

Abstract
Autophagy classically functions as a physiological process to degrade cytoplasmic components, protein aggregates, and/or organelles, as a mechanism for nutrient breakdown, and as a regulator of cellular architecture. Its biological functions include metabolic stress adaptation, stem cell differentiation, immunomodulation and diseases regulation, and so on. Current researches have proved that autophagy dysfunction may contribute to the pathogenesis of some myopathies through impairment of myofibres regeneration. Studies of autophagy inhibition also indicate the importance of autophagy in muscle regeneration, while activation of autophagy can restore muscle function in some myopathies. In this review, we aim to report the mechanisms of action of autophagy on muscle regeneration to provide relevant references for the treatment of regenerating defective myopathies by regulating autophagy. Results have shown that one key mechanism of autophagy regulating the muscle regeneration is to affect the differentiation fate of muscle stem cells (MuSCs), including quiescence maintenance, activation and differentiation. The roles of autophagy (organelle/protein degradation, energy facilitation, and/or other) vary at different myogenic stages of the repair process. When the muscle is in homeostasis, basal autophagy can maintain the quiescence state and stemness of MuSCs by renewing organelle and protein. After injury, the increased autophagy flux contributes to meet biological energy demand of MuSCs during activation and proliferation. By mitochondrial remodelling, autophagy during differentiation can promote the metabolic transformation and balance mitochondrial‐mediated apoptosis signals in myoblasts. Autophagy in mature myofibres is also essential for the degradation of necrotic myofibres, and may affect the dynamics of MuSCs by affecting the secretion spectrum of myofibres or the recruitment of supporting cells. Except for myogenic cells, autophagy also plays an important role in regulating the function of non‐myogenic cells in the muscle microenvironment, which is also essential for successful muscle recovery. Autophagy can regulate the immune microenvironment during muscle regeneration through the recruitment and polarization of macrophages, while autophagy in endothelial cells can regulate muscle regeneration in an angiogenic or angiogenesis‐independent manner. Drug or nutrition targeted autophagy has been preliminarily proved to restore muscle function in myopathies by promoting muscle regeneration, and further understanding the role and mechanism of autophagy in various cell types during muscle regeneration will enable more effective combinatorial therapeutic strategies.
Keywords: Autophagy, Myopathies, Skeletal muscle, Regeneration, MuSCs, Microenvironment
Introduction
Comprising about 30–40% of the whole‐body mass, skeletal muscle is the organ with the largest proportion in the body. 1 However, various genetic defects, substantial tissue damage, or exogenous signal changes all lead to muscle dysfunction and myopathies. 2 Myopathies are gradually becoming a serious public health problem in many countries, and this phenomenon will intensify with the ageing process. 3 , 4 These myopathies with high prevalence not only compromise quality of life and diminish functional capacity but also greatly increase social and economic burden. So the mechanism of myopathies has always been an intensely investigated area with the hope that an increased understanding will lead to therapies to restore skeletal muscle structure and function. Current studies suggest that regenerative dysfunction may contribute to the pathogenesis of myopathies. In this process, muscle stem cells (MuSCs) have attracted the most attention. In addition, successful muscle regeneration also requires the self‐degradation of the damaged myofibres and the regulation of other supporting cells in the microenvironment. Failure of muscle to regenerate properly can have a severe impact on the organism, which is particularly evident in some chronic injury myopathies, such as Duchenne muscular dystrophy (DMD). 5 Therefore, the process of regeneration following muscle injury has been an intensely investigated area to restore muscle structure and function when the regenerative process is impaired. But it is difficult to regulate this process due to the complexity of the dynamic changes of cell composition during muscle regeneration.
Autophagy refers to a series of tightly regulated catabolic processes, all of which transport cytoplasmic components to the lysosome for degradation. As a highly inducible dynamic metabolic process under the effect of the environment, hormones and other factors, autophagy can drive rapid cellular responses under conditions such as metabolic stress. 6 Recent researches have demonstrated that autophagy is activated and plays a crucial role in muscle regeneration. Moreover, drug activated autophagy has been proved to improve some myopathies by promoting muscle regeneration, indicating that autophagy has great clinical potential in the treatment of myopathies. 5 However, the detailed function of autophagy in muscle regeneration has not been summarized. Autophagy can promote myogenic differentiation and accelerate muscle recovery by removing excess/damaged organelles/protein or by regulating intracellular oxidative stress signals. 7 , 8 Autophagy can also regulate non‐myogenic cells in muscle microenvironment, such as immune cells and endothelial cells, thus affecting the regeneration of muscle. 9 , 10 These results highlight an emerging area of autophagy in the muscle regeneration. The studies of the molecular mechanism of autophagy regulation may serve as a target for future therapies to strengthen muscle function recovery in myopathies. The areas of focus for this review are the role of autophagy in muscle regeneration, specifically in the myogenic cells and non‐myogenic cells in the microenvironment.
Muscle regeneration in myopathies
Myopathies comprise a series of structurally abnormal and metabolically disturbed muscle diseases that are characterized by muscle weakness and motor dysfunction. As myopathies can occur owing to various causes, so they are challenging to treat. Among these myopathies, traumatic muscle injuries, hereditary muscular dysfunction and muscular atrophy induced by multiple factors (ageing, ischaemia, cancer cachexia, and immobilization) are being emphasized for their high incidence rate. 11 , 12 , 13 Besides, although toxic myopathies (induced by snake venom, barium chloride, chloroquine, etc.) are rare in humans, their serious injuries involving the whole body cannot be ignored. 4 , 14 In different types of myopathies, one thing in common is the process of muscle regeneration and repair caused by the abnormality of myofibres structure. For instance, traumatic and toxic muscle injuries usually lead to necrosis of myofibres, which requires the filling of regenerated myofibres. In the muscle atrophy caused by ageing, the age‐related reduction in muscle repair efficiency also contributes to the development of sarcopenia. 12 While in DMD patients, the lack of dystrophin protein results in the damage of myofibres during contraction. The chronic injury will cause inflammation, which in turn inhibits myofibres regeneration. 13 Abnormal muscle regeneration has been found in multiple kinds of myopathies, which may involve complex reasons, including stem cell aging, inflammation, fibrosis, and so on. 12 , 13 , 15 , 16 Regardless of the cause, enhancing the capacity of regeneration has proved as a potential therapeutic strategy. 17 Therefore, the process of muscle regeneration has been an intensely investigated area with the hope that an increased understanding will lead to therapies to restore optimal skeletal muscle structure and function. As an important biological process to maintain cell homeostasis under stress, autophagy is activated in the process of muscle regeneration and has been proved to be essential for successful myofibres recovery. A further understanding of the role of autophagy in muscle regeneration is required to define therapies in various myopathies.
Link between autophagy and muscle regeneration
Canonical degradative autophagy usually results from various stress conditions, such as starvation, hypoxia, oxidative stress, protein aggregation, endoplasmic reticulum (ER) stress and others. The common target of these stress conditions is the Unc‐51‐like kinase 1 (ULK1) complex, which then triggers nucleation of the phagophore by phosphorylating components of the class III PI3K (PI3KC3) complex I. The PI3KC3 complex I phosphorylates the lipid head group of phosphatidylinositol to generate phosphatidylinositol‐3‐phosphate (PI3P) in the rough endoplasmic reticulum, which is an essential early event in autophagy initiation. This nucleation process also requires ATG9A vesicles as seeds for membrane formation. 18 ATG8 family members are responsible for the subsequent phagophore expansion process. The nascent LC3 (mammalian homolog of yeast ATG8) is processed at its C‐terminal by the cysteine protease ATG4 to form cytoplasmic LC3‐I. LC3‐I exposes a glycine residue that binds to phosphatidylethanolamine (PE) and then converts to LC3‐II (the characteristic signature of autophagic membranes). ATG8s not only further attract components of the autophagic machinery that contain an LC3‐interacting region (LIR) but also are necessary for sealing the phagophore membrane. 19 ATG9A is also involved in lipid scrambling and maintaining plasma membrane integrity during autophagosome formation to promote membrane expansion. 18 , 20 Following expansion and sealing of the phagophore, the autophagosome undergoes maturation and fusion, which involves gradual clearance of ATGs from the nascent autophagosome outer membrane and fusion with lysosomes. This fusion process requires the concerted actions of multiple regulators of membrane dynamics, including SNAREs (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor), tethering proteins and RAB GTPases, and also transport of autophagosomes and late endosomes/lysosomes towards each other. 21 Finally, acidic hydrolases in lysosomes degrade the autophagic cargo, and salvaged nutrients are released back to the cytoplasm, where they are used again by cells and perform a variety of biological functions.
Autophagy is activated in response to biological stress and also plays a key role in the organ regeneration and the maintenance of cellular homeostasis, 24 which is extremely important for metabolically active tissues, such as skeletal muscle. First evidence linking autophagy to muscle regeneration comes from the study of response to strenuous exercise in myofibres. In 1984, Salminen et al. first found that many autophagic vacuoles were produced near necrotic myofibres and in regenerated myofibres after intense physical exercise. Therefore, a hypothesis has been proposed that the increased autophagic activity after muscle injury could be closely related to myofibres regeneration. 22 In subsequent decades, autophagy has been proven as an essential cellular process for timely muscle regeneration. 8 Among them, the most direct evidence is that drug inhibition of autophagy leads to incompletely skeletal muscle recovery from injury. Another evidence of the necessity of autophagy is the knockout mouse studies of autophagy proteins (Figure 1). By disrupting muscle regeneration, autophagy dysfunction is also responsible for the pathogenesis of some myopathies. The observed autophagy defects vary from disease to disease but have been proven to involve all steps of the autophagic cascade (from induction to lysosomal cargo degradation) and impair both non‐selective and selective autophagy. 14 The benefits of any autophagy‐modifying therapies—depend on the specific mechanism of autophagic dysfunction. As a result, optimal treatment strategies require further understanding of the mechanisms of autophagy in muscle regeneration.
Figure 1.
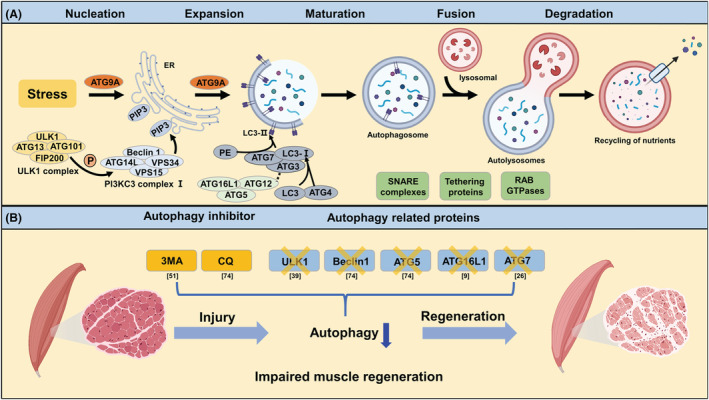
Link between autophagy and muscle regeneration. (A) Biological processes and related molecular mechanisms of classical degradative autophagy. (B) Inhibition of autophagy impairs skeletal muscle regeneration in animal models of muscle injury. As an inhibitor of PI3KC3, 3‐Methyladenine (3‐MA) can specifically block autophagosome formation. Treatment of myotoxic injury mice with 3‐MA resulted in decreased recovery of muscle strength and mitochondrial enzyme activity. Chloroquine (CQ) can inhibit the fusion of autophagosome and lysosome, thus blocking the autophagic flux. Similar to the results of 3‐MA, blocking the late process of autophagy with CQ also reduced the rate of regeneration with accumulation of sarcomere and nuclear debris. Skeletal muscle‐specific knockout of Ulk1, a kinase critical for autophagy initiation, attenuates the recovery of mitochondrial network and muscle strength in mice after muscle injury. In addition, genetic deletions of Atg5, Becn1, Atg16l, and Atg7 all lead to autophagy attenuation after muscle injury and impair muscle regeneration as well.
Autophagy in myogenic cells
Quiescence and function of muscle stem cells
Adult skeletal muscle almost has no turnover under normal conditions, so MuSCs are in quiescence (a G0 reversible arrested state). 23 Upon injury or be exposed to other stimuli, quiescent MuSCs can respond in time and then be activated to re‐enter the cell cycle to participate in the regeneration process. 24 Due to the characteristics of muscle regeneration, the state of MuSCs quiescence appears to be a reasonable way to maintain a low activity stem cell population in the absence of regenerative demand, yet functionally prepare for the future regeneration demands at any moment. 25 Previous evidence suggests that the changes in niche with cell‐autonomous signals (induced by aging and other factors) can destroy the quiescent state of MuSCs and blunt the capacity of muscle regeneration, thereby resulting in myopathies such as age‐related sarcopenia. 25 , 26 , 27 In these processes, the imbalance of autophagy may play a key role. 26
Evidence is accumulating that autophagy is essential for the maintenance of the quiescence state and normal function of stem cells, 28 and constitutive autophagic activity is also present in quiescent MuSCs. 26 In senescent MuSCs, the phosphorylation of AMPK and its downstream target P27Kip1 decreased, and the resulting stress of suppressed autophagy makes MuSCs more prone to apoptosis. 29 Further study found that quiescent MuSCs actively maintain organelle and protein turnover by autophagy. 26 During aging, the ability of MuSCs to clear damaged cellular materials (specially mitochondria) through autophagy declines, which will result in higher levels of reactive oxygen species (ROS). 26 Subsequently, p16INK4a (also called Cdkn2a, induced by ROS) can change quiescent MuSCs into an irreversible pre‐senescence state by inhibiting the expression of retinoblastoma (Rb) protein/E2F‐regulated genes (related to cell cycle). 26 , 30 Reactivating autophagy, either by genetically introducing Atg7 or via treatment with the autophagy activator rapamycin, can rescue the capacity of aged MuSCs to regeneration. 26 In addition to the induction of autophagosomes, the maintenance of MuSCs quiescence state also depends on eventual autophagosome clearance, and the hypothalamic–pituitary‐gonad (HPG) is involved in this process. With aging, the HPG axis activity gradually decreases, which will not affect the formation of autophagosome, but will lead to abnormal clearance of autophagosomes. 31 Mechanistically, androgen receptor was recruited to androgen response elements in the transcription of the transcription factor EB (Tfeb) promoter in MuSCs, thereby activating TFEB and its target genes. 31 As a master gene for lysosomal biogenesis, TFEB is required for autolysosome biosynthesis and autophagosomes degradation. 32 Therefore, the excessive accumulation of autophagosomes induced by aging also leads to the accumulation of ROS, which allows MuSCs to acquire a state of quiescence destruction analogous to that of senescence. Together, these findings indicate that autophagy is a key factor for the regeneration ability of muscle stem cells by maintaining quiescence and preventing senescence, both through induction and clearance of autophagosomes. However, whether there is a destruction of the quiescence state of MuSCs in other myopathies caused by genetic defects is still largely unknown.
Activation and proliferation of muscle stem cells
After muscle injury, MuSCs are activated to escape quiescence stage and start to proliferate; some daughter MuSCs continue to differentiate, whereas others return into quiescence to complete the self‐renewal of MuSCs. 33 With the changes of the myogenic transcription factors expression, differentiated MuSCs then form multinucleated myotubes and proceed through a stage of regeneration that is dominated by terminal differentiation and growth.
MuSCs activating out of the quiescence stage to generate proliferating progeny that will differentiate or self‐renew encounter different bioenergetic requirements at each juncture. 34 Previous researches have shown that autophagy was induced during MuSCs activation after muscle injury. Autophagy flux was increased upon SC activation from quiescence, both in vivo and in vitro, and was even increased to a higher level at the proliferation stage. 5 , 35 , 36 In Atg16l1 mutant mouse, where autophagy flux is reduced but still present, muscle regeneration was impaired due to a lack of MuSCs activation after damage. Atg16L1‐null mice had a greater proportion of MuSCs (Pax7+/MyoD−) and lower proportion of muscle precursors (Pax7+/MyoD+) or committed myoblasts (Pax7−/MyoD+) than wild type. 9 Compared with quiescent MuSCs in G0 (QSCs), activated MuSCs (ASCs) and proliferating MuSCs contained higher ATP content and mitochondrial activity. 37 Thus, increased autophagy flux may help support the cellular energy demands generated during MuSCs activation and proliferation. Inhibition of autophagy can significantly reduce the number of ASCs re‐entering the cell cycle, ATP production and mitochondrial activity during MuSCs activation, which can be partially remedied by supplementing exogenous metabolite sodium pyruvate. 35 Further studies found that Sirtuin1 (SIRT1) may mediate ATG7 deacetylation and AMPK phosphorylation during MuSCs activation, thereby activating autophagy and MuSCs. 35 These data suggest that SIRT1 may provide nutrients needs that meet biological energy demand by activating autophagy during this key transition from quiescence to activation. 35
Differentiation and fusion of myoblasts
Previous studies have reported that autophagic signals (e.g. decreased SQSTM1 and increased ATG7, BECN1, ULK1, LC3B levels) are further activated during myoblast differentiation, both in vivo and vitro. 7 , 35 , 38 , 39 During the differentiation of C2C12 myoblasts in vitro, Jon et al. found that although the expression of ATG5‐ATG12 complex gradually increased during myogenic differentiation, the autophagy flux shown by LC3 was activated only in the early stage of differentiation (first day of differentiation). 40 Therefore, they believe that autophagy is a transient phenomenon, which is robustly up‐regulated only during the early steps of differentiation. However, more evidence shows that autophagy is up‐regulated in the whole differentiation cycle of myoblasts in vitro. 7 , 38 , 39 In addition, muscle injury studies in LC3‐GFP mice also confirmed that autophagy was continuously activated during MuSCs activation and differentiation in vivo and showed a stronger autophagy level in the differentiated regenerated myofibres (the centre contains nucleus), which indicates that autophagy is also up‐regulated in the late stage of differentiation. 35 In the process of autophagy activation, myoblasts are often accompanied by the transformation of metabolic mode (glycolysis to oxidative phosphorylation) and the increase of the number of mitochondria. 41 With the deepening of research, the need for autophagy during myogenic differentiation has been well documented. Disruption of autophagy, whether at the initiation, cargo trafficking, or lysosomal fusion steps, inhibits myogenic differentiation and impairs strength recovery after muscle injury. For example, inhibition the formation of autophagosomes by treatment with 3‐MA or knockdown of Atg5 and Atg7 impairs myogenesis, accompanied by mitochondrial dysfunction. 7 , 40 Similar effects were seen when myoblasts were treated with Baf‐A1, an autophagy inhibitor that can interfere the fusion of autophagosome and lysosome. 40 Together, these findings show that autophagy is necessary for myogenic differentiation, and the regulatory role of autophagy in myoblast differentiation and fusion is summarized as follows.
Mitochondrial remodelling in differentiation and fusion
Myoblasts are primarily dependent on glycolysis to meet their quiescent metabolic needs, and therefore have a more sparse mitochondrial population. In contrast to myoblasts, the mature myotube is a metabolically active cell type that relies heavily on oxidative phosphorylation (OXPHOS). 42 To meet this high energetic demand, myotubes contain a large number of mitochondria arranged in a complex network. 43 Therefore, differentiation of primitive myoblasts into mature myotubes requires a metabolic transformation to support the increased energetic demand of skeletal muscle contractions. Autophagy is crucial for this shift in energy metabolism patterns. 44 Mitochondrial renewal during myogenic differentiation has been shown to be a necessary process, and the onset of regeneration of muscle is always accompanied by a marked activation of mitochondrial biogenesis. 45 These events lead to an increase in the amount of mitochondrial protein and the copy number of mitochondrial DNA. Current studies have found that instead of simply producing mitochondria and adding oxidative phosphorylation components, myoblasts first clear the original mitochondria of glycolysis mode through mitophagy and then regenerate mitochondria of new metabolic mode. Mitophagy is a conserved mechanism to selectively remove unwanted or damaged mitochondria, which has been shown to play an important role in mitochondrial function and biogenesis. 46 During early myogenic differentiation, autophagy was further up‐regulated, which was coincident with Dynamin 1 like (DNM1L)‐mediated fragmentation of mitochondrial networks, a decline in cellular mitochondrial protein, and an increase in mitochondrial LC3B‐II. 40 The high degree of co‐localization between autophagosome and mitochondria will help to eliminate the old mitochondrial network (glycolysis). As differentiation progresses, autophagy flux decreases, accompanied by an up‐regulation of mitochondrial fusion protein OPA1 and the key factor of mitochondrial production PGC1α. 7 , 40 At this point, mitochondrial content increased significantly and dense mitochondrial networks (OXPHOS). When autophagy is inhibited, such as by interference with Atg7, the colocalization of mitochondria and autophagic puncta is reduced, which impairs mitochondrial network remodelling and inhibits myoblast differentiation. Consistent with the effects observed in myoblasts interfering with Atg7, a similar phenomenon occurs when BCL2 interacting protein 3 (BNIP3), the critical regulator of mitophagy, 46 is knocked out. 7 Myoblasts treated with Baf‐A1 also underwent mitochondrial fragmentation early during differentiation but failed to reconstitute their networks. An abundance of autophagosomes remained until 6 days differentiation, while instances of elongated mitochondria were rare when compared with control. 40 Collectively, these data demonstrate that the induction of autophagy and intact autophagic flux during early differentiation appears to be required for mitochondrial network reconstruction and myotube formation.
The details of the mechanism of mammalian mitophagy have been summarized in several excellent reviews, 46 , 47 , 48 , 49 , 50 and the mitophagy involved in muscle regeneration is also gradually being revealed. Mice with muscle injury treated with 3‐MA showed decreased mitochondrial enzyme activity, suggesting that autophagy plays an important role in regulating mitochondrial remodelling during muscle regeneration. 51 Along with mitochondrial remodelling, ULK1 is also activated during muscle regeneration. 39 , 51 , 52 At initial stages of PINK1/Parkin‐mediated mitophagy, ULK1 complex is recruited to depolarized mitochondria. 53 Given the key role of ULK1 in autophagosome induction and mitophagy, 6 , 54 Jarrod A. Call et al. explored in depth the role and the mechanism of ULK1‐mediated mitophagy in muscle regeneration. Using muscle‐specific Ulk1 knockout mice, they found that Ulk1 is required for mitochondrial network remodelling during muscle regeneration. 39 , 52 In mammalian skeletal muscle, another key factor regulating mitochondrial autophagy is protein kinase CSNK2/CK2. Phosphorylation of translocase of outer mitochondrial membrane 22 (TOMM22, the central component of the translocase of outer mitochondrial membrane) by CSNK2/CK2 promoted the introduction of PINK1 into mitochondrial intima, where it was degraded by presenilin associated rhomboid like (PARL). While, lack of phosphorylated TOMM22 fosters PINK1 to accumulate on the outer membrane of mitochondria and induces mitophagy. 55 Although mitophagy mediated mitochondrial remodelling is necessary in myoblast differentiation and muscle regeneration, the role of various types of mitophagy in muscle regeneration is unclear. In addition, the induction mechanism of myoblast mitophagy also needs to be further studied in order to target autophagy to regulate mitochondrial function recovery during muscle regeneration.
Stress and apoptosis signals in differentiation and fusion
Myoblast differentiation is accompanied by significant levels of cellular stress and apoptosis, including elevated levels of ROS 56 and apoptotic signals. Moderate stress signals and proteolytic caspase (CASP) activation are required by skeletal muscle development. ROS is also essential mediator of skeletal muscle, 57 repressing mitochondrial ROS production results in the failure of myoblast differentiation. 58 AS a decisive myogenic transcription factor in myoblast differentiation, MyoD is also an activator of caspase‐3 and apoptosis, which also suggests the role of apoptotic signals in myoblast differentiation. 59 However, overexpression of ROS or apoptotic signals in myoblasts due to physiological dysfunction can also cause failure of myoblast differentiation and induce cell death. 60 , 61 So it is important that the level of ROS and CASP activation are tightly regulated and are maintained at a ‘sub‐apoptotic’ threshold in myogenic differentiation stage. While in some myopathies, this balance of MuSCs is broken, which makes them more prone to apoptosis. 29 , 62 Recent researches have found that autophagy is involved in regulating related signalling during skeletal muscle differentiation, and contributes to cell specialization rather than cell death. 63 Autophagy‐deficiency during myoblast differentiation augments apoptotic signalling, ER‐stress responses, and cell death. Knockdown of Atg7 increased transient CASP3 activation, DNA fragmentation and the apoptosis rate of myoblasts. Similarly, in addition to inhibit autophagy flux, 3‐MA treatment during differentiation also increased myoblasts apoptosis. 63 Furthermore, mitophagy plays an important role in removing damaged mitochondria before mitochondria‐mediated apoptotic signalling can be induced, thus decreasing cell stress and death. 64 Previous researches have found that opening of the mitochondrial permeability transition pore (mPTP) and loss of mitochondrial membrane integrity can result in CASP‐dependant and ‐independent apoptotic signalling via factors such as CYCS (cytochrome c) and AIFM1/AIF (apoptosis‐inducing factor, mitochondrion‐associated 1). 65 , 66 While cytosolic CYCS and AIFM1 levels were significantly greater in shAtg7 myoblasts during differentiation. Furthermore, differentiating shAtg7 myoblasts displayed CASP9 activation during differentiation. 7 The aforementioned results indicate that autophagy regulates mitochondria‐mediated apoptotic signalling during myoblast differentiation and protects differentiating myoblasts from apoptotic cell death.
Degradation and regeneration of myofibres
Skeletal muscle is mainly composed of myofibres, and single nucleus RNA‐seq (snRNA‐seq) have revealed that 68% of nuclei in adult skeletal muscle originates from multinuclear myofibres. 67 As the principle component of the MuSCs niche, myofibres are in direct contact with MuSCs, so they play an important role in the function of MuSCs. 68 Because skeletal muscle injury is mainly caused by mechanical movement or trauma, which will lead to the death of myofibres, 69 so the degradation of injured myofibres is the primary problem to be solved in the process of skeletal muscle regeneration. Decades of pathological observations have reached a consensus that regeneration of skeletal muscle usually starts with myofibres degeneration, a step that requires the initiation of myofibre necrosis. 70 Extracellular matrix remnants from degraded skeletal myofibres, also called “ghost fibres”, govern MuSCs divisions and migration during muscle regeneration. 71 Furthermore, the degraded myofibres not only provide space for the regenerated myofibres but also release factors into the MuSCs microenvironment. Damaged myofibres can induce MuSCs to enter the cell cycle by secretion of damaged‐myofibre‐derived factors (DMDFs). 72 By releasing Tenascin‐C (TNC), which was found to critical for MuSCs proliferation, necroptotic myofibres can also facilitate muscle regeneration. 70 Recent researches show that autophagy can expedite tissue regeneration by enhancing waste clearance after tissue injury. 73 Autophagy was activated early in the regenerative stage after muscle injury, while blocking autophagy flux by using chloroquine or Atg5/Becn1 knockdown slows the regeneration rate with accumulation of sarcomere and nuclear debris. 74 After muscle injury with CTX (cardiotoxin), attenuated autophagy flux makes myofibres more susceptible to infiltration by circulating immunoglobulins. These myofibres internalise dystrophin and nNOS (nitric oxide synthase one). Importantly, these myofibres have the ability to restore dystrophin and nNOS localisation without degeneration, this will delay the degradation of myofibres, thus slow skeletal muscle regeneration. 9 During muscle regeneration, Ulk1 deletion mediated autophagy abnormality in adult myofibres can also inhibit MuSCs proliferation. How does myofibres autophagy indirectly affect satellite cell dynamics? One possibility is that autophagy may be crucial for secretory factor expression and secretion in damaged myofibres. Another possibility is autophagy may contribute to the degeneration of damaged myofibres by autophagy‐induced cell death, which would be critical for setting the stage for satellite cell proliferation via timely recruitments of non‐muscle derived Sertoli cells, such as macrophages, endothelial cells, and FAPs (Fibroadipogenic progenitors). Therefore, regulation of myofibres autophagy may be a new therapeutically targeted in the future for improving muscle regeneration. However, these conjectures have not been rigorously tested, due to the lack of solid experimental data.
In conclusion, the importance of autophagy of muscle derived cells in the process of muscle regeneration is gradually being confirmed, which involves the whole differentiation cycle of MuSCs (Figure 2) and the degradation of myofibres. In addition to the myogenic cells, autophagy can also play a regulatory role by affecting the differentiation and function of non‐myogenic cells in the microenvironment of regeneration.
Figure 2.
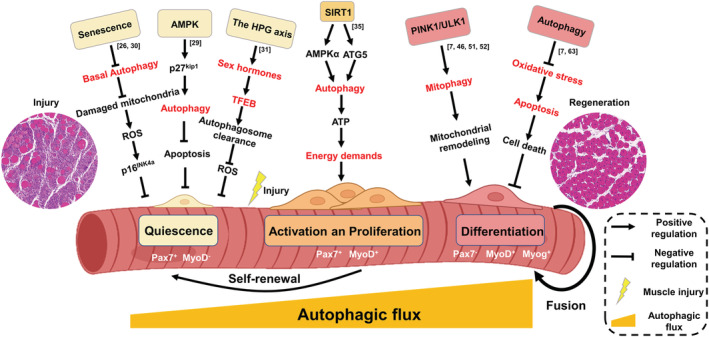
Function of autophagy in different stages of myogenic differentiation. (i) During aging, the ability of MuSCs to clear damaged cellular materials (specially mitochondria) through basal autophagy declines, which will result in higher levels of ROS. ROS can inhibit the expression of downstream target genes (related to cell cycle) by activating p16INK4a, so as to cause quiescent MuSCs to lose reversible quiescence by switching to an irreversible pre‐senescence state. (ii) The AMPK/p27Kip1 pathway may regulate the balance between autophagy and apoptosis in MuSCs, thus maintaining them in a steady‐state resting state. In senescent MuSCs, the phosphorylation of AMPK and its downstream target P27Kip1 decreased, and the resulting stress of suppressed autophagy makes MuSCs more prone to apoptosis. (iii) Sex steroid hormones controlled by the HPG axis transcriptionally induce the expression of Tfeb in MuSCs. TFEB systemically controls autophagosome clearance in MuSCs and reduces the accumulation of ROS. Accordingly, the HPG‐TFEB‐autophagosome pathway maintains stemness and quiescence of MuSCs. (iv) SIRT1 activates autophagy by mediating ATG7 deacetylation and AMPK phosphorylation. The increased level of autophagy promotes ATP production and mitochondrial activity to meet biological energy demand for MuSCs during this key transition from quiescence to activation. (v) In the process of differentiation, myoblasts first clear the original mitochondria of glycolysis mode through mitophagy, and then regenerate mitochondria of new metabolic mode (oxidative phosphorylation) to meet the increased energy demand of myotubes. PINK1 and ULK1 may be involved in the regulation of mitophagy. (vi) Autophagy regulates mitochondria‐mediated apoptotic signalling during myoblast differentiation, and protects differentiating myoblasts from apoptotic cell death. ROS, reactive oxygen species; AMPK, AMP‐activated protein kinase; HPG, hypothalamic–pituitary‐gonad; TFEB, transcription of the transcription factor EB; SIRT1, Sirtuin1; ATP, adenosine triphosphate; ATG, autophagy related; PINK1, PTEN induced putative kinase 1; ULK1, Unc‐51‐like kinase 1.
Autophagy in non‐myogenic cells
Skeletal muscle immune cells and regeneration
Alongside MuSCs, the innate immune system also plays an important role in skeletal muscle regeneration. 16 Following damage, muscle tissues exhibit traditional inflammatory. Macrophages play a dominate role in the inflammatory infiltrate of regenerating period. During the early proliferative stage of muscle regeneration, macrophages tend to be M1 phenotypes, and during the differentiation and growth phage of regeneration, macrophages tend to be M2 phenotypes. Inflammatory M1 macrophages first appear in the degenerative milieu. In this case, they secrete cytokines such as ADAMTS1, IFNγ, IGF‐1, IL‐6, IL‐1β, and TNFα to stimulate MuSCs proliferation while simultaneously restricting differentiation and fusion of MuSCs. 11 Following this phase, the anti‐inflammatory type, M2 macrophages become the dominant macrophage phenotype in the recovery tissue. Several compelling investigations have established that IL‐10 75 and fatty acid metabolism reprogram 76 may play a central role in regulating the switch of muscle macrophages from a M1 to M2 phenotype in injured muscle in vivo. If this switch is delayed and the M1 phenotype is prolonged, inflammatory cytokines could be produced continuously and myogenesis is impaired, indicating that the importance of this precisely timed phenotypic transformation. 77 Once M2 macrophages are present, they mark the beginning of the regenerative phase and support angiogenesis by initially secreting high levels of vascular endothelial growth factor (VEGF), which helps subsequent muscle recovery. 77 In addition, M2 macrophages can also directly foster MuSCs proliferation and myogenic differentiation through secretory factor IGF‐1, TNFα, TGFβ and growth differentiation factor 3 (GDF3). 11 Recent studies have shown that autophagy is extensively involved in monocyte differentiation and macrophage polarization. Following the activation by inflammatory signals, monocytes differentiate into macrophages or dendritic cells by means of autophagy. 78 Macrophagic differentiation and acquisition of phagocytic functions also require both Ulk1 and Atg7 dependent autophagy. 79 In addition to monocytes differentiate into macrophages, autophagy can also influence the polarization of the macrophages. Macrophage‐specific Atg7 knockout mice showed a change in the proportion to pro‐inflammatory M1 macrophages 80 ; thus, this may also influence the immune microenvironment during skeletal muscle regeneration. Besides the macrophages, injured muscle is also infiltrated by a specialized population of regulatory T (Treg) cells, which control both the inflammatory response, by promoting the M1‐to‐M2 switch, and the activation of satellite cells. 81 Although autophagy has been shown to enforce functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis, 82 direct evidence for its role in muscle regeneration has not been reported.
Vascular endothelial cells and regeneration
Endothelial cells (ECs) cover the inner wall of blood vessels and act as a gatekeeper of tissue regeneration by adapting oxygen and nutrient delivery to the metabolic needs of tissues through angiogenesis. Vascular homeostasis is largely dependent on proper behaviour of ECs, and therefore, not surprisingly that change of main EC's biological function caused by pathological insults is associated with a variety of diseases. EC‐mediated angiogenesis occurs in many physiological processes such as organ development, tissue regeneration and tumour growth. While except for angiogenesis‐dependent manner, ECs also have the ability to coordinate self‐renewal and differentiation of stem cells by providing membrane binding and secreted angiocrine factors, which is essential for tissue homeostasis and self‐renewal. 83 Interestingly, emerging evidence is unravelling a critical function of autophagy in ECs. 84 Autophagy supports angiogenesis during embryogenesis, while misregulations in autophagic pathways can led to angiodysplasia in the yolk sac. 85
Skeletal muscle is a highly vascularized tissue and is characterized by a remarkable capacity for regeneration. Therefore, ECs themselves and ECs‐mediated crosstalk with other cells are essential for skeletal muscle recovery. In various types of muscle injury, hypoxia inducible factor‐1α (HIF1α) is rapidly induced in the early stage due to disruption in the microvascular network. 10 , 86 Attenuated angiogenesis due to impaired normal function of ECs can increase hypoxic areas within the injured muscles and impair skeletal muscle regeneration. 87 Recent studies have suggested that autophagy may be a dynamic mechanism that plays a critical role in catabolic processes for cell survival under stressful conditions, 88 enabling ECs to adjust their bioenergetic and biosynthetic requirements in response to the angiogenesis following muscle injury. 10 , 77 Therefore, autophagy occur in ECs in response to hypoxia or serum depletion is necessary for angiogenic behaviour, 89 and activation of autophagy could improve ECs survival and function by alleviating oxidative stress. 90 Lack of endothelial the glucocorticoid receptor (GR) triggers autophagy flux, which will lead to activation of Wnt/β‐catenin signalling and promote angiogenesis. 91 In a muscle injury mouse model induced by hindlimb ischaemia, the autophagy pathway was induced under prolonged hypoxia in ECs, which could increase the angiogenic activities of ECs under tissue regeneration and consequently helps regeneration of surrounding tissues. 10 , 91 The transcription factor TFEB, demonstrated as a crucial regulator of lysosomal biogenesis and autophagy, 32 may mediate the link between endothelial autophagy and angiogenesis. Mechanistically, TFEB is up‐regulated in ischaemic skeletal muscle, subsequently activates AMPKα signalling and up‐regulates autophagy, which are essential for angiogenesis during skeletal muscle regeneration. 92 How autophagy exactly contributes to the maintenance of EC function remains to be further studied. Nevertheless, preventing mitochondrial dysfunction and metabolic stress‐induced endothelial injury by activating mitophagy (including PTEN‐induced kinase 1 and PARKIN) in response to metabolic stress in ECs may contribute to the intrinsic beneficial effect of EC‐associated autophagy. 93 Beyond the angiogenesis‐dependent way, ECs can support the muscle regenerative process through paracrine influence on MuSCs and additional cells in the local milieu, secreting factors such as VEGF, angiopoietin‐1 (Ang‐1) and lactate. 77 Autophagy in ECs appears to have a fundamental role in secretion, endothelial‐specific conditional deletion of Atg7 or Atg5 in mice exhibits impaired epinephrine‐stimulated von Willebrand factor (VWF) release. 94 Moreover, conditioned media from ECs that blocked autophagy could inhibit the proliferation of muscle cells, suggesting that autophagic stimulation in ECs has the capacity to affect the survival of adjacent cells by secretions. 10 Thus, autophagy may regulate ECs secretory profile thereby influencing the intercellular communication in the process of skeletal muscle regeneration, a research direction that needs to be experimentally validated in future studies.
Together, these studies confirmed that autophagy in ECs regulates skeletal muscle regeneration in an angiogenic and angiogenesis‐independent manner. Although most data suggest that endothelial autophagy is beneficial for skeletal muscle recovery, a connection that requires further experimental validation.
Regulation of regeneration‐defective myopathies by targeting autophagy
For cases in which the autophagic defects underlie myopathies, targeting autophagy in different cell populations constitutes an obvious therapeutic objective. Activation of autophagy by genetic, dietary, and pharmacological approaches has been proved to restore myofibre structure and ameliorate the muscle function in regeneration‐defective myopathies. The autophagy activator rapamycin can target defective autophagy in regeneration‐defective myopathies to rejuvenate muscle strength, including aging muscles, 26 muscular dystrophies, 95 and mitochondrial myopathies. 96 Exogenous supplement of the endogenous peptide apelin, induced by muscle contraction, can enhance the regeneration ability of aging muscle stem cells by triggering autophagy. 97 In spite of great potential, the limited specificity of these autophagy modulators often prevent the development for clinical use. For instance, the inhibition of mTORC1 using rapamycin not only activates autophagy but also affects cellular metabolism, growth, and survival. 98 Therefore, although the specificity is lower, the lower toxicity makes dietary regulation considered as a safer regulation measure targeting autophagy. Reactivating autophagy through starvation or a low‐protein diet can improve myofibres regeneration and function in collagen VI muscular dystrophies and DMD. 5 , 99 Plant extract polyphenol pterostilbene can promote muscle fibre remodelling in collagen VI deficient mice via enhancing autophagic flux. 100 Although increasing evidence has shown the enormous therapeutic potential of interventions autophagy, the final application still needs our further researches.
Prospects, challenges, and future directions
Through the analysis of systemic and tissue‐specific autophagy related genes knockout mice, great progress has been made in our understanding of the roles of autophagy in skeletal muscle regeneration after injury. Autophagy is a process necessary for efficient activity of MuSCs at different myogenic stages of the repair process, although its role (organelle/protein degradation, energy facilitation, and/or other) may vary at each stage (Figure 3). However, there are relatively few studies on the different stage of autophagy in MuSCs and myofibres, and the underlying mechanisms need to be further verified. When some of the proliferating MuSCs return to the quiescence stage to replenish the stem cell pool, the increased autophagy flux also returns to the resting level. How is the inactivation of autophagy orderly regulated in cells? In addition, asymmetric division of MuSCs is necessary to supplement the stem cell reservoir. Whether the difference in autophagy levels between ASCs and QSCs occurs during asymmetric division and determines the differentiation fate of cells is also a question worthy of consideration. Autophagy abnormalities are found in a variety of myopathies, and autophagy activation seems to improve muscle regeneration and most myopathies. However, stabilizing the Dishevelled‐2 by antagonizing selective autophagy can also promote muscle regeneration. 101 While in some myopathies, over activation of autophagy may also lead to muscle dysfunction, such as laminin α2 chain deficiency myopathies (also known as MDC1A). 102 Therefore, a deeper understanding of autophagy as a double‐edged sword in muscle regeneration and myopathies is required.
Figure 3.
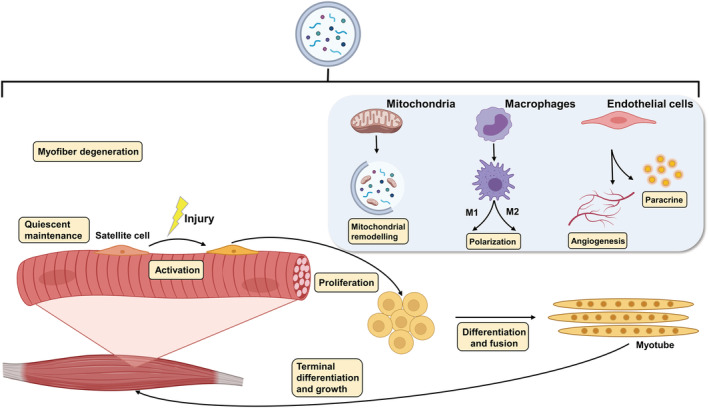
Autophagy is involved in several steps of skeletal muscle regeneration. (i) Degradation of necrotic myofibres and release of secretory factors; (ii) quiescent maintenance, activation, proliferation and differentiation of skeletal muscle stem cells; (iii) mitochondrial network remodelling mediated by mitophagy; (iv) differentiation and polarization of macrophages; (v) endothelial cell mediated angiogenesis and secretory factor release.
Single‐cell sequencing (scRNA‐seq) has analysed the temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. 103 Alongside the myogenic cells, other support cells are also involved in skeletal muscle development and regeneration. These non‐myogenic cells appear in skeletal muscle according a strictly programmed stage, and once the growth pattern of these support cells is changed, the normal function of skeletal muscles will be disrupted. 11 These studies emphasize the importance of supporting cell types and intercellular communication networks for normal skeletal muscle development and successful muscle regeneration. Autophagy has been shown to regulate the differentiation of a variety of stem cells and influence cell fate, while the deficiency of autophagy may lead to the failure of ordered differentiation. 104 In many previous studies on autophagy in muscle regeneration, muscle tissue is often deliberately confused with a single tissue containing only muscle derived cells. The dynamic cell composition during regeneration suggests that autophagy may be different in different cells. Whether autophagy can regulate the fate of supporting cells during skeletal muscle development and regeneration needs to be further investigated. Autophagy may regulate skeletal muscle metabolism by influencing the microenvironment of muscle tissue, rather than merely playing its role in myogenic cells, which is indeed a point worth thinking about.
A variety of animal injury models have been used to evaluate the factors affecting muscle regeneration, and the most commonly used acute injury models involve injection of myotoxins, 70 injection of barium chloride, 31 cryoinjury, 52 mechanical injury, 105 and ischaemia–reperfusion. 77 Despite similar initial necrosis and complete regeneration in different models, the cellular composition, inflammation, vascular reconstruction, and mitochondrial remodelling varied significantly among the groups over the time course of regeneration. 39 , 77 , 105 , 106 The convenient construction of muscle injury models helps us to study the molecular mechanism of muscle regeneration. However, these models are usually difficult to reflect the human pathology. Therefore, model mice corresponding to human myopathies should be developed to study the specific treatment, such as mdx mice, which have the same dystrophin mutation as human patients. In addition, due to the complexity of myopathies caused by autophagy deficiency, autophagy is rarely used in clinical treatment. Defects at every stage of the autophagy process (from induction to lysosomal cargo degradation) can affect the normal function of skeletal muscle. 14 However, different types of autophagy defects often cause different autophagy phenotypes. In some myopathies, failure of autophagy lysosomal degradation leads to impaired autophagy flux despite the high expression of ATG proteins. 14 Given the different autophagic defects that are observed in different skeletal myopathies, the pathogenesis of autophagy in various myopathy needs to be further understood to evaluate the benefits of autophagy‐modifying therapies on a disease‐by‐disease basis. In addition, clinically useful autophagy activators should be designed based on careful characterization of the autophagy defects associated with each specific myopathies.
In summary, an in‐depth understanding of the pathogenesis of autophagy in different myopathies, and the detection of the influence of autophagy on muscle microenvironment combined with scRNA‐seq will contribute to the clinical treatment of autophagy deficient myopathies.
Conflict of interest
The authors have declared that they have no conflict of interest.
Funding
This work was supported by the National Natural Science Foundation of China (U21A20249) and the Natural Science Foundation of Zhejiang Province (LZ22C170002).
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle. 107
Chen W., Chen Y., Liu Y., and Wang X. (2022) Autophagy in muscle regeneration: potential therapies for myopathies, Journal of Cachexia, Sarcopenia and Muscle, 13, 1673–1685, 10.1002/jcsm.13000
References
- 1. Janssen I, Heymsfield SB, Wang ZM, Ross R. Skeletal muscle mass and distribution in 468 men and women aged 18‐88 yr. J Appl Physiol 2000;89:81–88. [DOI] [PubMed] [Google Scholar]
- 2. Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004;84:209–238. [DOI] [PubMed] [Google Scholar]
- 3. Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, et al. Mortality, morbidity, and risk factors in China and its provinces, 1990‐2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019;394:1145–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qazi TH, Duda GN, Ort MJ, Perka C, Geissler S, Winkler T. Cell therapy to improve regeneration of skeletal muscle injuries. J Cachexia Sarcopenia Muscle 2019;10:501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fiacco E, Castagnetti F, Bianconi V, Madaro L, De Bardi M, Nazio F, et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ 2016;23:1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 2018;19:349–364. [DOI] [PubMed] [Google Scholar]
- 7. Baechler BL, Bloemberg D, Quadrilatero J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019;15:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Call JA, Nichenko AS. Autophagy: an essential but limited cellular process for timely skeletal muscle recovery from injury. Autophagy 2020;16:1344–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paolini A, Omairi S, Mitchell R, Vaughan D, Matsakas A, Vaiyapuri S, et al. Attenuation of autophagy impacts on muscle fibre development, starvation induced stress and fibre regeneration following acute injury. Sci Rep 2018;8:9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeong IH, Bae WY, Choi JS, Jeong JW. Ischemia induces autophagy of endothelial cells and stimulates angiogenic effects in a hindlimb ischemia mouse model. Cell Death Dis 2020;11:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wosczyna MN, Rando TA. A muscle stem cell support group: coordinated cellular responses in muscle regeneration. Dev Cell 2018;46:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Domingues‐Faria C, Vasson MP, Goncalves‐Mendes N, Boirie Y, Walrand S. Skeletal muscle regeneration and impact of aging and nutrition. Ageing Res Rev 2016;26:22–36. [DOI] [PubMed] [Google Scholar]
- 13. Verhaart IEC, Aartsma‐Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 2019;15:373–386. [DOI] [PubMed] [Google Scholar]
- 14. Margeta M. Autophagy defects in skeletal myopathies. Annu Rev Pathol 2020;15:261–285. [DOI] [PubMed] [Google Scholar]
- 15. Loell I, Lundberg IE. Can muscle regeneration fail in chronic inflammation: a weakness in inflammatory myopathies? J Intern Med 2011;269:243–257. [DOI] [PubMed] [Google Scholar]
- 16. Tidball JG. Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol 2017;17:165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saito Y, Chikenji TS, Matsumura T, Nakano M, Fujimiya M. Exercise enhances skeletal muscle regeneration by promoting senescence in fibro‐adipogenic progenitors. Nat Commun 2020;11:889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maeda S, Yamamoto H, Kinch LN, Garza CM, Takahashi S, Otomo C, et al. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat Struct Mol Biol 2020;27:1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He CW, Cui XF, Ma SJ, Xu Q, Ran YP, Chen WZ, et al. Membrane recruitment of Atg8 by Hfl1 facilitates turnover of vacuolar membrane proteins in yeast cells approaching stationary phase. BMC Biol 2021;19:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Claude‐Taupin A, Jia J, Bhujabal Z, Garfa‐Traore M, Kumar S, da Silva GPD, et al. ATG9A protects the plasma membrane from programmed and incidental permeabilization. Nat Cell Biol 2021;23:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol 2021;22:733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salminen A, Vihko V. Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch B Cell Pathol Incl Mol Pathol 1984;45:97–106. [DOI] [PubMed] [Google Scholar]
- 23. Schmidt M, Schuler SC, Huttner SS, von Eyss B, von Maltzahn J. Adult stem cells at work: regenerating skeletal muscle. Cell Mol Life Sci 2019;76:2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rayagiri SS, Ranaldi D, Raven A, Azhar NIFM, Lefebvre O, Zammit PS, et al. Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self‐renewal. Nat Commun 2018;9:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ancel S, Stuelsatz P, Feige JN. Muscle stem cell quiescence: controlling stemness by staying asleep. Trends Cell Biol 2021;31:556–568. [DOI] [PubMed] [Google Scholar]
- 26. Garcia‐Prat L, Martinez‐Vicente M, Perdiguero E, Ortet L, Rodriguez‐Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature 2016;529:37–42. [DOI] [PubMed] [Google Scholar]
- 27. Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature 2012;490:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dong S, Wang Q, Kao YR, Diaz A, Tasset I, Kaushik S, et al. Chaperone‐mediated autophagy sustains haematopoietic stem‐cell function. Nature 2021;591:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White JP, Billin AN, Campbell ME, Russell AJ, Huffman KM, Kraus WE. The AMPK/p27(Kip1) axis regulates autophagy/apoptosis decisions in aged skeletal muscle stem cells. Stem Cell Rep 2018;11:425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sousa‐Victor P, Gutarra S, Garcia‐Prat L, Rodriguez‐Ubreva J, Ortet L, Ruiz‐Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014;506:316–321. [DOI] [PubMed] [Google Scholar]
- 31. Kim JH, Park I, Shin HR, Rhee J, Seo JY, Jo YW, et al. The hypothalamic‐pituitary‐gonadal axis controls muscle stem cell senescence through autophagosome clearance. J Cachexia Sarcopenia Muscle 2021;12:177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feige P, Brun CE, Ritso M, Rudnicki MA. Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell 2018;23:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012;11:596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang AH, Rando TA. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J 2014;33:2782–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campanario S, Ramirez‐Pardo I, Hong X, Isern J, Munoz‐Canoves P. Assessing autophagy in muscle stem cells. Front Cell Dev Biol 2020;8:620409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert . Nature 2014;510:393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fortini P, Ferretti C, Iorio E, Cagnin M, Garribba L, Pietraforte D, et al. The fine tuning of metabolism, autophagy and differentiation during in vitro myogenesis. Cell Death Dis 2016;7:e2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Call JA, Wilson RJ, Laker RC, Zhang M, Kundu M, Yan Z. Ulk1‐mediated autophagy plays an essential role in mitochondrial remodeling and functional regeneration of skeletal muscle. Am J Physiol Cell Physiol 2017;312:C724–C732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sin J, Andres AM, Taylor DJR, Weston T, Hiraumi Y, Stotland A, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016;12:369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Remels AH, Langen RC, Schrauwen P, Schaart G, Schols AM, Gosker HR. Regulation of mitochondrial biogenesis during myogenesis. Mol Cell Endocrinol 2010;315:113–120. [DOI] [PubMed] [Google Scholar]
- 42. Paasuke R, Eimre M, Piirsoo A, Peet N, Laada L, Kadaja L, et al. Proliferation of human primary myoblasts is associated with altered energy metabolism in dependence on ageing in vivo and in vitro. Oxid Med Cell Longev 2016;2016:8296150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sala D, Cunningham TJ, Stec MJ, Etxaniz U, Nicoletti C, Dall'Agnese A, et al. The Stat3‐Fam3a axis promotes muscle stem cell myogenic lineage progression by inducing mitochondrial respiration. Nat Commun 2019;10:1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horie T, Kawamata T, Matsunami M, Ohsumi Y. Recycling of iron via autophagy is critical for the transition from glycolytic to respiratory growth. J Biol Chem 2017;292:8533–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duguez S, Feasson L, Denis C, Freyssenet D. Mitochondrial biogenesis during skeletal muscle regeneration. Am J Physiol Endocrinol Metab 2002;282:E802–E809. [DOI] [PubMed] [Google Scholar]
- 46. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor‐mediated mitophagy in yeast and mammalian systems. Cell Res 2014;24:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 2018;19:93–108. [DOI] [PubMed] [Google Scholar]
- 48. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol 2018;20:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J 2021;40:e104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 2018;16:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nichenko AS, Southern WM, Atuan M, Luan J, Peissig KB, Foltz SJ, et al. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am J Physiol Cell Physiol 2016;311:C190–C200. [DOI] [PubMed] [Google Scholar]
- 52. Nichenko AS, Southern WM, Tehrani KF, Qualls AE, Flemington AB, Mercer GH, et al. Mitochondrial‐specific autophagy linked to mitochondrial dysfunction following traumatic freeze injury in mice. Am J Physiol Cell Physiol 2020;318:C242–C252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kravic B, Harbauer AB, Romanello V, Simeone L, Vogtle FN, Kaiser T, et al. In mammalian skeletal muscle, phosphorylation of TOMM22 by protein kinase CSNK2/CK2 controls mitophagy. Autophagy 2018;14:311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu TZ, Dohl J, Elenberg F, Chen YF, Deuster P. Curcumin induces concentration‐dependent alterations in mitochondrial function through ROS in C2C12 mouse myoblasts. J Cell Physiol 2019;234:6371–6381. [DOI] [PubMed] [Google Scholar]
- 57. Le Moal E, Pialoux V, Juban G, Groussard C, Zouhal H, Chazaud B, et al. Redox control of skeletal muscle regeneration. Antioxid Redox Signal 2017;27:276–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yin WX, Yang L, Kong DL, Nie YZ, Liang Y, Teng CB. Guanine‐rich RNA binding protein GRSF1 inhibits myoblast differentiation through repressing mitochondrial ROS production. Exp Cell Res 2019;381:139–149. [DOI] [PubMed] [Google Scholar]
- 59. Hirai H, Verma M, Watanabe S, Tastad C, Asakura Y, Asakura A. MyoD regulates apoptosis of myoblasts through microRNA‐mediated down‐regulation of Pax3. J Cell Biol 2010;191:347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song J, Wang Y, Yuan X, Ji Q, Fan C, Zhao H, et al. Stretching magnitude‐dependent inactivation of AKT by ROS led to enhanced p53 mitochondrial translocation and myoblast apoptosis. Mol Biol Cell 2019;30:1182–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rajasekaran NS, Shelar SB, Jones DP, Hoidal JR. Reductive stress impairs myogenic differentiation. Redox Biol 2020;34:101492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, et al. Normal myogenesis and increased apoptosis in myotonic dystrophy type‐1 muscle cells. Cell Death Differ 2010;17:1315–1324. [DOI] [PubMed] [Google Scholar]
- 63. McMillan EM, Quadrilatero J. Autophagy is required and protects against apoptosis during myoblast differentiation. Biochem J 2014;462:267–277. [DOI] [PubMed] [Google Scholar]
- 64. Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012;111:1208–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2000;2:156–162. [DOI] [PubMed] [Google Scholar]
- 66. Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis‐inducing factor. Nature 1999;397:441–446. [DOI] [PubMed] [Google Scholar]
- 67. Dos Santos M, Backer S, Saintpierre B, Izac B, Andrieu M, Letourneur F, et al. Single‐nucleus RNA‐seq and FISH identify coordinated transcriptional activity in mammalian myofibers. Nat Commun 2020;11:5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lazure F, Blackburn DM, Corchado AH, Sahinyan K, Karam N, Sharanek A, et al. Myf6/MRF4 is a myogenic niche regulator required for the maintenance of the muscle stem cell pool. EMBO Rep 2020;21:e49499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Souza J, Gottfried C. Muscle injury: review of experimental models. J Electromyogr Kinesiol 2013;23:1253–1260. [DOI] [PubMed] [Google Scholar]
- 70. Zhou S, Zhang W, Cai G, Ding Y, Wei C, Li S, et al. Myofiber necroptosis promotes muscle stem cell proliferation via releasing Tenascin‐C during regeneration. Cell Res 2020;30:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Webster MT, Manor U, Lippincott‐Schwartz J, Fan CM. Intravital imaging reveals ghost fibers as architectural units guiding myogenic progenitors during regeneration. Cell Stem Cell 2016;18:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsuchiya Y, Kitajima Y, Masumoto H, Ono Y. Damaged myofiber‐derived metabolic enzymes act as activators of muscle satellite cells. Stem Cell Rep 2020;15:926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lutz AB, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, et al. Schwann cells use TAM receptor‐mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci U S A 2017;114:E8072–E8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saera‐Vila A, Kish PE, Louie KW, Grzegorski SJ, Klionsky DJ, Kahana A. Autophagy regulates cytoplasmic remodeling during cell reprogramming in a zebrafish model of muscle regeneration. Autophagy 2016;12:1864–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Deng B, Wehling‐Henricks M, Villalta SA, Wang Y, Tidball JG. IL‐10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol 2012;189:3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Giannakis N, Sansbury BE, Patsalos A, Hays TT, Riley CO, Han X, et al. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol 2019;20:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni‐Barrera R, Masschelein E, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2‐like macrophage polarization. Cell Metab 2020;31:1136–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte‐macrophage differentiation. Blood 2012;119:2895–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, et al. Autophagy is required for CSF‐1‐induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012;119:4527–4531. [DOI] [PubMed] [Google Scholar]
- 80. Kang YH, Cho MH, Kim JY, Kwon MS, Peak JJ, Kang SW, et al. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016;7:35577–35591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schiaffino S, Pereira MG, Ciciliot S, Rovere‐Querini P. Regulatory T cells and skeletal muscle regeneration. FEBS J 2017;284:517–524. [DOI] [PubMed] [Google Scholar]
- 82. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol 2016;17:277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ‐specific endothelial cells. Nature 2016;529:316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martello A, Lauriola A, Mellis D, Parish E, Dawson JC, Imrie L, et al. Trichoplein binds PCM1 and controls endothelial cell function by regulating autophagy. EMBO Rep 2020;21:e48192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lu WH, Shi YX, Ma ZL, Wang G, Liu LX, Chuai ML, et al. Proper autophagy is indispensable for angiogenesis during chick embryo development. Cell Cycle 2016;15:1742–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gutiérrez JM, Escalante T, Hernández R, Gastaldello S, Saravia‐Otten P, Rucavado A. Why is skeletal muscle regeneration impaired after myonecrosis induced by viperid snake venoms? Toxins 2018;10:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hardy D, Fefeu M, Besnard A, Briand D, Gasse P, Arenzana‐Seisdedos F, et al. Defective angiogenesis in CXCL12 mutant mice impairs skeletal muscle regeneration. Skelet Muscle 2019;9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shiiba I, Takeda K, Nagashima S, Ito N, Tokuyama T, Yamashita SI, et al. MITOL promotes cell survival by degrading Parkin during mitophagy. EMBO Rep 2021;22:e49097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sachdev U, Cui X, Hong G, Namkoong S, Karlsson JM, Baty CJ, et al. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J Vasc Surg 2012;55:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Carnevale R, Nocella C, Schiavon S, Cammisotto V, Cotugno M, Forte M, et al. Beneficial effects of a combination of natural product activators of autophagy on endothelial cells and platelets. Br J Pharmacol 2021;178:2146–2159. [DOI] [PubMed] [Google Scholar]
- 91. Liu B, Zhou H, Zhang T, Gao X, Tao B, Xing H, et al. Loss of endothelial glucocorticoid receptor promotes angiogenesis via upregulation of Wnt/beta‐catenin pathway. Angiogenesis 2021;24:631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fan Y, Lu H, Liang W, Garcia‐Barrio MT, Guo Y, Zhang J, et al. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ Res 2018;122:945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, et al. PINK1‐Parkin‐mediated mitophagy protects mitochondrial integrity and prevents metabolic stress‐induced endothelial injury. PLoS ONE 2015;10:e0132499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 2013;19:1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bibee KP, Cheng YJ, Ching JK, Marsh JN, Li AJ, Keeling RM, et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASB J 2014;28:2047–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C, et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med 2018;10:e8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Vinel C, Lukjanenko L, Batut A, Deleruyelle S, Pradere JP, Le Gonidec S, et al. The exerkine apelin reverses age‐associated sarcopenia. Nat Med 2018;24:1360–1371. [DOI] [PubMed] [Google Scholar]
- 98. Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. mTORC1 signaling: a double‐edged sword in diabetic beta cells. Cell Metab 2018;27:314–331. [DOI] [PubMed] [Google Scholar]
- 99. Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010;16:1313–1320. [DOI] [PubMed] [Google Scholar]
- 100. Metti S, Gambarotto L, Chrisam M, Baraldo M, Braghetta P, Blaauw B, et al. The polyphenol pterostilbene ameliorates the myopathic phenotype of collagen VI deficient mice via autophagy induction. Front Cell Dev Biol 2020;8:580933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang K, Zhang Y, Gu L, Lan M, Liu C, Wang M, et al. ISLR regulates canonical Wnt signaling‐mediated skeletal muscle regeneration by stabilizing Dishevelled‐2 and preventing autophagy. Nat Commun 2018;9:5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Carmignac V, Svensson M, Korner Z, Elowsson L, Matsumura C, Gawlik KI, et al. Autophagy is increased in laminin alpha2 chain‐deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet 2011;20:4891–4902. [DOI] [PubMed] [Google Scholar]
- 103. Oprescu SN, Yue F, Qiu J, Brito LF, Kuang S. Temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. iScience 2020;23:100993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chang NC. Autophagy and stem cells: self‐eating for self‐renewal. Front Cell Dev Biol 2020;8:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nichenko AS, Sorensen JR, Southern WM, Qualls AE, Schifino AG, McFaline‐Figueroa J, et al. Lifelong Ulk1‐mediated autophagy deficiency in muscle induces mitochondrial dysfunction and contractile weakness. Int J Mol Sci 2021;22, 1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thepenier C, et al. Comparative study of injury models for studying muscle regeneration in mice. PLoS ONE 2016;11:e0147198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. von Haehling S, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]