

Abstract
Background
Rhabdomyolysis (RM) is a common complication of exertional heat stroke (EHS) and constitutes a direct cause of death. However, the mechanism underlying RM following EHS remains unclear.
Methods
The murine EHS model was prepared by our previous protocol. RNA sequencing is applied to identify the pathological pathways that contribute to RM following EHS. Inhibition of the acyl‐CoA synthetase long‐chain family member 4 (ACSL4) was achieved by RNA silencing in vitro prior to ionomycin plus heat stress exposure or pharmacological inhibitors in vivo prior to heat and exertion exposure. The histological changes, the iron accumulation, oxidized phosphatidylethanolamines species, as well as histological evaluation and levels of lipid metabolites in skeletal muscle tissues were measured.
Results
We demonstrated that ferroptosis contributes to RM development following EHS. Ferroptosis inhibitor ferrostatin‐1 administration once EHS onset significantly ameliorated the survival rate of EHS mice from 35.357% to 52.288% within 24 h after EHS (P = 0.0028 compared with control) and markedly inhibited RM development induced by EHS. By comparing gene expression of between sham heat rest (SHR) (n = 3) and EHS (n = 3) mice in the gastrocnemius (Gas) muscle tissue, we identified that Acsl4 mRNA expression is elevated in Gas muscle tissue of EHS mice (P = 0.0038 compared with SHR), so as to its protein levels (P = 0.0001 compared with SHR). Followed by increase in creatine kinase (CK) and myoglobin (MB) levels, the labile iron accumulation, decrease in glutathione peroxidase 4 (GPX4) expression, and elevation of lipid peroxidation products. From in vivo and in vitro experiments, inhibition of Acsl4 significantly improves muscle cell death caused by EHS, thereby ameliorating RM development, followed by reduction in CK and MB levels by 30–40% (P < 0.0001; n = 8–10) and 40% (P < 0.0001; n = 8–10), restoration of GPX4 expression, and decrease in lipid peroxidation products. Mechanistically, ACSL4‐mediated RM seems to be Yes‐associated protein (YAP) dependent via TEA domain transcription factor1/TEA domain transcription factor4.
Conclusions
These findings demonstrate an important role of ACSL4 in mediating ferroptosis activation in the development of RM following EHS and suggest that targeting ACSL4 may represent a novel therapeutic strategy to limit the skeletal muscle cell death and prevent RM after EHS.
Keywords: Exertional heat stroke, Rhabdomyolysis, Ferroptosis, ACSL4, Lipid peroxidation
Introduction
Rhabdomyolysis (RM) is one of the most common complications of exertional heat stroke (EHS). 1 It is a clinical emergency caused by the rapid (rhabdo) skeletal muscle (myo) breakdown (lysis), resulting in the subsequent release of intracellular muscle components into the systemic circulation. The event could potentially lead to multiple organ injury and failure. 2 If the patients are not promptly treated, RM can result in considerable morbidity and mortality induced by EHS. 3 However, the mechanism underlying RM following EHS remains unclear.
Massive degenerative changes of skeletal muscle cells, including cell death, are the most common pathological change of EHS. 3 , 4 Apoptosis, 5 necroptosis, 6 and autophagy 7 , 8 represent extremes of the response of the muscle to injury and often coexisted in experimental models of acute muscle injury. 9 However, the above‐mentioned various deaths cannot clarify the mechanism of RM development following EHS. 10 Ferroptosis is a novel form of regulated cell death characterized by the iron‐dependent accumulation of lipid peroxides to lethal levels, which is morphologically, biochemically, and genetically distinct from apoptosis, necroptosis, and autophagy. 11 A recent study suggested that the activation of ferroptosis impeded the skeletal muscle regeneration in aged mice. 12 Although the role of ferroptosis in mouse skeletal muscles under ageing or injury conditions has been reported, 13 the role of cell ferroptosis in the RM following EHS has not yet been addressed.
ACSL4 has been identified as an essential component for ferroptosis execution. 14 ACSL4 enriches cellular membranes with long polyunsaturated ω 6 fatty acids, such as arachidonic acid (AA) and adrenic acid (AdA), which were able to sensitize the cells to undergo ferroptosis compared with the ω 3 fatty acids, 14 and suppression of ω 6 fatty acids esterification into PE by genetic or pharmacological inhibition of ACSL4 acts as a specific antiferroptotic rescue pathway. 15 ACSL4‐mediated ferroptosis has been reported to exacerbate the tissue injury, such as the intestinal injury caused by ischaemia/reperfusion 16 and the brain injury induced by ischemic stroke 17 ; however, there is no even research to clarify the effects of ACSL4 induced ferroptoisis on RM development after EHS.
In this study, using the previous established murine EHS model, 18 we demonstrate that YAP, acting through TEAD1/TEAD4 relevant Hippo signalling, mediated EHS‐induced upregulation of ACSL4 which in turn, increased lipid peroxiadation level, as well as skeletal muscle cell ferroptosis and subsequent augmented skeletal muscle tissue injury. Inhibition of YAP, pharmacological inhibition of ACSL4, or blocking lipid peroxidation prevented EHS‐induced ferroptosis and ameliorated skeletal muscle tissue injury. These findings suggest that targeting ACSL4 may represent a novel therapeutic strategy to limit cell death and prevent RM after EHS.
Materials and methods
Murine model of exertional heat stroke with rhabdomyolysis
Briefly, the animals were placed in the climate chamber in the absence of food and water. The chamber temperature was then increased to 39.5°C within 40 min, with a relative humidity (RH) 65%. The core temperatures (T c ) were measured at 10‐min intervals. The mice were allowed to run on the treadmill at a constant speed of 15 rpm/min. The time point of at which the T c start to reach 43°C or the mice appeared to be exhausted was taken as a reference point of EHS onset. Immediately after the onset of EHS, the mice were then removed from the climate chamber, weighed, and returned to their original cages with an ambient temperature of 25°C and with ad libitum access to food and water. 18
Results
Exertional heat stroke onset induces rhabdomyolysis development
To investigate the mechanism of RM development following EHS, we have established an EHS murine model with the high incidence rate for RM, which may truly reflect the clinical characteristics. 18 Based on the murine model, we observed that the CK and MB levels were powerful increased after EHS onset, and reached to the peak levels at 6 h after EHS (Figure S1A–S1B). Then, we focused on the 6 h after EHS as the monitoring point and measured the severity of skeletal muscle injury among the four different types: transverse abdominal, extensor digitorum longus, soleus (Figure S1C–S1D), and gastrocnemius (Gas) (Figures 1A and S1D). Figure 1A demonstrates the most severely histological changes in Gas at 6 h after EHS, as indicated by the markedly decreased muscle fibre density, remarkably disorganized muscle fibre arrangement, and significantly swelling degeneration. These phenomena indicated the most serious damage in Gas muscle. In order to investigate the potential death mode of skeletal muscle cells in RM following EHS, we performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology (GO) enrichment analysis based on the RNA sequencing data (Figures S2 and 1B–1C). Interestingly, as one of the pathways that affected cell death, ferroptosis pathway was suggested to play a significant role in RM following EHS (Figure 1B). A total of 14 genes with significant expression differences were enriched in ferroptosis pathway, of which 13 genes were significantly upregulated (Figure S2B). In addition, ferroptosis‐related lipid and iron metabolism were found dysregulated (Figure S2C–S2F). Ferroptosis is an iron‐dependent, lipid peroxidation‐driven cell death cascade, with special mitochondria characteristics. 19 Figure 1D shows the changes in the Gas muscle tissue ultrastructure at 6 h after EHS. The images reveal that the outer mitochondrial membrane has ruptured and mitochondrial cristae has disappeared, which is consistent with the ferroptosis ultrastructure features in intestinal ischaemia/reperfusion. 16 The prostaglandin‐endoperoxide synthase 2 (Ptgs2) mRNA, a universal marker of ferroptosis marker, 20 , 21 showed the maximal increase at 6 h after EHS (Figure 1E). The oxidized PE species, the key phospholipids that undergo oxidation and drive ferroptosis, 15 especially the PE (18:0/22:4 + 1[O]), was upregulated at 6 h after EHS (Figure 1F). Consistently, the lipid metabolites as potential markers of ferroptosis, including malondialdehyde (MDA), 12‐hydroxyeicosatetraenoic acid (12‐HETE), and 15‐hydroxyeicosatetraenoic acid (15‐HETE), were also markedly increased in the Gas muscle and reached to the peak levels at 6 h after EHS (Figure 1G–1I).
Figure 1.
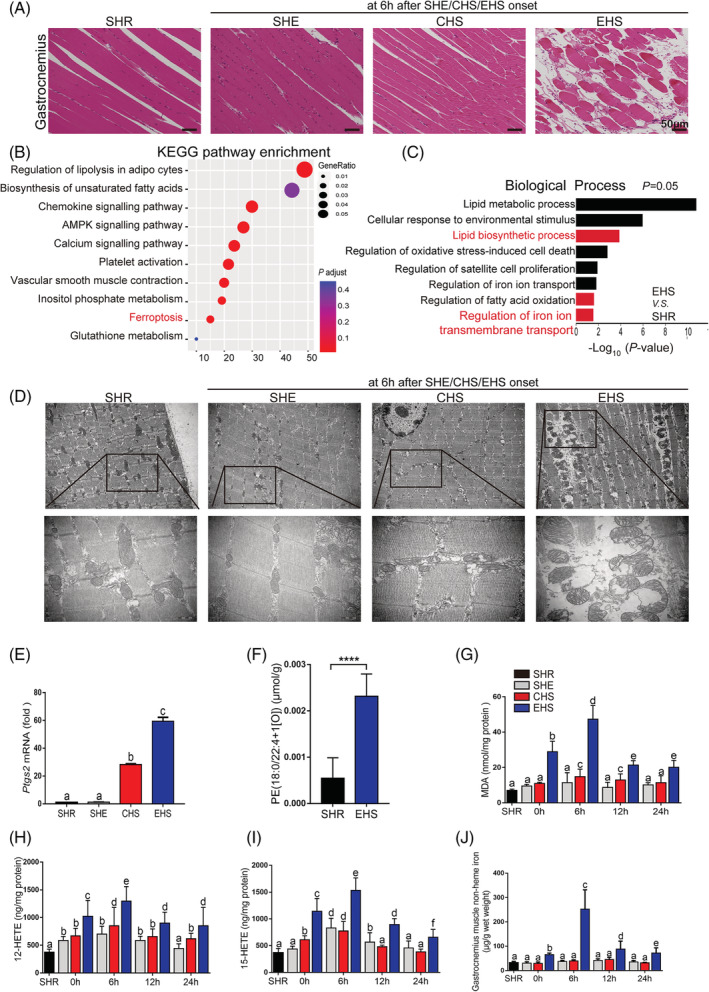
Distinguishing features of ferroptosis in RM after EHS. All samples of muscle tissues were collected at 6 h after SHE, CHS, and EHS onset, SHR as the control group (A–F). (A) Representative H&E staining of Gas muscle (SHR, SHE, CHS, and EHS, scale bar = 50 μm in H&E). (B) KEGG pathway enrichment analysis of upregulated differentially expressed genes (DEGs) identified ferroptosis‐related genes highly expressed in Gas muscle of EHS mice. (C) Gene ontology (GO: biological process) analysis against upregulated genes between SHR and EHS mice. (D) TEM representative images among SHR, SHE, CHS, and EHS groups. (E) Relative levels of Ptgs2 mRNA expression were measured in the Gas muscle suffering from SHE, CHS, EHS, or SHR (n = 6 mice/group). (F) LC–MS/MS assessment of pro‐ferroptotic PEox (PE (18:0/22:4 + 1[O])) (n = 6 mice/group). Gas muscle tissues were collected at intervals of 0, 6, 12, and 24 h following SHE, CHS, and EHS onset, SHR as the control group. (G–I) In parallel, the levels of MDA, 12‐HETE, and 15‐HETE from 0 to 24 h were assayed using the respective kits (n = 6 mice/group). (J) The non‐heme iron level in each group from 0 to 24 h (n = 6 mice/group). Significance in (F) was calculated using the Student's t‐test; ****P < 0.0001. Summary data are presented as the mean ± SEM. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labelled with different letters differed significantly (*P < 0.05).
The tissue non‐heme iron was accumulated in RM tissue following EHS, and archived to the peak levels at 6 h after EHS (Figure 1J). Furthermore, the iron accumulation in RM tissue was confirmed by the Prussia blue staining (Figure S3A), and this process is accompanied by significant degradation of heme (Figure S3B). Based on our KEGG pathway and GO enrichment analysis (Figure S2), we found that the expression of the iron homoeostasis‐related genes in Gas muscle tissue, such as the transferrin receptor (TFR), 5 SLC39A14, 22 and heme oxygenase 1 (Hmox1), 21 were dysregulated, and we performed the western blotting to determine the expression of the iron homoeostasis‐related genes in Gas muscle tissue, such as TFR, nuclear receptor coactivator 4 (NCOA4), 23 ferroportin1 (FPN), 24 SLC39A14, Hmox1, and ferritin heavy chain 1 (FTH1) 23 (Figure S3C). Our analysis demonstrate that the levels of TFR reached to the peak levels once EHS onset, the levels of NCOA4 and SLC39A14 significantly increased once EHS onset, and reached to the peak level at 6 h, then slightly decreased at 24 h after EHS. The level of FPN showed gradually increase once EHS onset, and reached to the peak levels at 12 h, thus decreased at 24 h after EHS (Figure S3C). The level of Hmox1 showed marked increase as early as EHS onset and further elevated in a time‐dependent manner (Figure S3C). However, the expression of FTH1 do not changed once EHS onset but decreased from 6 h after EHS (Figure S3C).
Based on the changes of several crucial iron homoeostasis‐related proteins, we speculate that the iron accumulation from 0 to 6 h after EHS onset is mainly due to the influx of extracellular iron mediated by TFR and SLC39A14. Consistently, the non‐heme iron level in the Gas muscle tissue was significantly upregulated after EHS (Figure 1J). In addition, the heme degradation by the increased Hmox1 might also contribute to the iron accumulation in Gas muscle after EHS, as indicated by the degradation of tissue heme (Figure S3B). Moreover, the increase in NCOA4 expression and the decrease in FTH1 demonstrated the ferritinophagy might be involved in the RM development following EHS. The decrease of the cellular iron levels in EHS group since 6 h after EHS might be due to the efflux of iron mediated by the upregulation of FPN. Taken together, the iron accumulation is due to the complex regulatory mechanism of iron homoeostasis‐related gene regulation after EHS.
Ferroptosis contributes to rhabdomyolysis development following exertional heat stroke
To determine the pathogenic role of ferroptosis in RM development following EHS, we treated a single dose of various inhibitors of cell death at 2 h prior to EHS experiment and measured the CK and MB levels at 6 h after EHS. The results demonstrate that the treatment of ferroptosis inhibitor ferrostatin‐1 (Fer‐1) significantly attenuated the Gas muscle injury in RM following EHS; in contrast, the injury was not significantly reversed in mice that received emricasan (an inhibitor of apoptosis), necrostatin‐1 (Nec‐1, a specific inhibitor of receptor interacting protein kinase 1‐mediated necroptosis), or 3‐methyladenine (3‐MA, an inhibitor of autophagy) (Figure S4).
Our previous study also demonstrated that the mortality rate of EHS model with RM could reached to 62.66% within 24 h after EHS. 18 To evaluate the therapeutic effects of ferroptosis activation on the mortality rate of EHS mice within 24 h, we administered the single dose of Fer‐1 at 0, 6, and 12 h to the mice separately after EHS and then measured the mortality rate within 24 h after EHS, respectively. Our analysis revealed that the single‐dose treatment of Fer‐1 at 0 h powerfully improved the survival rate from EHS within 24 h and remarkably blocked skeletal muscle injury induced by EHS, as indicated by the decrease in CK and MB levels at 6 h after EHS (Figure S5A–S5C). Furthermore, the Fer‐1 intervention at 6 h clearly ameliorated the survival from EHS within 24 h and inhibited the skeletal muscle injury at 12 h after EHS. However, the Fer‐1 intervention at 12 h has little effects on the survival but clearly inhibits the skeletal muscle injury at 24 h after EHS (Figure S5A–S5C). Additionally, the various forms of Fer‐1 intervention did not significantly influence the survival rate and the Gas tissue injury from CHS or SHE groups (Figure S5D–S5I).
The Gas muscle tissue injury at 6 h after EHS was shown with H&E staining, and the images demonstrate that the treatment of Fer‐1 2 h prior to EHS experiment significantly prevented the Gas muscle injury in RM following EHS, which is manifested by the decrease in Evans blue dye penetration, improvement in the grip strength, the reduction of the serum CK and MB levels, and the inhibition of lipid peroxidation, as indicated by the reduction of MDA, 12‐HETE, and 15‐HETE levels (Figures 2A–2G and S6A–S6B). Additionally, we found that the RM following EHS was involved in acute kidney injury (AKI), and the treatment of Fer‐1 significantly rescued the AKI, as confirmed by histological and serological measurements, such as the H&E staining, Paller score, and blood urea nitrogen levels (Figure S6C–S6E). Briefly, ferroptosis activation played an essential role in the RM associated AKI following EHS onset.
Figure 2.
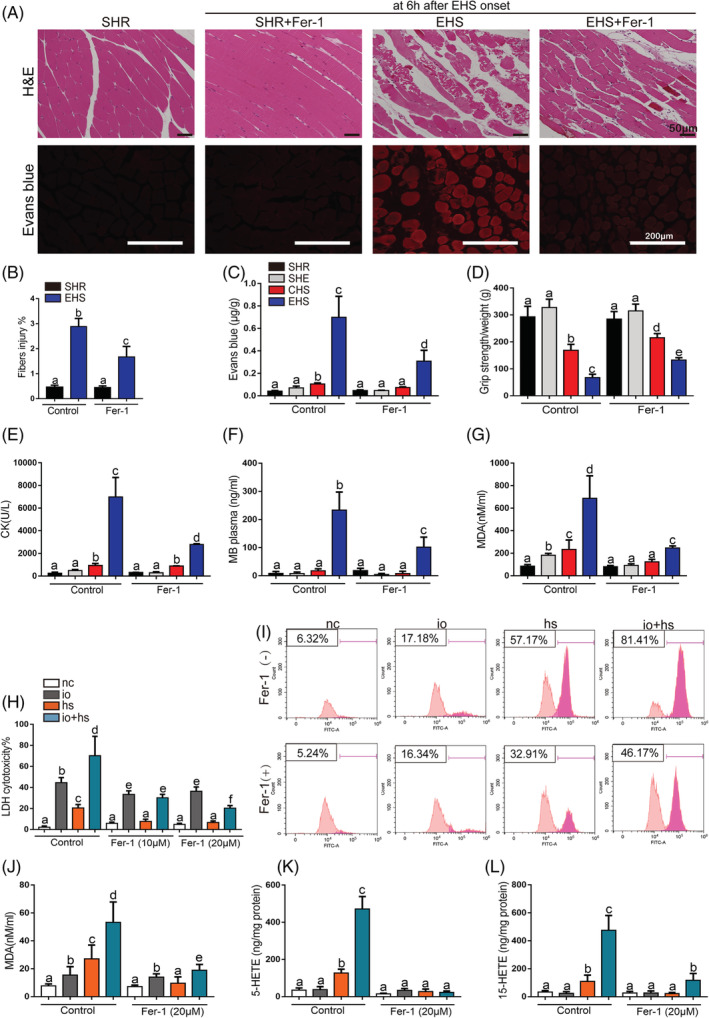
The effects of Fer‐1 on the skeletal muscle injury of EHS mice in vivo and on the viability and lipid peroxidation of primary myoblast cells exposed to io + hs in vitro. Mice were treated with ferrostatin‐1 (10 mg/kg) by intraperitoneal injection 2 h before experiment. All samples of Gas muscle were collected at 6 h following SHE, CHS, and EHS onset, SHR as the control. (A) Representative H&E staining and EBD fluorescence of Gas muscle slices were imaged by microscopy (scale bar = 50 μm in H&E, 200 μm in EBD testing image). (B) The fibres injury score in EHS mice with or without Fer‐1 treatment (n = 6 mice/group). (C) The degree of permeability in muscle cells was detected by measuring the quality of EBD permeating into muscle cells under standard concentration curve (n = 6 mice/group). (D) The grip strength test in each group (n = 10 mice/group). (E–G) In parallel, serum creatine kinase (CK) levels and MB level were measured at 6 h after SHE, CHS, and EHS onset, SHR as the control (n = 6 mice/group); before io + hs induction experiment (containing 5 μM of ionomycin in medium, heat stress, 43°C 5% CO2 incubator heat stress for 2 h), primary myoblasts pretreatment with ferropstatin‐1 (20 μM) for 6 h. All samples were collected at 6 h following io + hs exposure. (H) LDH cytotoxicity percent was assayed at 6 h after io + hs induction (n = 6). (I) Cell lipid peroxidation signal was detected by C11 BODIPI 581/591 staining by flow cytometry. (J–L) In parallel, the levels of MDA, 5‐HETE, and 15‐HETE were assayed at 6 h following io + hs exposure (n = 6). Summary data are presented as the mean ± SEM. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labelled with different letters differed significantly (*P < 0.05).
To investigate the activation of ferroptosis in RM in vitro, the primary myoblasts cells were exposed to ionomycin plus heat stress (io + hs) for simulating the effects of exercise and heat stress. 18 For the treatment of ionomycin leads to calcium overload, which is frequently accompanied by RM following EHS. 20 The treatment of Fer‐1 protect against the primary myoblasts death induced by io + hs, as shown by the decrease in LDH release (Figure 2H); the inhibited lipid peroxidation, as indicated by C11 BODIPY 581/591 fluorescence (Figure 2I); and the reduced lipid metabolites levels of MDA, 5‐HETE, and 15‐HETE (Figure 2J–2L). The effects of Fer‐1 treatment were further confirmed by the other two ferroptosis inhibitors treatment, such as liproxstatin‐1 and deferoxamine (Figure S7A–S7C), as well as the results in primary myoblasts were validated in C2C12 cells (Figure S7D–S7H).
ACSL4‐mediated ferroptosis contributes to rhabdomyolysis development following exertional heat stroke
Lipid peroxidation is the main cause of ferroptosis. 25 To explore the regulatory mechanism of the lipid peroxidation induced by EHS (Figure 1F–1I), the KEGG pathway enrichment analysis and GO analysis were performed and the analysis showed that Acsl4 was significantly enriched in the ferroptosis pathway, lipid biosynthetic process, and fatty acid metabolism (Figure S2). ACSL4 has been reported to dictate ferroptosis sensitivity by shaping cellular lipid composition. 14 In current study, the ACSL4 expression was upregulated after EHS and reached to the peak levels at 6 h after EHS (Figure 3A). The treatment of ACSL4 inhibitor rosiglitazone (Rosi) significantly attenuated the skeletal muscle injury induced by EHS, as indicated by the decrease in Evans blue dye penetration, the improvement of the grip strength and the reduction of the serum CK and MB levels (Figure 3B–3E and Figure 3G–3H). Moreover, the treatment of Rosi restored the GPX4 expression (Figure 3A), which is a universal inhibitor of ferroptosis 20 ; reduced the cyclooxygenase‐2 (COX2, Ptgs2 gene) expression (Figure 3F); and diminished the lipid metabolites levels, such as MDA, 5‐HETE (Figure 3I–3J), 12‐HETE, and 15‐HETE (Figure S8A–S8B). The effects of Rosi on the RM following EHS were further confirmed by the other ACSL4 inhibitor pioglitazone (Piog) (Figure S8C–S8K).
Figure 3.
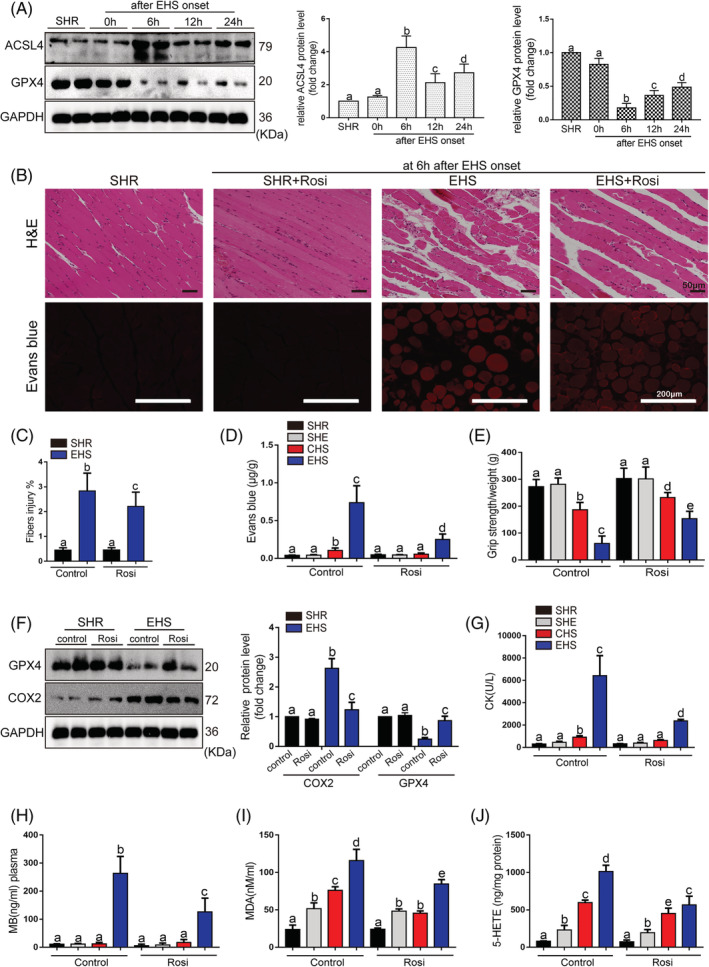
The effects of pharmacological inhibition of ACSL4 by Rosi on the development of RM after EHS onset. (A) The expression levels of ACSL4 and GPX4 in EHS mice were detected using the western blotting test. The representative western blotting images were from three independent experiments. Rosi (0.4 mg/kg, intravenous injection, 2 h before EHS experiment) was administered to mice. All samples were collected at 6 h after EHS onset. (B) Representative H&E staining and Evans blue fluorescence of Gas muscle slices were imaged by microscopy (scale bar = 50 μm in H&E, 200 μm in Evans blue testing image). (C) The fibre injury score in EHS mice with or without Rosi treatment (n = 6 mice/group). (D) The degree of permeability in muscle cells was detected by measuring the quality of EBD permeating into muscle cells under standard concentration curve (n = 6 mice/group). (E) The grip strength test in each group (n = 10 mice/group). (F) GPX4 and COX2 protein levels were assessed by western blotting, muscle tissues were collected at 6 h after EHS onset, the representative images of western blotting were from three independent experiments. (G–J) In parallel serum CK levels and MB levels, Gas muscles levels of MDA and 5‐HETE were measured in each groups (n = 6 mice/group). Summary data are presented as the mean ± SEM. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labelled with different letters differed significantly (*P < 0.05).
The elevated ACSL4 expression was observed in primary myoblasts cells induced by io + hs (Figure 4A). The treatment of si‐Acsl4 markedly decreased the io + hs induced ACSL4 upregulation, restored GPX4 expression, inhibited COX2 expression, and rescued the io + hs induced cell death, as shown by the decrease in LDH release (Figure 4B–4D). Furthermore, the si‐Acsl4 intervention significantly inhibited the lipid peroxidation and diminished the levels of lipid metabolites, as indicated by the levels of MDA, 5‐HETE, 12‐HETE, and 15‐HETE (Figure 4E–4I). The results in primary myoblasts cells were also validated in C2C12 cells (Figure S9).
Figure 4.
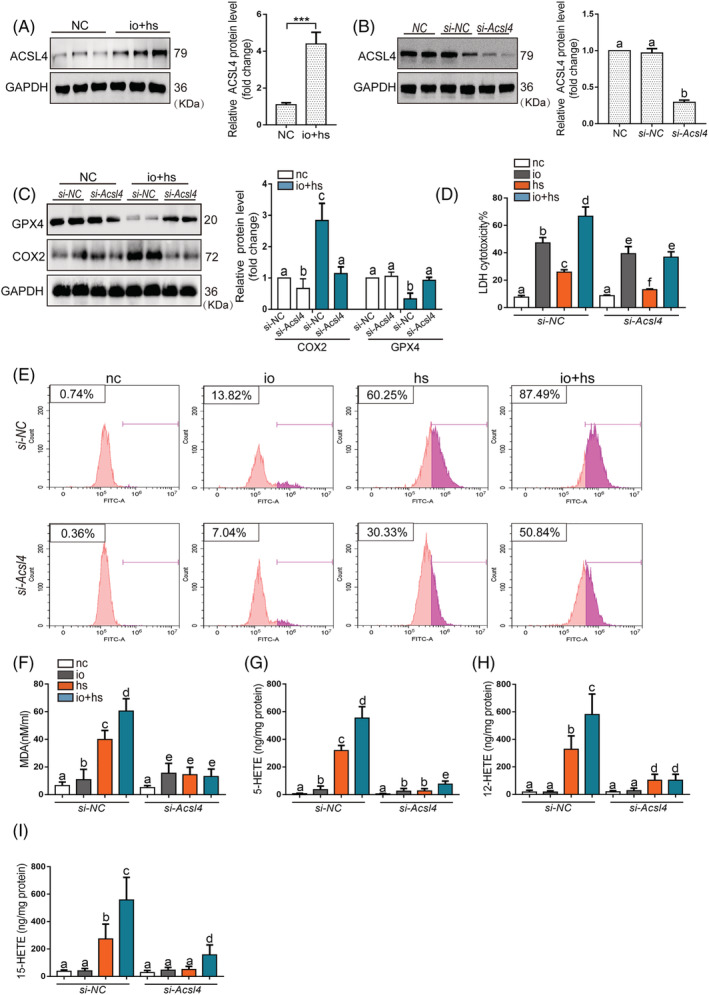
The effects of genetic inhibition of Acsl4 on the viability and lipid peroxidation of primary myoblasts cells exposed to io + hs in vitro. (A) Western blotting was used to determine ACSL4 expression of primary myoblasts in the presence or absence of io + hs exposure, the representative images of western blotting were from three independent experiments. (B) Primary myoblast were transfected with si‐NC or si‐Acsl4 for 2 days before io + hs induction. All samples were collected at 6 h after io + hs induction. ACSL4 expression level under io + hs induction for 2 h after siRNA transfection was assessed by western blotting, the representative images of western blotting were from three independent experiments. (C) GPX4 and ACSL4 protein levels were determined by western blotting after io + hs induction, the representative images of western blotting were from three independent experiments. (D) The LDH cytotoxicity percent was assayed at 6 h after io + hs induction (n = 6). (E) Cell lipid peroxidation signal was detected by C11 BODIPI 581/591 staining by flow cytometry. (F–I) In parallel, the levels of MDA, 5‐HETE, and 15‐HETE were assayed at 6 h following io + hs exposure (n = 6). Significance was calculated using the Student's t‐test; *P < 0.05. Summary data are presented as the mean ± SEM. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labelled with different letters differed significantly (*P < 0.05).
YAP‐dependent upregulation of ACSL4‐mediated ferroptosis in rhabdomyolysis following exertional heat stroke
We performed the KEGG pathway enrichment analysis to find the mechanism by which EHS upregulated ACSL4 expression, and our analysis suggested that the Hippo signalling may play a significant role in RM following EHS (Figure 5A), the strong and rapid YAP activation in cancer cells was induced by heat stress for the expression of heat shock proteins. 26 This is consistent with our analysis, and the Hspb1, Hsp90aa1, and Hspa1a were significantly upregulated in EHS groups (Figure S10A), and the Yap expression was significantly increased in the EHS group (Figure S10B). The primary myoblasts were treated with io + hs to simulate the exercise and heat stress in vitro, and the YAP in primary myoblasts showed clear colocalization with the nucleus (Figure 5B–5C). The mRNA levels of Acsl4 and Yap were both increased in primary myoblasts exposed to io + hs and Gas muscle tissue of EHS mice, respectively (Figure 5D–5E). The treatment of si‐Yap protects against the primary myoblasts cells death induced by io + hs, as revealed by the decreased LDH release (Figure S10C). The si‐Yap intervention inhibits the lipid peroxidation and the levels of lipid metabolites, such as MDA, 5‐HETE, induced by the treatment of io + hs (Figure S10D–S10F). The results with si‐Yap were further confirmed by results with YAP–TEAD interaction inhibitor verteporfin in vivo (Figure S11). In primary myoblasts, overexpression of Yap mRNA elevated the ACSL4 expression in the absence or presence of io + hs (Figure 5F–5H). Inversely, si‐Yap inhibited the ACSL4 expression (Figure 5I–5K). The results in primary myoblasts were also validated in the C2C12 cells (Figure S12).
Figure 5.
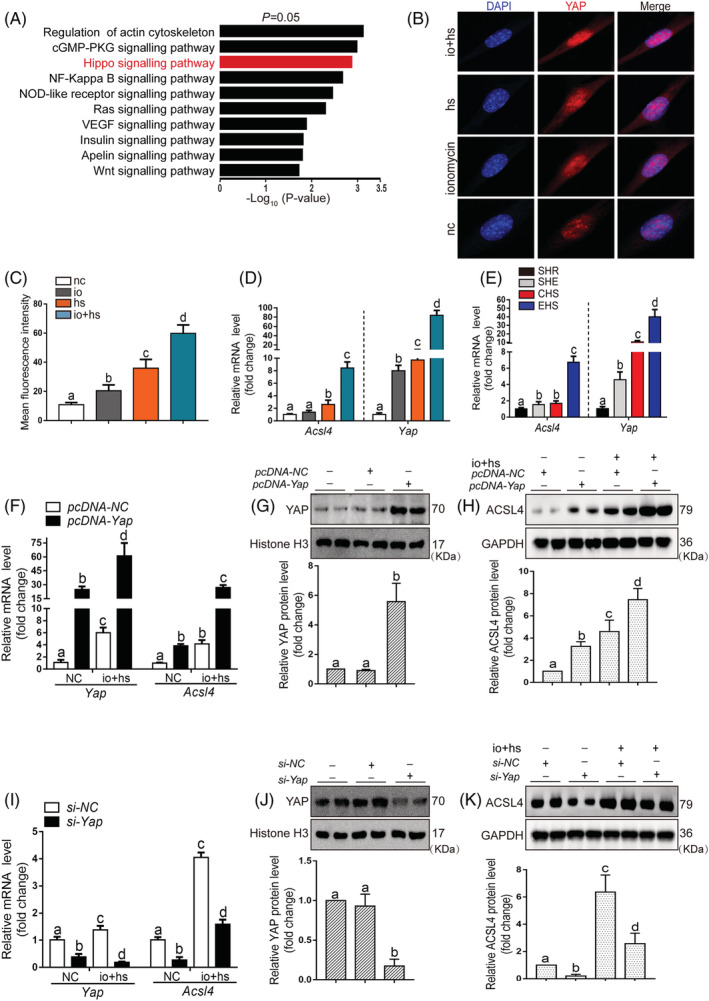
The regulation of the transcription and expression of ACSL4 by YAP entry into the nucleus in primary myoblasts. (A) KEGG analyses of RNA‐seq data showing the top 10 enriched pathways in EHS mice compared with SHR mice. (B–C) The location of YAP in nuclear was determined by laser scanning confocal microscopy (n = 6). (D–E) In parallel, QPCR analysis of Acsl4 and Yap mRNA levels in primary myoblast subjected to io + hs and Gas muscle tissues of mice with EHS, samples were collected at 6 h after EHS onset or after io + hs induction during recovery time (n = 6 mice/group or n = 6). (F–K) Primary myoblast cells were transfected with Yap overexpression plasmid or siRNA for 2 days, and then subjected to io + hs induction. The YAP protein levels and the mRNA and protein levels of ACSL4 were determined after io + hs induction. The representative western blotting images were from three independent experiments. Summary data are presented as the mean ± SEM. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labelled with different letters differed significantly (*P < 0.05).
TEAD1/TEAD4 binding to the promoter of Acsl4 regulates its expression
We have proved that YAP promotes the Acsl4 mRNA expression. Thus, we performed a database analysis to predict the binding motif(s) on the Acsl4 promoter region and confirm the binding position. The fragments (−700 to 0 bp relative to the transcription to start site [TSS]) in Acsl4 promoter sequence was validated as the effective binding target compared with other fragments (Figure 6A–6B). YAP‐dependent gene induction requires the TEAD family transcription factors. 27 The si‐TEAD1, si‐TEAD2, si‐TEAD3, and si‐TEAD4 were used to determine their regulatory role in the expression of Acsl4, and the si‐TEAD1 and si‐TEAD4 significantly diminish the ACSL4 mRNA and protein levels (Figure 6C–6G). Moreover, the ChIP assay showed two effective binding sites for TEAD1 (one binding site, located −190 to −220 bp relative to the TSS, and another binding site, located −540 to −560 bp relative to the TSS), as confirmed by the Mut plasmids (Figure 6H–6I). And another two effective binding sites for TEAD4 (one binding site, located −68 to −90 bp relative to the TSS, and another binding site, located −170 to −180 bp relative to the TSS) (Figure 6J–6L), as confirmed by the Mut and Del plasmids.
Figure 6.
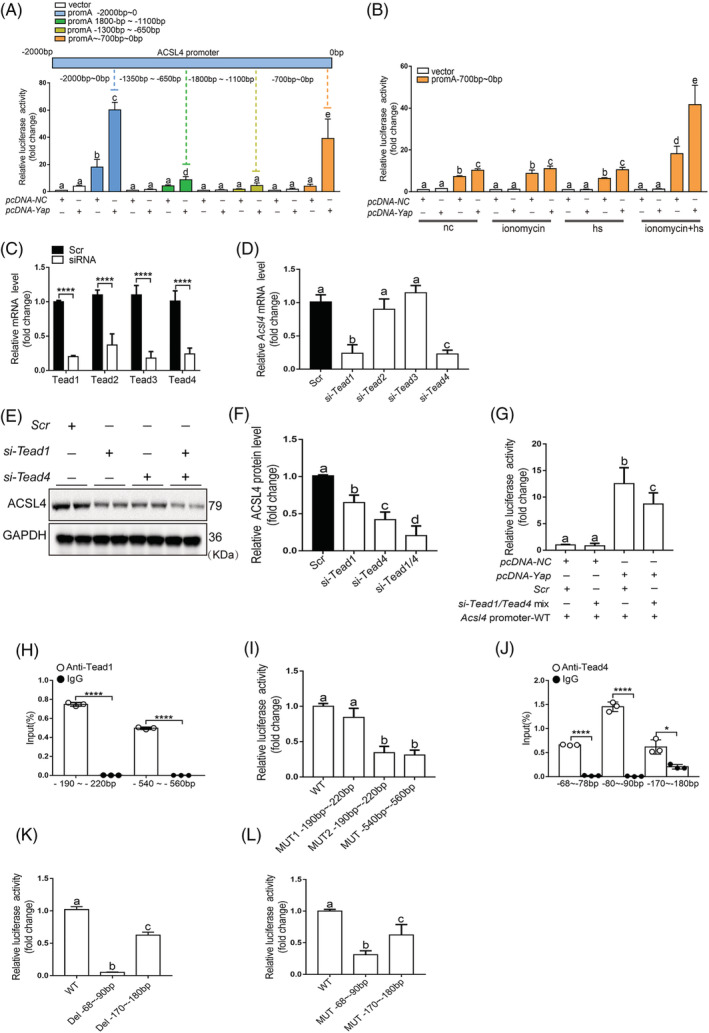
TEAD1/TEAD4 acting through binding to the ACSL4 promoter region for its expression. (A) Regions of the 2000‐bp proximal promoter sequence of Acsl4 gene via distinct promoter‐luciferase reporters are identified. Luciferase activity measurements of designated promoter‐reporter constructs in C2C12 cells were transfected with Yap plasmids (n = 3). The region −700‐bp upstream of the translation start site of the Acsl4 promoter was essential for YAP‐induced promoter‐reporter induction. (B) C2C12 cells were collected for assay in four conditions. Cells cotransfected with the Yap and WT (−700 bp) plasmids were collected and luciferase activity was analysed (n = 3). (C) siRNA transfection significantly decreased the expression of Tead1, Tead2, Tead3, and Tead4. N = 3 independent experiments (n = 6). (D) C2C12 cells were transfected with Tead1, Tead2, Tead3, and Tead4 siRNA or scramble siRNA for 48 h, respectively. Total RNA was isolated analysis of mRNA expression of Acsl4. (E–F) C2C12 cell were transfected with Tead1, Tead4, and Tead1 + Tead4 siRNA or scrambled siRNA (Scr) for 48 h, and then, western blotting for ACSL4 expression was performed, the representative images of western blotting were from three independent experiments. (G) Luciferase reporter constructs harbouring the WT (0–700 bp) Acsl4 promoter were cotransfected with the Yap overexpression plasmid and scramble siRNA or Tead1/4 siRNA into C2C12 cells. Forty‐eight post‐transfection, the cells were harvested for dual luciferase assays (n = 3). N = 3 independent experiments. (H) TEAD1 binding to the promoter region of Acsl4 was analysed by ChIP monitoring the occupancy of Tead1 on the Acsl4 promoters in two different part of promoter (n = 3). (I) Cells transfected with three Mut plasmid were used in luciferase assay and the results were normalized to those of the WT group (n = 3). (J) TEAD4 binding to the promoter region of Acsl4 was analysed by ChIP monitoring the occupancy of TEAD4 on the Acsl4 promoters in three different part of promoter (n = 3). (K–L) Cells transfected with two Del plasmid or Mut plasmid were used in the luciferase assay and the results were normalized to those of the WT group (n = 3). Significance in (C), (H), and (J) were calculated using the Student's t‐test; *P < 0.05, ****P < 0.0001. Summary data are presented as the mean ± SEM. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labelled with different letters differed significantly (*P < 0.05).
Discussion
Rhabdomyolysis, a common complication of EHS, causes significant modification of the tissue and blood parameters, which in turn challenge multiple organs function and subsequent patients' death. 10 The mechanism by which EHS induces RM remains unclear. In the current study, using a murine EHS model, 18 we uncovered an important role of ACSL4 in mediating EHS‐induced RM. We demonstrated that EHS‐induced YAP acting through TEAD1/TEAD4 upregulates ACSL4, which, in turn, promotes lipid peroxidation, skeletal muscle cells ferroptosis, thereby, leading to loss of functional skeletal muscle cells and subsequent RM (Figure 7).
Figure 7.
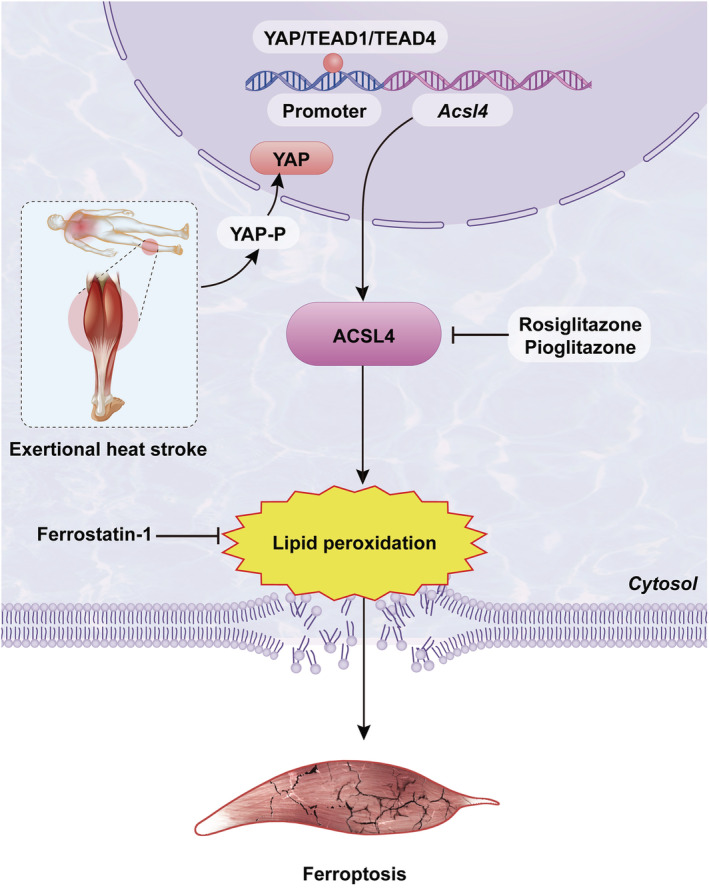
The mechanism of RM mediated by ferroptosis following EHS.
According to the murine EHS model with RM we established previously, 18 EHS markedly induced RM as early as the onset of EHS and reached to the peak levels at 6 h after EHS. The Gas muscle tissue showed the mostly severe damage, as indicated by the markedly decreased muscle fibre density, remarkably disorganized muscle fibre arrangement, and significantly swelling degeneration. And it might be due to the increased fibre work done of the Gas muscle, as the mice suffered from EHS via running on a rotating running wheel in the hot and humid environment which considered as the uphill running, mice performed uphill running mainly with the fore foot strike pattern, which is characterized by a higher step frequency, increased internal mechanical work, shorter swing/aerial phase duration, and greater duty factor. 28 Moreover, the heat production of the Gas muscle is more as the murine fast‐twitch muscle than that of the slow‐twitch soleus muscles. 2 Collectively, the types of cell death in Gas muscle tissue were still not addressed. Our bioinformatics analysis showed that the activation of ferroptosis potentially play an important role in the RM development following EHS. We found the hallmarks of ferroptosis were induced following EHS, such as ultrastructural changes in mitochondrial membrane, iron accumulation, and the increase in lipid peroxidation. The data suggest a direct effect of ferroptosis on EHS‐induced RM development.
It is well known that the standard mortality ratio, a key element in intensive care unit (ICU), is calculated based on the data available within the first 24 h of ICU stay. 29 Recent studies have reported that the content of the CK and MB levels were powerfully elevated in the nonsurvival group of EHS patients within 24 h in ICU, which indicated that RM has an important contribution to the mortality of EHS patients. 30 , 31 Fer‐1 was identified as a small molecule inhibitor of ferroptosis. 11 In the current study, the treatment of Fer‐1 significantly attenuated the skeletal muscle injury in RM following EHS compared with other forms of cell death inhibitors, which is manifested by diminishing the increased serum CK and MB levels induced by EHS. Then, the mice with Fer‐1 treatment markedly ameliorated survival rate from EHS within 24 h, and the earlier the treatment, the better the effect. Briefly, the current study demonstrates for the first time that EHS induces skeletal muscle cell ferroptosis, which in turn promotes RM development.
Additionally, ferroptosis activation has been reported to induce acute kidney failure and early death in mice caused by the disruption of GPX4. 26 In addition, a recent study demonstrated that ferroptosis is involved in RM associated kidney damage in vivo and in vitro. 32 RM causes the release of MB and other muscle cell components to the bloodstream. MB is freely filtered by glomeruli and reabsorbed by proximal tubules, promoting ferroptosis‐mediated cell death, thus leading to AKI, the treatment of Fer‐1 strongly inhibits the severity of AKI via decreasing the MB‐derived iron accumulation and lipid peroxidation. 32 This is consistent with the results in current study, RM following the EHS could induce remarkable AKI and the severity of AKI induced by the RM after EHS were strongly ameliorated by the Fer‐1 administration in vivo.
Lipid peroxidation, the key characteristics of ferroptosis, is the process by which oxygen combines with lipids to generate lipid hydroperoxides via intermediate formation of peroxyl radicals. 25 The accumulation of lipid peroxidation products in skeletal muscle tissue was induced by EHS, and reducing the lipid peroxidation products markedly prevents the EHS development. 10 Consistently, our analysis revealed that the increased lipid peroxidation and the elevated lipid metabolites, such as oxidized PE, MDA, 12‐HETE, and 15‐HETE, were induced by EHS, and inhibiting the lipid peroxidation levels significantly block the RM development, which in turn improve the survival from EHS. However, the regulatory mechanism of lipid peroxidation by EHS remains unclear. Then, the KEGG and GO analysis were performed to investigate how EHS onset regulates the lipid peroxidation process, and Acsl4 may play a potential vital role in the RM. ACSL4 is one of the most widely recognized mediator of ferroptosis. 14 , 15 We observed that ACSL4 expression paralleled the RM development, and pharmacological inhibition of ACSL4 with Rosi and Piog blocked RM following EHS in vivo, respectively. Genetic inhibition of Acsl4 ameliorated primary myoblasts and C2C12 cells ferroptosis following EHS in vitro. In addition, the reduction of GPX4 in presence of ASCL4 upregulation is very interesting, as suggested by previous studies demonstrating upregulation of ACSL4 induced by iron‐uptake be required for GPX4 degradation. 33 GPX4 has been proved to be an essential regulator of ferroptosis, and the insufficiency of GPX4 is thought to lead to increased levels of uncontrolled lipid peroxidation, culminating in ferroptotic cell death in vitro and in vivo. 20 , 26 However, the lipid oxidation upon GPX4 inhibition requires ACSL4, which catalyses the addition of coenzyme A to the long‐chain polyunsaturated bonds of AA or AdA, thereby promoting the esterification of polyunsaturated fatty acids to phospholipids, specifically towards the PE. 14 The oxidized AA and AdA PEs were found to be more abundant in Gpx4 knockout (KO) cells, 15 in line with this, the free AA or AdA preferentially sensitize Acsl4 KO cells to undergo ferroptosis. 14 Compared with the Gpx4 KO cells, the Acsl4 and Gpx4 double‐KO cells were viable and proliferated normally in cell culture for an extended period, underscoring an important functional interplay between GPX4 and ACSL4. In current study, we found the expression of ACSL4 was significantly upregulated since 6 h after EHS onset, even at the same time the expression of GPX4 was markedly downregulated (Figure 3A), so the GPX4 expression seems to be dependent on ACSL4 expression, which is suggested by the published research demonstrating that the treatment of the ACSL4‐specific inhibitor Rosi can significantly block the brain injury and improve the neurological function after stroke. 34 Additionally, iron‐uptake induced upregulation of ACSL4 was required for GPX4 degradation. 33 In brief, the current study suggests a key pathologic role of ACSL4 in mediating EHS‐induced skeletal muscle cell ferroptosis activation via lipid peroxidation.
Our KEGG enrichment analysis suggest that the Hippo signalling may play a significant role in RM following EHS, and the Yap expression was significantly increased in the EHS group. The strong and rapid YAP activation in cancer cells was induced by heat stress for the expression of heat shock proteins, 26 which is also suggested by our analysis. Additionally, YAP is involved in the heat stress mediated testicular damage. 35 In addition, the Hippo signalling pathway in skeletal muscle is involved in the regulation of exercise adaptation. 28 However, both the exercise (physiological limitation) and heat stress (environmental factor) are the most importantly predisposing factors for EHS. 36 In current study, genetic inhibition of Yap was able to suppress the primary myoblasts ferroptosis following EHS in vitro, and pharmacological inhibition of YAP with verteporfin blocks the RM development following EHS in vivo; overexpression of Yap mRNA can upregulate the expression of ACSL4, while genetic inhibition of YAP can downregulate the expression of ACSL4 in vitro. YAP‐dependent gene induction requires the TEAD family transcription factors. 37 In current study, we proved that the TEAD1/TEAD4 acts through binding to the Acsl4 promoter region for its expression. We further validated two effective binding sites for TEAD1 and TEAD4, respectively. Taken together, the Hippo signalling pathway played an essential role in the upregulation of ACSL4 expression by EHS.
In summary, this study demonstrates a novel mechanism by which YAP mediates ACSL4 upregulation and the consequent skeletal muscle cell ferroptosis, in turn, promotes RM development following EHS. Targeting ACSL4 and ferroptosis may serve as effective approaches to terminate the injury‐promoted mechanism and a new therapeutic strategy of post‐EHS RM.
Limitation of study
The current research mainly focuses on the study of EHS‐induced lipid peroxidation that causes ferroptosis. We have observed the accumulation of iron and changes in some proteins that are closely related to iron homoeostasis, but the mechanism of iron accumulation has not been studied in depth. In addition, we have also observed the correlation between GPX4 and ACSL4. This interesting phenomenon is also worthy of further exploration.
Conflict of interests
The authors declare that they have no conflict of interest.
Supporting information
Table S1 siRNA sequence and Primers
Table S2 Acsl4 promoter sequence
Figure S1. The EHS mice undergo rhabdomyolysis (RM) and reach to the peak level around 6 h after EHS onset
Figure S2. The gene expression profile related to Figure1b‐c
Figure S3. The changes in iron‐related metabolism protein in EHS mice
Figure S4. The effects of different death inhibitors on the skeletal muscle injury induced by EHS in vivo
Figure S5. The effects of Fer‐1 on the survival rate and skeletal muscle injury of EHS mice
Figure S6. The effects of Fer‐1 on kidney injury caused by RM following EHS
Figure S7. The effects of Fer‐1 on the viability and lipid peroxidation of C2C12 cells exposed to io+hs
Figure S8. The effects of pharmacological inhibition of ACSL4 by pioglitazone on the development of RM after EHS
Figure S9. The effects of genetic inhibition of Acsl4 on the viability and lipid peroxidation of C2C12 cells exposed to io+hs in vitro.
Figure S10. The effects of genetic inhibition of Yap on the viability and lipid peroxidation of primary myoblasts exposed to io+hs in vitro.
Figure S11. The effects of pharmacological inhibition of YAP‐TEAD interaction by verteporfin (VP) on RM development following EHS
Figure S12. The regulation of the transcription and expression of ACSL4 by YAP entry into the nucleus
Acknowledgements
This work was supported by the following funding sources: the National Natural Science Foundation of China (81772133 and 82072100 to Q.M.), the Guangdong Natural Science Fund (2020A1515011367 to Q.M.), the National Key Research and Development Program of China (2018YFA0507802 to F.W.), the National Natural Science Foundation of China (31930057 to F.W.). The authors thank Professor Jin‐qiang Guo (Department of Heat Environmental Medicine, Southern Medical University) for his assistance in the use of the artificial climate chamber. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 38
He S., Li R., Peng Y., Wang Z., Huang J., Meng H., Min J., Wang F., and Ma Q. (2022) ACSL4 contributes to ferroptosis‐mediated rhabdomyolysis in exertional heat stroke, Journal of Cachexia, Sarcopenia and Muscle, 13, 1717–1730, 10.1002/jcsm.12953
Contributor Information
Fudi Wang, Email: fwang@zju.edu.cn.
Qiang Ma, Email: mq@smu.edu.cn.
References
- 1. Epstein Y, Yanovich R. Heatstroke. N Engl J Med 2019;380:2449–2459. [DOI] [PubMed] [Google Scholar]
- 2. Laitano O, Oki K, Leon LR. The role of skeletal muscles in exertional heat stroke pathophysiology. Int J Sports Med 2021;42:673–681. [DOI] [PubMed] [Google Scholar]
- 3. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve 2002;25:332–347. [DOI] [PubMed] [Google Scholar]
- 4. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med 2007;18:90–100. [DOI] [PubMed] [Google Scholar]
- 5. Widner DB, Liu C, Zhao Q, Sharp S, Eber MR, Park SH, et al. Activated mast cells in skeletal muscle can be a potential mediator for cancer‐associated cachexia. J Cachexia Sarcopenia Muscle 2021;12:1079–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou S, Zhang W, Cai G, Ding Y, Wei C, Li S, et al. Myofiber necroptosis promotes muscle stem cell proliferation via releasing Tenascin‐C during regeneration. Cell Res 2020;30:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. You JS, Singh N, Reyes‐Ordonez A, Khanna N, Bao Z, Zhao H, et al. ARHGEF3 regulates skeletal muscle regeneration and strength through autophagy. Cell Rep 2021;34:108594. 10.1016/j.celrep.2020.108594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. You JS, Chen J. Autophagy‐dependent regulation of skeletal muscle regeneration and strength by a RHOGEF. Autophagy 2021;17:1044–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sciorati C, Rigamonti E, Manfredi AA, Rovere‐Querini P. Cell death, clearance and immunity in the skeletal muscle. Cell Death Differ 2016;23:927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guarnier FA, Michelucci A, Serano M, Pietrangelo L, Pecorai C, Boncompagni S, et al. Aerobic training prevents heatstrokes in calsequestrin‐1 knockout mice by reducing oxidative stress. Oxid Med Cell Longev 2018;2018:1–14, 4652480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding H, Chen S, Pan X, Dai X, Pan G, Li Z, et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J Cachexia Sarcopenia Muscle 2021;12:746–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Yu R, Wu L, Yang G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell Signal 2021;78:109870. 10.1016/j.cellsig.2020.109870 [DOI] [PubMed] [Google Scholar]
- 14. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia‐induced ACSL4 activation contributes to ferroptosis‐mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 2019;26:2284–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cui Y, Zhang Y, Zhao X, Shao L, Liu G, Sun C, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis‐induced brain injury and neuroinflammation. Brain Behav Immun 2021;93:312–321. [DOI] [PubMed] [Google Scholar]
- 18. He SX, Li R, Yang HH, Wang ZQ, Peng YM, Huang JH, et al. Optimization of a rhabdomyolysis model in mice with exertional heat stroke mouse model of EHS‐rhabdomyolysis. Front Physiol 2020;11:642. 10.3389/fphys.2020.00642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021;22:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 2019;116:2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu Y, Jiang L, Wang H, Shen Z, Cheng Q, Zhang P, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020;136:726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res 2016;26:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bao WD, Pang P, Zhou XT, Hu F, Xiong W, Chen K, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell Death Differ 2021;28:1548–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol 2016;26:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014;16:1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gabriel BM, Hamilton DL, Tremblay AM, Wackerhage H. The Hippo signal transduction network for exercise physiologists. J Appl Physiol (1985) 2016;120:1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vernillo G, Giandolini M, Edwards WB, Morin JB, Samozino P, Horvais N, et al. Biomechanics and physiology of uphill and downhill running. Sports Med 2017;47:615–629. [DOI] [PubMed] [Google Scholar]
- 29. Strand K, Flaatten H. Severity scoring in the ICU: a review. Acta Anaesthesiol Scand 2008;52:467–478. [DOI] [PubMed] [Google Scholar]
- 30. Wu M, Wang C, Liu Z, Liu Z. Sequential organ failure assessment score for prediction of mortality of patients with rhabdomyolysis following exertional heatstroke: a longitudinal cohort study in Southern China. Front Med (Lausanne) 2021;8:724319. 10.3389/fmed.2021.724319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang MM, Wang L, Zhang Y, Yuan R, Zhao Y, Hu J, et al. Establishment and effectiveness evaluation of a scoring system for exertional heat stroke by retrospective analysis. Mil Med Res 2020;7:40. 10.1186/s40779-020-00269-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang LN, Tian H, Zhou XL, Tian SC, Zhang XH, Wu TJ. Upregulation of microRNA‐351 exerts protective effects during sepsis by ameliorating skeletal muscle wasting through the Tead‐4‐mediated blockade of the Hippo signaling pathway. FASEB J 2018;32:6934–6947. [DOI] [PubMed] [Google Scholar]
- 33. Zhang L, Wang F, Li D, Yan Y, Wang H. Transferrin receptor‐mediated reactive oxygen species promotes ferroptosis of KGN cells via regulating NADPH oxidase 1/PTEN induced kinase 1/acyl‐CoA synthetase long chain family member 4 signaling. Bioengineered 2021;12:4983–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J, Yang L, Geng L, He J, Chen L, Sun Q, et al. Inhibition of acyl‐CoA synthetase long‐chain family member 4 facilitates neurological recovery after stroke by regulation ferroptosis. Front Cell Neurosci 2021;15:632354. 10.3389/fncel.2021.632354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Badr G, Abdel‐Tawab HS, Ramadan NK, Ahmed SF, Mahmoud MH. Protective effects of camel whey protein against scrotal heat‐mediated damage and infertility in the mouse testis through YAP/Nrf2 and PPAR‐gamma signaling pathways. Mol Reprod Dev 2018;85:505–518. [DOI] [PubMed] [Google Scholar]
- 36. Leon LR, Bouchama A. Heat stroke. Compr Physiol 2015;5:611–647. [DOI] [PubMed] [Google Scholar]
- 37. Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov 2020;19:480–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 siRNA sequence and Primers
Table S2 Acsl4 promoter sequence
Figure S1. The EHS mice undergo rhabdomyolysis (RM) and reach to the peak level around 6 h after EHS onset
Figure S2. The gene expression profile related to Figure1b‐c
Figure S3. The changes in iron‐related metabolism protein in EHS mice
Figure S4. The effects of different death inhibitors on the skeletal muscle injury induced by EHS in vivo
Figure S5. The effects of Fer‐1 on the survival rate and skeletal muscle injury of EHS mice
Figure S6. The effects of Fer‐1 on kidney injury caused by RM following EHS
Figure S7. The effects of Fer‐1 on the viability and lipid peroxidation of C2C12 cells exposed to io+hs
Figure S8. The effects of pharmacological inhibition of ACSL4 by pioglitazone on the development of RM after EHS
Figure S9. The effects of genetic inhibition of Acsl4 on the viability and lipid peroxidation of C2C12 cells exposed to io+hs in vitro.
Figure S10. The effects of genetic inhibition of Yap on the viability and lipid peroxidation of primary myoblasts exposed to io+hs in vitro.
Figure S11. The effects of pharmacological inhibition of YAP‐TEAD interaction by verteporfin (VP) on RM development following EHS
Figure S12. The regulation of the transcription and expression of ACSL4 by YAP entry into the nucleus