

Abstract
Primaquine (PQ), an 8-aminoquinoline antimalarial drug, has been widely used for the eradication of hypnozoites from the liver and, therefore, recognized as the radical cure of malaria. However, the clinical applications of PQ are restricted to patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency due to severe dose-related hemolytic side effects. Nanoparticle carriers have shown great potential in achieving higher PQ concentrations in the target site, thereby reducing dose-related systemic toxicity caused by non-specific exposure. This work aims to develop, compare, and evaluate three PQ-loaded lipid-based drug carriers including solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and nano-emulsions (NE). The optimized PQ-SLN, PQ-NLC, and PQ-NE had a particle size of 250 nm, a PDI range of 0.1 to 0.3, a zeta potential of −30 mV, and entrapment efficiency of ∼90 %. All lipid formulations showed sustained release in both simulated gastric and intestinal fluids over 6 hours. Four empirical models - including zero-order, Higuchi, Korsmeyer-Peppas, and Hixson-Crowell models - were tested to understand the drug release mechanisms of PQ-SLN, PQ-NLC, and PQ-NE. The model fitness was found to be the highest in the Korsmeyer-Peppas model for all the PQ-loaded lipid formulations (R2: 0.88–0.94). No significant changes were observed in the entrapment efficiency, particle size, and PDI of lipid formulations throughout 1 month of storage at 4 °C and 25 °C. PQ-SLN and PQ-NLC can be further lyophilized with cryoprotectants to improve long-term stability. Finally, the treatment of erythrocytes with PQ-SLN, PQ-NLC, and PQ-NE reduced erythrocyte hemolysis by approximately 4.5-fold compared to the free drug solution.
Keywords: Primaquine, Lipid-based formulation(s), Particle size, In vitro release, Stability, Ex vivo hemolysis
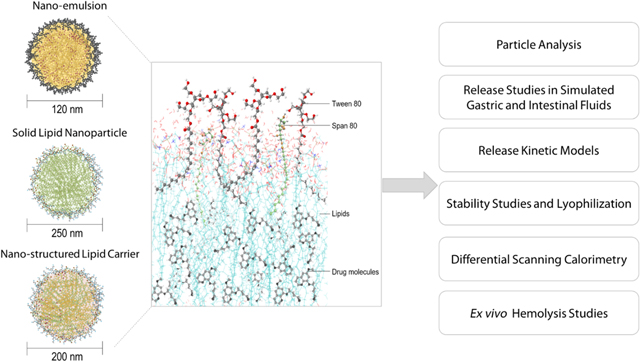
Introduction
Malaria, a disease caused by Plasmodium parasites, has been a severe public health problem that causes an estimated 1.5 billion cases and 7.6 million deaths from 2000–2019 [1]. Among the six Plasmodium parasite species known to infect humans only two species, P. vivax and P. ovale, can form dormant liver-stage parasites (hypnozoites) and cause frequent relapses of malaria. A proportion of the parasites growing in the liver transform into hypnozoites and later become a persistent reservoir of infection. Hypnozoites lie dormant for weeks to months after the primary infection until they develop into mature liver-stage schizonts containing thousands of merozoites. Eventually, the schizonts burst and release merozoites into the bloodstream to infect red blood cells (RBCs), resulting in the relapse of malaria [2, 3].
Primaquine (PQ), an 8-aminoquinoline drug approved by the FDA in 1952, has been widely used ever since for the eradication of liver-stage parasites [4]. Despite more than 70 years of clinical applications, the mechanism of toxicity and efficacy are still not entirely understood. A recent study, for example, suggested that hydrogen peroxide generated from the spontaneous oxidation of hydroxylated metabolites of PQ is believed to kill dormant and active liver-stage Plasmodium parasites [5]. Additionally, biotransformation of PQ hydroxylated metabolites in the RBCs is believed to be associated with PQ-induced hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency, the most common enzyme deficiency affecting over 400 million people worldwide [6]. The degree of hemolysis is dependent on the total dose and genetic variability of G6PD, therefore, the dosing regimens are often limited to 15 mg or 30 mg daily for two weeks, resulting in poor patient compliance to drug therapy [4, 7].
In oral drug delivery, lipid-based formulations have shown several advantages over other types of delivery systems for improving solubilization and bioavailability of poorly water-soluble drugs, protecting drugs from degradation in the gastrointestinal tract, controlled release profiles, and high drug load potential [8–10]. Previous studies have shown that encapsulating PQ into liposomes, chylomicron emulsions, and dendrimers can significantly improve the therapeutic performance by achieving higher PQ concentrations in the liver in comparison to other organs (e.g., blood, lungs, kidneys, hearts, etc.), through intravenous route [11–13]. However, intravenous administration has several disadvantages compared with the oral route: trained medical staff is required as well as there being an increased cost, the risk of infection, and pain. Drugs given by the intravenous route without constant monitoring for adverse effects can cause significant harm. The oral route is considered to be safer, more effective, and convenient. Oral lipid nanoparticles have been reported to enhance the accumulation of PQ in the liver [14, 15], however, these reports did not explore, compare, or contrast the characteristics of various lipid-based formulations to examine how structural differences could affect the efficiency of drug loading as well as the ability to reduce hemolytic toxicity.
The present study aimed to develop, optimize, and compare three PQ-loaded oral lipid-based formulations namely, solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and nano-emulsion (NE). The influence of lipid excipients, surfactants, and drug loading on the physicochemical characteristics of lipid formulations was evaluated through particle size, polydispersity index, drug content, entrapment efficiency, and differential scanning calorimetry. The release kinetics of the drug from SLN, NLC, and NE was explored in simulated gastric and intestinal conditions. Four empirical release kinetic models — including zero-order, Higuchi, Korsmeyer-Peppas, and Hixson-Crowell models — were employed to understand the drug release mechanisms from the lipid-based formulations. The storage stability of lipid nanoparticles was examined at 25 °C and 4 °C. Finally, the ability of SLN, NLC, and NE to protect erythrocytes from PQ-induced cell lysis was investigated in an ex vivo hemolysis toxicity study.
2. Materials and Methods
2.1. Materials
PQ phosphate was obtained from Sigma-Aldrich, USA. Tween® 80 and Span® 80 were supplied by Acros Organics, USA. Compritol® 888 ATO, Precirol® ATO 5, Geleol™ Mono and Diglycerides NF, Lauroglycol™ FCC and Gelucire® 43/01 were obtained from Gattefossé, USA. Solutol® HS 15 was supplied by BASF, USA. Poloxamer® 188, Miglyol® 829, olive oil, soybean oil, corn oil, sesame oil, oleic acid, and castor oil were purchased and used as received. Amicon® Ultra centrifugal filter units (Molecular weight cut off (MWCO) 100 kDa) were purchased from Millipore, USA. Fasted-state simulated gastric fluid (FaSSGF) and fasted-state simulated intestinal fluid (FaSSIF) powder were purchased from Biorelevant, UK. Sprague Dawley rat red blood cells (catalog number: IRTSDRBC 10 ML) were purchased from Innovative Research, USA. All other chemicals and solvents were of analytical or high-performance liquid chromatography (HPLC) grade.
2.2. Production of PQ base
PQ phosphate was converted into PQ base following procedures as described by Kamalinder et al [13]. PQ base was obtained by alkalinization of PQ phosphate with ammonium hydroxide at pH 12.0, which was then followed by two extractions with chloroform. The organic phase was washed twice with water and twice with a saturated solution of sodium chloride. Before evaporation, the chloroform was dried with anhydrous sodium sulfate.
2.3. HPLC analysis of PQ
Samples were analyzed using a Waters chromatographic system consisting of Waters 717 plus autosampler and Waters 2487 dual λ absorbance detector. A mixture of water, acetonitrile, tetrahydrofuran, and trifluoroacetic acid (900:90:10:1) was used as the mobile phase on a Phenomenex Luna® C8(2) column (4.6 mm x 30 mm, 3.0 µm). The mobile phase was filtered through a 0.22 μm Millipore membrane filter and degassed for 15 min before use. All samples were injected and analyzed under isocratic elution at the flow rate of 1.2 mL/min. UV detection was set at 254 nm wavelength. Linear regression analysis of PQ peak area as a function of concentration exhibited linearity (R2 > 0.999) in the concentration ranges of 1 and 100 µg/mL. The average percentage of recoveries for PQ at three different concentrations (1 µg/mL, 10 µg/mL, 25 µg/mL) was found to be 99.4± 1.7 %. The intra-day and inter-day RSD of standard solutions at three concentration levels were lower than 1.2 and 1.6 %, respectively.
2.4. Screening of lipids
Five hundred milligram of solid lipids were heated to 80 °C and then small amounts of PQ was added gradually under continuous stirring until the mixture became turbid and precipitation was observed [16]. Additionally, a drop of the supernatant was transferred onto a microscope slide covered with a glass coverslip and examined using light microscopy. In the case of liquid lipids, PQ was gradually added into 500 mg of liquid lipids and then the mixture was stirred in the shaker for 24 h, filtered through 0.22 µm filters, and analyzed by HPLC [17].
2.5. Preparation and characterization of PQ-SLN, PQ-NLC, and PQ-NE
Three different types of lipid-based formulations (PQ-SLN, PQ-NLC, and PQ-NE) were prepared by homogenization and ultrasonication methods [18]. Castor oil and Compritol® 888 ATO were used as liquid lipid and solid lipid, respectively. The lipid phase containing PQ, lipids, and Span® 80 was heated at 80 °C. The aqueous solution containing Tween® 80 was heated to the same temperature and then added into the lipid phase under stirring at 1,000 rpm. The premix was homogenized at 16,000 rpm for 5 min using T 25 digital Ultra-Turrax® and further sonicated at 40 % amplitude for 15 min using a probe sonicator (Sonics, USA). The hot nanoemulsion was cooled down to room temperature to obtain lipid nanoparticles.
The intensity weighted average particle size (z-average) and polydispersity index (PDI) of SLN, NLC, and NE formulations were determined by photon correlation spectroscopy (Malvern Zetasizer Nano ZS Zen3600, Malvern Instruments, USA). Samples were diluted at 1:10 (v/v) with ultra-pure water before measurement. All measurements were performed in triplicate at 25 °C. The surface charge of lipid nanoparticles was determined by using Malvern Zetasizer Nano ZS Zen3600 (Malvern Instruments, USA).
For the measurement of entrapment efficiency, a 500 µL aliquot of the formulation was centrifuged at 5,000 rpm for 15 min in a centrifugal filter unit (MWCO 100 kDa). After centrifugation, the amount of PQ in the filtrate was quantified using the HPLC method mentioned earlier. Entrapment efficiency was calculated using the equation below:
| (equation 1) |
Drug loading in nanoparticles was calculated using the following equation:
| (equation 2) |
2.6. Lyophilization of PQ-SLN and PQ-NLC
PQ-loaded SLN and NLC formulations (5 mL) were diluted at a ratio of 1:1 (v/v) with 20 % (w/v) of mannitol solutions before lyophilization [19]. Blank SLN and NLC formulations were diluted with distilled water at a ratio of 1:1 (v/v) as controls. The samples were frozen at −80 °C overnight and then lyophilized using a Labconco FreeZone freeze-drier (Labconco, Kansas City, USA). For the primary drying of the dispersion, the temperature was increased and maintained at −5 °C for 12 h at a pressure of 0.2 mbar. During the secondary drying, the shelf temperature was increased to 25 °C at +1 °C/min. The temperature was maintained at 25 °C for 24 h. The lyophilized powders in glass vials were reconstituted to the original volume (5 mL) with ultra-pure water for further characterization.
2.7. Scanning electron microscopy (SEM)
SEM enables high resolution imaging of nanoparticles and quantitative/qualitative analysis of nanoparticles. SEM images of PQ-SLN and PQ-NLC were obtained using a JSM-7200 FLV Field Emission Scanning Electron Microscope (JOEL, USA) operated at 5 and 10 kV acceleration voltage. The samples were coated with Platinum under an argon atmosphere in an auto coater before imaging.
2.8. Differential scanning calorimetry (DSC)
Thermal studies were performed using a DSC (TA Instruments, USA) equipped with an automated refrigerated cooling system. Pure PQ, pure Compritol® 888 ATO, and lyophilized PQ-SLN and PQ-NLC samples (3–5 mg) were sealed in standard aluminum pans and heated at a rate of 10 °C per min under a nitrogen gas flow of 50 mL/min. An empty sealed pan was used as a reference pan. DSC thermograms were collected and analyzed using DSC Trios software.
2.9. In vitro drug release studies and release kinetics
In vitro release studies of selected formulations were performed through the dialysis diffusion method. Dialysis membrane bags (MWCO 10 kDa) containing 500 µL of 0.3 % (w/v) PQ solution, PQ-SLN, PQ-NLC, and PQ-NE formulations were added in scintillation vials containing 20 mL of FaSSGF and FaSSIF. The mediums were maintained at 37 °C under with continuous magnetic stirring. Samples were collected at predetermined time intervals in centrifugal filter units and centrifuged at 5,000 rpm for 10 min. After centrifugation, samples were analyzed using the HPLC method mentioned earlier. Each study was carried out in triplicates. In vitro release study data of lipid formulations were fitted to four mathematical models − including zero-order, Higuchi, Korsmeyer-Peppas, and Hixon-Crowell models − to understand release mechanisms [20, 21]. The goodness-of-fit of the models was determined and quantified by the coefficient of determination (R2).
| (equation 3) |
Where Wt is the amount of drug released on time t, W0 is the initial amount of drug, and k is the release constant.
| (equation 4) |
Where Wt is the amount of drug released on time t, and k is the release constant.
| (equation 5) |
Where Wt / W∞ is the fraction of drug released on time t, n is the release exponent, and k is the release constant.
| (equation 6) |
Where Wt is the remaining amount of drug on time t, W0 is the initial amount of drug, and k is the release constant.
2.10. Ex vivo erythrocyte hemolytic studies
The hemolytic studies were performed using a previously reported method [22]. The erythrocyte suspension was diluted with phosphate-buffered saline (PBS) at pH 7.4 and used immediately after preparation. PQ in PBS (PQ solution), PQ-SLN, PQ-NLC, and PQ-NE formulations were co-incubated with the erythrocyte suspension at 37 °C. After 2 h of incubation, samples were centrifuged and the supernatant was added to 96-well plates. The release of hemoglobin was monitored by measuring the absorbance of the supernatant at 540 nm with a microplate reader. Hemolysis induced with Triton X-100 (1 % w/v) in PBS was taken as a positive control. Percentage hemolysis was calculated according to the following formula:
| (equation 7) |
Amax, Amin, and As represent the absorbance value of the hemoglobin released from erythrocytes treated with Triton X-100 solution, PBS buffer, and each sample, respectively.
2.11. Stability upon storage
The physical and chemical stability of PQ-SLN, PQ-NLC, and PQ-NE formulations were evaluated for one month at 4 °C and 25 °C. Samples were analyzed after one month for changes in particle size, PDI, entrapment efficiency, and drug content in addition to physical appearance.
2.12. Statistical analysis
Data analysis was performed using GraphPad Prism. ANOVA analysis was used to compare the means of different measurement groups. A p-value less than 0.05 was considered significant. Data are expressed as mean ± standard deviation from triplicate measurements
3. Results and Discussion
3.1. Solubility of PQ in lipids
The PQ base was a viscous, yellowish-brown liquid with a reported boiling point of 176 oC [14]. The purity of PQ base, molecular weight 259.35, was evaluated using mass spectrometry and NMR techniques (see supplementary material Figures S1-S3). Lipids of various grades generally recognized as safe (GRAS) were selected for the preparation of lipid-based formulations. To achieve the highest drug loading efficiency, while minimizing the number of excipients required to fabricate the lipid-based formulations, the solubility of PQ in the selected lipids was first investigated. As seen in Figure. 1A, Compritol® 888 ATO exhibited the highest solubilizing capacity compared to the other solid lipids tested (Precirol® ATO 5, Geleol® Mono and Diglycerides NF, and Gelucire® 43/01). In the case of liquid lipids tested, PQ was most soluble in castor oil, followed by corn oil, sesame oil, olive oil, oleic acid, Miglyol® 829, and soybean oil (Figure. 1B). Due to the highest solubility of PQ, Compritol® 888 ATO and castor oil were selected as the solid and liquid lipids, respectively, for preparation of SLN and NE formulations. In the case of NLC, the miscibility of the solid and liquid lipid, at different ratios ranging from 9:1 to 1:1, was examined visually under a microscope. Compritol® 888 ATO and castor oil (3:1) did not reveal oil droplets on the microscope slide, indicating that there was good miscibility between the two at this ratio. The optimized solid/liquid lipid ratio not only helps to achieve high drug loading but maintains the structure consistency at room temperature. Thus, the mixture of Compritol® 888 ATO and castor oil (3:1) was selected for the development of NLC.
Figure 1.

Solubility profile of PQ in (A) solid lipids and (B) liquid lipids; (C) The effect of drug loading on the particle size and entrapment efficiency of SLN, NLC, and NE formulations (Mean ± SD, n=3). (** significant at p≤0.01, *** significant at p≤0.001, **** significant at p≤0.0001).
3.2. Emulsification efficiency of surfactants
Surfactants play a crucial role in stabilizing the nanoparticle/emulsion formulations by avoiding aggregation as well as determining the properties of the nanoparticles/emulsion droplets, such as particle size, zeta potential, and polymorphic phase transitions. Six surfactants, including Poloxamer® 188, Solutol® HS 15, Tween® 80, Lauroglycol™ FCC, Span® 80, and sodium cholate, either alone or in combination, were investigated for their emulsification effect at the same concentration (1.5 % w/v). The hydrophilic-lipophilic balance (HLB) number of surfactant mixtures was calculated according to Equation (8):
| (equation 8) |
where X1, X2, and X3 are the weight fractions of the three surfactants with HLB1, HLB2, and HLB3
As shown in Table 1, blank NLC prepared using hydrophilic surfactant Tween® 80 (HLB 15) displayed the smallest particle size, at 188 nm, and with a PDI of 0.18. In contrast, large particle size was formed using lipophilic surfactants Span® 80 (412 nm) and Lauroglycol™ FCC (427 nm). For lipid nanoparticles prepared by oil-in-water (o/w) emulsification method, hydrophilic surfactants (HLB>12) such as Tween® 80, Poloxamer® 188, Solutol® HS 15, and sodium cholate were found to have better emulsification efficiency compared to lipophilic surfactants (HLB<12). Using surfactants containing different proportions of hydrophilic and hydrophobic surfactants was reported to form a complex interfacial film between the oil and water phases, which may yield a synergetic effect in reducing particle size and stabilizing the formulations [23, 24]. It was observed that the combination of Span® 80 and Tween® 80 at a 1:4 ratio can significantly increase the encapsulation efficiency of PQ and improve suspension stability. The large oxyethylene hydrophilic headgroup of Tween® 80 provides a layer of steric hindrance and hence prevents oil droplets from coalescence or nanoparticle agglomeration. A small amount of Span® 80 (0.5 %) was mixed with the lipid phase. The lipophilic surfactant is less likely to desorb into the aqueous phase, therefore, it serves as an anchor to stabilize the oil droplets during formulation preparation and prevent drug molecules from diffusing into the aqueous phase [25].
Table 1:
The effect of surfactants on average particle size and PDI of blank NLC.
| Surfactants (1.5 % w/v) | HLB | Particle size (nm) | PDI |
|---|---|---|---|
|
| |||
| Poloxamer® 188 | 29 | 222 | 0.15 |
| Sodium cholate | 18 | 266 | 0.19 |
| Solutol® HS 15 | 15 | 265 | 0.19 |
| Tween® 80 | 15 | 188 | 0.18 |
| Lauroglycol™ FCC | 4 | 427 | 0.35 |
| Span® 80 | 4.3 | 412 | 0.4 |
| Poloxamer® 188: Solutol® HS 15=1:1 | 22 | 289 | 0.2 |
| Tween® 80: Solutol® HS 15=1:1 | 15 | 231 | 0.21 |
| Tween® 80: Poloxamer® 188=1:1 | 22 | 202 | 0.15 |
| Tween® 80: Span® 80=4:1 | 12.9 | 180 | 0.21 |
| Tween® 80: Poloxamer® 188: Span® 80=1:1:1 | 16.1 | 270 | 0.22 |
| Tween® 80: Solutol® HS 15: Span® 80=1:1:1 | 11.4 | 311 | 0.21 |
| Poloxamer® 188: Solutol® HS 15: Span® 80=1:1:1 | 16.1 | 356 | 0.24 |
3.3. Drug loading
SLN, NLC, and NE with PQ concentrations ranging from 0.15 % to 0.6 % w/v were developed while keeping the other excipient concentrations constant. All formulations were prepared under the same processing conditions. The compositions of lipid formulations are shown in Table 2. Figure. 1C shows, for the blank SLN, NLC, and NE nanoparticles, the average particle size was 210.0±11.0 nm, 123.0±5.0 nm, and 96.0±3.0 nm, respectively, indicating that replacing part of solid lipid with liquid oil would result in a decrease in particle size. Blank SLN composed of a crystalline core of long-chain Compritol® 888 ATO (C22) produces a bigger particle size compared to the amorphous core of blank NLC and blank NE which were comprised of liquid/solid or just liquid matrices. When PQ was added in the formulations with concentration up to 0.6 % w/v, the mean diameter of the PQ-SLN particles increased significantly from 210.0±7.0 nm to 1066.0±11.0 nm (5.1 folds), while the PQ-NLC and PQ-NE formulation displayed a particle/droplet size change in the range of 123.0±5.0 to 707.0±18.0 nm (5.7 folds) and 96.0±3.0 to 220.0±9.0 nm (2.2 folds), respectively.
Table 2:
Composition of primaquine loaded SLN, NLC and NE formulations and evaluation results. All formulations contain 2.0 % (w/v) Tween® 80 and 0.5 % (w/v) Span® 80
| Formulations | PQ (w/v %) | Compritol® 888 ATO (w/v %) | Castor oil (w/v %) | Mannitol (w/v %) | Particle size (nm) | Entrapment efficiency (%) |
|---|---|---|---|---|---|---|
|
| ||||||
| Blank SLN | 0 | 3 | - | - | 210 | - |
| PQ-SLN-1 | 0.15 | 3 | - | - | 215 | 87 |
| PQ-SLN-2 | 0.3 | 3 | - | - | 249 | 90 |
| PQ-SLN-3 | 0.45 | 3 | - | - | 730 | 87 |
| PQ-SLN-4 | 0.6 | 3 | - | - | 1066 | 84 |
| Post-lyophilized PQ-SLN 2 | 0.3 | 3 | - | 10 | 417 | 75 |
|
| ||||||
| Blank NLC | 0 | 2.25 | 0.75 | - | 123 | - |
| PQ-NLC-1 | 0.15 | 2.25 | 0.75 | - | 118 | 89 |
| PQ-NLC-2 | 0.3 | 2.25 | 0.75 | - | 197 | 90 |
| PQ-NLC-3 | 0.45 | 2.25 | 0.75 | - | 318 | 82 |
| PQ-NLC-4 | 0.6 | 2.25 | 0.75 | - | 707 | 78 |
| Post-lyophilized PQ-NLC 2 | 0.3 | 2.25 | 0.75 | 10 | 460 | 85 |
| Blank NE | 0 | - | 3 | - | 96 | - |
| PQ-NE-1 | 0.15 | - | 3 | - | 107 | 83 |
| PQ-NE-2 | 0.3 | - | 3 | - | 117 | 89 |
| PQ-NE-3 | 0.45 | - | 3 | - | 137 | 84 |
| PQ-NE-4 | 0.6 | - | 3 | - | 231 | 82 |
Lipid composition significantly affects the amount of drug loaded in the formulations. Based on the data of entrapment efficiency and particle size shown in Fig 1C, the estimated amount of drug incorporated in PQ-NE-4, PQ-NLC-2, and PQ-SLN-1, which have particle sizes around 200 nm, was 50.0±0.3 mg, 27.0±0.8 mg, and 13.0±0.1 mg, respectively.
It indicated that NLC and NE may carry more drugs due to the presence of amorphous liquid lipid, which can distort the perfect crystallization of solid lipids, thus increasing the drug loading capacity [26]. In other words, the solid lipids in SLN may form perfect lipid crystals and limit their capacity to accommodate drug molecules.
The optimized PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 formulations had an entrapment efficiency of ∼90 %, a particle size of <250 nm, a PDI range of 0.1 to 0.3, the zeta potential of ∼ −30 mV. The drug loading is approximately 4.9 %, 5 %, and 4.8 % of the lipid content for PQ-SLN-2, PQ-NLC-2, and PQ-NE-2, respectively. SEM was used to study the morphology of PQ-SLN-2 and PQ-NLC-2. SEM Images of PQ-SLN-2 and PQ-NLC-2 show spherical shapes in the size range of 200–500 nm (Figure. 2). Particle aggregation was not observed.
Figure 2.

SEM images of (A) PQ-SLN-2 and (B) PQ-NLC-2
The developed formulations with desired particle size ranged ∼100–250 nm could be absorbed by the intestine via endocytosis or lymphatic pathway [27]. When PQ concentration exceeds 0.45 %, an unstable suspension with drug precipitation and gelation phenomenon was observed. This may be due to the drug concentrations going beyond the saturation point of the lipid-based nanoparticles, or insufficient amounts of surfactants that were unable to stabilize the nanoparticle systems.
3.4. DSC
DSC analysis was undertaken to investigate the melting and crystallinity state of lipids in the nanoparticles. Figure 3 shows the DSC thermograms of Compritol® 888 ATO, PQ, and lyophilized PQ-SLN-2, and PQ-NLC-2 formulations. Melting, decomposition, or glass transition events were not observed in the DSC thermograms of PQ base (Supplementary S4). Pure Compritol® 888 ATO alone exhibited an endothermic peak at 72.45 °C. This peak was observed in PQ-SLN-2 and PQ-NLC-2 formulations, with a slight shift to a lower temperature of 71.51 °C for PQ-SLN-2 and 71.73 °C for PQ-NLC-2. This shift in temperatures might be explained by the decrease in the crystalline structure of SLN and NLC due to the dispersion of amorphous PQ and castor oil within the nanoparticles. It was noted that the enthalpy of bulk material, PQ-SLN-2, and PQ-NLC-2 was 180.30 J/g, 103.71 J/g, and 48.57 J/g, respectively. The decrease in enthalpy of NLC indicated that lipids were arranged in a less-ordered state within the nanoparticles due to the presence of liquid lipid; thus, less energy was required to overcome the crystal lattice.
Figure 3.

DSC thermograms of PQ base, Compritol® 888 ATO, lyophilized PQ-SLN-2 and PQ-NLC-2.
3.5. In vitro drug release studies and release kinetics
To study the effect of pH and lipid composition on the drug-release behavior of the optimized lipid formulations (PQ-SLN-2, PQ-NLC-2, and PQ-NE-2), an in vitro release study was performed for 6 h in FaSSGF (pH 1.2) and FaSSIF (pH 6.8) at 37±0.5 °C using the dialysis bag method. Free PQ was released rapidly from the dialysis bag and reached 100 % in 1 h, which indicated that a significant amount of free PQ was able to diffuse out of the dialysis bag in a short time. The release of PQ from SLN, NLC, and NE formulations shows an initial burst release over the first 30 min, followed by a slower release rate over 6 h (Figure. 4A, 4B). The burst release of PQ may be attributed to the unentrapped drugs which are adsorbed on the surface of lipid nanoparticles rather than the drugs entrapped in the lipid phase. The cumulative release of PQ from SLN, NLC and NE was 69.5 ± 0.6 %, 68.9 ± 0.2 %, 66.1± 0.7 % at 6 h in FaSSGF, respectively. In FaSSIF, the release of PQ from SLN, NLC, and NE was 63.6±1.7 %, 63.7±1.8 %, and 66.2±0.7 %, respectively. Around 30 % to 36 % of PQ remained encapsulated in the lipid nanoparticle may deliver the drug to the liver while protecting RBCs from being exposed to the PQ-induced hemolysis.
Figure 4.

In vitro drug release profiles of PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 in (A) FaSSIF (pH 6.8) and (B) FaSSGF (pH 1.2). Experimental data vs. predicted data plot of (C) zero order model, (D) Higuchi model, (E) Korsmeyer-Peppas model, and (F) Hixson-Crowell model.
To understand the mechanism of drug release from the lipid nanoparticles, data were fitted to several empirical models, including zero-order, Higuchi, Korsmeyer-Peppas, and Hixson-Crowell models. The higher value of R2 indicates a better fit of data. Table 3 shows the R2, k, and n obtained from the analysis of the drug-release data for nano-formulations with different fit models. The model fitness was found to be the highest in the Korsmeyer-Peppas model followed by the Higuchi, zero-order, and Hixson-Crowell models. The goodness-of-fit was also visualized using the bivariate plot of the predicted and experimental release data (Figure. 4C–4F). For a sphere geometry, the n value of 0.43 corresponds to Fickian diffusion. The value of n > 0.43 but < 0.85 is considered as anomalous (non-Fickian) transport. The n value of 0.85 corresponds to the Case-II transport [28, 29]. The n value of PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 was calculated to be between 0.57 to 0.65, showing that both diffusion and lipid erosion are involved in controlling the rate of drug release from the lipid core and surfactant interfacial films.
Table 3:
Drug-release kinetics of PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 formulations.
| Formulation | Zero order model | Higuchi model | Korsmeyer-Peppas model | Hixon–Crowell model | |||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| R2 | k | R2 | k | R2 | n | k | R2 | k | |
|
| |||||||||
| PQ-SLN-2 at pH 1.2 | 0.4 | 31.69 | 0.5 | 38.67 | 0.88 | 0.57 | 1.03 | 0.39 | 0.99 |
| PQ-NLC-2 at pH 1.2 | 0.47 | 29.11 | 0.59 | 37.37 | 0.9 | 0.57 | 0.95 | 0.4 | 0.94 |
| PQ-NE-2 at pH 1.2 | 0.44 | 29.20 | 0.55 | 36.72 | 0.91 | 0.59 | 0.96 | 0.35 | 0.94 |
| PQ-SLN-2 at pH 6.8 | 0.73 | 16.09 | 0.9 | 30.13 | 0.92 | 0.67 | 0.6 | 0.65 | 0.34 |
| PQ-NLC-2 at pH 6.8 | 0.64 | 19.62 | 0.82 | 31.84 | 0.92 | 0.65 | 0.7 | 0.5 | 0.39 |
| PQ-NE-2 at pH 6.8 | 0.6 | 22.53 | 0.77 | 33.91 | 0.94 | 0.65 | 0.79 | 0.45 | 0.49 |
3.6. Stability studies and lyophilization of lipid nanoparticles
PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 were stored at 25±1 °C and 4±1 °C for one month. There was no significant change in mean particle size, PDI, drug content, and entrapment efficiency of the nano-formulations stored at 25±1 °C and 4±1 °C (Figure. 5).
Figure 5.

Particle size, PDI of PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 after 30 days of storage at (A) 25 °C and (B) 4 °C. Assay and entrapment efficiency of PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 after 30 days of storage at (C) 25 °C and (D) 4 °C. Circle, triangle, and square dots represent PDI for SLN, NLC, and NE, respectively. Bars represent particle size. (Mean ±SD, n=3).
PQ-SLN-2 and PQ-NLC-2 were further lyophilized with and without cryoprotectants. Without the presence of cryoprotectants, the lyophilized formulations formed sticky and large clumps. The physical-chemical properties could not be analyzed for these preparations. In contrast, lyophilized formulations with cryoprotectants could be reconstituted and redispersed. In the preliminary studies, four cryoprotectants (5–10 % w/v of mannitol, trehalose, lactose, and glucose) were tested. 10 % w/v of mannitol was found to be the most suitable in preventing particles aggregation. Generally, lyophilization helps in enhancing the physical stability of lipid nanoparticles by removing water, however, the extreme pressure and temperature produced during freeze-drying might affect the physical-chemical properties of the formulations. Aggregation of particles and a decrease in entrapment efficiency were observed after lyophilization (Figure. 6). PQ-SLN-2 has a higher level of particle growth and lower entrapment efficiency after reconstitution compared to PQ-NLC-2. This can be attributed to the drugs expelled from solid lipid lattice. Water was crystallized upon freezing, leading to increased drug concentration in the dispersion medium. The accumulation of protonated molecules could reduce zeta potential and cause particle aggregation [30]. Optimization of concentration of the cryoprotectants and lyophilization cycle can improve the overall product quality.
Figure 6.

Effect of lyophilization on (A) particle size and PDI, and (B) assay and entrapment efficiency of PQ-SLN-2 and PQ-NLC-2 (Mean ±SD, n=3).
3.7. Ex vivo erythrocyte hemolysis studies
The ex vivo hemolysis assay was performed to evaluate the hemolytic toxicity of PQ lipid nanocarriers. Erythrocytes were isolated from Sprague Dawley rats and co-incubated at 37 oC for 2 h with 1 µg/mL of PQ solution and PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 formulations. We observed that PQ-related hemolysis is dose-dependent, as increased drug concentration correlated with higher levels of hemolysis. Triton X-100 (1 % w/v) and PBS were used as positive and negative controls, respectively. As shown in Figure 7, the hemolysis level for free PQ solution, PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 formulations, was 77.3±1.6 %, 17.5±0.6 %, 17.7 %±0.3 %, and 16.7±1.3 %, respectively. PQ solution showed significant hemolysis levels compared to PQ lipid nanoparticles (p<0.0001). Encapsulation of PQ in the lipid-based formulations reduced hemolytic toxicity by approximately 4.5-folds compared to PQ solution. The results were in accordance with the previous reports that hemolytic toxicity of PQ could be reduced by formulating PQ into an appropriate drug delivery system [31].
Figure 7.

Ex vivo erythrocyte hemolysis studies in the presence of the free drug, PQ-SLN-2, PQ-NLC-2, and PQ-NE-2 (Mean ±SD, n=3). (**** significant at p≤0.0001; free drug – PQ in PBS solution).
Conclusion
This study presents optimization and comparison of three nano-carrier PQ drug delivery systems namely, PQ-SLN, PQ-NLC, and PQ-NE, and their exhaustive pharmaceutical characterization. The physicochemical properties of the drug, lipid matrix, and surfactant HLB values play a crucial role in the early formulation development stage. For the preparation of lipid nanoparticle dispersions, hydrophilic surfactants such as Tween® 80 and Poloxamer® 188 were proved to have higher emulsification efficiency and smaller particles compared to lipophilic surfactants. Synergistic combinations of hydrophilic and lipophilic surfactants improved the stability and entrapment efficiency of drugs in the lipid formulations. Furthermore, replacing solid lipid with liquid lipid resulted in smaller particles and a higher drug load. The presence of liquid lipid in NLC can distort the perfect crystallization of solid lipids, thus increasing the drug loading capacity. In vitro release studies demonstrated prolonged PQ release from the lipid nanoformulations, compared with PQ solution, in simulated gastric and intestinal fluids. The release mechanisms of drugs from SLN, NLC, and NE can be explained by the Korsmeyer-Peppas model in which both drug diffusion and lipid erosion are involved in the drug release behaviors. Finally, all lipid formulations can successfully protect erythrocytes from PQ-induced cell lysis. PQ-NE appears to be a better formulation choice in terms of small particle size, high drug loading, and ease of scalability and manufacturing. Around 10 mL of the PQ-NE formulation can achieve the current therapeutic dose of 15 or 30 mg PQ. All three formulations, however, will be evaluated in vivo to determine the effect of the particulate system on absorption, liver targeting, and hemolytic toxicity.
Supplementary Material
{kind=link}
Acknowledgments
Authors acknowledge Chemistry and Drug Metabolism and Pharmacokinetics core, Center of Biomedical Research Excellence (COBRE), (P30GM122733–01A1) funded by the National Institute of General Medical Sciences (NIGMS) a component of the National Institutes of Health (NIH). Scanning electron microscopy images presented in this study were generated using the instruments and services at the Microscopy and Image Center, the University of Mississippi. This facility is supported in part by the National Science Foundation (Grant # 1726880).
Funding
This work was supported by the National Institute of Allergy and Infectious Disease (NIAID), National Institute of Health (NIH; Grant# 1R01AI132579). The contents are the sole responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.World malaria report 2020: 20 years of global progress and challenges. World Health Organization; 2020. [Google Scholar]
- 2.Baird JK, Hoffman SL. Primaquine therapy for malaria. Clin Infect Dis. 2004;39(9):1336–45. [DOI] [PubMed] [Google Scholar]
- 3.Baird JK, Rieckmann KH. Can primaquine therapy for vivax malaria be improved? Trends in parasitology. 2003;19(3):115–20. [DOI] [PubMed] [Google Scholar]
- 4.Fernando D, Rodrigo C, Rajapakse S. Primaquine in vivax malaria: an update and review on management issues. Malar J. 2011;10(1):351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camarda G, Jirawatcharadech P, Priestley RS, Saif A, March S, Wong MHL, et al. Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat Commun. 2019;10(1):3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fasinu PS, Nanayakkara NPD, Wang YH, Chaurasiya ND, Herath HMB, McChesney JD, et al. Formation primaquine-5,6-orthoquinone, the putative active and toxic metabolite of primaquine via direct oxidation in human erythrocytes. Malar J. 2019;18(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg. 2006;75(3):402–15. [PubMed] [Google Scholar]
- 8.Das S, Chaudhury A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech. 2011;12(1):62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauss DJ. Oral lipid-based formulations. Advanced drug delivery reviews. 2007;59(7):667–76. [DOI] [PubMed] [Google Scholar]
- 10.Rahman A, Harwansh R, Mirza A, Hussain S, Hussain A. Oral lipid based drug delivery system (LBDDS): formulation, characterization and application: a review. Current drug delivery. 2011;8(4):330–45. [DOI] [PubMed] [Google Scholar]
- 11.Pirson P, Steiger R, Trouet A. The disposition of free and liposomally encapsulated antimalarial primaquine in mice. Biochemical pharmacology. 1982;31(21):3501–7. [DOI] [PubMed] [Google Scholar]
- 12.Bhadra D, Yadav A, Bhadra S, Jain N. Glycodendrimeric nanoparticulate carriers of primaquine phosphate for liver targeting. International journal of pharmaceutics. 2005;295(1–2):221–33. [DOI] [PubMed] [Google Scholar]
- 13.Dierling AM, Cui Z. Targeting primaquine into liver using chylomicron emulsions for potential vivax malaria therapy. Int J Pharm. 2005;303(1–2):143–52. [DOI] [PubMed] [Google Scholar]
- 14.Singh KK, Vingkar SK. Formulation, antimalarial activity and biodistribution of oral lipid nanoemulsion of primaquine. Int J Pharm. 2008;347(1–2):136–43. [DOI] [PubMed] [Google Scholar]
- 15.Omwoyo WN, Ogutu B, Oloo F, Swai H, Kalombo L, Melariri P, et al. Preparation, characterization, and optimization of primaquine-loaded solid lipid nanoparticles. Int J Nanomedicine. 2014;9:3865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoubari G, Staufenbiel S, Volz P, Alexiev U, Bodmeier R. Effect of drug solubility and lipid carrier on drug release from lipid nanoparticles for dermal delivery. Eur J Pharm Biopharm. 2017;110:39–46. [DOI] [PubMed] [Google Scholar]
- 17.Azeem A, Rizwan M, Ahmad FJ, Iqbal Z, Khar RK, Aqil M, et al. Nanoemulsion components screening and selection: a technical note. AAPS PharmSciTech. 2009;10(1):69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–77. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Ji Z, Zou M, Nie X, Shi Y, Cheng G. Preparation, characterization, pharmacokinetics and tissue distribution of solid lipid nanoparticles loaded with tetrandrine. AAPS PharmSciTech. 2011;12(3):1011–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–33. [DOI] [PubMed] [Google Scholar]
- 21.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010;67(3):217–23. [PubMed] [Google Scholar]
- 22.Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li H, et al. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J Vis Exp. 2013(73):e50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong W-T, Tan C-P, Cheah Y-K, Lajis BAF, Habi Mat Dian NL, Kanagaratnam S, et al. Optimization of process parameters in preparation of tocotrienol-rich red palm oil-based nanoemulsion stabilized by Tween80-Span 80 using response surface methodology. PloS one. 2018;13(8):e0202771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lv G, Wang F, Cai W, Zhang X. Characterization of the addition of lipophilic Span 80 to the hydrophilic Tween 80-stabilized emulsions. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2014;447:8–13. [Google Scholar]
- 25.Athas JC, Jun K, McCafferty C, Owoseni O, John VT, Raghavan SR. An effective dispersant for oil spills based on food-grade amphiphiles. Langmuir. 2014;30(31):9285–94. [DOI] [PubMed] [Google Scholar]
- 26.Muller RH, Shegokar R, Keck CM. 20 years of lipid nanoparticles (SLN and NLC): present state of development and industrial applications. Curr Drug Discov Technol. 2011;8(3):207–27. [DOI] [PubMed] [Google Scholar]
- 27.Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–22. [DOI] [PubMed] [Google Scholar]
- 28.Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. Journal of controlled release. 1987;5(1):37–42. [PubMed] [Google Scholar]
- 29.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Advanced drug delivery reviews. 2012;64:163–74. [DOI] [PubMed] [Google Scholar]
- 30.Schwarz C, Mehnert W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). International journal of pharmaceutics. 1997;157(2):171–9. [DOI] [PubMed] [Google Scholar]
- 31.Kumar H, Gothwal A, Khan I, Nakhate KT, Alexander A, Singh V, et al. Galactose-anchored gelatin nanoparticles for primaquine delivery and improved pharmacokinetics: A biodegradable and safe approach for effective antiplasmodial activity against p. Falciparum 3d7 and in vivo hepatocyte targeting. Molecular pharmaceutics. 2017;14(10):3356–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.