


Abstract
Although recent regulatory approval of splice-switching oligonucleotides (SSOs) for the treatment of neuromuscular disease such as Duchenne muscular dystrophy has been an advance for the splice-switching field, current SSO chemistries have shown limited clinical benefit due to poor pharmacology. To overcome limitations of existing technologies, we engineered chimeric stereopure oligonucleotides with phosphorothioate (PS) and phosphoryl guanidine-containing (PN) backbones. We demonstrate that these chimeric stereopure oligonucleotides have markedly improved pharmacology and efficacy compared with PS-modified oligonucleotides, preventing premature death and improving median survival from 49 days to at least 280 days in a dystrophic mouse model with an aggressive phenotype. These data demonstrate that chemical optimization alone can profoundly impact oligonucleotide pharmacology and highlight the potential for continued innovation around the oligonucleotide backbone. More specifically, we conclude that chimeric stereopure oligonucleotides are a promising splice-switching modality with potential for the treatment of neuromuscular and other genetic diseases impacting difficult to reach tissues such as the skeletal muscle and heart.
INTRODUCTION
Exon-skipping oligonucleotides, a type of a splice-switching technology, are designed to bind through complementary base pairing to pre-mRNA, causing the splicing machinery to bypass exon(s) to restore the translational reading frame and protein production for out-of-frame mutations (1). The recent FDA approval of phosphorodiamidate morpholino oligomers (PMOs) for the treatment of Duchenne muscular dystrophy has been an important advance for the exon-skipping field. Despite this enthusiasm for PMOs, they have demonstrated only limited disease-modifying benefit. Because they restore low levels of dystrophin protein and have poor pharmacology (2) (https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation; https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation; https://www.nspharma.com/pdfs/NSPharma_Long-term_Data_PPMD_New.pdf), there continues to be substantial unmet clinical need (3,4). Duchenne muscular dystrophy is the most common genetic muscular dystrophy caused by mutations in the gene encoding dystrophin (DMD) (5). Duchenne remains a relentless disease (2), where lack of dystrophin protein results in severe, progressive muscle atrophy, eventual loss of ambulation, respiratory deficiency, cardiomyopathy and premature death (3,6). Many investigational exon-skipping oligonucleotides with a variety of chemistries have failed in the clinic, including the first-generation compound suvodirsen (2,7). Delivery to and efficacy in heart and diaphragm muscles, which have been notoriously difficult to reach in preclinical studies (8–12), will be necessary to provide survival benefit in patients (3,6), so there remains a high bar for achieving desired outcomes in this indication.
We set out to develop an exon-skipping technology that rises to this challenge. With this aim in mind, we build on previous findings that phosphorothioate (PS)-modified stereopure oligonucleotides (Figure 1A)—with precisely controlled chirality of inter-nucleotide PS backbone linkages—can improve the thermal and metabolic stability, lipophilicity and biodistribution of oligonucleotides (13–15) and apply them to exon-skipping oligonucleotides. Unlike stereorandom mixtures, stereopure oligonucleotides are dominated by a single stereoisomer with known pharmacological properties; thus, in addition to simplifying the composition of the oligonucleotide, control over chirality enables interrogation of the pharmacology associated with an individual isomer. In this study, we expand our investigations of chiral control over the backbone beyond PS modifications to investigate the impact of phosphoramidate-containing backbone linkages such as those generated with phosphoryl guanidine (PN) (16–22) (Figure 1A). PN linkages are neutral and more stable to nuclease degradation (23,24); hence, they decrease the overall charge of an oligonucleotide and may improve pharmacology. Here we report on the impact of PN backbone linkages on the application of stereopure splice-switching oligonucleotides in mouse models of Duchenne muscular dystrophy, including the severely affected double knockout mouse model.
Figure 1.

Controlling backbone chemistry and stereochemistry for synthesis of exon-skipping oligonucleotides. (A) Images of backbone chemistry and stereochemistry used in this work. (B) The L chiral auxiliary leads to Rp configuration of a PN linkage and Sp configuration of a PS linkage with identical geometry. (C) Crystal structures for the PN-fCfC dimers in the Rp (left) and Sp (right) configurations. (D) Ratio of Rp to Sp linkages from reverse-phase (RP)-UPLC analysis post-dimer synthesis illustrating diastereoselectivity.
MATERIALS AND METHODS
Materials
Anhydrous organic solvents were purchased and prepared by appropriate procedures prior to use. The other organic solvents were reagent grade and used as received. NMR spectra (1H NMR, 31P NMR and 19F NMR) were recorded with the appropriate reference on a Bruker Ascend Avance III HD 400 MHz or Avance NEO 600 MHz NMR spectrometer.
Chemical synthesis
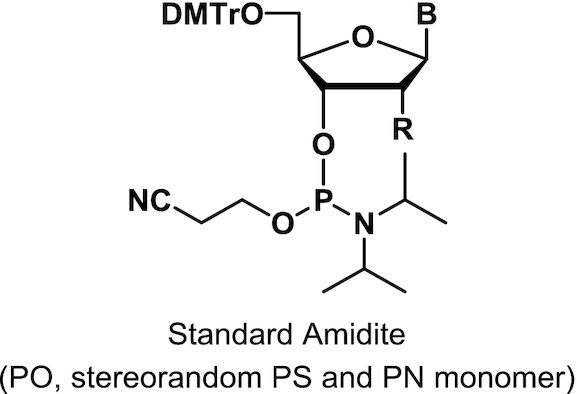
DMTr: 4,4′-dimethoxytrityl; B: base; CN: cyano
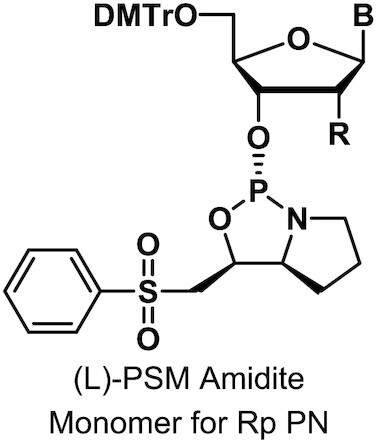
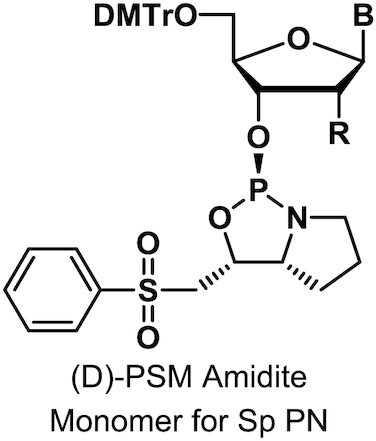
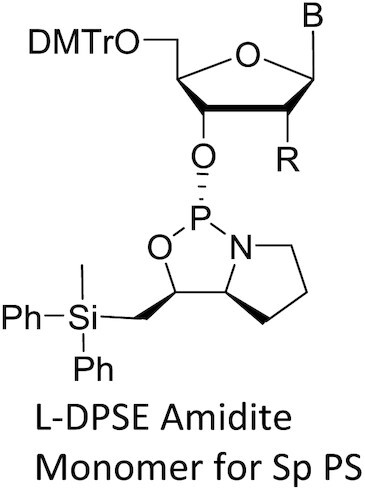
Monomers were synthesized and characterized as described (WO2019200185, Supplementary Materials and Methods).
Dimers
Automated solid-phase synthesis of dimers was performed with regular phosphoramidite cycle for stereorandom PN linkages (25) or with PSM amidite cycle for chiral PN linkages using mU-CPG (WO2019200185). After completion of the synthesis, the Controlled Pore Glass (CPG) solid support was dried and transferred into 15 ml plastic tube. We added concentrated NH4OH (4.8 ml; 200 μmol/μl) and heated for 16 h at 37°C. The reaction mixture was cooled to room temperature and the CPG was filtered and then washed with 10 ml of H2O. The crude material (filtrate) was analyzed by single quadrupole mass detector (SQD) and RP-UPLC (Supplementary Figure S8).
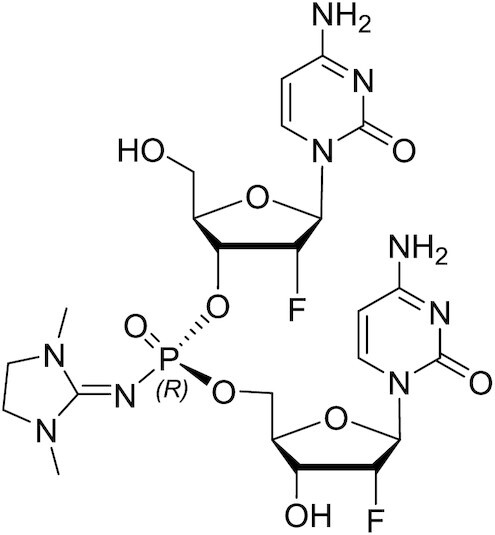
PN-Rp-fCfC dimer
1H NMR (600 MHz, CD3OD): δ 7.98 (d, J = 7.5 Hz, 1H), 7.80 (d, J = 7.5 Hz, 1H), 5.90 (dd, J = 17.8, 1.4 Hz, 2H), 5.85 (d, J = 7.5 Hz, 1H), 5.81 (d, J = 7.5 Hz, 1H), 5.23 (dd, J = 4.3, 1.6 Hz, 1H), 4.95 (dd, J = 4.2, 1.2 Hz, 1H), 4.90–4.82 (m, 4H), 4.36 (ddd, J = 11.8, 5.0, 2.1 Hz, 1H), 4.27 (dd, J = 8.7, 4.3 Hz, 1H), 4.26–4.16 (m, 4H), 4.11 (dq, J = 8.3, 2.6 Hz, 1H), 3.95 (dd, J = 12.7, 2.3 Hz, 1H), 3.76 (dd, J = 12.6, 2.9 Hz, 1H), 3.43 (s, 4H), 3.30 (s, 1H), 2.89 (s, 6H). 31P NMR (243 MHz, CD3OD): δ –2.16 19F NMR (565 MHz, CD3OD): δ –201.46, –202.71. LCMS: m/z calculated for C23H32F2N9O9P [M + H]+) 648.20, found: 648.81; m/z calculated for C23H32F2N9O9P [M-H]–) 646.20, found: 646.49.
Crystallography
The crystal was a colorless block with the following dimensions: 0.06 × 0.02 × 0.02 mm3. The symmetry of the crystal structure was assigned the monoclinic space group P21 with the following parameters: a = 11.1446(2) Å, b = 7.74300(10) Å, c = 17.4849(3) Å, α = 90°, β = 107.047(2)°, γ = 90°, V = 1442.53(4) Å3, Z = 2, Dc = 1.574 g/cm3, F(000) = 716, μ(Cu Kα) = 1.645 mm–1 and T = 100.00(11) K.
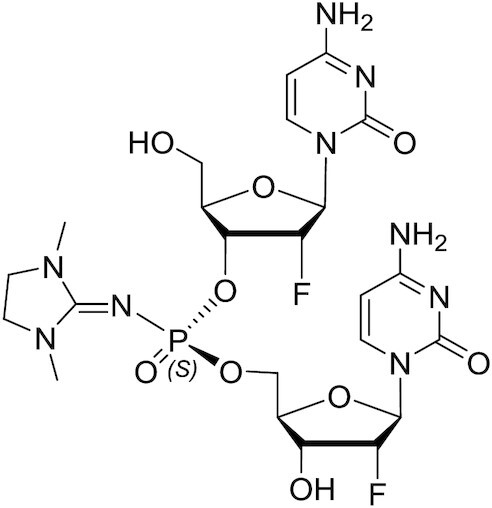
PN-Sp-fCfC dimer
1H NMR (600 MHz, CD3OD): δ 8.00 (d, J = 7.6 Hz, 1H), 7.81 (d, J = 7.6 Hz, 1H), 5.94 (d, J = 1.3 Hz, 1H), 5.91 (dd, J = 2.9, 1.4 Hz, 1H), 5.88 (d, J = 1.5 Hz, 1H), 5.83 (dd, J = 10.8, 7.5 Hz, 2H), 5.26 (dd, J = 4.3, 1.5 Hz, 1H), 4.96 (dd, J = 4.4, 1.3 Hz, 1H), 4.93–4.83 (m, 4H), 4.41 (ddd, J = 11.7, 5.0, 2.1 Hz, 1H), 4.25 (ddd, J = 22.4, 8.6, 4.3 Hz, 1H), 4.21–4.15 (m, 3H), 4.16–4.09 (m, 1H), 3.96 (dd, J = 12.7, 2.2 Hz, 1H), 3.75 (dd, J = 12.6, 2.9 Hz, 1H), 3.42 (s, 4H), 3.30 (s, 1H), 2.89 (s, 6H). 31P NMR (243 MHz, CD3OD): δ –2.25 ppm. 19F NMR (565 MHz, CD3OD): δ –200.81, –203.21. LCMS: m/z calculated for C23H32F2N9O9P [M + H]+) 648.20, found: 648.38; m/z calculated for C23H32F2N9O9P [M–H]–) 646.20, found: 646.85
Crystallography
The crystal was a colorless block with the following dimensions: 0.10 × 0.10 × 0.10 mm3. The symmetry of the crystal structure was assigned the triclinic space group P1 with the following parameters: a = 8.52280(10) Å, b = 10.06160(10) Å, c = 10.62870(10) Å, α = 95.5040(10)°, β = 102.5670(10)°, γ = 113.5290(10)°, V = 798.449(16) Å3, Z = 1, Dc = 1.347 Mg/m3, F(000) = 338, μ(Cu Kα) = 1.410 mm–1 and T = 99.99(10) K. (Supplementary Tables S2, S3, Supplementary Datasets 1, 2).
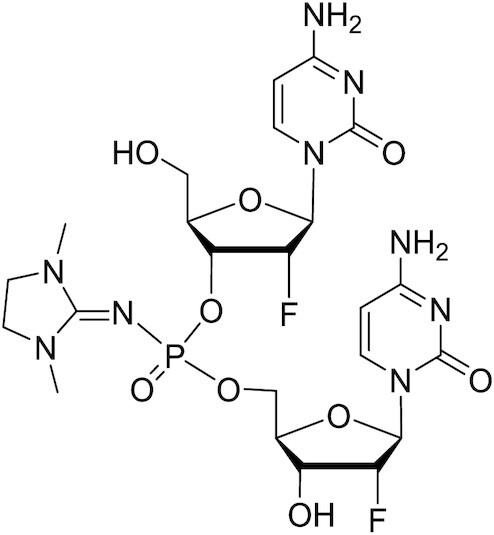
PN-stereorandom-fCfC dimer
1H NMR (600 MHz, CD3OD) δ 7.99 (dd, J = 11.3, 7.5 Hz, 1H), 7.84 – 7.78 (m, 1H), 5.95 – 5.87 (m, 2H), 5.87–5.78 (m, 2H), 5.24 (ddd, J = 15.5, 4.3, 1.6 Hz, 1H), 5.16 (ddd, J = 15.6, 4.3, 1.6 Hz, 1H), 4.96 (ddd, J = 5.9, 4.3, 1.2 Hz, 1H), 4.93–4.82 (m, 4H), 4.38 (dddd, J = 26.9, 11.7, 5.0, 2.1 Hz, 1H), 4.25 (dddd, J = 22.4, 8.6, 4.4, 1.7 Hz, 1H), 4.22 – 4.15 (m, 3H), 4.12 (tq, J = 8.4, 2.6 Hz, 1H), 3.95 (dt, J = 12.7, 2.6 Hz, 1H), 3.76 (ddd, J = 12.6, 5.2, 2.9 Hz, 1H), 3.43 (d, J = 5.3 Hz, 4H), 3.30 (s, 1H), 2.89 (d, J = 2.9 Hz, 6H). 31P NMR (243 MHz, CD3OD): δ –2.16, –2.25. 19F NMR (565 MHz, CD3OD): δ –200.81, –201.45, –202.70, –203.22. LCMS: m/z calculated for C23H32F2N9O9P [M + H]+) 648.20, found: 648.74; m/z calculated for C23H32F2N9O9P [M-H]–) 646.20, found: 646.71
Oligonucleotides
We obtained phosphodiester-based and stereorandom PS-modified oligonucleotides from Integrated DNA Technologies or by using a standard solid-phase oligonucleotide synthesis protocol. We synthesized and purified chemically modified, stereopure oligonucleotides as described (13) (WO2018237194, WO2014012081) (Supplementary Materials in Methods). We characterized stereopure oligonucleotides by LC-HRMS and HPLC. Synthesis of oligonucleotides containing PN linkages was automated in solid phase following standard amidite cycle for stereorandom PN (22,25) and adapted cycle for stereopure PN (WO2019200185). The sequences of oligonucleotides used to identify the new targeting site in exon 23 are shown in Supplementary Table S1.The reference sequence for mouse exon 23 (5′-GGCCAAACCUCGGCUUACCU-3′) is based on a previously published report (26). The sequences for oligonucleotides targeting human exon 51 (5'-UCAAGGAAGAUGGCAUUUCU-3'), mouse exon 23 (5′-UCACUCAGAUAGUUGAAGCC-3′) and human exon 44 (5′-CUUAAGAUACCAUUUGUAUU-3′) are indicated. DMD-17 is complementary to mouse exon 23 but served as a non-targeting control in human exon 51 experiments (5′-GGCCAAACCUCGGCUUACCU-3′).
Crystallography
For PN-Rp-fCfC dimer, the compound was dissolved in 1 mL acetonitrile:methanol (4:1) and kept in a half sealed 4-ml vial. The solution was allowed to evaporate slowly at 4°C. The crystal was obtained in the sixth day. Diffraction data was collected on Rigaku Oxford Diffraction XtaLAB Synergy four-circle diffractometer equipped with a HyPix-6000HE area detector (λ = 1.54184 Å, 50W, Micro focus source with multilayer mirror (μ-CMF)) using Oxford Cryostream 800 cryogenic system. Data were collected with the crystal 35 mm from CCD detector with tube voltage = 50 kV and tube current = 1 mA.
We collected a total of 24 453 reflections in the 2Θ range from 5.286 to 133.198. The limiting indices were: –10 ≤ h ≤ 13, –9 ≤ k ≤ 9, –20 ≤ l ≤ 20; which yielded 5049 unique reflections (Rint = 0.0791). The structure was solved using SHELXT(27) and refined using SHELXL (against F2)(28). The total number of refined parameters was 445, compared with 5049 data. All reflections were included in the refinement. The goodness of fit on F2 was 1.055 with a final R value for [I > 2σ (I)] R1 = 0.0503 and wR2 = 0.0917. The largest differential peak and hole were 0.36 and –0.45 eÅ–3, respectively.
For PN-Sp-fCfC dimer, the compound was dissolved in 1 ml acetonitrile:methanol (1:2) and kept in a half sealed 4 ml vial. The solution was allowed to evaporate slowly at 60°C. A crystal was observed in the second day. Its transparency was checked by microscope, and then the crystal was sent for analysis. Diffraction data were collected as above.
We collected a total of 28,207 reflections in the 2Θ range from 8.708 to 133.172. The limiting indices were: –10 ≤ h ≤ 10, –11 ≤ k ≤ 11, –12 ≤ l ≤ 12; which yielded 5160 unique reflections (Rint = 0.0535). The structure was solved using SHELXT(27) and refined using SHELXL (against F2)(28). The total number of refined parameters was 401, compared with 5160 data. All reflections were included in the refinement. The goodness of fit on F2 was 1.055 with a final R value for [I > 2σ (I)] R1 = 0.0468 and wR2 = 0.1221. The largest differential peak and hole were 1.21 and –0.31 Å–3, respectively.
RP-UPLC
Dimer analysis was performed on Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm) at 55°C (A: 0.1% formic acid in water; B: MeCN) with ultraviolet detection at 260 nm with the following conditions.
| Time | Flow rate (ml/min) | %A | %B | Curve |
| Initial | 0.80 | 95.0% | 5.0% | |
| 0.30 | 0.80 | 95.0 | 5.0 | 6 |
| 3.30 | 0.80 | 85.0 | 15.0 | 6 |
| 3.80 | 0.80 | 5.0 | 95.0 | 1 |
| 4.50 | 0.80 | 95.0 | 5.0 | 1 |
| 5.00 | 0.80 | 95.0 | 5.0 | 1 |
Oligonucleotide analysis was performed on Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm) at 55°C (A: 100 mM HFIP, 10 mM TEA in water; B: MeCN) with ultraviolet detection at 260 nm with the following conditions.
| Time | Flow rate (ml/min) | %A | %B | Curve |
| initial | 0.80 | 99.0 | 1.0 | |
| 1.00 | 0.80 | 99.0 | 1.0 | 6 |
| 10.00 | 0.80 | 85.0 | 15.0 | 6 |
| 12.00 | 0.80 | 50.0 | 50.0 | 6 |
| 13.00 | 0.80 | 50.0 | 50.0 | 1 |
| 14.00 | 0.80 | 99.00 | 1.00 | 1 |
Cell culture conditions
We obtained H2K mdx23 cells (29) from Professor Jenny Morgan. H2K cells were counted using cell Countess and plated at 40 000 cells/well in 24-well plates coated with Matrigel (Scientific Laboratory Supplies, Cat. No. 354234). For gymnotic delivery, growth media [DMEM, 20% FBS, 2% Chick embryo extract (Sera Laboratories, Cat. No. CE-650-J), 1% penicillin/streptomycin, mouse IFN-γ (1:10 000; Millipore, Cat. No. IF005)] was replaced the next day with 500 μl differentiation media [DMEM, 1% penicillin/streptomycin, 5% horse serum inactivated (Thermo Fisher, Cat. No. 26050–088)] containing 1–10 μM of oligonucleotide. For transfection, we used RNAiMax (Thermo Fisher, 0.3 μl RNAiMax per well with 1, 3 or 10 nM oligonucleotide) according to manufacturer's protocol. After 96 h, cells were harvested in 500 μl Trizol. We obtained human DMD Δ48–50 myoblasts (ID6594-I) from Dr Vincent Mouly (Center for Myology, GH Pitié-Salpétrière, Paris, France). DMD Δ48–50 myoblasts were plated at 15 000 cells/well in 96-well plates coated with Matrigel (Scientific Laboratory Supplies, Cat. No. 354234). The next day cells were pre-differentiated for 4 days by replacing growth media [complete Skeletal Muscle Growth Medium (Promocell) supplemented with 5% FBS, 1× penicillin–streptomycin and 1× l-glutamine] with 200 μl of differentiation media [DMEM, 1% penicillin/streptomycin, 5% horse serum inactivated (Thermo Fisher, Cat. No. 26050–088), 10 μg/ml Insulin (ThermoFisher Scientific, Cat No. 12585014)]. For gymnotic delivery, media were removed and replaced with 200 μl of differentiation media containing desired concentration of oligonucleotide. After 5 days of treatment, cells were harvested in 100 μl of Trizol for RNA extraction.
RNA extraction and quantification of exon skipping
RNA was extracted from human or mouse myoblasts using Promega SV96 Total RNA isolation kits (Promega, Cat. No. Z3500). Briefly, lysate (in Trizol) was transferred to a 96-well plate, extracted with chloroform for 10 min prior to phase-separation by centrifugation (4000 rpm for 10 min, 4°C). The aqueous phase was combined with 1 volume of 70% ethanol, mixed and transferred to the column. After vacuum application, the column was washed with RNA wash buffer, treated with DNase, washed with 200 μl DNase stop and then RNA wash buffer. After centrifugation to remove residual buffer, pre-heated (60°C) RNA elution buffer was added to the column for a 5–10 min incubation at 60°C prior to elution by centrifugation.
For H2K cells, RNA was converted to cDNA, followed by pre-amplification using 3 μl cDNA and Taqman PreAmp Master Mix (Thermo Fisher Cat. No. 4384266). Pre-amp was diluted 1:5 with water, and 3 μl of the dilution was used for qPCR TaqMan analysis using total and skipped TaqMan probes (see below) and BioRad iQ Multiplex Powermix (BioRad, Cat. No. 1725849) using the following thermocycler parameters: 95°C 3 min; 95°C 10 s, 60°C 30 s (40 cycles). Absolute quantification of exon skipping was determined using a standard curve (calculated with Bio-Rad CFX Maestro software) generated from gBlocks, which are diluted to 125,000 copies/μl and sequentially diluted 1:1 for 12 point-curve. Gblocks and cDNA are used at 3 μl per well (up to 375 000 copies). Experiments were performed as biological triplicates (separate wells); qPCR reactions were performed as technical duplicates for each sample. Total probes: mDMD20-21 FAM/ZEN/IABkFQ (custom, IDT) (Primer 1 5′-AGATGACAACTACTGCCGAA; Primer 2 5′-GAAGAGCTGACAATCTGTTGAC; Probe 5′-AGTCTACCACCCTATCAGAGCCAACA). Skipped probes: mDMD22-24 HEX/ZEN/IABkFQ (custom, IDT) (Primer 1 5′-CTGAATATGAAATAATGGAGGAGAGACTCG; Primer 2 5′-CTTCAGCCATCCATTTCTGTAAGGT; Probe 5′-ATGTGATTCTGTAATTTCC).
For human myoblasts, RNA was converted to cDNA, cDNA was diluted 1:1 with water, and 4 μl of the dilution was used for qPCR TaqMan analysis using unskipped, skipped and SFSR9 (internal control) TaqMan probes (see below) and BioRad iQ Multiplex Powermix (BioRad, Cat. No. 1725849) using the amplification parameters delineated above. Relative quantification was determined using Bio-Rad CFX Maestro software. Unskipped and skipped DMD transcripts were normalized to SFSR9 internal control. Percentage skipping was determined by calculating skipped/(skipped + unskipped)x100. Experiments were performed as biological duplicates (separate wells); qPCR reactions were performed as technical duplicates for each sample. Skipped probes: DMD 43–45-FAM/ZEN/IABkFQ (custom, IDT) (Primer 1 5′-GCAGATTCAGGCTTCCCAAT; Primer 2 5′-CAAGGACCGACAAGGGAAC; Probe 5′-TTCTGACAACAGTTTGCCGCTGC). Unskipped probes: DMD 44–45 FAM/ZEN/IABkFQ (custom, IDT) (Primer 1 5′-CAGTGGCTAACAGAAGCTGAA; Primer 2 5′-TCCTGGAGTTCCTTAAGATACCA; Probe 5′ ACA CAAATTCCTGAGAATTGGGAACATGC). SFRS9 probes: HEX/ZEN/IABkFQ (custom, IDT) (Primer 1 5′-TGGTGCTTCTCTCAGGATAAAC; Primer 2 5′-TGGAATATGCCCTGCGTAAA; Probe 5′-TGGATGACACCAAATTCCGCTCTCA).
Capillary western immunoassay
The protocol was adapted from previous reports (30). Protein lysates from muscle tissue were prepared by cutting off small pieces of frozen tissue (20–30 mg) and placing in a Precellys tissue lysing vial (Precellys, Cat. No. P000910-LYSK0-A), adding 20 volumes of tissue homogenization buffer [125 mM Tris pH 8.0, 12% SDS, 4% glycerol, 50 mM DTT, 5 mM EDTA, 1× protease inhibitor (Roche, Cat No. 05892970001)]. The samples were then homogenized by 2–4 cycles (3 × 20 s; 6800 rpm) in the Precellys Evolution Tissue Homogenizer (Bertin Technologies) and spun down for 10 min at 14 000 rpm. The supernatants were carefully transferred into new tubes. To measure total protein concentration, 2 μl of a 20× dilution of the lysates was quantified using the Qubit protein assay kit (Thermo Fisher Scientific, Cat. No. Q33211) according to the manufacturer's protocol.
Dystrophin quantitation was performed on a capillary immunoassay (Wes) system (ProteinSimple, Cat. No. 004-600), according to the manufacturer's instructions using a 66–440 kDa Separation Module (ProteinSimple, Cat. No. SM-W008), the Anti-Rabbit Detection Module (ProteinSimple, Cat No. DM-001) and the Anti-Mouse Detection Module (ProteinSimple, Cat. No. DM-002), depending on the primary antibody used. Samples were diluted to 0.5 μg/μl in 0.1× Sample Buffer (10× Sample Buffer from the Separation Module), mixed with Fluorescent Master Mix (from the Separation Module) and denatured at 95°C for 5 min. A 7-point calibration curve was generated using pooled lysate from C57BL10 (healthy tissue)/mdx23 (DMD tissue) and was included with each assay. The samples, blocking reagent (antibody diluent), primary antibodies [1:50 Anti-Dystrophin antibody (AbCam, Cat. No. ab15277), 1:1000 Anti-Vinculin antibody (ThermoFisher Scientific, Cat. No. MA5-11690) in antibody diluent], HRP-conjugated secondary antibodies (ready to use anti-mouse diluted 1:10 in ready to use anti-rabbit) plus chemiluminescent substrate were pipetted into the plate (part of Separation Module). We used the instrument's default settings with stacking and separation performed at 475 V for 30 min; blocking for 5 min, primary and secondary antibody for 30 min each; luminol/peroxide chemiluminescence detection for 15 min with exposures of 1, 2, 4, 8, 16, 32, 64, 128 and 512 s. We quantified chemiluminescence as area under the curve (AUC) of detected peaks using Compass software, and it is displayed herein as a virtual blot-like image. Standard curves were generated by plotting percentage standard versus the ratio of AUCdystrophin/AUCvinculin. We calculated concentrations of dystrophin in oligonucleotide-treated samples by interpolation from lines generated by these standards.
Subcellular fractionation
Cells were grown to 70–80% confluence in growth media before being treated with oligonucleotide in differentiation media for 24 h. Cells were washed 3× in PBS before being treated with trypsin until cells dissociate from the flask. Trypsin was then quenched with differentiation media, the cells were separated: 15% for whole cell lysate, 15% for RIPA lysate and 70% for cell fractionation. Cells were then pelleted and lysed in a suitable buffer. For whole cell lysates, cells were transferred to tubes and pelleted. Weight of cellular pellet was recorded. Pellet was resuspended in proteinase K lysis buffer (10 mM Tris–HCl, 100 mM NaCl, 5 mM EDTA, 0.5% NP40 and 2 mg/ml proteinase K), incubated at 55°C for 1 h, and stored at –80°C until used. Nuclear and cytoplasmic extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents following manufacturer's instructions (Thermo Fisher Scientific, Cat. No. 78835). Fractionated lysates were normalized to 500 μg/ml and used directly in the hybridization ELISA, in place of Proteinase K prepared lysates or further diluted for RTqPCR (see below).
Tissue processing for transcript analyses by PCR and oligonucleotide quantification
We dissected and fresh-froze tissues in pre-weighed Eppendorf tubes. We calculated tissue weight by re-weighing. For lysis, we added Trizol or lysis buffer (10 mM Tris, 100 mM NaCl, 5 mM EDTA, 0.5% NP-40 supplemented with 2 mg/ml proteinase K) and homogenized tissue at 4°C using Precellys until all the tissue pieces were dissolved. 30–50 μl of tissue lysates were saved in 96-well plates for oligonucleotide quantification by hybridization ELISA. The remaining lysates were either stored at -80°C (when in lysis buffer) or used for RNA extraction (when in Trizol).
Thermal denaturation (Tm)
Equimolar amounts of surrogate RNA (5′- GGCUUCAACUAUCUGAGUGA) and oligonucleotide were dissolved in 1× PBS to obtain a final concentration of 1 μM of each strand (3 ml). Duplex samples were then annealed by heating at 90°C, followed by slow cooling to 4°C and storage at 4°C. UV absorbance at 260 nm was recorded at intervals of 30 s as the temperature was raised from 15°C to 95°C at a rate of +0.5°C per min, using a Cary Series UV–Vis spectrophotometer (Agilent Technologies). Absorbance was plotted against the temperature and the Tm values were calculated by taking the first derivative of each curve.
Hybridization ELISA
Flash frozen tissues were weighed. Lysis buffer (0.5% IGEPAL, 100 mM NaCl, 5 mM EDTA, 10 mM Tris-HCl, pH7.5, 2 mg/ml proteinase K) was added to frozen tissues (4 volumes: 1 weight) directly (4 μl of lysis buffer to 1 mg of tissue). Tissues were lysed in Precellys Evolution homogenizer with 1 mm Zirconium Oxide beads, at 6000 rpm for 5 min. The lysates were incubated at 55°C for 1 h in thermomixer at 250 rpm, followed by Precellys homogenizing at 6000 rpm for 5 min. The lysates were used directly in Hybridization ELISA assay or frozen at –80°C for storage. No extraction step was needed for hybridization ELISA. We utilized the following probes to selectively quantify the oligonucleotides by hybridization ELISA: Capture probe: /5AmMC12/GGCTTCAACT; Detection probe: A + T + CT + GAGTGA/3BioTEG/ (Integrated DNA Technologies, Coralville, IA). We coated maleic anhydride-activated 96-well plates (Pierce 15110) with 50 μl of capture probe at 500 nM in 2.5% NaHCO3 (Gibco, 25080-094) for 2 h at 37°C. The plate was then washed 3 times with PBST (PBS + 0.1% Tween-20), blocked with 5% fat free milk-PBST at 37°C for 1 h. Payload oligonucleotide (800 μg/ml) was serially diluted (2-fold) into matrix. A separate standard is generated for every oligonucleotide for quantification. 20 μl of diluted samples were mixed with 180 μl of 333 nM detection probe diluted in PBST, then denatured (65°C, 10 min, 95°C, 15 min, 4°C ∞). 50 μl of the denatured samples were distributed in blocked ELISA plates in duplicates, and incubated overnight at 4°C. After three washes with PBST, 50 μl of 1:2000 streptavidin-AP (Southern Biotech, 7100-04) in PBST was added, 50 μl per well and incubated at room temperature for 1 h. After extensive washes with PBST, 100 μl of AttoPhos (Promega S1000) was added, incubated at room temperature in the dark for 10 min and read on the plate reader (Molecular Device, M5) fluorescence channel: Ex435 nm, Em555 nm. The oligonucleotide in samples were calculated according to standard curve by 4-parameter regression. The lower limit of detection was 1.25 μg oligonucleotide per gram of tissue.
RT-qPCR oligonucleotide quantification in cellular extracts
Lysates from the subcellular fractionation were diluted 1:10 000 in ddH2O and used directly for reverse transcription. Reverse transcription was performed using TaqMan™ MicroRNA Reverse Transcription Kit (Applied Biosystems, 4366596) according to manufacturer's instructions, supplementing purified total RNA with the diluted lysate. We used Thermo Fisher Custom TaqMan Small RNA Assay Design Tool to create stem loop primers and probes for oligonucleotides. For absolute quantification, a 16-point, 2-fold dilution series was prepared using the oligonucleotide of interest, beginning with 5 × 106 copies. RT-qPCR was performed using Taqman Fast Advanced Master Mix (Applied Biosystems, 4444557) according to manufacturer's instruction.
Oligonucleotide uptake time course
H2K cells were plated at a density of 75 000 in Matrigel coated wells of a 12-well dish and differentiated for 96 h. Cells were then dosed with 1 μM oligonucleotide in differentiation media (See Cell Culture section). After 3 h, the cells were washed three times in PBS. The 0-h time point was collected while all others were refreshed with differentiation media until the specified time point. At time of collection, cells were trypsinized, quenched with differentiation media and pelleted before being washed a final time in PBS. Cell pellets were resuspended in lysis buffer (10 mM Tris, 10 mM NaCl, 5 mM EDTA, 0.5% NP-40) supplemented with 2 mg/ml Proteinase K and incubated at 55°C for 1 h before being used directly in the hybridization ELISA assay.
Animals
We obtained mdx23 mice (Stock No.: 001801 C57BL/10ScSn-Dmdmdx/J) from Jackson Laboratory (Bar Harbor, ME). All mdx23 work was performed under an approved IACUC protocol of Biomedical Research Models, Inc. (Worcester, MA). We obtained dKO (mdx/utr–/+) mice breeders from Jackson Laboratory (Stock no: 014563) to obtain homozygous mdx/utr–/– ‘double knockout’ (dKO) mice. All dKO work was carried out at the Biomedical Sciences Unit, University of Oxford, in accordance with procedures approved by the UK Home Office (PPL PDFEDC6F0).
ViewRNA in situ hybridization (ISH) Assay
We adapted ViewRNA ISH Tissue 1-Plex Assay (Thermo Fisher Scientific, Cat. No. QVT0051) for detection of ASOs in situ. Briefly, muscle biopsies were fixed in 10% neutral buffered formalin overnight at 2–8°C, processed and embedded in paraffin. Paraffin sections (5 μm) were prepared and stored at room temperature until use. After baking slides at 60°C for at least 1 h, we dewaxed in xylene (VWR Chemicals) for 10 min and rinsed in 100% ethanol (Thermo Fisher Scientific). After air drying slides for at least 30 min at room temperature, we created a hydrophobic barrier before continuing with ViewRNA ISH standard protocol. We treated rehydrated slides with pre-heated target retrieval reagent for 5 min at 95°C, followed by protease digestion (Protease QF 1:100 in 1× PBS, pre-warmed) at 37°C for 10 min. We rinsed slides in 1X PBS with agitation and then treated them with DVM1-10702 probe sets for exon 23 skipping oligos, PPIB (positive control), and/or dapB (negative control) (Thermo Fisher Scientific) diluted to 12.5 nM in pre-warmed Probe Set Diluent QT (300 μl per section) for 2 h at 40°C. We stored rinsed slides at room temperature for up to 24 h. For signal amplification and detection, we incubated slides in working PreAmp1 QF solution diluted at 1:200 in prewarmed Amplifier Diluent QF for 30 min at 40°C; we rinsed in wash buffer with agitation, which was followed by incubation in working Amp1 QF solution (1:200) in prewarmed Amplifier Diluent QF for 20 min at 40°C. After rinsing, we incubated the slides in Label Probe-AP working solution (1:1,000 in Label Probe Diluent QF) for 20 min at 40°C and rinsed in wash buffer with agitation. We added AP-Enhancer Solution and incubated for 5 min at room temperature before adding Fast Red Substrate and incubating a further 30 min at 40°C to develop red color deposit. Afterwards, we counterstained DNA with Hematoxylin and/or Hoechst 33342 dye. The slides were mounted with ProLong Gold Antifade mounting medium (Molecular Probes, Cat. No. P36930) and covered with a thin glass coverslip. For each section, the representative digital images were generated using a Zeiss Axio Observer microscope (Zeiss, Thornwood, NY, USA) under brightfield or fluorescent field.
Immunofluorescence
Mouse muscle biopsies were snap frozen in 2-methylbutane cooled in liquid nitrogen as described previously (31). Using a cryotome (CM3050 S, Leica Biosystems, Buffalo Grove, IL, USA), 10 μm muscle cross-sections were cut and placed on positively charged Superfrost Ultra Plus Microscope Slides (Thermo Fisher Scientific, Cat No. 22-037-246). After drying at room temperature for 2 h, the slides were placed in a -80°C freezer and used within 2 months. The staining procedure was performed at room temperature, unless otherwise stated. Frozen slides were removed from the freezer, directly fixed with ice-cold 1:1 acetone/methanol for 5 min, washed with PBS, followed by permeabilization for 15 min with 0.5% Triton X-100 in PBS, and subsequently washed with PBS. No antigen retrieval step was performed prior to staining. Non-specific protein-protein interactions were blocked with 5% normal goat serum in PBS for 1 h at room temperature. All endogenous biotin, biotin receptors, and streptavidin binding sites present in tissues were further blocked with Streptavidin/Biotin blocking kits (Vector laboratories, Cat. No. SP-2002) for 15 min. Sections were incubated in a humidified chamber with a polyclonal rabbit anti-dystrophin antibody (Abcam, Cat. No. ab15277, 1:400 diluent) and a monoclonal rat anti-laminin antibody (Abcam, Cat No. ab11576, 1:400 diluent) prepared in antibody dilution buffer (Invitrogen, Cat. No. 003218) overnight at +4°C. The sections were then rinsed and treated with secondary Biotin-SP (long spacer) AffiniPure Goat anti-Rabbit IgG (H + L) at 1:200 diluent (Jackson ImmunoResearch, Cat. No. 111-065-144) and Goat anti-rat IgG (H + L) at 1:400 diluent (ThermoFisher, Cat. No. SA5-10021, DyLight 650) for 1 h. After washing again with PBS, the slides were fluorescently developed with Alexa Fluor™ 555 conjugated Streptavidin (Life Technologies, Cat. No. S32355) at 1:500 diluent for 30 min to signal dystrophin. DNA was counterstained with Hoechst 33342 dye. The slides were mounted with ProLong Gold Antifade mounting medium (Molecular Probes, Cat. No. P36930), and covered with a thin glass coverslip. Negative controls omitted the primary antibodies. For each muscle cross-section, representative images at 10× magnification were acquired using a Fluorescent Microscope (Zeiss Axio Observer, Thornwood, NY, USA).
Muscle function analysis
Specific force generation and force drop following repeat eccentric contraction, were measured from dissected extensor digitorum longus muscle of control and treated mice as previously described (32).
Serum biomarker analysis
Methodology for miRNA analysis was detailed previously (33). Briefly, blood was collected from jugular vein upon sacrifice, and spun to isolate serum. 50 μl of serum was spiked with cel-miR39 prior to RNA extraction. miRNA levels were quantified relative to cel-miR39 levels using Small RNA Taqman Assay (Applied Biosystems, Assay IDs: miR-1 002222, miR-133a 002246, miR-206 000510, cel-miR-39 000200). Serum creatine kinase levels were quantified on a Beckman Coulter AU680 clinical chemistry analyzer at Mary Lyon Centre, Medical Research Centre, Harwell, UK.
Whole-body plethysmography
Respiratory function was evaluated using non-invasive whole-body plethysmography to measure respiratory frequency (number of breaths per minute), tidal volume (the volume at start of expiration minus the volume at the start of inspiration), minute volume (the rate of ventilation as the product of the tidal volume and frequency), estimated peak inspiratory flow, and estimated peak expiratory flow. Values for these derived signals are calculated on a breath-to-breath basis for each respiratory cycle. Data for each individual mouse were collected at stated time points over a 30 min period, following 5 min acclimatization, using a Buxco Small Animal Whole Body Plethysmography unit (Data Sciences International, St. Paul, MN, USA).
Statistics
One-way and two-way ANOVAs as well as unpaired t tests were performed using GraphPad Prism software version 8.4.3. Kaplan–Meier survival analyses were conducted using R version 3.6.3.
RESULTS
Chiral control of PN backbone linkages
To assess the impact of introducing PN linkages to stereopure PS-modified oligonucleotides, we developed a method and the synthetic capabilities necessary to engineer oligonucleotides containing stereopure PN linkages (Figure 1A) (WO2019200185). The synthesis of various stereorandom phosphoramidate (mono substituted)-modified oligonucleotides using phosphoramidite and H-phosphonate chemistries have been reported (23,24,34–40). We were particularly interested in exploring phosphoryl guanidine-based backbones (PN) (16–21,23) in combination with stereopure phosphorothioate backbone (PS/PN) to study the pharmacological properties of these chimeric backbone oligonucleotides. Towards this end, we synthesized stereopure PN linkages and their derivatives and successfully synthesized chimeric PS/PN or PS/PN/PO stereopure backbone-containing oligonucleotides.
To validate the fidelity of synthesis, we first assessed diastereoselectivity from synthesis of C-C PN dimers with 2′-deoxyfluoro (2′-F) ribose modifications (PN-fCfC). For these syntheses, we used standard phosphoramidite, which yielded the stereorandom dimer, or L-PSM ((S)-2-(phenylsulfonyl)-1-((S)-pyrrolidin-2-yl)ethan-1-ol) and D-PSM ((R)-2-(phenylsulfonyl)-1-((R)-pyrrolidin-2-yl)ethan-1-ol) amidites, which yielded Rp and Sp PN dimers, respectively. We designed the PSM chiral auxiliary, which gives high diastereoselectivity (Figure 1), to undergo beta elimination during deprotection similar to the deprotection of the cyanoethyl protecting group in standard oligonucleotide synthesis. Thus, the Rp configuration of a PN linkage is comparable in chirality to the Sp configuration of a PS linkage (Figure 1B). The dimers were characterized by RP-UPLC and 31P NMR (Supplementary Materials and Methods). The predicted backbone stereochemistry based on the chiral auxiliary was confirmed by crystallography (Figure 1C). We evaluated diastereoselectivity from synthesis of additional PN dimers, containing 2′-O-methyl ribose (2′-OMe) U in the first position with various 2′-F-modified nucleotides in the second position (Figure 1D). Following routine purification, this method allows for the synthesis of stereopure oligonucleotides containing chimeric PS/PO/PN backbones in high yields with both high chemical and stereochemical purity (WO2019200185).
Optimizing chemistry and stereochemistry of PS-modified exon-skipping oligonucleotides in vitro
Our design for stereopure PS-modified oligonucleotides was based on work performed to develop oligonucleotides for skipping of human DMD exon 51. For these efforts, we began with a known exon 51 skipping sequence (5′-UCAAGGAAGAUGGCAUUUCU-3′) (41) on a fully PS-modified stereorandom backbone with 2′-OMe ribose modifications (DMD-16). In human Δ48–50 myoblasts treated gymnotically with 10 μM DMD-16, we observed ∼2% mean exon 51 skipping. For all in vitro experiments in this study, we have relied on gymnotic delivery, which have been shown to better predict in vivo activity than transfection conditions (42,43). We evaluated the impact of replacing the 2′-OMe modifications with 2′-F on in vitro skipping activity. As 2′-F modifications are known to increase duplex stability (23), we added these modifications to the 5′- and 3′-ends of DMD-16 (Figure 2A). The introduction of up to four 2′-F modifications on the ends had a modest impact on exon skipping activity. With the introduction of five or six 2′-F modifications on the ends, exon-skipping activity was notably increased (Figure 2A, mean skipping: DMD-103 4.7%; DMD-100, 12.8%), so we included at least six 2′-F modifications per end going forward.
Figure 2.

Characterization and optimization of exon skipping oligonucleotide chemistry for human DMD exon 51. Quantification of exon 51 skipping in human Δ48–50 myoblasts treated gymnotically with the indicated oligonucleotide (10 μM A-C; dose response 0.1, 0.3, 1, 3 or 10 μM D,E). (A) Comparison of activities for oligonucleotides with identical sequence and backbone stereochemistry (stereorandom PS at all positions) with variable numbers of 2′-F modifications in termini. DMD-17 is complementary to mouse Dmd exon 23 and serves as non-targeting control in this experiment. (B) Evaluation of the effect of PO linkages on activity of oligonucleotides with identical sequence and 2′ ribose modification pattern **** P < 0.0001, * P < 0.05 One-way ANOVA with post-hoc comparison to DMD-16 (panel A) or to DMD-100 (panel B), Mean ± s.e.m., n = 2. (C) Evaluation of relationship between PO linkages and 2′-ribose modifications in the center of the oligonucleotide and its activity. **** P < 0.0001, ns, non-significant, One-way ANOVA with post-hoc comparison to DMD-16, Mean ± s.e.m., n = 2. (D) Application of Sp PS backbone stereochemistry and exploration of various 2′-ribose modification patterns in the center of the oligonucleotide. **** P < 0.0001, *** P < 0.001, **P < 0.01,* P < 0.05 Two-way ANOVA with post-hoc comparison to DMD-16, Mean ± s.e.m., n = 2. (E) Impact of PO linkages at various backbone positions on activity. **** P < 0.0001, *** P < 0.001, **P < 0.01,* P < 0.05 Two-way ANOVA with post-hoc comparison to DMD-16, Mean ± s.e.m., n = 2.
Next, we evaluated the impact of adding PO backbone modifications to the oligonucleotide. Beginning with DMD-100, a fully stereorandom PS-modified oligonucleotide with 2′-F and 2′-OMe ribose modifications, we generated a new series of oligonucleotides that included PO linkages initially at the junction between the distinct 2′-modified nucleosides and adding one PO linkage per end moving through the 2′-F-modified region of the oligonucleotide (Figure 2B). The introduction of PO linkages had a net negative impact on in vitro exon skipping activity (Figure 2B). To further explore the impact of PO backbone substitutions, we generated oligonucleotides with just one PO linkage in an otherwise fully stereorandom PS-modified backbone. DMD-360 harbored only 2′-OMe modifications and DMD-367 contained predominantly 2′-F modifications with two 2′-OMe modifications in the center of the oligonucleotide (Figure 2C). DMD-367 exhibited more exon skipping activity in vitro than any of the previously tested oligonucleotides with PO linkages (Figure 2C, mean skipping: 5.9%).
Next, we introduced stereopure PS-modified backbone linkages to DMD-367 to create DMD-335 and optimized the 2′-ribose pattern for exon skipping activity. We initially included Sp linkages at all PS-modified backbone positions, as the Sp configuration is known to confer metabolic stability (13,44). The introduction of all-Sp linkages at PS-modified positions in DMD-335 had a large beneficial effect on overall exon skipping activity in vitro (mean exon skipping 10 μM DMD-335: 29.4%). In DMD-332, we replaced the central PO linkage in DMD-335 with an Rp PS linkage, and this change had nominal impact on exon skipping activity (Figure 2D). In DMD-486 and DMD-487, we varied the position of the 2′-OMe modifications and the associated PO linkages and found that DMD-486 yielded the most robust activity (Figure 2D, mean exon skipping 10 μM DMD-486: 34.7%). In DMD-506–508, we explored additional variations on the position and number of 2′-OMe modifications and the associated PO linkages, and none of these oligonucleotides led to mean exon skipping levels (mean skipping at 10 μM: DMD-506 3.8%; DMD-507 20.0%, DMD-508 28.3%) (Figure 2E) that matched those observed with DMD-486, which contained four 2′-OMe modifications and two PO linkages optimally placed for the sequence.
Based on the data generated for exon 51, efficient exon skipping can be achieved in vitro using oligonucleotides containing a stretch of at least 5 to 6 2′-F modifications at the 3′ and 5′ ends with stereopure Sp phosphorothioate backbone linkages and a few 2′-OMe modifications in the middle with a chimeric stereopure Sp PS and PO backbone. This general chemical modification pattern forms the foundation for all subsequent oligonucleotides.
Application of rational design principles to exon 23
Before applying our learnings from the design of exon 51 skipping oligonucleotides to the mouse Dmd transcript, we first identified a suitable targeting sequence to yield productive skipping of Dmd exon 23, which would allow us to evaluate oligonucleotides in model systems such as mdx23 mice and mdx23-derived H2K cells, which contain a nonsense mutation in Dmd exon 23 (45). We generated a series of stereorandom oligonucleotides, using the chemical modification pattern used for DMD-100, that span the region in and around exon 23 of mouse Dmd (Figure 3A, Supplementary Figure S1) and evaluated them in exon skipping assays in differentiated H2K myoblasts under gymnotic conditions (Figure 3B). Activity was benchmarked against a previously published comparator sequence (DMD-91, mean skipping 1.0%) (35). Several sequences yielded significantly more exon skipping activity than DMD-91, including DMD-898 (2.5%), DMD-890 (3.9%), DMD-876 (4.0%) and DMD-865 (7.0%). DMD-865 yielded the highest mean skipping with this chemical modification pattern, so we used this sequence going forward.
Figure 3.

Identification of exon 23 skipping site. (A) To identify an exon 23 skipping targeting sequence, we screened oligonucleotides (depicted by staggered bars) complementary to sequences in and around exon 23 (yellow) of the mouse Dmd gene (cytogenetic view at top) using oligonucleotides with the chemistry mask identified from preliminary studies (shown in panel B). (B) Exon 23 skipping percentages in H2K cells treated gymnotically with 10 μM of the indicated oligonucleotides (shown in Supplementary Table S1) that differ in sequence but have the same chemical modifications (illustrated to the right). Analysis revealed a new targeting sequence (light blue) that is suitable for mediating efficient exon-23 skipping. DMD-91 (black) targets a previously published comparator sequence (5′- GGCCAAACCUCGGCUUACCU -3′) (26). ****P < 0.0001, **P < 0.01, *P < 0.05 One-way ANOVA with post-hoc comparisons to DMD-91. Mean ± s.e.m., n ≥ 3.
PN chemistry and optimization enhance the activity of stereopure oligonucleotides in vitro
To evaluate the impact of PN chemistry on exon 23 skipping oligonucleotides, we applied a pattern of 2′-ribose modifications, including 2′-F and 2′-OMe to develop a benchmark comparator sequence (DMD-3034), that closely matched the modification pattern identified for exon 51 (Figure 2). We generated a series of oligonucleotides based on DMD-3034 (DMD-4062–4076) to test the impact of individual stereopure Rp PN linkages at all positions along the stereopure Sp PS backbone. In H2K cells, the introduction of a single Rp PN linkage at multiple backbone positions (e.g. DMD-4063, DMD-4066, DMD-4075) increased mean exon skipping in vitro compared with DMD-3034 (Figure 4A).
Figure 4.

Application of PN chemistry to exon skipping oligonucleotides. (A) Placement of PN linkages was evaluated by sequentially replacing each PS linkage with a stereopure PN linkage (Rp configuration). The resulting oligonucleotide series is shown on the left (legend for modifications is shown on right). The graph depicts percentage of exon 23 skipping detected after 4 days of treatment with 0.3 μM (beige), 1 μM (green), or 3 μM (red) of the indicated oligonucleotide (n = 2 per concentration per oligonucleotide). Data are represented as mean ± s.d. (B) Number and position of PN linkages was evaluated in the series of oligonucleotides. The graph depicts percentage of exon 23 skipping detected as shown in panel A. (C) Schematic representation of oligonucleotides evaluated (left). The graph depicts percentage of exon 23 skipping detected as shown in panel A. Dotted gray line separates data from oligonucleotides with and without PN-backbone linkages. Data are presented as mean ± s.e.m., n = 3. Two-way ANOVA with multiple comparisons: * P < 0.05, ** P < 0.01, *** P < 0.001. (D) Oligonucleotide walk across human DMD exon 44 to compare average exon-skipping activity (n = 2) in Δ48–50 primary human myoblasts for oligonucleotides with all-Sp PS backbone to those with inclusion of a few stereorandom PN linkages. PS oligonucleotides, which differ from each other in sequence, are ranked along the x axis with the most active oligonucleotides on the right. Sequence-matched PN-containing oligonucleotides are plotted at the same position (x axis) as their PS counterpart. The chemical modifications for the PS and PS-PN oligonucleotides are shown. DMI, 1,3-dimethylimididazolidine-2-imine
To determine whether the inclusion of multiple PN linkages could further boost exon skipping activity, we generated another series of oligonucleotides containing two to six Rp PN linkages while retaining the same pattern of 2′-ribose modifications. We evaluated exon skipping activity of these oligonucleotides in differentiated H2K myoblasts under gymnotic conditions using DMD-3034 as a benchmark (Figure 4B). The incorporation of two non-consecutive PN linkages in DMD-4417, DMD-4418, DMD-4419 and DMD-4425 dramatically improved exon skipping activity in vitro compared with DMD-3034. The inclusion of multiple consecutive PN linkages in the 5′-half (DMD-4420), the 3′-half (DMD-4421) or at the center (DMD-4423) of the oligonucleotide was less beneficial. The inclusion of three (DMD-2788, DMD-3038), four (DMD-4427) or five (DMD-4426) PN linkages was comparably beneficial, but the addition of more than three PN linkages did not provide any additional activity benefits. The inclusion of three consecutive PN linkages in both the 5′-half of the same oligonucleotide (DMD-4422) supported exon skipping activity comparable to the benchmark stereopure oligonucleotide, DMD-3034.
We next evaluated additional backbone configurations for both PS and PN modifications on exon-skipping activity (Figure 4C). We generated a new series of oligonucleotides targeting the same exon 23 sequence, once again applying the same pattern of ribose modifications from DMD-3034. We placed PN linkages at positions 3, 6 and 17, which showed beneficial activity in our previous analysis. The series included oligonucleotides with PS-modified stereorandom (DMD-3839) backbone; stereopure PS all-Rp (DMD-4009); stereopure PS all-Sp (DMD-3034); as well as mixed PS/PN stereorandom (DMD-3840) backbone; stereopure PS all-Rp, stereorandom PN (DMD-4010); stereopure PS all-Sp, stereorandom PN (DMD-3715); stereopure PS all-Sp, stereopure PN all-Rp (DMD-2788); and stereopure PS all-Sp, stereopure PN all-Sp (DMD-3724).
We evaluated the activity of these oligonucleotides in differentiated H2K myoblasts in vitro under gymnotic conditions (Figure 4C). For PS-modified oligonucleotides, Sp PS linkages (DMD-3034) but not Rp linkages (DMD-4009) increased exon-skipping activity compared with the stereorandom PS oligonucleotide (DMD-3839). The introduction of stereorandom PN linkages to PS-modified oligonucleotides increased activity for all PS configurations, but the combination of stereorandom PN with stereopure PS all-Sp in DMD-3715 was most beneficial. For the stereopure configurations, the combination of Rp PN linkages and Sp PS linkages in DMD-2788 was most potent, inducing the highest percentage of exon skipping for any oligonucleotide tested. To confirm that the inclusion of PN linkages increased exon-skipping activity independent of sequence, we performed a screen targeting human DMD exon 44 where we compared all-Sp PS-modified oligonucleotides with sequence-matched PS-PN-containing oligonucleotides (Figure 4D). In general, the inclusion of three PN linkages in an otherwise stereopure PS-modified oligonucleotide, increased exon skipping.
PN modifications promote exon skipping and dystrophin restoration in vivo
To investigate whether the activities observed in vitro translate in vivo, we evaluated exon-skipping activity of a subset of oligonucleotides in mdx23 mice. One week after a single dose (mdx-1 study, Figure 5A), all oligonucleotides containing PN linkages in combinations with stereopure PS backbone modifications yielded statistically significantly increased levels of exon skipping compared with PBS (P < 0.05, one-way ANOVA, Figure 5B). DMD-3840, which contains PN linkages and a stereorandom PS backbone, did not elicit significant exon skipping compared with PBS-treated controls (P = 0.99, One-way ANOVA). In a 6-week multi-dose study (mdx-2 study, Figure 5A), a similar trend was observed (Figure 5C). DMD-2788 was the best performing molecule, leading to a significant increase in exon skipping in all muscles tested, including gastrocnemius, quadricep, diaphragm and heart (P < 0.01, one-way ANOVA, Figure 5C, Supplementary Figure S1). DMD-3034, the stereopure all-Sp PS molecule supported significant activity in gastrocnemius and quadricep but not in diaphragm or heart (Figure 5C, Supplementary Figure S1, P < 0.01 One-way ANOVA). Stereorandom oligonucleotides, with or without PN linkages, did not induce substantial exon skipping in any tissue. Importantly, these exon-skipping data are positively correlated with dystrophin protein restoration data quantified by capillary immunoassay (Figure 5D, Supplementary Figures S2, S3; Spearman's rank correlation P < 0.01). Taken together, these data indicate that PN linkages can increase the activity of stereopure PS-modified exon-skipping oligonucleotides in vitro and in vivo, with a chimeric all-Sp PS, all-Rp PN and PO backbones most effective for skipping Dmd exon 23.
Figure 5.
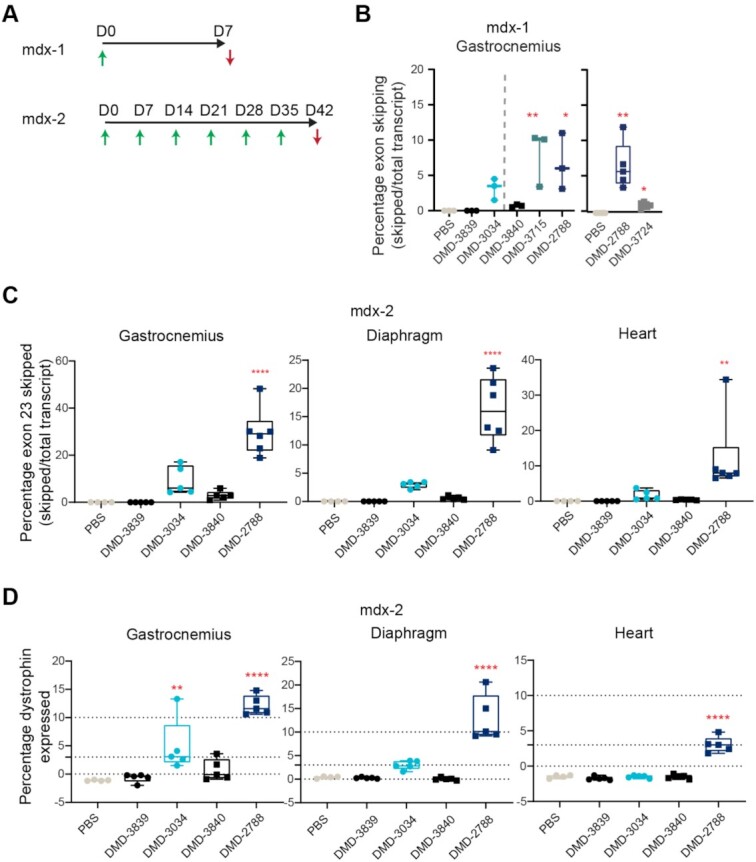
Relative exon 23-skipping activity in skeletal muscle in mdx23 mouse correlates with in vitro activity. (A) Dosing paradigms for in vivo studies mdx-1 and mdx-2. Green arrows indicate intravenous (IV) administration of 30 mg/kg (mdx-1) or 75 mg/kg (mdx-2) of oligonucleotide to 6–8-week-old mice. Red arrows indicate sample collection. (B) Percentage of exon 23-skipping detected in mdx23 mouse gastrocnemius muscle at day 7 (D7) in mdx-1 study. n ≥ 3. Data from two separate experiments are shown. (C) Percentage of exon 23-skipping in gastrocnemius, heart, and diaphragm at day 42 (D42) in mdx-2. n ≥ 4. (D) Percentage of dystrophin expression detected at D42 in the gastrocnemius, diaphragm, and heart in mdx-2. WES quantification protocol is outlined in Supplementary Figure S2, images of blots are shown in Supplementary Figure S3, n ≥ 4. For C-D, scales of y axes differ across the graphs; for B-D, box and whisker plots show min to max; data points are individual animals; stats: One-way ANOVA with multiple comparisons to PBS **** P < 0.0001; ** P < 0.01; * P < 0.05.
PN modifications increase muscle exposure
Activity differences were not driven by thermal stability, as the most active oligonucleotide in vivo, DMD-2788, had a relatively lower melting temperature (Supplementary Figure S1). To investigate why PN-containing oligonucleotides exhibit improved activity, we quantified oligonucleotide in muscles of treated animals in the mdx-2 study (Figure 6A, Supplementary Figure S1). One week after the last dose, we observed the highest tissue exposure for PN-containing molecules, DMD-2788 and DMD-3840. Muscle exposures for DMD-3034 and DMD-3839 were much lower. For DMD-3034, these observations are consistent with activity observed in skeletal muscles, including gastrocnemius (Figure 5) and quadriceps (Supplementary Figure S1). To understand the relationship between tissue exposure and activity for these oligonucleotides, we plotted oligonucleotide exposure with respect to exon-skipping activity (Figure 6B, Supplementary Figure S1). For DMD-2788, high tissue exposure correlated with high activity across muscle types. For DMD-3840, however, increased tissue exposure did not correspond with activity. Indeed, activity benefits of PN modifications were only observed in vivo when they were combined with a stereopure PS-modified backbone.
Figure 6.

PN backbone chemistry enhances muscle exposure. (A) Concentration of the indicated oligonucleotide detected in gastrocnemius, diaphragm, and heart muscle in mdx-2 study. Dots represent individual mice; box and whiskers show min to max. Mean ± s.d. n ≥ 4. Stats performed with pairwise comparisons to PBS. (B) Relationship between tissue exposure and exon-skipping activity for the indicated oligonucleotides in gastrocnemius, diaphragm, and heart muscle in mdx-2 study. (C) Images showing DMD-3034 and DMD-2788 in gastrocnemius, diaphragm, and cardiac muscle after ViewRNA staining from mdx-1 study at day 7. Tissues treated with PBS are also shown. Arrows demarcate myoblast nuclei that are positive for DMD-2788. Scale bars are 20 μm. (D) Concentration of DMD-3034 or DMD-2788 detected in whole cell (left), nuclear (middle), or cytoplasmic (right) extracts from differentiated myoblasts treated for 24 h as quantified by hybridization ELISA. n = 3 mean ± s.e.m. Unpaired, two-tailed t test *P < 0.05, **P < 0.01, *** P < 0.001. (E) Concentration of DMD-3034 or DMD-2788 in whole cell extracts from myoblasts treated for 3 h and washed for the indicated time. n = 3 mean ± s.e.m. * P < 0.05, ** P < 0.01, **** P < 0.0001 Two-way ANOVA with multiple comparisons.
We further investigated tissue exposure by visualizing oligonucleotides in muscle sections from the mdx-1 animals using ViewRNA, an in situ hybridization method used to detect oligonucleotide. These studies corroborated the findings that PN-containing oligonucleotides had improved muscle exposure (Figure 6C, Supplementary Figure S4) and demonstrated that DMD-2788 distributed to nuclei, which is necessary for target engagement with dystrophin pre-mRNA.
To further explore differences in tissue exposure and exon-skipping activity, we quantified the gross uptake and intracellular distribution of oligonucleotides in vitro in differentiated myoblasts using multiple methods, including hybridization ELISA with variably stringent wash conditions (Figure 6D, Supplementary Figure S4) and qRT-PCR (Supplementary Figure S4). In these experiments, we detected a 2–3-fold increase in the amount of DMD-2788, the PN-containing oligonucleotide, compared with DMD-3034 in extracts from whole cells, nuclei, and cytoplasm (Figure 6D, Supplementary Figure S4; P < 0.05 unpaired t test). To explore whether the increase in intracellular oligonucleotide resulted from an improvement in cellular uptake or improved intracellular stability, we evaluated intracellular oligonucleotide levels over time after a short exposure to oligonucleotide (3 h) with a variable wash-out period (Figure 6E). Surprisingly, at the shortest time interval (0 h, no wash out), we detected comparable amounts of DMD-3034 and DMD-2788. With wash out, the amount of DMD-3034 in cells steadily declined, whereas the amount of DMD-2788 in cells remained constant. The effect of time on the amount of oligonucleotide detected was statistically significant (P < 0.001, Two-way ANOVA). These data suggest that both oligonucleotides are readily taken up by myoblasts but that DMD-2788 persists longer than DMD-3034 once inside of the cells. This is consistent with previous in vitro findings that Sp and Rp configurations of PN linkages have comparable nuclease stability, and both are more stable than PS linkages (46).
PN modified oligonucleotides are well tolerated in mice
We performed two additional studies in mdx23 mice to evaluate the tolerability of DMD-3034 and DMD-2788 as well as their distribution to other tissues. For the tolerability study, we evaluated serum samples of mdx23 mice after treatment with 60 mg/kg oligonucleotide or PBS delivered once weekly for six weeks intravenously (Figure 7A, mdx-3). We first evaluated creatine kinase levels, which are known to be elevated in mdx23 mice (47). Compared with PBS-treated controls, DMD-3034 and DMD-2788 both significantly decreased serum creatine kinase levels (Figure 7A). We next evaluated serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), which are common markers for hepatotoxicity but are also known to be elevated in mdx23 mice due to muscle damage (48). Upon treatment with DMD-3034 or DMD-2788, ALT and AST levels dropped significantly compared with controls (Figure 7A). We also evaluated the ratios of CK: ALT and CK: AST, as these ratios provide insight into hepatotoxicity independent of muscle damage (48). There was no statistically significant change in these ratios for treated animals compared with controls (Figure 7B), indicating that neither DMD-3034 nor DMD-2788 led to hepatoxicity.
Figure 7.

In vivo tolerability studies. (A) Dosing paradigms for in vivo studies mdx-3 and mdx-4. Green arrows indicate intravenous (IV) administration of 60 mg/kg (mdx-3) or 75 mg/kg (mdx-4) of oligonucleotide to 10-week-old male mdx23 mice. Red arrows indicate sample collection. Serum CK (left) serum ALT (middle) and serum AST (right) are shown for animals treated as indicated in the mdx-3 study. Dots represent individual mice. Data are presented as mean ± s.d. n = 3. (B) Ratios of serum CK:ALT (left) and CK:AST (right) are shown for the mdx-3 study. mean ± s.d. are indicated. (C) Concentration of the indicated oligonucleotide detected in liver and kidney in mdx-4 study at days 7 (D7) and 28 (D28). Dots represent individual mice; box and whiskers show min to max. Data are presented as mean ± s.d. n = 4. Stats for all panels by One-way ANOVA with Dunnett's post-hoc test for multiple comparisons. ns non-significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
To evaluate the distribution of DMD-3034 and DMD-2788, we quantified oligonucleotide concentrations in liver and kidney after treatment with a single 75 mg/kg IV dose of oligonucleotide or PBS (Figure 7A, mdx-4). Mean oligonucleotide levels were highest 1-week post-injection for both DMD-3034 and DMD-2788 in both tissues (Mean concentration liver: DMD-3034 23.4 μg/g, DMD-2788 97.3 μg/g; Mean concentration kidney: DMD-3034 107.4 μg/g, DMD-2788 130.6 μg/g) (Figure 7C). These observations are consistent with liver and kidney serving to clear these molecules from blood by filtration and excretion (49). Although DMD-2788 concentrations were higher in liver and kidney than DMD-3034, we detected no signs of toxicity in these tolerability studies.
PN modification improves muscle function and increases lifespan of severely dystrophic mice
We next sought to assess the benefits of DMD-3034 and DMD-2788 in the double knockout (dKO) mouse model for Duchenne muscular dystrophy, which lacks both utrophin and dystrophin and develops a severe muscular dystrophy phenotype comparable to that observed in patients (50). The mice exhibit abnormal gait, reduced movement and premature death (50,51). We assessed if there was an improvement in muscle biomarkers, including dystrophin, or muscle function in a 6-week study (Figure 8A, dKO-1 study). In the dKO-1 study, age-matched untreated C57Bl10 mice served as controls, and dKO mice were treated with PBS or 75 mg/kg DMD-3034 or DMD-2788 weekly by subcutaneous injection for 6 weeks and evaluated at day 47. Specific force production in the extensor digitorum longus (EDL) muscle, following normalization to muscle cross-sectional area, was significantly improved over PBS-treated controls in mice treated with DMD-3034 (22% increase, P < 0.05, two-way ANOVA) or DMD-2788 (55% increase, P < 0.0001, Two-way ANOVA) (Figure 8A). To evaluate resistance to exercise-induced damage, we measured maintenance of muscle force following repeated eccentric contractions. PBS-treated dKO mice experienced a force drop of ∼80% following 10 repeat contractions. Neither DMD-3034 nor DMD-2788 substantially impacted this phenotype at day 47 (Figure 8A).
Figure 8.

DMD-3034 and DMD-2788 improve biomarker profiles and muscle function in dKO mice. (A) Schematic representation of weekly subcutaneous dosing regimen (75 mg/kg) for study dKO-1. First administration corresponds to 10-day old mice. Mice were dosed weekly (green arrows) for 6 weeks starting on day 10 of age and assessed for functional assays before sacrifice 2 days after the last injection (red arrow). Specific force (middle) production over increasing stimulation frequency and force drop as a percentage of initial eccentric force (right) following repeated contraction for C57Bl10 mice and dKO mice treated with PBS or the indicated oligonucleotide. Mean ± s.d., n ≥ 4. Two-way ANOVA with comparisons to PBS. * P < 0.05,** P < 0.01, ***P < 0.001, **** P < 0.0001. All significant differences increased or were maintained at later experimental points. For example, by contraction 3 (bottom), untreated C57Bl10 mice are significantly different from PBS-treated dKO mice (P < 0.0001), and this level of significance persists through the end of the experiment. (B) Percentage exon skipping measured in the gastrocnemius (left), diaphragm (middle), and heart (right) in dKO-1 study at day 47. Mean ± s.d., n = 4. ***P < 0.001, **** P < 0.0001. (C) Percentage dystrophin restoration measured in the same tissues as in panel B. Dystrophin quantification as shown in Supplementary Figure S2. Green arrow denotes dystrophin; yellow arrow, vinculin. (D) Serum creatine kinase levels for mice from dKO-1 study at day 47. Mean ± s.e.m., C57BL10 n = 6; dKO n = 8. (E) Serum miRNA levels for dKO-1 study at day 47. Mean ± s.e.m., C57Bl10 n = 6; dKO n = 8. Panels B–D: one-way ANOVA with comparison to PBS (B), PBS and C57Bl10 (C) or C57Bl10 (D), ** P < 0.01, *** P < 0.001, **** P < 0.0001.
In the dKO-1 study, mice treated with DMD-3034 had small increases in exon 23 skipping and dystrophin compared with PBS-treated controls, whereas in DMD-2788-treated animals, dystrophin levels reached 5–15% of wild-type levels in skeletal muscle, 12–20% in diaphragm and ∼10% in heart (Figure 8B,C, Supplementary Figure S5). Dystrophin expression levels were well correlated with exon skipping (Spearman's rank correlations, P < 0.0001). We also assessed serum biomarkers for muscle damage (creatine kinase) and dystrophic muscle [microRNA (miR)-1, miR-133a, miR-206, also known as myomiRs (52)]. In PBS- and DMD-3034-treated dKO mice, serum creatine kinase levels were elevated compared with C57Bl10 controls (Figure 8D). In the DMD-2788-treated dKO cohort, serum creatine kinase levels were restored towards levels matching C57Bl10 control (Figure 8D). MyomiRs, which were significantly elevated in PBS and DMD-3034 groups compared to C57Bl10 controls, were decreased by DMD-2788 treatment to levels that were not significantly different from those detected in C57Bl10 serum (Figure 8E, miR-1 P = 0.62; miR-133a P = 0.48; miR-206 P = 0.46, one-way ANOVA). Collectively, these data clearly demonstrate that DMD-2788 treatment induces a measurable improvement in dystrophin production, biomarkers of muscle damage, and muscle function in the dKO model. Although DMD-3034 also yielded detectable dystrophin protein, the levels were relatively low and did not correspond with improvements in muscle function or serum biomarkers.
To assess whether treatment with DMD-3034 or DMD-2788 could impact the lethality observed in dKO mice, we performed a survival study (Figure 9A, dKO-2 study). In the first dosing regimen (R1) evaluated in this study, dKO mice were administered a weekly subcutaneous 150 mg/kg dose of oligonucleotide or PBS and compared with untreated mdx23 littermates. As expected, dKO mice treated with PBS had a short lifespan, with a median survival of 49 days, and untreated mdx23 mice survived for the duration of the study (Figure 9A). DMD-3034 improved this phenotype, increasing median survival to 86 days. With the same dosing regimen, DMD-2788 resolved the severe phenotype exhibited by dKO mice, with behavior and gait matching mdx23 littermates (Supplementary videos S1A,B). Indeed, all DMD-2788 treated dKO mice survived with no obvious pathological or behavioral defects for a median of 274 days, when we terminated the survival portion of the study to assess muscle function and dystrophin restoration (P = 2 × 10–11, χ2 analysis). During this survival study, we also assessed body weight (Figure 9B); after an initial period with similar growth rates, PBS control and DMD-3034-treated groups showed a sharp decline in weight gain prior to death. By contrast, weights for DMD-2788-treated animals plateaued around 8 weeks, and mdx23 littermates gained weight for the duration of the study. At terminal time points, which differed for each cohort, we evaluated dystrophin levels and other biomarkers in the mice. Dystrophin levels significantly increased in the DMD-2788 group, reaching 60% of wild-type levels in gastrocnemius and diaphragm and 10% in heart (Figure 9C, P < 0.0001, One-way ANOVA, Supplementary Figure S6). Low but detectable levels of dystrophin were also detected with DMD-3034 although these were not significantly different than PBS-treated controls in any muscle evaluated. In these terminal samples, we confirmed that dystrophin was localized to the muscle sarcolemma with immunostaining (Figure 9D, Supplementary Figure S6). For treated animals, serum creatine kinase and myomiR levels also improved dramatically compared with PBS-treated dKO mice and mdx23 controls (Supplementary Figure S7).
Figure 9.

DMD-2788 rescues early fatality in dKO mice. (A) Schematic representation of dosing regimens for dKO-2 study, with 10-day old mice receiving weekly subcutaneous 150 mg/kg doses of oligonucleotide or PBS in regimen 1 (R1, top) (n = 8 per group). Mice receiving DMD-2788 survived beyond the study window and were euthanized for analysis between 38 (day 266) and 41 weeks (day 287). Kaplan-Meier survival curves showing probability of survival over time for mice in R1. Hash marks indicate time points when mice were culled (some hash marks represent more than 1 mouse), and shaded red box indicates window for study termination. (B) Body weights (g) over time for males (left) and females (right) in R1. Data are presented as mean ± s.e.m., n = 8. (C) Dystrophin expression detected in gastrocnemius, diaphragm, and heart in mice from R1. Dystrophin quantification as shown in Supplementary Figure S2. Tissues from PBS- and DMD-3034-treated animals were taken at terminal time points when they were no longer healthy. Dots represent individual mice; box and whiskers show min to max. n = 2, PBS control; n = 4, treated; ns non-significant, **** P < 0.0001, one-way ANOVA with multiple comparisons to PBS. (D) Visualization of restored dystrophin co-localized with laminin in gastrocnemius muscle of dKO mice treated with DMD-2788. 50 μm scale bar is shown. (E) Percentage of initial specific force following repeat eccentric contraction (top) and specific force at increasing stimulation frequency (bottom) for dKO mice treated with DMD-2788 (150 mg/kg) for ∼38 weeks and comparably aged and untreated mdx23 mice from dKO-1 R1. Mean ± s.d., n ≥ 4. Two-way ANOVA with comparisons to PBS. * P < 0.05,** P < 0.01, ***P < 0.001, **** P < 0.0001. All significant differences increased or were maintained at later experimental points.
Because survival was so dramatically impacted in this longer study, we also assessed EDL force production and exercise-induced damage in older DMD-2788-treated mice and mdx23 mice at the termination of the survival study (dKO mice treated with PBS or DMD-3034 do not survive to this stage) (Figure 9E). DMD-2788 increased specific force production by 58% compared to mdx23 mice (P < 0.0001, two-way ANOVA), and these mice exhibited a force drop of only 25% following repeated contraction compared to ∼80% for the mdx23 cohort (Figure 9E), suggesting that prolonged DMD-2788 treatment can restore near-normal muscle function in dKO mice.
Finally, we sought to determine whether extended survival and other benefits could be observed with a more clinically relevant dosing regimen (R2) for DMD-2788. In dKO-2 R2, we decreased both the dose and frequency of DMD-2788 dosing by 50%, resulting in an overall 75% decrease in total dose (Figure 10A). We administered 75 mg/kg DMD-2788 bi-weekly starting at postnatal day 10. Similar to observations in the dKO-2 R1 cohort, DMD-2788 resolved the severe phenotype exhibited by dKO mice, with behavior and gait matching mdx23 littermates and mice surviving for a median of 280 days with no obvious behavioral defects before the study was terminated (Figure 10A, Supplementary video S2). DMD-2788-treated mice in the R2 cohort exhibited body weight profiles more similar to mdx23 mice than those from the higher-dose R1 cohort (Figure 10B), and while dystrophin levels were lower than observed in the 150 mg/kg dKO-1 R1 DMD-2788 cohort, they were significantly improved compared with PBS-treated mice (gastrocnemius 12% dystrophin, P = 0.0098; diaphragm 30% dystrophin, P = 0.0003; heart 3% dystrophin, P = 0.0095 one-way ANOVA) (Figure 10C). Serum biomarkers including creatine kinase and myomiRs were also improved in the low-dose regimen (Figure 10D–F). Taken together, these data demonstrate that a much lower dose of DMD-2788 conveys a substantial survival benefit as well as other functional and biomarker improvements in the severely dystrophic dKO mouse model.
Figure 10.

Clinically relevant doses of DMD-2788 rescue early fatality in dKO mice. (A) Schematic representation of dosing regimen for dKO-2 study, with 10-day old mice receiving biweekly subcutaneous 75 mg/kg doses in regimen 2 (R2) (n = 9 per group). Kaplan-Meier survival curves showing probability of survival over time for mice in R2. Hash marks indicate time points when mice were culled (some hash marks represent more than 1 mouse), and shaded red box indicates window for study termination. (B) Body weights (g) over time for males (left) and females (right) in R2, respectively. Data are presented as mean ± s.e.m., n = 8. (C) Dystrophin expression detected in gastrocnemius, diaphragm, and heart in mice from dKO-2 R1 (150 mg/kg) and R2 (75 mg/kg). Dystrophin quantification as shown in Supplementary Figure S2. Tissues from PBS- and DMD-3034-treated animals were taken at terminal time points when they were no longer healthy. Dots represent individual mice; box and whiskers show min to max. n = 2, PBS control; n = 4, treated; ns non-significant, **** P < 0.0001, one-way ANOVA with multiple comparisons to PBS. (D) Percentage of initial specific force following repeat eccentric contraction (top) and specific force at increasing stimulation frequency (bottom) for dKO mice treated with DMD-2788 (75 mg/kg) for ∼38 weeks and comparably aged and untreated mdx23 mice from dKO-2. Data are presented as mean ± s.e.m., n ≥ 4. Two-way ANOVA with comparison to mdx. ns nonsignificant, **** P < 0.0001. (E) Serum creatine kinase levels for dKO mice from dKO-2 R1 and R2 and mdx23 controls. Box and violin plots show min to max, dKO (DMD-3034) n = 4, dKO (DMD-2788 150 mpk) n = 8, (DMD-2788 75 mpk) n = 9, mdxn = 6. ** P = 0.0016, *** P = 0.0003, **** P < 0.0001, one-way ANOVA with multiple comparisons to PBS-treated dKO mice. (F) Serum miRNA levels for dKO-2 study R1 and R2 and mdx23 controls. Box and violin plots show min to max, C57Bl10 n = 6; DMD-3034 n = 8, DMD-2788 n = 8, mdx n = 8. ** P < 0.01, *** P < 0.001, **** P < 0.0001, one-way ANOVA with multiple comparisons to mdx23 mice.
DMD-2788 rescues respiratory function in dKO mice
To further explore physiological drivers of survival benefit observed in dKO mice, we evaluated respiratory function in these mice after 6 weeks of treatment with a weekly, 150 mg/kg subcutaneous dose of DMD-2788 (Figure 11A, study dKO-3). Respiratory function was evaluated at days 21, 35 and 47. All measures of respiratory function were improved by treatment of dKO mice with DMD-2788 compared with PBS except respiration frequency, which differed nominally between cohorts (Figure 11B). For all other measures, including tidal volume, minute volume, peak inspiratory flow and peak expiratory flow, respiratory function in dKO mice treated with DMD-2788 tracked better with wild-type C57Bl/6 mice than PBS-treated dKO mice, and the effect of treatment was statistically significant (P < 0.0001, two-way ANOVA). These data demonstrate that DMD-2788 improves respiratory function of dKO mice.
Figure 11.
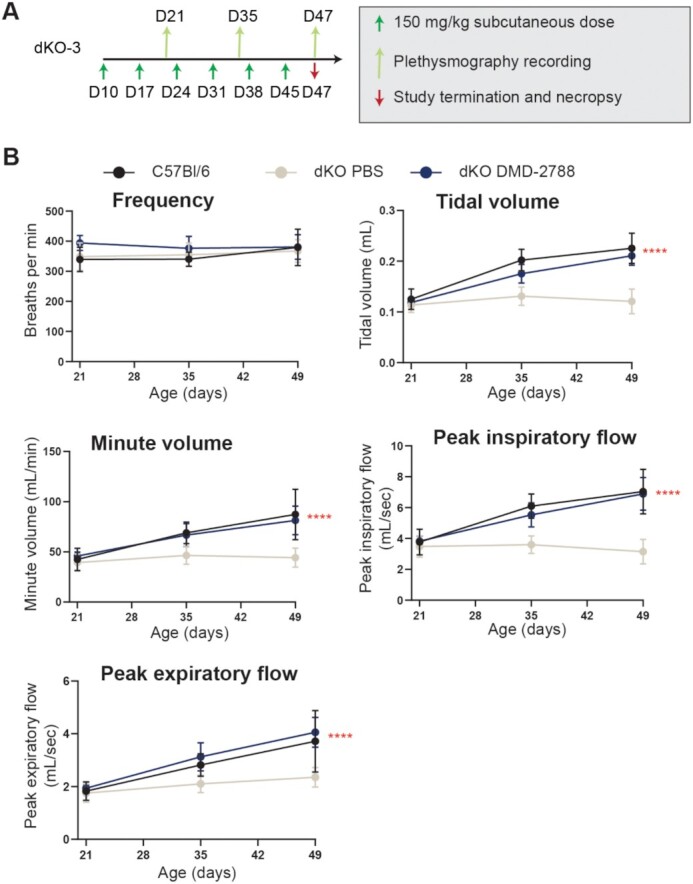
DMD-2788 rescues respiratory function in dKO mice. (A) Schematic representation of dKO-3 study with 10-day old mice receiving weekly subcutaneous 150 mg/kg doses of DMD-2788 (n = 8) or PBS (n = 9). Age-matched C57Bl/6 wild-type mice (n = 8) were also included in the study. Green arrows (below black line) indicate dosing, light green arrows (above black line) indicated whole-body non-invasive plethysmography over a 30 min recording period, and red arrow indicates the end of the study. (B) Graphs depict frequency of respiration, tidal volume, minute volume, peak inspiratory flow, or peak expiratory flow at days 21, 35 and 49 of age. Data are presented as mean ± s.d. Stats from two-way ANOVA **** P < 0.0001.
DISCUSSION
We developed the methods necessary to engineer stereopure PN-containing oligonucleotides with chimeric backbones comprising a combination of PS, PO and PN linkages. These methods are efficient, yielding ∼99% stereochemical selectivity for each chiral backbone linkage. We applied this chemistry to investigate the impact of PN-backbone modifications in combination with PS backbone stereochemistry and 2′-ribonucleotide modifications on the activity and pharmacology of exon-skipping oligonucleotides. Using Duchenne muscular dystrophy as an example, we found that PN-containing oligonucleotides have superior pharmacology and activity in vivo compared with oligonucleotides harboring only PS and PO linkages, whether they are stereopure or stereorandom.
We found that oligonucleotides containing 2′-F-modifications, which impart increased binding efficiency and nuclease stability (23,53,54) are both well tolerated in vivo and support the activity of exon-skipping oligonucleotides. Prior reports considering the application of this 2′ chemistry have been inconsistent. In one report, 2′-F modifications enhanced exon-skipping activity in vitro, but this activity did not translate in vivo, and oligonucleotides incorporating this modification were not well tolerated (55). In another report, 2′-F modifications enhanced exon skipping, and the position of the modification impacted how well the oligonucleotides were tolerated (56). The fact that 2′-F-modified oligonucleotides reported herein rescued multiple phenotypes in severely dystrophic mice in long-term experiments at multiple doses and led to no signs of hepatotoxicity suggests they were well tolerated, and these data lend credence to the notion that context for 2′-F modification (e.g. sequence and presence of other chemical modifications) matters.
The superior pharmacology of PN-containing chimeric molecules is best explained by benefits observed in vivo in muscle exposure assays, where they achieved considerable distribution, exposure and function across muscle types. Our in vitro investigations comparing intracellular oligonucleotide concentrations in differentiated myoblasts suggest that increased muscle exposure observed with PN-containing molecules derives at least in part from increased intracellular and intranuclear exposure. Because the PN-containing oligonucleotides persisted longer than the PS-PO oligonucleotides, the PN modification likely drives this effect, potentially creating a more stable reservoir of oligonucleotide inside the cell. The fact that this improved pharmacological profile and superior in vivo activity can be engineered into the same molecule is encouraging, as poor cellular uptake and intracellular exposure have been widely recognized as barriers that have limited the translation of exon-skipping and other oligonucleotide-based technologies and was the most apparent explanation for the failure of the first-generation stereopure PS-PO molecule suvodirsen (7,57–62) (https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-discontinuation-suvodirsen).
PN-containing chimeric oligonucleotides prevented premature death and improved both muscle and respiratory function in a severely dystrophic dKO mouse model (50,51) at clinically relevant doses (63). Although high doses (150 mg/kg, weekly) of the suvodirsen-like molecule DMD-3034 also provided survival benefits, albeit more modest, the dose corresponds to a weekly human equivalent dose (HED) of ∼12 mg (63), which was not achieved in the suvodirsen trial. By contrast, the 75 mg/kg biweekly dose of PN-containing DMD-2788 provided a profound survival benefit in mice. This 75% lower total dose corresponds to a biweekly HED ∼6 mg. Although the introduction of a few PN-backbone linkages improved the activity of PS-modified exon-skipping oligonucleotides in vitro in human and mouse myoblasts independent of oligonucleotide sequence and backbone stereochemistry, this enhanced activity only translated in vivo when PN linkages were combined with stereopure PS modification. Because increased muscle exposure was observed for all PN-containing molecules but increased activity was observed only in the context of a stereopure PS backbone, the stereopure PS linkages must affect additional properties of these oligonucleotides, such as those that impact target engagement or intracellular trafficking. Future investigations will interrogate mechanisms that underly these benefits.
To our knowledge, this is the first application of stereopure backbone chemistry to exon-skipping oligonucleotides. The realization of benefits observed with stereopure oligonucleotides for the RNase H modality (13–15) for exon skipping suggests that control over backbone stereochemistry has enabling potential across multiple oligonucleotide modalities. The benefits of PN-backbone chemistry highlight that there is untapped potential in optimizing oligonucleotide pharmacology by controlling the chemistry and stereochemistry of the backbone. Importantly, these findings also show that chemical modifications to the oligonucleotide alone can be sufficient to elicit critical activity benefits in the absence of tissue-targeting conjugates. Herein, we illustrate that chimeric stereopure oligonucleotides may be suitable for therapeutic development in Duchenne muscular dystrophy, where restoration of dystrophin to levels sufficient to improve muscle function are expected to provide clinical benefit (64,65). The levels of dystrophin achieved in diaphragm and heart and rescue of respiratory function of severely affected dKO mice are particularly encouraging, as these muscles have been notoriously difficult to reach in preclinical studies (8–12) and may be essential to prolong function and improve survival in Duchenne muscular dystrophy (3,6). Overall, improvements in cellular uptake and exposure as well as activity gains in exon skipping and protein restoration in hard-to-reach tissues suggest this splice-switching technology could be amenable to a multitude of disease-causing RNA mis-splicing mutations.
DATA AVAILABILITY
Source data for figures Figures 2A–E, 3B, 4A–D, 5B–D, 6A–B, D–E, 7A–C, 8A–E, 9A–C, E, 10A–F and 11B are provided with the paper.
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the CCDC with the refcode 2113502.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Amy Donner for support with writing and graphics; Lauren Norwood, Jake Metterville and Kris Taborn for help with in vivo studies; Chris Francis for establishing the collaboration between Wave and Oxford and design ideas during early stages of this work; Keith Bowman and Stephany Standley for overseeing process development for and synthesis of all oligonucleotides used in this work; Khoa Luu for the technical assistance of thermal denaturation (Tm) experiment: Tracy Chen for project management. We are also grateful to Professor Jenny Morgan (Dubowitz Neuromuscular Centre, UCL, London, UK) for H2K cells and Dr. Vincent Mouly (Center for Myology, GH Pitié-Salpétrière, Paris, France) for human Δ48–50 myoblasts (ID6594-I).
Contributor Information
Pachamuthu Kandasamy, Wave Life Sciences, Cambridge, MA, USA.
Graham McClorey, Department of Paediatrics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK.
Mamoru Shimizu, Wave Life Sciences, Cambridge, MA, USA.
Nayantara Kothari, Wave Life Sciences, Cambridge, MA, USA.
Rowshon Alam, Wave Life Sciences, Cambridge, MA, USA.
Naoki Iwamoto, Wave Life Sciences, Cambridge, MA, USA.
Jayakanthan Kumarasamy, Wave Life Sciences, Cambridge, MA, USA.
Gopal R Bommineni, Wave Life Sciences, Cambridge, MA, USA.
Adam Bezigian, Wave Life Sciences, Cambridge, MA, USA.
Onanong Chivatakarn, Wave Life Sciences, Cambridge, MA, USA.
David C D Butler, Wave Life Sciences, Cambridge, MA, USA.
Michael Byrne, Wave Life Sciences, Cambridge, MA, USA.
Katarzyna Chwalenia, Department of Paediatrics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK.
Kay E Davies, Department of Physiology, Anatomy and Genetics, University of Oxford, South Parks Road, Oxford OX1 3PT, UK.
Jigar Desai, Wave Life Sciences, Cambridge, MA, USA.
Juili Dilip Shelke, Wave Life Sciences, Cambridge, MA, USA.
Ann F Durbin, Wave Life Sciences, Cambridge, MA, USA.
Ruth Ellerington, Department of Paediatrics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK.
Ben Edwards, Department of Physiology, Anatomy and Genetics, University of Oxford, South Parks Road, Oxford OX1 3PT, UK.
Jack Godfrey, Wave Life Sciences, Cambridge, MA, USA.
Andrew Hoss, Wave Life Sciences, Cambridge, MA, USA.
Fangjun Liu, Wave Life Sciences, Cambridge, MA, USA.
Kenneth Longo, Wave Life Sciences, Cambridge, MA, USA; MDUK Oxford Neuromuscular Centre, University of Oxford, Oxford OX2 9DU, UK.
Genliang Lu, Wave Life Sciences, Cambridge, MA, USA.
Subramanian Marappan, Wave Life Sciences, Cambridge, MA, USA.
Jacopo Oieni, Department of Paediatrics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK.
Ik-Hyeon Paik, Wave Life Sciences, Cambridge, MA, USA.
Erin Purcell Estabrook, Wave Life Sciences, Cambridge, MA, USA.
Chikdu Shivalila, Wave Life Sciences, Cambridge, MA, USA.
Maeve Tischbein, Wave Life Sciences, Cambridge, MA, USA.
Tomomi Kawamoto, Wave Life Sciences, Cambridge, MA, USA.
Carlo Rinaldi, Department of Paediatrics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK; MDUK Oxford Neuromuscular Centre, University of Oxford, Oxford OX2 9DU, UK.
Joana Rajão-Saraiva, Department of Physiology, Anatomy and Genetics, University of Oxford, South Parks Road, Oxford OX1 3PT, UK.
Snehlata Tripathi, Wave Life Sciences, Cambridge, MA, USA.
Hailin Yang, Wave Life Sciences, Cambridge, MA, USA.
Yuan Yin, Wave Life Sciences, Cambridge, MA, USA.
Xiansi Zhao, Wave Life Sciences, Cambridge, MA, USA.
Cong Zhou, Wave Life Sciences, Cambridge, MA, USA.
Jason Zhang, Wave Life Sciences, Cambridge, MA, USA.
Luciano Apponi, Wave Life Sciences, Cambridge, MA, USA.
Matthew J A Wood, Department of Paediatrics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK; MDUK Oxford Neuromuscular Centre, University of Oxford, Oxford OX2 9DU, UK.
Chandra Vargeese, Wave Life Sciences, Cambridge, MA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wave Life Sciences. Funding for open access charge: Wave Life Sciences.
Conflict of interest statement. P.K., L.A., G.B., A.B., M.S., R.A., D.C.D.B., M.B., O.C., J.D., J.D.S., A.D., J.G., A.H., N.I., T.K., N.K., J.K., F.L., K.L., G.L., S.M., I.H.P., E.P.E., C.S., M.T., H.Y., Y.Y., J.Z., A.Z., C.Z. and C.V. were employees of Wave Life Sciences during completion of this work. M.J.A.W.’s laboratory received funding support from Wave Life Sciences, and M.J.A.W. is a cofounder of PepGen, a company developing peptide delivery of oligonucleotide therapeutics for treatment of neuromuscular disease. P.K., M.S., N.K., R.A., D.C.D.B., N.I., J.K., G.B., J.D., A.D., J.G., A.H., K.L., G.L., S.M., I.H.P., C.S., S.T., H.Y., J.Z., L.A. and C.V. are inventors on patents based on this work.
This paper is linked to: doi:10.1093/nar/gkac037.
REFERENCES
- 1. Hoffman E.P., McNally E.M.. Exon-skipping therapy: a roadblock, detour, or bump in the road?. Sci. Transl. Med. 2014; 6:230fs214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Verhaart I.E.C., Aartsma-Rus A.. Therapeutic developments for duchenne muscular dystrophy. Nat. Rev. Neurol. 2019; 15:373–386. [DOI] [PubMed] [Google Scholar]
- 3. Mah J.K. Current and emerging treatment strategies for duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 2016; 12:1795–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birnkrant D.J., Bushby K., Bann C.M., Apkon S.D., Blackwell A., Brumbaugh D., Case L.E., Clemens P.R., Hadjiyannakis S., Pandya S.et al.. Diagnosis and management of duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet. Neurology. 2018; 17:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blake D.J., Weir A., Newey S.E., Davies K.E.. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002; 82:291–329. [DOI] [PubMed] [Google Scholar]
- 6. Boland B.J., Silbert P.L., Groover R.V., Wollan P.C., Silverstein M.D.. Skeletal, cardiac, and smooth muscle failure in duchenne muscular dystrophy. Pediatr. Neurol. 1996; 14:7–12. [DOI] [PubMed] [Google Scholar]
- 7. Phan H., Cripe L., Eagle M., Muntoni F., Niks E., Straub V., Servais L., Gidaro T., Boespflug-Tanguy O., Goemans G.et al.. MDA Clinical & Scientific Conference. 2020; [Google Scholar]
- 8. Wu B., Lu P., Benrashid E., Malik S., Ashar J., Doran T.J., Lu Q.L.. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010; 17:132–140. [DOI] [PubMed] [Google Scholar]
- 9. Tanganyika-de Winter C.L., Heemskerk H., Karnaoukh T.G., van Putten M., de Kimpe S.J., van Deutekom J., Aartsma-Rus A.. Long-term exon skipping studies with 2′-O-Methyl phosphorothioate antisense oligonucleotides in dystrophic mouse models. Mol. Ther. Nucleic Acids. 2012; 1:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Putten M., Hulsker M., Young C., Nadarajah V.D., Heemskerk H., van der Weerd L., Hoen P.A., van Ommen G.J., Aartsma-Rus A.M.. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. 2013; 27:2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu B., Xiao B., Cloer C., Shaban M., Sali A., Lu P., Li J., Nagaraju K., Xiao X., Lu Q.L.. One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol. Ther. 2011; 19:576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goyenvalle A., Griffith G., Babbs A., El Andaloussi S., Ezzat K., Avril A., Dugovic B., Chaussenot R., Ferry A., Voit T.et al.. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015; 21:270–275. [DOI] [PubMed] [Google Scholar]
- 13. Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena, Standley S.M., Lu G., Apponi L.H.et al.. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:845–851. [DOI] [PubMed] [Google Scholar]
- 14. Byrne M., Vathipadiekal V., Apponi L., Iwamoto N., Kandasamy P., Longo K., Liu F., Looby R., Norwood L., Shah A.et al.. Stereochemistry enhances potency, efficacy, and durability of malat1 antisense oligonucleotides in vitro and in vivo in multiple species. Transl. Vis. Sci. Technol. 2021; 10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y., Dodart J.C., Tran H., Berkovitch S., Braun M., Byrne M., Durbin A.F., Hu X.S., Iwamoto N., Jang H.G.et al.. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 2021; 12:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lomzov A.A., Golyshev V.M., Dyudeevaa E.S., Kuprushkin M.S., Pyshnyi D.V.. Book of abstracts. Albany 2019: the 20th conversation: structure and hybridization properties of phosphorylguanidine oligonucleotides. J. Biomol. Struct. Dyn. 2019; 37:83. [Google Scholar]
- 17. Golyshev V.M., Pyshnyi D.V., Lomzov A.A.. Effects of phosphoryl guanidine modification of phosphate residues on the structure and hybridization of oligodeoxyribonucleotides. J. Phys. Chem. B. 2021; 125:2841–2855. [DOI] [PubMed] [Google Scholar]
- 18. Lomzov A.A., Kupryushkin M.S., Shernyukov A.V., Nekrasov M.D., Dovydenko I.S., Stetsenko D.A., Pyshnyi D.V.. Data for isolation and properties analysis of diastereomers of a mono-substituted phosphoryl guanidine trideoxyribonucleotide. Data Brief. 2019; 25:104148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dyudeeva E.S., Kupryushkin M.S., Lomzov A.A., Pyshnaya b.I.A., Pyshnyi D.V.. Physicochemical properties of the phosphoryl guanidine oligodeoxyribonucleotide analogs. Russ. J. Bioorg. Chem. 2019; 45:709–718. [Google Scholar]
- 20. Zhukov S.A., Pyshnyi D.V., Kupryushkin M.S.. Synthesis of novel representatives of phosphoryl guanidine oligonucleotides. Russ. J. Bioorg. Chem. 2021; 47:380–389. [Google Scholar]
- 21. Bazhenov M., Shernyukov A., Kupryushkin M., Pyshnyi D.. Study of the staudinger reaction and reveal of key factors affecting the efficacy of automatic synthesis of phosphoryl guanidinic oligonucleotide analogs. Russ. J. Bioorg. Chem. 2019; 45:699–708. [Google Scholar]
- 22. Kupryushkin M.S., Pyshnyi D.V., Stetsenko D.A.. Phosphoryl guanidines: a new type of nucleic acid analogues. Acta Naturae. 2014; 6:116–118. [PMC free article] [PubMed] [Google Scholar]
- 23. Maier M.A., Guzaev A.P., Manoharan M.. Synthesis of chimeric oligonucleotides containing phosphodiester, phosphorothioate, and phosphoramidate linkages. Org. Lett. 2000; 2:1819–1822. [DOI] [PubMed] [Google Scholar]
- 24. Lennox K.A., Sabel J.L., Johnson M.J., Moreira B.G., Fletcher C.A., Rose S.D., Behlke M.A., Laikhter A.L., Walder J.A., Dagle J.M.. Characterization of modified antisense oligonucleotides in xenopus laevis embryos. Oligonucleotides. 2006; 16:26–42. [DOI] [PubMed] [Google Scholar]
- 25. Skvortsova Y.V., Salina E.G., Burakova E.A., Bychenko O.S., Stetsenko D.A., Azhikina T.L.. A new antisense phosphoryl guanidine Oligo-2′-O-methylribonucleotide penetrates into intracellular mycobacteria and suppresses target gene expression. Front. Pharmacol. 2019; 10:1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dickson G., Hill V., Graham I.R.. Screening for antisense modulation of dystrophin pre-mRNA splicing. Neuromusc. Disord.: NMD. 2002; 12(Suppl. 1):S67–S70. [DOI] [PubMed] [Google Scholar]
- 27. Sheldrick G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. A, Found. Adv. 2015; 71:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheldrick G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C, Struct. Chem. 2015; 71:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morgan J.E., Beauchamp J.R., Pagel C.N., Peckham M., Ataliotis P., Jat P.S., Noble M.D., Farmer K., Partridge T.A.. Myogenic cell lines derived from transgenic mice carrying a thermolabile t antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev. Biol. 1994; 162:486–498. [DOI] [PubMed] [Google Scholar]
- 30. Beekman C., Janson A.A., Baghat A., van Deutekom J.C., Datson N.A.. Use of capillary western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with becker and duchenne muscular dystrophy. PLoS One. 2018; 13:e0195850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Deutekom J.C., Janson A.A., Ginjaar I.B., Frankhuizen W.S., Aartsma-Rus A., Bremmer-Bout M., den Dunnen J.T., Koop K., van der Kooi A.J., Goemans N.M.et al.. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007; 357:2677–2686. [DOI] [PubMed] [Google Scholar]
- 32. Goyenvalle A., Babbs A., Powell D., Kole R., Fletcher S., Wilton S.D., Davies K.E.. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol. Ther. 2010; 18:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roberts T.C., Coenen-Stass A.M., Betts C.A., Wood M.J.. Detection and quantification of extracellular microRNAs in murine biofluids. Biol. Proced. Online. 2014; 16:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dagle J.M., Littig J.L., Sutherland L.B., Weeks D.L.. Targeted elimination of zygotic messages in xenopus laevis embryos by modified oligonucleotides possessing terminal cationic linkages. Nucleic Acids Res. 2000; 28:2153–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jain M.L., Bruice P.Y., Szabó I.E., Bruice T.C.. Incorporation of positively charged linkages into DNA and RNA backbones: a novel strategy for antigene and antisense agents. Chem. Rev. 2012; 112:1284–1309. [DOI] [PubMed] [Google Scholar]
- 36. Chelobanov B.P., Burakova E.A., Prokhorova D.V., Fokina A.A., Stetsenko D.A.. New oligodeoxynucleotide derivatives containing N-(methanesulfonyl)-phosphoramidate (mesyl phosphoramidate) internucleotide group. Russ. J. Bioorg. Chem. 2017; 43:664–668. [Google Scholar]
- 37. Vlaho D., Fakhoury J.F., Damha M.J.. Structural studies and gene silencing activity of siRNAs containing cationic phosphoramidate linkages. Nucleic Acid Ther. 2018; 28:34–43. [DOI] [PubMed] [Google Scholar]
- 38. Burakova E.A., Derzhalova A.S., Chelobanov B.P., Fokina A.A., Stetsenko D.A.. New oligodeoxynucleotide derivatives containing N-(Sulfonyl)-phosphoramide groups. Russ. J. Bioorg. Chem. 2019; 45:662–668. [Google Scholar]
- 39. Derzhalova A., Markov O., Fokina A., Shiohama Y., Zatsepin T., Fujii M., Zenkova M., Stetsenko D.. Novel lipid-oligonucleotide conjugates containing long-chain sulfonyl phosphoramidate groups: synthesis and biological properties. Appl. Sci. 2021; 11:1174. [Google Scholar]
- 40. Hammond S.M., Sergeeva O.V., Melnikov P.A., Goli L., Stoodley J., Zatsepin T.S., Stetsenko D.A., Wood M.J.A.. Mesyl phosphoramidate oligonucleotides as potential splice-switching agents: impact of backbone structure on activity and intracellular localization. Nucleic Acid Ther. 2021; 31:190–200. [DOI] [PubMed] [Google Scholar]
- 41. Arechavala-Gomeza V., Graham I.R., Popplewell L.J., Adams A.M., Aartsma-Rus A., Kinali M., Morgan J.E., van Deutekom J.C., Wilton S.D., Dickson G.et al.. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum. Gene Ther. 2007; 18:798–810. [DOI] [PubMed] [Google Scholar]
- 42. Gonzalez-Barriga A., Nillessen B., Kranzen J., van Kessel I.D.G., Croes H.J.E., Aguilera B., de Visser P.C., Datson N.A., Mulders S.A.M., van Deutekom J.C.T.et al.. Intracellular distribution and nuclear activity of antisense oligonucleotides after unassisted uptake in myoblasts and differentiated myotubes in vitro. Nucleic Acid Ther. 2017; 27:144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stein C.A., Hansen J.B., Lai J., Wu S., Voskresenskiy A., Hog A., Worm J., Hedtjarn M., Souleimanian N., Miller P.et al.. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010; 38:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krieg A.M., Guga P., Stec W.. P-chirality-dependent immune activation by phosphorothioate CpG oligodeoxynucleotides. Oligonucleotides. 2003; 13:491–499. [DOI] [PubMed] [Google Scholar]
- 45. Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J.. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989; 244:1578–1580. [DOI] [PubMed] [Google Scholar]
- 46. Lomzov A.A., Kupryushkin M.S., Shernyukov A.V., Nekrasov M.D., Dovydenko I.S., Stetsenko D.A., Pyshnyi D.V.. Diastereomers of a mono-substituted phosphoryl guanidine trideoxyribonucleotide: isolation and properties. Biochem. Biophys. Res. Commun. 2019; 513:807–811. [DOI] [PubMed] [Google Scholar]
- 47. Han G., Lin C., Ning H., Gao X., Yin H.. Long-Term morpholino oligomers in hexose elicits long-lasting therapeutic improvements in mdx mice. Mol. Ther. Nucleic Acids. 2018; 12:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang L., Chen M., Xu M., Li J., Feng P., He R., Zhu Y., Li H., Lin J., Zhang C.. Ratio of creatine kinase to alanine aminotransferase as a biomarker of acute liver injury in dystrophinopathy. Dis. Markers. 2018; 2018:6484610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geary R.S., Norris D., Yu R., Bennett C.F.. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug. Deliv. Rev. 2015; 87:46–51. [DOI] [PubMed] [Google Scholar]
- 50. Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M., Davies K.E.. Utrophin-dystrophin-deficient mice as a model for duchenne muscular dystrophy. Cell. 1997; 90:717–727. [DOI] [PubMed] [Google Scholar]
- 51. Grady R.M., Teng H., Nichol M.C., Cunningham J.C., Wilkinson R.S., Sanes J.R.. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for duchenne muscular dystrophy. Cell. 1997; 90:729–738. [DOI] [PubMed] [Google Scholar]
- 52. Cacchiarelli D., Legnini I., Martone J., Cazzella V., D’Amico A., Bertini E., Bozzoni I.. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011; 3:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Freier S.M., Altmann K.H.. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997; 25:4429–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawasaki A.M., Casper M.D., Freier S.M., Lesnik E.A., Zounes M.C., Cummins L.L., Gonzalez C., Cook P.D.. Uniformly modified 2′-deoxy-2′-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J. Med. Chem. 1993; 36:831–841. [DOI] [PubMed] [Google Scholar]
- 55. Jirka S.M., Tanganyika-de Winter C.L., Boertje-van der Meulen J.W., van Putten M., Hiller M., Vermue R., de Visser P.C., Aartsma-Rus A.. Evaluation of 2′-Deoxy-2′-fluoro antisense oligonucleotides for exon skipping in duchenne muscular dystrophy. Mol. Ther. Nucleic Acids. 2015; 4:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen S., Le B.T., Chakravarthy M., Kosbar T.R., Veedu R.N.. Systematic evaluation of 2′-Fluoro modified chimeric antisense oligonucleotide-mediated exon skipping in vitro. Sci. Rep. 2019; 9:6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vila M.C., Klimek M.B., Novak J.S., Rayavarapu S., Uaesoontrachoon K., Boehler J.F., Fiorillo A.A., Hogarth M.W., Zhang A., Shaughnessy C.et al.. Elusive sources of variability of dystrophin rescue by exon skipping. Skeletal Muscle. 2015; 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aartsma-Rus A., Janson A.A., Kaman W.E., Bremmer-Bout M., van Ommen G.J., den Dunnen J.T., van Deutekom J.C.. Antisense-induced multiexon skipping for duchenne muscular dystrophy makes more sense. Am. J. Hum. Genet. 2004; 74:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McClorey G., Moulton H.M., Iversen P.L., Fletcher S., Wilton S.D.. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006; 13:1373–1381. [DOI] [PubMed] [Google Scholar]
- 60. Lu Q.L., Rabinowitz A., Chen Y.C., Yokota T., Yin H., Alter J., Jadoon A., Bou-Gharios G., Partridge T.. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Nat. Acad. Sci. U.S.A. 2005; 102:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alter J., Lou F., Rabinowitz A., Yin H., Rosenfeld J., Wilton S.D., Partridge T.A., Lu Q.L.. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006; 12:175–177. [DOI] [PubMed] [Google Scholar]
- 62. Dowdy S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017; 35:222–229. [DOI] [PubMed] [Google Scholar]
- 63. Nair A.B., Jacob S.. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016; 7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Flanigan K.M. Duchenne and becker muscular dystrophies. Neurol. Clin. 2014; 32:671–688. [DOI] [PubMed] [Google Scholar]
- 65. Wilton S.D., Veedu R.N., Fletcher S.. The emperor's new dystrophin: finding sense in the noise. Trends Mol. Med. 2015; 21:417–426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for figures Figures 2A–E, 3B, 4A–D, 5B–D, 6A–B, D–E, 7A–C, 8A–E, 9A–C, E, 10A–F and 11B are provided with the paper.
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the CCDC with the refcode 2113502.