

Abstract
Background
Cancer patients at advanced stages experience a severe depletion of skeletal muscle compartment together with a decrease in muscle function, known as cancer cachexia. Cachexia contributes to reducing quality of life, treatment efficiency, and lifespan of cancer patients. However, the systemic nature of the syndrome is poorly documented. Here, we hypothesize that glucocorticoids would be important systemic mediators of cancer cachexia.
Methods
To explore the role of glucocorticoids during cancer cachexia, biomolecular analyses were performed on several tissues (adrenal glands, blood, hypothalamus, liver, and skeletal muscle) collected from Apc Min/+ male mice, a mouse model of intestine and colon cancer, aged of 13 and 23 weeks, and compared with wild type age‐matched C57BL/6J littermates.
Results
Twenty‐three‐week‐old Apc mice recapitulated important features of cancer cachexia including body weight loss (−16%, P < 0.0001), muscle atrophy (gastrocnemius muscle: −53%, P < 0.0001), and weakness (−50% in tibialis anterior muscle force, P < 0.0001), increased expression of atrogens (7‐fold increase in MuRF1 transcript level, P < 0.0001) and down‐regulation of Akt–mTOR pathway (3.3‐fold increase in 4EBP1 protein content, P < 0.0001), together with a marked transcriptional rewiring of hepatic metabolism toward an increased expression of gluconeogenic genes (Pcx: +90%, Pck1: +85%), and decreased expression of glycolytic (Slc2a2: −40%, Gk: −30%, Pklr: −60%), ketogenic (Hmgcs2: −55%, Bdh1: −80%), lipolytic/fatty oxidation (Lipe: −50%, Mgll: −60%, Cpt2: −60%, Hadh: −30%), and lipogenic (Acly: −30%, Acacb: −70%, Fasn: −45%) genes. The hypothalamic pituitary–adrenal axis was activated, as evidenced by the increase in the transcript levels of genes encoding corticotropin‐releasing hormone in the hypothalamus (2‐fold increase, P < 0.01), adrenocorticotropic hormone receptor (3.4‐fold increase, P < 0.001), and steroid biosynthesis enzymes (Cyp21a1, P < 0.0001, and Cyp11b1, P < 0.01) in the adrenal glands, as well as by the increase in corticosterone level in the serum (+73%, P < 0.05), skeletal muscle (+17%, P < 0.001), and liver (+24%, P < 0.05) of cachectic 23‐week‐old Apc mice. A comparative transcriptional analysis with dexamethasone‐treated C57BL/6J mice indicated that the activation of the hypothalamic–pituitary–adrenal axis in 23‐week‐old Apc Min/+ mice was significantly associated with the transcription of glucocorticoid‐responsive genes in skeletal muscle (P < 0.05) and liver (P < 0.001). The transcriptional regulation of glucocorticoid‐responsive genes was also observed in the gastrocnemius muscle of Lewis lung carcinoma tumour‐bearing mice and in KPC mice (tibialis anterior muscle and liver).
Conclusions
These findings highlight the role of the hypothalamic–pituitary–adrenal‐glucocorticoid pathway in the transcriptional regulation of skeletal muscle catabolism and hepatic metabolism during cancer cachexia. They also provide the paradigm for the design of new therapeutic strategies.
Keywords: Cancer cachexia, Glucocorticoid, Hypothalamic–pituitary–adrenal axis, Liver, Metabolism, Skeletal muscle
Introduction
Cancer cachexia is a devastating hypo‐anabolic and catabolic syndrome characterized by a progressive intentional loss of body mass, essentially due to a severe depletion of skeletal muscle compartment, that cannot be reversed by conventional nutritional supports. 1 The prevalence of cancer cachexia is quite variable, affecting 15% of prostate or breast cancer patients to 50% of colorectal cancer patients, and even 70% of pancreatic cancer patients. 2 It has been estimated that 15.8 subjects per 10 000 of the total general population in European Union suffered from cancer cachexia in 2013, while it affected approximately 16.5 individuals per 10 000 people in the USA in 2014. 3 Skeletal muscle depletion has very strong adverse effects for cancer patient health care as it increases the risk of surgical complications, and chemotherapy toxicity, ultimately reducing cancer patient's quality of life and contributing to mortality. 1
Along with skeletal muscle depletion and functional related complications, an often‐overlooked aspect of cancer cachexia is its systemic nature. Although skeletal muscle plays a central role in the syndrome, cancer cachexia also involves multiple tissues and organs with important metabolic functions, including brain, adipose tissue, and liver. 4 To provide new insights into the mechanisms involved in cancer cachexia and to understand as a whole the diversity and complexity this syndrome, we looked for circulating molecular determinants of cancer cachexia. Our attention turned out to glucocorticoids (GC), a class of steroid hormones that play important metabolic functions in multiple organs and tissues. GC are synthesized and secreted by the adrenal cortex under the control of the hypothalamic–pituitary–adrenal (HPA) axis. GC bind to GC receptor, which then binds to GC response elements in the promoters of target genes to activate or inhibit their transcription. In skeletal muscle, GC administration has long been known to decrease protein synthesis 5 and increase protein degradation, 5 an effect that is mediated by a transcriptional program that decreases the expression of genes promoting protein synthesis and increases those promoting protein degradation. 6 , 7 , 8 , 9 , 10 , 11 Accordingly, skeletal muscle mass loss is abrogated in LLC‐tumour bearing mice with targeted deletion of GC receptor in skeletal muscle. 12 Another target metabolic tissue of GC is the liver. Molecular studies have indicated that GC stimulated the transcription of genes encoding gluconeogenic enzymes to increase glucose production. 13 , 14 Accordingly, clinical studies show that hepatic glucose metabolism is markedly impaired in cancer patients as exemplified by decreased hepatic glycogen store 15 and heightened hepatic gluconeogenesis from lactate, 15 glycerol, 16 or alanine. 17 Finally, an increase in circulating GC level has also been reported in cancer patients, 18 , 19 , 20 as well as in animal models of cancer. 21 , 22 , 23 , 24 , 25 , 26 , 27 However, it remains largely unsolved whether the HPA axis is activated during cancer cachexia to coordinate GC‐dependent transcriptional responses in skeletal muscle and liver.
In the present study, we show that HPA axis activation is associated with an increase in GC concentration in the serum, skeletal muscle, and liver of 23‐week‐old Apc mice, a mouse model of intestine and colon cancer. This response was associated with the transcription of GC‐responsive genes involved in skeletal muscle proteostasis and liver metabolism.
Materials and methods
Animals
Animal experiments were conducted in accordance with the European Community guidelines and with the authorization of the Ministère français de l'Enseignement Supérieur, de la Recherche, et de l'Innovation. Mice were co‐housed with one to three other mice in filter‐top cages with bedding and nest building enrichment in a temperature‐controlled (20–21°C) with a 12‐h light/dark cycle. Animals had free access to food and water.
Apc male mice 28 were purchased from the Jackson Laboratory. Mice showing signs of distress (sores, weight loss greater than 20%, rectal bleeding) were removed from the study. For dexamethasone treatment, C57BL/6J male mice (87.4 ± 0.7 days) were randomly allocated to dexamethasone‐treated (3 mg/kg daily i.p. injection of a water‐soluble form of dexamethasone for 5 days) or vehicle‐treated (PBS) groups. KPC female and male mice (LSLKrasG12D/+; LSLTrp53R172H/+; Ptf1a‐p48‐Cre/+) 29 were obtained by breeding [ LSL Kras G12D/+ ; LSL Trp53 R172H/+ ] founding couples with [Ptf1a‐p48‐Cre /+ ] mice. Age‐matched and sex‐matched control mice were [WT], [ LSL Kras G12D/+ ], [ LSL Trp53 R172H/+ ], or [p48‐Cre /+ ] mice. All mice were maintained on a C57BL/6J background and were used as they became available, allocated by genotype and paired with corresponding wild‐type (WT) or control littermates.
Anaesthesia and tissue removal
Tissues from 13‐ and 23‐week‐old wild type and Apc mice, dexamethasone‐treated mice, and KPC mice were removed on anaesthetized mice (90 mg/kg ketamine and 10 mg/kg xylazine) between 10 am and 12 am. Due the heterogeneity of the disease progression, KPC and their control littermates were euthanized between 3 and 6 months of age. KPC mice were categorized as severe (SEV) upon reaching at least one of the following endpoints, body weight loss (based on the kinetic analysis of body weight), adipose tissue loss, abdominal distension, and/or lethargy. Moderate (MOD KPC) were euthanized before the onset of one of these endpoints. All mice were euthanized by cervical dislocation.
Blood analyses and serum assays
Haematocrit, glycaemia, and lactatemia were measured during tissue collection using a Mini Compur M1100 microspin, FreeStyle Optium Neo H glucometer (Abbott Diabetes Care), and Accutrend® Plus lactate analyser (Roche Diagnostics GmbH), respectively. Sera were prepared from blood samples removed from the tail vein under isoflurane (3%) anaesthesia between 10 am and 12 am in the fed state, and used for the measurement of corticosterone (MyBiosource) and β‐hydroxybutyrate (APExBIO) concentrations.
Analysis of pancreatic lesions in KPC mice, image acquisition, and analysis
Pancreatic tissue was fixed in 4% formalin (Electron Microscopy Science); 4 μm slides of paraffin‐embedded pancreatic tumours tissue were deparaffinized and rehydrated, stained with Mayer haematoxylin and eosin, dehydrated and mounted. Images were acquired with a Zeiss Imager M2 AX10 microscope and processed using Zeiss Zen. Pancreatic neoplasm lesions were assessed and classified according to the Baltimore consensus.
Immunofluorescence analysis of myofibre cross‐sectional area, image acquisition, and analysis
Gastrocnemius muscle sections (10 μm) were prepared (Leica CM1950 cryostat microtome) and then immuno‐stained (mouse anti‐laminin, Sigma‐Aldrich) using an Alexa Fluor 488 goat anti‐mouse secondary antibody (Thermo Fisher Scientific). Slides were mounted with antifade mounting medium with DAPI (Vector Laboratories). Images were acquired (Zeiss Axio Imager 2 Microscope) and used for the quantification of muscle cross‐sectional area (ImageJ).
In situ force measurement
The distal tendon of the tibialis anterior muscle of anaesthetized (90 mg/kg ketamine and 10 mg/kg xylazine) 23‐week‐old WT and Apc mice was attached to the force‐position transducer apparatus, while mice knee was anchored and the sciatic nerve was stimulated. After determination of optimum muscle length (L0), maximum rate of force development to peak twitch and maximum rate of relaxation to the twitch were analysed (Aurora Scientific). Maximum absolute isometric tetanic force (P0) was determined as the maximal force recorded during a stimulation of 0.5 s at 100 Hz. Maximum specific isometric tetanic force (sP0) was calculated as the maximum absolute isometric tetanic force divided by tibialis anterior muscle weight. Muscle fatigue was determined by recording force production during a 120 s stimulation protocol at 50 Hz.
Isolation of RNA and reverse transcription‐quantitative polymerase chain reaction analysis
RNA was extracted (NucleoSpin RNA Set for NucleoZOL, Macherey‐Nagel) from adrenal glands, hypothalamus, liver, and quadriceps muscle in Apc mice, gastrocnemius muscle, liver, and hypothalamus in dexamethasone‐treated mice, and liver and tibialis anterior muscle in KPC mice; 200 ng of RNA were reverse transcribed (Reverse Transcriptase Core kit, Eurogentec). qPCR was carried out on 2 μL of cDNA (Takyon™ No Rox SYBR MasterMix dTTP Blue, Eurogentec) in a total reaction volume of 10 μL. Primer sequences are provided in Supporting information, Table S1. Fluorescence intensity was recorded using a CFX96 Real‐Time PCR detection system (Bio‐Rad). Data were analysed using the ∆∆CT method of analysis. Reference genes (Ppia and Hprt for quadriceps muscle, Actb and Hprt for liver and adrenal glands, Actb and Tuba1 for hypothalamus) were used for normalization.
Protein extraction and immunoblotting, data acquisition, and analysis
Powdered quadriceps muscle and liver were homogenized (1:20 w:v) in a RIPA buffer (Cell Signalling Technology) supplemented with protease and phosphatase inhibitors (Roche). Samples were centrifuged (10 min, 12 000 g, 4°C). Protein concentration of supernatants was determined (DC protein assay, Bio‐Rad); 30 μg of protein in Laemmli buffer (Bio‐Rad) were subjected to 8–16% gel electrophoresis (Bio‐Rad) and transferred onto nitrocellulose membrane (Trans‐Blot Turbo Transfer System, Bio‐Rad). Membranes were then blocked with a Tris Buffered Saline‐0.1% Tween 20 solution containing 5% non‐fat dried milk for 2 h at room temperature and incubated overnight at 4°C with primary antibody against rpS6Ser235/236 (1:1000), DDIT4 (1:1000), 4EBP1 (1:1000), PGC1A (1:1000), CRTC2Ser171 (1:1000), PCK1 (1:500), CREBSer133 (1:1000) and CREB (1:1000), followed by probing with HRP‐conjugated secondary antibody (Agilent, 1:2000). Membranes were incubated with ECL (Bio‐Rad) and imaged (ChemiDoc MP Imaging System, Bio‐Rad). The protein band intensity was determined using Image Lab™ 6.0. Stain‐free technology (Bio‐Rad) was used for protein normalization.
Skeletal muscle and liver corticosterone content
A solid‐phase enzyme‐linked immunosorbent assay (MyBiosource, San Diego, USA) was used for the measurement of corticosterone content in the quadriceps muscle and liver protein extracts of Apc mice.
Statistical analysis
Data are expressed as means ± SEM. Statistical analyses were performed using GraphPad Prism 9.0. Grubb's test was performed to identify outliers. Data were tested for normal distribution using Shapiro–Wilk test. Mann–Whitney or two‐tailed unpaired t‐test with Welch's correction if necessary, one‐way ANOVA followed by Tukey's multiple comparison test, Kruskal–Wallis test followed by Dunn's multiple comparison test, and two‐way ANOVA followed by Sidak's multiple comparison test were used. Pearson correlation coefficient and log‐rank (Mantel–Cox) test were also used. Gene expression overlapping significance, odds ratio, and Jaccard index were calculated with Fisher's exact test using GeneOverlap package (http://shenlab‐sinai.github.io/shenlab‐sinai/) in R (version 3.6.3). Statistical significance was considered when P < 0.05.
Results
Apc mice recapitulate the main features of cancer cachexia
To investigate the mechanisms driving cancer cachexia, we used the Apc mouse, a mouse model of intestinal and colon cancer that harbours a heterozygous mutation in the adenomatous polyposis coli (APC) gene. 28 Mutations in the APC gene activate the β‐catenin‐Tcf signalling and are responsible for familial adenomatous polyposis, an inherited cancer predisposition syndrome. 30 The APC gene is mutated in more than half of colorectal cancer patients 30 and the risk of colorectal cancer by the age of 40 is almost 100% in patients. 30 Here, Apc mice experienced a progressive loss in body mass that began 15 weeks after birth, reaching −16% of their peak body mass at the age of 23 weeks (Figure 1A). Apc mice fed normally (Figure S1A; Data S1) and had similar tibia length (Figure S1B; Data S1) than WT age‐matched littermates, indicating that the lower body mass of Apc mice was not due to a slower growth. Apc mice developed polyposis (Figure 1B), as well as anaemia (Figure 1C). The median survival of Apc mice was 26 weeks (Figure 1D).
Figure 1.
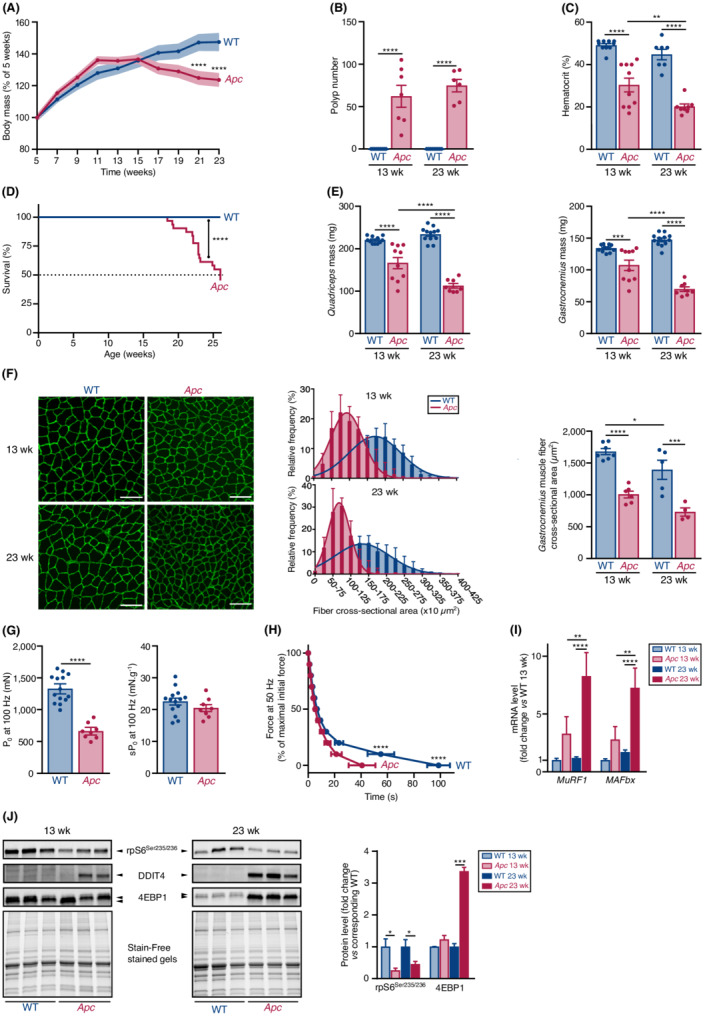
Apc mice recapitulate the main features of cancer cachexia. (A) Relative changes in body mass in WT (n = 9) and Apc (n = 24 at 5 weeks) mice from 5 to 23 weeks after birth. (B) Polyp number in small intestine and colon of WT and Apc mice at 13 (WT n = 9; Apc n = 7) and 23 (WT n = 10; Apc n = 6) weeks. (C) Haematocrit in WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 7; Apc n = 8) weeks. (D) Kaplan–Meier survival curve of WT (n = 31) and Apc (n = 31) mice indicates that half‐life survival of Apc mice was 26 weeks. (E) Quadriceps (left) and gastrocnemius (right) muscle mass of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 8) weeks. (F) Representative laminin immunostaining (left) of gastrocnemius muscle transverse sections in WT and Apc mice at 13 and 23 weeks. Scale bar, 100 μm. Frequency distribution of gastrocnemius muscle fibre cross‐sectional area (centre) in WT and Apc mice at 13 (WT n = 7; Apc n = 6) and 23 (WT n = 5; Apc n = 4) weeks. Lines represent the Gaussian curve fit. Quantification of gastrocnemius muscle fibre cross‐sectional area (right) of WT and Apc mice at 13 (WT n = 7; Apc n = 6) and 23 (WT n = 5; Apc n = 4) weeks. A mean of 540 ± 247 fibres per muscle were counted. (G) Absolute maximal isometric tetanic force (P0, left) of tibialis anterior muscle recorded in 23‐week‐old WT (n = 14) and Apc (n = 8) mice. Specific maximal isometric tetanic force (sP0, right) of tibialis anterior muscle recorded at 100 Hz in 23‐week‐old WT (n = 14) and Apc (n = 8) mice. (H) Tibialis anterior muscle force during a 120 s fatigue protocol at 50 Hz in 23‐week‐old WT (n = 12) and Apc (n = 8) mice. (I) RT‐qPCR analysis of MuRF1 and MAFbx mRNA levels in the quadriceps muscle of WT and Apc mice at 13 (WT n = 11; Apc n = 9) and 23 (WT n = 12; Apc n = 7) weeks. (J) Representative immunoblot (left) of phosphorylated ribosomal protein S6 (rpS6Ser235/236), DDIT4 protein, and 4EBP1 protein content in the quadriceps muscle of WT and Apc mice at 13 and 23 weeks. The hypophosphorylated active form of 4EBP1 migrates faster than the hyperphosphorylated inactive form. Quantitative analysis (right) of rpS6Ser235/236 and 4EBP1 protein content in the quadriceps muscle of WT and Apc mice at 13 (WT n = 8; Apc n = 8) and 23 (WT n = 8; Apc n = 8) weeks. DDIT4 protein level was not quantified as expression level in WT mice was barely detectable. Data are means ± SEM. (A, B, C, E, F right, H, I) Data were analysed by a two‐way ANOVA with Sidak's multiple comparison test. (D) Data were analysed by a log‐rank (Mantel–Cox) test. (G, J) Data were analysed by a two‐tailed unpaired t‐test with Welch's correction when necessary. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Marked reductions in adipose tissue store (Figure S1C; Data S1) and skeletal muscle compartment were also observed. Cachexia indifferently affected skeletal muscles with mixed contractile and metabolic phenotypes, such as quadriceps (−24% at 13 weeks and −52% at 23 weeks) and gastrocnemius (−20% at 13 weeks and −53% at 23 weeks) muscles (Figure 1E), and those with a predominance of fast‐twitch glycolytic fibres such as extensor digitorum longus (−24% at 13 weeks and −52% at 23 weeks) and tibialis anterior (−19% at 13 weeks and −49% at 23 weeks) muscles (Figure S1D; Data S1). Soleus muscle, a slow‐twitch oxidative postural muscle, which has not developed atrophy yet at 13 weeks, became atrophic at 23 weeks (Figure S1D; Data S1). The reduction in gastrocnemius muscle mass was associated with a progressive decrease in muscle fibre cross‐sectional area (Figure 1F). Finally, skeletal muscle depletion was also correlated with body mass loss (Figure S1E; Data S1).
Functionally, the absolute maximal isometric tetanic force of tibialis anterior muscle (P0) was significantly reduced in 23‐week‐old Apc mice compared with WT mice (Figure 1G). Specific muscle force (sP0) was not different (Figures 1G and S1F, Data S1), indicating that the loss in skeletal muscle mass strongly contributed to the loss in force. The maximum rates of force development and relaxation were also reduced during a single twitch response (Figure S1G; Data S1). Finally, muscle fatigue was also increased as shown by the faster and greater decrease in muscle force during a fatigue protocol (Figure 1H).
Molecularly, the mRNA levels of Trim63 (MuRF1) and Fbxo32 (MAFbx) encoding E3‐ubiquitin ligases involved in ubiquitin‐proteasome dependent proteolysis 31 were increased in the quadriceps muscle of 23‐week‐old Apc mice compared with WT mice (Figure 1I). This was associated with an increase in the protein content of the translational repressors, DDIT4 32 and 4EBP1 33 (Figure 1J). The fast migrating hypo‐phosphorylated active form of 4EBP1 was also increased (Figure 1J). Finally, the active phosphorylated form of ribosomal protein S6 was decreased (Figure 1J). Together, these data support the existence of an unbalance between protein synthesis and degradation that progressively contribute to the loss in muscle mass and function in Apc mice.
Transcriptional rewiring of hepatic metabolism in 23‐week‐old Apc mice
Hepatic gluconeogenesis is increased in cancer patients, 15 , 16 , 17 suggesting that molecular adaptations should occur to sustain hepatic glucose production. In the present study, transcript levels of key gluconeogenic enzymes, Pcx and Pck1, were increased in 23‐week‐old cachectic mice (Figure 2A and 2C). The increased expression of Pck1 was also confirmed at the protein level (Figure 2D). Conversely, the mRNA levels of Mlxipl (ChREBP), a transcription factor that drives the expression of glycolytic genes such as Gk and Pklr, as well as that of the glucose transporter Slc2a2, 34 were decreased in 23‐week‐old Apc mice (Figure 2B and 2C). Transcript level of Gpd2, a component of the glycerol phosphate shuttle that boots glucose oxidation, 35 was also down‐regulated (Figure 2B and 2C). Expression of gluconeogenic genes can be activated in response to glucagon by stimulation of CREB transcription factor. 14 Here, the active phosphorylated form of CREB was decreased in the liver of Apc mice and phosphorylation of the CREB‐regulated transcription coactivator 2 (CRTC2) remained unchanged (Figure 2D), suggesting that this mechanism was not activated. The expression of gluconeogenic genes can be also activated by the coordinate interplay of the transcriptional co‐activator PPARGC1A (PGC1A) and FOXO1 transcription factor. 36 The mRNA levels of Pgc1a and Foxo1, but also that of Foxo3, were increased in the liver of 23‐week‐old Apc mice (Figure 2A). The increased expression of Pgc1a was also confirmed at the protein level (Figure 2D), suggesting an activation of a PGC1A/FOXO1 axis in cachectic mice. These adaptations in 23‐week‐old Apc mice were accompanied by a decrease in blood glucose concentration and an increase in blood lactate concentration (Figure 2E).
Figure 2.

Transcriptional rewiring of gluconeogenic and glycolytic gene expression in Apc mice. (A) Transcript level of gluconeogenic genes in liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. (B) Transcript level of glycolytic genes in liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. (C) Schematic view of glycolysis and gluconeogenesis. Decreased, unchanged, or increased transcript levels are indicated in blue, grey, and red, respectively. (D) Representative immunoblot (left) and quantitative analysis (right) of PGC1A, phosphorylated CRTC2 (CRTC2Ser171), PCK1, phosphorylated CREB (CREBSer133), and total CREB protein content in the quadriceps muscle of WT and Apc mice at 13 (WT n = 8; Apc n = 8) and 23 (WT n = 8; Apc n = 8) weeks. (E) Blood glucose concentration (left) in WT and Apc mice at 13 (WT n = 12; Apc n = 12) and 23 (WT n = 12; Apc n = 12) weeks. Blood lactate concentration (right) in WT and Apc mice at 13 (WT n = 11; Apc n = 11) and 23 (WT n = 11; Apc n = 11) weeks. Data are means ± SEM. (A, B, E) Data were analysed by two‐way ANOVA with Sidak's multiple comparison test. § indicates a main genotype effect. (D) Data were analysed by a two‐tailed unpaired t‐test with Welch's correction when necessary. * or §P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. DHAP, dihydroxyacetone phosphate; F‐1,6‐BP, fructose‐1,6‐bisphosphate; F‐6‐P, fructose‐6‐phosphate; G‐3‐P, glycerol‐3‐phosphate; G‐6‐P, glucose‐6‐phosphate; GAP, glyceraldehyde‐3‐phosphate; OAA, oxaloacetic acid; PEP, phosphoenolpyruvate.
Transcriptional rewiring of hepatic metabolism was not limited to glucose metabolism. Ketone bodies, which are produced by the liver from acetyl‐CoA during excessive fatty acid oxidation, become important substrates for energy production by the brain, kidneys, and skeletal muscle when others substrates are missing or less used. Serum concentration of β‐hydroxybutyrate, the most prevalent circulating ketone body, was decreased in 23‐week‐old Apc mice (Figure 3A). Accordingly, the transcript level of Ppara, a master transcriptional regulator of fatty acid oxidation and ketogenic gene expression, 37 , 38 was down‐regulated in 23‐week‐old Apc mice, as well as the transcript level of ketogenesis rate‐limiting enzymes (Hmgcs2 and Bdh1) and fatty acid oxidation genes (Figure 3B). Therefore, 23‐week‐old Apc mice display a hypo‐ketonemic phenotype associated with a reduction in the expression of hepatic fatty acid oxidation genes, consistent with the reduction in PPARα gene expression. Expression of genes encoding lipolytic (Lipe, Mgll) and lipogenesis (Acly, Acacb, Fasn, Scd1) proteins were also down‐regulated (Figure 3C–3D). Finally, disturbed expression of transcripts encoding urea cycle enzymes (Cps1, Otc, Ass1, Asl) was also observed (Figure 3E). Collectively, these data indicate that the liver of Apc mice experience a major transcriptional rewiring that affects glucose, ketone body, lipid, and nitrogen metabolisms.
Figure 3.
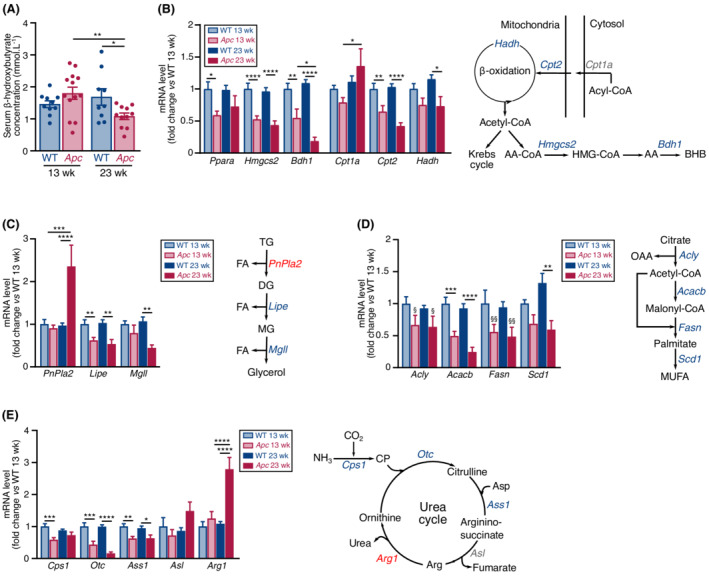
F Transcriptional rewiring of ketogenic, lipid, and nitrogen metabolisms in Apc mice. (A) Serum β‐hydroxybutyrate concentration in WT and Apc mice at 13 (WT n = 10; Apc n = 13) and 23 (WT n = 9; Apc n = 11) weeks. (B) Transcript level of ketogenic genes and fatty acid oxidation genes (left) in the liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. Schematic view of ketogenesis in the liver (right). Decreased and unchanged transcript levels are indicated in blue and grey, respectively. (C) Transcript level of lipolytic genes (left) in the liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. Schematic representation of triglyceride hydrolysis (right). Decreased or unchanged transcript levels are indicated in blue and grey, respectively. (D) Transcript level of lipogenic genes (left) in the liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. Schematic view of lipogenesis (right). Decreased and increased transcript levels are indicated in blue and red, respectively. (E) Transcript level of genes encoding urea cycle enzymes (left) in liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. Schematic view of the urea cycle (right). Decreased, unchanged, and increased transcript levels are indicated in blue, grey, and red, respectively. Data are means ± SEM. (A, B, C, D, E) Data were analysed by two‐way ANOVA with Sidak's multiple comparison test. § indicates a main genotype effect. (B) Data were analysed by a two‐tailed unpaired t‐test with Welch's correction when necessary. * or §P < 0.05, §§ or **P < 0.01, ***P < 0.001, and ****P < 0.0001. AA, acetoacetate; AA‐CoA, acetoacetyl‐CoA; Arg, arginine; asp, aspartate; BHB, beta‐hydroxybutyrate; CP, carbamoyl phosphate; DG, diglyceride; FA, fatty acid; HMG‐CoA, beta‐hydroxy beta‐methylglutaryl‐CoA; MG, monoglyceride; MUFA, monounsaturated fatty acid; OAA, oxaloacetic acid; TG, triglyceride.
The HPA axis is activated in 23‐week‐old Apc mice
The secretion of GC is regulated by the HPA axis. Hypothalamic mRNA level of corticotropin‐releasing hormone (CRH) that activates the pituitary secretion of adrenocorticotropic hormone (ACTH) was increased in 23‐week‐old Apc mice (Figure 4A). Accordingly, hypothalamic expression of Fkbp5, indicative of the hyperactive function of the hypothalamus in response to GC injection, 39 was also increased (Figure 4A). ACTH stimulates the release of GC by adrenal glands. mRNA levels of ACTH receptor (Mc2r), as well as those of Cyp21a1 and Cyp11b1, which encode enzymes involved in adrenal corticosterone biosynthesis, 40 were increased in 23‐week‐old Apc mice (Figure 4B). Adrenomegaly was also evident (Figure 4C), further supporting an hyperactivation of adrenal glands.42 Accordingly, the circulating level of corticosterone, the predominant GC in mouse, was higher in Apc mice than in WT littermates (Figure 4D). Hepatic transcript levels encoding steroid detoxifying enzymes, such as Papss2, Srd5a1, Ugt2b35, and Ugt2b36, 40 were also significantly decreased in 23‐week‐old Apc mice compared with WT mice (Figure 4E–4F). Finally, in agreement with the hypogonadotropic hypogonadism found in male Cushing's syndrome,43 testis mass was also significantly decreased in Apc mice (Figure 4G). Together, these data indicate that an activation of the HPA axis and a lower capacity of hepatic GC detoxification increase circulating corticosterone level in 23‐week‐old Apc mice.
Figure 4.
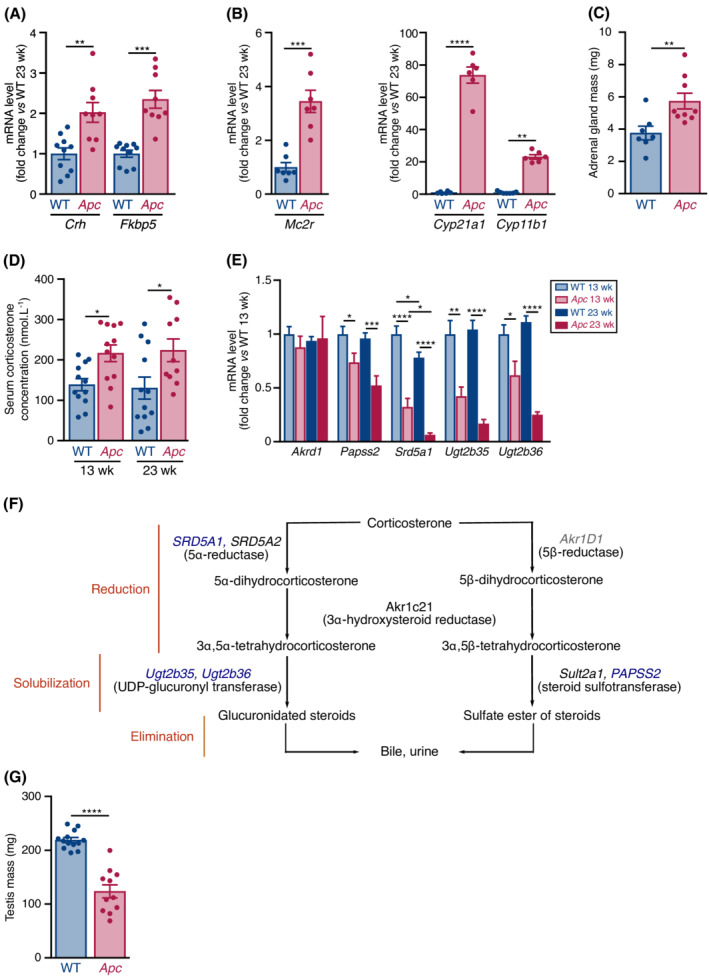
The hypothalamic–pituitary–adrenal axis is activated in 23‐week‐old Apc mice. (A) RT‐qPCR analysis of Crh and Fkbp5 mRNA levels in the hypothalamus of 23‐week‐old WT (n = 10) and Apc (n = 9) mice. (B) RT‐qPCR analysis of Mc2r (left), Cyp21a1 and Cyp11b1 (right) mRNA levels in the adrenal glands of 23‐week‐old WT (n = 6–7) and Apc (n = 6–7) mice. (C) Mass of adrenal glands in 23‐week‐old WT (n = 7) and Apc (n = 9) mice. (D) Serum corticosterone concentration in WT and Apc mice at 13 (WT n = 12; Apc n = 12) and 23 (WT n = 12; Apc n = 10) weeks. (E) RT‐qPCR analysis of transcripts encoding steroid detoxification enzymes in the liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. (F) Schematic view of liver corticosterone detoxification. Corticosterone is reduced, oxidized (not represented), or hydroxylated (not represented), and the products of these reactions are made water soluble by conjugation with glucuronic (glucuronidation) or sulfate (sulfonation) acid to facilitate their excretion in urine. Decreased and unchanged transcript levels are indicated in blue and grey, respectively. (G) Testis mass in 23‐week‐old WT (n = 13) and Apc (n = 11) mice. Data are means ± SEM. (A, B, C, G) Data were analysed by Mann–Whitney test or two‐tailed unpaired t‐test with Welch's correction when necessary. (D, E) Data were analysed by two‐way ANOVA with Sidak's multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
The expression of GC‐responsive genes is regulated in skeletal muscle and liver of 23‐week‐old Apc mice
Prompted by the finding of HPA axis activation, we pursued our investigations to determine whether expression of GC‐responsive genes is regulated in skeletal muscle and liver of Apc mice. Although the transcript level encoding GC receptor (Nr3c1) remained unchanged, mRNA level encoding HSD11B1, a dehydrogenase that converts the inactive 11‐dehydrocorticosterone to the active corticosterone44 was increased in the quadriceps muscle of 23‐week‐old Apc mice (Figure 5A), suggesting an increased conversion of inactive 11‐dehydrocorticosterone into active corticosterone. Accordingly, corticosterone content was increased in the quadriceps muscle of 23‐week‐old Apc mice (Figure 5B). Muscle corticosterone level also negatively correlated with body mass (Figure 5C). Previous studies indicated that administration of dexamethasone, a synthetic GC, triggers a typical transcriptional response in skeletal muscle characterized by an increase in the transcript level of Bnip3, 8 Ddit4, 7 , 8 Fkbp5, 7 , 8 Foxo1, 7 , 8 , 10 Foxo3, 7 , 8 Klf15, 7 , 8 Lc3b, 8 MuRF1, 7 , 8 , 10 , 31 MAFbx, 7 , 8 , 10 , 31 Mstn, 6 , 7 4ebp1, 10 Cebpd, 11 Mt1, 9 and Mt2, 9 , 10 whereas the transcript levels of other genes, such as Foxo4 7 , 8 and Bcat2, 7 remain unchanged. We therefore reasoned that this transcriptional signature should be reproduced in skeletal muscle of Apc mice. Slc39a14 (Zip14), whose expression is highly induced during cancer cachexia45 was also included in this analysis, suspecting that its expression could be also induced by dexamethasone. Intraperitoneal administration of dexamethasone for 5 days in WT mice reproduced this transcriptomic signature (Figure 5D and Table S2). Most importantly, the quadriceps muscle of 23‐week‐old Apc mice also displayed an increase in the expression of these GC‐responsive genes, as evidenced by the marked and significant increases in the transcript level of 4ebp1, Cebpd, Ddit4, Fkbp5, Foxo1, Foxo3, Klf15, MAFbx, Mt1, Mt2, MuRF1, and Slc39a14 (Figure 5D and Table S2). Out of 15 genes that were either up‐regulated or down‐regulated in dexamethasone‐treated mice, 13 behaved similarly in 23‐week‐old Apc mice. Overall, the overlap of the transcriptional signature in skeletal muscle of dexamethasone‐treated and 23‐week‐old Apc mice was significant (Figure 5E). Importantly, the GC‐dependent transcriptional response was partially shared by skeletal muscle of 13‐week‐old Apc mice.
Figure 5.
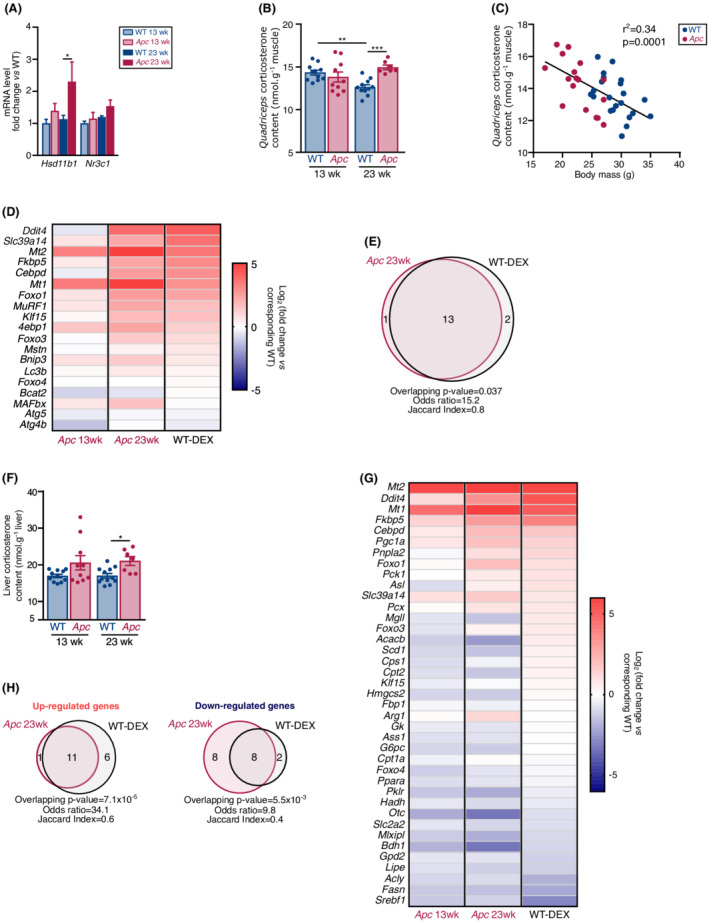
The expression of GC‐responsive genes is regulated in the quadriceps muscle and liver of Apc mice during cancer cachexia. (A) RT‐qPCR analysis of Hsd11b1 and Nr3c1 mRNA levels in the quadriceps muscle of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. (B) Corticosterone level in the quadriceps muscle of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 11; Apc n = 7) weeks (left). (C) Pearson correlation between quadriceps muscle corticosterone level and body mass in WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 11; Apc n = 7) weeks. (D) Heatmap showing a GC‐responsive transcriptional signature in the quadriceps muscle of 13‐week‐old (n = 10) and 23‐week‐old (n = 7) Apc mice, and dexamethasone‐treated (WT‐DEX n = 10) mice. Data were obtained by RT‐qPCR analysis and normalized to corresponding WT mice (13‐week‐old WT mice n = 11; 23‐week‐old WT mice n = 12, and vehicle‐treated WT mice n = 10). For Log2(fold change) > 5, cells are set by default in the most intense red. (E) Venn diagram of differentially expressed genes in the quadriceps muscle of 23‐week‐old Apc and WT‐DEX mice. (F) Corticosterone level in the liver of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 7) weeks. (G) Heatmap displaying a GC‐responsive transcriptional signature in the liver of 13‐week‐old (n = 10) and 23‐week‐old (n = 7) Apc mice, and dexamethasone‐treated WT mice (WT‐DEX n = 10). Data were obtained by RT‐qPCR analysis and normalized to corresponding WT mice (13‐week‐old WT mice n = 11; 23‐week‐old WT mice n = 12, and vehicle‐treated mice n = 10). For Log2(fold change) > 6, cells are set by default in the most intense red. (H) Venn diagram of up‐regulated (left) and down‐regulated (right) genes in the liver of 23‐week‐old Apc and WT‐DEX mice. Data are means ± SEM. (A, B, F) Data were analysed by two‐way ANOVA with Sidak's multiple comparison test. (C) Data were analysed with a Pearson correlation test. (D, G) Differentially expressed genes were identified after Mann–Whitney test or two‐tailed unpaired t‐test with Welch's correction if necessary. (E, H) Overlapping significance, odds ratio, and Jaccard index were calculated with Fisher's exact test. *P < 0.05, **P < 0.01, and ***P < 0.001.
We next hypothesized that if GC are systemic regulators of cancer cachexia, hepatic metabolic rewiring observed in 23‐week‐old Apc mice should sign a GC action and should be therefore reproduced in the liver of dexamethasone‐treated mice. First, liver corticosterone content was increased in 23‐week‐old Apc mice compared with age‐matched WT mice (Figure 5F). Expression levels of genes involved in glucose, nitrogen, ketone body, and lipid metabolisms described in Apc mice were then compared with those of dexamethasone‐treated mice. We also included genes that were identified as GC‐responsive (Cebpd, Ddit4, Fkbp5, Klf15, Mt1, Mt2, and Slc39a14) and non‐responsive (Foxo4) in skeletal muscle. Nineteen out of 28 genes that were differentially expressed in the liver of 23‐week‐old Apc mice were similarly regulated in dexamethasone‐treated mice (Figure 5G and Table S3). The overlap between transcription profiles was significant both for up‐regulated and down‐regulated transcripts (Figure 5H). Of note, only seven genes were similarly expressed in 13‐week‐old Apc mice and dexamethasone‐treated mice (Figure 5G), suggesting that the transcriptional regulation of GC‐responsive genes in the liver progressively takes place during the progression through the disease.
The expression of GC‐responsive genes is regulated in other animal models of cancer
We next wanted to further establish and strengthen the biological relevance of our observations in other models of cancer cachexia. Lewis lung carcinoma (LLC) tumour‐bearing mice, a fast tumour burden model of cancer,46 were first used (Data S1). Thirty‐five days after LLC cell inoculation, tumour‐bearing mice displayed pronounced weight loss (Figure S2A; Data S1) and marked decrease in skeletal muscle mass (Figure S2B; Data S1). The expression pattern of GC‐responsive genes in the gastrocnemius muscle of LLC‐tumour bearing mice was similar to the one reported in 23‐week‐old Apc mice, as shown by the increased expression of 4ebp1, Cebpd, Ddit4, Fkbp5, Foxo1, MAFbx, MuRF1, Mt1, Mt2, and Slc39a14 (Figure S2C).
The expression of GC‐responsive genes was also investigated in the established KPC mouse model of pancreatic ductal adenocarcinoma (PDAC). 29 As KPC mice showed heterogeneity in disease progression and mortality, mice were stratified as moderate (MOD) and severe (SEV) KPC animals. SEV KPC mice were euthanized at age ranging from 98 to 198 days (Figure 6A) upon reaching at least one of the following endpoints, body weight loss (based on the kinetic analysis of body weight), adipose tissue loss, abdominal distension, and/or lethargy. MOD KPC mice were euthanized before the onset of one of these cachectic features at age ranging from 96 to 146 days (Figure 6A). Despite heterogeneity in body weight at analysis partly due to the presence of large pancreatic tumours in some SEV KPC mice, body weight was significantly decreased in SEV KPC mice compared with control mice (Figure 6B). MOD and SEV KPC mice develop pancreatic lesions ranging from pancreatic intraepithelial neoplasia (PanIN) to PDAC (Figure 6C). No difference in the relative occurrence of PanIN and PDAC was observed between MOD and SEV KPC mice, indicating that the severity of the pancreatic lesions was not necessarily associated with the severity of the disease. SEV KPC mice exhibited lower masses of the tibialis anterior and gastrocnemius skeletal muscles compared with control mice (Figure 6D). Consistent with the data obtained in 23‐week‐old APC mice, a significant increase in the transcript level of Cebpd, Ddit4, Fkbp5, Foxo1, Foxo3, Klf15, MAFbx, Mstn, Mt1, Mt2, MuRF1, and Slc39a14 (Figure 6E) was also observed in the tibialis anterior muscle of SEV KPC mice. Importantly, this was not observed in MOD KPC mice, confirming that the signature was associated with the severity of the disease. In the liver, and with the notable exception of the transcripts of genes encoding gluconeogenic proteins whose expression remained unchanged (Pcx, Pck1, Fbp1) or even decreased (G6pc), the expression profiles of GC‐responsive genes (Cebpd, Ddit4, Fkbp5, Mt1, Mt2, Slc39a14) (Figure 6F), as well as that of glycolytic (Mlxipl, G6pd2, Pklr), ketogenic (Hmgcs2, Bdh1), and fatty acid synthesis (Acly, Fasn) genes (Figure 6G) were similar in SEV KPC mice and 23‐week‐old Apc mice. This response was not observed in MOD KPC mice.
Figure 6.
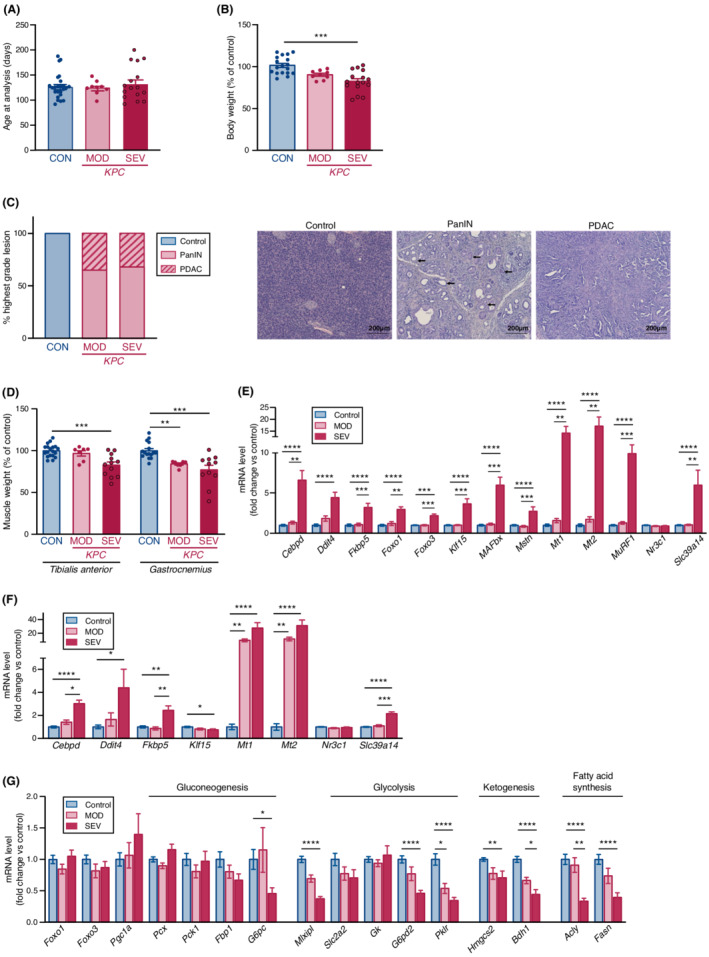
F The expression of GC‐responsive genes is also observed in KPC mice during pancreatic cancer cachexia. (A) Age at analysis of control (CON, n = 24), moderate (MOD, n = 9), and severe (SEV, n = 16) KPC mice. (B) SEV KPC mice (n = 16) have lower body weights than control littermates (n = 18) mice. MOD KPC mice (n = 9). (C) MOD (n = 9) and SEV (n = 15) KPC mice displayed similar percentage of pancreatic intraepithelial neoplasia (PanIN) and pancreatic ductal adenocarcinoma (PDAC) lesions (left). Representative pancreatic histopathology of lesions (right). Arrows indicate PanIN. Control littermates (n = 23) did not show any histological feature. (D) SEV KPC mice (n = 11–12) have lower tibialis anterior (TA) and gastrocnemius (GAS) muscle weights than littermate controls (n = 16–19) mice. (E) RT‐qPCR analysis of transcript levels of GC‐responsive genes in the tibialis anterior muscle of control (n = 17), MOD (n = 9) and SEV (n = 16) KPC mice. (F) RT‐qPCR analyses of transcript levels of GC‐responsive genes in the liver of control (n = 19), MOD KPC (n = 9), and SEV KPC (n = 14) mice. (G) RT‐qPCR analyses of transcript levels of gluconeogenic genes, glycolytic genes, ketogenic genes, and genes encoding fatty acid synthesis enzymes in the liver of control (n = 19), MOD KPC (n = 9), and SEV KPC mice (n = 14). Data are means ± SEM. (A, B, D, E, F, G) Data were analysed by Kruskal–Wallis test followed by Dunn's multiple comparison test. Closed symbols represent individual data points. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Discussion
In the present study, we demonstrate a unique phenotype in 23‐week‐old Apc mice characterized by an activation of the HPA axis, a lower potential for the clearance of GC by the liver, and elevated GC levels in the serum, skeletal muscle, and liver. Using molecular, biological, and biochemical assays, we show that HPA axis activation was associated with a transcriptional response of GC‐responsive genes involved in skeletal muscle catabolism and hepatic metabolism.
The hepatic expression of glucose metabolism‐related genes in 23‐week‐old Apc mice was rearranged in the direction of increased gluconeogenesis and reduced glycolysis, what with previous studies showing marked depletion in liver glycogen store in cancer cachectic mice, 27 ,47,48 indicate a greater reliance on hepatic glucose production for gluconeogenesis. Collectively, with the changes in glycaemia and lactatemia, these data are consistent with an increased systemic demand for glucose. Amino acids coming from skeletal muscle catabolism and the lactate produced by tumour metabolism can be used as substrates for hepatic gluconeogenesis to produce glucose for tumour growth. 4 Supporting this hypothesis, increased expression of glucose transporter 149 and increased lactate content50 has been reported in the tumour of cancer mice, suggesting active glucose uptake and increased lactate production by the tumour. As the cost of partial glucose oxidation into lactate by glycolysis (2 ATP) is less than the cost of glucose synthesis by gluconeogenesis (6 ATP), this metabolic cycle could contribute to energy loss in cachectic mice. One may also consider metabolic cycles between skeletal muscle and tumour, or between skeletal muscle and liver, whereby lactate produced by skeletal muscle from glucose could be used by the tumour (lactate oxidation for tumour growth) or by the liver for gluconeogenesis. However, data indicate a reduction in the whole energy metabolism of cachectic skeletal muscle,47,50–52 suggesting that these metabolic cycles would be less relevant in cancer mouse.
As previously described, 27 cachectic cancer mice displayed a hypo‐ketonemic phenotype characterized by a decrease in β‐hydroxybutyrate circulating level. Production of ketone bodies increases when acetyl‐CoA production by fatty acid oxidation exceeds the capacity of citrate to be incorporated into the tricarboxylic acid cycle. Here, the complete exhaustion of adipose tissue store and the decreased expression of fatty oxidation genes (Cpt1, Hadh1), Ppara, and PPARA ketogenic target genes (Hmgcs2, Bdh1) in the liver of 23‐week‐old Apc mice agree with the hypo‐ketonemic phenotype. Conversely, one may expect that ketone body production has transitorily increased during the time course of adipose tissue store depletion earlier during the development of cachexia. Accordingly, increased ketogenesis has been reported during the time course of the disease in C26 tumour‐bearing mice,48 a fast tumour growth model. One may also question the fate of ketogenic amino acids produced by skeletal muscle catabolism. However, as most of the ketogenic amino acids (phenylalanine, isoleucine, threonine, tryptophan, and tyrosine) are also gluconeogenic, our data suggest that amino acids coming from skeletal muscle would be mainly used to sustain liver gluconeogenesis. In support of this assumption, an increase in the expression of many genes involved in the conversion of amino acids into pyruvate has been reported in the liver of cachectic cancer mice. 27 Therefore, hypo‐ketonemia would be a hallmark of an advanced cachectic state characterized by depleted adipose tissue store. Our findings complement previous works, 27 showing that fenofibrate, a PPARA agonist, restores β‐hydroxybutyrate circulating level and prevents cancer cachexia in tumour‐bearing mice, and that ketone bodies attenuate skeletal muscle atrophy in cachectic mice.53 Maintaining ketone production could be thus favourable to limit the progression of cancer cachexia.
Our findings demonstrate that HPA axis activation in 23‐week‐old Apc mice was associated with a decrease in the transcript level of liver GC detoxifying enzymes, an increase in corticosterone content (serum, skeletal muscle, and liver), and the induction of a typical transcriptional response in skeletal muscle and liver that was recapitulated by dexamethasone injection in WT mice. In skeletal muscle, this signature was more specifically associated with the regulation of GC metabolism (Cebpd, Fkbp5, Klf15), the activation of metal cation transport and binding (Slc39a14, Mt1, Mt2), and the negative regulation of proteostasis (Bnip3, Ddit4, Foxo1, Foxo3, Lc3b, MuRF1, 4ebp1). We also showed that this transcriptional signature was shared by other models of cancer cachexia (LLC tumour‐bearing mice and KPC mice). Finally, the analysis of published transcriptomic lists of differentially expressed genes indicate that the transcriptional signature of GC‐responsive genes in skeletal muscle was also largely shared by multiple animal models of cancer cachexia (Figure S3; Data S1), further establishing the relevance of our observations. Although not investigated in the present study, GC may also exert indirect effects on skeletal muscle proteostasis by modulating testosterone biosynthesis. Indeed, GC are known to inhibit testosterone biosynthesis by Leydig cells,54 and circulating testosterone level is decreased in Apc mice,55 together with a decrease in gonad size55 (also observed in the present study). GC may thus indirectly contribute to muscle loss by decreasing testosterone production/secretion and its subsequent anabolic action. In the liver, GC‐responsive genes were involved in glycolysis and gluconeogenesis (Foxo1, Gpd2, Mlxipl, Pck1, Pcx, Pgc1a, Slc2a2), ketogenesis (Bdh1), lipid metabolism (Acly, Fasn, Hadh, Lipe, Pnpla2), GC metabolism (Cebpd, Fkbp5), and metal cation transport and binding (Slc39a14, Mt1, Mt2). Some transcripts, such as Arg1, Asl, Ass1, and Cps1 encoding urea cycle enzymes, were not coordinately regulated between 23‐week‐old Apc mice and dexamethasone‐treated mice, indicating that GC‐independent mechanisms also transcriptionally regulated liver metabolism. This transcriptional signature was also shared by SEV KPC mice, with the exception of genes encoding gluconeogenic proteins. The non‐responsiveness of gluconeogenic genes could be explained by the selective erosion of β‐cells (the endocrine cells that produce insulin) that occur in KPC mice, whereas the pool of alpha‐cells (the endocrine cells that produce glucagon) appear to be relatively preserved.56 This is expected to create an imbalance between the production of insulin and glucagon, which should conceivably alter the regulation of hepatic glucose metabolism in this mouse model. Overall, our data strongly support a role for GC in targeting transcriptional responses in skeletal muscle and liver of cachectic Apc mice. Importantly, animal models of cancer cachexia have been shown to only partially recapitulate the phenotype of human cancers.57 Therefore, a challenging aspect will be then to translate these advances to the clinic and determine whether this response occurs in human cancer patients, and if so, for what type of cancers and at what stage of the disease.
HPA axis activation and increased GC level may have potential impact on the development of insulin resistance, a condition that is frequently observed in human cancer patients and in animal models of cancer cachexia.58 Physiological doses of GC have thus been shown to inhibit the insulin signal through genomic and non‐genomic effects,59,60 suggesting that HPA axis activation in Apc mice may also contribute to insulin resistance. Corollary, GC‐induced impaired insulin signalling may also contribute to increase protein degradation and stimulate muscle wasting.61 Characterizing the mechanisms that contribute to the regulation of insulin pathway in relation to the HPA axis will help to better understand systemic metabolic issues of cancer cachexia, but also to delineate the genomic and non‐genomic effects of GC during cancer cachexia.
One may also question the existence of a gender effect in the regulation of HPA axis in Apc mice. Female mice have been described to produce a more robust neuroendocrine response to acute stress, evidenced by higher ACTH and corticosterone levels compared with males.62 Whether or not this can contribute to a gender difference in the sensibility to cancer cachexia is currently unknown, but clearly needs to be investigated.
Our data raise an important question: How is the HPA axis regulated to control the transcription of GC‐responsive genes during cancer cachexia? Previous studies indicated that intracerebroventricular injection of IL‐1β activated the HPA axis, increased the circulating corticosterone concentration, triggered the expression of GC‐responsive genes in skeletal muscle, and skeletal muscle atrophy.63 Accordingly, a hypothalamic inflammation has also been consistently reported in animal models of cancer cachexia,64–67 together with the invasion of circulating immune cells68,69 and astrogliosis,69 which also testify of hypothalamic inflammation. Although the existence of central inflammation in cancer cachectic patients is currently unknown, a chronic systemic inflammation state has also been associated with skeletal muscle mass loss in human cancer patients,70 and the intravenous injection of cytokines in cancer patients increases circulating cortisol level by activating the HPA axis.71,72 Tumour‐induced production of cytokines, together with the host immune response to tumour growth, can be thus centrally sensed and relayed within the hypothalamus to drive the activation of the HPA axis and increase the circulating level of GC during disease progression. In light of our findings, this mechanism may also function under other pathological situations associated with inflammation, such as sepsis. Thus, the physiological and molecular framework that we propose in the context of cancer cachexia could also be investigated in other cachexia‐associated diseases.
As the syndrome is firmly established in 23‐week‐old animals, data obtained at this time point only testify to the activation of the HPA axis. Similar ascertainments can also be drawn from the data obtained with SEV KPC mice. But one may question whether HPA axis activation occurs early during the pathogenic process to trigger cachexia, or later to maintain the driving force of cancer cachexia. In the first case, GC will be drivers of cancer cachexia, whereas in the second case they will be amplifiers of cancer cachexia. In an attempt to answer this question, we performed a subgroup analysis on 13‐week‐old mice. Indeed, 13‐week‐old Apc mice displayed heterogeneity in the syndrome. Comparative analysis with age‐matched WT mice allowed us to stratify 13‐week‐old Apc mice into pre‐cachectic (Pre‐CX) and cachectic (CX) Apc mice (Figure S4A–F; Data S1). Whereas all 13‐week‐old Apc mice experienced polyposis, CX Apc mice only displayed a GC‐responsive gene signature in skeletal muscle and liver (Figure S4J–M; Data S1). These data indicate that a GC‐dependent gene response is elicited as soon as 13‐weeks of age in a subset of Apc mice at a time when skeletal muscle atrophy is still ongoing. They also suggest that polyp growth would not appear to be a prerequisite for increased corticosterone levels and the occurrence of a GC‐dependent transcriptional response. However, this last observation must be qualified because Pre‐CX Apc mice had fewer polyps and a greater haematocrit than CX Apc mice (Figure S4B and S4C; Data S1), and that body weight and muscle mass were positively correlated with haematocrit (Figure S4N–O; Data S1), indicating that skeletal muscle mass loss is functionally linked to the development of polyposis. To get further in establishing the role of GC, targeting HPA‐GC pathway during the time course of the disease would clarify the time frame of HPA axis activation and GC‐dependent transcriptional response in relation with the development of cancer cachexia and tumour growth. Considering the systemic nature of the cachectic syndrome that involves multiple circulating factors that are regulated within a specific time frame during the progression of the disease, we believe that the response we identified here is part of a more complex systemic response that occurs during the progression of the disease.
Finally, our data also question the relevance of GC‐inhibiting strategies to combat cancer cachexia. Pioneering studies indicate that adrenalectomy of tumour‐bearing animals did not attenuate cancer cachexia. 26 ,73 However, this surgical approach to the treatment of cancer cachexia induces weight loss by itself.74 The steroid hormone inhibitor RU486 has provided contrasting results, with some positive effects on the attenuation of body weight loss, 24 or not. 22 , 23 However, in addition to having anti‐GC effects, RU486 also exerts antiprogestogen and antiandrogenic effects, which may mitigate the potential anticatabolic effects of GC inhibition. By contrast, the potential relevance of GC‐based therapy has been convincingly reported in LLC‐tumour bearing mice with targeted deletion of GC receptor in skeletal muscle. 12 This single targeted tissue approach showed that skeletal muscle mass loss was abrogated in tumour‐bearing mice. One may thus speculate that targeting GC at the systemic level by specific molecular tools may be promising in preclinical models of cancer cachexia.
While we highlight the implication of HPA axis activation and GC in the course of cancer cachexia, the current work does not investigate the upstream mechanisms by which the HPA axis is activated and does not determine whether GC are drivers and/or amplifiers of cancer cachexia. Anyhow, it is likely that additional local or systemic signals also contribute to skeletal muscle wasting and liver transcriptional rewiring. Finally, while previous works have shown that circulating GC level in human patients are associated with the severity of cachexia, 18 , 19 the direct human applicability of our findings remains to be established.
In summary, our results demonstrate that HPA axis activation and increased corticosterone levels in Apc mice are associated with the transcription of GC‐responsive genes in skeletal muscle and liver of cachectic Apc mice. These results highlight a neuroendocrine mechanism that involves the HPA‐GC pathway in contributing to the transcriptional regulation of skeletal muscle catabolism and hepatic metabolism during cancer cachexia in Apc mice. Understanding the mechanisms regulating the HPA axis and GC secretion during cancer cachexia, together with the knowledge of the molecular basis of the GC‐induced effects in skeletal muscle and liver will offer the potential to develop strategies that may contribute to minimize the debilitating effects of cancer cachexia.
Conflict of interest
The authors declare no competing interest.
Funding
A. M. was financially supported by the Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation. D. F. was supported by the Fondation ARC pour la Recherche sur le Cancer and the Ligue contre le Cancer (Comité Loire). The AFM‐Téléthon is also acknowledged for its constant support. L. B. and L. S. were supported by the Fondation ARC pour la Recherche sur le Cancer and the Ligue contre le Cancer (Comité Rhône). A. E. and V. C. were financially supported by the Agence Nationale de la Recherche. V. C.‐R. was financially supported by the Ligue Nationale contre le Cancer (Comité Saône et Loire).
Supporting information
Figure S1. Apc mice recapitulate the main features of cancer cachexia.
(A) Food intake in WT (n = 14) and Apc (n = 14 at 15 weeks) mice.
(B) Tibia length of WT and Apc mice at 13 (WT n = 10; Apc n = 10) and 23 (WT n = 12; Apc n = 8) weeks.
(C) Adipose tissue mass of WT and Apc mice at 13 (WT n = 11; Apc n = 9) and 23 weeks (WT n = 11; Apc n = 8).
(D) Extensor digitorum longus (left), tibialis anterior (center) and soleus (right) muscle mass of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 6–8) weeks.
(E) Pearson correlation between body mass and quadriceps muscle mass in WT (n = 23) and Apc (n = 18) mice at 13 and 23 weeks.
(F) Pearson correlation between P0 and the mass of tibialis anterior muscle in 23‐week‐old WT (n = 14) and Apc (n = 8) mice.
(G) Maximum rate of force development (left) and maximum rate of force relaxation (right) during a single twitch of tibialis anterior muscle in 23‐week‐old WT (n = 8) and Apc (n = 5) mice.
Data are means ± SEM. (A, B, C, D) data were analyzed by a two‐way ANOVA followed by a Sidak's multiple comparison test. (E, F) Data were analyzed with a Pearson correlation test. (G) data were analyzed by a two‐tailed unpaired t‐test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Figure S2. Tumor‐free body weight, gastrocnemius muscle weight, and RT‐qPCR analysis of GC‐responsive genes in the gastrocnemius muscle of Lewis lung carcinoma (LLC)‐tumor bearing mice.
(A) Body weight in wild type (n = 9) and LLC‐tumor bearing mice (n = 11) 5 weeks after LLC inoculation, as previously published in 54.
(B) Gastrocnemius muscle weight in wild type (n = 9) and LLC‐tumor bearing mice (n = 12) 5 weeks after LLC inoculation, as previously published in 54.
(C) RT‐qPCR analysis of GC‐responsive gene mRNA level in the gastrocnemius muscle of WT (n = 6–7) and LLC‐tumor bearing mice (WT‐LLC, n = 5) 5 weeks after LLC inoculation.
Data are means ± SEM. Data were analyzed by a two‐tailed unpaired t‐test with Welch's correction when necessary. * p < 0.05, **p < 0.01, and ***p < 0.001.
Figure S3. GC‐responsive gene expression in skeletal muscle of cachectic cancer animals relative to controls in published transcriptomic studies.
Transcriptomic data from referenced studies were specifically screened for the expression of GC‐responsive genes in skeletal muscle. A red box indicates a significant increase of the transcript level. A white box indicates unchanged transcript level.
Figure S4. Morphometric, biochemical, and transcriptional analyses in 13‐week‐old WT mice, pre‐cachectic (Pre‐CX) and cachectic (CX) 13‐week‐old Apc mice.
(A) Body weight, (B) polyp number, (C) hematocrit, (D) adipose tissue weight, (E) quadriceps, and (F) gastrocnemius muscle weight in WT mice (n = 11), Pre‐CX (n = 4–5) and CX (n = 3–5) Apc mice.
(G) Serum corticosterone concentration, (H) quadriceps and (I) liver corticosterone content of WT mice (n = 11–12), Pre‐CX (n = 7–5) and CX (n = 5) Apc mice.
(I) RT‐qPCR analyses and (K) protein level of 4EBP1 and phosphorylated ribosomal protein S6 (rpS6Ser235/236) in the quadriceps muscle of Pre‐CX (n = 4) and CX (n = 4) Apc mice.
(L and M) RT‐qPCR analyses in the liver of Pre‐CX (n = 4–5) and CX (n = 4–5) Apc mice.
(N) Pearson correlation between body mass and hematocrit in WT (n = 10), Pre‐CX Apc (n = 5), and CX Apc (n = 5) mice.
(O) Pearson correlation between quadriceps muscle mass and hematocrit in WT (n = 10), Pre‐CX Apc (n = 5), and CX Apc (n = 5) mice.
Data are means ± SEM. (A to M) Data were analyzed by a Kruskal‐Wallis test followed by Dunn's multiple comparison test. (N, O) Data were analyzed with a Pearson correlation test. *p < 0.05, **p < 0.01, and *** p < 0.001. a and b indicate a significant difference with 13‐week‐old WT mice and 13‐week‐old Pre‐CX Apc mice, respectively. Dotted lines indicate the transcript level of WT mice.
Table S1. List of primers used for RT‐qPCR analysis.
Table S2. Transcript levels analyzed by RT‐qPCR in the quadriceps muscle of 13‐ and 23‐week‐old Apc mice, and dexamethasone‐treated mice.
Table S3. Transcript levels analyzed by RT‐qPCR in the liver of 13‐ and 23‐week‐old Apc mice, and dexamethasone‐treated mice.
Data S1. Supporting Information
Data S2. Supporting Information
Acknowledgements
We thank the Plexan animal facility (Université Jean Monnet Saint Etienne), AniCan animal facility (Centre de Recherche en Cancérologie de Lyon) and the SFR Santé Lyon‐Est (UCBL, UAR3453/CNRS, US7/Inserm) animal facility ANIPHY for animal care. GIMAP laboratory (Université Jean Monnet Saint Etienne) is acknowledged for the use of cryostat microtome, and SAINBIOSE Laboratory (Université Jean Monnet Saint Etienne) for the use of microplate reader. Thomas Simonet is acknowledged for his help in bioinformatic analyses. Rémi Mounier is gratefully acknowledged for helpful and stimulating discussions. The authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia Sarcopenia and Muscle. 41
Martin A., Castells J., Allibert V., Emerit A., Zolotoff C., Cardot‐Ruffino V., Gallot Y. S., Vernus B., Chauvet V., Bartholin L., Schaeffer L., Durieux A.‐C., Hourdé C., Favier F. B., Mazelin L., and Freyssenet D. (2022) Hypothalamic–pituitary–adrenal axis activation and glucocorticoid‐responsive gene expression in skeletal muscle and liver of Apc mice, Journal of Cachexia, Sarcopenia and Muscle, 13, 1686–1703, 10.1002/jcsm.12939
REFERENCES
- 1. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013;10:90–99. [DOI] [PubMed] [Google Scholar]
- 2. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 3. Anker MS, Holcomb R, Muscaritoli M, von Haehling S, Haverkamp W, Jatoi A, et al. Orphan disease status of cancer cachexia in the USA and in the European Union: a systematic review. J Cachexia Sarcopenia Muscle 2019;10:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt SF, Rohm M, Herzig S, Berriel Diaz M. Cancer cachexia: more than skeletal muscle wasting. Trends Cancer 2018;4:849–860. [DOI] [PubMed] [Google Scholar]
- 5. Goldberg AL. Protein turnover in skeletal muscle. II. Effects of denervation and cortisone on protein catabolism in skeletal muscle. J Biol Chem 1969;244:3223–3229. [PubMed] [Google Scholar]
- 6. Ma K, Mallidis C, Bhasin S, Mahabadi V, Artaza J, Gonzalez‐Cadavid N, et al. Glucocorticoid‐induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab 2003;285:E363–E371. [DOI] [PubMed] [Google Scholar]
- 7. Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 2011;13:170–182. [DOI] [PubMed] [Google Scholar]
- 8. Shimizu N, Maruyama T, Yoshikawa N, Matsumiya R, Ma Y, Ito N, et al. A muscle‐liver‐fat signalling axis is essential for central control of adaptive adipose remodelling. Nat Commun 2015;6:6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Summermatter S, Bouzan A, Pierrel E, Melly S, Stauffer D, Gutzwiller S, et al. Blockade of metallothioneins 1 and 2 increases skeletal muscle mass and strength. Mol Cell Biol 2017;37:e00305–e00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab 2012;302:E1210–E1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang H, Mammen J, Wei W, Menconi M, Evenson A, Fareed M, et al. Expression and activity of C/EBPbeta and delta are upregulated by dexamethasone in skeletal muscle. J Cell Physiol 2005;204:219–226. [DOI] [PubMed] [Google Scholar]
- 12. Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, et al. Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J 2013;27:3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC‐1. Nature 2001;413:131–138. [DOI] [PubMed] [Google Scholar]
- 14. Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC‐1. Nature 2001;413:179–183. [DOI] [PubMed] [Google Scholar]
- 15. Holroyde CP, Skutches CL, Boden G, Reichard GA. Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res 1984;44:5910–5913. [PubMed] [Google Scholar]
- 16. Lundholm K, Bennegard K, Eden E, Svaninger G, Emery PW, Rennie MJ. Efflux of 3‐methylhistidine from the leg in cancer patients who experience weight loss. Cancer Res 1982;42:4807–4811. [PubMed] [Google Scholar]
- 17. Waterhouse C, Jeanpretre N, Keilson J. Gluconeogenesis from alanine in patients with progressive malignant disease. Cancer Res 1979;39:1968–1972. [PubMed] [Google Scholar]
- 18. Cala MP, Agullo‐Ortuno MT, Prieto‐Garcia E, Gonzalez‐Riano C, Parrilla‐Rubio L, Barbas C, et al. Multiplatform plasma fingerprinting in cancer cachexia: a pilot observational and translational study. J Cachexia Sarcopenia Muscle 2018;9:348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor‐induced IL‐6 reprograms host metabolism to suppress anti‐tumor immunity. Cell Metab 2016;24:672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knapp ML, al‐Sheibani S, Riches PG, Hanham IW, Phillips RH. Hormonal factors associated with weight loss in patients with advanced breast cancer. Ann Clin Biochem 1991;28:480–486. [DOI] [PubMed] [Google Scholar]
- 21. Costelli P, Carbo N, Tessitore L, Bagby GJ, Lopez‐Soriano FJ, Argiles JM, et al. Tumor necrosis factor‐alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J Clin Invest 1993;92:2783–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Llovera M, Garcia‐Martinez C, Costelli P, Agell N, Carbo N, Lopez‐Soriano FJ, et al. Muscle hypercatabolism during cancer cachexia is not reversed by the glucocorticoid receptor antagonist RU38486. Cancer Lett 1996;99:7–14. [DOI] [PubMed] [Google Scholar]
- 23. Rivadeneira DE, Naama HA, McCarter MD, Fujita J, Evoy D, Mackrell P, et al. Glucocorticoid blockade does not abrogate tumor‐induced cachexia. Nutr Cancer 1999;35:202–206. [DOI] [PubMed] [Google Scholar]
- 24. Russell ST, Tisdale MJ. The role of glucocorticoids in the induction of zinc‐alpha2‐glycoprotein expression in adipose tissue in cancer cachexia. Br J Cancer 2005;92:876–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanaka Y, Eda H, Tanaka T, Udagawa T, Ishikawa T, Horii I, et al. Experimental cancer cachexia induced by transplantable colon 26 adenocarcinoma in mice. Cancer Res 1990;50:2290–2295. [PubMed] [Google Scholar]
- 26. Tessitore L, Costelli P, Baccino FM. Pharmacological interference with tissue hypercatabolism in tumour‐bearing rats. Biochem J 1994;299:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goncalves MD, Hwang SK, Pauli C, Murphy CJ, Cheng Z, Hopkins BD, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci U S A 2018;115:E743–E752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992;256:668–670. [DOI] [PubMed] [Google Scholar]
- 29. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–483. [DOI] [PubMed] [Google Scholar]
- 30. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 2009;361:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 32. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004;18:2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC Jr, et al. Insulin‐dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′‐cap function. Nature 1994;371:762–767. [DOI] [PubMed] [Google Scholar]
- 34. Poungvarin N, Chang B, Imamura M, Chen J, Moolsuwan K, Sae‐Lee C, et al. Genome‐wide analysis of ChREBP binding sites on male mouse liver and white adipose chromatin. Endocrinology 2015;156:1982–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langston PK, Nambu A, Jung J, Shibata M, Aksoylar HI, Lei J, et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat Immunol 2019;20:1186–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin‐regulated hepatic gluconeogenesis through FOXO1‐PGC‐1alpha interaction. Nature 2003;423:550–555. [DOI] [PubMed] [Google Scholar]
- 37. Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator‐activated receptor alpha‐inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 2000;275:28918–28928. [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez JC, Gil‐Gomez G, Hegardt FG, Haro D. Peroxisome proliferator‐activated receptor mediates induction of the mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA synthase gene by fatty acids. J Biol Chem 1994;269:18767–18772. [PubMed] [Google Scholar]
- 39. Scharf SH, Liebl C, Binder EB, Schmidt MV, Muller MB. Expression and regulation of the Fkbp5 gene in the adult mouse brain. PLoS One 2011;6:e16883. 10.1371/journal.pone.0016883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schiffer L, Barnard L, Baranowski ES, Gilligan LC, Taylor AE, Arlt W, et al. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: a comprehensive review. J Steroid Biochem Mol Biol 2019;194:105439. 10.1016/j.jsbmb.2019.105439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. von Haehling S, Coats AJS, Anker SD Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Apc mice recapitulate the main features of cancer cachexia.
(A) Food intake in WT (n = 14) and Apc (n = 14 at 15 weeks) mice.
(B) Tibia length of WT and Apc mice at 13 (WT n = 10; Apc n = 10) and 23 (WT n = 12; Apc n = 8) weeks.
(C) Adipose tissue mass of WT and Apc mice at 13 (WT n = 11; Apc n = 9) and 23 weeks (WT n = 11; Apc n = 8).
(D) Extensor digitorum longus (left), tibialis anterior (center) and soleus (right) muscle mass of WT and Apc mice at 13 (WT n = 11; Apc n = 10) and 23 (WT n = 12; Apc n = 6–8) weeks.
(E) Pearson correlation between body mass and quadriceps muscle mass in WT (n = 23) and Apc (n = 18) mice at 13 and 23 weeks.
(F) Pearson correlation between P0 and the mass of tibialis anterior muscle in 23‐week‐old WT (n = 14) and Apc (n = 8) mice.
(G) Maximum rate of force development (left) and maximum rate of force relaxation (right) during a single twitch of tibialis anterior muscle in 23‐week‐old WT (n = 8) and Apc (n = 5) mice.
Data are means ± SEM. (A, B, C, D) data were analyzed by a two‐way ANOVA followed by a Sidak's multiple comparison test. (E, F) Data were analyzed with a Pearson correlation test. (G) data were analyzed by a two‐tailed unpaired t‐test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Figure S2. Tumor‐free body weight, gastrocnemius muscle weight, and RT‐qPCR analysis of GC‐responsive genes in the gastrocnemius muscle of Lewis lung carcinoma (LLC)‐tumor bearing mice.
(A) Body weight in wild type (n = 9) and LLC‐tumor bearing mice (n = 11) 5 weeks after LLC inoculation, as previously published in 54.
(B) Gastrocnemius muscle weight in wild type (n = 9) and LLC‐tumor bearing mice (n = 12) 5 weeks after LLC inoculation, as previously published in 54.
(C) RT‐qPCR analysis of GC‐responsive gene mRNA level in the gastrocnemius muscle of WT (n = 6–7) and LLC‐tumor bearing mice (WT‐LLC, n = 5) 5 weeks after LLC inoculation.
Data are means ± SEM. Data were analyzed by a two‐tailed unpaired t‐test with Welch's correction when necessary. * p < 0.05, **p < 0.01, and ***p < 0.001.
Figure S3. GC‐responsive gene expression in skeletal muscle of cachectic cancer animals relative to controls in published transcriptomic studies.
Transcriptomic data from referenced studies were specifically screened for the expression of GC‐responsive genes in skeletal muscle. A red box indicates a significant increase of the transcript level. A white box indicates unchanged transcript level.
Figure S4. Morphometric, biochemical, and transcriptional analyses in 13‐week‐old WT mice, pre‐cachectic (Pre‐CX) and cachectic (CX) 13‐week‐old Apc mice.
(A) Body weight, (B) polyp number, (C) hematocrit, (D) adipose tissue weight, (E) quadriceps, and (F) gastrocnemius muscle weight in WT mice (n = 11), Pre‐CX (n = 4–5) and CX (n = 3–5) Apc mice.
(G) Serum corticosterone concentration, (H) quadriceps and (I) liver corticosterone content of WT mice (n = 11–12), Pre‐CX (n = 7–5) and CX (n = 5) Apc mice.
(I) RT‐qPCR analyses and (K) protein level of 4EBP1 and phosphorylated ribosomal protein S6 (rpS6Ser235/236) in the quadriceps muscle of Pre‐CX (n = 4) and CX (n = 4) Apc mice.
(L and M) RT‐qPCR analyses in the liver of Pre‐CX (n = 4–5) and CX (n = 4–5) Apc mice.
(N) Pearson correlation between body mass and hematocrit in WT (n = 10), Pre‐CX Apc (n = 5), and CX Apc (n = 5) mice.
(O) Pearson correlation between quadriceps muscle mass and hematocrit in WT (n = 10), Pre‐CX Apc (n = 5), and CX Apc (n = 5) mice.
Data are means ± SEM. (A to M) Data were analyzed by a Kruskal‐Wallis test followed by Dunn's multiple comparison test. (N, O) Data were analyzed with a Pearson correlation test. *p < 0.05, **p < 0.01, and *** p < 0.001. a and b indicate a significant difference with 13‐week‐old WT mice and 13‐week‐old Pre‐CX Apc mice, respectively. Dotted lines indicate the transcript level of WT mice.
Table S1. List of primers used for RT‐qPCR analysis.
Table S2. Transcript levels analyzed by RT‐qPCR in the quadriceps muscle of 13‐ and 23‐week‐old Apc mice, and dexamethasone‐treated mice.
Table S3. Transcript levels analyzed by RT‐qPCR in the liver of 13‐ and 23‐week‐old Apc mice, and dexamethasone‐treated mice.
Data S1. Supporting Information
Data S2. Supporting Information