

Abstract
Background
Mitochondrial oxidative phosphorylation (OxPhos) is a critical regulator of skeletal muscle mass and function. Although muscle atrophy due to mitochondrial dysfunction is closely associated with bone loss, the biological characteristics of the relationship between muscle and bone remain obscure. We showed that muscle atrophy caused by skeletal muscle‐specific CR6‐interacting factor 1 knockout (MKO) modulates the bone marrow (BM) inflammatory response, leading to low bone mass.
Methods
MKO mice with lower muscle OxPhos were fed a normal chow or high‐fat diet and then evaluated for muscle mass and function, and bone mineral density. Immunophenotyping of BM immune cells was also performed. BM transcriptomic analysis was used to identify key factors regulating bone mass in MKO mice. To determine the effects of BM‐derived CXCL12 (C–X–C motif chemokine ligand 12) on regulation of bone homeostasis, a variety of BM niche‐resident cells were treated with recombinant CXCL12. Vastus lateralis muscle and BM immune cell samples from 14 patients with hip fracture were investigated to examine the association between muscle function and BM inflammation.
Results
MKO mice exhibited significant reductions in both muscle mass and expression of OxPhos subunits but increased transcription of mitochondrial stress response‐related genes in the extensor digitorum longus (P < 0.01). MKO mice showed a decline in grip strength and a higher drop rate in the wire hanging test (P < 0.01). Micro‐computed tomography and von Kossa staining revealed that MKO mice developed a low mass phenotype in cortical and trabecular bone (P < 0.01). Transcriptomic analysis of the BM revealed that mitochondrial stress responses in skeletal muscles induce an inflammatory response and adipogenesis in the BM and that the CXCL12–CXCR4 (C–X–C chemokine receptor 4) axis is important for T‐cell homing to the BM. Antagonism of CXCR4 attenuated BM inflammation and increased bone mass in MKO mice. In humans, patients with low body mass index (BMI = 17.2 ± 0.42 kg/m2) harboured a larger population of proinflammatory and cytotoxic senescent T‐cells in the BMI (P < 0.05) and showed reduced expression of OxPhos subunits in the vastus lateralis, compared with controls with a normal BMI (23.7 ± 0.88 kg/m2) (P < 0.01).
Conclusions
Defects in muscle mitochondrial OxPhos promote BM inflammation in mice, leading to decreased bone mass. Muscle mitochondrial dysfunction is linked to BM inflammatory cytokine secretion via the CXCL12–CXCR4 signalling axis, which is critical for inducing low bone mass.
Keywords: Mitochondria, Inflammation, Bone marrow, Bone loss
Introduction
Mitochondria quality control pathways regulating mitochondrial morphology/function have been implicated in the homeostatic control of muscle mass. 1 Gradual loss of muscle mass and strength results from an inflammatory state brought about by cytokines, apoptosis, alteration in mitochondrial respiration, and oxidative stress. 2 Decrease or dysfunction of skeletal muscle mitochondria is involved in loss of muscle mass and muscle strength in humans. 3 Progressive loss of muscle mass and function has also been implicated as a critical risk factor for osteoporosis through reduction of bone strength caused by decreasing mechanical loading on the skeleton. 4 Moreover, mitochondrial DNA‐mutator mice showed premature aging‐related phenotypes including muscle dysfunction and osteoporosis. 5 However, little information is available on the mechanical link between mitochondrial dysfunction‐mediated loss of muscle mass and strength, and bone loss.
Owing to their proximities, bone marrow (BM) immune cells contribute to the regulation of osteoblasts and osteoclasts, leading to modulation of bone remodelling. For example, ovariectomy induces T‐cell activation and proliferation in BM, which promotes osteoclastogenesis 6 due to increases in the populations of CD4+ and CD8+ T‐cells, and in T‐cell‐derived tumour necrosis factor alpha (TNF‐α). Additionally, ovariectomy‐induced bone loss does not occur in mice lacking T‐cells or lacking the T‐cell receptor CD40 ligand. 7 Germ‐free mice have a smaller population of CD4+ T‐cells and reduced levels of proinflammatory cytokines in the BM in combination with increased bone mass. 8 Moreover, BM CD4+ T‐cells producing interleukin (IL)‐17 and TNF‐α, but not interferon (IFN)‐γ, activate bone resorption by inducing osteoclast differentiation in animal models of inflammatory bowel disease. 9 However, although previous investigations revealed several factors promoting BM inflammatory‐mediated bone loss, the role of mitochondrial function in skeletal muscle on the BM immune micro‐environment and bone mass remains to be elucidated.
CR6‐interacting factor 1 (CRIF1), as a critical mitoribosomal protein, is essential for the translation of mitochondrial oxidative phosphorylation (OxPhos) subunits and their insertion in the mitochondrial inner membrane. 10 Deficiency of CRIF1 leads to impaired formation of the OxPhos complex and to a reduction in mitochondrial respiration and the mitochondrial stress response in skeletal muscle‐specific Crif1 knockout (MKO) mice, which have smaller gastrocnemius and EDL muscles. 11 Thus, we hypothesized that MKO mice could be used as a model for studying the mechanisms underlying muscular mitochondrial dysfunction‐induced bone loss.
Herein, we investigated whether skeletal muscle dysfunction caused by muscle‐specific mitoribosomal defects changes the population of inflammatory T‐cells and the levels of proinflammatory cytokines in the BM, thereby resulting in bone loss. We also examined whether key mitokines produced in response to mitochondrial stress regulate bone mass and bone mineral density (BMD). Fibroblast growth factor 21 (FGF21) is a representative mitokine that is responsive to mitochondrial diseases and mtDNA deletions. 12 FGF21 released from skeletal muscle is an important endocrine regulator of whole‐body energy metabolism. 11 , 13 Although FGF21 promotes bone loss by potentiating the activity of peroxisome proliferator‐activated receptor γ, 14 the significance of FGF21 as a myo‐mitokine during crosstalk between muscle and bone has not been studied extensively. Therefore, the aim of the present study was to identify the mechanism underlying deterioration in bone mass and quality caused by loss of muscular mitochondrial function and, consequently, to identify a potential therapeutic target. We also define the distinct roles of FGF21 during muscular mitochondrial dysfunction‐induced bone loss in MKO mice.
Materials and methods
Animals
To generate skeletal muscle‐specific Crif1 deficiency in mice, floxed Crif1 mice were crossed with Mlc1f‐Cre mice on a C57BL/6 background (provided by S.J. Burden, New York University, Baltimore, MD, USA) expressing a Cre‐recombinase gene under the control of the myosin light chain 1 fast promoter. 15 All of the experiments were performed in homozygous male mice, and Crif1 f/f littermates were used as controls. MKO mice were crossed with a global Fgf21 knockout on a C57BL/6 background (a kind gift from N. Itoh, Kyoto University Graduate School of Pharmaceutical Sciences, Kyoto, Japan) to generate MKFO mice as a mitokine double knockout mouse model. All animal experiments used male mice that were maintained in a controlled environment (12 h light/12 h dark cycle; humidity, 50–60%; ambient temperature, 22 ± 2°C) and fed a normal chow diet. All experimental procedures involving mice were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Chungnam National University School of Medicine (CNUH‐017‐A0048, Daejeon, Korea).
Flow cytometry analysis
Isolated BM cells were filtered through a 70 μm cell strainer, washed in phosphate‐buffered saline, and resuspended in a 40% Percoll (GE Healthcare, Chalfont St Giles, UK) gradient. The cell suspension was centrifuged at 1128 g for 30 min at 4°C. Then, the cells were incubated with directly fluorochrome‐conjugated monoclonal antibodies for 40 min at 4°C. The antibodies used in this study are listed in Supporting Information, Table S2. For blocking non‐specific antibody binding, cells were pre‐incubated with anti‐mouse CD16/32 mouse Fc blocker (BD Biosciences, San Jose, CA, USA) prior to staining with the antibodies. For intracellular staining, BM cells were stimulated for 5 h with Cell Stimulation Cocktail comprising phorbol myristate acetate, ionomycin, brefeldin A, and monensin (eBioscience, San Diego, CA, USA). The cells were fixed and permeabilized using a Fixation/Permeabilization Buffer kit (eBioscience), washed and resuspended in 1% formaldehyde, and then stained with anti‐IFN‐γ‐PE‐Cy7, anti‐TNF‐α‐APC, or anti‐IL‐17A‐APC. Multicolour flow cytometry was performed using a BD LSRFortessa flow cytometer (BD Biosciences), and the data were analysed using FlowJo software (Tree Star, Ashland, OR, USA). Results are expressed as cell frequency (%).
Bone histological and morphological analysis
Mice were euthanized at 14 weeks and the tibia and femur were removed and fixed at 4°C overnight in 4% paraformaldehyde (Biosesang, Seongnam, Korea). To evaluate dynamic histomorphometry, mice were injected intraperitoneally with alizarin red (Sigma‐Aldrich, Dorset, UK; 30 mg/kg) and calcein (Sigma‐Aldrich; 10 mg/kg) on Days 8 and 3 prior to CO2 asphyxiation, respectively. For bone histological analysis, femurs and tibias were harvested, skinned, and fixed at 4°C overnight in 4% paraformaldehyde. The samples were then decalcified in 14% ethylenediaminetetraacetic acid (Junsei, Japan) for 4 weeks at room temperature. The buffer was changed every 3–4 days until complete decalcification. The tissues were embedded in paraffin, and 4 μm sagittal‐oriented sections were prepared and stained with tartrate‐resistant acid phosphatase (TRAP) for histological analysis using standard protocols. For von Kossa staining, undecalcified bones were embedded in methyl‐methacrylate (Sigma) and sectioned at a thickness of 6 μm, as previously described. 16 For Oil Red O staining, decalcified bones were embedded in FSC 22 Frozen Media (Leica Biosystems, IL, USA). Transverse (8 μm) bone sections were stained with 0.3% Oil Red O solution and lightly stained nuclei with alum haematoxylin. Bone histomorphometric analysis was performed with the Bioquant Osteo II program (Bio‐Quant, Inc., Nashville, TN, USA). All nomenclature related to bone histomorphometry follows the guidelines of the ASBMR. 17
Statistical analysis
Results are expressed as mean values ± standard error of the mean. Data were analysed using Prism (Version 8, GraphPad Software Inc., San Diego, CA, USA). A two‐tailed, unpaired t‐test with Welch's correction was used to assess statistical significance between two groups. A one‐way analysis of variance (ANOVA) with Bonferroni's correction for multiple comparisons was used to examine differences between more than two groups. A P‐value < 0.05 was considered statistically significant.
Results
Crif1 deficiency induces defective mitochondrial oxidative phosphorylation function and stress response in skeletal muscle
To assess the effect of skeletal muscle‐specific Crif1 deficiency on the mitochondrial function and physical performance of the mice, we generated MKO mice by crossing Mlc1f‐Cre mice with Crif1 flox/flox mice (Figure S1A). CRIF1 protein expression was markedly lower in the EDL and gastrocnemius of the MKO mice than in the wild‐type (WT) controls (Figure 1A and 1B). Consistent with the reduction in CRIF1 expression, the EDL and gastrocnemius in MKO mice showed significantly lower expression of mitochondrial OxPhos complex subunits, including complex II (SDHA), III (UQCRC2), and V (ATP5A), indicating that deficiency of Crif1 induces abnormal mitochondrial proliferation and function in skeletal muscles (Figure 1A and 1B and Figure S1B and S1C). Moreover, blue native polyacrylamide gel electrophoresis (BN‐PAGE) analysis revealed a decrease in assembly of complex I, III, and V in MKO mouse EDL and gastrocnemius muscle compared with the WT (Figure 1C). MKO mice also exhibited lower succinate dehydrogenase (SDH) activity in the EDL, gastrocnemius, and soleus muscles than the WT (Figure 1D and 1E). In addition, immunohistochemical analysis of laminin and myosin heavy chain Type IIB (MyHC2b) expression demonstrated that the size of the muscle fibres in MKO mice decreased (Figure 1F). Electron microscopy analysis of the gastrocnemius muscle of MKO mice also revealed accumulation of abnormal, swollen mitochondria with disrupted cristae (Figure S1D). In addition, EDL from MKO mice expressed higher levels of mitochondrial stress response‐related genes, such as Lonp1, Clpp, Hspd1, and Atf4 (Figure 1G). To exclude off‐target effects in MKO mice, we examined expression of CRIF1 and OxPhos complex subunits in the BM and femur of MKO mice and controls. There was no significant difference in expression of CRIF and mitochondrial OxPhos complex subunits in the BM or cortical bone of control and MKO mice (Figure S1E and S1F).
Figure 1.
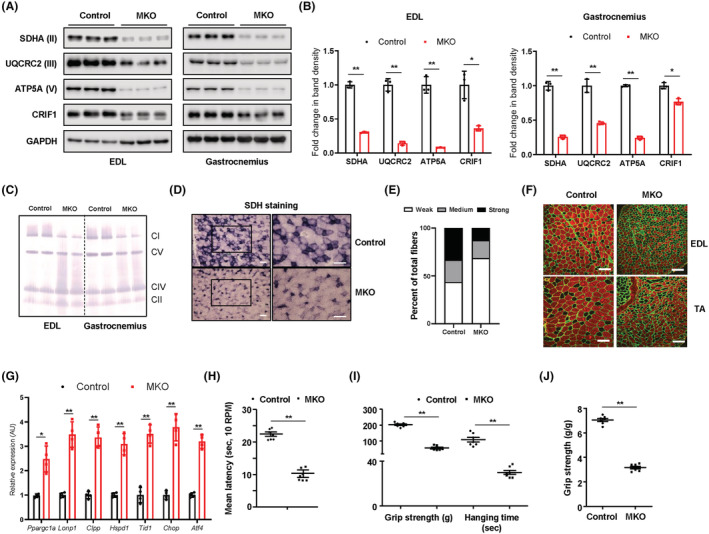
MKO mice show impairment in OxPhos and deterioration in physical performance. (A,B) Representative western blots and band density measurements for OxPhos complex subunits and CRIF1 in the EDL and gastrocnemius of chow‐fed control and MKO mice at 14 weeks of age; n = 3. (C) Representative blots showing BN‐PAGE of the assembled OxPhos complex in EDL and gastrocnemius from chow‐fed control and MKO mice at 14 weeks of age. (D) Transverse EDL sections were stained histochemically for SDH to identify oxidative muscle fibres at 14 weeks of age. Scale bar, 100 μm. (E) Quantification of unstained fibres in the EDL of controls and MKO mice at 14 weeks of age; n = 5. (F) Cross‐sections of the EDL and TA muscle from 10‐week‐old control and MKO mice were subjected to immunohistochemical staining for MyHC2b (red) and laminin (green). Scale bar, 100 μm. (G) Relative expression of mRNA encoding genes related to mitochondrial stress response from EDL in 14‐week‐old control and MKO mice; n = 4. (H) Latency to fall in rotarod test at 0.00336 g; n = 7. (I) Forelimb grip strength and time to fall in the wire hanging assay for control and MKO mice; n = 10. (J) Grip strength normalized to the body weight of control and MKO mice; n = 10. Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Next, we measured body weight and characterized the physical performance of MKO mice fed a normal chow diet. MKO mice showed a significant decrease in body mass relative to controls after 11 weeks of age (Figure S1G). Motor coordination was assessed using rotarod apparatus. At fixed rotarod speed (0.00336 g), a significant difference between control and MKO mice was observed in latency to fall (Figure 1H). Furthermore, unlike the controls, MKO mice showed a decline in grip strength and a higher drop rate in the wire hanging test at 13 weeks of age (Figure 1I and 1J). Collectively, these findings indicate that Crif1 deficiency not only adversely affects mitochondrial OxPhos function but also reduces muscle strength and endurance in mice.
Mitochondrial oxidative phosphorylation dysfunction in muscle leads to low bone mass in mice
Loss of muscle mass and function is closely associated with increased bone loss and fracture incidence. Thus, to examine the link between mitochondria in skeletal muscle and bone mass, we investigated bone mass and BMD of control and MKO mice (aged 14 weeks) fed a normal chow diet (Figure 2A and Figure S2A). Femurs from MKO mice showed a decrease in the number of trabeculae (Tb.N), trabecular bone volume/total volume (Tb.BV/TV), and trabecular thickness (Tb.Th.), all of which are associated with an increase in trabecular separation (Tb.Sp.). With respect to cortical bone, MKO mice also showed a decrease in cortical volume (Ct.V), although cortical thickness (Ct.Th) was not significantly different from that in controls (Figure 2B and 2C). Quantification and statistical analysis of the bone phenotypes characterized by micro‐computed tomography (CT) are shown in Figure 2C and Figure S2B.
Figure 2.
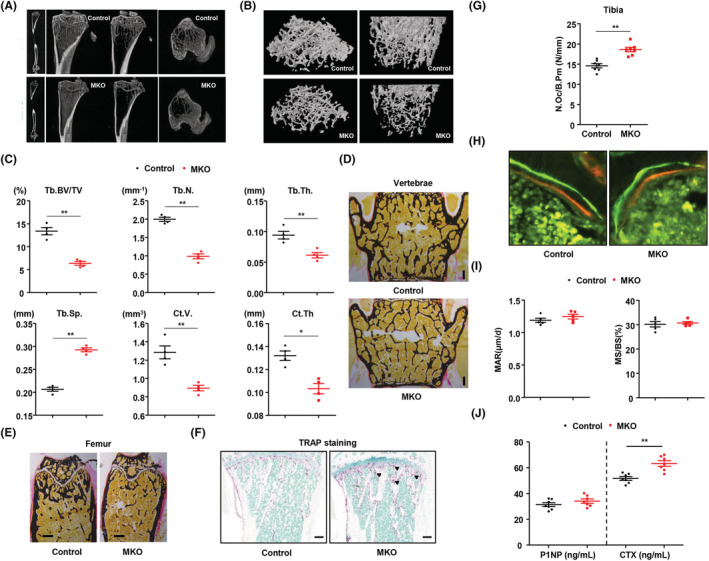
Mitochondrial stress response in skeletal muscle promotes lower bone mass compared with wild‐type control. (A,B) Representative images of micro‐CT of cortical and trabecular regions in the distal femur from control and MKO mice at 14 weeks of age. (C) Measurement of Tb.Th., trabecular number (Tb.N.), BV/TV, BS/TV, BS/BV, cortical volume (Ct.V.), Tb.Sp., and total bone volume (TBV) using micro‐CT analysis. (D,E) Von Kossa staining of undecalcified sections of vertebrae and femurs of control and MKO mice at 14 weeks of age. Scale bars, 250 μm. (F) TRAP staining to reveal osteoclasts. Scale bars: 100 μm. (G) The number of osteoclasts per bone perimeter [N.Oc/B.Pm (N/mm)] is indicated. (H) Histomorphometric analysis of calcein/alizarin red double‐stained sections was conducted to quantify bone formation in vertebrae; n = 5 per group. Green indicates calcein, and red indicates alizarin red. (I) Measurement of endocortical MAR and MS/BS (%) of vertebrae in control and MKO mice. (J) Serum levels of P1NP and CTX in 14‐week‐old control and MKO mice; n = 7. Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Bone histomorphometry verified that MKO mice developed a low bone mass phenotype in cortical and trabecular bone, as shown by von Kossa staining (Figure 2D and 2E and Figure S2C and S2D), but they showed a high number of osteoclasts per bone surface, as indicated by TRAP‐stained sections of tibia (Figure 2F and 2G). The bone eroded surfaces and the osteoclast surface per bone surface were also significantly increased in the MKO mice (Figure S2E and S2F). Next, to quantify the extent of increased osteoblast activity, calcein and alizarin red were sequentially given to control and MKO mice 3 and 8 days prior to sacrifice. Analysis of undecalcified, unstained sections showed no notable difference in vertebral bone formation rates between control and MKO mice (Figure 2H and 2I). Additionally, measurement of the serum level of procollagen type I N‐terminal propeptide (P1NP), a marker for bone formation, and of C‐terminal telopeptide of collagen (CTX), a marker for bone resorption, indicated no significant change in bone formation, but higher bone resorption in MKO mice than in controls (Figure 2J). To exclude the effect of other endocrine hormones on bone loss, we checked the serum levels of parathyroid hormone, testosterone, thyroxine, and triiodothyronine in the control and MKO mice at 14 weeks of age. There was no significant difference in the serum level of each of these hormones between the control and MKO mice (Figure S3A–S3D). Taken together, these data provide direct evidence that lower mitochondrial OxPhos‐mediated muscle dysfunction in MKO mice results in reduced bone mass compared with controls and that this occurs via activation of osteoclasts.
Low bone mass in MKO mice is independent of FGF21
The phenotypic changes observed so far suggested that MKO mice express circulating factors involved in signalling from muscle to bone. Therefore, to study the effects of muscular mitochondrial dysfunction‐induced secretory factors on low bone mass in MKO mice, we performed RNA sequencing of EDL transcripts from control and MKO mice, focusing on genes encoding secreted proteins. A large number of transcripts were altered in the EDL from MKO mice (Figure S4A and S4B). In particular, we found that the expression of Fgf21, which is known as a potent regulator of skeletal homeostasis, was much higher in the EDL of MKO mice than in that of the controls (Figure S4B–S4D). As shown in Figure S4E, the serum levels of FGF21 were also markedly elevated in the MKO mice.
Next, to investigate the role of FGF21 on the skeletal phenotype in MKO mice, we generated global Fgf21 knockout mice and MKO mice with global Fgf21 deletion (MFKO), both on a C57BL/6J background (Figure S4F and S4G). Analysis of femurs by micro‐CT revealed that cortical thickness, Tb.Th., and trabecular number were markedly lower in MKO mice compared with the controls, but global Fgf21 knockout did not have any effect on the bone phenotype of WT control or MKO mice at 14 weeks of age (Figure S5A and S5B). These findings suggest that a reduction of BMD in MKO mice is independent of FGF21 production caused by muscular mitochondrial OxPhos dysfunction.
MKO mice exhibit inflammatory responses in the bone marrow
In the next set of experiments, we asked whether mitochondrial OxPhos dysfunction in muscle affects BM inflammation, which is an important factor for instigating bone resorption. To address this issue, we investigated the expression of proinflammatory cytokines in the BM cells from control and MKO mice. The expression of Tnf, Rankl, Rorat, and Il17a was significantly increased in the BM from MKO mice compared with the controls (Figure 3A). Seeking further evidence in support of the concept that mitochondrial OxPhos dysfunction in skeletal muscle induces inflammation in BM, we measured the populations of diverse T‐cells in the BM from control and MKO mice using flow cytometry analysis. We found that at 14 weeks of age, mature CD3+ T‐cells were markedly elevated in the BM from MKO mice compared with those from the controls (Figure 3B and 3C). Moreover, the population of TNF‐α‐producing CD4+ and CD8+ T‐cells was significantly larger in the BM of MKO mice (Figure 3D–3F). The populations of CD44+TNF‐α+ and CD44+IL‐17A+ cells among the CD4+ and CD8+ T‐cells in the BM of MKO mice were also larger (Figure 3G and 3H). However, there was no difference in the size of the BM CD4+CD25+Foxp3+ regulatory T‐cell populations between control and MKO mice (Figure 3I). Furthermore, to exclude systemic inflammation underlying low bone mass in MKO mice, we measured TNF‐α and IL‐17A production in CD4+ T‐cells from the spleen of control and MKO mice at 14 weeks of age. The expression of proinflammatory cytokines by CD4+ T‐cells was not different in the spleens of control and MKO mice (Figure 3J–3L). In addition, there was no notable difference in the serum level of TNF‐α or IL‐17A between control and MKO mice at 14 weeks of age (Figure S6A). Thus, these data revealed that the muscular mitochondrial dysfunction‐mediated reduction of bone mass observed in MKO mice was associated with BM inflammation rather than with a systemic inflammatory response.
Figure 3.
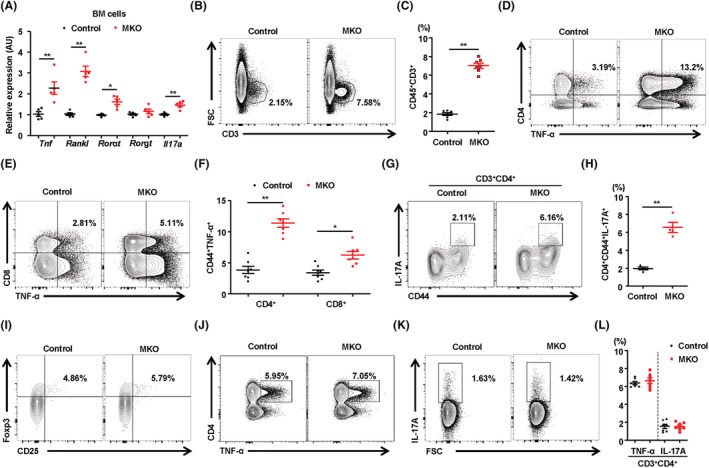
MKO mice exhibit local induction of TNF‐α and IL‐17 in BM without a systemic inflammatory response. (A) BM cells isolated from control and MKO mice at 14 weeks of age were subjected to real‐time PCR analysis of osteoclastogenic genes; n = 5. (B) Representative flow cytometry contour plots are presented for CD3 expression by BM cells in control and MKO mice at 14 weeks of age; n = 7. (C) Statistical analysis of the population of CD3+ T‐cells in BM cells in the two groups. (D–H) The number of TNF‐α‐secreting or IL‐17A‐secreting cells in the populations of BM CD4+CD44+ and CD8+C44+ T‐cells was compared between the two groups; n = 7. (I) Population of regulatory T‐cells (CD4+CD25+FOXP3+) in the BM of 14‐week‐old control and MKO mice; n = 7. (J–L) TNF‐α‐producing or IL‐17A‐producing cells among CD4+ and CD8+ T‐cells from the spleens of control and MKO mice at 14 weeks of age; n = 7. Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
MKO mice show adipogenesis and an inflammatory response in the bone marrow
Based on the results from the flow cytometry analysis of the BM of control and MKO mice, we further investigated the transcripts of whole BM cells using RNA sequencing. Macroscopic analyses showed that the long bones dissected from MKO mice were more reddish than those of the controls. This is because the colour of the BM is visible in MKO mice due to cortical thinning or reduced cortical BMD (Figure 4A). After Oil Red O staining, we also discovered a higher number of adipocytes in the BM of MKO mice (Figure 4B and Figure S6B). Thus, we conducted RNA sequencing to identify genes in BM cells differentially expressed between control and MKO mice at 14 weeks of age. Enrichment analysis with Network2Canvas revealed that genes associated with myopathy, osteoporosis, signalling in the immune system, T‐cells, and IL‐12 and IL‐17 pathways were enriched in the BM cells from MKO mice (Figure 4C and Figure S7A and S7B). Moreover, gene set enrichment analysis (GSEA) indicated that the gene sets involved in adipogenesis or adipocyte maturation were enriched in the BM cells of MKO mice compared with the controls (Figure 4D and 4E). In addition, consistent with the long bones appearing more reddish in MKO mice, genes related to T‐cell activation and co‐stimulation were markedly increased in the BM cells of MKO mice, which may be closely associated with the observed BM inflammation and decreased bone mass (Figure 4F and 4G). A volcano plot based on differential expression and statistical significance of the difference showed that, in addition to that of the adipogenesis and T‐cell activation gene sets, expression of Cxcl12 as well as of Fabp4, Adipoq, and Cd3e was also changed in the BM cells of MKO mice (Figure 4H). We also found that CXCL12 protein expression was higher in the PDGFRβ+VCAM‐1+ stromal cells (CXCL12‐rich cells) among BM cells from MKO mice using flow cytometry analysis (Figure 4I and Figure S7C). Furthermore, we searched genes potentially associated with Cxcl12 by applying Gene‐Module Association Determination (G‐MAD) to mouse expression data sets using GeneBridge tools. 18 As a result, genes annotated in the inflammatory response and T‐cell activation functional clusters were strongly enriched (Figure S7D). These results implicate the distinct effects of muscular mitochondrial dysfunction on BM inflammation and low bone mass in mice.
Figure 4.
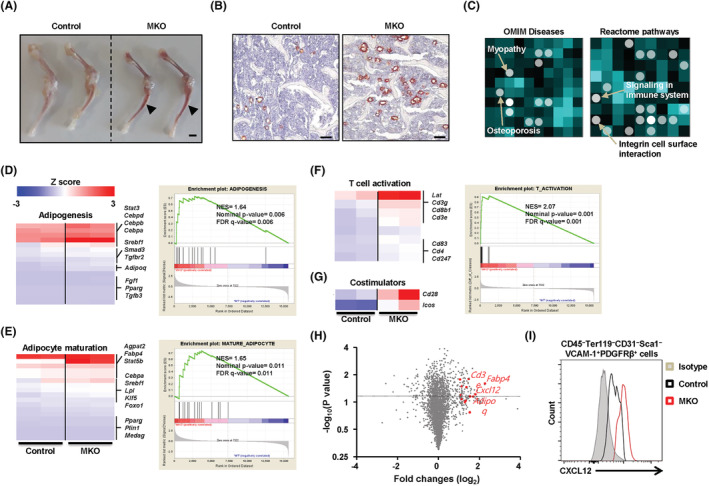
Transcriptome analysis shows adipogenesis and inflammatory response in the BM of MKO mice. (A) Femur and tibia of a 14‐week‐old control and MKO mouse. BM is more visible through the thin cortical bone of MKO mice. (B) A transverse section of femur from a 14‐week‐old control and MKO mouse stained with Oil Red O. Adipocyte‐rich BM (arrowhead) are visible in the MKO femur. (C) Using RNA sequencing data, genes that were significantly up‐regulated in the BM cells of control and MKO mice were analysed for gene‐list enrichment with gene set libraries created from Level 4 of the MGI mouse phenotype ontology using Network2Canvas. (D–G) The diagram shows the results of gene set (adipogenesis, adipocyte maturation, T‐cell activation, and co‐stimulators) enrichment analysis, including the enrichment scores. (H) Volcano plot based on the differential expression and significance of the differences in the data from RNA sequencing of BM cells of control and MKO mice at 14 weeks of age. Red dots represent genes associated with adipogenesis and T‐cell activation with a P‐value < 0.05 and a fold change > 1 log2. (I) In the flow cytometry analysis, total BM cells were gated on a population negative for CD45, Ter119, CD31, and Sca1 and positive for VCAM and PDGFRβ, the phenotype of CXCL12‐abundant reticular cells. Intracellular levels of CXCL12 are shown as amount of protein in CAR cells in the BM of MKO mice relative to CAR cells in the BM of the controls at 14 weeks of age, n = 4 mice/genotype. Scale bars: 2 mm (A) and 100 μm (B).
Recombinant CXCL12 activates osteoclasts and promotes T‐cell activation in vitro
The abundant expression of CXCL12 in reticular cells associated with the perivascular niches of the BM suggests that CXCL12 may have cell type‐specific roles in BM‐resident cells. Therefore, to determine the effect of BM‐derived CXCL12 on regulation of bone homeostasis, we treated a variety of BM niche‐resident cells with recombinant CXCL12. Treatment with recombinant CXCL12 during BM‐derived osteoclast differentiation increased expression of mRNA transcripts encoding osteoclast‐specific genes (Figure 5A). TRAP staining showed that the number of TRAP‐positive multinucleated cells increased after treatment with recombinant CXCL12 (Figure 5B and 5C). Although recombinant CXCL12 increased expression of RUNX2 modestly in pre‐osteoblastic MC2T3‐E1 cells (Figure 5D), neither expression of genes related to osteogenic differentiation nor the number of alkaline phosphatase‐stained cells changed markedly (Figure 5E and 5F). Next, to determine whether the inflammatory response in the BM of MKO mice is dependent on the level of CXCL12, we treated BM immune cells with recombinant CXCL12 in vitro. We found that CXCL12 increased the percentages of TNF‐α‐producing BM CD4+ and CD8+ T‐cells (Figure 5G–5I).
Figure 5.
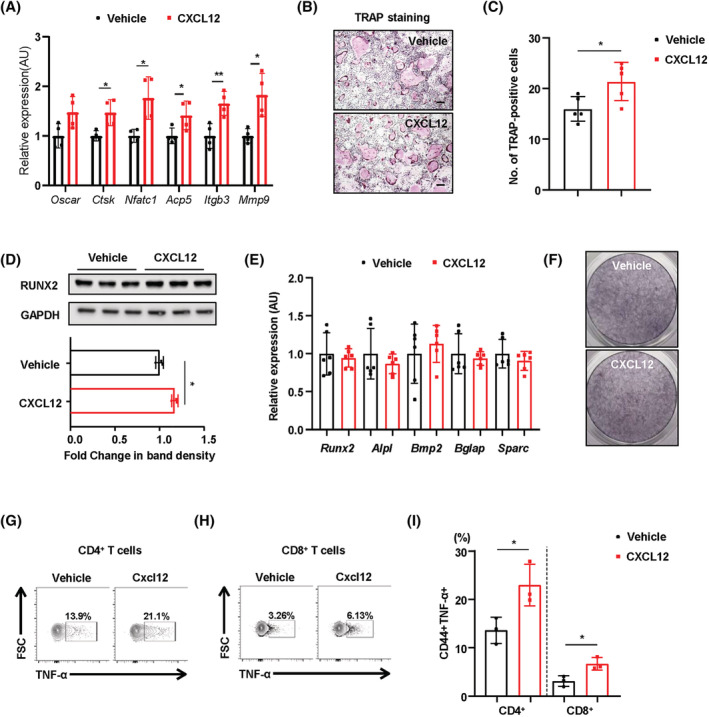
Treatment with recombinant CXCL12 increases activation of osteoclasts and promotes production of TNF‐α by BM T‐cells in vitro. (A) Transcript levels of osteoclast‐specific genes in differentiated osteoclasts treated with or without recombinant CXCL12 (5 ng/mL). (B) TRAP staining of osteoclasts. Scale bars: 100 μm. (C) The number of TRAP‐positive multinucleated cells was counted. (D) Representative western blots and band density of RUNX2 in MC3T3‐E1 cells treated with or without recombinant CXCL12 (5 ng/mL). (E, F) MC3T3‐E1 cells treated with or without recombinant CXCL12 (5 ng/mL) were subjected to real‐time PCR analysis of genes related to osteoblastogenesis. Cells were also stained for alkaline phosphatase activity. (G–I) TNF‐α‐producing cells within the BM CD4+ and CD8+ T‐cell populations treated with or without recombinant CXCL12 (5 ng/mL). Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
CXCR4 antagonism attenuates bone marrow inflammation in MKO mice
CXCL12 is also strongly expressed in reticular cells located adjacent to sinusoids in BM, which express adiponectin and are targetable with an adiponectin‐Cre transgene. 19 In addition, differentiated adipocytes induce CXCL12 secretion, which recruits proinflammatory macrophages into the adipose tissue of diet‐induced obese mice. 20 The chemokine CXCL12 binds primarily to CXCR4, leading to activation of intracellular signalling in multiple cell types including lymphocytes. 21 Moreover, the CXCL12–CXCR4 axis is involved in many physiological and pathological processes. 21 Thus, we asked whether migration of CXCR4+ immune cells to the MKO mouse BM contributes to the inflammatory response and proinflammatory cytokine production therein. As shown in Figure 6A, G‐MAD revealed that genes annotated in the inflammatory response and T‐cell activation functional clusters were strongly enriched. Next, to reveal the function of the CXCL12–CXCR4 axis on BM inflammation, we administered the CXCR4 receptor antagonist AMD3100 (5 mg/kg/day) intraperitoneally to 10‐week‐old control and MKO mice for 3 weeks. To determine whether AMD3100 can reduce infiltration of proinflammatory immune cells into the BM of MKO mice, we performed flow cytometry analysis to evaluate the proportions of diverse types of immune cells in the BM. AMD3100 induced a significant decrease in CD3+ T‐cells of the BM in MKO mice, but it caused little change in the populations of BM CD4+ and CD8+ T‐cells in either WT or MKO mice (Figure 6B and Figure S8A). Moreover, the production of proinflammatory cytokines including IFN‐γ, TNF‐α, and IL‐17A in CD4+ and CD8+ T‐cells by BM cells was significantly reduced in the MKO mice treated with AMD3100 (Figure S8B–S8E). Additionally, the expression of proinflammatory cytokines by the effector CD4+ and CD8+ T‐cells was significantly decreased in the BM cells from MKO mice treated with AMD3100 (Figure 6C–6H and Figure S8F). These data indicate that CXCR4 antagonism attenuates BM infiltration by inflammatory T‐cells in the MKO mouse model of muscular mitochondrial dysfunction.
Figure 6.
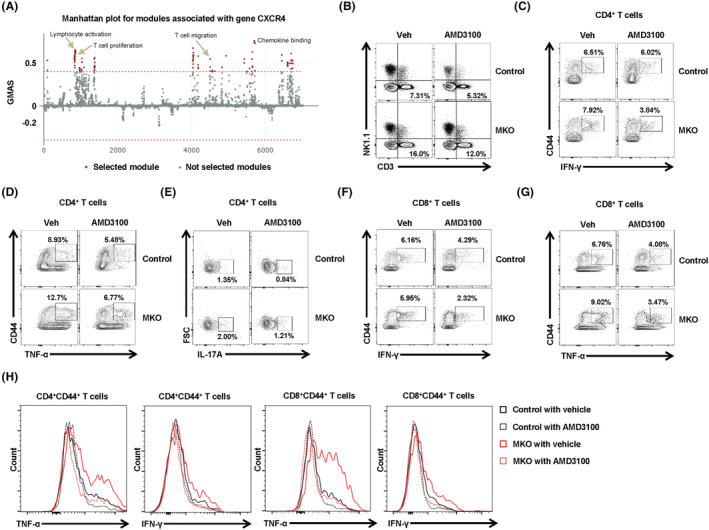
Treatment with CXCR4 antagonist reduces BM inflammation in MKO mice. (A) G‐MAD analysis shows that CXCR4 is associated with T‐cell migration and proliferation, and with chemokine‐binding modules, in mice. The threshold of significant gene‐module association is indicated by the red dashed line. Modules are organized by module similarities. Known modules connected to CXCR4 are highlighted in red. GMAS, gene‐module association score. (B) BM cells were subjected to flow cytometry for mature T‐cells, natural killer, and natural killer T‐cell phenotypes. (C,D) Populations of BM CD44+IFN‐γ+ or CD44+TNF‐α+ among CD4+ T‐cells from control and MKO mice treated with AMD3100 or vehicle at 14 weeks of age. (E) IL‐17A production by BM CD4+ T‐cells was analysed by flow cytometry. (F,G) Populations of BM CD44+IFN‐γ+ or CD44+TNF‐α+ among CD8+ T‐cells from control and MKO mice treated with AMD3100 or vehicle at 14 weeks of age. (H) Production of IFN‐γ or TNF‐α by BM CD4+CD44+ or CD8+CD44+ T‐cells of control and MKO mice treated with AMD3100 or vehicle at 14 weeks of age. Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by one‐way ANOVA.
Treatment with CXCR4 antagonists partially increases bone mass in MKO mice
To investigate the effect of CXCR4 antagonism on bone mass in MKO mice, micro‐CT analysis was conducted to assess BMD, BV, and cortical and trabecular bone architecture in the control and MKO mice treated with or without AMD3100. Consistent with previous results, MKO mice showed lower cortical and trabecular BV, BMD, trabecular number, and Tb.Th. compared with the controls (Figure 7A–7C). Intriguingly, AMD3100 significantly increased cortical and trabecular BV, trabecular number, and cortical bone area fraction in MKO mice but not in the controls (Figure 7A–7C and Figure S9A and S9B), without any changes in markers indicative of liver injury or perturbed lipid metabolism (Figure S9C–S9F). Taken together, these findings demonstrate that the CXCL12–CXCR4 axis is important for the BM inflammatory response and bone mass in the MKO mouse model with muscle dysfunction caused by reduced mitochondrial OxPhos.
Figure 7.
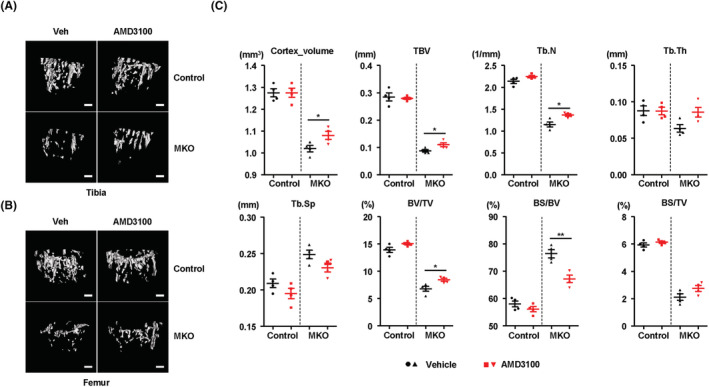
Treatment with AMD3100 attenuates bone loss in MKO mice. (A,B) At 9 weeks of age, control and MKO mice were injected intraperitoneally with AMD3100 (5 mg/kg, three times per week) for 3 weeks. Tibial and femoral trabeculae of control and MKO mice were measured by micro‐CT. Scale bars, 250 μm. (C) Measurement of Tb.N., Tb.Th., BV/TV, BS/TV, BS/BV, Ct.V., Tb.Sp., and TBV in the tibiae from control and MKO mice using micro‐CT analysis. Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by one‐way ANOVA. *, P < 0.05 and **, P < 0.01 compared with the indicated group. DW, distilled water.
Stronger inflammatory response in the bone marrow of hip fracture patients with low body mass index
Muscle mitochondrial impairment is an important contributor to age‐related frailty in humans. It is also well known that body mass index (BMI) shows a positive correlation with muscle strength and mass in humans. 22 As shown in Figure S10A, grip strength was significantly lower in hip fracture patients with lower BMI compared with age‐matched and gender‐matched control subjects (Table S1). Consistent with these data, we found that the vastus lateralis muscle of patients with a lower BMI showed markedly lower expression of mitochondrial OxPhos complex subunits, including complex I (NDUF8), II (SDHA), III (UQCRC2), and V (ATP5A), than that in patients with a normal BMI (Figure 8A and 8B). However, BN‐PAGE analysis revealed no significant differences in assembly of OxPhos complexes in the vastus lateralis muscle of patients with a lower BMI or a normal BMI (Figure S10B). Next, we investigated the immunophenotype of T‐cells in the BM of hip fracture patients with lower or normal BMI. To compare the different subsets of T‐cells in the BM of patients with a lower BMI with those with a normal BMI, we evaluated the frequency of CD4+ and CD8+ BM T‐cells expressing naïve/memory markers (CD45RA+/RO+). The patients with lower BMI had larger and smaller populations of CD8+ T‐cells and CD4+ T‐cells, respectively (Figure S10C). In the subset analysis of CD4+ and CD8+ T‐cells, in both populations, the proportion of CD45RA−CD45RO+ memory T‐cells was significantly increased, while that of CD45RA+CD45RO− naïve T‐cells was decreased in patients with lower BMI (Figure 8C and 8D and Figure S10D and S10E). Similar to the role of T‐cell senescence in chronic inflammatory conditions, 23 the population of BM CD57+ senescent CD4+ and CD8+ T‐cells was also larger in the patients with lower BMI (Figure 8E and 8F and Figure S10F). Furthermore, the expression of bone resorptive cytokines, including TNF‐α and IL‐17A, was markedly increased in the BM CD4+ and CD8+ T‐cells of patients with lower BMI (Figure 8G–8J). The expression of inflammation‐related genes including CXCL12, CD44, TNF, and IL17A was also higher in the BM of patients with lower BMI (Figure S10G–S10J). Consistent with MKO mice, serum levels of FGF21 and growth differentiation factor 15 (GDF15) tended to be higher in patients with a lower BMI (Figure S11A and S11B). Collectively, these data suggest that lower BMI and muscle function predict a larger population of proinflammatory and cytotoxic senescent T‐cells in the BM of patients with hip fracture.
Figure 8.
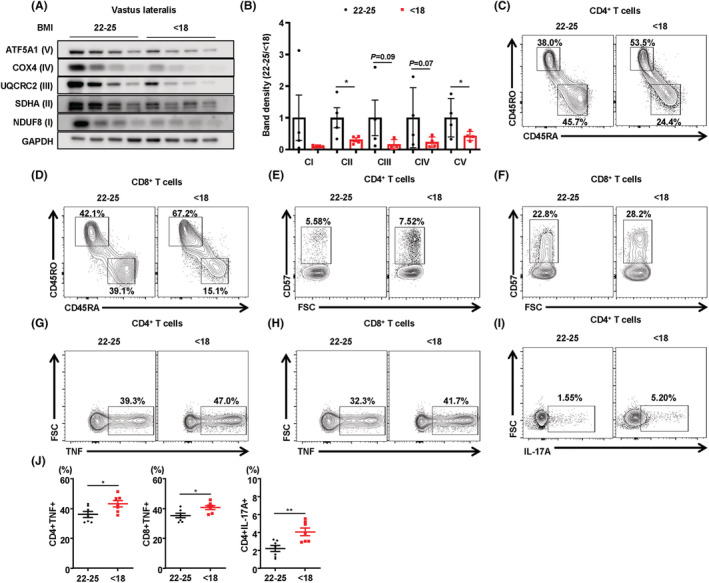
Immunophenotyping of BM T‐cells in patients with hip fracture according to BMI. (A) Representative western blots of OxPhos complex subunits in the vastus lateralis muscle of patients with a normal (22–25 kg/m2) or low (<18 kg/m2) BMI. (B) Band density measurement of OxPhos complex subunits in vastus lateralis muscle from the patients with low or normal BMI. (C, D) Representative flow cytometry contour plots are presented for CD4 and CD8 expression by CD3+ T‐cells in hip fracture patients with normal (22–25 kg/m2) and low (<18 kg/m2) BMI. Representative plots of CD45RO and CD45RA expression among CD4+ or CD8+ T‐cells in the BM from hip fracture patients with a low (<18 kg/m2) or normal (22–25 kg/m2) BMI (each n = 7). (E,F) CD57+ senescent population in CD4+ and CD8+ T‐cells of the BM from hip fracture patients with a low (<18) or normal (22–25) BMI, each n = 7. (G–I) Frequency of TNF‐α‐producing or IL‐17A‐producing cells among the BM CD4+ and CD8+ cells were evaluated by flow cytometry, each n = 7. (J) Statistical analysis of the population of TNF‐α‐producing or IL‐17A‐producing cells among the BM CD4+ and CD8+ cell populations in the two groups, n = 7 per group. Data are expressed as the mean ± standard error of the mean. Statistical significance was analysed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Discussion
In the present work, we investigated the impact of the mitoribosomal function in skeletal muscle on the maintenance of bone mass in mice. Our observations indicate that lower mitochondrial OxPhos function characterizes a reduction in muscle mass and physical activity, which contribute to the bone fragility of MKO mice. In addition, we report that mitochondrial stress in skeletal muscle induces BM inflammation, wherein the CXCL12–CXCR4 axis is involved in the migration and activation of T‐cells. Antagonism of CXCR4 increased bone mass in the MKO mouse model with muscular mitochondrial dysfunction via attenuation of BM inflammation.
Mitochondria are important organelles regulating critical cellular processes in the physiology and pathology of skeletal muscle. Consistent with this, our mouse model harbouring skeletal muscle‐specific mitoribosomal defects showed lower muscle mass and worse physical performance (Figure 1), making it suitable for use as a model of muscle atrophy. Moreover, it has been suggested that appendicular muscle mass and strength are closely associated with BMD. 24 In line with this report, we have demonstrated that MKO mice also exhibit lower bone mass, as well as a reduction in cortical and trabecular BMD (Figure 2). Previous reports also indicate a positive correlation between bone mass and the mass and function of skeletal muscle, which may be due to reduced physical loading or a diminished response to loading. However, the biological mechanism underlying loss of bone mass, and fragility due to muscle dysfunction, has not yet been elucidated. To this end, in the current study, we showed through an osteoimmunologic analysis of MKO mice that BM inflammation plays an essential role in muscle dysfunction‐induced low bone mass and structural deterioration of bone tissue. Thus, it is possible that the improvement of muscle mitochondrial function by exercise training and/or diet may enhance bone mass and quality.
Inflammation is regarded as playing a causal role in bone loss in humans and mice with sex steroid deprivation. TNF‐α‐producing T‐cells are increased in the BM of mice and humans through natural or surgical menopause, contributing to bone loss and fragility. 25 , 26 In addition, oestrogen deficiency induces high levels of serum IL‐17 and promotes Th17 cell differentiation in ovariectomized mice. 27 IL‐17 is increased in patients with primary hyperparathyroidism and also mediates parathyroid hormone‐induced bone loss. 28 Moreover, systemic inflammation caused by sepsis produces osteoblast ablation and bone loss without any change in osteoclast function. 29 In the current work, we show that, in MKO mice, muscular mitochondrial OxPhos dysfunction induces an increase in TNF‐α‐producing and IL‐17A‐producing T‐cells of the BM. The hip fracture patients with a markedly lower BMI also exhibited a higher inflammatory response, as measured by proinflammatory cytokine production, in the BM. Collectively, the present findings indicate that BM inflammation is required for loss of muscle mass and function‐mediated reduction of bone mass and fragility.
CXCR4 stimulates signal transduction for T‐cell chemotaxis and gene expression via association with the T‐cell receptor. 30 CXCR4 is required for the migration of pathogenic CD4+ and CD8+ T‐cells to the BM in a mouse model of aplastic anaemia. 31 In addition, CXCR4 overexpression by CD8+ T‐cells increases their migration towards the BM micro‐environment, leading to memory T‐cell differentiation and production of effector cytokines including IFN‐γ, TNF‐α, and IL‐2. 32 Adoptively transferred central memory T‐cells accumulated more efficiently in BM cavities via their higher level of CXCR4 expression compared with naïve and effector T‐cells. 33 One of the most prominent features of the present work is the observation that CXCR4 antagonism protected MKO mice against BM inflammation and increased bone mass. Consistent with these findings, the CXCR4 antagonist AMD3100 has been shown to improve ovariectomy‐induced bone loss by facilitating mobilization of haematopoietic progenitor cells. 34 Moreover, treatment with vascular endothelial growth factor and AMD3100 can mobilize mesenchymal stem cells towards fracture healing, leading to bone formation in a delayed union osteotomy model. 35 CXCL12, a major ligand for CXCR4, is not only linked to the severity of postmenopausal osteoporosis 36 but also acts as a proinflammatory factor during progression of collagen‐induced osteoarthritis by attracting inflammatory cells to joints and by activating osteoclasts. 37 On the other hand, genetic disruption of CXCR4 enhances osteoclastogenesis and leads to increased osteolytic tumour growth in bone. 38 Discrepancies in findings between different studies could be attributed to the use of different mouse or disease models and/or to the distinct role of CXCR4 in many different kinds of cell. Therefore, further investigation is required to establish the role of CXCR4 in the diverse context of bone loss using a cell type‐specific Cxcr4 knockout mouse model.
In this study, we analysed two kinds of RNA sequencing data derived from EDL muscle and whole BM cells from control and MKO mice. We observed that mitochondrial stress via OxPhos dysfunction increases local expression and serum levels of FGF21, which do not have any effects on bone mass and quality in MKO mice (Figures S4 and S5). On the other hand, GSEA indicated that BM cells from MKO mice showed prominent expression differences in inflammatory response and adipogenesis gene sets compared with the controls (Figure 4). Ectopic fat, defined as storage of triglyceride in tissues other than adipose tissue, is associated with metabolic deterioration in humans as well as rodents. Although the pathogenesis of ectopic fat deposition is largely unknown, free fatty acids released by adipocyte hypertrophy and inflammatory response are important in the development of ectopic fat‐induced organ dysfunction in liver, skeletal muscle, and heart. 39 Excessive fat deposition in non‐adipose tissues recruits immune cells including macrophages and activated T‐cells, thereby promoting chronic inflammation in the metabolic organs. 40 The BM is also susceptible to fat deposition through anorexia nervosa or weight loss surgery. 41 , 42 Although multiple studies have investigated ectopic adipocyte accumulation in BM cavities, our understanding of its role in the inflammatory response in BM is incomplete. Ectopic fat accumulation in BM is associated with activation of immune cells and bone loss. 43 Taken together, these data suggest that muscular mitochondrial dysfunction‐mediated bone loss is, at least partially, caused by the changes in the BM micro‐environment.
In conclusion, mitochondrial OxPhos function in skeletal muscles play a pivotal role in the regulation of BM inflammation and bone loss in mice. Inhibition of BM inflammation by a CXCR4 antagonist increases bone mass in a mouse model with muscle mitochondrial dysfunction. However, the human relevance of muscular mitochondrial OxPhos dysfunction and BM inflammation in the regulation of skeletal homeostasis needs to be clarified.
Funding
This work was supported by the Basic Science Research Program, through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT, and Future Planning, Korea (NRF‐2019M3E5D1A02068575 and NRF‐2021R1A5A8029876). H.S.Y. was supported by the Chungnam National University Hospital Research Fund (2019) and by a Korean Endocrine Society of Hyangseol Young Investigator Award (2020). M.S. was supported by the NRF (NRF‐2017R1E1A1A01075126).
Conflict of interests
The authors declare no competing interests.
Supporting information
Figure S1. Generation of the skeletal muscle‐specific mitochondrial oxidative phosphorylation (OxPhos) dysfunction mouse model. (A) Strategy for generating MKO mice through disrupting Crif1 in skeletal muscle using theMLC1f (myosin light chain 1f)‐Cre mice through Cre‐LoxP system. (B, C) Immunoblotting and band density measurement of OxPhos complex subunits in soleus muscle from 14‐ week‐old control and MKO mice, n = 3. (D) Mitochondrial morphology of controls and MKO mice, visualized by electron microscopy; scale bars: 1 μm. (E, F) Immunoblotting and band density measurement of CRIF1 and OxPhos; complex subunits in BM and cortical bone of femur from 14‐ week‐old control and MKO mice, n = 3. (G) Body weight evolution of control and MKO mice fed a chow diet for 8 weeks, n = 10 per group. Data are expressed as the mean ± SEM. Statistical significance was analyzed by unpaired t‐tests. **, P < 0.01 compared with the indicated group.
Figure S2. Micro‐CT analysis of vertebrae and femurs of control and MKO mice at 14 weeks‐of‐age. (A) The cortical area in femurs of control and MKO mice were measured by micro‐CT. (B) Cortical bone area/total tissue area (Ct.Ar/Tt.Ar; %) was measured in femurs of control and MKO mice. (C, D) Measurement of bone volume (BV)/total volume (TV) using von Kossa staining of vertebrae and femurs from 14‐week‐old control and MKO mice, n = 5. (E, F) Quantitative analysis of eroded surface per bone surface (%) and osteoclast surface (%) of tibiae from 14‐week‐old control and MKO mice, n = 4. Data are expressed as the mean ± SEM. Statistical significance was analyzed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Figure S3. Measurement of serum levels of hormones affecting bone metabolism in control and MKO mice. (A‐D) Serum concentrations of intact parathyroid hormone (iPTH), testosterone, T4, and T3 in 14‐week‐old control and MKO mice, n = 7/group. Data are expressed as the mean ± SEM.
Figure S4. FGF21 is highly induced by mitochondrial stress in skeletal muscle of MKO mice. (A) Scatterplots of RNA sequencing data, displaying transcript levels in EDL of control (x‐axis) and MKO (y‐axis) mice at 10 weeks‐of‐age. The text indicates that mitokines show much higher fold change in MKO mice. (B, C) Volcano plot and heat map showing upregulated genes in EDL from normal chow diet‐fed control and MKO mice at 10 weeks‐of‐age. (D) Relative expression of mRNA encoding Fgf21 in EDL from 14‐week‐old control and MKO mice, n = 6. (E) Serum levels of FGF21 in 14‐week‐old control and MKO mice, n = 6. (F) Strategy for generating FGF21/CRF1 double knockout mice (MFKO) and genotype analysis using real‐time PCR. (G) Crif1 and Fgf21 expression in the extensor digitorum longus muscle of MKO and MFKO mice at 6 weeks‐of‐age. Data are expressed as the mean ± SEM. **, P < 0.01 compared with the corresponding controls.
Figure S5. Lower bone mass in MKO mice is independent of FGF21 production caused by mitochondrial stress in skeletal muscle. (A) Micro‐CT images of the trabecular bone (Tr.b) near the distal femoral metaphyseal region of control and MKO mice at 14 weeks‐of‐age. Scale bar for the front view and 3D image of the Tr.b = 1000 and 500 μm, respectively; n = 4. (B) Measurement of Tb.Th., Tb.N., BV/TV, BS/TV, BS/BV, Ct.V., Tb.Sp., and TBV using micro‐CT analysis. Data are expressed as the mean ± SEM. Statistical significance was analyzed by unpaired t‐tests.
Figure S6. Measurement of serum levels of proinflammatory cytokines in 14‐week‐old control and MKO mice. (A) Serum TNF‐α and IL‐17A concentrations in control (n = 7) and MKO (n = 7) mice at 14 weeks‐of‐age. (B) A representative section of tibia from a 14‐week‐old control and MKO mouse stained with H&E. Adipocyte‐rich BM (arrowhead) are visible in the MKO mice. Scale bars, 100 μm. Quantification of the number of adipocytes per bone marrow surface area (mm2) in control (n = 4) and MKO (n = 4) mice at 14 weeks‐of‐age. Data are expressed as the mean ± SEM. **, P < 0.01 compared with the corresponding controls.
Figure S7. Analysis of RNA sequencing data from BM cells of control and MKO mice. (A, B) The analysis was performed using Network2Canvas. Genes that were significantly upregulated in the BM cells of control and MKO mice were analyzed for gene‐list enrichment, with gene set libraries created from level 4 of the MGI mouse phenotype ontology using Network2Canvas. (C) Cxcl12 mRNA expression by BM cells from control and MKO mice at 14 weeks‐of‐age. (D) In the G‐MAD analysis, CXCL12 associates with T‐cell proliferation and CXCR chemokine receptor binding modules in mice. The threshold of significant gene‐module association is indicated by the red dashed line. Modules are organized by module similarities. Known modules connected to CXCL12 are highlighted in red. Data are expressed as mean ± SEM. *, P < 0.05 and **, P < 0.01 compared with the corresponding controls.
Figure S8. Flow cytometry analysis of BM cells from 12‐week‐old control and MKO mice treated with or without AMD3100. (A) Populations of CD4+ and CD8+ T‐cells from control and MKO mice treated with or without AMD3100 for 3 weeks. (B‐E) IFN‐γ‐ and TNF‐α‐producing CD4+ and CD8+ T‐cells in the BM from control and MKO mice treated with or without AMD3100 for 3 weeks. (F) Statistical analysis of phenotypes defined by flow cytometry in the BM from control and MKO mice treated with or without AMD3100 for 3 weeks. Data are expressed as mean ± SEM. Statistical significance was analyzed by one‐way ANOVA. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Figure S9. Micro‐CT analysis and measurement of serum markers of liver injury and lipid metabolism. (A,B) Cortical bone area/total tissue area (Ct.Ar/Tt.Ar; %) was measured in tibia and femurs of control and MKO mice treated with or without AMD3100. (C‐F) Serum levels of aspartate transaminase (AST), alanine aminotransferase (ALT), total cholesterol (CHOL), and triglyceride (TG) from 12‐week‐old control and MKO mice treated with or without AMD3100. Data are expressed as mean ± SEM. Statistical significance was analyzed by one‐way ANOVA. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Figure S10. Grip strength and flow cytometry analysis of BM cells of patients with hip fracture. (A) Measurement of grip strength of the hip fracture patients using a handheld dynamometer. (B) Representative blots showing BN‐PAGE of the assembled OxPhos complex in vastus lateralis muscle from patients with lower (<18 kg/m2) or normal (22–25 kg/m2) BMI. (C) Populations of CD4+ and CD8+ T‐cells from the hip fracture patients with lower (<18 kg/m2) or normal (22–25 kg/m2) body mass index (BMI). (D, E) Naïve (CD45RA+CD45RO‐) and memory (CD45RA‐CD45RO+) CD4+ and CD8+ T‐cells in the BM from the hip fracture patients with lower or normal BMI. (F) BM senescent (CD28‐CD57+) CD4+ and CD8+ T‐cells from hip fracture patients with lower (<18) or normal (22–25) BMI. (G‐J) Transcript levels of CXCL12, CD44, TNF, and IL17A in the BM from hip fracture patients with lower (<18) or normal (22–25) BMI. Data are expressed as mean ± SEM. *, P < 0.05 and **, P < 0.01 compared with the corresponding controls.
Figure S11. Serum levels of mitokines in patients with hip fracture. (A, B) Serum FGF21 and GDF15 levels were measured in hip fracture patients with lower (<18 kg/m2) or normal (22–25 kg/m2) BMI. Data are expressed as the mean ± SEM. *, P < 0.05 compared with the corresponding controls.
Table S1. Demographics and baseline characteristics of the human subjects
Table S2. Antibodies used for flow cytometry analysis
Table S3. Primers used for real‐time PCR (mouse)
Table S4. Primers used for real‐time PCR (human)
Acknowledgements
All mouse and human experiments were performed in accordance with local and national ethical regulations. All authors of this manuscript complied with the guidelines of ethical authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 44
Tian J., Chung H. K., Moon J. S., Nga H. T., Lee H. Y., Kim J. T., Chang J. Y., Kang S. G., Ryu D., Che X., Choi J.‐Y., Tsukasaki M., Sasako T., Lee S.‐H., Shong M., and Yi H.‐S. (2022) Skeletal muscle mitoribosomal defects are linked to low bone mass caused by bone marrow inflammation in male mice, Journal of Cachexia, Sarcopenia and Muscle, 13, 1785–1799, 10.1002/jcsm.12975
Jingwen Tian and Hyo Kyun Chung contributed equally to this work.
References
- 1. Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol 2015;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, et al. Identification of a molecular signature of sarcopenia. Physiol Genomics 2005;21:253–263. [DOI] [PubMed] [Google Scholar]
- 3. Johannsen DL, Conley KE, Bajpeyi S, Punyanitya M, Gallagher D, Zhang Z, et al. Ectopic lipid accumulation and reduced glucose tolerance in elderly adults are accompanied by altered skeletal muscle mitochondrial activity. J Clin Endocrinol Metab 2012;97:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Verschueren S, Gielen E, O'Neill TW, Pye SR, Adams JE, Ward KA, et al. Sarcopenia and its relationship with bone mineral density in middle‐aged and elderly European men. Osteoporos Int 2013;24:87–98. [DOI] [PubMed] [Google Scholar]
- 5. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004;429:417–423. [DOI] [PubMed] [Google Scholar]
- 6. Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, et al. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN‐gamma‐induced class II transactivator. Proc Natl Acad Sci U S A 2003;100:10405–10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li JY, Tawfeek H, Bedi B, Yang X, Adams J, Gao KY, et al. Ovariectomy disregulates osteoblast and osteoclast formation through the T‐cell receptor CD40 ligand. Proc Natl Acad Sci U S A 2011;108:768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sjogren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, et al. The gut microbiota regulates bone mass in mice. J Bone Miner Res 2012;27:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciucci T, Ibanez L, Boucoiran A, Birgy‐Barelli E, Pene J, Abou‐Ezzi G, et al. Bone marrow Th17 TNFα cells induce osteoclast differentiation, and link bone destruction to IBD. Gut 2015;64:1072–1081. [DOI] [PubMed] [Google Scholar]
- 10. Kim SJ, Kwon MC, Ryu MJ, Chung HK, Tadi S, Kim YK, et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab 2012;16:274–283. [DOI] [PubMed] [Google Scholar]
- 11. Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J Cell Biol 2017;216:149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forsström S, Jackson CB, Carroll CJ, Kuronen M, Pirinen E, Pradhan S, et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab 2019;30:1040, e7–1054. [DOI] [PubMed] [Google Scholar]
- 13. Keipert S, Ost M, Johann K, Imber F, Jastroch M, van Schothorst EM, et al. Skeletal muscle mitochondrial uncoupling drives endocrine cross‐talk through the induction of FGF21 as a myokine. Am J Physiol Endocrinol Metab 2014;306:E469–E482. [DOI] [PubMed] [Google Scholar]
- 14. Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator‐activated receptor γ. Proc Natl Acad Sci U S A 2012;109:3143–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis (New York, NY: 2000) 2000;26:165–166. [PubMed] [Google Scholar]
- 16. Erben RG. Embedding of bone samples in methylmethacrylate: an improved method suitable for bone histomorphometry, histochemistry, and immunohistochemistry. J Histochem Cytochem 1997;45:307–313. [DOI] [PubMed] [Google Scholar]
- 17. Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2013;28:2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Rukina D, David FPA, Li TY, Oh CM, Gao AW, et al. Identifying gene function and module connections by the integration of multispecies expression compendia. Genome Res 2019;29:2034–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukohira H, Hara T, Abe S, Tani‐Ichi S, Sehara‐Fujisawa A, Nagasawa T, et al. Mesenchymal stromal cells in bone marrow express adiponectin and are efficiently targeted by an adiponectin promoter‐driven Cre transgene. Int Immunol 2019;31:729–742. [DOI] [PubMed] [Google Scholar]
- 20. Kim D, Kim J, Yoon JH, Ghim J, Yea K, Song P, et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 2014;57:1456–1465. [DOI] [PubMed] [Google Scholar]
- 21. Teicher BA, Fricker SP. CXCL12 (SDF‐1)/CXCR4 pathway in cancer. Clin Cancer Res 2010;16:2927–2931. [DOI] [PubMed] [Google Scholar]
- 22. Pasdar Y, Darbandi M, Mirtaher E, Rezaeian S, Najafi F, Hamzeh B. Associations between muscle strength with different measures of obesity and lipid profiles in men and women: results from RaNCD cohort study. Clin Nutr Res 2019;8:148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bektas A, Schurman SH, Sen R, Ferrucci L. Human T cell immunosenescence and inflammation in aging. J Leukoc Biol 2017;102:977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blain H, Jaussent A, Thomas E, Micallef JP, Dupuy AM, Bernard PL, et al. Appendicular skeletal muscle mass is the strongest independent factor associated with femoral neck bone mineral density in adult and older men. Exp Gerontol 2010;45:679–684. [DOI] [PubMed] [Google Scholar]
- 25. Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, et al. Sex steroid deficiency‐associated bone loss is microbiota dependent and prevented by probiotics. J Clin Invest 2016;126:2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adeel S, Singh K, Vydareny KH, Kumari M, Shah E, Weitzmann MN, et al. Bone loss in surgically ovariectomized premenopausal women is associated with T lymphocyte activation and thymic hypertrophy. J Invest Med 2013;61:1178–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL‐17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS ONE 2012;7:e44552, 10.1371/journal.pone.0044552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li JY, D'Amelio P, Robinson J, Walker LD, Vaccaro C, Luo T, et al. IL‐17A is increased in humans with primary hyperparathyroidism and mediates PTH‐induced bone loss in mice. Cell Metab 2015;22:799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terashima A, Okamoto K, Nakashima T, Akira S, Ikuta K, Takayanagi H. Sepsis‐induced osteoblast ablation causes immunodeficiency. Immunity 2016;44:1434–1443. [DOI] [PubMed] [Google Scholar]
- 30. Kumar A, Humphreys TD, Kremer KN, Bramati PS, Bradfield L, Edgar CE, et al. CXCR4 physically associates with the T cell receptor to signal in T cells. Immunity 2006;25:213–224. [DOI] [PubMed] [Google Scholar]
- 31. Arieta Kuksin C, Gonzalez‐Perez G, Minter LM. CXCR4 expression on pathogenic T cells facilitates their bone marrow infiltration in a mouse model of aplastic anemia. Blood 2015;125:2087–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khan AB, Carpenter B, Santos ESP, Pospori C, Khorshed R, Griffin J, et al. Redirection to the bone marrow improves T cell persistence and antitumor functions. J Clin Invest 2018;128:2010–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mazo IB, Honczarenko M, Leung H, Cavanagh LL, Bonasio R, Weninger W, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 2005;22:259–270. [DOI] [PubMed] [Google Scholar]
- 34. Im JY, Min WK, Park MH, Kim N, Lee JK, Jin HK, et al. AMD3100 improves ovariectomy‐induced osteoporosis in mice by facilitating mobilization of hematopoietic stem/progenitor cells. BMB Rep 2014;47:439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meeson R, Sanghani‐Keri A, Coathup M, Blunn G. VEGF with AMD3100 endogenously mobilizes mesenchymal stem cells and improves fracture healing. J Orthop Res 2019;37:1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang XW, Huang HX, Wang F, Zhou QL, Huang YQ, Qin RZ. Elevated plasma CXCL12/SDF‐1 levels are linked with disease severity of postmenopausal osteoporosis. Innate Immun 2020;26:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Klerck B, Geboes L, Hatse S, Kelchtermans H, Meyvis Y, Vermeire K, et al. Pro‐inflammatory properties of stromal cell‐derived factor‐1 (CXCL12) in collagen‐induced arthritis. Arthritis Res Ther 2005;7:R1208–R1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hirbe AC, Rubin J, Uluckan O, Morgan EA, Eagleton MC, Prior JL, et al. Disruption of CXCR4 enhances osteoclastogenesis and tumor growth in bone. Proc Natl Acad Sci U S A 2007;104:14062–14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Snel M, Jonker JT, Schoones J, Lamb H, de Roos A, Pijl H, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol 2012;2012:983814–18, 10.1155/2012/983814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Byun JS, Yi HS. Hepatic immune microenvironment in alcoholic and nonalcoholic liver disease. Biomed Res Int 2017;2017:6862439, 10.1155/2017/6862439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim TY, Schwartz AV, Li X, Xu K, Black DM, Petrenko DM, et al. Bone marrow fat changes after gastric bypass surgery are associated with loss of bone mass. J Bone Miner Res 2017;32:2239–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bredella MA, Fazeli PK, Miller KK, Misra M, Torriani M, Thomas BJ, et al. Increased bone marrow fat in anorexia nervosa. J Clin Endocrinol Metab 2009;94:2129–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miggitsch C, Meryk A, Naismith E, Pangrazzi L, Ejaz A, Jenewein B, et al. Human bone marrow adipocytes display distinct immune regulatory properties. EBioMedicine 2019;46:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Generation of the skeletal muscle‐specific mitochondrial oxidative phosphorylation (OxPhos) dysfunction mouse model. (A) Strategy for generating MKO mice through disrupting Crif1 in skeletal muscle using theMLC1f (myosin light chain 1f)‐Cre mice through Cre‐LoxP system. (B, C) Immunoblotting and band density measurement of OxPhos complex subunits in soleus muscle from 14‐ week‐old control and MKO mice, n = 3. (D) Mitochondrial morphology of controls and MKO mice, visualized by electron microscopy; scale bars: 1 μm. (E, F) Immunoblotting and band density measurement of CRIF1 and OxPhos; complex subunits in BM and cortical bone of femur from 14‐ week‐old control and MKO mice, n = 3. (G) Body weight evolution of control and MKO mice fed a chow diet for 8 weeks, n = 10 per group. Data are expressed as the mean ± SEM. Statistical significance was analyzed by unpaired t‐tests. **, P < 0.01 compared with the indicated group.
Figure S2. Micro‐CT analysis of vertebrae and femurs of control and MKO mice at 14 weeks‐of‐age. (A) The cortical area in femurs of control and MKO mice were measured by micro‐CT. (B) Cortical bone area/total tissue area (Ct.Ar/Tt.Ar; %) was measured in femurs of control and MKO mice. (C, D) Measurement of bone volume (BV)/total volume (TV) using von Kossa staining of vertebrae and femurs from 14‐week‐old control and MKO mice, n = 5. (E, F) Quantitative analysis of eroded surface per bone surface (%) and osteoclast surface (%) of tibiae from 14‐week‐old control and MKO mice, n = 4. Data are expressed as the mean ± SEM. Statistical significance was analyzed by unpaired t‐tests. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Figure S3. Measurement of serum levels of hormones affecting bone metabolism in control and MKO mice. (A‐D) Serum concentrations of intact parathyroid hormone (iPTH), testosterone, T4, and T3 in 14‐week‐old control and MKO mice, n = 7/group. Data are expressed as the mean ± SEM.
Figure S4. FGF21 is highly induced by mitochondrial stress in skeletal muscle of MKO mice. (A) Scatterplots of RNA sequencing data, displaying transcript levels in EDL of control (x‐axis) and MKO (y‐axis) mice at 10 weeks‐of‐age. The text indicates that mitokines show much higher fold change in MKO mice. (B, C) Volcano plot and heat map showing upregulated genes in EDL from normal chow diet‐fed control and MKO mice at 10 weeks‐of‐age. (D) Relative expression of mRNA encoding Fgf21 in EDL from 14‐week‐old control and MKO mice, n = 6. (E) Serum levels of FGF21 in 14‐week‐old control and MKO mice, n = 6. (F) Strategy for generating FGF21/CRF1 double knockout mice (MFKO) and genotype analysis using real‐time PCR. (G) Crif1 and Fgf21 expression in the extensor digitorum longus muscle of MKO and MFKO mice at 6 weeks‐of‐age. Data are expressed as the mean ± SEM. **, P < 0.01 compared with the corresponding controls.
Figure S5. Lower bone mass in MKO mice is independent of FGF21 production caused by mitochondrial stress in skeletal muscle. (A) Micro‐CT images of the trabecular bone (Tr.b) near the distal femoral metaphyseal region of control and MKO mice at 14 weeks‐of‐age. Scale bar for the front view and 3D image of the Tr.b = 1000 and 500 μm, respectively; n = 4. (B) Measurement of Tb.Th., Tb.N., BV/TV, BS/TV, BS/BV, Ct.V., Tb.Sp., and TBV using micro‐CT analysis. Data are expressed as the mean ± SEM. Statistical significance was analyzed by unpaired t‐tests.
Figure S6. Measurement of serum levels of proinflammatory cytokines in 14‐week‐old control and MKO mice. (A) Serum TNF‐α and IL‐17A concentrations in control (n = 7) and MKO (n = 7) mice at 14 weeks‐of‐age. (B) A representative section of tibia from a 14‐week‐old control and MKO mouse stained with H&E. Adipocyte‐rich BM (arrowhead) are visible in the MKO mice. Scale bars, 100 μm. Quantification of the number of adipocytes per bone marrow surface area (mm2) in control (n = 4) and MKO (n = 4) mice at 14 weeks‐of‐age. Data are expressed as the mean ± SEM. **, P < 0.01 compared with the corresponding controls.
Figure S7. Analysis of RNA sequencing data from BM cells of control and MKO mice. (A, B) The analysis was performed using Network2Canvas. Genes that were significantly upregulated in the BM cells of control and MKO mice were analyzed for gene‐list enrichment, with gene set libraries created from level 4 of the MGI mouse phenotype ontology using Network2Canvas. (C) Cxcl12 mRNA expression by BM cells from control and MKO mice at 14 weeks‐of‐age. (D) In the G‐MAD analysis, CXCL12 associates with T‐cell proliferation and CXCR chemokine receptor binding modules in mice. The threshold of significant gene‐module association is indicated by the red dashed line. Modules are organized by module similarities. Known modules connected to CXCL12 are highlighted in red. Data are expressed as mean ± SEM. *, P < 0.05 and **, P < 0.01 compared with the corresponding controls.
Figure S8. Flow cytometry analysis of BM cells from 12‐week‐old control and MKO mice treated with or without AMD3100. (A) Populations of CD4+ and CD8+ T‐cells from control and MKO mice treated with or without AMD3100 for 3 weeks. (B‐E) IFN‐γ‐ and TNF‐α‐producing CD4+ and CD8+ T‐cells in the BM from control and MKO mice treated with or without AMD3100 for 3 weeks. (F) Statistical analysis of phenotypes defined by flow cytometry in the BM from control and MKO mice treated with or without AMD3100 for 3 weeks. Data are expressed as mean ± SEM. Statistical significance was analyzed by one‐way ANOVA. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Figure S9. Micro‐CT analysis and measurement of serum markers of liver injury and lipid metabolism. (A,B) Cortical bone area/total tissue area (Ct.Ar/Tt.Ar; %) was measured in tibia and femurs of control and MKO mice treated with or without AMD3100. (C‐F) Serum levels of aspartate transaminase (AST), alanine aminotransferase (ALT), total cholesterol (CHOL), and triglyceride (TG) from 12‐week‐old control and MKO mice treated with or without AMD3100. Data are expressed as mean ± SEM. Statistical significance was analyzed by one‐way ANOVA. *, P < 0.05 and **, P < 0.01 compared with the indicated group.
Figure S10. Grip strength and flow cytometry analysis of BM cells of patients with hip fracture. (A) Measurement of grip strength of the hip fracture patients using a handheld dynamometer. (B) Representative blots showing BN‐PAGE of the assembled OxPhos complex in vastus lateralis muscle from patients with lower (<18 kg/m2) or normal (22–25 kg/m2) BMI. (C) Populations of CD4+ and CD8+ T‐cells from the hip fracture patients with lower (<18 kg/m2) or normal (22–25 kg/m2) body mass index (BMI). (D, E) Naïve (CD45RA+CD45RO‐) and memory (CD45RA‐CD45RO+) CD4+ and CD8+ T‐cells in the BM from the hip fracture patients with lower or normal BMI. (F) BM senescent (CD28‐CD57+) CD4+ and CD8+ T‐cells from hip fracture patients with lower (<18) or normal (22–25) BMI. (G‐J) Transcript levels of CXCL12, CD44, TNF, and IL17A in the BM from hip fracture patients with lower (<18) or normal (22–25) BMI. Data are expressed as mean ± SEM. *, P < 0.05 and **, P < 0.01 compared with the corresponding controls.
Figure S11. Serum levels of mitokines in patients with hip fracture. (A, B) Serum FGF21 and GDF15 levels were measured in hip fracture patients with lower (<18 kg/m2) or normal (22–25 kg/m2) BMI. Data are expressed as the mean ± SEM. *, P < 0.05 compared with the corresponding controls.
Table S1. Demographics and baseline characteristics of the human subjects
Table S2. Antibodies used for flow cytometry analysis
Table S3. Primers used for real‐time PCR (mouse)
Table S4. Primers used for real‐time PCR (human)