), effect of the common Rifr mutations on RNA synthesis is modest. Therefore, for reliable detection of transcription defects we have chosen the least fit βQ513P and βT563P mutants. All the experiments were performed at 37°C and 20°C since the latter is an average temperature for natural habitats where E. coli is able to proliferate (43). In addition, by conducting the tests at these two temperatures, we intended to investigate the nature of cold sensitivity for the above mutants, which has not been addressed so far.
First, we determined the relative growth rate of the selected Rifr mutants by measuring their duplication time in liquid medium. In parallel, the bacterial fitness (defined as relative growth rate) was estimated qualitatively by comparing the colony size on agar plates. These two approaches yielded consistent results. Thus, the colony size determined upon plating was proportional to the growth rate observed for the same strains in the liquid medium.
In agreement with previous studies (9) βQ513P and βT563P mutants displayed significantly reduced relative growth rate (Supplementary Table S1 and Figure 2). Thus, in liquid medium at 20°C the relative growth rate was about 20% for the first, and about 40% for the second strain (Figure 2). Raising the growth temperature to 37°C enhanced the fitness to 70% and 67% of WT, respectively (Figure 2A). The same trend was observed for the bacterial growth on agar plates (Figure 2B and C). Thus, no growth for βQ513P and βT563P mutants was detected after 84 h incubation at 20°C, while WT strain produced visible colonies. At this temperature detectable colonies were observed only after 135 h for βT563P and 410 h βQ513P mutant (Figure 2B), while at 37°C the colonies were seen after 22 h incubation. Notably, for some bacterial strains smaller colonies were observed in population. However, they constituted relatively small fraction of the total. Therefore, we believe this factor did not contribute significantly to our results. Additional study is required to address this phenomenon.
Figure 2.

Growth rates for RNAP mutant strains. (A) Relative growth rates measured in liquid medium. Error bars represent standard deviation from three independent experiments in which different colonies of the same strain were used. The growth curves are presented in the Figure 1 of the Supplemental Material. (B, C). Growth for the same strains as in (A) determined by plating. Growth temperature, incubation time, and colony size are indicated where appropriate.
Compensatory mutations increase bacterial proliferation rate
To select suppressor mutations that would compensate for the fitness loss, we took advantage of the slow growth of the βQ513P and βT563P mutants at 20°C. Thus, we grew a large load of bacteria in the absence of Rif and plated them on agar medium lacking the drug. Some fast growing colonies were spotted and recovered. As followed from the sequence analysis, in the case of βT563P mutant, along with original Rifr substitution, the selected fast-growing cells contained additional βD516N or βH554Y mutations in the same RNAP subunit (rpoB gene). The βQ513P- bearing fast growing strains displayed a single secondary mutation in one of core RNAP subunits: αR191C (rpoA gene), βD142G (rpoB gene), or β′D1051N (rpoC gene). Secondary substitutions improved bacterial relative growth rate (Figure 2). The effect was temperature-dependent. In all cases it was more pronounced at 20°C compared to 37°C. Thus, βD516N, βH554Y, αR191C, βD142G and β′D1051N mutations increased the relative growth rates of the ancestral strains about 1.6-, 1.75-, 4-, 5.5- and 4.8-fold, respectively at 20°C. At 37°C βD516N, βH554Y mutations marginally increased the growth rate of the original βT563P mutant (1.05- and 1.2-fold, respectively), while the relative effect of αR191C, βD142G and β′D1051N compensatory mutations on the growth of βQ513P was greater at this temperature (1.7-, 1.64- and 1.47-fold, respectively). The growth-stimulating effect of the compensatory substitutions was observed when the bacteria were grown in liquid (Figure 2A), or solid media (Figure 2B, C). Thus, detectable colonies for parental βQ513P mutant were seen only after 400 h incubation at 20°C and after 130 h for βT563P mutant, while all strains with compensatory mutations produced visible colonies after 84 h growth in both cases (Figure 2B, C). Remarkably, the magnitude of compensatory effect observed for βQ513P and βT563P Rifr strains was much more significant than that previously reported for other Rifr mutants with less severe growth defects (11,12,18).
Compensatory mutations affect catalysis and product retention at the active center in abortive RNA synthesis
To assess the effect of the primary and compensatory mutations on basal RNAP transcription we purified the enzyme from relevant bacterial strains and performed in vitro transcription assays.
Since the bacterial relative growth rate was highly temperature-dependent, we tested the enzymes at 20°C and 37°C to determine if this steep dependence reflects the basal transcription activity. First, we tested the enzymes in the simplest RNAP reaction, dinucleotide synthesis, which proceeds in the TIC in the presence of two nucleotide substrates, specified by promoter sequence. Initially, we performed the assay in the multiple-round mode with T7 phage A2 (T7A2) promoter-bearing DNA fragment using GTP and [α-32P]CTP substrates that yield dinucleotide pppGpC as the reaction product. Since the synthesized product dissociates from the active center, in the excess of NTPs the synthesis can repeat multiple times. As seen in Figure 3A at 20°C the βT563P and βQ513P substitutions displayed the opposite effects on the reaction product accumulation (200% and 70% of WT, respectively). Compared to WT, increased reaction rate was observed for both compensatory variants for βT563P mutation (βT563P/βD516N and βT563P/βH554Y) as well as for βD516N control mutant. The same effect was seen for all mutations compensating for βQ513P catalytic defect (αR191C, βD142G and β′D1051N). Raising the reaction temperature to 37°C moderately (1.3-fold) increased the relative performance of βT563P mutant (normalized to WT RNAP), whereas compensatory variants for this mutant as well as βD516N substituted RNAP were stimulated to the same extent as WT enzyme. The temperature raise enhanced the βQ513P mutant activity (about 2-fold) to the level of WT enzyme. As βQ513P mutant, all compensatory variants for this enzyme were stimulated to a greater extent than WT RNAP by the temperature raise.
Figure 3.

Activity of the WT and mutant enzymes in dinucleotide synthesis (A, B) and promoter clearance (C) assays. (A) Multiple-round conditions. Top, TLC analysis of the reaction products. Bottom, quantitation of the results of the top panel. (B) Single-round conditions. Top, TLC analysis of the reaction products. Bottom, quantitation of the results of the top panel. (C) Compensatory effect of secondary mutations on promoter escape by RNA polymerase. Top. Separation of the reaction products by gel-electrophoresis (representative gel). Relative amount of the elongated products and elongated-to-abortive products ratio for each enzyme are indicated. Bottom. Graphical representation of the results of the top panel. Error bars represent standard deviation from three independent experiments.
The enhanced reaction rate seen for some enzyme variants described above could be due to improved catalytic performance or due to destabilization of the synthesized dinucleotide at the active center (10). The latter would promote product dissociation, increasing the turnover, and therefore, the apparent reaction rate. Decreased reaction rate could be the result of slower nucleotidyl transfer reaction, or reflect stronger binding of the reaction product to the active center, which would retard turnover. To distinguish between these possibilities, we carried out the same reaction, but in single-round conditions by using concentrations of radiolabeled CTP substrate lower than that of the RNAP transcription complex. For the absence of turnover factor, at these conditions, variation in the yield of the dinucleotide product would reflect the change in enzymes’ catalytic performance. Surprisingly, in this setup the pppGpC yield for βT563P was significantly higher (6-fold, compared to WT) than in multiple round conditions (2-fold), suggesting that the mutation simultaneously increased the rate of nucleotidyl transfer reaction and also suppressed dinucleotide dissociation from TIC (Figure 3B). The same conclusion can be drawn for the mutations alleviating the defects of βQ513P substitution. Relative activity of the βT563P/βD516N and βT563P/βH554Y double mutants was nearly the same in both assays. Therefore, the enhanced dinucleotide production for these mutants compared to βT563P can be accounted for increased catalytic performance. Notably, as each of single βT563P and βD516N substitutions significantly improved catalysis in single-round assay (6–7-fold compared to WT RNAP), double βT563P/βD516N mutant was only about 2-fold more active than WT enzyme. The absence of synergistic effect of the mutations reflects complex mechanism for the mutations interactions and their effect on catalysis. While at 20°C the βQ513P enzyme displayed reduced reaction rate at multiple-round conditions, in single-round mode its activity was equal to that of WT RNAP at the same temperature, suggesting that smaller dinucleotide yield (compared to WT enzyme) in the first test was due to stronger product retention at the active center. Compensatory substitutions αR191C, βD142G and β′D1051N moderately increased the relative reaction rate (compared to that of primary βQ513P mutant) at multiple-round conditions. At the same time, the effect of these compensatory mutations in the single-round assay was more pronounced (especially at 20°C) reflecting strong positive influence of the substitutions on the catalytic activity at this temperature. These results are consistent with stronger influence of the same mutations on bacterial relative growth rate at lower temperature. In multiple-round conditions, the temperature increase stimulated the dinucleotide synthesis in βQ513P enzyme and in all βQ513P compensatory variants to a greater extent than in WT RNAP. This suggests that compensatory mutations, while increasing general activity, did not eliminate temperature-sensitive defect of the primary mutation. However, in single-round assay the temperature increase had the opposite effect on the relative yield of the reaction products with all compensatory RNAP mutants (Figure 3B). This can be explained by enhanced dissociation of the product from the active center at higher temperature in multiple-round assay, which would increase turnover, and therefore, the product accumulation.
Overall, the above tests revealed that primary Rifr as well as compensatory mutations affected enzyme performance in nucleotidyl transfer reaction and product retention at the active center, thus reflecting complex mutations effect on catalysis.
Compensatory substitutions enhance initiation-to-elongation transition by RNAP
Promoter clearance efficiency defined as the elongated-to-aborted products ratio is an important characteristic reflecting the ability of the enzyme to escape the promoter and carry out productive RNA synthesis (44). Since both primary Rifr substitutions rpoB βT563P and βQ513P reside in RNA exit channel (Figure 1, panel II) it was expected that they would display the most prominent effect at the above step of transcription due to high sensitivity of the TIC to the nucleic acid-protein interactions retaining RNA product.
As seen in Figure 3C, transcription from the T7A2 promoter template yields abortive and elongated products. The latter include partially elongated RNAs, run-off transcript as well as longer products, whose size exceeds that of the full length run-off RNA. These products originate due to template switch (45) whereby RNAP relocates to another template molecule when the enzyme approaches the end of the first DNA template upon RNA synthesis, without losing the transcript. Remarkably, at 20°C βT563P substitution decreased the yield of elongated RNA transcripts (about 60% of WT) consistent with reduced relative growth rate of the corresponding mutant strain. As follows from quantitation of the reaction products (Figure 3C, bottom), this was due to the increase in the production of short abortive RNAs, so that the elongated-to-aborted products ratio dropped about 3.5-fold compared to WT enzyme. Strikingly, both compensatory substitutions βD516N and βH554Y alleviated the negative effect of the primary βT563P mutation by enhancing significantly the yield of elongated transcripts (about 7-fold), thus producing the enzymes more active than WT (about 400% of WT). Surprisingly, the control βD516N mutant (lacking primary Rifr mutation) was more active in promoter escape producing ∼9-fold more elongated products (at 20°C) and less abortive RNAs compared to the ancestral WT enzyme. This can account for the compensatory effect of the βD516N substitution when it is coupled to the βT563P primary Rifr mutation. The bulk of the compensatory effect of the secondary βD516N and βH554Y mutations can be attributed to suppression of RNA abortion, since the elongated-to-aborted products ratio for both mutants (2.15- and 2.6-fold, respectively) was close to that for WT enzyme (2.0-fold). Quantitation of the reaction products suggests that the secondary mutations also improved general catalytic activity of the ancestral βT563P enzyme variant. The testing of these enzymes in transcription elongation assay (see below) also supports the latter conclusion.
When assayed at 20°C, βQ513P substitution had significant negative effect on transcription by decreasing the yield of elongated RNAs to 50% of WT level and increasing the relative amount of abortive products (Figure 3C). All three compensatory mutations αR191C, βD142G and β′D1051N strongly (about 4-fold) improved the yield of elongated transcripts at this temperature. In the case of αR191C and β′D1051N this was achieved by increasing the general catalytic activity without changing the elongated-to-aborted products ratio, while βD142G mutation simultaneously suppressed abortive products release and improved the catalysis. These results are consistent with enhanced catalytic activity of these compensatory mutants observed in the dinucleotide synthesis reaction. The positive effect of the all of the above compensatory mutations in this test accounts for the increased relative growth rate of the corresponding bacterial strains.
To determine whether strong temperature-sensitive growth phenotype of the βT563P and βQ513P mutations reflects the change in basal activity of RNAP we repeated the above test at 37°C. It is seen that raising the reaction temperature significantly increased the abortive pathway for WT and all mutant enzymes, which indicates stronger negative effect of the temperature shift on short RNAs retention in TIC than on stimulation of RNA elongation. Strikingly, increasing the reaction temperature essentially levelled the amount of elongated transcripts for βT563P, βQ513P and WT enzymes, which is consistent with smaller difference in relative growth rates for the corresponding bacterial strains at the elevated temperature (Figure 2A). While in the case of βT563P Rifr mutation relative increase in the activity of RNAP (about 2-fold) can account for the growth rate increase upon the same temperature shift (1.6-fold), the growth rate change for βQ513P strain (3.5-fold) is greater than the temperature-induced enzyme activation (about 2-fold) as seen from Figures 2A and 3C. These data suggest that in the case of βQ513P mutation the bulk of the relative growth rate reduction at lower temperature was not due to the temperature-depended malfunction of the core transcription apparatus at this step of the transcription cycle. Notably, at 37°C the compensatory effect of the substitutions was less pronounced than at 20°C (Figure 3C (top), lanes 10–18, and Figure 3C, bottom) consistent with the relative growth rates values for the corresponding bacterial strains (Figure 2A) at this temperature.
Compensatory mutations improve RNAP activity in full transcription cycle assay
Next, we tested the mutant enzymes in a whole transcription cycle assay by measuring the amount of the RNA synthesized on circular plasmid DNA bearing Bla and T7 phage A1 promoters in multiple rounds of transcription. The enzymes’ activity was determined based on the incorporation of the radioactive material from NTP into acid-insoluble product, which constitutes the pool of transcripts whose size exceeds 8–10 nt. As seen from Figure 4A, at 20°C βT563P and βQ513P mutants possessed about 60% and 20% of WT activity, respectively. As in the previous assay, compensatory mutations significantly improved the performance of the original enzyme variants. Thus, additional βH554Y and βD516N substitutions elevated the βT563P enzyme activity to the level superior to that of WT (2.25 and 2.9-fold, respectively). Compensatory αR191C, βD142G and β′D1051N substitutions increased the activity of βQ513P enzyme by 5.5, 3, and 6-fold respectively. Elevating the assay temperature to 37°C moderately increased the activity of βT563P and βQ513P mutants (80% and 33% of that of WT), while relative effect of the suppressing mutations at this temperature was only slightly lower than at 20°C (1.63 and 2-fold for βH554Y and βD516N, respectively and 3.3-, 2.5- and 5-fold for αR191C, βD142G and β′D1051N, respectively). This is in contrast to the effect of the compensatory mutations in the promoter escape assay, in which the magnitude of the compensatory action was much greater at the lower temperature. In addition, in promoter clearance assay βH554Y and βD516N compensatory substitutions displayed generally stronger stimulatory effect than αR191C, βD142G and β′D1051N substitutions, while the opposite was observed in present test. The above differences can be due to: (i) involvement of the other events of transcription cycle (36) on the plasmid template; (ii) promoter-specific effects and (iii) the influence of plasmid supercoiling on RNA transcription. However, the general effect of the primary Rifr and compensatory mutations on transcription efficiency was the same in both assays.
Figure 4.

Performance of the mutant enzymes in whole transcription cycle and elongation assays. (A) Top, phosphoimage of the acid-insoluble radioactive RNA material accumulated during incubation of WT and mutant enzymes with circular plasmid template and NTP substrates. Bottom, quantitation of the results of the top panel. (B) Top, elongation of the RNA oligonucleotide by NTP substrates in TEC complexes with WT and mutant RNAP. Bottom, quantitation of the products of the top panel. Error bars represent standard deviation from three independent experiments.
Remarkably, as in the previous test on T7 A2 promoter template, temperature stimulation of βT563P RNAP in this assay (1.33-fold) can account for the increased growth rate of the corresponding mutant strain upon the temperature shift (1.6-fold), while temperature stimulation of the βQ513P RNAP enzyme (1.65-fold) was much lower that the growth rate increase for the mutant strain (3.5-fold). Collectively, these data strongly suggest that the bulk of the reduced relative growth rate for the βQ513P mutant at lower temperature was not due to the temperature-sensitive malfunction of the core transcription machinery.
Compensatory RNAP mutations increase RNA elongation rate and hydrolytic transcript degradation
To elucidate the influence of the primary Rifr mutations and compensatory substitutions on the elongation step of RNA synthesis we selected βT563P, βT563P/βD516N, βT563P/βH554Y, and βD516N subset of mutants, since it includes a primary Rifr mutation, the same mutation with compensatory substitution and compensatory substitution on its own. For the absence of transcript abortive dissociation, the activity of RNAP at this step of transcription cycle reflects the enzyme's true catalytic performance. In this assay we assembled the TECs from RNAP and synthetic oligonucleotides (46), thereby bypassing initiation. The RNA product in the reconstituted complexes was labeled by extension with the next cognate radioactive substrate, CTP, followed by incubation of the TECs with complete set of NTPs at 20°C. Generally, the activity patterns of mutant and WT enzymes were similar as in the promoter clearance and full transcription cycle assays. Thus, compared to WT enzyme, βT563P substitution reduced the amount of elongation products, whereas additional compensatory mutations elevated the activity of the βT563P enzyme essentially to the level of WT enzyme (Figure 4B). Control βD516N variant was about 2-fold more active than WT RNAP, thus accounting for the compensatory action of the mutation at this stage of transcription cycle. Remarkably, βD516V Rifr mutation (E. coli numbering) in M. tuberculosis also produced enzyme more active in elongation (36), suggesting the relevance of our data to MTB.
As seen in Figure 4B, along with elongated transcripts, RNA degradation products accumulate during the TEC incubation. These products originate due to RNAP intrinsic transcript hydrolytic cleavage activities (30,47) as explained in Introduction. Notably, βT563P substitution reduced the amount of both elongated and cleaved RNA products indicating that both activities were affected. Remarkably, compensatory substitutions improved the enzyme's performance in both reactions, consistent with the notion that these reactions occur at the same active center following the same catalytic mechanism (33).
Overall, the above results suggest that the negative effect of primary drug-resistance substitution as well as alleviating action of compensatory mutations extend to RNA elongation phase of transcription and to intrinsic RNA degradation reactions.
DISCUSSION
In the present study, we demonstrate that compensatory mutations in RNAP directly increase the enzyme's catalytic performance that is compromised by primary mutations conferring Rif resistance, thus restoring the bacterial fitness. In this way we generated a panel of Rifr bacterial mutants and determined their fitness (defined as relative growth rate) in the absence of the drug. In light of our previous study (10) it was expected that the effect of the selected Rifr mutations on RNAP transcription would be rather small. Therefore, for further studies we’ve chosen mutants with the most severe growth defects (βT563P and βQ513P), that rendered the bacteria temperature-sensitive. We also reasoned that compensatory substitutions efficiently alleviating the defects of the above highly deleterious mutations would identify the principal structural features and side chains interactions in RNAP that are the most critical for the catalysis augmentation.
In vitro testing of RNAP purified from βT563P and βQ513P mutants strains revealed significant malfunctions at various steps of transcription cycle (for summary see Figure 5). As expected, the most severe defect of these mutations was observed at the promoter clearance step, which is highly sensitive to RNA-protein interactions, impaired by the amino acid changes. The negative effect of these mutations on elongation was smaller (Figure 5). Conceivably, βT563P and βQ513P mutations could affect interactions of RNAP core enzyme with sigma subunit. However, this seems unlikely, since sigma subunit resides at considerable distance (13–14 Ǻ) from the side chains of the above loci. This conclusion is also supported by the fact that both WT and mutant RNAP enzymes purified from bacterial strains displayed nearly the same saturation with sigma subunit.
Figure 5.

Relative growth rate of E.coli strains tested in this study and performance of RNAP purified from these strains in various in vitro transcription assays at 20°C.
Overall, our results revealed multiple catalytic defects of primary Rifr mutations in the steps of the transcription cycle. Compensatory mutations strongly (2–9-fold) increased the activity of the above Rifr RNAP in the same assays, thus accounting for the enhanced growth rates of the corresponding bacterial strains (Figure 5). We find that compensatory substitutions counteract Rifr RNAP deficiencies at each step, thereby displaying pleotropic action.
Testing the enzyme purified from the strain containing a single βD516N substitution, which meant to be a ‘control’ for the βT563P/βD516N compensatory variant, revealed an unexpectedly strong positive effect of the mutation on RNA synthesis (Figure 5). These properties of the mutant enzyme can account for both improved in vitro transcription of the βT563P/βD516N enzyme and for increased relative growth rate of the corresponding mutant strain. Notably, the relative stimulatory effect by βD516N substitution in the promoter clearance, elongation and full transcription cycle assays in WT background RNAP was about of the same magnitude as that exerted by the above substitution on βT563P background enzyme, reflecting independent action of the primary Rifr and compensatory mutations on RNA synthesis.
Strikingly, the βD516N compensatory mutation on its own represents a common substitution conferring strong rifampicin resistance as evidenced by high minimal inhibitory concentration, MIC, 100–200 μg/ml for the corresponding bacterial strain (5,11,35). As such, in combination with high Rif resistance of βT563P substitution (MIC = 100 μg/ml (48)) double βT563P/βD516N substitution is expected to produce exceptionally Rifr strain. This conclusion is supported by consideration of the crystallographic image of Rif in binary complex with RNAP (Figure 6). It is seen that βD516 residue makes direct contact with Rif, thus accounting for the Rif resistance of the mutants at this position. The effect of βT563P mutation is more complex. Even though βT563 side chain does not contact Rif, as follows from our modeling, this mutation induces protein backbone rearrangement (as show in Figure 7) that disrupts the contact of the neighboring residue βP564 with the bound drug, thus explaining Rif resistance. In agreement with this proposed mechanism, mutational substitutions at βP564 locus produce Rifr bacteria (10,11). Importantly, polypeptide chain's segments bearing the above β563 and β516 loci do not interact with each other (Figure 6), strongly suggesting that each mutation contributes independently to the reduction in Rif retention. Therefore, the βD516N change is expected to exert a dual effect by enhancing both bacterial fitness and drug resistance, thus creating more virulent species.
Figure 6.
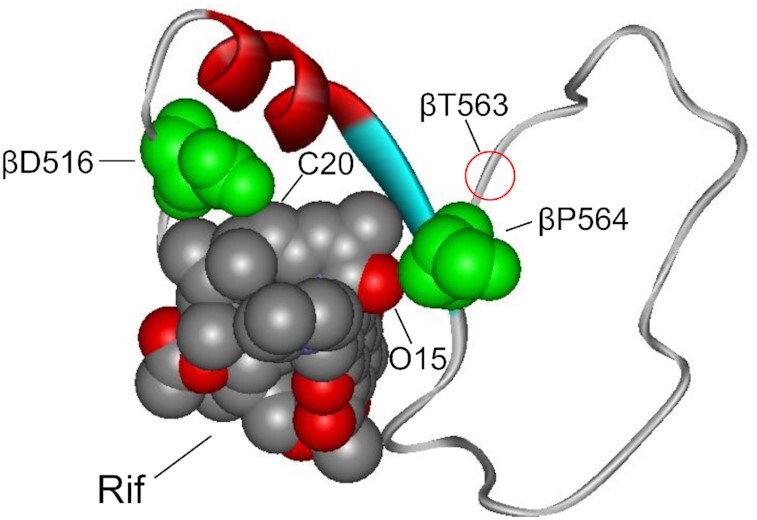
Interactions of βD516 and βP564 residues with rifampicin at the drug-binding pocket of RNAP. The interacting residues (green) and Rif molecule are shown in space-fill rendition and marked. The polypeptide segment of the β subunit (residues 513–566) involved in the formation of Rif binding pocket is shown in solid ribbon rendition. Position of βT563 residue is indicated.
Figure 7.
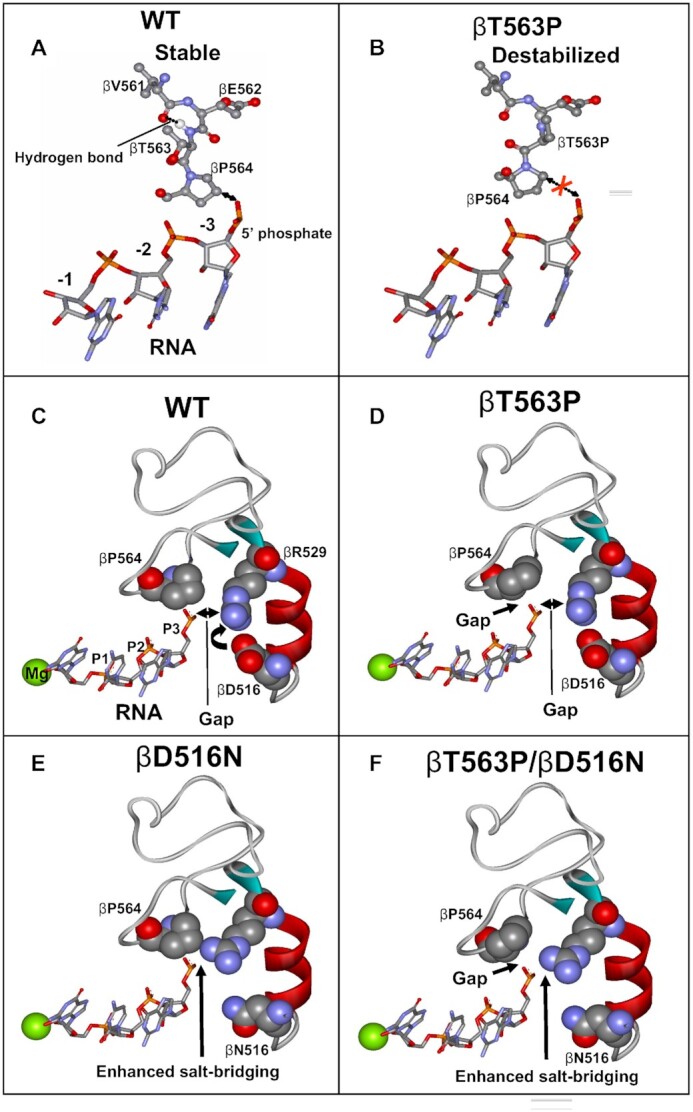
Molecular modeling explaining the effect of βT563P substitution and βD516N compensatory mutation on RNA retention in transcription complex. The change in the position of βP564 resulting from the rotation around the bound connecting the Cα and carbonyl carbon of βE562 residue (which eliminates steric hindrance between γ carbon of the βP563 side chain and the carbonyl oxygen of βV561) is shown. (A) relative arrangement of RNA transcript and V561-P564 β-subunit segment in TIC. (B) Destabilizing effect of βT563P mutation on RNA retention. The gap between the RNA transcript and β564P residue is indicated. (C) spatial arrangement of RNA, βP564, βD516, and βR529 in TIC. Salt bridge between βD516 and R529 is indicated. (D) βT563P substitution creates a gap between RNA and βP564 causing transcript destabilization. (E) βD516N substitution eliminates the salt bridge with βR529 thus allowing more efficient electrostatic interaction of this residue with RNA backbone phosphodiester group. (F) βD516N compensatory substitution suppresses the negative effect of βT563P mutation by reinforcing βR529 salt-bridging with RNA. The structure used for the modeling is 2O5J.
In light of the above consideration, compensatory effect of a βD516G mutation (which according to our modeling (Figure 7) should have the same effect as βD516N) can account for clinical prevalence (49) of MTB strains containing double Rifr substitution βL533P and βD516G (E. coli numbering). In the above view, bacterial strains carrying other double Rifr substitutions, found in E. coli (11), S. aureus (17) and S. enterica (18) represent compensatory strains. Similar to our findings, second mutations of Rif resistance displayed a significant compensatory effect in the above bacteria, producing species with equal or even superior fitness compared to an ancestral WT strain. This can account for the selection mechanism of such strains with double Rifr mutations, which otherwise would be puzzling, considering the absence of the selection pressure due to high resistance of the ancestral single-substitution mutants to the drug. In addition, proposed selection mechanism for the secondary mutations of Rif resistance is strongly supported by the fact that in our case βD516N compensatory substitution was selected in the absence of rifampicin.
To explain the effect of the RNAP mutations we took advantage of crystallographic structures and molecular modeling. One of the possible changes in RNAP structure caused by βT563P mutation shown in Figure 7 suggests that βT563P substitution disrupts the Rif binding pocket by eliminating a stabilizing H- bond between hydrogen of the amido group connecting βE562 and βT563 residues and a carbonyl oxygen of the βV561. In addition, Pro substitution is expected to create a steric clash between γ carbon of the Pro side chain and the carbonyl oxygen of βV561 in the arrangement depicted by the X-ray structure of this segment. One of the possible conformational changes that would resolve the clash is shown in the Figure 7B. As suggested by the modeling, the change would cause the distancing of the adjacent βP564 residue (which is a part of RNA holding clamp in transcription complex as shown in Figure 1B, II and IV) from the phosphate group of the RNA backbone at position −3 (Figure 7A and B). This structural rearrangement is consistent with: i) Rif resistance (as described above), ii) increased RNA abortion by RNAP and therefore, growth defect caused by βT563P substitution, iii) high evolutional conservation of β564 locus, and iv) significantly reduced bacterial fitness of βP564 mutants (11).
As follows from consideration of the crystallographic structures, the positive effect of the βD516N mutation on RNA synthesis as well as suppressive action of the same substitution on βT563P catalytic defect in RNAP can be rationalized by the involvement of the nearby βR529 residue of Rif binding pocket in transcript retention. This residue is within salt-bridging distance to phosphate group of RNA transcript at −3 register (Figure 7C). However, proximal negatively charged βD516 side chain competes with the phosphate group of the RNA backbone for salt-bridging with βR529, thereby weakening transcript retention. βD516N substitution eliminates the salt bridge (Figure 7E), which should enhance the βR529 hold on RNA transcript and therefore alleviate the negative effect of the primary mutation as shown in Figure 7F. This consideration also accounts for the observed enhancing effect of βD516N substitution on its own on transcription in respect to WT enzyme (lanes 1 and 5, Figure 3C). In addition, the proposed mechanism is supported by: i) strong involvement of the βR529 residue in transcript stabilization observed in our previous study (10); ii) dramatic effect of the βR529 substitutions on bacterial fitness (8,9,18); and iii) exceptional evolutionary conservation of this residue (Figure 1A). Remarkably, βD516/ βR529 salt-bridging sets these residues for interaction with bound Rif. Therefore, mutational substitutions of either of the residues eliminating the salt bridge account for their strong effect on the drug retention.
The nature of the compensatory effect of the βH554Y substitution on βT563P mutant is not as obvious, since the side chain of βH554Y is not involved in interactions with either RNA, or the surrounding protein residues. As seen from Figure 1 panel II, this residue is in close proximity to the β563/564 loci. Therefore, it can be envisioned that bulkier Tyr side chain exerts some pressure on the neighboring protein segment contacting RNA, which could narrow the gap between the βP564 residue and RNA created by the primary Rifr mutation, thus stabilizing the transcript in TIC. It is also conceivable that βH554Y substitution directly affects nucleotidyl transfer at some step of catalysis (e.g. conformational transition accompanying the reaction), which escapes the resolution of crystallographic analysis. This would explain fair evolutionary conservation of βH554 locus (Figure 1A) in the absence of its interactions in available crystallographic structures.
βQ513 residue (shown in Figure 1B, panels II, III, and IV) is part of RNAP Main Channel, which accommodates RNA:DNA hybrid of transcription complex through multiple interactions. In particular, βQ513 side chain hydrogen-bonds to RNA. Therefore, βQ513P mutation is expected to affect promoter escape by RNAP through loosening the enzyme's grip on RNA/DNA hybrid by: (i) elimination the side-chain contact with RNA and (ii) re-shaping the peptide backbone, due to ‘rigid’ nature of the Pro substitution. These variations can explain the mutation's effect on RNA synthesis and therefore, bacterial growth.
The β513 and β142 loci are in close proximity to each other in the enzyme's structure (Figure 1, panel III). Hence, the compensatory action of the βE142G substitution on the βQ513P enzyme variant can be explained by the direct influence of the former on catalysis through distorting the neighboring protein segments. Indeed, the βE142 side chain is involved in hydrogen-bonding with nearby residues βN133 and βN517 (Figure 1, panel III) thus stabilizing the structure of the hybrid-holding setup. Gly substitution, which eliminates these interactions and renders the segment more flexible, is expected to distort the structure, thus conferring the neighboring polypeptide chain holding the hybrid (blue in Figure 1, panel III) more adaptable. As such, this substitution could compensate for the catalytic defect by restoring the protein domain grasp on RNA/DNA hybrid affected by βQ513P substitution. Notably, compensatory change at βE142 locus detected in (20) in response to βΔ504–513 deletion mutation can be accounted for the same effect.
Two other compensatory mutations, αR191C and β′D1051N are far away from the locus of the βQ513P primary Rifr substitution, and therefore must work by different mechanism. As seen in Figure 1B (panel V) and Figure 8B, the αR191 residue salt-bridges to β′D410 and β′D413 side chains of the ψβ-barrel structure, which is a protein domain bearing active center carboxylate residues that coordinate catalytic Mg2+ ions. Therefore, the αR191C substitution that eliminates the bridge can render the active center domain more mobile, thus affecting catalysis. This is strongly supported by compensatory action of the β′D410N (E. coli numbering) substitution in MTB (12) that eliminates the same salt bridge with αR191 counterpart.
The β′D1051N compensatory mutation resides in the structural domain constituting a ‘trigger loop’ (TL), which is directly involved in catalysis (51). It is suggested that re-folding of the TL segments adjacent to the tips of G and G’ α-helixes of the β′-subunit (Figure 1B, panel VI) resulting in extension of the α-helixes, executes substrate loading to the active center from the pre-insertion to the catalytically active insertion state. In various bacteria TL size is highly variable and species-specific (52). Thus, in T. aquaticus TL is small (about 10–15 residues), while in E. coli it is about 230 residues-long. In the latter bacterium the TL domain is a subject of transcriptional regulation by protein factors (for review see (52)), which apparently target the loop's conformational transitions, and therefore nucleotidyl transfer catalysis. The β′D1051 side chain is engaged in a salt bridge with β′R1123 and β′K1104. In addition, it is hydrogen-bound to β′T1054 (Figure 1B, panel VI). The β′D1051N substitution eliminating these interactions can affect TL transitions accompanying the catalysis and as such, nucleotidyl transfer. Notably, some compensatory mutations in the TL have been previously mapped in Salmonella (18), suggesting similar action mode and therefore, general involvement of this RNAP feature in the compensatory mechanism.
In conclusion, structural consideration and the molecular modeling suggest that compensatory substitutions can enhance the transcription by: (i) reinforcing the enzyme's hold on RNA transcript through direct influence on the site affected by a primary Rifr mutation (βD516N, βD142G and βH554Y), (ii) promoting catalysis through readjustment of the active center domain (αR191C), and iii) facilitating active center rearrangement that executes NTP substrate loading (β′D1051N).
Analysis of the compensatory mutations identified in the previous studies (12,15,18) reveals that large group of these substitutions reside in the aforementioned active center ψβ -barrel structural domain (Figure 8). Remarkably, the sites for the mutated residues pave the barrel's segment exposed to the enzyme's inner core (Figure 8B). These residues participate in interactions apparently responsible for the presentation of the barrel domain for catalysis. Therefore, the compensatory mutations affecting these interactions can act by the mechanism analogous to that of αR191C through rendering the active center more adjustable. Other compensatory substitutions found in MTB RNAP α-subunit reside in close proximity to αR191 locus (Figure 8B), therefore their effect can be accounted for the same mechanism as well. Consistent with the above concept, a study on Rifr MTB RNAP compensatory evolution (36) demonstrated direct effect of some compensatory substitutions in ψβ –barrel on the enzyme's in vitro performance.
The loci for compensatory mutations (Figures 1A and 8A) are highly conserved among bacteria. Moreover, some of the loci, are also identical in the related enzymes from chloroplasts and eukaryotes, reflecting general functional significance of the side chains. Therefore, substitutions at these loci exerting compensatory action is a drastic measure that affects conserved side chain interactions optimal for WT enzyme.
Consideration of the present and previously published data reveals two important aspects of compensatory action. Thus, catalytic defects of different nature exerted by various primary Rifr mutations can be alleviated by the same amino acid change. This is illustrated by the fact that a mutation at the position equivalent to αR191 in MTB can compensate for the growth defect of a Rifr substitution in this bacterium (12), suggesting that this mutation displays the same compensatory mechanism. However, in MTB this substitution is associated with βS531L mutation of Rif resistance, while in our study αR191C change emerged from compensatory evolution of βQ513P Rifr variantant. In addition, αR191C substitution can compensate for the growth defect of βR529C Rifr mutation in S. enterica (18). On the other hand, we demonstrate that various substitutions can compensate for the defects associated with the same Rifr mutation. Thus, we show that αR191C, βE142G and β′D1051N compensatory substitutions are selected in response to βQ513P Rifr mutation, while βD516N and βH554Y variants originate from evolution of βT563P Rifr strain. This observation is also supported by the fact that various substitutions in S. enterica RNAP genes can alleviate negative effect of βR529C (18) and βS531L (19) Rifr mutations.
By conducting the same transcription tests in vitro at 20°C and 37°C we set out to address the question about the strong influence of the temperature on the growth of βT563P and βQ513P bacterial strains chosen for compensatory evolution studies and relevance of the temperature effect to compensatory mechanism. Consistent with growth rate trend, we demonstrate that the temperature rise narrowed the gap between WT and the primary Rifr mutant RNAPs activity in vitro transcription assays. Our data suggest that the growth stimulation for βT563P strain upon 20–37°C temperature raise can be explained by increase in the RNAP activity observed in promoter clearance and whole transcription cycle assays. However, with βQ513P substitution, the temperature shift affected the growth rate of the corresponding mutant strain to much greater extent than the activity of the mutant enzyme. The latter is indicative for complex temperature sensitivity mechanism, suggesting that the βQ513P mutation’s in vivo effect extends beyond the basal transcription machinery, and might include temperature-sensitive interactions with external regulatory protein factors. Notably, all compensatory substitutions essentially eliminated strong temperature effect on bacterial growth conferred by primary Rifr mutations. Remarkable example of interplay of the enzyme's temperature-sensitive loci is displayed by αR191C compensatory mutation. Thus, in E. coli this mutation on its own confers inability of the mutant strain to grow at 37°C (50). Here, we demonstrate that if combined with the cold-sensitive βQ513P variant, the αR191C substitution eliminates the growth defect of the βQ513P mutation at 20°C. In turn, βQ513P mutation in βQ513P/αR191C variant restores the bacterial growth at 37°C impaired by αR191C substitution.
Our data shed light on the relationship between the basal transcriptional activity of RNAP and bacterial growth rate. It is thought that transcription rate is a limiting factor determining bacterial growth (11,53). Consistent with this paradigm, we show that single Rifr mutations in RNAP reduce the performance of the enzyme in vitro assays and retard the cellular growth (present data and (10)). Indeed, βT563P mutation decreased both RNAP activity and relative growth rate at 20 °C, whereas both these characteristics were restored by βD516N compensatory substitution that accelerated the rate of transcription. However, βD516N substitution, while significantly increasing basal transcription activity of RNAP compared to WT, on its own failed to elaborate faster growing bacterial strain (Figure 5), strongly suggesting that RNAP basal activity displayed by WT enzyme is optimal for cellular growth. The above results can be explained by involvement of cellular regulatory factors that could retard fast-transcribing RNAP, thus counteracting the positive effect of the βD516N substitution on RNA synthesis observed in vitro. In addition, some other steps of gene expression (e.g. translation) can limit the cell growth, when the rate of RNA synthesis exceeds a certain limit. Finally, fast-transcribing enzyme is less prone to termination, which can alter the gene expression and therefore, affect the relative growth rate.
Analysis of the published data suggest the relevance of our results on E. coli to other bacteria, which is evidenced by the same compensatory substitutions in these species. Thus, mutations at the positions equivalent to αR191 and βD516 in MTB (12,49) as well as βE142 in Salmonella (20) can compensate for the growth defect of Rifr mutations.
In regard to RNAP catalytic function, this study identified loci involved in interactions networks that tune the enzyme to significantly increase its performance (2 – 9-fold) in response to severe catalytic defects imposed by primary Rifr substitutions. Some of these loci (βD516, βH554, βE142 and αR191) have been identified previously, while to the best of our knowledge β′D1051was mapped for the first time.
Our approach allowed explanation in some cases at the atomic level, how mutations affect the enzyme's activity and as such, the growth rates of the corresponding bacterial strains. However, additional studies are required to elucidate the compensatory mechanism in details. Thus, further biochemical testing aided by direct structural analysis and molecular dynamic simulations could address (i) the exact structural changes in RNAP caused by primary and compensatory mutations and their influence on catalysis; (ii) promoter identity effect on compensatory action, and (iii) possible involvement of the intracellular environment in compensatory mechanism.
This study demonstrates how systematic exploration of the compensatory effects can provide insights into the mechanisms of an enzyme action and fine features of cell biology. Comprehensive exploration of compensatory evolution and compensatory mechanism are mandatory to understand this phenomenon in details in order to assess its impact on epidemiology of drug resistant infections as well as to develop strategies to deal with the issue.
Supplementary Material
ACKNOWLEDGEMENTS
Regretfully, VN passed away as the manuscript was in preparation.
Contributor Information
Natalia Kurepina, Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, NJ 07110, USA.
Maxim Chudaev, Public Health Research Institute, and Department of Microbiology, Biochemistry & Molecular Genetics, Rutgers New Jersey Medical School, Rutgers Biomedical and Health Sciences, Newark, NJ 07103, USA.
Barry N Kreiswirth, Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, NJ 07110, USA.
Vadim Nikiforov, Public Health Research Institute, and Department of Microbiology, Biochemistry & Molecular Genetics, Rutgers New Jersey Medical School, Rutgers Biomedical and Health Sciences, Newark, NJ 07103, USA; Institute of Molecular Genetics, Russian Academy of Sciences, Moscow 123182, Russia.
Arkady Mustaev, Public Health Research Institute, and Department of Microbiology, Biochemistry & Molecular Genetics, Rutgers New Jersey Medical School, Rutgers Biomedical and Health Sciences, Newark, NJ 07103, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
PHRI Internal Support Fund (to A.M.). Funding for open access charge: Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, NJ, USA.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hancock R. The end of an era. Nat. Rev. Drug Discov. 2007; 6:28. [Google Scholar]
- 2. Theuretzbacher U., Toney J.. Nature's clarion call of antibacterial resistance: are we listening. Curr. Opin. Invest. Drugs. 2006; 7:158–166. [PubMed] [Google Scholar]
- 3. Projan S. Why is big pharma getting out of antibacterial drug discovery. Curr. Opin. Microbiol. 2003; 6:427–430. [DOI] [PubMed] [Google Scholar]
- 4. Ovchinnikov Y.A., Monastyrskaya G.S., Gubanov V.V., Lipkin V.M., Sverdlov E.D., Kiver I.F., Bass I.A., Mindlin S.Z., Danilevskaya O.N., Khesin R.B.. Primary structure of Escherichia coli RNA polymerase nucleotide substitution in the beta subunit gene of the rifampicin resistant rpoB255 mutant. Mol. Gen. Genet. 1981; 184:536–538. [DOI] [PubMed] [Google Scholar]
- 5. Ovchinnikov Y.A., Monastyrskaya G.S., Guriev S.O., Kalinina N.F., Sverdlov E.D., Gragerov A.I., Bass I.A., Kiver I.F., Moiseeva E.P., Igumnov V.et al.. RNA polymerase rifampicin resistance mutations in Escherichia coli: sequence changes and dominance. Mol. Gen. Genet. 1983; 190:344–348. [DOI] [PubMed] [Google Scholar]
- 6. Jin D.J., Gross C.. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J. Mol. Biol. 1988; 202:45–58. [DOI] [PubMed] [Google Scholar]
- 7. Jin D.J., Gross C.. RpoB8, a Rifampicin-resistant termination-proficient RNA polymerase has an increased Km for purine nucleotides during transcription elongation. J. Biol. Chem. 1991; 266:14478–14485. [PubMed] [Google Scholar]
- 8. Jin D.J., Turnbough C.. An Escherichia coli RNA polymerase defective in transcription due to overproduction of abortive initiation products. J. Mol. Biol. 1994; 236:72–80. [DOI] [PubMed] [Google Scholar]
- 9. Jin D.J., Gross C.. Characterization of the pleiotropic phenotypes of rifampicin-resistant rpoB mutants of Escherichia coli. J. Bacteriol. 1989; 171:5229–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yurieva O., Nikiforov V. Jr, Nikiforov V., O’Donnell M., Mustaev A.. Insights into RNA polymerase catalysis and adaptive evolution gained from mutational analysis of a locus conferring rifampicin resistance. Nucleic Acids Res. 2017; 45:11327–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reynolds M. Compensatory evolution of rifampicin-resistant Escherichia coli. Genetics. 2000; 156:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Comas I., Borrell S., Roetzer A., Rose G., Malla B., Kato-Maeda M., Galagan J., Niemann S., Gagneux S.. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2012; 44:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gagneux S., Long C.D., Small P.M., Van T., Schoolnik G.K., Bohannan B.. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006; 312:1944–1946. [DOI] [PubMed] [Google Scholar]
- 14. Wang S., Zhou Y., Zhao B., Ou X., Xia H., Zheng Y., Song Y., Cheng. Q., Wang X., Zhao Y. Characteristics of compensatory mutations in the rpoC gene and their association with compensated transmission of Mycobacterium tuberculosis. Front. Med. 2020; 14:51–59. [DOI] [PubMed] [Google Scholar]
- 15. Vargas A., Rios A., Grandjean L., Kirwan D., Gilman R., Sheen P., Zimic M.. Determination of potentially novel compensatory mutations in rpoC associated with rifampin resistance and rpoB mutations in Mycobacterium tuberculosis clinical isolates from peru. Int. J. Mycobacteriol. 2020; 9:121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Q-J., Jiao W-W., Yin Q-Q., Xu F., Li J-Q., Sun L., Xiao J., Li Y-J., Mokrousov I., Huang H.R.et al.. Compensatory mutations of rifampin resistance are associated with transmission of multidrug-resistant Mycobacterium tuberculosis Beijing genotype strains in China. Antimicrob. Agents Chemother. 2016; 60:2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O’Neill A.G., Huovinen T., Fishwick C.W.G., Chopra I.. Molecular genetic and structural modeling studies of Staphylococcus aureus RNA polymerase and the fitness of rifampin resistance genotypes in relation to clinical prevalence. Antimicrob. Agents Chemother. 2006; 60:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brandis G., Wrande M., Lijas L., Hughes D.. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol. Microbiol. 2012; 85:142–151. [DOI] [PubMed] [Google Scholar]
- 19. Brandis G., Hughes D.. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. Antimicrob. Chemother. 2013; 68:2493–2497. [DOI] [PubMed] [Google Scholar]
- 20. Brandis G., Hughes D.. Mechanisms of fitness cost reduction for rifampicin-resistant strains with deletion or duplication mutations in rpoB. Sci. Rep. 2018; 8:17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Enne V.I., Delsol A.A., Roe J.M., Bennet P.M.. Rifampicin resistance and its fitness cost in Enterococcus faecium. J. Antimicrob. Chemother. 2004; 53:203–207. [DOI] [PubMed] [Google Scholar]
- 22. Qi Q., Toll-Riera M., Heilbron K., Preston G.M., MacLean R.C.. The genomic basis of adaptation to the fitness cost of rifampicin resistance in Pseudomonas aeruginosa. Proc. R. Soc. B. 2016; 283:20152452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hartman G., Honikel K., Nuesch J.. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochem. Biophys. Acta. 1967; 145:843–844. [DOI] [PubMed] [Google Scholar]
- 24. Chopra I. Bacterial RNA polymerase: a promising target for the discovery of new antimicrobial agents. Curr. Opin. Investig. Drugs. 2007; 8:600–607. [PubMed] [Google Scholar]
- 25. Darst S.A. New inhibitors targeting bacterial RNA polymerase. Trends Biochem. Sci. 2004; 29:159–160. [DOI] [PubMed] [Google Scholar]
- 26. Villain-Guillot P., Bastide L., Gualtieri M., Leonetti J.P.. Progress in targeting bacterial transcription. Drug Discov. Today. 2007; 12:200–208. [DOI] [PubMed] [Google Scholar]
- 27. Mustaev A., Goldfarb A.. Lennarz W., Lane M.. RNA polymerase reaction in bacteria. Encyclopedia of Biological Chemistry. 2004; Academic Press, Elsevier Inc; 775–780. [Google Scholar]
- 28. Nudler E., Mustaev A., Lukhtanov E., Goldfarb A.. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997; 89:33–41. [DOI] [PubMed] [Google Scholar]
- 29. Komissarova N., Kashlev M.. RNA polymerase switches between inactivated and activated states by translocating back and forth along the DNA and the RNA. J. Biol. Chem. 1997; 272:15329–15338. [DOI] [PubMed] [Google Scholar]
- 30. Orlova M., Newlands J., Das A., Goldfarb A., Borukhov S.. Intrinsic transcript cleavage activity of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:4596–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borukhov S., Sagitov V., Goldfarb A.. Transcript cleavage factors from E. coli. Cell. 1993; 72:459–466. [DOI] [PubMed] [Google Scholar]
- 32. Sosunova E., Sosunov V., Kozlov M., Nikiforov V., Goldfarb A., Mustaev A.. Donation of catalytic residues to RNA polymerase active center by transcription factor Gre. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15469–15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sosunov V., Zorov S., Sosunova E., Nikolaev A., Zakeyeva I., Bass I., Goldfarb A., Nikiforov V., Severinov K, Mustaev A.. The involvement of the aspartate triad of the active center in all catalytic activities of multisubunit RNA polymerase. Nucleic Acids Res. 2005; 33:4202–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Campbell E.A., Korzheva N., Mustaev A., Murakami K., Nair S., Goldfarb A., Darst S.. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001; 104:901–912. [DOI] [PubMed] [Google Scholar]
- 35. Feklistov A., Mekler V., Jiang Q., Westblade L., Irschik H., Jansen R., Mustaev A., Darst S., Ebright R.. Rifamycins do not function by allosteric modulation of binding of Mg2+ to the RNA polymerase active center. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:14820–14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stefan M.A., Ugur F.S., Garcia G.A.. Source of the fitness defect in rifamycin-resistant mycobacterium tuberculosis RNA polymerase and the mechanism of compensation by mutations in the β' subunit. Antimicrob. Agents Chemother. 2018; 62:e00164-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joo D.M., Nolte A., Calendar R., Zhou Y.N., Jin D.J.. Multiple regions on the Escherichia coli heat shock transcription factor ς32 determine core RNA polymerase binding specificity. J. Bacteriol. 1998; 180:1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burgess R., Hendrisak J.. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975; 14:4634–4638. [DOI] [PubMed] [Google Scholar]
- 39. Kashlev M., Martin E., Polyakov A., Severinov K., Nikiforov V., Goldfarb A.. Histidine-tagged RNA polymerase: dissection of the transcription cycle using immobilized enzyme. Gene. 1993; 130:9–14. [DOI] [PubMed] [Google Scholar]
- 40. Serpinski O., Karginova E., Mikryukov N., Kravchenko V., Zaychikov E., Maksimova T., Onikienko A., Pletnev A., Mitina Yu.. Cloning of bacteriophage T7 fragment containing T2 promoter. Bioorgan. Chimia (Russ.). 1982; 8:840–846. [Google Scholar]
- 41. Nudler E., Goldfarb A., Kashlev M.. Discontinuous mechanism of transcription elongation. Science. 1994; 265:793–796. [DOI] [PubMed] [Google Scholar]
- 42. Vitiello C.L., Kireeva M.L., Lubkowska L., Kashlev M., Gottesman M.. Coliphage HK022 Nun protein inhibits RNA polymerase translocation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2368–E2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Elsas J.D., Semenov A.V., Costa R., Trevors J.T.. Survival of Escherichia coli in the environment: fundamental and public health aspects. ISME J. 2011; 5:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McClure W.R. Mechanism and control of transcription initiation in eukaryotes. Ann. Rev. Biochem. 1985; 54:171–204. [DOI] [PubMed] [Google Scholar]
- 45. Nudler E., Avetissova E., Markovtsov V., Goldfarb A.. Transcription processivity: protein-DNA interactions holding together the elongation complex. Science. 1996; 273:211–217. [DOI] [PubMed] [Google Scholar]
- 46. Komissarova N., Kireeva M., Becker J., Sidorenkov I., Kashlev M.. Engineering of elongation complexes of bacterial and yeast RNA polymerases. Method. Enzymol. 2003; 371:233–251. [DOI] [PubMed] [Google Scholar]
- 47. Sosunov V., Sosunova E., Mustaev A., Bass I., Nikiforov V., Goldfarb A.. Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J. 2003; 22:2234–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trautinger B.W., Lloyd R.G.. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002; 21:6944–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brown T.S., Challagundla L., Baugh E.H., Omar S.V., Mustaev A., Auld S.C., Sarita Shah N., Kreiswirth B.N., Brust J.C.M., Nelson K.N.et al.. Pre-detection history of extensively drug-resistant tuberculosis in KwaZulu-Natal, South Africa. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:23284–23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Igarashi K., Fujita N., Ishihama A.. Sequence analysis of two temperature-sensitive mutations in the alpha subunit gene (rpoA) of Escherichia coli RNA polymerase. Nucleic Acids Res. 1990; 18:5945–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vassylyev D.G., Vassylyeva M.N., Zhang J., Palangat M., Artsimovitch I., Landick R.. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007; 448:163–168. [DOI] [PubMed] [Google Scholar]
- 52. Chlenov M., Masuda S., Murakami K.S., Nikiforov V., Darst S.A., Mustaev A.. Structure and function of lineage-specific sequence insertions in the bacterial RNA polymerase beta' subunit. J. Mol. Biol. 2005; 353:138–154. [DOI] [PubMed] [Google Scholar]
- 53. Condon C., Squires C., Squires C.L.. Control of rRNA transcription in Escherichia coli. Microbiol. Rev. 1995; 59:623–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Opalka N., Brown J., Lane W.J., Twist K.F., Landick R., Asturias F.J., Darst S.A.. Complete structural model of Escherichia coli RNA polymerase from a hybrid approach. PLoS Biol. 2010; 8:e1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.