

Abstract
Background
Human endogenous retroviruses (HERVs) result from ancestral infections caused by exogenous retroviruses that became incorporated into the germline DNA and evolutionarily fixed in the human genome. HERVs can be transmitted vertically in a Mendelian fashion and be stably maintained in the human genome, of which they are estimated to comprise approximately 8%. HERV-K (HML1-10) transcription has been confirmed to be associated with a variety of diseases, such as breast cancer, lung cancer, prostate cancer, melanoma, rheumatoid arthritis, and amyotrophic lateral sclerosis. However, the poor characterization of HML-9 prevents a detailed understanding of the regulation of the expression of this family in humans and its impact on the host genome. In light of this, a precise and updated HERV-K HML-9 genomic map is urgently needed to better evaluate the role of these elements in human health.
Results
We report a comprehensive analysis of the presence and distribution of HERV-K HML-9 elements within the human genome, with a detailed characterization of the structural and phylogenetic properties of the group. A total of 23 proviruses and 47 solo LTR elements were characterized, with a detailed description of the provirus structure, integration time, potential regulated genes, transcription factor binding sites (TFBS), and primer binding site (PBS) features. The integration time results showed that the HML-9 elements found in the human genome integrated into the primate lineage between 17.5 and 48.5 million years ago (mya).
Conclusion
The results provide a clear characterization of HML-9 and a comprehensive background for subsequent functional studies.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12977-022-00596-2.
Keywords: Human endogenous retrovirus, HML-9, BLAT, GRCh38/hg38, Gene regulation
Background
Approximately 45% of the human genome is composed of transposable elements (TEs) [1–3]. Of these, a fraction of TEs are retroelements (REs), which move via a ‘copy and paste’ mechanism involving the reverse transcription of an RNA intermediate and insertion of its cDNA copy at a new position within the host genome [3, 4]. One class of REs, human endogenous retroviruses (HERVs), result from ancestral infections by exogenous retroviruses that became incorporated into the germline DNA and evolutionarily fixed in the genome; HERVs are estimated to comprise approximately 8% of the human genome [5]. HERVs can be transmitted vertically as proviruses in a Mendelian fashion but are not inherently infectious [6–8]. HERVs are structurally similar to the proviruses of common retroviruses, in which the gag, pol, and env genes are flanked by two long terminal repeats (LTRs) that act as promoters [5]. Most HERV families have the pro genes, but some families, such as HERV-K HML10, have none [9]. These elements are usually inactive due to the accumulation of substitutions, deletions, and insertions [10, 11]. However, integrated LTR elements have been shown to influence gene regulation by providing regulatory elements such as enhancers, promoters, and splice- and polyadenylation sites for various host genes [4].
HERVs have been divided into three classes, namely, Class I (gamma retrovirus-like), Class II (beta retrovirus-like), and Class III (vaguely spuma retrovirus-like) [12]. The classification of HERVs is complex, with several different classification systems in use. In addition to a system based on pol sequence identity, a system based on the tRNA molecule used by retroviruses as a primer during replication is also used. The primer binding site (PBS) regions of Class II HERVs are complementary to lysine (K) tRNA molecules; thus, these HERVs have been designated HERV-Ks [13]. HERV-K proviruses appeared approximately 30–35 million years ago (mya) and are divided into subfamilies from HML-1 through HML-10 [14, 15]. HML-2 of HERV-K, the clade of beta retrovirus-like endogenous retroviruses, is recognized as the most biologically active subgroup, and many of its members still have transcriptional activity [16–20].
The distribution of HERV elements is usually enriched outside transcription units in the human genome. In addition, the few HERV elements within transcription units exhibit a strong orientation bias, such that the orientation of the viral genome is usually opposite to that of host gene transcription. Both of these trends in the location of HERV distribution are likely to be the result of purifying selection. In this case, the harmful HERV provirus within a transcription unit is subject to negative selection and disappears over the course of evolution [12, 15, 21–24]. Because the splicing and poly(A) addition signals of HERV are present in the antisense direction, HERV transcription in the opposite direction to that of the gene may be the least disruptive to mRNA synthesis [15, 21, 22, 25]. A recent study proposed a correlation between silencing mechanisms and the evolutionary age of HERVs. CpG-rich young LTRs were found to be repressed by DNA methylation, while middle-aged LTRs were silenced mainly by posttranslational histone modifications such as H3K9me3 [26].
The first study of the relationship between the expression of the reverse transcriptase (RT) protein of HERV-K and cancer was reported in the early 1970s [27, 28]. Correlations between HERVs and human cancers such as melanoma, breast cancer, germ cell tumors, and ovarian cancer have been described, with significant differences in the protein expression of HERV-K (HML-2/HML-6) in cancer tissues compared to normal tissues. In addition, HERV-K (HML-2) is associated with autoimmune diseases and motor neuron diseases, such as rheumatoid arthritis and amyotrophic lateral sclerosis [29–36]. HERV-K (HML-2) proviruses are classified as type 1 or type 2 based on the presence or absence of a 292 nt deletion at the pol-env junction [37]. Type 2 proviruses without these deletions encode Rec or Env. The type 1 provirus with the 292 nt deletion encodes the Np9 protein [38, 39]. Env protein can act as a tumor-specific antigen that impacts both innate and adaptive immune responses, leading to B- and T-cell stimulation and activation and inducing antibody production and cytotoxic T-cell responses [40]. Elevated levels of the Rec or Np9 protein have been observed in breast cancer, ovarian cancer, and leukemia [18, 41, 42]. HERV-H is transcriptionally repressed in adult tissues through DNMT1-dependent cytosine methylation, which contributes to blocks its transcription and translation, potentially triggering an autoimmune response [43, 44]. However, histone deacetylation alone is not responsible for the repression of HERV family members (HERV-K (HML-2), HERV-W, HERV-FRD), and HDACi treatment did not significantly upregulate HERVs in either latent cell lines or primary T cells infected with HIV-1 [45].
Characterization of the genomic distribution of the HML-9 group is critical to understanding the regulation of the expression of this family and its relationship with human health and disease. To date, there has been only limited characterization of and research on HML-9. In light of this, a precise and updated HML-9 genomic map is urgently needed to better evaluate the role of these elements in human health.
Here, we report a comprehensive analysis of the presence and distribution of HERV-K HML-9 elements within the human genome, with a detailed characterization of the structural and phylogenetic properties of the group. Additionally, we analyzed the provirus integration time and the genes that may be regulated by these elements. Overall, the results provide a clear characterization of HML-9 and a comprehensive background for subsequent functional studies.
Materials and methods
HML-9 identification, localization, and genomic distribution
To evaluate the HML-9 provirus and solo LTR distribution in the human genome, we performed HML-9 identification by using the Genome Reference Consortium assembly GRCh38/hg38 (released Dec. 2013) as the human genome reference. A traditional BLAT search tool [46] in the UCSC Genome Browser database [47] was used to identify the integrated HML-9 elements. DNA BLAT works by keeping an index of the entire genome in its memory. The index consists of all overlapping 11-mers stepped by 5, except for those heavily involved in repeats (http://genome.ucsc.edu/cgi-bin/hgBlat). The assembled LTR14C-HERVK14C-LTR14C sequence was used as a query. Generally, there are two resources that can be selected as references: consensus representatives or single best representative strains. The major advantage of consensus representatives is their much broader representation [9, 48]. Therefore, they are used as references or queries in most studies. The assembled LTR14C-HERVK14C-LTR14C in the current work is from the Dfam database. Additionally, the expected distribution of the HML-9 loci on each chromosome was predicted according to the formula e = Cl × n/Tl (e is the number of integrations expected in the chromosome, Cl represents the ungapped length of the chromosome, n is the total number of actual HML-9 loci identified in the human genome, and Tl represents the sum ungapped length of all chromosomes) [49]. The difference between the expected number of integrations and the actual number of HML-9 loci was analyzed via the chi-square (χ2) test, and statistical significance was estimated according to the p value.
Structural characterization
The identified HML-9 elements were aligned to the proviral reference LTR14C-HERVK14C-LTR14C. Alignments were analyzed on the BioEdit software platform [50]. All insertions and deletions were annotated.
Phylogenetic analyses
Maximum likelihood (ML) phylogenetic trees were built with MEGA7 [51] to confirm the assignment of the identified HML-9 elements. The 44 out of 47 solo LTR sequences that were longer than 90% of LTR14C and the 5 out of 23 proviral sequences that were longer than 80% of LTR14C-HERVK14C-LTR14C were used to construct phylogenetic trees. The best-fit models of nucleotide substitution for solo LTRs and full-length proviruses were calculated as K2 + G and GTR + G + I by the Model Selection function in MEGA7, respectively. For the 4 coding regions, elements longer than 90% of the corresponding section of HML-9 were screened to construct phylogenetic trees. According to the model selection function of MEGA7, the best-fit models of nucleotide substitution for gag, pro, pol, and env analysis were HKY + G + I, GTR + G + I, GTR + G, and HKY + G, respectively. Tree topologies were searched using the nearest neighbor interchange (NNI) procedure. The confidence of each node in phylogenetic trees was determined using bootstrap testing with 500 bootstrap replicates. The final ML trees were visualized using iTOL [52].
Estimation of the integration time of HML-9
To estimate the time of integration, we used the substitution rate of 0.2%/nucleotide/million years to assess the effect of divergence on each HML-9 element [53]. D is the percentage of divergent nucleotides, and the D of each HML-9 member was estimated between (1) the 5′ and 3′ LTRs of each provirus and (2) each HML-9 internal element (gag, pro, pol, and env genes) and its generated consensus. The divergence values were estimated with MEGA7. For the 4 internal regions, the integration time was calculated based on the formula T = D/0.2, in which T represents the estimated time of integration (in million years). For the flanking LTR regions, the provirus integration time was calculated based on the formula T = D/0.2/2.
Functional prediction of cis-regulatory regions and enrichment analysis
Noncoding regions typically lack biological function annotations. To understand the biological significance of both HML-9 solo LTRs and proviral LTRs, an analysis of the annotations of genes adjacent to LTRs was performed based on the Genomic Regions Enrichment of Annotations Tool (GREAT) [54]. The association rule was as follows: basal + extension: 5000 bp upstream, 1000 bp downstream; 1,000,000 bp max extension; curated regulatory domains included. After identifying potential regulatory genes, the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) [55] was used to analyze their functional enrichment (http://www.webgestalt.org), which is crucial for interpreting the list of genes of interest. The enrichment method used in the current work was over-representation analysis (ORA). The parameters for the enrichment analysis were as follows: minimum number of IDs in the category: 5; maximum number of IDs in the category: 2000; FDR Method: Benjamini–Hochberg (BH); and significance level: top 10.
In silico examination of conserved transcription factor binding sites
The transcription factor binding sites of the HML-9 LTR consensus reference sequence were predicted from the JASPAR (https://jaspar.genereg.net/) database. The taxon was vertebrates, and the species was Homo sapiens. The data chosen for the prediction of transcription factors were ChIP-seq data in JASPAR with a relative profile score threshold of 95%. The alignment and annotation of the HML-9 LTR reference sequence with the 4 proviral sequences (length of 5′LTR > 90%) were performed using Geneious software [56].
Primer binding site feature representation
The composition of the primer binding sites (PBSs) of 11 near-full-length proviruses (LTR length > 80%) and the HML-9 reference sequence were all analyzed using MEGA7 and BioEdit. The degree of conservation at each position was represented by a logo built from WebLogo at http://weblogo.berkeley.edu [57]. Then, the PBS type was identified with tRNAdb (http://trna.bioinf.uni-leipzig.de/) [58].
Results
HML-9 element identification, localization, and distribution in hg38
According to the BLAT results for LTR14C-HERVK14C-LTR14C in GRCh38/hg38, we characterized a total of 23 HERV-K HML-9 proviral elements. Each HML-9 element was named according to the genomic locus of integration, as a previously proposed nomenclature for HERV-Ks [16] (Table 1). Element length analysis indicated that 6 elements were longer than 70% of the full length of the reference, 9 elements were between 40 and 70% of the reference length, and the remaining 8 elements were between 17.11 and 34.26% of the reference length. Moreover, a total of 47 solo LTR elements of HERV-K HML-9 were characterized. Of these, 44 solo LTRs (93.62%) were longer than 90% of the representative reference LTR14C. The nucleotide sequence of each element is shown in Additional file 1: Dataset S1, Additional file 2: Dataset S2. The overall HML-9 element distribution is displayed based on the data representation obtained from the Ensemble website (www.ensembl.org) (Fig. 1A).
Table 1.
HML-9 provirus distribution
| Number | Locus | Chromosome | Strand | Position start | Position end | Length (bp) | Match + mismatch (bp)/full length (bp) (%) | Range (%) | Qgap (bp)/match + mismatch + Qgap (bp) (%) | Insertion or deletion | Intergenic/intron/exon | Gene including the region |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 16p12.3 | chr16 | − | 19393581 | 19402152 | 8572 | 96.00 | (90–100) | 1.01 | NA | Exon_intron | AC130456.2 |
| 2 | 2p12 | chr2 | + | 82022660 | 82031279 | 8620 | 95.91 | (90–100) | 1.13 | NA | Intergenic | NA |
| 3 | 15q21.1 | chr15 | − | 45234477 | 45243073 | 8597 | 95.34 | (90–100) | 1.85 | NA | Exon_intron | AC051619.4 |
| 4 | 8p11.1 | chr8 | − | 43694016 | 43702583 | 8568 | 95.10 | (90–100) | 2.14 | NA | Intergenic | NA |
| 5 | 13q31.1 | chr13 | + | 84869526 | 84877320 | 7795 | 86.84 | (80–90) | 6.67 | NA | Exon_intron | AL445588.1 |
| 6 | 4q33 | chr4 | − | 170126345 | 170133883 | 7539 | 70.03 | (70–80) | 0.79 | Insertion | Intergenic | NA |
| 7 | 6p12.3 | chr6 | + | 48873675 | 48879725 | 6051 | 64.84 | (60–70) | 34.48 | Deletion | Intergenic | NA |
| 8 | Yp11.2 | chrY | − | 9273707 | 9279611 | 5905 | 59.83 | (50–60) | 39.23 | Deletion | Intergenic | NA |
| 9 | 8q24.3 | chr8 | + | 145019974 | 145032719 | 12,746 | 57.06 | (50–60) | 0.79 | Insertion | Intergenic | NA |
| 10 | Yq11.223 | chrY | + | 21580120 | 21585551 | 5432 | 57.04 | (50–60) | 38.30 | Deletion | Exon_intron | TTTY13 |
| 11 | 19q13.2 | chr19 | + | 40954172 | 40959178 | 5007 | 56.66 | (50–60) | 41.93 | Deletion | Intergenic | NA |
| 12 | Yp11.2 | chrY | − | 8121821 | 8126768 | 4948 | 54.57 | (50–60) | 44.92 | Deletion | Intergenic | NA |
| 13 | Yp11.2 | chrY | + | 8996062 | 9000755 | 4694 | 50.80 | (50–60) | 41.95 | Deletion | Intergenic | NA |
| 14 | Yq11.222 | chrY | − | 18622534 | 18626952 | 4419 | 47.33 | (40–50) | 52.23 | Deletion | Intergenic | NA |
| 15 | Yq11.223 | chrY | − | 21845475 | 21850069 | 4595 | 43.18 | (40–50) | 49.78 | Deletion/insertion | Exonic_intergenic | AC024236.1 |
| 16 | 21q21.1 | chr21 | − | 18563368 | 18566735 | 3368 | 34.26 | (30–40) | 8.19 | NA | Exon_intron | MIR548XHG |
| 17 | 5q33.3 | chr5 | − | 156660448 | 156663815 | 3368 | 34.14 | (30–40) | 8.50 | NA | Intron | SGCD |
| 18 | 1q22 | chr1 | − | 155629408 | 155632775 | 3368 | 33.75 | (30–40) | 9.56 | NA | Intron | AL353807.5 |
| 19 | 7q36.1 | chr7 | − | 150561277 | 150563994 | 2718 | 27.92 | (20–30) | 10.27 | Deletion | Intergenic | NA |
| 20 | 8q21.13 | chr8 | + | 78652302 | 78654820 | 2519 | 26.60 | (20–30) | 0.30 | NA | Intron | AC068700.2 |
| 21 | 10q24.2 | chr10 | − | 99822511 | 99825532 | 3022 | 25.36 | (20–30) | 24.65 | Deletion/insertion | Intron | ABCC2 |
| 22 | 12q13.11 | chr12 | + | 48509228 | 48511681 | 2454 | 18.44 | (10–20) | 33.18 | Deletion/insertion | Intergenic | NA |
| 23 | Yq11.222 | chrY | − | 17669948 | 17671523 | 1576 | 17.11 | (10–20) | 12.69 | Deletion | Intergenic | NA |
Fig. 1.

Chromosomal distribution of HML-9 loci. A All HML-9 elements (red arrows) are displayed on the human karyotype (www.ensembl.org). The number of HML-9 proviral elements (B) and solo LTRs (C) integrated into each human chromosome was determined and compared to the expected number of insertion events. The expected number of sequences in each chromosome is marked in blue, and the actual number of sequences detected is marked in orange
Next, the expected number of integrations of HML-9 elements per chromosome was predicted and compared with the number of actually detected sites to assess whether HML-9 is randomly distributed in the human genome. The number of HML-9 integration events observed was often inconsistent with the expected number (Fig. 1B, C). For the proviral elements, the number of HML-9 insertions on chromosomes 8, 13, 15, 16, 19, 21, and Y was higher than expected. In particular, the number of proviral elements on the Y chromosome was significantly higher than that predicted by the chi-square test (p < 0.05). On chromosomes 1, 2, 4, 5, 6, 7, 10, and 12, the actual numbers identified were lower than expected (Fig. 1B). Notably, no HML-9 provirus integration was detected on chromosomes 3, 9, 11, 14, 17, 18, 20, 22, and X. With respect to the solo LTR elements, the number of HML-9 solo LTRs on chromosomes 2, 3, 14, 15, 18, 21, X, and Y was higher than expected. On chromosomes 1, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17, and 20, the actual numbers identified were lower than expected. In particular, no HML-9 solo LTR integration was detected on chromosomes 9, 16, 19, and 22 (Fig. 1C). This analysis revealed that HML-9 provirus and solo LTR integration is nonrandom among human chromosomes.
Furthermore, all 23 identified proviral elements and 47 solo LTRs were analyzed to determine their locations in intergenic regions, introns, or exons (Tables 1, 2). The results showed that 13 proviral elements were located in intergenic regions, accounting for 56.52% of all proviral elements. Four proviral elements (17.39%) were located in introns. Six proviral elements (26.09%) were located in both introns and exons (Table 1). With respect to solo LTRs, 28 (59.57%) were located in intergenic regions, and the remaining 19 (40.43%) were located in introns (Table 2). The results revealed an apparent distribution preference for intergenic regions and introns. Previously, Brady et al. [15] demonstrated that the accumulation of HML-2 proviruses in introns and intergenic regions is not a result of integration bias but selection against proviruses that integrate into exons and genic regions. This conclusion also applies to the current study. The proviruses in genes and their relative transcriptional orientation are presented in Additional file 3: Table S1, Additional file 4: Table S2.
Table 2.
HML-9 solo LTR tracks distribution
| Number | Locus | Chromosome | Strand | Position start | Position end | Length (bp) | Percentage of LTR14C in length (%) | Match + mismatch/full length (%) | Range (%) | Qgap (bp)/match + mismatch + Qgap (bp) (%) | Insertion or deletion | Intergenic/intron/exon | Gene including the region |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 14q21.1 | chr14 | + | 38011040 | 38012012 | 973 | 101.36 | 6.91 | (0–10) | 35.61 | Deletion, insertion | Intron | TTC6 |
| 2 | Xq21.32 | chrX | − | 93273183 | 93274197 | 1015 | 100.85 | 6.88 | (0–10) | 2.47 | Insertion | Intergenic | NA |
| 3 | 2q31.1 | chr2 | + | 180236847 | 180237437 | 591 | 100.34 | 6.84 | (0–10) | 0.34 | NA | Intergenic | NA |
| 4 | 18p11.31 | chr18 | – | 4527618 | 4528209 | 592 | 100.17 | 6.83 | (0–10) | 0.00 | NA | Intergenic | NA |
| 5 | 2q11.2 | chr2 | + | 97964920 | 97965508 | 589 | 100.00 | 6.82 | (0–10) | 0.00 | NA | Intron | TMEM131 |
| 6 | 15q14 | chr15 | + | 39011033 | 39011621 | 589 | 100.00 | 6.82 | (0–10) | 0.00 | NA | Intron | LINC02694 |
| 7 | 2p12 | chr2 | – | 81304430 | 81305068 | 639 | 99.83 | 6.81 | (0–10) | 0.00 | NA | Intergenic | NA |
| 8 | 2q32.3 | chr2 | – | 194256159 | 194256746 | 588 | 99.83 | 6.81 | (0–10) | 0.00 | NA | Intergenic | NA |
| 9 | 3q26.1 | chr3 | – | 163283189 | 163283777 | 589 | 99.83 | 6.81 | (0–10) | 0.00 | NA | Intron | LINC01192 |
| 10 | 4q26 | chr4 | + | 116980222 | 116980809 | 588 | 99.83 | 6.81 | (0–10) | 0.34 | NA | Intergenic | NA |
| 11 | 4p15.31 | chr4 | + | 19556097 | 19556684 | 588 | 99.83 | 6.81 | (0–10) | 0.17 | NA | Intron | AC024230.1 |
| 12 | 7p21.2 | chr7 | + | 14509240 | 14509827 | 588 | 99.83 | 6.81 | (0–10) | 0.00 | NA | Intron | DGKB |
| 13 | 8q11.21 | chr8 | – | 51178592 | 51179179 | 588 | 99.83 | 6.81 | (0–10) | 0.00 | NA | Intergenic | NA |
| 14 | 11q12.3 | chr11 | – | 62185237 | 62185824 | 588 | 99.83 | 6.81 | (0–10) | 0.00 | NA | Intergenic | NA |
| 15 | 2q21.3 | chr2 | + | 135521883 | 135522470 | 588 | 99.66 | 6.80 | (0–10) | 0.17 | NA | Intron | ZRANB3 |
| 16 | 3p12.2 | chr3 | – | 81329902 | 81330488 | 587 | 99.66 | 6.80 | (0–10) | 0.17 | NA | Intergenic | NA |
| 17 | 3p12.1 | chr3 | – | 83618409 | 83618995 | 587 | 99.66 | 6.80 | (0–10) | 0.17 | NA | Intergenic | NA |
| 18 | 5q21.3 | chr5 | – | 105998962 | 105999549 | 588 | 99.66 | 6.80 | (0–10) | 0.34 | NA | Intergenic | NA |
| 19 | 10p12.31 | chr10 | – | 18856645 | 18857233 | 589 | 99.66 | 6.80 | (0–10) | 0.17 | NA | Intergenic | NA |
| 20 | Yq11.23 | chrY | – | 25974734 | 25975320 | 587 | 99.66 | 6.80 | (0–10) | 0.17 | NA | Intergenic | NA |
| 21 | 6q14.1 | chr6 | – | 82297755 | 82298498 | 744 | 99.49 | 6.78 | (0–10) | 0.34 | Insertion | Intergenic | NA |
| 22 | Xp22.2 | chrX | + | 11033746 | 11034330 | 585 | 99.32 | 6.77 | (0–10) | 0.51 | NA | Intron | AC073529.1 |
| 23 | 2p12 | chr2 | + | 77807602 | 77808185 | 584 | 99.15 | 6.76 | (0–10) | 0.68 | NA | Intron | AC012494.1 |
| 24 | 1q23.3 | chr1 | + | 162419359 | 162419942 | 584 | 98.98 | 6.75 | (0–10) | 0.17 | NA | Intergenic | NA |
| 25 | Xq27.2 | chrX | – | 142767872 | 142768454 | 583 | 98.98 | 6.75 | (0–10) | 0.00 | NA | Intergenic | NA |
| 26 | 2q31.1 | chr2 | + | 171365032 | 171365617 | 586 | 98.64 | 6.73 | (0–10) | 1.19 | NA | Intron | METTL8 |
| 27 | 5q13.3 | chr5 | + | 75859521 | 75860102 | 582 | 98.64 | 6.73 | (0–10) | 0.17 | NA | Intergenic | NA |
| 28 | Xq27.3 | chrX | – | 144791258 | 144791846 | 589 | 98.47 | 6.71 | (0–10) | 1.37 | NA | Intergenic | NA |
| 29 | 12q12 | chr12 | + | 38144469 | 38145052 | 584 | 98.30 | 6.70 | (0–10) | 1.03 | NA | Intergenic | NA |
| 30 | 15q21.3 | chr15 | – | 54594796 | 54595373 | 578 | 98.13 | 6.69 | (0–10) | 2.04 | NA | Intron | UNC13C |
| 31 | 21q11.2 | chr21 | – | 14080466 | 14081052 | 587 | 97.79 | 6.67 | (0–10) | 2.05 | NA | Intron | AP001347.1 |
| 32 | 4q28.2 | chr4 | + | 129080872 | 129081454 | 583 | 97.61 | 6.66 | (0–10) | 2.22 | NA | Intron | SCLT1 |
| 33 | 3q25.2 | chr3 | – | 154944330 | 154944911 | 582 | 97.44 | 6.64 | (0–10) | 2.56 | NA | Intergenic | NA |
| 34 | 11q24.2 | chr11 | + | 124270705 | 124271275 | 571 | 96.93 | 6.61 | (0–10) | 0.00 | NA | Intergenic | NA |
| 35 | 2q14.3 | chr2 | – | 125024208 | 125024792 | 585 | 96.76 | 6.60 | (0–10) | 3.07 | NA | Intergenic | NA |
| 36 | 6q27 | chr6 | + | 169084226 | 169084808 | 583 | 96.76 | 6.60 | (0–10) | 3.07 | NA | Intergenic | NA |
| 37 | 13q13.3 | chr13 | – | 38319721 | 38320300 | 580 | 96.76 | 6.60 | (0–10) | 3.24 | NA | Intron | LINC00571 |
| 38 | 7q35 | chr7 | + | 143472173 | 143472744 | 572 | 96.08 | 6.55 | (0–10) | 3.75 | NA | Intron | EPHA1-AS1 |
| 39 | 14q21.3 | chr14 | + | 48011215 | 48011780 | 566 | 95.91 | 6.54 | (0–10) | 0.53 | NA | Intergenic | NA |
| 40 | 3p21.31 | chr3 | + | 44534488 | 44535059 | 572 | 95.74 | 6.53 | (0–10) | 3.77 | NA | Intergenic | NA |
| 41 | 2q22.1 | chr2 | – | 138860917 | 138861512 | 596 | 95.06 | 6.48 | (0–10) | 3.79 | NA | Intergenic | NA |
| 42 | 12p13.32 | chr12 | – | 4720007 | 4720593 | 587 | 94.89 | 6.47 | (0–10) | 4.95 | NA | Intron | AC005833.1 |
| 43 | 3p14.2 | chr3 | – | 59469489 | 59470030 | 542 | 91.82 | 6.26 | (0–10) | 3.23 | NA | Intron | AC126121.3 |
| 44 | 1q24.2 | chr1 | + | 168457190 | 168457732 | 543 | 90.80 | 6.19 | (0–10) | 0.37 | NA | Intron | AL023755.1 |
| 45 | 20p13 | chr20 | – | 2809052 | 2809886 | 835 | 88.42 | 6.03 | (0–10) | 0.38 | Insertion | Intergenic | NA |
| 46 | 18q21.33 | chr18 | + | 63648105 | 63648555 | 451 | 76.49 | 5.22 | (0–10) | 0.22 | NA | Intron | SERPINB11 |
| 47 | 17q22 | chr17 | + | 52961655 | 52962071 | 417 | 70.87 | 4.83 | (0–10) | 0.24 | NA | Intergenic | NA |
Structural characterization
To define the structural characteristics of HML-9 elements, the 23 proviruses were further analyzed by comparing them with the reference LTR14C-HERVK14C-LTR14C. According to the annotation information summarized in the Dfam database (https://www.dfam.org/family/DF0000193/features), the complete HML-9 reference exhibits a typical proviral structure, containing 4 open reading frames (ORFs) and 2 flanking LTRs. Specifically, the 5′ LTR is located from nucleotides 1 to 587, the CDS range of the HERVK14C_gag protein is from nucleotides 758 to 2548, the CDS range of the HERVK14C_pro protein is from nucleotides 2548 to 3435, the CDS range of the HERVK14C_pol protein is from nucleotides 3411 to 6060, the CDS range of the HERVK14C_env protein is from nucleotides 5975 to 8020, and the 3′ LTR is from nucleotides 8022 to 8608. We aligned the 23 HML-9 proviral sequences and annotated the position of the single retroviral component and deletions to describe the structure of each HML-9 provirus element (Fig. 2). HML-9 16p12.3, 2p12, 15q21.1, 8p11.1, 13q31.1 and 4q33 are longer than 70% of the complete reference sequence in length. Furthermore, the integrity of 6 separate regions (5′ LTR, gag, pro, pol, env, and 3’ LTR) is summarized in Table 3.
Fig. 2.

HML-9 provirus structural characterization. Each HML-9 provirus element was analyzed and compared to the Dfam reference sequence. The LTRs and the gag, pro, pol, and env genes were annotated. Black lines represent deleted regions
Table 3.
The integrity of 6 separate regions relative to the corresponding sections of reference
| Number | Locus | Provirus regions | 5′LTR (%) | gag (%) | pro (%) | pol (%) | env (%) | 3′LTR (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 16p12.3 | chr16 19393581 19402152 | 100.00 | 99.83 | 99.89 | 99.39 | 99.17 | 99.66 |
| 2 | 2p12 | chr2 82022660 82031279 | 98.98 | 99.72 | 99.89 | 99.43 | 99.90 | 99.15 |
| 3 | 15q21.1 | chr15 45234477 45243073 | 99.83 | 99.27 | 100.00 | 99.66 | 99.56 | 99.83 |
| 4 | 8p11.1 | chr8 43694016 43702583 | 99.83 | 99.44 | 98.31 | 99.39 | 99.66 | 99.83 |
| 5 | 13q31.1 | chr13 84869526 84877320 | 35.78 | 99.55 | 52.20 | 99.77 | 99.80 | 99.32 |
| 6 | 4q33 | chr4 170126345 170133883 | 99.66 | 99.89 | 99.77 | 99.70 | 13.93 | 0.00 |
| 7 | 6p12.3 | chr6 48873675 48879725 | 99.15 | 92.79 | 65.84 | 6.13 | 99.85 | 99.66 |
| 8 | Yp11.2 | chrY 9273707 9279611 | 88.42 | 90.73 | 63.81 | 6.36 | 98.83 | 99.49 |
| 9 | 8q24.3 | chr8 145019974 145032719 | 0.00 | 0.00 | 0.79 | 99.77 | 99.90 | 95.23 |
| 10 | Yq11.223 | chrY 21580120 21585551 | 88.25 | 91.74 | 64.83 | 6.25 | 99.36 | 15.84 |
| 11 | 19q13.2 | chr19 40954172 40959178 | 98.98 | 98.94 | 64.26 | 6.17 | 67.40 | 77.51 |
| 12 | Yp11.2 | chrY 8121821 8126768 | 99.66 | 76.49 | 14.09 | 6.40 | 99.17 | 98.81 |
| 13 | Yp11.2 | chrY 8996062 9000755 | 0.00 | 80.23 | 64.37 | 6.47 | 99.80 | 95.55 |
| 14 | Yq11.222 | chrY 18622534 18626952 | 96.76 | 75.21 | 0.00 | 0.00 | 84.75 | 98.47 |
| 15 | Yq11.223 | chrY 21845475 21850069 | 0.00 | 70.85 | 64.60 | 6.40 | 99.32 | 99.15 |
| 16 | 21q21.1 | chr21 18563368 18566735 | 0.00 | 0.00 | 95.26 | 96.12 | 0.00 | 0.00 |
| 17 | 5q33.3 | chr5 156660448 156663815 | 0.00 | 0.00 | 95.26 | 96.12 | 0.00 | 0.00 |
| 18 | 1q22 | chr1 155629408 155632775 | 0.00 | 0.00 | 95.26 | 96.12 | 0.00 | 0.00 |
| 19 | 7q36.1 | chr7 150561277 150563994 | 0.00 | 0.00 | 0.00 | 16.91 | 99.02 | 54.51 |
| 20 | 8q21.13 | chr8 78652302 78654820 | 0.00 | 0.00 | 0.00 | 0.00 | 87.34 | 100.00 |
| 21 | 10q24.2 | chr10 99822511 99825532 | 0.00 | 0.00 | 52.29 | 95.43 | 0.00 | 0.00 |
| 22 | 12q13.11 | chr12 48509228 48511681 | 0.00 | 0.00 | 88.72 | 63.52 | 0.00 | 0.00 |
| 23 | Yq11.222 | chrY 17669948 17671523 | 52.81 | 62.09 | 0.00 | 0.00 | 0.00 | 0.00 |
Phylogenetic analyses
To characterize the phylogenetic relationships among the HML-9 group, 5 proviral sequences (longer than 80% of the HML-9 reference length) were screened together with Dfam HERV-K groups (HML-1–10) and exogenous betaretroviruses as representatives to generate ML trees. The 5 identified proviruses all clustered with the Dfam HML-9 reference by 100% of bootstrap support (Fig. 3A). Subsequently, phylogenetic trees of a total of 44 solo LTRs identified to be longer than 90% of LTR14C were constructed together with the LTR reference (Fig. 3B). Next, 4 ML trees were constructed for subregions whose lengths were longer than 90% of the corresponding section of the reference sequence, including 10 gag elements, 8 pro elements, 11 pol elements, and 13 env elements (Fig. 3C–F). These phylogenetic groups of different regions of HML-9 all clustered together and were clearly separated from the other HERV-K groups (HML1-8, 10). Two distinct clusters in the pro and pol groups were observed. They were statistically supported by 100% of bootstrap values and were named HML-9 type a and type b. The results showed that HML-9 21q21.1, HML-9 1q22, and HML-9 5q33.3 were included in HML-9 type a, whereas HML-9 15q21.1, HML-9 16p12.3, HML-9 4q33, HML-9 8p11.1, and HML-9 2p12 were included in HML-9 type b. HML-9 type b sequences included the Dfam HML-9 reference, whereas HML-9 type a elements showed more divergence relative to the group references.
Fig. 3.

Phylogenetic analysis of the HML-9 near-full-length proviruses, solo LTRs, and 4 subregions by the maximum likelihood method. Phylogenetic analyses of 5 HML-9 proviral elements (A), 44 solo LTRs (B), 10 gag elements (C), 8 pro elements (D), 11 pol elements (E), and 13 env elements (F), together with references. The two intragroup clusters of the pro and pol genes (types a and b) were annotated and depicted with brown and orange background colors, respectively. The resulting phylogeny was tested by the bootstrap method with 500 replicates. The branch length indicates the number of substitutions per site
Estimated time of integration
The HML-9 proviral age in the human genome was estimated based on the LTRs and the gag, pro, pol, and env regions. Each region whose length exceeded 90% of the corresponding reference sequence was selected to calculate the integration time. For LTRs, the 5′ and 3′ LTRs of a given provirus are identical at the time of integration and then accumulate random substitutions in an independent manner [53]; therefore, the T value was estimated by the relation T = D/0.2/2. For the gag, pro, pol, and env regions, the ancestral sequences of each region were generated via MEGA7 following the ML method based on multiple alignments of all elements. The T value (integration time) was estimated by the relation T = D/0.2, where 0.2 represents the human genome neutral mutation rate expressed in substitutions/nucleotide/million years. For each provirus region, we provide details on the period of provirus formation in Table 4. Overall, the estimated time of integration based on LTR elements is later than that estimated based on the four regions (gag, pro, pol, and env). The LTRs integrated between 17.5 and 48.5 mya. The average time of integration was 28.83 mya. However, the majority of HML-9 elements (gag, pro, pol, and env) found in the human genome integrated between 37.5 and 151.5 mya. The average time of integration was 76 mya. There exists a very large discrepancy between the two analyses. A reasonable explanation for the difference between the two methods is as follows. The two flanking LTRs (5' LTR and 3' LTR) were identical when the provirus was integrated into the host genome. However, the internal regions contain multiple sequence differences due to the mutations accumulated during viral replication cycles, with a much higher error rate. This difference would inevitably lead to LTRs being a more accurate timing starting point for integration time estimation.
Table 4.
Estimated time of HML-9 elements integration
| Locus | Provirus regions | Divergence from consensus sequence | Mean divergences | T = D/0.2 | Age/million years (gene vs consensus) | Divergence between 2 LTRs | T = D/0.2/2 | Age/million years (LTR vs LTR) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| gag | pro | pol | env | ||||||||
| 16p12.3 | chr16 19393581 19402152 | 0.059 | 0.158 | 0.206 | 0.089 | 0.128 | 0.64 | 64.00 | 0.082 | 0.20500 | 20.50000 |
| 2p12 | chr2 82022660 82031279 | 0.061 | 0.182 | 0.204 | 0.101 | 0.137 | 0.685 | 68.50 | 0.070 | 0.17500 | 17.50000 |
| 15q21.1 | chr15 45234477 45243073 | 0.051 | 0.126 | 0.206 | 0.099 | 0.121 | 0.6025 | 60.25 | 0.080 | 0.20000 | 20.00000 |
| 8p11.1 | chr8 43694016 43702583 | 0.091 | 0.177 | 0.231 | 0.121 | 0.155 | 0.775 | 77.50 | 0.107 | 0.26750 | 26.75000 |
| 13q31.1 | chr13 84869526 84877320 | 0.054 | NA | 0.208 | 0.103 | 0.122 | 0.608333333 | 60.83 | NA | NA | NA |
| 4q33 | chr4 170126345 170133883 | 0.058 | 0.172 | 0.214 | NA | 0.148 | 0.74 | 74.00 | NA | NA | NA |
| 6p12.3 | chr6 48873675 48879725 | 0.063 | NA | NA | 0.106 | 0.085 | 0.4225 | 42.25 | 0.110 | 0.27500 | 27.50000 |
| Yp11.2 | chrY 9273707 9279611 | 0.114 | NA | NA | 0.139 | 0.127 | 0.6325 | 63.25 | 0.141 | 0.35250 | 35.25000 |
| 8q24.3 | chr8 145019974 145032719 | NA | NA | 0.513 | 0.093 | 0.303 | 1.515 | 151.50 | NA | NA | NA |
| Yq11.223 | chrY 21580120 21585551 | 0.105 | NA | NA | 0.140 | 0.123 | 0.6125 | 61.25 | NA | NA | NA |
| 19q13.2 | chr19 40954172 40959178 | 0.075 | NA | NA | 0.075 | 0.375 | 37.50 | 0.097 | 0.24250 | 24.25000 | |
| Yp11.2 | chrY 8121821 8126768 | NA | NA | NA | 0.125 | 0.125 | 0.625 | 62.50 | 0.157 | 0.39250 | 39.25000 |
| Yp11.2 | chrY 8996062 9000755 | NA | NA | NA | 0.133 | 0.133 | 0.665 | 66.50 | NA | NA | NA |
| Yq11.222 | chrY:18622534–18626952 | NA | NA | NA | NA | NA | NA | NA | 0.194 | 0.48500 | 48.50000 |
| Yq11.223 | chrY 21845475 21850069 | NA | NA | NA | 0.143 | 0.143 | 0.715 | 71.50 | NA | NA | NA |
| 21q21.1 | chr21 18563368 18566735 | NA | 0.266 | 0.190 | NA | 0.228 | 1.14 | 114.00 | NA | NA | NA |
| 5q33.3 | chr5 156660448 156663815 | NA | 0.215 | 0.160 | NA | 0.188 | 0.9375 | 93.75 | NA | NA | NA |
| 1q22 | chr1 155629408 155632775 | NA | 0.212 | 0.156 | NA | 0.184 | 0.92 | 92.00 | NA | NA | NA |
| 7q36.1 | chr7 150561277 150563994 | NA | NA | NA | 0.197 | 0.197 | 0.985 | 98.50 | NA | NA | NA |
| 10q24.2 | chr10 99822511 99825532 | NA | NA | 0.169 | NA | 0.169 | 0.845 | 84.50 | NA | NA | NA |
Functional prediction of cis-regulatory regions and enrichment analysis
GREAT analysis can predict possible regulated genes based on spatial proximity. The results describing the associations between each solo LTR and its putative regulated gene(s) are shown in Additional file 5: Table S3. A total of 69 genes were predicted. Among these, 5 solo LTRs were not associated with any genes, 15 solo LTRs were associated with 1 gene, and 27 solo LTRs were associated with 2 genes (Fig. 4A). The absolute distances of 3 genes to the transcription start site (TSS) were less than 5 kb. The absolute distances of 13 genes to the TSS were between 5 and 50 kb. The absolute distances of 34 genes to the TSS were between 50 and 500 kb. The absolute distances of 19 genes to the TSS were more than 500 kb (Fig. 4B, C).
Fig. 4.

The genes associated with solo LTRs and GO Slim summaries. A The number of associated genes per solo LTR. B Binned by orientation and distance to TSS. C Binned by absolute distance to TSS. Biological process (D), cellular component (E), and molecular function (F) summaries are represented by red, blue, and green bars, respectively. The height of the bar represents the number of IDs in the gene list and in the category
To analyze the biological classification of key genes related to solo LTRs, GO Slim summaries were generated. The biological processes (BP) summary revealed that these genes were mainly enriched in biological regulation, metabolic process, multicellular organismal process, response to stimulus, cell communication, developmental process, localization, and cellular component organization (Fig. 4D). The GO Slim cellular component (CC) summary showed that these genes were significantly enriched in the membrane and nucleus, and the GO Slim molecular function (MF) summary showed that these genes were significantly enriched in protein binding and ion binding (Fig. 4E, F).
Furthermore, these potential regulatory genes were all annotated to the selected functional categories and subjected to enrichment analysis. The top 10 most significant GO terms according to FDR value for BPs included the regulation of endothelial cell chemotaxis, the regulation of natural killer cell-mediated immunity, the positive regulation of synapse assembly, natural killer cell-mediated immunity, the regulation of synapse assembly, the positive regulation of chemotaxis, synapse assembly, the regulation of synapse organization, the regulation of synapse structure or activity, and synapse organization (Fig. 5A). The bar chart shows the enrichment ratio of the results. Bars representing categories with an FDR ≤ 0.05 are shown in a darker shade (Fig. 5A). The volcano plot in Fig. 5B shows the log2 of the FDR versus the enrichment ratio for all the functional categories in the database, highlighting the degree to which the significant categories are separated from the background. The size and color of a dot are proportional to the number of overlaps (for ORA). The significantly enriched categories are labeled, and the labels are positioned automatically by a force field-based algorithm at startup. The enrichment results for the CC and MF categories are illustrated in Fig. 5C–F. It must be noted that these results are entirely prediction-based and that future research is required to confirm any of the implied associations between solo LTRs and nearby genes.
Fig. 5.
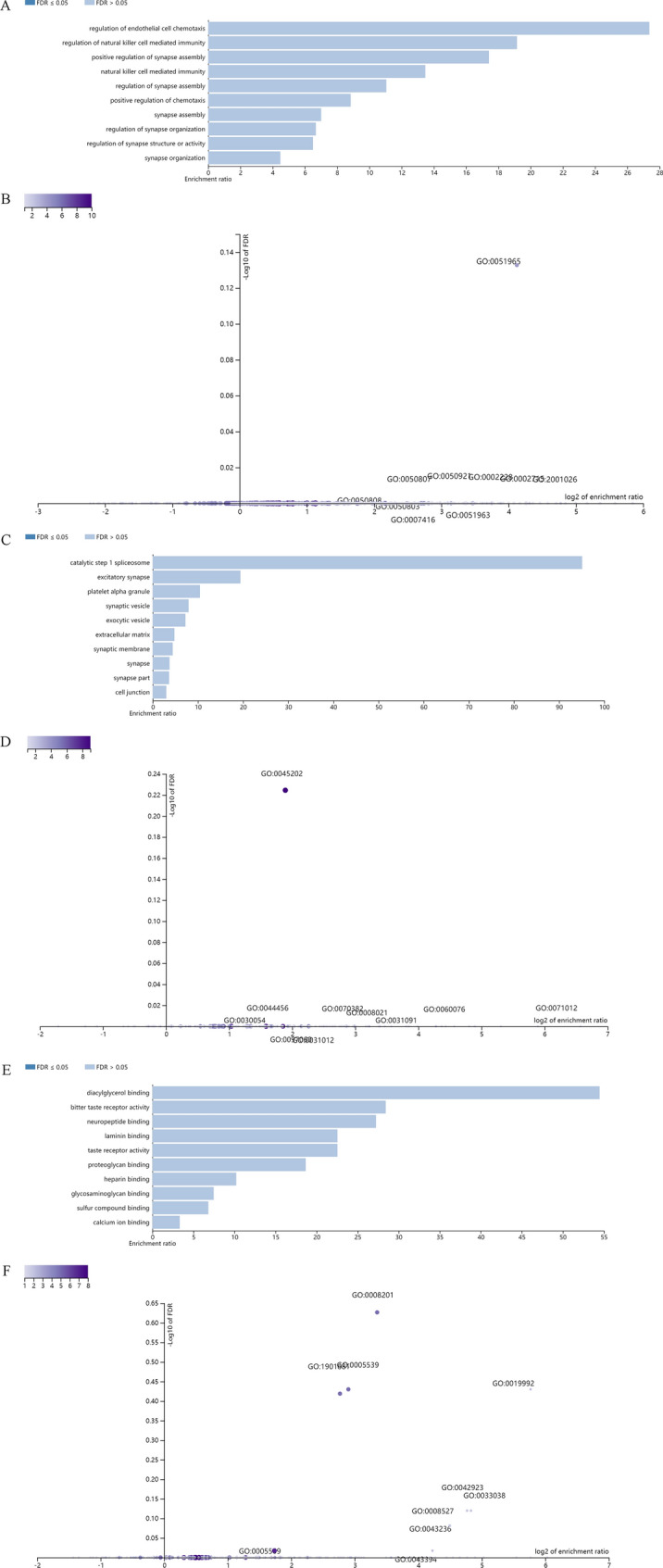
Enrichment result categories binned by biological process, cellular component, and molecular function. A, B Bar chart and customizable volcano plot of the biological process enrichment results. C and D, Bar chart and customizable volcano plot of the cellular component enrichment results. E and F, Bar chart and customizable volcano plot of molecular function enrichment results
Similar to the approach used for solo LTRs, GREAT prediction of genes putative regulated by proviral LTRs was also performed. Enrichment analysis was carried out as described for solo LTRs. The results describing the associations between each proviral LTR and its putative regulated gene(s) are shown in Additional file 6: Table S4. A total of 36 genes were predicted. Among these, 4 proviral LTRs were not associated with any genes, 6 proviral LTRs were associated with 1 gene, and 15 proviral LTRs were associated with 2 genes (Additional file 8: Figure S1A). Of these, the 4 proviral LTRs associated with no genes belong to two pairs of 5′ and 3′ LTRs of the same provirus, 2p12 and Yp11.2, respectively. In particular, 2p12 is a rather complete provirus. No genes with an absolute distance of less than 5 kb from the LTR to the TSS were found. The absolute distances to the TSSs were between 5 and 50 kb for 13 genes. The absolute distances to the TSSs were between 50 and 500 kb for 15 genes. The absolute distances to the TSSs were more than 500 kb for 8 genes (Additional file 8: Figure S1B, C). The GO Slim summaries for the biological classification of key genes related to proviral LTRs are shown in Additional file 8: Figure S1D–F. The enrichment results for BP, CC, and MF categories are shown in Additional file 9: Figure S2, Additional file 10: Figure S3, Additional file 11: Figure S4.
In silico examination of the conserved transcription factor binding sites
Specific base insertions in HML-9 elements may influence the complexity of LTR transcriptional regulation [16]. A complete view of the putative transcription factor binding sites within the HML-9 LTR is shown in Fig. 6A. A total of 22 human transcription binding sites were predicted for 19 transcription factors: EHF, SOX10, FOS, FOSL1, FOSL2, JUNB, JUND, ETV4, KLF1, KLF5, KLF7, ZNF263, THAP1, SP4, RBPJ, HAND2, MAZ, NEUROG2, and NEUROD1. The motifs are marked on the sense strand and antisense strand of the consensus sequence.
Fig. 6.

In silico examination of the conserved transcription factor binding sites and logos representing the PBSs of HML-9. A The forward arrow indicates the sense strand, and the reverse arrow indicates the antisense strand. Different transcription factors are marked with different colors. B PBS nucleotide sequence
PBS type of HML-9 sequences
Traditionally, HERVs have been named according to the tRNA that binds their RT enzyme and PBS [59]. Thus, HERV-K is named after the lysine-tRNA. In the 11 proviral and consensus sequences of HML-9 elements analyzed, the PBS was located approximately 3–20 nucleotides downstream of the 5′LTR. To summarize the general variation of the PBS sequence within the HML-9 group, we generated a logo in which the letter height is proportional to the nucleotide conservation at each position (Fig. 6B). The results showed that the TGG starting nucleotides were the most conserved among the 18 bases. However, only the 15q21.1 and 8p11.1 PBSs belong to lysine, confirming the relatively low taxonomic value of this feature (Additional file 7: Table S5) [61, 62].
Discussion
At present, the HML1-8 and HML10 groups have been characterized and identified [9, 16, 48, 49, 60–64]. However, existing research on HML-9 elements is very limited [44]. A descriptive study of HML-9 elements, especially the characterization and description of their features, is still lacking. Characterization of the genomic distribution of the HML-9 group is critical to understanding the regulation of the expression of this family in healthy humans and its relationship with diseases. Therefore, it is necessary to perform a systematic and comprehensive characterization of HML-9.
Our research followed the approach carried out in previously published studies [9, 48], completely mapping out the HML-9 proviruses and solo LTRs in the human genome and thus providing an exhaustive characterization of this group, including genomic distribution, structural characterization, phylogenesis, integration time analysis and regulatory function prediction. A total of 23 HERV-K HML-9 proviruses and 47 solo LTR elements were characterized. The chromosomal distribution of these proviruses and the solo LTRs revealed a nonrandom integration pattern. HERV-K HML-9 elements are usually enriched outside transcription units in the human genome [15, 65]. The results showed that these elements are mainly distributed in intergenic regions and introns. This may be because the integration of a HERV provirus within the transcription unit is harmful and therefore subject to negative selection and elimination during evolution [12, 15, 21–24]. In particular, the number of proviruses on the Y chromosome was significantly different from that predicted by the chi-square test (p = 0.01), which indicates that the male-specific region of the Y chromosome (MSY) accumulates higher densities of HERVs and associated sequences, consistent with previous studies [65].
Phylogenetic analyses showed that 5 sequences of HML-9 near-full-length proviruses as well as 10 gag elements, 8 pro elements, 11 pol elements, and 13 env sequences formed a unique monophyletic cluster, clearly divided from other HML groups, supported by the maximum bootstrap value. The phylogenetic trees of the pro and pol regions both revealed the presence of two well-supported clusters, identified here as HML-9 types a and b, which were statistically supported by bootstrap values of 100. The HML-9 type b cluster included the Dfam HML-9 reference, whereas the HML-9 type a cluster showed more divergence relative to the group references. In addition, the integration time of HML-9 proviruses was calculated using the LTR, gag, pro, pol, and env regions. The results indicated that the LTRs integrated between 17.5 and 48.5 mya. However, the main period of HML-9 integration based on 4 internal regions is between 37.5 and 151.5 mya. The difference in estimated integration time between the two methods likely occurred because internal coding regions can accumulate mutations during every replication cycle, while two identical LTRs integrate into the host genome during the integration phase [66]. Therefore, it is more reasonable to use LTRs to evaluate the integration time.
Furthermore, we performed prediction and cluster analysis of potential regulatory genes for both the HML-9 provirus and solo LTRs. A total of 69 genes were predicted. BP and MF analyses showed that these genes were associated with synapses. Previous studies have shown that HERV-W can interfere with neuronal protrusions and alter N-methyl-D-aspartate receptor (NMDAR)-mediated synaptic organization and plasticity through glia- and cytokine-dependent changes [67]. Here, our work suggested that HML-9 LTR-regulated genes may also be widely involved in the function of synapses. Furthermore, the prediction of TFBSs in HML-9 elements by JASPAR also indicated that HML-9 is likely to play a role in regulating downstream genes. In addition, for the PBS analysis of HML-9 elements, the results showed that the TGG starting nucleotides were the most conserved among the 18 bases. Similar to previous work [68, 69], we identified 11 proviral PBS sequences and confirmed that this nomenclature is imprecise because although HML-9 belongs to the HERV-K subgroup, only the PBSs of 15q21.1 and 8p11.1 belong to lysine. It should be noted that these results are entirely prediction based. Experimental validation studies are required to confirm the associations between these elements and these genes.
Conclusion
A previous study of HML-9 (HERVK14C) indicated that HML-9 could exert its effects in different tissues under physiological conditions as well as during disease development, possibly contributing to immune regulation and antiviral defense [44]. To systematically study the important role of HML-9 in pathological and physiological processes, the current work provides a clear and detailed description of all HML-9 elements integrated into the human genome, which could contribute to better defining the real impact of these elements and their contribution to the genome.
Supplementary Information
Additional file 1: Dataset S1. The nucleotide sequence of HML-9 proviral elements.
Additional file 2: Dataset S2. The nucleotide sequence of HML-9 solo LTR elements.
Additional file 3: Table S1. HML-9 proviral sequences colocalized with genes.
Additional file 4: Table S2. HML-9 solo LTR sequences colocalized with genes.
Additional file 5: Table S3. The associations between each solo LTR and its putative regulated gene(s).
Additional file 6: Table S4. The associations between each proviral LTR and its putative regulated gene(s).
Additional file 7: Table S5. PBS types in HML-9.
Additional file 8: Figure S1. The genes associated with proviral LTRs and GO Slim summaries. A The number of associated genes per proviral LTR. B Binned by orientation and distance to TSS. C Binned by absolute distance to TSS. Biological process (D), cellular component (E), and molecular function (F) summaries are represented by red, blue, and green bars, respectively. The height of the bar represents the number of IDs in the gene list and in the category.
Additional file 9: Figure S2. The enrichment results for the biological process. A The bar chart plots the enrichment results vertically with the bar width equal to the enrichment ratio in ORA. B Customizable volcano plot. The inset shows an initial layout for comparison.
Additional file 10: Figure S3. The enrichment results for cellular component. A The bar chart plots the enrichment results vertically with the bar width equal to the enrichment ratio in ORA. B Customizable volcano plot. The inset shows an initial layout for comparison.
Additional file 11: Figure S4. The enrichment results for molecular function. A The bar chart plots the enrichment results vertically with the bar width equal to the enrichment ratio in ORA. B Customizable volcano plot. The inset shows an initial layout for comparison.
Acknowledgements
This study was supported by NSFC (31900157).
Abbreviations
- HERVs
Human endogenous retroviruses
- TEs
Transposable elements
- REs
Retroelements
- LTRs
Long terminal repeats
- PBS
Primer binding site
- HML
Human MMTV-like
- MSY
Male-specific regions of the Y chromosome
- RT
Reverse transcriptase
- ML
Maximum likelihood
- NNI
Nearest neighbor interchange
- GREAT
Genomic regions enrichment of annotations tool
- WebGestalt
WEB-based Gene SeT AnaLysis Toolkit
- ORA
Over-representation analysis
- GSEA
Gene set enrichment analysis
- ORFs
Open reading frames
- Mya
Million years ago
- TSS
Transcription start site
- BP
Biological processes
- CC
Cellular component
- MF
Molecular function
- FDR
False discovery rate
Author contributions
Research design: LL and CY. Performed the analysis: LJ, ML, CQY, HL, YL, JH, XZ, XW, TL, BZ and JL. Contributed to the composition of the manuscript: LJ, ML, and LL. All authors read and approved the final manuscript.
Funding
This study was supported by NSFC (31900157).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Lei Jia and Mengying Liu contributed equally to this work
Contributor Information
Lei Jia, Email: 15001193408@163.com.
Mengying Liu, Email: ccbbpxx@sina.com.
Caiqin Yang, Email: 2945656576@qq.com.
Hanping Li, Email: hanpingline@163.com.
Yongjian Liu, Email: yongjian325@sina.com.
Jingwan Han, Email: hanjingwan@outlook.com.
Xiuli Zhai, Email: zhaixiuli2021@126.com.
Xiaolin Wang, Email: woodsxl@163.com.
Tianyi Li, Email: litianyi1983@yahoo.com.
Jingyun Li, Email: lijyjk@163.com.
Bohan Zhang, Email: zbhforjob@163.com.
Changyuan Yu, Email: yucy@mail.buct.edu.cn.
Lin Li, Email: dearwood@sina.com.
References
- 1.Griffiths DJ. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001;2:REVIEWS1017. doi: 10.1186/gb-2001-2-6-reviews1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Gogvadze E, Buzdin A. Retroelements and their impact on genome evolution and functioning. Cell Mol Life Sci. 2009;66:3727–3742. doi: 10.1007/s00018-009-0107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannert N, Kurth R. The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genom Hum Genet. 2006;7:149–173. doi: 10.1146/annurev.genom.7.080505.115700. [DOI] [PubMed] [Google Scholar]
- 6.Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14572–14579. doi: 10.1073/pnas.0404838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boller K, Schönfeld K, Lischer S, Fischer N, Hoffmann A, Kurth R, Tönjes RR. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J Gen Virol. 2008;89:567–572. doi: 10.1099/vir.0.83534-0. [DOI] [PubMed] [Google Scholar]
- 8.Schiavetti F, Thonnard J, Colau D, Boon T, Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Can Res. 2002;62:5510–5516. [PubMed] [Google Scholar]
- 9.Grandi N, Cadeddu M, Pisano MP, Esposito F, Blomberg J, Tramontano E. Identification of a novel HERV-K(HML10): comprehensive characterization and comparative analysis in non-human primates provide insights about HML10 proviruses structure and diffusion. Mob DNA. 2017;8:15. doi: 10.1186/s13100-017-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson WE. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol. 2019;17:355–370. doi: 10.1038/s41579-019-0189-2. [DOI] [PubMed] [Google Scholar]
- 11.Benachenhou F, Sperber GO, Bongcam-Rudloff E, Andersson G, Boeke JD, Blomberg J. Conserved structure and inferred evolutionary history of long terminal repeats (LTRs) Mob DNA. 2013;4:5. doi: 10.1186/1759-8753-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medstrand P, van de Lagemaat LN, Mager DL. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res. 2002;12:1483–1495. doi: 10.1101/gr.388902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andersson ML, Lindeskog M, Medstrand P, Westley B, May F, Blomberg J. Diversity of human endogenous retrovirus class II-like sequences. J Gen Virol. 1999;80(Pt 1):255–260. doi: 10.1099/0022-1317-80-1-255. [DOI] [PubMed] [Google Scholar]
- 14.Medstrand P, Mager DL. Human-specific integrations of the HERV-K endogenous retrovirus family. J Virol. 1998;72:9782–9787. doi: 10.1128/JVI.72.12.9782-9787.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brady T, Lee YN, Ronen K, Malani N, Berry CC, Bieniasz PD, Bushman FD. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 2009;23:633–642. doi: 10.1101/gad.1762309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011;8:90. doi: 10.1186/1742-4690-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue B, Sechi LA, Kelvin DJ. Human endogenous retrovirus K (HML-2) in health and disease. Front Microbiol. 2020;11:1690. doi: 10.3389/fmicb.2020.01690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Montojo M, Doucet-O'Hare T, Henderson L, Nath A. Human endogenous retrovirus-K (HML-2): a comprehensive review. Crit Rev Microbiol. 2018;44:715–738. doi: 10.1080/1040841X.2018.1501345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace TA, Downey RF, Seufert CJ, Schetter A, Dorsey TH, Johnson CA, Goldman R, Loffredo CA, Yan P, Sullivan FJ, et al. Elevated HERV-K mRNA expression in PBMC is associated with a prostate cancer diagnosis particularly in older men and smokers. Carcinogenesis. 2014;35:2074–2083. doi: 10.1093/carcin/bgu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vargiu L, Rodriguez-Tomé P, Sperber GO, Cadeddu M, Grandi N, Blikstad V, Tramontano E, Blomberg J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology. 2016;13:7–7. doi: 10.1186/s12977-015-0232-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/S0959-437X(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 22.van de Lagemaat LN, Medstrand P, Mager DL. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol. 2006;7:R86. doi: 10.1186/gb-2006-7-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2006;2:e2. doi: 10.1371/journal.pgen.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cutter AD, Good JM, Pappas CT, Saunders MA, Starrett DM, Wheeler TJ. Transposable element orientation bias in the Drosophila melanogaster genome. J Mol Evol. 2005;61:733–741. doi: 10.1007/s00239-004-0243-0. [DOI] [PubMed] [Google Scholar]
- 25.Mager DL. Human endogenous retroviruses and pathogenicity: genomic considerations. Trends Microbiol. 1999;7:431. doi: 10.1016/S0966-842X(99)01615-7. [DOI] [PubMed] [Google Scholar]
- 26.Ohtani H, Liu M, Zhou W, Liang G, Jones PA. Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 2018;28:1147–1157. doi: 10.1101/gr.234229.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhdanov VM, Soloviev VD, Bektemirov TA, Ilyin KV, Bykovsky AF, Mazurenko NP, Irlin IS, Yershov FI. Isolation of oncornaviruses from continuous human cell cultures. Intervirology. 1973;1:19–26. doi: 10.1159/000148828. [DOI] [PubMed] [Google Scholar]
- 28.Sarngadharan MG, Sarin PS, Reitz MS, Gallo RC. Reverse transcriptase activity of human acute leukaemic cells: purification of the enzyme, response to AMV 70S RNA, and characterization of the DNA product. Nat New Biol. 1972;240:67–72. doi: 10.1038/newbio240067a0. [DOI] [PubMed] [Google Scholar]
- 29.Matteucci C, Balestrieri E, Argaw-Denboba A, Sinibaldi-Vallebona P. Human endogenous retroviruses role in cancer cell stemness. Semin Cancer Biol. 2018;53:17–30. doi: 10.1016/j.semcancer.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Barth M, Groger V, Cynis H, Staege MS. Identification of human endogenous retrovirus transcripts in Hodgkin Lymphoma cells. Mol Biol Rep. 2019;46:1885–1893. doi: 10.1007/s11033-019-04640-x. [DOI] [PubMed] [Google Scholar]
- 31.Grabski DF, Hu Y, Sharma M, Rasmussen SK. Close to the bedside: a systematic review of endogenous retroviruses and their impact in oncology. J Surg Res. 2019;240:145–155. doi: 10.1016/j.jss.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi M, Kuang Y, Zhang Y, Gjini E, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018;24:1143–1150. doi: 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arru G, Mameli G, Deiana GA, Rassu AL, Piredda R, Sechi E, Caggiu E, Bo M, Nako E, Urso D, et al. Humoral immunity response to human endogenous retroviruses K/W differentiates between amyotrophic lateral sclerosis and other neurological diseases. Eur J Neurol. 2018;25:1076–1071e1084. doi: 10.1111/ene.13648. [DOI] [PubMed] [Google Scholar]
- 34.Mameli G, Erre GL, Caggiu E, Mura S, Cossu D, Bo M, Cadoni ML, Piras A, Mundula N, Colombo E, et al. Identification of a HERV-K env surface peptide highly recognized in Rheumatoid Arthritis (RA) patients: a cross-sectional case-control study. Clin Exp Immunol. 2017;189:127–131. doi: 10.1111/cei.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arru G, Galleri G, Deiana GA, Zarbo IR, Sechi E, Bo M, Cadoni MPL, Corda DG, Frau C, Simula ER, et al. HERV-K modulates the immune response in ALS patients. Microorganisms. 2021 doi: 10.3390/microorganisms9081784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue B, Zeng T, Jia L, Yang D, Lin SL, Sechi LA, Kelvin DJ. Identification of the distribution of human endogenous retroviruses K (HML-2) by PCR-based target enrichment sequencing. Retrovirology. 2020;17:10. doi: 10.1186/s12977-020-00519-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Löwer R, Löwer J, Tondera-Koch C, Kurth R. A general method for the identification of transcribed retrovirus sequences (R-U5 PCR) reveals the expression of the human endogenous retrovirus loci HERV-H and HERV-K in teratocarcinoma cells. Virology. 1993;192:501–511. doi: 10.1006/viro.1993.1066. [DOI] [PubMed] [Google Scholar]
- 38.Mayer J, Ehlhardt S, Seifert M, Sauter M, Müller-Lantzsch N, Mehraein Y, Zang KD, Meese E. Human endogenous retrovirus HERV-K(HML-2) proviruses with Rec protein coding capacity and transcriptional activity. Virology. 2004;322:190–198. doi: 10.1016/j.virol.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 39.Büscher K, Hahn S, Hofmann M, Trefzer U, Ozel M, Sterry W, Löwer J, Löwer R, Kurth R, Denner J. Expression of the human endogenous retrovirus-K transmembrane envelope, Rec and Np9 proteins in melanomas and melanoma cell lines. Melanoma Res. 2006;16:223–234. doi: 10.1097/01.cmr.0000215031.07941.ca. [DOI] [PubMed] [Google Scholar]
- 40.Kraus B, Fischer K, Büchner SM, Wels WS, Löwer R, Sliva K, Schnierle BS. Vaccination directed against the human endogenous retrovirus-K envelope protein inhibits tumor growth in a murine model system. PLoS ONE. 2013;8:e72756. doi: 10.1371/journal.pone.0072756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armbruester V, Sauter M, Krautkraemer E, Meese E, Kleiman A, Best B, Roemer K, Mueller-Lantzsch N. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin Cancer Res. 2002;8:1800–1807. [PubMed] [Google Scholar]
- 42.Chen T, Meng Z, Gan Y, Wang X, Xu F, Gu Y, Xu X, Tang J, Zhou H, Zhang X, et al. The viral oncogene Np9 acts as a critical molecular switch for co-activating β-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia. 2013;27:1469–1478. doi: 10.1038/leu.2013.8. [DOI] [PubMed] [Google Scholar]
- 43.Antony JM, van Marle G, Opii W, Butterfield DA, Mallet F, Yong VW, Wallace JL, Deacon RM, Warren K, Power C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- 44.Tie CH, Fernandes L, Conde L, Robbez-Masson L, Sumner RP, Peacock T, Rodriguez-Plata MT, Mickute G, Gifford R, Towers GJ, et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018 doi: 10.15252/embr.201745000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hurst T, Pace M, Katzourakis A, Phillips R, Klenerman P, Frater J, Magiorkinis G. Human endogenous retrovirus (HERV) expression is not induced by treatment with the histone deacetylase (HDAC) inhibitors in cellular models of HIV-1 latency. Retrovirology. 2016;13:10. doi: 10.1186/s12977-016-0242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002 doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pisano MP, Grandi N, Cadeddu M, Blomberg J, Tramontano E. Comprehensive characterization of the human endogenous retrovirus HERV-K(HML-6) group: overview of structure, phylogeny, and contribution to the human genome. J Virol. 2019 doi: 10.1128/JVI.00110-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grandi N, Pisano MP, Pessiu E, Scognamiglio S, Tramontano E. HERV-K(HML7) integrations in the human genome: comprehensive characterization and comparative analysis in non-human primates. Biology. 2021 doi: 10.3390/biology10050439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nuclei Acids. 1999;41:95–98. [Google Scholar]
- 51.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lebedev YB, Belonovitch OS, Zybrova NV, Khil PP, Kurdyukov SG, Vinogradova TV, Hunsmann G, Sverdlov ED. Differences in HERV-K LTR insertions in orthologous loci of humans and great apes. Gene. 2000;247:265–277. doi: 10.1016/S0378-1119(00)00062-7. [DOI] [PubMed] [Google Scholar]
- 54.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, Pütz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cohen M, Larsson E. Human endogenous retroviruses. BioEssays. 1988;9:191–196. doi: 10.1002/bies.950090603. [DOI] [PubMed] [Google Scholar]
- 60.Flockerzi A, Burkhardt S, Schempp W, Meese E, Mayer J. Human endogenous retrovirus HERV-K14 families: status, variants, evolution, and mobilization of other cellular sequences. J Virol. 2005;79:2941–2949. doi: 10.1128/JVI.79.5.2941-2949.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mayer J, Meese EU. The human endogenous retrovirus family HERV-K(HML-3) Genomics. 2002;80:331–343. doi: 10.1006/geno.2002.6839. [DOI] [PubMed] [Google Scholar]
- 62.Seifarth W, Baust C, Murr A, Skladny H, Krieg-Schneider F, Blusch J, Werner T, Hehlmann R, Leib-Mosch C. Proviral structure, chromosomal location, and expression of HERV-K-T47D, a novel human endogenous retrovirus derived from T47D particles. J Virol. 1998;72:8384–8391. doi: 10.1128/JVI.72.10.8384-8391.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lavie L, Medstrand P, Schempp W, Meese E, Mayer J. Human endogenous retrovirus family HERV-K(HML-5): status, evolution, and reconstruction of an ancient betaretrovirus in the human genome. J Virol. 2004;78:8788–8798. doi: 10.1128/JVI.78.16.8788-8798.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu M, Jia L, Li H, Liu Y, Han J, Zhai X, Wang X, Li T, Li J, Zhang B, et al. Identification and characterization of the HERV-K (HML-8) group of human endogenous retroviruses in the genome. bioRxiv. 2022 doi: 10.1101/2022.02.10.479833. [DOI] [PubMed] [Google Scholar]
- 65.Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 66.Belshaw R. What is special about RNA viruses? Trends Ecol Evol. 2010;25:264–265. doi: 10.1016/j.tree.2010.01.001. [DOI] [Google Scholar]
- 67.Johansson EM, Bouchet D, Tamouza R, Ellul P, Morr AS, Avignone E, Germi R, Leboyer M, Perron H, Groc L. Human endogenous retroviral protein triggers deficit in glutamate synapse maturation and behaviors associated with psychosis. Sci Adv. 2020;6:eabc0708. doi: 10.1126/sciadv.abc0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jern P, Stoye JP, Coffin JM. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007;3:2014–2022. doi: 10.1371/journal.pgen.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuraguchi M, Ohene-Baah NY, Sonkin D, Bronson RT, Kucherlapati R. Genetic mechanisms in Apc-mediated mammary tumorigenesis. PLoS Genet. 2009;5:e1000367. doi: 10.1371/journal.pgen.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Dataset S1. The nucleotide sequence of HML-9 proviral elements.
Additional file 2: Dataset S2. The nucleotide sequence of HML-9 solo LTR elements.
Additional file 3: Table S1. HML-9 proviral sequences colocalized with genes.
Additional file 4: Table S2. HML-9 solo LTR sequences colocalized with genes.
Additional file 5: Table S3. The associations between each solo LTR and its putative regulated gene(s).
Additional file 6: Table S4. The associations between each proviral LTR and its putative regulated gene(s).
Additional file 7: Table S5. PBS types in HML-9.
Additional file 8: Figure S1. The genes associated with proviral LTRs and GO Slim summaries. A The number of associated genes per proviral LTR. B Binned by orientation and distance to TSS. C Binned by absolute distance to TSS. Biological process (D), cellular component (E), and molecular function (F) summaries are represented by red, blue, and green bars, respectively. The height of the bar represents the number of IDs in the gene list and in the category.
Additional file 9: Figure S2. The enrichment results for the biological process. A The bar chart plots the enrichment results vertically with the bar width equal to the enrichment ratio in ORA. B Customizable volcano plot. The inset shows an initial layout for comparison.
Additional file 10: Figure S3. The enrichment results for cellular component. A The bar chart plots the enrichment results vertically with the bar width equal to the enrichment ratio in ORA. B Customizable volcano plot. The inset shows an initial layout for comparison.
Additional file 11: Figure S4. The enrichment results for molecular function. A The bar chart plots the enrichment results vertically with the bar width equal to the enrichment ratio in ORA. B Customizable volcano plot. The inset shows an initial layout for comparison.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.