


Abstract
Attaining sufficient tissue exposure at the site of action to achieve the desired pharmacodynamic effect on a target is an important determinant for any drug discovery program, and this can be particularly challenging for oligonucleotides in deep tissues of the CNS. Herein, we report the synthesis and impact of stereopure phosphoryl guanidine-containing backbone linkages (PN linkages) to oligonucleotides acting through an RNase H-mediated mechanism, using Malat1 and C9orf72 as benchmarks. We found that the incorporation of various types of PN linkages to a stereopure oligonucleotide backbone can increase potency of silencing in cultured neurons under free-uptake conditions 10-fold compared with similarly modified stereopure phosphorothioate (PS) and phosphodiester (PO)-based molecules. One of these backbone types, called PN-1, also yielded profound silencing benefits throughout the mouse brain and spinal cord at low doses, improving both the potency and durability of response, especially in difficult to reach brain tissues. Given these benefits in preclinical models, the incorporation of PN linkages into stereopure oligonucleotides with chimeric backbone modifications has the potential to render regions of the brain beyond the spinal cord more accessible to oligonucleotides and, consequently, may also expand the scope of neurological indications amenable to oligonucleotide therapeutics.
INTRODUCTION
Antisense oligonucleotides can be designed to promote degradation of a targeted transcript through an RNase H-dependent mechanism. RNase H binds a heteroduplex formed between a DNA-containing oligonucleotide and the targeted RNA and cleaves the RNA (1). Unmodified antisense oligonucleotides have poor pharmacokinetic (PK) properties, so chemical modifications are needed to enable their use in preclinical research and as therapeutics. Phosphorothioate (PS) modification of the phosphodiester (PO) backbone is one of the most common backbone modifications used in research and to improve the properties of oligonucleotide therapeutics (1,2). A consequence of PS modification is the creation of a chiral center, where the molecule can adopt either an Rp or Sp configuration (1,3). We have previously demonstrated that PS-modified stereopure oligonucleotides—with precisely controlled chirality of inter-nucleotide PS backbone linkages—can outperform stereorandom oligonucleotides—those racemic mixtures derived from traditional chemistries for which the chiral configuration of the backbone is not controlled (4–6). New backbone modifications and methods to control their stereochemistry continue to emerge (3,4,7–9).
With the FDA’s 2016 approval of nusinersen, an intrathecally (IT) administered antisense oligonucleotide for the treatment of spinal muscular atrophy (SMA) (https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy), interest in developing oligonucleotide therapeutics for diseases of the CNS has increased. Numerous preclinical reports have now demonstrated application of antisense technology to achieve RNase H-mediated degradation of disease-associated transcripts in the CNS of mouse models with encouraging results (6,10–18). Despite these successes, challenges remain. Achieving oligonucleotide distribution throughout the CNS, beyond the spinal cord and other brain tissues that are readily accessed upon intracerebroventricular (ICV) or IT administration, remains challenging. Likewise, achieving durable target knockdown in these harder-to-reach regions of the CNS is difficult. Preclinical mouse studies that have reported success with oligonucleotide distribution and/or durable transcript knockdown in the brain have typically used very high doses of oligonucleotide (≥300 μg) (10–14,16). Because such high doses may not translate to clinically relevant dosing regimens, we aimed to identify chemical modifications that could improve oligonucleotide pharmacology in the CNS, making broad tissue distribution and durable activity achievable at lower doses.
The synthesis of various stereorandom phosphoramidate (mono substituted)-modified oligonucleotides using phosphoramidite and H-phosphonate chemistries have been reported (19–27). We were particularly interested in exploring phosphoryl guanidine-based backbones (PN) (28–32) in combination with stereopure phosphorothioate backbone (PS/PN) to study the pharmacological properties of these chimeric backbone oligonucleotides. Towards this end, we synthesized stereopure PN linkages and their derivatives (Supplementary Figure S1) and successfully synthesized chimeric PS/PN or PS/PN/PO stereopure backbone-containing oligonucleotides (Figure 1). We recently applied this backbone chemistry to splice switching oligonucleotides in muscle (9) and found that chimeric oligonucleotides, with a stereopure PN/PS/PO backbone, yielded profound phenotypic benefits over comparable stereopure PS/PO molecules, with activity gains deriving at least in part from improved pharmacology. We have also applied this backbone chemistry to our RNA editing oligonucleotides (Monian et al., in press). Herein, we applied PN chemistry to gapmer oligonucleotides acting through an RNase H-mediated mechanism and found that it can also enhance their pharmacology in the CNS.
Figure 1.

Stereopure oligonucleotide linkages with chiral backbone modifications. (A) Images depicting backbones used in this work, including phosphodiester (PO), phosphorothioate (PS), where a sulfur replaces a non-bridging oxygen, and phosphoryl guanidine diester (PN), where phosphoryl guanidine-containing moiety or derivative replaces a non-bridging oxygen. (B) The chemical structures for L-PSM and D-PSM amidites. (C) Staudinger reaction mechanism for chirally controlled PN backbone linkage formation.
MATERIALS AND METHODS
Synthesis of 2-azido-(1,3-dimethyl-3,4,5,6-tetrahydropyrimidinium) hexafluorophosphate (1d)
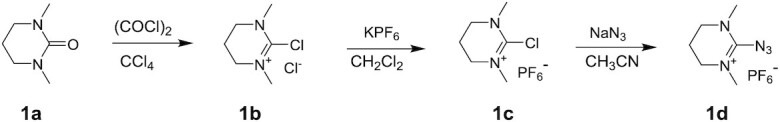
Synthesis of 2-chloro-1,3-dimethyl-3,4,5,6-tetrahydro pyrimidinium chloride (1b)
To a stirred solution of 1,3-dimethyltetrahydropyrimidin-2(1H)-one, 1a (25 g, 0.195 mol) in anhydrous carbon tetrachloride (375 ml) was added freshly distilled oxalyl chloride (25.0 ml, 0.292 mol) using an additional funnel over a period of 20 min. The reaction mixture was heated to 65°C for 48 h. After completion of the reaction (TLC – 5% CH3OH:CH2Cl2; TLC charring – phosphomolybdic acid), the mixture was cooled to room temperature and diethyl ether (300 ml) was added. After 5 min at room temperature, the obtained precipitate was then filtered, and washed with diethyl ether (3 × 500 ml). The compound was dried under high vacuum to give 2-chloro-1,3-dimethyl-3,4,5,6-tetrahydropyrimidinium chloride 1b as a brown solid (31 g, 87%).
1H NMR (400 MHz, CDCl3): δ 3.97 (t, 4H, J = 5.8 Hz), 3.51 (s, 6H), 2.37–2.31 (m, 2H). MS: m/z calculated for C6H12Cl2N2 [M−Cl]+: 147.06; found: 146.95.
Synthesis of 2-chloro-1,3-dimethyl-3,4,5,6-tetrahydro pyrimidinium hexafluorophosphate (1c)
To a stirred solution of 2-chloro-1,3-dimethyl-3,4,5,6-tetrahydropyrimidinium chloride, 1b (31 g, 0.169 mol) in CH2Cl2 (310 ml), KPF6 was added (31.16 g, 0.169 mol) over a period of 10 min. The reaction mixture was then stirred at room temperature for 1.5 h. After completion of the reaction (TLC – 5% CH3OH:CH2Cl2; TLC charring – phosphomolybdic acid), the reaction mixture was filtered through celite and the filter cake was washed with CH2Cl2 (150 ml). The filtrate was concentrated to dryness under reduced pressure to obtain crude product, which was then dissolved in CH2Cl2 (25 ml). The compound was precipitated by dropwise addition of diethyl ether (25 ml) and the solvent was decanted to get the product, which was dried under vacuum to give 2-chloro-1,3-dimethyl-3,4,5,6-tetrahydropyrimidinium hexafluorophosphate (1c), as a white solid (45 g, 91%).
1H NMR (500 MHz, CDCl3): δ 3.84 (s, 4H), 3.47 (s, 6H), 2.30 (s, 2H). 19F NMR (471 MHz, CDCl3): δ −73.02 and −74.54.
Synthesis of 2-azido-(1,3-dimethyl-3,4,5,6-tetrahydro pyrimidinium) hexafluorophosphate (1d)
To a stirred solution of 2-chloro-1,3-dimethyl-3,4,5,6-tetrahydropyrimidinium hexafluorophosphate (1c), (45 g, 0.154 mol) in anhydrous acetonitrile (450 ml) under argon atmosphere, sodium azide (14.99 g, 0.231 mol) was added over the period of 10 min. The reaction mixture was stirred at room temperature for 8 h. After completion of reaction (TLC – 5% CH3OH:CH2Cl2; TLC charring – ninhydrin), reaction mixture was filtered through a pad of celite and washed with CH3CN (30 ml). The obtained filtrate was dried under reduced pressure to get the crude product. The crude compound was dissolved in CH3CN (150 ml). Product was precipitated by dropwise addition of diethyl ether:hexane mixture. After complete precipitation, the solvent was decanted and the solid was dried under vacuum. The above precipitation procedure was repeated two more times to get pure 2-azido-(1,3-dimethyl-3,4,5,6-tetrahydropyrimidinium) hexafluorophosphate 1d as white solid (26 g, 57%).
1H NMR (400 MHz, CDCl3): δ 3.59 (t, 4H, J = 6.0 Hz), 3.33 (s, 6H), 2.26–2.20 (m, 2H). 19F NMR (377 MHz, CDCl3): δ −72.99 and −74.88. MS: m/z calcd for C6H12F6N5P [M-PF6]+: 154.11; found: 154.29. IR (KBr pellet): N3 (2184 cm–1).
Synthesis of 2-azido-(1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium) hexafluoro phosphate (2f)
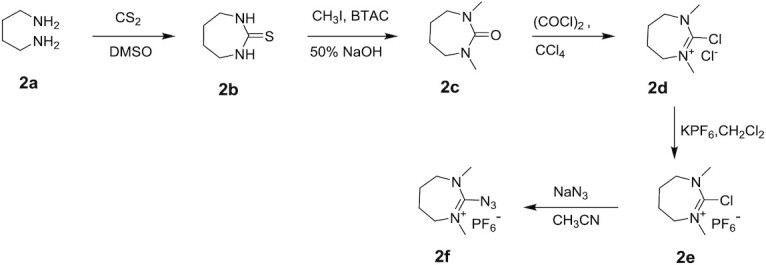
Synthesis of 1,3-diazepane-2-thione (2b)
To a stirred solution of butane-1,4-diamine 2a (50 g, 0.567 mol) in DMSO (500 ml) at 0°C, carbon disulphide (41.2 ml, 0.682 mol) was added by using addition funnel. Then, the reaction mixture was heated to 70°C for 16 h. After completion of the reaction (TLC – 5% CH3OH:CH2Cl2), the reaction mixture was cooled to room temperature. The precipitated solid was filtered and dried under high vacuum to get 32.0 g of product. The obtained filtrate was diluted with water (1.0 l) followed by extraction with CH2Cl2 (3 × 1000 ml), dried over sodium sulphate and the solvent was evaporated under reduced pressure to get the crude product. The crude product was dissolved in minimum volume of CH2Cl2 then precipitated by the dropwise addition of hexane. The precipitate was filtered and dried under high vacuum to give 3-diazepane-2-thione 2b (18.0 g), as a white solid and the combined yield was 68% (50 g).
1H NMR (400 MHz, CDCl3): δ 6.69 (s, 2H), 3.28–3.24 (m, 4H), 1.77–1.74 (m, 4H).
Synthesis of 1,3-dimethyl-1,3-diazepan-2-one (2c)
To a mixture of 1,3-diazepane-2-thione 2b (21 g, 0.161 mol) and benzyl trimethylammonium chloride (BTAC, 1.49 g, 0.008 mol) at 0°C, methyl iodide (65.0 ml, 1.044 mol) and 50% aq. NaOH solution (58.68 ml) were added drop wise. The reaction mixture was heated to 100°C for 8 h. After completion of the reaction (TLC – 5% CH3OH:CH2Cl2), the reaction mixture was cooled to room temperature. The reaction mixture was extracted with chloroform (3 × 1000 ml), dried over sodium sulphate, and the solvent was removed under reduced pressure to get crude product. The crude product was purified by column chromatography (30–80% ethyl acetate in hexanes) to give 1,3-dimethyl-1,3-diazepan-2-one, 2c as a light-yellow oil (9 g, 39% yield).
1H NMR (500 MHz, CDCl3): δ 3.13–3.11 (m, 4H), 2.84 (s, 6H), 1.68–1.65 (m, 4H).
Synthesis of 2-chloro-1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium chloride (2d)
To stirred solution of 1,3-dimethyl-1,3-diazepan-2-one 2c, (25 g, 0.176 mol) in anhydrous carbon tetrachloride (250 ml) freshly distilled oxalyl chloride (22.6 ml, 0.264 mol) was added using addition funnel over a period of 20 min. The reaction mixture was heated to 70°C for 16 h. After completion of the reaction (TLC – 10% CH3OH:CH2Cl2), the reaction mixture was cooled to room temperature, then diluted with diethyl ether (500 ml) and stirred for 5 min. The precipitate was collected after filtration and washed with diethyl ether (2 × 500 ml). The obtained crude product was dissolved in minimum amount of solvent and precipitated by the addition of 50% ethyl acetate in hexanes. The compound was collected by filtration and dried under vacuum to give 2-chloro-1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium chloride, 2d as a white solid (30 g). The crude compound was directly used for next reaction without any further purification.
Synthesis of 2-chloro (1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium) hexafluoro phosphate (2e)
To a stirred solution of 2-chloro-1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium chloride, 2d (30 g, 0.152 mol) in CH2Cl2 (300 ml), KPF6 (42.02 g, 0.228 mol) was added over a period of 10 min. The reaction mixture was stirred at room temperature for 4.5 h. After completion of reaction (TLC – 10% CH3OH:CH2Cl2), the reaction mixture was filter over celite, the filter cake was washed with CH2Cl2 (150 ml), and the filtrate was concentrated to dryness. The crude compound was dissolved in CH2Cl2 and washed with water (2 × 500 ml). The organic layer was dried over sodium sulphate and the solvent was removed under reduced pressure to give 2-chloro-(1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium) hexafluoro phosphate, 2e as white solid (25 g, 54%).
1H NMR (500 MHz, CDCl3): δ 3.90 (t, 4H, J = 5.9 Hz), 3.38 (s, 6H), 2.09–2.07 (m, 4H). 19F NMR (471 MHz, CDCl3): δ −72.66 and −74.16.
Synthesis of 2-azido-(1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium) hexafluoro phosphate (2f)
To a stirred solution of 2-chloro-1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium hexafluoro phosphate 2e (25 g, 0.081 mol) in anhydrous CH3CN (250 ml), sodium azide (7.95 g, 0.122 mol) was added over a period of 10 min and the reaction mixture was stirred at room temperature for 4 h. After completion of the reaction (TLC – 10% CH3OH:CH2Cl2; TLC charring – ninhydrin), the reaction mixture was filtered through a pad of celite and washed with CH3CN (30 ml). The organic layer was evaporated under reduced pressure to give crude product. The crude product was dissolved in CH3CN (50 ml) and the product was precipitated by adding diethyl ether at −78°C. The solvent was removed, and the solid obtained was dried under vacuum. The above precipitation procedure was repeated two more times to give 2-azido-(1,3-dimethyl-4,5,6,7-tetrahydro-1H-1,3-diazepinium) hexafluoro phosphate 2f, as pale yellow solid (21 g, 82%).
1H NMR (500 MHz, CDCl3): δ 3.63 (t, 4H, J = 5.5 Hz), 3.51 (d, 4H, J = 25.5 Hz), 3.25 (s, 6H), 3.15 (s, 6H), 2.02–1.96 (m, 4H), 1.89 (s, 4H). 19F NMR (471 MHz, CDCl3): δ −72.15, −72.56, −73.67 and −74.08. MS: m/z calcd for C6H14F6N5P [M-PF6]+: 168.22; found: 168.15. IR (KBr pellet): N3 (2162 cm–1).
Synthesis of 1-azido(pyrrolidin-1-yl) methylene) pyrrolidinium) hexafluorophosphate, (3e)
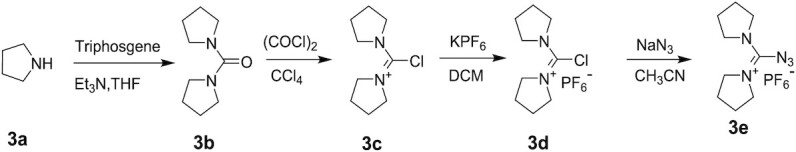
Synthesis of di(pyrrolidin-1-yl)methanone (3b)
To a stirred solution of pyrrolidine 3a (117 ml, 1.424 mol) in anhydrous THF (1380 ml), triethylamine (212 ml, 1.521 mol) was added, and the reaction mixture was cooled to 0°C by using an ice bath. To the above reaction mixture, a solution of triphosgene (70.0 g, 0.236 mol, in 224 ml THF) was added dropwise using dropping funnel over a period of 30 min. The resulting precipitated mixture was heated at 70°C for 2 h. Then the reaction mixture was cooled to room temperature and stirred for another 2 h. TLC showed the reaction was complete (TLC − 5% CH3OH:CH2Cl2; TLC charring – KMnO4). The reaction mixture was filtered through Buckner funnel. The obtained cake was washed with THF (250 ml) and the solvent was removed under reduced pressure to give di(pyrrolidin-1-yl)methanone, 3b as a brown liquid (124 g, 52%).
1H NMR (500 MHz, CDCl3): δ 3.37 (t, 8H, J = 6.9 Hz), 1.81–1.84 (m, 8H). MS: m/z calcd for C9H16N2O [M + H]+: 169.24; found: 169.11.
Synthesis of 1-(chloro(pyrrolidin-1-yl) methylene) pyrrolidinium chloride (3c)
To a stirred solution of di(pyrrolidin-1-yl)methanone 3b (124 g, 0.737 mol) in dry CH2Cl2 (1340 ml), a solution of oxalyl chloride (63.2 ml, 0.737 mol) in dry CH2Cl2 (520 ml) was added dropwise using dropping funnel at room temperature for over a period of 40 min. The reaction mixture was heated to 60°C for 5 h. TLC showed the reaction was complete (TLC – 5% CH3OH:CH2Cl2; TLC charring – KMnO4). The solvent was evaporated to dryness to get 1-(chloro(pyrrolidin-1-yl)methylene)pyrrolidinium chloride 3c as a brown liquid (160 g). The crude material was directly used without purification for the next step.
Synthesis of 1-(chloro(pyrrolidin-1-yl) methylene) pyrrolidinium hexafluorophosphate (3d)
To a stirred solution of 1-(chloro(pyrrolidin-1-yl)methylene) pyrrolidinium chloride 3c (160 g, 0.717 mol) in water (1525 ml) at room temperature, solution of KPF6 (158.9 g, 0.863 mol, dissolved in 326 ml water) was added a saturated dropwise using dropping funnel over a period of 20 min. While adding, some of the product precipitated. Stirring continued for another 10 min at room temperature. Then the reaction mixture was filtered through Buckner funnel using Whatman filter paper. The solid was washed with water (1500 ml) and dried under high vacuum to get crude product. The crude product was dissolved in acetone (110 ml) and the product was precipitated by dropwise addition of diethyl ether (1000 ml). The above precipitation method was repeated one more time to give 1-(chloro(pyrrolidin-1-yl) methylene)pyrrolidinium hexafluorophosphate, 3d as a cream-colored solid. (142.1 g, 60%).
1H NMR (500 MHz, CDCl3): δ 3.92 (t, 8H, J = 6.2 Hz), 2.10 (t, 8H, J = 6.5 Hz).
Synthesis of 1-(azido(pyrrolidin-1-yl) methylene) pyrrolidinium hexafluorophosphate (3e)
To a stirred solution of 1-(chloro(pyrrolidin-1-yl)methylene)pyrrolidinium hexafluorophosphate, 3d (71.0 g, 0.213 mol) in anhydrous CH3CN (213 ml) under argon atmosphere, sodium azide (3.58 g, 0.055 mol) was added and the reaction mixture was stirred for 3 h at 30°C. After completion of the reaction (TLC – 5% CH3OH:CH2Cl2; TLC charring – ninhydrin), the reaction mixture was filtered through a pad of celite and washed with CH3CN (50 ml). The organic layer was removed under reduced pressure to get the crude product. The solid obtained was dissolved in CH3CN (60 ml) and the product was precipitated by adding dropwise diethyl ether (850 ml). The above precipitation method was repeated one more time to give 3e as an off white solid (65.1 g, 89%).
1H NMR (400 MHz, CDCl3): δ 3.77 (t, 8H, J = 6.5 Hz), 2.03–2.06 (m, 8H). 19F NMR (377 MHz, CDCl3): δ −73.36 and −75.26. MS: m/z calcd for C9H16N5PF6 [M-PF6]+: 194.26; found: 194.16. IR (KBr pellet): N3 (2153 cm–1)
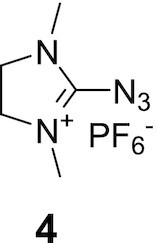
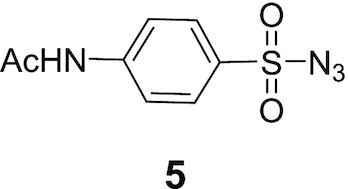
The following 2-azido-1,3-dimethylimidazolinium hexafluorophosphate in salt form, (4) 98% purity and 4-aetamidobenzenesulfonyl azide (5) 95% purity were purchased from TCI America and Sigma Aldrich, USA, respectively.
Oligonucleotides
We synthesized phosphodiester-based and stereorandom PS-modified oligonucleotides using a standard solid-phase oligonucleotide synthesis protocol. We synthesized and purified chemically modified, stereopure oligonucleotides as described with minor modifications (4) (WO2014012081, WO2018237194) (Supplementary Materials and Methods, Supplementary Tables S2 and S3). We characterized all oligonucleotides by LC–MS and HPLC (Supplementary Tables S1 and S4). All oligonucleotides designed to target Malat1 (5′-UGCCAGGCTGGTTATGACUC-3′) or C9orf72 (5′ ACTCACCCACTCGCCACCGC-3′) have the same sequences.
Thermal denaturation (Tm)
Equimolar amounts of surrogate Malat1 RNA (5′-GAGUCAUAACCAGCCUGGCA) or C9orf72 RNA (5′-GCGGUGGCGAGUGGGUGAGU-3′) and oligonucleotide were combined in 1× PBS (pH 7.2) to obtain a final concentration of 1 μM of each strand (3 ml). Duplex samples were then annealed by heating at 90°C, followed by slow cooling to 4°C and storage at 4°C. UV absorbance at 254 nm was recorded at intervals of 30 s as the temperature was raised from 15°C to 95°C at a rate of +0.5°C per min, using a Cary Series UV–Vis spectrophotometer (Agilent Technologies). Absorbance was plotted against the temperature and the Tm values were calculated by taking the first derivative of each curve.
RNase H assays
For RNase H assays, we incubated heteroduplexes with human RNase HC (prepared as described (4)) at 37°C. Duplexes were prepared by mixing equimolar solutions of oligonucleotide and RNA (Malat1 RNA 5′-GAGUCAUAACCAGCCUGGCA) yielding a final concentration of 20 μM. Each reaction contained 5 μM ASO-RNA, in RNase H buffer (75 mM KCl, 50 mM Tris–HCl, 3 mM MgCl2, 10 mM dithiothreitol, pH 8.3) in a reaction volume of 50 μl. The pre-mixtures were incubated at 37°C for 10 min prior to the addition of enzyme with a final concentration ratio of 500:1 (substrate:RNase HC). We quenched the reactions at 5, 10, 15, 20, 30, 45, and 60 min using 7.0 μl of 250 mM EDTA disodium solution in water. For the 0 min-time point, we added EDTA to the reaction mixture before enzyme. UV absorbance was recorded at 254, 210 and 280 nm for each reaction after injection onto an Agilent Poroshell 120 EC-C18 column (2.7 μm, 2.1 × 50 mm) at 60°C using a gradient of buffer A (200 mM HFIP and 8 mM triethylamine) and buffer B (methanol). We integrated the peak areas from the chromatograms, corresponding to full-length RNA oligomer, normalized them compared to the antisense strand. The percentage RNA remaining was plotted, with the 0 min-time point defined as 100%, to show relative rates of RNA cleavage.
We characterized the products of the RNase H assay by LC–MS to determine the cleavage sites. The extent of cleavage at each site was determined by relative ratio of the UV peak area of a fragment to the extinction coefficient (http://www.scripps.edu/california/research/dna-protein-research/forms/biopolymercalc2.html) of that fragment normalized to that of oligonucleotide (ratio of UV peak area of oligonucleotide to extinction coefficient of the oligonucleotide). Normalization to oligonucleotide controls was performed to standardize for variation in the amount of duplex used across reactions. The normalized ratio of each RNA fragment was then expressed as a percentage of the total normalized RNA (RNA fragments and uncleaved RNA) present in the reaction. The percentage amount of fragment (preferably 3′-OH RNA cleavage product or 5′-phosphorylated RNA cleavage product if former was not detected) was used to determine extent of cleavage at a particular site.
Cell lines
iCell neurons
iCell neurons (Cellular Dynamics, Madison, WI) were seeded at 4 x 104 cells/well. After 24 h, fresh medium was added containing 1.0 pM to 10 μM of oligonucleotide in duplicate. Four days after treatment, cells were lysed, RNA was extracted following RNeasy 96 kit (Qiagen) protocol, and MALAT1 expression was determined by quantitative polymerase chain reaction (qPCR) (see below).
iCell astrocytes
iCell Astrocytes (Cellular Dynamics, Madison, WI) were seeded at 4 x 104 cells/well in DMEM/F-12, HEPES (ThermoFisher Cat. No. 11330) with 2% FBS (Fetal bovine serum, GE Health life sciences #SH30071.03) and N-2 supplement (ThermoFisher Cat. No. 17502048). After 24 h, fresh medium was added containing 1.0 pM to 10 μM of oligonucleotide in duplicate. Four days after treatment, cells were lysed, RNA was extracted following RNeasy 96 kit (Qiagen) protocol, and MALAT1 expression was determined by qPCR.
ALS motor neurons
Spinal motor neurons were generated from a human ALS iPSC line harbouring a C9ORF72 mutation (Target ALS ID: TALS9-9.3; Rutgers ID: 150000002; NINDS ID: ND50000). Directed differentiation was performed by BrainXell as described (33). Briefly, iPSCs were treated with small molecules CHIR99021, DMH-1 and SB431542 for 6 days to induce SOX1+ neuroepithelial progenitors (NEPs). The NEPs were split and treated with CHIR99021, DMH-1, SB431542, retinoic acid and purmorphamine for another 6 days to generate OLIG2 + motor neuron progenitors (MNPs). These OLIG2+ MNPs were expanded and frozen at the point of early differentiation to motor neurons. ALS motor neurons were seeded in 12-well plates (2.8 x 105 cells per well) from frozen stocks and treated gymnotically on day 7 and harvested on day 14. 50 μl of a growth factor cocktail containing 10 ng BDNF, 10 ng of GDNF, and 1 ng of TGF-β1were added on day 10 without changing the medium.
HEK293 cells
HEK293 cells were seeded at 8 x 104 cells/well in 96-well plates and cultured in DMEM, 4.5 g/l glucose, 2 mM l-glutamine, 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 100 ug/ml normocin. Cells were treated with oligonucleotides for 16–20 h before processing for survival.
RNA Isolation and cDNA synthesis
Frozen mouse tissue was added to 700 μl of TRIzol and homogenized for 3 min. Bromochloropropane was added to each sample, which was shaken vigorously and centrifuged at 4000g for 5 min. Supernatant (250 μl) was transferred to the binding plate from SV96 total RNA extraction kit (Promega) or RNeasy 96 kit (Qiagen) and RNA was extracted per protocol. The gDNA was removed with DNase I (New England Biolabs, M0303L). cDNA was synthesized by adding 2 μg of total RNA to a 20 μl reverse-transcription reaction using the High-Capacity cDNA Reverse-Transcription Kit (Thermo Fisher #4368814).
In ALS motor neurons, total RNA was extracted using TRIzol (Invitrogen) according to manufacturer's protocol. For each sample, total RNA was eluted in 29.5 μl of RNase-free water followed by the addition of 2 μl (4U) of DNase I (New England Biolabs, M0303L) and 3.5 μl of 10× reaction buffer. Samples were incubated at 37°C for 15 min for gDNA removal. EDTA was added to 5 mM final concentration, and DNase I was heat inactivated at 75°C for 10 min. RNA was reverse transcribed with High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems) according to manufacturer's instructions.
In iCell neurons, 4 days after treatment, RNA is extracted in one of two ways. Cells are lysed with 500 μl Trizol and 100 μl of bromochloropropane was added to each well, supernatant was transferred to the binding plate from RNeasy 96-well plate kit (Qiagen) following the manufacturer's protocol of ‘the purification of total RNA from cells using vacuum/spin technology’ with on-column DNase I treatment. Alternatively, RNA extraction was performed with SV40 total RNA extraction kit (Promega) according to manufacturer's protocols. RNA was reverse transcribed with High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems).
Quantification of RNA
qPCR was performed in a reaction mixture including diluted sample (4 μl from a mix of 20 μl of cDNA and 30 μl of water) using either mouse Malat1 primers (Thermo Fisher Mm01227912_s1) for rodent samples or human MALAT1 primers (Thermo Fisher Hs00273907-s1) for human samples in addition to the normalizer primers for human SRSF9 (forward primer TGG AAT ATG CCC TGC GTA AA, probe sequence /5HEX/TG GAT GAC A/Zen/C CAA ATT CCG CTC TCA/3IABkFQ/, reverse primer TGG TGC TTC TCT CAG GAT AAA C) (Integrated DNA Technologies). The thermocycling conditions for PCR include 95°C for 3 min; and 40 cycles of 95°C for 10 s and 60°C for 30 s.
C9orf72 transcripts were quantified using Taqman assays. We used the following Taqman probes: Hs00376619_m1 (FAM) (Catalog # 4351368, ThermoFisher) for C9orf72 All Transcripts (common on V1, V2 and V3); Hs00948764_m1(FAM) (Catalog # 4351368, ThermoFisher) for C9orf72 V3 transcripts; Hs02800695_m1 for human HPRT1 transcripts (Catalog# 4448486, ThermoFisher). Mouse Hprt1 transcripts were quantified with the following primers: forward primer: CAA ACT TTG CTT TCC CTG GTT; reverse primer: TGG CCT GTA TCC AAC ACT TC; probe sequence:/5HEX/AC CAG CAA G/Zen/C TTG CAA CCT TAA CC/3IABkFQ/ (Integrated DNA Technologies).
Relative EC50 calculations
We perform four-parameter log-logistic curve fit to data from dose-response experiments using R and the drc package (https://www.r-project.org/foundation/). We employ the LL2.4 function for the calculation of 95% confidence intervals (CIs, top and bottom), EC50s and Hill slope coefficients. Because cell culture conditions vary slightly between experiments, there is some variation in numeric value of EC50s across experiments. These experiments were each replicated 2–4 times, and the relative performance of the oligonucleotides with respect to other oligonucleotides in the same experiment was consistent across experiments.
Cell viability assays
Cell viability experiments in iCell Neurons and iCell Astrocytes were performed using CellTiter-Blue Cell Viability Assay according to manufacturer's protocol (Promega). Cell viability experiments in HEK293 cells were performed using CellTiter-Glo Cell Viability Assay according to manufacturer's protocol (Promega).
Toll-like receptor 9 (TLR9) activation assays
HEK-Blue™ hTLR9 cells that are stably transfected with human TLR9 gene, and a NF-κB inducible SEAP reporter gene were purchased (InvivoGen, CA). 8 × 104 HEK-Blue™ TLR9 cells/well were seeded in 96 well plates in HEK-Blue Detection Media (InvivoGen #hb-det2). Oligonucleotides (in water) were added to the cells for a final concentration of 30 μM (1 μM TLR9 agonist) and cells were incubated for 16 h at 37°C in a humidified incubator. SEAP was quantified in the media indirectly by measuring the absorbance at 655 nm (OD655). Results were plotted as fold-change in NF-κB activation with respect to controls treated with phosphate buffered saline (PBS) using GraphPad Prism software. The TLR9 agonist was designed based on ODN2006 (34), a fully PS-modified stereorandom deoxyoligonucleotide 5′-TCGTCGTTTTGTCGTTTTGTCGTT-3′.
Cytokine activation assays
The pro-inflammatory potential of oligonucleotides was investigated in multiple species using ex vivo multiplex cytokine assays. Peripheral blood mononuclear cells (PBMCs) from three human donors (AllCell Inc. (Cat# LP, FR, MNC, 100M; Donor IDs: 3033038, 3033468, 3033043) and three cynomolgus monkeys (Biomedical Research Models, Inc. dba Biomere; Animal ID: 289231, 289216, 382711), and pooled splenocytes from C57Bl/6 mice (Biomedical Research Models, Inc dba Biomere) were used. 4 × 105 human PBMCs/well were assayed in 96 well plates containing IMDM (Life technology, Cat#: 12440-053) supplemented with 10% heat-inactivated (HI) FBS (Life technology, Cat#:10082–147) and 50 U/ml Penicillin-Streptomycin (Lonza, Cat#17-602E). Monkey PBMCs and mouse splenocytes (1 × 106 cells/well) were assayed in 96-well plates containing RPMI medium (Lonza, Cat # 12-115Q) supplemented with 10% HI FBS and 50 U/ml penicillin–streptomycin.
Oligonucleotides (in water) (final concentration: 0.1, 0.3, 1.0, 3.0, 10 or 30 μM) were added to the cells and allowed to incubate at 37°C, 95% humidity and 5% CO2 with constant agitation. At the conclusion of treatment, cells were spun down before supernatant was collected. The Multiplex cytokine assay was performed following manufacturer's protocol (EMD Cat. No. HCYTMAG-60K) with modifications. Fluorescence intensity was read on FLEXMAP3D system (Luminex, TX). Homogenous cell suspensions (10 μl) were collected and mixed with equal volume of Cell Titer-Glow to determine ATP levels (CellTiter-Glo® 2.0, Promega, Cat. No. G9242). Cytokine levels were determined using a 5-parameter non-linear regression fit. Pro-inflammatory and neutral control oligonucleotides were based on drisapersen (35) and eteplirsen (36), respectively, and were synthesized in house.
Stability in mouse brain homogenate
Stability of oligonucleotides in mouse brain homogenate was performed as described (6). Briefly, we determined the stability of the oligonucleotides in mouse brain homogenate by adding 5 μl of each oligo solution (200 μM) to 45 μl of mouse brain homogenate (prepared in-house, 20 mg/ml). We incubated each reaction at 37°C while shaking at 400 rpm. We terminated reactions by adding 50 μl of Stop buffer (2.5% IGEPAL, 0.5 M NaCl, 10 mM EDTA, 50 mM Tris, pH 8.0). We than added 20 μl of internal standard (50 μM: 5′-GCGTTTGCTCTTCTTCTTGCGTTTTTT-3′), 250 μl of 2% ammonium hydroxide and 100 μl of phenol:chloroform:isoamyl alcohol (25:24:1) to each tube. After extraction, we dried and then reconstituted each sample with water in a volume of 100 μl. 2 μl of the mixture was injected to Waters Xevo QTOF (Waters) using Agilent Poroshell column (120, EC-C18 2.7 μm, 2.1 × 50 mm) and mobile Phase A (400 mM HFIP, 15 mM TEA in water) and Mobile Phase B (Methanol). We used Waters Xevo QTOF (Waters) for data capture and to calculate peak areas and peak area ratios of analytes to the internal standard. Reduction in analyte amount was used to evaluate the extent of in vitro stability.
Animals and ICV Injections
Unless otherwise noted, animal experiments were performed at Biomedical Research Models, Inc. dba Biomere (Worcester, MA) in compliance with Biomere's Institutional Animal Care and Use Committee guidelines for care and use of animals. Mice were on a 12-h light–dark cycle. Food (lab diet 5001) and water were available ad libitum. Housing rooms were maintained at 20–26°C and relative humidity was 30–70%. Male and female 10–12 week old wild-type C57Bl/6 mice (Jackson Laboratories, No. 000664) or C9BAC transgenic mice (37), (Tg(C9orf72_3) No. 023099, Jackson Laboratories), which have several tandem copies of the C9orf72 transgene, with each copy having between 100 and 1000 hexanucleotide repeats, were used in these studies. For intracerebroventricular cannulation, mice were anesthetized using Avertin, were placed on a rodent stereotaxic apparatus and were implanted with a stainless-steel guide cannula in the right lateral ventricle (coordinates: −0.3 mm posterior, +1.0 mm lateral and −2.2 mm vertically from bregma), which was secured in place using dental cement. Mice were allowed a one-week recovery period prior to dosing.
In the Malat1 dose-escalation study, 5, 10 or 20 μg of oligonucleotide was administered in 2.5 μl on day 1, and mice were sacrificed 7 days after injection. For the Malat1 duration study, 100 μg oligonucleotide was administered in 2.5 μl on day 1, and mice were sacrificed 4 or 10 weeks later. For the C9orf72 duration study, 50 μg doses of oligonucleotide were administered in 2.5 μl on days 0 and 7, and mice were sacrificed 2, 4 or 8 weeks later. At necropsy, all mice were transcardially perfused with phosphate buffered saline (PBS) under isoflurane anaesthesia. Brains were rapidly removed from the skull; one hemisphere was processed for pharmacokinetic (PK) analyses and the other for pharmacodynamic (PD) analysis. Hemispheres were dissected into cortex, hippocampus, striatum and cerebellum and flash frozen on dry ice. Spinal cord was removed and flash frozen on dry ice.
Wild-type tolerability study
This study was performed by Charles River Laboratories, South San Francisco (SSF). Adult male C57Bl/6 mice (Charles River Laboratories) were used for this study. Animals were individually housed in polycarbonate cages and acclimated for at least 4 days prior to the beginning of the study. Animals were maintained in a 12 h light/dark cycle with constant room temperature (22 ± 2°C) and humidity (∼50%). Animals had access to food and water ad libitum. Experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Charles River Laboratories, SSF. Mice were anesthetized using isoflurane (2%, 800 ml/min O2). Bupivacaine was used for local analgesia and carprofen was used for peri-/post-operative analgesia. The animals were placed in a stereotaxic frame (Kopf instruments, USA). Surgeries were performed using aseptic technique. Anterior-posterior (AP), medial-lateral (ML) and dorsal-ventral (DV) were zeroed on the bregma. The following coordinates were used for the ICV infusion cannula (Plastics One): AP −0.3 mm, lateral −1.0, DV −2.2 mm from dura. After surgery, animals were provided food and water ad libitum. Animals were observed and scored at 30 min, 1 h, 2 h, 24 h, and 1, 2, 3, 4, 5, 6, 7 and 8 weeks following ICV dosing. Body weights and overall health observations were also recorded at each timepoint. Brains were collected post-mortem and were immersed in 10% neutral-buffered formalin.
Brains were sectioned and evaluated for expression of immune markers: Glial Fibrillary Acidic Protein (Gfap), a marker for activated astrocytes, and Ionized calcium-binding adaptor molecule 1 (Iba1), a marker for activated microglia.
Quantification of oligonucleotides by hybridization ELISA
Antisense oligonucleotides were specifically quantified by hybridization ELISA utilizing the following probes: Malat1-Univ-Capture probe /5AmMC12/G + A + G + T + C + A + T + AA; Malat1-Univ-Detection probe CC + AGC + CT + GG/3BioTEG/ (Integrated DNA Technologies, Coralville, IA). Capture probe: ‘C9-Intron-Cap’: /5AmMC12/GCGG+TGGCG+A; Detection probe: ‘C9-Intron-Det’: G+TGGG+TG+AGT/3BioTEG/. The maleic anhydride-activated 96-well plates (Pierce 15110) were coated with 50 μl of capture probe at 500 nM in 2.5% NaHCO3 (Gibco, 25080-094) for 2 h at 37°C. The plate was then washed 3 times with PBST (PBS + 0.1% Tween-20), blocked with 5% fat free milk-PBST at 37°C for 1 h. Payload oligonucleotide was serially diluted into tissue lysate matrix. This standard together with original samples were diluted with lysis buffer so that the oligonucleotide amount in all samples was less than 50 ng/ml. 20 μl of diluted samples were mixed with 180 μl of 333 nM detection probe diluted in PBST, then denatured (65°C, 10 min; 95°C, 15 min; 4°C). 50 μl of the denatured samples were distributed in blocked ELISA plates in duplicates and incubated overnight at 4°C. After 3 washes with PBST, 50 μl of 1:2000 streptavidin-AP (Southern Biotech, 7100-04) in PBST was added, and incubated at room temperature for 1 h. After extensive washes with PBST, 100 μl of AttoPhos (Promega S1000) was added, incubated at room temperature in the dark for 10 min and read on the plate reader (Molecular Device, M5) fluorescence channel: Excitation: 435 nm, Emission: 555 nm. The oligonucleotide in samples were calculated according to standard curve by 4-parameter regression. The lower limit of detection was 1.25 μg oligonucleotide per gram of tissue.
Pharmacokinetic (PK)–pharmacodynamic (PD) analysis
For PK–PD analyses, the concentration of oligonucleotide detected in a specific tissue as measured by hybridization ELISA (see above) were plotted with respect to quantified and normalized expression of RNA (see above) in the same tissue at the same time.
Poly-GP quantification
Brain and spinal cord samples were homogenized in four volumes RIPA (50 mM Tris, 150 mM NaCl, 0.5% DOC, 1% NP40, 0.1% SDS and Complete protease inhibitor, pH 8.0) by shaking in a Precellys instrument with 1.4 mm zirconium oxide beads. Samples were centrifuged at 12 100 g for 10 min at 4°C, and total protein concentration of clarified lysate was determined with 600 nm Protein Assay Reagent (Pierce). MSD Small-Spot plates were coated with 1 μl of a 10 μg/ml solution of a polyclonal capture antibody (rabbit anti-poly-GP; AB1358, Millipore) and incubated at 4°C overnight. The next day, plates were washed with PBST, blocked with a 10% FBS/PBST solution for 1 h at room temperature, and then washed with PBST and incubated with 50–120 μg of brain lysate (diluted 1:4 or 1:5 into 10% FBS/PBST) for 2–4 h. Plates were washed with PBST and incubated with Sulfo-tag-conjugated detection antibody (rabbit anti-poly-GP; AB1358, Millipore) for 1 h at room temperature. Plates were washed with PBST and incubated with 150 μl of MSD Read Buffer T 1× and read in an MSD QuickPlex SQ 120 plate reader with Discovery Workbench 4.0.12 (LSR_4_0_12) software. A standard curve of recombinant purified poly-GPx30 was prepared in a matrix of wild-type mouse cortex or spinal cord homogenate. After subtracting the background signal measured from empty-wells, a linear best-fit regression line for the standard curve was used to interpolate the concentration of poly-GP per microgram of tissue. Data are reported as relative fold-change in poly-GP concentration, reflecting amount of poly-GP detected compared with PBS-treated samples.
Capillary western immunoassay
C9orf72 protein analysis was performed using capillary western immunoassay (WES). Protein lysates from spinal cord and cortex tissue were prepared by adding 10X weight in volume of RIPA buffer with tablets and a scoop of lysis beads. The samples were then homogenized for 2–4 cycles (3 × 20 s) on the Precellys Evolution Tissue Homogenizer and spun down for 10 min at 14,000 rpm at 4°C. The supernatants were carefully transferred into new tubes. We quantified total protein using the Pierce BCA protein assay kit with BSA standards according to the manufacturer's protocol. Lysates were normalized to 0.5 μg/μl in 0.1× sample buffer. C9orf72 protein quantitation was performed on a Wes system, according to the manufacturer's instructions using a 12–230 kDa Separation Module, the Anti-Rabbit Detection Module and the Anti-Mouse Detection Module. Lysates were mixed with Fluorescent Master Mix and denatured at 95°C for 5 min. The samples, blocking reagent (antibody diluent), primary antibodies (1:100 anti-C9orf72, 1:250 anti-Hprt in antibody diluent), HRP-conjugated secondary antibodies (ready to use anti-mouse combined with ready to use anti-rabbit in 1:1 ratio) plus chemiluminescent substrate were combined in the plate. We utilized instrument default settings with stacking and separation at 475 V for 30 min; blocking 5 min, primary and secondary antibody both for 30 min; luminol/peroxide chemiluminescence detection for ∼15 min (with exposures of 1, 2, 4, 8, 16, 32, 64, 128 and 512 s). Chemiluminescence is automatically quantified (area under the curve or ‘AUC’ of detected peaks) by the Compass software and is displayed as an electropherogram or as a virtual blot-like image. The calculated concentrations were analysed by dividing the AUC of the C9orf72 peak by the AUC of the Hprt peak. All data points were normalized to that average PBS value.
Statistical analyses
Unless otherwise indicated, data were analysed by a one-way or two-way analysis of variance (ANOVA) followed by post-hoc analyses for multiple comparisons with appropriate test using GraphPad Prism software.
RESULTS
Stereopure oligonucleotides with PN chemistry
We have previously reported a method to generate oligonucleotides containing stereopure PN linkages based on (1,3-dimethylimidazolidin-2-ylidene) phosphoramidates (Figure 1ASupplementary Figure S1) with 2′-O-methyl (2′-OMe) and 2′-deoxyfluoro (2′-F) ribose modifications, and we have applied these methods to the synthesis and evaluation of splice switching oligonucleotides (9). To apply this chemistry to antisense oligonucleotides that promote RNase H-mediated RNA degradation, we expanded on this capability to incorporate additional 2′-ribose modifications, such as 2′-O-methoxyethyl (2′-MOE). To synthesize PN linkages, we used standard phosphoramidites, which yield the stereorandom dimer, or L-PSM ((S)-2-(phenylsulfonyl)-1-((S)-pyrrolidin-2-yl) ethan-1-ol) and D-PSM ((R)-2-(phenylsulfonyl)-1-((R)-pyrrolidin-2-yl) ethan-1-ol) amidites) (Figure 1B), which yield Rp and Sp PN dimers, respectively (Figure 1C). To validate the fidelity of synthesis with 2′-MOE modifications, we assessed diastereoselectivity after synthesis of PN-1 dimers in solution, containing 2′-MOE-G or -5-methyl-C (5MeC) in the first position and 2′-OMe-U in the second position (Supplementary Figure S1). We also measured diastereoselectivity for synthesis of PN dimers in solution containing 2′-OMe-G or -U in the first position and 2′-OMe-U in the second position. For all dimer combinations tested, we detected >99% diastereoselectivity. This method allows for the synthesis of stereopure oligonucleotides containing chimeric PS/PO/PN backbones in high yields with both high chemical and stereochemical purity. We have also explored alternate synthetic methods for generating these PN-containing oligonucleotides (Supplementary Materials and Methods).
PN modifications increases potency of antisense oligonucleotides in neurons
Metastasis-associated lung adenocarcinoma transcript 1 (Malat1, denoted MALAT1 for human) encodes a ubiquitously expressed (including brain tissues) long noncoding RNA that has been used as a surrogate target to evaluate the impact of chemical modifications to oligonucleotides acting through an RNase H mechanism (38,39). Malat1 RNA is primarily localized in the nucleus (40); therefore, its depletion provides confirmation that an antisense oligonucleotide entered the cell, then entered the nucleus and executed its activity by the expected RNase H-dependent mechanism of action. For these reasons, we opted to use Malat1 as a surrogate transcript to evaluate the impact of backbone chemistry and stereochemistry on the pharmacology of oligonucleotides in the CNS.
We have previously reported a stereopure Malat1-targeting PS/PO oligonucleotide (MALAT1-200) that demonstrated superior activity in the eye than a stereorandom PS/PO oligonucleotide (MALAT1-181) with the same sequence and 2′-ribose modification pattern (5). To assess whether incorporation of the PN backbone linkages based on (1,3-dimethylimidazolidin-2-ylidene) phosphoramidate (1), (denoted PN-1, Supplementary Figure S1) can further improve on this activity, we generated a series of chimeric oligonucleotides derived from MALAT-200 containing three consecutive stereopure or stereorandom PN-1 backbone linkages in the 5′-wing, the 3′-wing, or both (Figure 2A). We initially positioned the PN-1 linkages in the wings where PO is often incorporated (5,6).
Figure 2.

PN-1 linkages in wings increase potency of MALAT1 oligonucleotides in neurons. (A) Schematic representation of oligonucleotides designed to promote RNase H-mediated cleavage of MALAT1 RNA. Legend delineates representations for backbone and other chemical modifications to oligonucleotides. Measured Tm in °C in 1× PBS and relative EC50 with 95% confidence interval in human iCell neurons is shown for each oligonucleotide. (B) The mean percentage of full-length MALAT1 RNA remaining after addition of RNase H to preformed heteroduplex is plotted with respect to time (minutes) for the indicated oligonucleotide. Data are represented as mean ± SEM, n = 3 per time point. In cases where error bars are not shown, the error was smaller than the symbol. Data were fit to non-linear, one-phase decay using GraphPad Prism software. (C) Schematic representations of the MALAT1 RNA (green, shown 5′ to 3′)-oligonucleotide (shown in 3′–5′ orientation) heteroduplexes illustrating positions of MALAT1 RNA cleavage in biochemical RNase H experiments (red arrows). (D) Dose–response curves showing percentage of MALAT1 RNA expression (normalized to SFRS9) with increasing concentrations of the indicated oligonucleotide in human iCell neurons. Data are presented as mean ± SD, n = 2 per concentration per oligonucleotide. Lines depict four-parameter non-linear least squares fit of data. EC50s shown in panel A are derived from these experiments.
To assess the impact of PN-1 backbone chemistry on RNase H activity, we evaluated this series of oligonucleotides in biochemical assays measuring thermal stability, relative RNase H activity and cleavage position, as well as MALAT1 depletion in cultured human iCell neurons. The inclusion of PN-1 linkages generally does not substantially alter the thermal stability of RNA-oligonucleotide heteroduplexes compared with stereorandom and stereopure PS/PO-modified oligonucleotides with the same sequence and 2′-ribose modification pattern (Figure 2A), which is consistent with other reports (21,23). As expected based on our previous work (5), stereopure MALAT1-200 was more active in biochemical RNase H assays than stereorandom MALAT1-181 (Figure 2B). The introduction of PN-1 linkages in the wings did not alter on this biochemical activity. MALAT1-200 also showed a more specific RNase H cleavage pattern than the stereorandom control, MALAT1-181, yielding cleavage in the Malat1 RNA only as expected based on the positioning of an 3′-SpSpRp-5′ stereochemical motif in the PS-modified gap (Figure 2C, Supplementary Figure S2) (4). The introduction of PN-1 backbone linkages in the wings did not change this cleavage pattern (Figure 2C). In cellular assays performed under gymnotic (i.e., free uptake) conditions in iCell neurons, stereopure MALAT1-200 (EC50= 0.18 μM) was ∼10-fold more potent than stereorandom MALAT1-181 (EC50= 1.4 μM) (Figure 2A, D). MALAT1-244 (EC50= 0.02 μM), a chimeric oligonucleotide based on MALAT1-200 with three consecutive stereorandom PN-1 linkages in the 5′-wing, was ∼10-fold more potent than stereopure MALAT1-200 and 70-fold more potent than the stereorandom MALAT1-181 (Figure 2A, D). All other PN-1-containing oligonucleotides in this series including MALAT1-384 and MALAT1-385 which are the all-Rp and all-Sp stereopure PN versions of MALAT1-244, had silencing activity in neurons comparable to MALAT1-244 (Figure 2A, D, Supplementary Figure S2).
In another series of oligonucleotides, we assessed additional wing configurations that split the PN-1 linkages apart from each other and distributed them between the 5′- and 3′-wings. For this series, we evaluated oligonucleotides with two non-consecutive PN-1 linkages in the 5′-wing and either one or two non-consecutive linkages in the 3′-wing. We also varied the stereochemistry of these linkages, assessing stereorandom, all-Rp and all-Sp for each wing configuration. Once again, we found that the PN-1-containing oligonucleotides in this series had potencies in neurons comparable to that of MALAT1-244 (Supplementary Figure S2). Together these data suggest that introduction of three or four PN-1 backbone linkages to the wings of stereopure MALAT1-200 increases its silencing potency in cultured iCell neurons, likely without directly affecting RNase H.
Multiple types of PN backbone modifications can improve the activity of stereopure oligonucleotides
Next, we explored the impact of ring size in five (PN-1), six and seven membered rings, as well as exocyclic rings and sulfonamide to test whether other types of PN backbone linkages could have the same impact as PN-1 on the activity of stereopure antisense oligonucleotides in vitro. We synthesized a series of stereopure oligonucleotides having the same sequence, chemistry, and pattern of backbone stereochemistry. The only thing that changed from one oligonucleotide to the next was the chemical moiety deployed as the PN modification. Since we did not observe substantial in vitro activity differences in PN-1 (Figure 2A) based on the stereochemical configuration of the backbone, we focused on the compounds having Rp PN linkages as they are geometrically equivalent to the Sp PS linkage.
In the first series of oligonucleotides, we compared the activity of benchmark stereopure PS/PO and PN-1 backbones to a sulfonamide-containing backbone denoted PN-2. These backbones were tested in MALAT1-200, MALAT1-384 and MALAT1-489, respectively (Figure 3A). In dose-dependent, MALAT1 silencing experiments in cultured human iCell neurons, MALAT1-200 and MALAT1-384 performed as expected and consistent with data reported in Figure 2. MALAT1-489, which contained the sulfonamide modification, did not show the same potency benefits that we observed with MALAT1-384.
Figure 3.

Assessment of multiple PN backbone chemistries. (A) Schematic representation of oligonucleotides designed to promote RNase H-mediated cleavage of MALAT1 RNA (left). Legend delineates representations for backbone and other chemical modifications to oligonucleotides. The position of PN linkages is the same for all PN-containing oligonucleotides, but the moiety that replaces a non-bridging oxygen varies. Measured relative EC50 (μM) with 95% confidence interval in human iCell neurons is shown for each oligonucleotide. Dose–response curves showing percentage of MALAT1 RNA expression (normalized to SFRS9) with increasing concentrations of the indicated oligonucleotide in human iCell neurons (right). Chemical structures for the backbones tested are also shown. Data are presented as mean ± SD, n = 3 per concentration per oligonucleotide. Lines depict four-parameter non-linear least squares fit of data. Relative EC50s are derived from these experiments. (B) Schematic representation of oligonucleotides (left). Legend is shown in panel A. The PN backbone for each oligonucleotide is indicated. Measured relative EC50 (μM) with 95% confidence interval in human iCell neurons is also shown. Dose-response curves showing percentage of MALAT1 RNA expression (normalized to SFRS9) with increasing concentrations of the indicated oligonucleotide in human iCell neurons (right). Chemical structures for the backbones tested are also shown. Data are presented as mean ± SD, n = 3 per concentration per oligonucleotide. Lines depict four-parameter non-linear least squares fit of data. Relative EC50s are derived from these experiments.
We next explored the impact of changing the size of the 5-membered imidazolidine ring in PN-1 by converting it to a 6-membered hexahydropyrimidine ring in PN-3 or a 7-membered 1,3-diazepane ring in PN-4. We also evaluated a backbone with exo-cyclic guanidine in PN-5. These new backbones were tested in MALAT1-1044, MALAT1-1045 and MALAT1-1043, respectively, and they were compared to MALAT1-384 as a benchmark (Figure 3B). In dose-dependent, MALAT1 silencing experiments in cultured human iCell neurons, the PN variants (PN-3─PN-5) performed comparably to PN-1, at least in this configuration. Given this comparable activity in vitro and easy access to commercially available azide reagent 4 in salt form, we opted to further evaluate oligonucleotides based on the PN-1 backbone in vivo in mice.
Before initiating in vivo studies, we performed cellular cytotoxicity assays (Supplementary Figure S3), which showed that the profiles for MALAT1-200 and MALAT1-244 were comparable in both iCell neurons and iCell astrocytes, and neither oligonucleotide exhibited a cytotoxic profile at concentrations up to 10 μM.
PN linkages increase potency and durability of activity in mouse CNS tissue
To investigate whether the increased potency observed in cultured iCell neurons with PN-1 chemistry translates in vivo to the CNS, we evaluated Malat1 expression in wild-type mice administered MALAT1-200 or MALAT1-244 in a single-dose, dose-escalation experiment, assessing activity in the spinal cord, a readily accessible tissue, and cortex, a more challenging region to access, 1-week after dosing. In the spinal cord, a single 10 or 20 μg dose of MALAT1-200, the stereopure control, decreased the expression of Malat1 by 50% or more compared with PBS treatment. By contrast, a single 5 μg dose of MALAT1-244 was sufficient to decrease expression to the same 50% threshold (Figure 4A), and a 20 μg dose decreased mean Malat1 expression to just 8% of PBS-treated controls. In the cortex, a 20 μg dose of MALAT1-200 was needed to decrease expression to approximately the 50% threshold (PBS: 100% mean Malat1 expression; 20 μg: 61% mean expression), whereas a 5 μg dose of MALAT1-244 decreased Malat1 expression comparably (PBS: 100% mean Malat1 expression; 5 μg: 63% mean expression). In both spinal cord and cortex, the chimeric PN-1-containing MALAT1-244 yielded ∼50% Malat1 knockdown with a lower dose than MALAT1-200, illustrating a potency benefit from the addition of PN-1 linkages to the wings of a stereopure oligonucleotide in the CNS in vivo.
Figure 4.

Oligonucleotides with chimeric PN-containing backbones have improved potency and durability in CNS. (A) Schematic representation of dosing regimen with black arrow indicating administration of intracerebroventricular dose (day 0, D0) and blue arrow indicating day of analysis (day 7, D7). Relative Malat1 expression (normalized to Hprt1) in spinal cord (left) and cortex (right) one-week post treatment with PBS (beige), stereopure PS oligonucleotide (Malat1-200, light blue) or chimeric PN containing oligonucleotide (Malat1-244, navy blue) at the indicated dose. Data are presented as box and whisker plots with box from min to max with data from individual mice shown, n = 8 * P< 0.05, ** P< 0.01, *** P< 0.001 mixed-effects model with multiple comparisons. (B) Schematic representation of dosing regimen with black arrow indicating administration of ICV dose (day 0, D0) and blue arrow indicating day of analysis (day 28, D28). Relative Malat1 expression (normalized to Hprt1) in spinal cord (top left) and cortex (top right) 4 weeks post treatment with PBS (beige), stereopure PS oligonucleotide (Malat1-200, light blue) or chimeric PN containing oligonucleotide (Malat1-244, navy blue). Concentration of oligonucleotide detected in spinal cord (bottom left) and cortex (bottom right) 4-weeks post treatment with PBS (beige), stereopure PS oligonucleotide (Malat1-200, light blue) or chimeric PN containing oligonucleotide (Malat1-244, navy blue). Data are presented as in panel A, n = 4, **** P< 0.0001 one-way ANOVA with multiple comparisons. (C) Schematic representation of dosing regimen with black arrow indicating administration of intracerebroventricular dose (day 0, D0) and blue arrow indicating day of analysis (day 70, D70). Relative Malat1 expression (normalized to Hprt1) in the indicated tissue in CNS 10-weeks post treatment with PBS (beige), stereopure PS oligonucleotide (Malat1-200, light blue) or chimeric PN containing oligonucleotide (Malat1-244, navy blue). Data are presented as in panel A, n = 3 one-way ANOVA with multiple comparisons. Red asterisks show comparison of MALAT1-244 to PBS; blue asterisks MALAT1-244 to MALAT1-200.
Many aspects of oligonucleotide pharmacology, such as tissue distribution and durability of activity, are difficult to model in vitro. To investigate whether the presence of PN-1 linkages in antisense oligonucleotides impacts these parameters, we performed additional in vivo assessments in wild-type mice. Our previous work on splice-switching oligonucleotides indicated that PN chemistry can improve oligonucleotide distribution to muscle in vivo upon systemic administration (9). To assess distribution in CNS, we measured oligonucleotide concentrations and Malat1 expression levels in the spinal cord and cortex 4-weeks after a single 100 μg ICV injection. In this experiment both MALAT1-200 and MALAT1-244 decreased Malat1 RNA expression in spinal cord and cortex by ∼80% (spinal cord PBS mean 100% expression, MALAT1-200 mean 21%, MALAT1-244 mean 21%; cortex PBS mean 100% expression, MALAT1-200 mean 15%, MALAT1-244 mean 18%) (Figure 4B). For MALAT1-200, the robust Malat1 knockdown in cortex was surprising, as similarly designed stereopure PS/PO oligonucleotides targeting C9orf72 did not achieve 50% transcript knockdown in the cortex at 4 weeks post-injection (6). Nevertheless, at 4 weeks, there was no difference in Malat1 expression in cortex or spinal cord in animals treated with MALAT1-200 or MALAT1-244.
We also evaluated oligonucleotide concentrations in spinal cord and cortex from the same single dose experiment and found that the average concentration of MALAT1-244 was ∼2-fold higher than MALAT1-200 in both tissues (spinal cord: 2.0 and 0.9 μg/g, respectively; cortex: 7.7 and 4.5 μg/g, respectively) (Figure 4B). MALAT1-244 concentrations were significantly greater than PBS-treated negative controls in both tissues (spinal cord P = 0.0009, cortex P = 0.0078, one-way ANOVA), whereas MALAT1-200 was not (spinal cord P = 0.93, cortex P= 0.12, one-way ANOVA). Because MALAT1-244 was more prevalent in both spinal cord and cortex than MALAT1-200, we suspected it might also be more durable.
Because MALAT1-200 achieved better than expected activity particularly in cortex at 4 weeks, we evaluated Malat1 expression in CNS from wild-type mice 10 weeks after receiving a single 100 μg ICV dose. In this longer experiment, we observed profound differences in the activities of MALAT1-200 and MALAT1-244 throughout the CNS. By 10-weeks post-injection in MALAT1-200-treated animals, Malat1 expression had recovered so it was above the 50% threshold in all tissues (spinal cord mean 83% expression, cortex 112%, hippocampus 65%, cerebellum 70%, striatum 68%) and was statistically significantly different from PBS-treated controls only in the hippocampus (Figure 4C; spinal cord P= 0.89, cortex P= 0.75, hippocampus P= 0.02, cerebellum P= 0.29, striatum P= 0.07). By contrast in MALAT1-244-treated animals, Malat1 expression remained very low, below 20% of normal expression levels in all tissues evaluated and below 10% in striatum, cerebellum and hippocampus (spinal cord mean 20% expression, P= 0.17; cortex 19%, P= 0.005; hippocampus 6.8%, P= 0.0001; cerebellum 10%, P= 0.006; striatum 10%, P= 0.0005, one-way ANOVA), indicating durable silencing. Importantly, the 80% Malat1 knockdown observed in cortex and spinal cord with MALAT1-244 at 4 weeks was unchanged at 10 weeks. Taken together, these data indicate the PN-1-containing chimeric Malat1 oligonucleotide is more potent, achieves higher tissue exposure and is more durable than a stereopure PS-modified molecule with the same sequence and 2′-ribose modifications in vivo in CNS.
PN linkages preserve variant-selective and potent activity against C9orf72
To test whether the introduction of PN-1 chemistry can enhance the activity and pharmacology of antisense oligonucleotides in general, we assessed its impact on C9orf72-targeting oligonucleotides. C9orf72 is a more challenging target than Malat1, because the goal is to achieve variant-selective activity on a subset of the transcriptional variants. We have previously reported that stereopure PS/PO oligonucleotides targeting C9orf72 at the exon 1b-intron 1 boundary (denoted splice-site 1b, SS1b) yield superior activity to stereorandom oligonucleotides with the same sequence and 2′-ribose modification pattern (6). Stereopure oligonucleotides with asymmetric 2′-ribose modifications in their 5′- and 3′-wings acting through SS1b executed the desired mechanism of action on C9orf72; that is, they led to the selective depletion of transcripts harbouring the hexanucleotide-repeat expansion mutation (e.g. V3), but they spared expression of V2, a transcriptional variant unaffected by the expansion mutation (6) (Figure 5A).
Figure 5.

Application of PN backbone chemistry improves potency of C9orf72-targeting oligonucleotides. (A) (Left) C9orf72 allele with hexanucleotide-repeat expansion mutation (G4C2, beige box) in intron 1 upstream of exon 1b (green box). (Right) Transcriptional pre-mRNA variants (V1, V2 and V3) produced by the mutated allele. V2 uses exon 1b as the first exon, whereas V1 and V3 use variations of exon 1a. Splice site 1b (SS1b, orange star) is at the exon 1b-intron 1 boundary. SS1b is accessible to antisense oligonucleotide in transcripts that use exon 1a (V1 and V3) but protected from degradation by the spliceosome in transcripts that use exon 1b (V2) (6). (B) Schematic representation of oligonucleotides (left) evaluated for activity against C9orf72 V3 in ALS motor neurons, including non-targeting control (NTC, 5′- CCTTCCCTGAAGGTTCCUCC-3′, gray), stereorandom control (C9orf72-1341, black), stereopure control (C9orf72-1124, light blue) and chimeric oligonucleotides with various combinations of PN linkages in the wings (navy blue). Oligonucleotides were delivered to ALS motor neurons under gymnotic conditions with concentrations increasing (0.08, 0.4, 2 μM) from bottom to top. Data are presented as mean ± sem, n = 3 per oligonucleotide per concentration. two-way RM ANOVA * P < 0.05, ** P< 0.01. (C) Schematic representation of oligonucleotides (left) evaluated for activity against C9orf72 V3 in ALS motor neurons, including non-targeting control (NTC, 5′-CCTTCCCTGAAGGTTCCTCC-3′, gray), fully stereorandom C9orf72-1377 (black) and fully stereopure C9orf72-1130 (navy). Oligonucleotides were delivered to ALS motor neurons under gymnotic conditions with concentrations increasing (0.08, 0.4, 2 μM) from bottom to top. Data are presented as mean ± sem, n = 3 per oligonucleotide per concentration.
We synthesized a series of PN-1-containing oligonucleotides and compared their activity in cultured motor neurons to stereorandom (C9orf72-1341) and stereopure (C9orf72-1124) PS/PO controls. All oligonucleotides feature the same asymmetric pattern of 2′-ribose modifications and the same sequence (Figure 5B). Because these oligonucleotides are designed to selectively deplete transcripts containing the C9orf72 repeat-expansion mutation, we evaluated their activity in human iPSC-derived motor neurons generated from the cells from a patient with C9orf72-ALS, which contain the hexanucleotide repeat-expansion mutation (denoted ALS motor neurons).
As expected based on our prior publication (6), we found that a stereopure (PS/PO) oligonucleotide was more potent than a comparable stereorandom (PS/PO) oligonucleotide. 0.4 μM C9orf72-1124 (stereopure, PS/PO) yielded >50% knockdown (∼60%) of the C9orf72 V3 transcript, whereas 2 μM C9orf72-1341 (stereorandom, PS/PO) yielded comparable knockdown (∼57%) (Figure 5B). Consistent with our observations from the Malat1 oligonucleotide series, we also observed that chimeric oligonucleotides (PS/PO/PN) were more potent than their stereopure and stereorandom (PS/PO) counterparts. For most chimeric oligonucleotides (PS/PO/PN), 0.08 μM yielded ∼50% knockdown of the C9orf72 V3 transcript (Figure 5B, mean knockdown 50%), with C9orf72-1376 yielding the most knockdown at 65%. At 2 μM, the PN-1-containing oligonucleotides led to an average 95% knockdown of V3. In a parallel series of experiments, we compared the activity of one of the stereopure PN-1-modified oligonucleotides, C9orf72-1130, with a fully stereorandom compound with the same pattern of backbone PN modifications, C9orf72-1377. As expected, the stereopure compound exhibited more potent silencing activity at all concentrations than the stereorandom molecule (Figure 5C).
To execute the desired mechanism of action, oligonucleotides targeting the C9orf72-repeat expansion must also be selective, sparing expression of V2, the most abundant C9orf72 transcript. To ensure that chimeric (PS/PO/PN) oligonucleotides preserved selectivity, we assessed their activity against all C9orf72 transcriptional variants (All V) in ALS motor neurons (Supplementary Figure S4). C9orf72-1130 and C9orf72-1129 preserved >50% of total C9orf72 transcript expression at all concentrations tested, while the remaining chimeric oligonucleotides (C9orf72-1381, -1376 and -1378) were deemed too potent against all variants for further analysis. C9orf72-1130 and C9orf72-1129 were potent against V3 (93% of V3 was depleted at the highest concentrations tested) and selective for V3.
We also assessed their thermal stability and stability in mouse brain homogenate. As noted for the Malat1 oligonucleotide series, the introduction of PN chemistry did not substantially impact the thermal stability of C9orf72 RNA-oligonucleotide heteroduplexes (Supplementary Figure S4), indicating that activity gains with the inclusion of PN chemistry are not Tm-driven. The PN modifications did not substantially change oligonucleotide stability ex vivo in mouse brain homogenate, (Supplementary Figure S4), indicating that the PN linkages do not substantially alter susceptibility to nucleases. We selected C9orf72-1130 for in vivo evaluation.
PN-1 linkages are not pro-inflammatory in vitro
Before evaluating efficacy of PN-1-modified oligonucleotides in mice, we performed preliminary in vitro and in vivo safety assessments. As with the Malat1 oligonucleotides, we evaluated C9orf72-targeting oligonucleotides in HEK293 cell viability assays to assess cytotoxicity of the PN-containing oligonucleotide. Cellular viability in the presence of PN-modified C9orf72-1130 was not affected at concentrations up to 30 μM and was comparable to C9of72-1124 (Supplementary Fig S3).
Because chemically modified oligonucleotides have the potential to activate innate immune receptors, especially TLR9 (41–43), we also evaluated C9orf72 oligonucleotides in TLR9 reporter assays. In contrast to an oligonucleotide that was designed to be a strong TLR9 agonist (34), C9orf72-targeting oligonucleotides showed no detectable activity when tested at 30 μM in this assay (Supplementary Figure S5). To further explore potential impact of these oligonucleotides on immune response, we evaluated their cytokine-activation profiles in PBMCs from humans and monkeys and splenocytes from mice at concentration ranging from 0.1 to 30 μM. To benchmark these profiles, we incorporated control oligonucleotides with known neutral or pro-inflammatory profiles. We considered cytokine activation by two complementary approaches. In the first approach, we compared the maximum-fold induction of IL-6, TNF-α and MIP-1β secretion in response to increasing doses of PS-modified C9orf72-1124 or PN-modified C9orf72-1129 and C9orf72-1130 to the pro-inflammatory and neutral oligonucleotide controls. All C9orf72-targeting oligonucleotides exhibited lower maximum-fold cytokine activation than the pro-inflammatory control, and PN-modified oligonucleotides showed less maximum-fold activation than C9orf72-1124, indicating that the PN-1 modification may alleviate cytokine activation (Supplementary Figure S5). In the second approach, we compared levels of cytokine secretion induced by the PS- and PN-modified oligonucleotides to the maximum amount of secretion observed for neutral and pro-inflammatory controls. Similar to the approach above, the C9orf72-targeting oligonucleotides induced a cytokine secretion profile more like the neutral than the pro-inflammatory oligonucleotide. In assays where the PS-modified C9orf72-1124 showed some pro-inflammatory activity (e.g. mouse IL-6, mouse MIP-1β), the PN-modified oligonucleotides showed a less inflammatory profile (Supplementary Figure S5), again supporting the notion that the PN-1 modification may alleviate inflammatory effects of PS-modified oligonucleotides.
PN-1 linkages are well tolerated in mice following a single ICV injection
We evaluated the C9orf72-targeting oligonucleotides in a series of 8-week single dose ICV tolerability studies in wild-type C57Bl/6 mice. In these studies, following a single 100 μg ICV injection, the body weight of mice treated with C9orf72-1124, C9orf72-1129 or C9orf72-1130 was similar to those treated with PBS (Supplementary Figure S6). A modified Irwin test was used to observe changes in health, activity levels or motor function post-dose. Minor acute changes were noted; oligonucleotide-treated mice did not differ from PBS-treated mice after 24-h acute post-dose period (Supplementary Figure S6). We evaluated brains from treated animals for signs of immune activation (Gfap and Iba1 expression) or histological abnormalities. Brain tissues from mice receiving oligonucleotides did not notably differ from PBS-treated animals in expression of immune-activation markers or in appearance upon histological examination (Supplementary Figure S7), indicating that stereopure PN-1-containing oligonucleotides were well tolerated in mice following a single 100 μg ICV injection.
Stereopure PN-1-containing oligonucleotides have potency benefit in mouse CNS
We initially compared the activity of C9orf72-1130 in the CNS of C9BAC transgenic mice (37) to treatment with PBS, a chemistry matched non-targeting control (NTC) or C9orf72-1377, an oligonucleotide with the same sequence and chemical modification pattern but that has a fully stereorandom backbone (Figure 6A). In mice administered a 100 μg total dose and evaluated two weeks after treatment onset, the NTC tracked with PBS treated animals, indicating that the chemical modifications are not driving non-specific silencing activity. In all tissues evaluated, C9orf72-1130 (mean C9orf72 V3 expression cortex: 51%; spinal cord: 30%; striatum: 47%; hippocampus: 32%; cerebellum: 47%) outperformed controls and C9orf72-1377 (mean C9orf72 V3 expression cortex: 83%; spinal cord: 67%; striatum: 82%; hippocampus: 85%; cerebellum: 62%), indicating that the stereopure backbone enhanced silencing across the CNS (Figure 6B). We compared activity profiles with respect to measured tissue exposure for the oligonucleotides at 2 weeks. The NTC showed a relatively flat response with nominal activity across tissue concentrations (Figure 6C, top row), indicating again that neither treatment nor the chemical modifications are driving non-specific silencing activity. In most tissues, C9orf72-1377 and C9orf72-1130 showed increased silencing activity as tissue exposure increased. In all tissues, data for C9orf72-1130 was shifted slightly rightward, indicating increased tissue exposure, and substantially downward, indicating enhanced silencing, compared with C9orf72-1377 (Figure 6C, bottom row). However, tissue exposure benefits for the stereopure molecule are modest, indicating that activity gains of the PN-1-containing oligonucleotide did not just depend on increased tissue concentrations.
Figure 6.

Stereopure oligonucleotides exhibit enhanced silencing compared with stereorandom across the CNS. (A) Schematic representation of oligonucleotides used in this study and the dosing regimen (top). Black arrows indicate dosing; light blue arrows indicate evaluation. (B) Relative expression of C9orf73 V3 at 2 weeks is shown for each treatment group in cortex, striatum, spinal cord, cerebellum, and hippocampus. Data are presented as box and whisker plots with box from min to max with data from individual mice shown, n = 7, **P< 0.01, *** P< 0.001, ****P< 0.0001; P values calculated by one-way ANOVA. (C) Relationship between fold change in C9orf72 V3 expression and measured tissue exposure at 2 weeks is shown for NTC (top) and C9orf72 oligonucleotides C9-1377 and C9-1130 (bottom) in cortex, striatum, spinal cord, cerebellum, and hippocampus. Each point represents one mouse. Lines represent best-fit for simple linear regression analysis.
To evaluate the durability of this response, we assessed mice treated the same way 8 weeks after the first dose (Figure 7A). In all tissues evaluated, C9orf72-1130 (mean C9orf72 V3 expression cortex: 46%; spinal cord: 15%; striatum: 37%; hippocampus: 25%; cerebellum: 42%) outperformed the stereorandom C9orf72-1377 (mean C9orf72 V3 expression cortex: 73%; spinal cord: 49%; striatum: 64%; hippocampus: 63%; cerebellum: 80%), with expression in the control rising above the 50% mark in all tissues except spinal cord for C9orf72-1377, indicating that the stereopure backbone enhanced durability throughout the brain (Figure 7B). In all tissues, the percentage of C9orf72 V3 expression was lower for C9orf72-1130 at 8 weeks than 2 weeks, suggesting that silencing activity increases with time beyond two weeks. For C9orf72-1377, C9orf72 V3 expression increased in cerebellum from a mean of 62% at 2 weeks to 80% at 8 weeks, indicating that V3 expression in animals treated with C9orf72-1377 is recovering. We next compared activity profiles with respect to measured tissue exposure at 8 weeks.
Figure 7.

Stereopure oligonucleotides exhibit enhanced silencing durability compared with stereorandom across the CNS. (A) Schematic representation of oligonucleotides used in this study and the dosing regimen (top). Black arrows indicate dosing; light blue arrows indicate evaluation. (B) Relative expression of C9orf73 V3 at 8 weeks is shown for each treatment group in cortex, striatum, spinal cord, cerebellum, and hippocampus. Data are presented as box and whisker plots with box from min to max with data from individual mice shown, n = 7, *** P< 0.001, ****P< 0.0001; P values calculated by one-way ANOVA. (C) Relationship between fold change in C9orf72 V3 expression and measured tissue exposure at 8 weeks is shown for C9orf72 oligonucleotides C9-1377 and C9-1130 (bottom) in cortex, striatum, spinal cord, cerebellum, and hippocampus. Each point represents one mouse. Lines represent best-fit for simple linear regression analysis.
Once again data for C9orf72-1130 was shifted rightward and downward, indicating increased tissue exposure and enhanced silencing, respectively compared with C9orf72-1377 (Figure 7C). Once again, tissue exposure benefits for the stereopure molecule were modest, indicating that activity gains of the PN-1-containing oligonucleotide did not depend on increased tissue concentrations. These data indicate that chimeric stereopure oligonucleotides outperform their sequence and chemistry matched stereorandom counterparts in the CNS.
PN-1 linkages support sustained activity against C9orf72 V3 in mouse cortex
To evaluate the impact of PN-1 chemistry compared with a stereopure oligonucleotide lacking the modification, we compared the activity of C9orf72-1124 (stereopure PS/PO) and C9orf72-1130 (stereopure PS/PO/PN) in mouse CNS focusing on tissues impacted by C9orf72-repeat expansion-associated pathology in ALS and FTD. To assess activity, we measured C9orf72 V3 expression in the spinal cord and cortex 2, 4 and 8 weeks after ICV injection. C9orf72-1124 significantly depleted V3 expression compared with PBS-treated control in spinal cord at all time points (mean C9orf72 V3 expression 2 weeks: 31%, 4 weeks: 28%, 8 weeks: 67%; P < 0.001, two-way ANOVA with multiple comparisons). C9orf72-1130 yielded more effective and durable silencing in the spinal cord, with significant knockdown compared with PBS at all time points (mean C9orf72 V3 expression 2 weeks: 18%, 4 weeks: 14%, 8 weeks: 16%; P < 0.0001, two-way ANOVA with multiple comparisons) (Figure 8A). As expected from our prior studies (6), the PS/PO molecule C9orf72-1124 was not as effective in depleting C9orf72 V3 transcripts in cortex as it was in the spinal cord, yielding significant knockdown compared with PBS through 4 weeks (mean C9orf72 V3 expression 2 weeks: 80%, 4 weeks: 68% (P < 0.05), 8 weeks: 88%; two-way ANOVA with multiple comparisons) but with <50% knockdown at all evaluated time points (Figure 8B). By contrast C9orf72-1130 yielded significant V3 knockdown in cortex compared with PBS at all time points (mean C9orf72 V3 expression 2 weeks: 54%, 4 weeks: 48% (P < 0.05), 8 weeks: 30% P < 0.0001, two-way ANOVA with multiple comparisons), with the magnitude of knockdown increasing over time (Figure 8A). These data indicate that the addition of PN-1 linkages improved activity in the CNS.
Figure 8.

Application of PN backbone linkages to C9orf72 increases potency and durability of activity in spinal cord and cortex. (A) Relative fold expression of C9orf72 V3 (normalized to HPRT) after treatment with PBS (beige), stereopure PS oligonucleotide (C9orf72-1124, light blue) or chimeric PN containing oligonucleotide (C9orf72-1130, navy blue) in spinal cord (top) and cortex (bottom) at 2, 4 or 8 weeks post dose. Box and whisker plots show minimum to maximum, and data points represent individual mice, n = 7. two-way ANOVA with multiple comparisons to PBS *P< 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (B) Relative poly-GP levels detected (relative to PBS) after treatment with PBS (beige), stereopure PS oligonucleotide (C9orf72-1124, light blue) or chimeric PN containing oligonucleotide (C9orf72-1130, navy blue) in spinal cord (top) and cortex (bottom) at 2, 4 and 8 weeks post-dose. Data representation and stats are as in panel A, n = 7. (C) PK-PD relationship for stereopure PS oligonucleotide (C9orf72-1124, light blue) or chimeric PN containing oligonucleotide (C9orf72-1130, navy blue) in spinal cord (left) and cortex (right) at 8 weeks post-dose. The percentage of C9orf72 V3 expression remaining (PD effect) is plotted with respect to the concentration of oligonucleotide detected in the tissue (PK effect). Shading depicts 95% confidence intervals. Each point represents data from one mouse. (D) Relative fold expression of C9orf72 protein (normalized to Hprt) after treatment with PBS (beige), stereopure PS oligonucleotide (C9orf72-1124, light blue) or chimeric PN containing oligonucleotide (C9orf72-1130, navy blue) in spinal cord (left) and cortex (right) at 8 weeks post-dose. Data are presented as mean ± sd. Each point represents data from one mouse, n = 7.
To confirm this activity against V3 with a secondary biomarker, we also evaluated poly-glycine-proline (poly-GP) levels in the spinal cord and cortex (Figure 8B). Poly-GP is a DPR protein produced from both sense and antisense C9orf72 transcripts (44–47) and is a biomarker for C9orf72-target engagement (48,49). In the spinal cord, where C9orf72-1124 was most effective, poly-GP levels were very low at all time points (mean poly-GP levels 2 weeks: 7%; 4 weeks: 0%; 8 weeks: 6%). The persistence of poly-GP depletion beyond that observed for V3 is consistent with prior reports (11). C9orf72-1130 had similarly potent and durable effects up to 8 weeks in the spinal cord (mean poly-GP levels 2 weeks: 7%; 4 weeks: 0%; 8 weeks: 0%) (Figure 8B). In the cortex, where the activity differences between C9orf72-1124 and C9orf72-1130 against V3 were more profound, we also detected more substantial differences in poly-GP. C9orf72-1124 significantly decreased the amount of poly-GP in the cortex at 4 weeks (mean poly-GP levels 2 weeks: 60%; 4 weeks: 27.5%; 8 weeks: 53%; P < 0.05, one-way ANOVA), but by 8 weeks, poly-GP levels had begun to rise. By contrast, C9orf72-1130 significantly decreased the amount of poly-GP at all time points (mean poly-GP levels 2 weeks: 31%; 4 weeks: 15%; 8 weeks: 3%; P < 0.01, one-way ANOVA), and mean levels were lower at 8 weeks than at earlier time points (Figure 8B). These data are consistent with the conclusion that C9orf72-1130 has a more durable impact on V3 transcript expression and downstream biomarkers than C9orf72-1124, especially in the cortex.
To investigate the source of the improved potency and durability of C9orf72-1130, we evaluated the relationship between oligonucleotide concentration in a tissue (pharmacokinetic effect, PK) and the amount of V3 knockdown detected in that tissue (pharmacodynamic effect, PD) for this in vivo experiment (Figure 8C). To visualize this relationship, we generated the PK-PD plots shown in Figure 8C. Although some improvement in tissue exposure was observed for C9orf72-1130 in the cortex (Figure 8C, right graph, rightward shift of navy dots), improved exposure was not observed in the spinal cord. In both tissues, the knockdown of V3 transcripts obtained with C9orf72-1130 was superior to that of C9orf72-1124 even when tissue exposure levels were comparable, indicating that activity gains of the PN-1-containing oligonucleotide did not depend on increased tissue concentrations of the oligonucleotides.
To confirm these oligonucleotides spare V2 and execute our desired variant-selective mechanism of action, we evaluated the expression of all C9orf72 transcriptional variants (All V) as well as C9orf72 protein expression, as preserving C9orf72 protein at levels sufficient to avoid haploinsufficiency is the main reason for pursuing a variant-selective approach (6). In the 8-week experiment in C9BAC transgenic mice, we detected >50% expression of all C9orf72 transcriptional variants at all time points evaluated in spinal cord and cortex after treatment with either C9orf72-1124 or C9orf72-1130 (Supplementary Figure S8), indicating that both oligonucleotides are selective for expansion-containing variants. These levels of C9orf72 transcript expression were sufficient to preserve expression of C9orf72 protein, as we did not detect significant changes in protein expression in the spinal cord (C9orf72-1124 P = 0.83; C9orf72-1130 P = 0.86 at 8 weeks) or cortex (C9orf72-1124 P = 0.82; C9orf72-1130 P = 0.80 at 8 weeks) of treated mice compared with PBS controls (Figure 8D). Taken together, these data support the notion that PN-1 linkages drive activity gains for stereopure C9orf72-1130 in the CNS and preserve a selective mechanism of action against C9orf72 transcriptional variants.
DISCUSSION
Herein, we report the methods necessary to synthesize various PN-containing stereopure oligonucleotides with chimeric backbones comprising a combination of PS, PO and/or PN linkages. We demonstrate that this chemistry is compatible with an expanding repertoire of ribose-modified nucleotides, including 2′-MOE, 2′-H, 2′-OMe and 2′-F. This method is efficient, yielding high stereochemical selectivity at each chiral backbone position. We applied this chemistry to investigate the impact of PN-backbone modifications on the activity and pharmacology of RNase H-active stereopure antisense oligonucleotides, using Malat1 and C9orf72 as benchmarks. We found that various types of PN-containing oligonucleotides have superior potency in vitro in cultured neurons, and oligonucleotides incorporating one of these backbone types, PN-1, were well tolerated in mice following a single ICV administration of 100 μg, and improved pharmacology and silencing in vivo in CNS compared with oligonucleotides harbouring only PS and PO linkages.
Application of PN backbone modifications increases the complexity of oligonucleotide synthesis but also creates a new opportunity to optimize the silencing and pharmacology of these chimeric molecules. Herein, we identify multiple types of PN modifications with potential to improve on the activity achievable with the PS/PO backbone. For these investigations, we have focused on the application of the PN-1 modification to the wings of these molecules. Given the number of PN backbone types that show some benefit in vitro and the number of backbone positions in an antisense oligonucleotide, investigations to optimize the position and stereochemical configuration of these PN modifications have the potential to be vast in scope. The advent of the chiral PS backbone increased the complexity of oligonucleotide synthesis from a single isomer, where every backbone linkage was a prochiral PO, to 2n diastereomers, where n is the number of backbone positions, with synthesis yielding up to 524 288 diastereomers for a 20mer (1). With the introduction of PN-1, another chiral backbone, the number of possible permutations of PO, PS and PN backbone configurations in an oligonucleotide further increases this calculation. Because of this complexity, we believe that to gain insight into the relationship between these new chemistries and their impact on biological activity, it is advantageous to evaluate them in a stereopure or largely stereopure format.
The inclusion of PN-1 in the wings of antisense oligonucleotides increased silencing likely through an RNase H-independent mechanism, as the PN-1-containing molecules did not differ from a stereopure PS/PO control in biochemical RNase H experiments assessing their activity levels or their cleavage patterns. This potency increase is also unlikely to be Tm driven, as PN modifications do not substantially alter the Tm of RNA-oligonucleotide heteroduplexes for Malat1 or C9orf72. This observation distinguishes PN linkages from other chemical modifications (e.g., constrained ethyl and locked nucleic acid) that have been shown to improve antisense activity (2,50). Improved cellular uptake, intracellular stability or trafficking could also drive the observed activity gains, and others have observed cellular uptake benefits by modulating the charge of the oligonucleotide backbone (51). This increased potency translated in vivo in CNS, especially in difficult to reach regions of the brain, as the impact was more evident in the cortex than the spinal cord for both Malat1 and C9orf72. This improved potency is likely driven in part by tissue exposure benefits, as PN-1-containing molecules were more abundant in some brain tissues. However, this is unlikely the only explanation, as potency benefits were observed in these analyses even when tissue exposure was comparable between PN-1-containing and PS/PO molecules. In addition to potency and tissue exposure improvements, the PN-1-containing molecules exhibited profound improvements in durability, extending duration of Malat1 knockdown throughout the brain at ≥80% for at least 10 weeks. Comparably profound durability benefits were also observed for the more challenging C9orf72 target, where >50% knockdown of C9orf73 V3 was maintained for at least 8 weeks throughout the brain, while selectivity for expansion-containing variants was preserved. Based on these observations, we believe PN-1 backbone chemistry and its application to generate chimeric stereopure oligonucleotides represents a major advance for the field that could have important implications for oligonucleotide therapeutics designed to address CNS disease.
Achieving sufficient bioavailability and tissue exposure at the site of action to achieve the desired pharmacodynamic effect on the target is an important determinant for any drug discovery program (52), and this can be particularly challenging in CNS (53). Currently, successful application of antisense oligonucleotides to diseases that impact the CNS depends on administration directly to the CSF, which enables rapid distribution to most regions of the brain (39,54). Early clinical success with intrathecally administered antisense oligonucleotides was achieved by concentrating on pathologies that affect tissues that are readily accessible from CSF (e.g. spinal cord) (54–56). To build on this success, we need to increase oligonucleotide exposure in more difficult to reach regions of the CNS as well as to prolong their activity, which would help minimize risks associated with their frequent administration (57). We believe PN-1 linkages have potential to advance the pharmacology of oligonucleotide therapeutics on both fronts, and our application to C9orf72-associated ALS and FTD best illustrates this point. Despite substantial overlap in the clinical manifestation of C9orf72-ALS and C9orf72-FTD (58–60), differences in regional manifestation in the brain may impact outcomes in these indications. For those on the ALS end of the spectrum, neurodegeneration impacts upper and lower motor neurons of the motor cortex and spinal cord (61–63). These are more readily accessible tissues for traditional oligonucleotides. By contrast, neurodegeneration in FTD primarily impacts cortical neurons of the frontal and anterior temporal lobes (62). We believe the pharmacological benefits of PN-1 backbone chemistry on stereopure antisense oligonucleotides make FTD a more addressable indication with an oligonucleotide therapeutic.
DATA AVAILABILITY
Source data for figures Figures 2B, D, 3A, B, 4A–C, 5B, C, 6B, C, 7A–C, 8A, B, and Supplementary Figures S1, S2A–C, S3A, B, S4A, B, S6A, S8 are provided with the paper.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Professors Gregory Verdine and Takeshi Wada for their continued support and inspiration to explore the impact of backbone chirality on oligonucleotide pharmacology. The authors are also grateful to Jeff Rossi, Timea Kolozsvary, Cong Zhou, Ben Lagan, Karley Bussow and Boris Lamberg for synthesis, purification, characterization and formulation of some of the oligonucleotides used in this study; and Kuldeep Singh for the images shown in Supplementary Figure S7. The authors are also grateful to Amy Donner for writing, editorial and graphical support for this paper.
Contributor Information
Pachamuthu Kandasamy, Wave Life Sciences, Cambridge, MA 02138, USA.
Yuanjing Liu, Wave Life Sciences, Cambridge, MA 02138, USA.
Vincent Aduda, Wave Life Sciences, Cambridge, MA 02138, USA.
Sandheep Akare, Wave Life Sciences, Cambridge, MA 02138, USA.
Rowshon Alam, Wave Life Sciences, Cambridge, MA 02138, USA.
Amy Andreucci, Wave Life Sciences, Cambridge, MA 02138, USA.
David Boulay, Wave Life Sciences, Cambridge, MA 02138, USA.
Keith Bowman, Wave Life Sciences, Cambridge, MA 02138, USA.
Michael Byrne, Wave Life Sciences, Cambridge, MA 02138, USA.
Megan Cannon, Wave Life Sciences, Cambridge, MA 02138, USA.
Onanong Chivatakarn, Wave Life Sciences, Cambridge, MA 02138, USA.
Juili Dilip Shelke, Wave Life Sciences, Cambridge, MA 02138, USA.
Naoki Iwamoto, Wave Life Sciences, Cambridge, MA 02138, USA.
Tomomi Kawamoto, Wave Life Sciences, Cambridge, MA 02138, USA.
Jayakanthan Kumarasamy, Wave Life Sciences, Cambridge, MA 02138, USA.
Sarah Lamore, Wave Life Sciences, Cambridge, MA 02138, USA.
Muriel Lemaitre, Wave Life Sciences, Cambridge, MA 02138, USA.
Xuena Lin, Wave Life Sciences, Cambridge, MA 02138, USA.
Kenneth Longo, Wave Life Sciences, Cambridge, MA 02138, USA.
Richard Looby, Wave Life Sciences, Cambridge, MA 02138, USA.
Subramanian Marappan, Wave Life Sciences, Cambridge, MA 02138, USA.
Jake Metterville, Wave Life Sciences, Cambridge, MA 02138, USA.
Susovan Mohapatra, Wave Life Sciences, Cambridge, MA 02138, USA.
Bridget Newman, Wave Life Sciences, Cambridge, MA 02138, USA.
Ik-Hyeon Paik, Wave Life Sciences, Cambridge, MA 02138, USA.
Saurabh Patil, Wave Life Sciences, Cambridge, MA 02138, USA.
Erin Purcell-Estabrook, Wave Life Sciences, Cambridge, MA 02138, USA.
Mamoru Shimizu, Wave Life Sciences, Cambridge, MA 02138, USA.
Pochi Shum, Wave Life Sciences, Cambridge, MA 02138, USA.
Stephany Standley, Wave Life Sciences, Cambridge, MA 02138, USA.
Kris Taborn, Wave Life Sciences, Cambridge, MA 02138, USA.
Snehlata Tripathi, Wave Life Sciences, Cambridge, MA 02138, USA.
Hailin Yang, Wave Life Sciences, Cambridge, MA 02138, USA.
Yuan Yin, Wave Life Sciences, Cambridge, MA 02138, USA.
Xiansi Zhao, Wave Life Sciences, Cambridge, MA 02138, USA.
Elena Dale, Wave Life Sciences, Cambridge, MA 02138, USA.
Chandra Vargeese, Wave Life Sciences, Cambridge, MA 02138, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wave Life Sciences. Funding for open access charge: Wave Life Sciences.
Conflict of interest statement. All authors were employees of Wave Life Sciences during completion of this work.
This paper is linked to: doi:10.1093/nar/gkac018.
REFERENCES
- 1. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014; 24:374–387. [DOI] [PubMed] [Google Scholar]
- 2. Khvorova A., Watts J.K.. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017; 35:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knouse K.W., deGruyter J.N., Schmidt M.A., Zheng B., Vantourout J.C., Kingston C., Mercer S.E., McDonald I.M., Olson R.E., Zhu Y.et al.. Unlocking P(V): reagents for chiral phosphorothioate synthesis. Science (New York, N.Y.). 2018; 361:1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena, Standley S.M., Lu G., Apponi L.H.et al.. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:845–851. [DOI] [PubMed] [Google Scholar]
- 5. Byrne M., Vathipadiekal V., Apponi L., Iwamoto N., Kandasamy P., Longo K., Liu F., Looby R., Norwood L., Shah A.et al.. Stereochemistry enhances potency, efficacy, and durability of malat1 antisense oligonucleotides in vitro and in vivo in multiple species. Transl. Vis. Sci. Technol. 2021; 10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Y., Dodart J.C., Tran H., Berkovitch S., Braun M., Byrne M., Durbin A.F., Hu X.S., Iwamoto N., Jang H.G.et al.. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 2021; 12:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu D., Rivas-Bascón N., Padial N.M., Knouse K.W., Zheng B., Vantourout J.C., Schmidt M.A., Eastgate M.D., Baran P.S.. Enantiodivergent formation of C-P bonds: synthesis of P-Chiral phosphines and methylphosphonate oligonucleotides. J. Am. Chem. Soc. 2020; 142:5785–5792. [DOI] [PubMed] [Google Scholar]
- 8. Featherston A.L., Kwon Y., Pompeo M.M., Engl O.D., Leahy D.K., Miller S.J.. Catalytic asymmetric and stereodivergent oligonucleotide synthesis. Science (New York, N.Y.). 2021; 371:702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kandasamy P., McClorey G., Shimizu M., Kothari N., Alam R., Iwamoto I., Kumarasamy J., Bommineni G.R., Bezigian A., Chivatakarn O.et al.. Control of backbone chemistry and chirality boost oligonucleotide splice switching activity. Nucleic Acids Res. 2022; gkac018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao H.T., John N., Delic V., Ikeda-Lee K., Kim A., Weihofen A., Swayze E.E., Kordasiewicz H.B., West A.B., Volpicelli-Daley L.A.. LRRK2 antisense oligonucleotides ameliorate α-Synuclein inclusion formation in a parkinson's disease mouse model. Mol. Ther. Nucleic Acids. 2017; 8:508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang J., Zhu Q., Gendron T.F., Saberi S., McAlonis-Downes M., Seelman A., Stauffer J.E., Jafar-Nejad P., Drenner K., Schulte D.et al.. Gain of toxicity from ALS/FTD-Linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016; 90:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hagemann T.L., Powers B., Mazur C., Kim A., Wheeler S., Hung G., Swayze E., Messing A.. Antisense suppression of glial fibrillary acidic protein as a treatment for alexander disease. Ann. Neurol. 2018; 83:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elitt M.S., Barbar L., Shick H.E., Powers B.E., Maeno-Hikichi Y., Madhavan M., Allan K.C., Nawash B.S., Gevorgyan A.S., Hung S.et al.. Suppression of proteolipid protein rescues pelizaeus-merzbacher disease. Nature. 2020; 585:397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raymond G.J., Zhao H.T., Race B., Raymond L.D., Williams K., Swayze E.E., Graffam S., Le J., Caron T., Stathopoulos J.et al.. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 2019; 4:e131175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Becker L.A., Huang B., Bieri G., Ma R., Knowles D.A., Jafar-Nejad P., Messing J., Kim H.J., Soriano A., Auburger G.et al.. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017; 544:367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLoughlin H.S., Moore L.R., Chopra R., Komlo R., McKenzie M., Blumenstein K.G., Zhao H., Kordasiewicz H.B., Shakkottai V.G., Paulson H.L.. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018; 84:64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scoles D.R., Meera P., Schneider M.D., Paul S., Dansithong W., Figueroa K.P., Hung G., Rigo F., Bennett C.F., Otis T.S.et al.. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017; 544:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lenk G.M., Jafar-Nejad P., Hill S.F., Huffman L.D., Smolen C.E., Wagnon J.L., Petit H., Yu W., Ziobro J., Bhatia K.et al.. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and dravet syndrome. Ann. Neurol. 2020; 87:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maier M.A., Guzaev A.P., Manoharan M.. Synthesis of chimeric oligonucleotides containing phosphodiester, phosphorothioate, and phosphoramidate linkages. Org. Lett. 2000; 2:1819–1822. [DOI] [PubMed] [Google Scholar]
- 20. Dagle J.M., Littig J.L., Sutherland L.B., Weeks D.L.. Targeted elimination of zygotic messages in xenopus laevis embryos by modified oligonucleotides possessing terminal cationic linkages. Nucleic Acids Res. 2000; 28:2153–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lennox K.A., Sabel J.L., Johnson M.J., Moreira B.G., Fletcher C.A., Rose S.D., Behlke M.A., Laikhter A.L., Walder J.A., Dagle J.M.. Characterization of modified antisense oligonucleotides in xenopus laevis embryos. Oligonucleotides. 2006; 16:26–42. [DOI] [PubMed] [Google Scholar]
- 22. Jain M.L., Bruice P.Y., Szabó I.E., Bruice T.C.. Incorporation of positively charged linkages into DNA and RNA backbones: a novel strategy for antigene and antisense agents. Chem. Rev. 2012; 112:1284–1309. [DOI] [PubMed] [Google Scholar]
- 23. Chelobanov B.P., Burakova E.A., Prokhorova D.V., Fokina A.A., Stetsenko D.A.. New oligodeoxynucleotide derivatives containing N-(methanesulfonyl)-phosphoramidate (mesyl phosphoramidate) internucleotide group. Russ. J. Bioorg. Chem. 2017; 43:664–668. [Google Scholar]
- 24. Vlaho D., Fakhoury J.F., Damha M.J.. Structural studies and gene silencing activity of siRNAs containing cationic phosphoramidate linkages. Nucleic Acid Ther. 2018; 28:34–43. [DOI] [PubMed] [Google Scholar]
- 25. Burakova E.A., Derzhalova A.S., Chelobanov B.P., Fokina A.A., Stetsenko D.A.. New oligodeoxynucleotide derivatives containing N-(sulfonyl)-phosphoramide groups. Russ. J. Bioorg. Chem. 2019; 45:662–668. [Google Scholar]
- 26. Derzhalova A., Markov O., Fokina A., Shiohama Y., Zatsepin T., Fujii M., Zenkova M., Stetsenko D. Novel lipid-oligonucleotide conjugates containing long-chain sulfonyl phosphoramidate groups: synthesis and biological properties. Applied Sciences. 2021; 11:1174. [Google Scholar]
- 27. Hammond S.M., Sergeeva O.V., Melnikov P.A., Goli L., Stoodley J., Zatsepin T.S., Stetsenko D.A., Wood M.J.A.. Mesyl phosphoramidate oligonucleotides as potential splice-switching agents: impact of backbone structure on activity and intracellular localization. Nucleic Acid Ther. 2021; 31:190–200. [DOI] [PubMed] [Google Scholar]
- 28. Lomzov A.A., Golyshev V.M., Dyudeevaa E.S., Kuprushkin M.S., Pyshnyi D.V.. Book of abstracts. Albany 2019: the 20th conversation: structure and hybridization properties of phosphorylguanidine oligonucleotides. J. Biomol. Struct. Dyn. 2019; 37:83. [Google Scholar]
- 29. Golyshev V.M., Pyshnyi D.V., Lomzov A.A.. Effects of phosphoryl guanidine modification of phosphate residues on the structure and hybridization of oligodeoxyribonucleotides. J. Phys. Chem. B. 2021; 125:2841–2855. [DOI] [PubMed] [Google Scholar]
- 30. Lomzov A.A., Kupryushkin M.S., Shernyukov A.V., Nekrasov M.D., Dovydenko I.S., Stetsenko D.A., Pyshnyi D.V.. Data for isolation and properties analysis of diastereomers of a mono-substituted phosphoryl guanidine trideoxyribonucleotide. Data Brief. 2019; 25:104148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dyudeeva E.S., Kupryushkin M.S., Lomzov A.A., Pyshnaya b.I.A., Pyshnyi D.V.. Physicochemical properties of the phosphoryl guanidine oligodeoxyribonucleotide analogs. Russ. J. Bioorg. Chem. 2019; 45:709–718. [Google Scholar]
- 32. Zhukov S.A., Pyshnyi D.V., Kupryushkin M.S.. Synthesis of novel representatives of phosphoryl guanidine oligonucleotides. Russ. J. Bioorg. Chem. 2021; 47:380–389. [Google Scholar]
- 33. Du Z.W., Chen H., Liu H., Lu J., Qian K., Huang C.L., Zhong X., Fan F., Zhang S.C.. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015; 6:6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hartmann G., Battiany J., Poeck H., Wagner M., Kerkmann M., Lubenow N., Rothenfusser S., Endres S.. Rational design of new CpG oligonucleotides that combine b cell activation with high IFN-alpha induction in plasmacytoid dendritic cells. Eur. J. Immunol. 2003; 33:1633–1641. [DOI] [PubMed] [Google Scholar]
- 35. Goemans N., Mercuri E., Belousova E., Komaki H., Dubrovsky A., McDonald C.M., Kraus J.E., Lourbakos A., Lin Z., Campion G.et al.. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in duchenne muscular dystrophy. Neuromusc. Disord.: NMD. 2018; 28:4–15. [DOI] [PubMed] [Google Scholar]
- 36. Mendell J.R., Rodino-Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J.et al.. Eteplirsen for the treatment of duchenne muscular dystrophy. Ann. Neurol. 2013; 74:637–647. [DOI] [PubMed] [Google Scholar]
- 37. O’Rourke J.G., Bogdanik L., Muhammad A., Gendron T.F., Kim K.J., Austin A., Cady J., Liu E.Y., Zarrow J., Grant S.et al.. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015; 88:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hung G., Xiao X., Peralta R., Bhattacharjee G., Murray S., Norris D., Guo S., Monia B.P.. Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid Ther. 2013; 23:369–378. [DOI] [PubMed] [Google Scholar]
- 39. Jafar-Nejad P., Powers B., Soriano A., Zhao H., Norris D.A., Matson J., DeBrosse-Serra B., Watson J., Narayanan P., Chun S.J.et al.. The atlas of RNase h antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. 2021; 49:657–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hutchinson J.N., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A.. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007; 8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K.et al.. A Toll-like receptor recognizes bacterial DNA. Nature. 2000; 408:740–745. [DOI] [PubMed] [Google Scholar]
- 42. Levin A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019; 380:57–70. [DOI] [PubMed] [Google Scholar]
- 43. Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., Koretzky G.A., Klinman D.M.. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995; 374:546–549. [DOI] [PubMed] [Google Scholar]
- 44. Ash P.E., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W. 3rd, Rademakers R.et al.. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013; 77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gendron T.F., Bieniek K.F., Zhang Y.J., Jansen-West K., Ash P.E., Caulfield T., Daughrity L., Dunmore J.H., Castanedes-Casey M., Chew J.et al.. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013; 126:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C.et al.. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science (New York, N.Y.). 2013; 339:1335–1338. [DOI] [PubMed] [Google Scholar]
- 47. Zu T., Liu Y., Banez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T.M., Harms M.B., Falchook A.E., Subramony S.H.et al.. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Nat. Acad. Sci. U.S.A. 2013; 110:E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Meeter L.H.H., Gendron T.F., Sias A.C., Jiskoot L.C., Russo S.P., Donker Kaat L., Papma J.M., Panman J.L., van der Ende E.L., Dopper E.G.et al.. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann. Clin. Transl. Neurol. 2018; 5:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gendron T.F., Chew J., Stankowski J.N., Hayes L.R., Zhang Y.J., Prudencio M., Carlomagno Y., Daughrity L.M., Jansen-West K., Perkerson E.A.et al.. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017; 9:eaai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dieckmann A., Hagedorn P.H., Burki Y., Brügmann C., Berrera M., Ebeling M., Singer T., Schuler F.. A sensitive in vitro approach to assess the hybridization-dependent toxic potential of high affinity gapmer oligonucleotides. Mol. Ther. Nucleic Acids. 2018; 10:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Su Y., Bayarjargal M., Hale T.K., Filichev V.V.. DNA with zwitterionic and negatively charged phosphate modifications: formation of DNA triplexes, duplexes and cell uptake studies. Beilstein J. Org. Chem. 2021; 17:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cook D., Brown D., Alexander R., March R., Morgan P., Satterthwaite G., Pangalos M.N.. Lessons learned from the fate of astrazeneca's drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discov. 2014; 13:419–431. [DOI] [PubMed] [Google Scholar]
- 53. Benn C.L., Gibson K.R., Reynolds D.S.. Drugging DNA damage repair pathways for trinucleotide repeat expansion diseases. J. Huntington's Dis. 2021; 10:203–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bennett C.F., Krainer A.R., Cleveland D.W.. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu. Rev. Neurosci. 2019; 42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E.et al.. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017; 377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 56. Mercuri E., Darras B.T., Chiriboga C.A., Day J.W., Campbell C., Connolly A.M., Iannaccone S.T., Kirschner J., Kuntz N.L., Saito K.et al.. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018; 378:625–635. [DOI] [PubMed] [Google Scholar]
- 57. Vickers A., Donnelly J.P., Moore J.X., Barnum S.R., Schein T.N., Wang H.E.. Epidemiology of lumbar punctures in hospitalized patients in the united states. PLoS One. 2018; 13:e0208622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J.et al.. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011; 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Renton A.E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L.et al.. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011; 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Majounie E., Renton A.E., Mok K., Dopper E.G., Waite A., Rollinson S., Chio A., Restagno G., Nicolaou N., Simon-Sanchez J.et al.. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet. Neurol. 2012; 11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Caron N.S., Southwell A.L., Brouwers C.C., Cengio L.D., Xie Y., Black H.F., Anderson L.M., Ko S., Zhu X., van Deventer S.J.et al.. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of huntington disease. Nucleic Acids Res. 2020; 48:36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Starr A., Sattler R.. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 2018; 1693:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grad L.I., Rouleau G.A., Ravits J., Cashman N.R.. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017; 7:a024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for figures Figures 2B, D, 3A, B, 4A–C, 5B, C, 6B, C, 7A–C, 8A, B, and Supplementary Figures S1, S2A–C, S3A, B, S4A, B, S6A, S8 are provided with the paper.