

Abstract
Treatment of bleeding disorders using transfusion of donor-derived platelets faces logistical challenges due to their limited availability, high risk of contamination, and short (5 to 7 days) shelf life. These challenges could be potentially addressed by designing platelet mimetics that emulate the adhesion, aggregation, and procoagulant functions of platelets. To this end, we created liposome-based platelet-mimicking procoagulant nanoparticles (PPNs) that can expose the phospholipid phosphatidylserine on their surface in response to plasmin. First, we tested PPNs in vitro using human plasma and demonstrated plasmin-triggered exposure of phosphatidylserine and the resultant assembly of coagulation factors on the PPN surface. We also showed that this phosphatidylserine exposed on the PPN surface could restore and enhance thrombin generation and fibrin formation in human plasma depleted of platelets. In human plasma and whole blood in vitro, PPNs improved fibrin stability and clot robustness in a fibrinolytic environment. We then tested PPNs in vivo in a mouse model of thrombocytopenia where treatment with PPNs reduced blood loss in a manner comparable to treatment with syngeneic platelets. Furthermore, in rat and mouse models of traumatic hemorrhage, treatment with PPNs substantially reduced bleeding and improved survival. No sign of systemic or off-target thrombotic risks was observed in the animal studies. These findings demonstrate the potential of PPNs as a platelet surrogate that should be further investigated for the management of bleeding.
INTRODUCTION
Platelets stanch bleeding by binding to von Willebrand factor (vWF) through platelet glycoprotein GPIbα and to collagen through platelet glycoproteins GPIa/IIa and GPVI at the vascular injury site. Concomitantly, platelets activate and then aggregate at the injury site through the binding of fibrinogen to platelet surface integrin αIIbβ3. A subset of these activated aggregated platelets exposes the anionic phospholipid, phosphatidylserine (PS), at the platelet surface, facilitating the assembly of coagulation factors and resulting in thrombin amplification. This leads to locally enhanced fibrin generation from fibrinogen and stable hemostatic clot formation (fig. S1) (1, 2). Transfusion of platelets is used clinically for prophylactic treatment of bleeding in thrombocytopenia (TCP), surgery, etc. and for emergency management of severe bleeding due to, for example, traumatic injury (3–7). However, the limited availability, portability, special storage requirements, high risk of bacterial contamination, and short shelf life (~5 days at room temperature) of platelets present logistical challenges for their effective use in hospital and prehospital settings (8–10). These challenges have led to robust efforts to improve the shelf life of platelets while reducing bacterial contamination risks and conserving hemostatic capabilities. These include ultraviolet (UV) irradiation to reduce the risk of bacterial contamination and storing platelets under low temperature conditions (1° to 6°C or lyophilized) (11–16). Such approaches have improved platelet shelf life from ~5 days to several weeks (17, 18), which is still not optimal. Also, cold-stored or lyophilized platelets reportedly undergo partial loss of their surface functions and express activation markers that increase their rapid clearance in vivo (17, 19–21). Nonetheless, cold-stored or lyophilized platelets hold promise for the emergency management of bleeding in patients (12, 13), but such platelet products are still dependent on donors (22). These issues can be potentially addressed by designing synthetic platelet mimetics that can selectively perform the hemostatic functions of platelets (23–25).
The phospholipid composition of the resting platelet membrane predominantly contains the neutral zwitterionic lipid phosphatidylcholine on the outer leaflet, whereas the inner leaflet contains a higher amount of anionic PS (26, 27). Upon platelet adhesion and activation at the site of vascular injury, action of translocase enzymes causes transport of PS to the outer leaflet of the platelet membrane (fig. S1) (26, 28–30). Activated platelets with a PS-enriched surface are “procoagulant,” given that PS allows coagulation factors to form complexes with Ca2+ ions resulting in formation of the tenase complex (FX + FIXa + FVIIIa), which then leads to formation of the prothrombinase complex (FXa + FVa + FII). This prothrombinase complex results in the conversion of prothrombin to thrombin, leading to locally amplified thrombin generation, which then augments the conversion of fibrinogen to fibrin leading to hemostatic clot formation (31).
Here, we have developed platelet-mimicking procoagulant nanoparticles (PPNs) (figs. S1 and S2) (32–34) that mimic the procoagulant function of native platelets. We used a hybrid liposomal nanoparticle system where the liposome membrane contained distearoyl PS (DSPS) together with other lipopeptide components that enabled injury site–specific adhesion and aggregation. Given that exposed PS would present a procoagulant risk in the circulation and also could elicit rapid clearance by macrophages, PPNs were modified with cholesterol-tethered polyethylene glycol (PEG) that cloaked the PS in circulation but could be cleaved off by plasmin specifically at the injury site to expose PS. This exposed PS could then promote platelet-mimetic, site-specific thrombin amplification and fibrin generation resulting in improved hemostatic clot formation and stability. We first evaluated the ability of PPNs to enable plasmin-triggered PS exposure and resultant assembly of coagulation factors at the PPN surface. We then established that PS-exposed PPNs could amplify thrombin generation in human plasma depleted of native platelets and that this thrombin could generate fibrin resulting in stable clot formation. Next, we evaluated prophylactic administration of PPNs in thrombocytopenic (TCP) mice and assessed their hemostatic efficacy compared to syngeneic platelets for curbing tail bleeding. PPNs were then evaluated in rat and mouse traumatic hemorrhage models to assess their effect on blood loss and animal survival.
RESULTS
Design and characterization of PPNs
PPNs were manufactured by the thin-film rehydration and extrusion technique (35, 36) through coassembly of distearoylphosphatidylcholine (DSPC), distearoylphosphatidylethanolamine-PEG2000-peptides, cholesterol, DSPS, and cholesterol-KTFKC-PEG2000 (with KTFKC being a plasmin-cleavable peptide sequence) (figs. S1 and S2) (37). When needed, dihexadecanoylphosphoethanolamine rhodamine B (DHPE-RhB) at 1 mole % was incorporated into the particles for fluorescent labeling. Figure 1A shows the synthesis scheme for cholesterol-KTFKC-PEG; the mass spectrometry characterization of its synthesis and plasmin-triggered cleavage is shown in Fig. 1B. Size characterization of PPNs was conducted by cryo–transmission electron microscopy (cryo-TEM), atomic force microscopy (AFM), and dynamic light scattering (DLS), which together indicated a PPN diameter of ~100 to 150 nm (Fig. 1C and table S1).
Fig. 1. Design and characterization of PPNs.
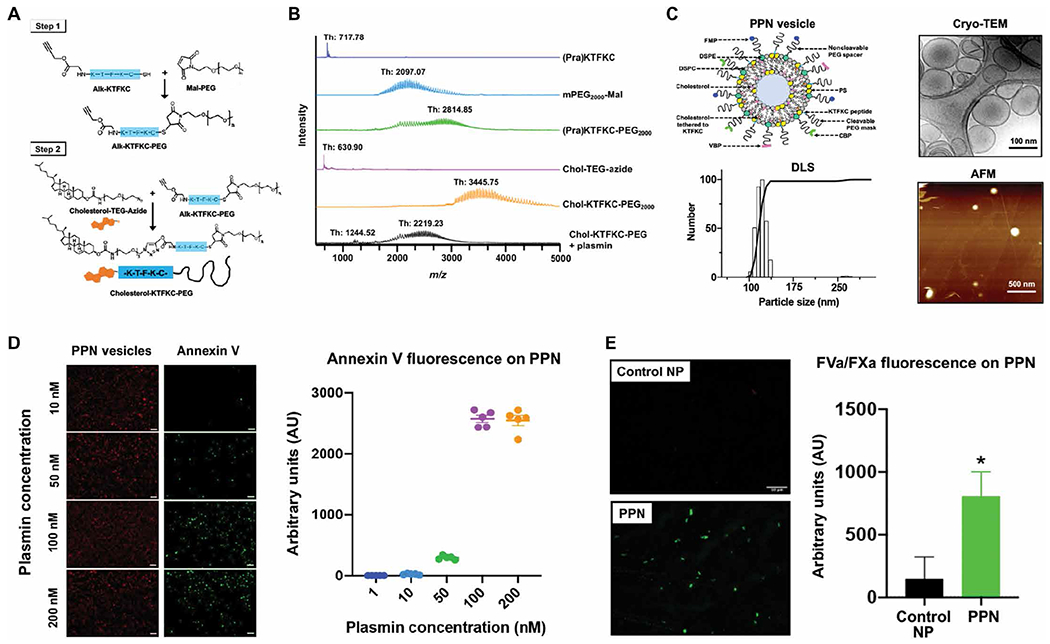
(A) Shown is the two-step process for synthesis of cholesterol-KTFKC-PEG using alkyne-terminated plasmin-cleavable peptide (Alk-KTFKC), maleimide-terminated polyethylene glycol (Mal-PEG), and azide-terminated cholesterol-triethylene glycol (cholesterol-TEG-azide). Cholesterol-KTFKC-PEG was incorporated into PPNs providing a PEG cloak that could be cleaved by plasmin. (B) Matrix-assisted laser desorption ionization–time-of-flight mass spectrometry was used to characterize synthesized cholesterol-KTFKC-PEG and its plasmin-induced degradation (Th, theoretical mass). (C) Shown is the design of PPN vesicles and their size characterization by dynamic light scattering (DLS), cryo–transmission electron microscopy (cryo-TEM), and atomic force microscopy (AFM). The PPN diameter ranged from 100 to 150 nm. (D) Shown are representative images and fluorescence intensity quantification of fluorescently labeled annexin V that was bound to exposed phosphatidylserine at the surface of immobilized PPNs after exposure to plasmin. (E) Shown are representative images and fluorescence intensity quantification of Alexa Fluor 488–labeled antibody bound to factors FVa and FXa (green fluorescence) of the prothrombinase complex that was assembled at the surface of PPNs with exposed phosphatidylserine but not on control nanoparticles (Control NP) without exposed phosphatidylserine. *P ≤ 0.05, two tailed t test.
Additional batches of DHPE-RhB–labeled PPNs were made to incorporate DSPE-PEG-biotin (1 mol %), such that they could be immobilized on avidin-coated glass slides (fig. S3). These immobilized PPNs were exposed to various concentrations of human plasmin (0 to 200 nM, 30 min) and then were incubated with fluorescent (Alexa Fluor 647) annexin V, which bound to exposed PS (38). The representative images and corresponding fluorescence analysis (Fig. 1D and table S2) showed that immobilized PPNs incubated with low (1 and 10 nM) concentrations of human plasmin had minimal annexin V staining, whereas PPNs incubated with ≥50 nM human plasmin had increased annexin V staining. Additional images are shown in fig. S3. Annexin V fluorescence studies also established that PS could be efficiently incorporated into PPNs as the DSPS component of the liposomal membrane (fig. S4).
Next, biotinylated PPNs were immobilized on avidin-coated glass slides, exposed to human plasmin (200 nM, 30 min), and then were incubated with platelet-free human plasma to assess the ability of the exposed PS to render the assembly of FVa and FXa. Here, immobilized biotinylated liposomes containing all other lipid components of PPNs except DSPS and cholesterol-KTFKC-PEG (which ensured that there was no plasmin-triggered exposure of PS) were used as control nanoparticles (Control NP). Plasmin-incubated PPNs showed greater (P ≤ 0.05) FVa and FXa assembly compared to control nanoparticles (Fig. 1E and table S3).
PPNs rescue thrombin generation and improve clot formation in vitro
Thrombin generation assays (39, 40) were performed with PPNs in the presence of platelet-depleted plasma and tissue factor (TF) (Fig. 2, A and D). Platelet-rich human plasma (PRP) incubated with TF was used as a positive control and was compared to platelet-depleted plasma containing only ~5000 platelets/μl (designated in Fig. 2 as thrombocytopenic plasma TCP5K). Liposomes containing all other lipid components of PPNs except DSPS and cholesterol-KTFKC-PEG were used as control nanoparticles (Control NP in Fig. 2). Raw data from these studies are shown in fig. S5. The time to reach peak thrombin (tPeak) was ~15 min for PRP and increased to ~30 min for thrombocytopenic plasma. Adding control nanoparticles in thrombocytopenic plasma only slightly reduced tPeak to ~25 min. Addition of DSPS-containing PPNs—where the PS on the surface was cloaked by cholesterol-KTFKC-PEG (“PS-cloaked PPN”; Fig. 2)—to thrombocytopenic plasma showed effects similar to control nanoparticles. In contrast, addition of DSPS-containing PPNs with PS exposed at the surface (“PS-exposed PPN”; Fig. 2) to thrombocytopenic plasma significantly reduced tPeak to values comparable to PRP (P ≤ 0.01) (Fig. 2A and table S4). Correspondingly, the thrombin lag time in thrombocytopenic plasma was greater compared to PRP; addition of control nanoparticles or PS-cloaked PPNs only slightly shortened the thrombin lag time (Fig. 2B and table S5). In contrast, addition of PS-exposed PPNs significantly shortened the thrombin lag time comparable to that for PRP (P ≤ 0.01) (Fig. 2B). Analyses of peak thrombin generation (Fig. 2C and table S6) and endogenous thrombin potential (Fig. 2D and table S7) showed that these characteristics of thrombin production were substantially reduced in thrombocytopenic plasma compared to PRP; addition of PS-exposed PPNs significantly rescued these parameters (P ≤ 0.01).
Fig. 2. PPNs rescue thrombin generation and clot quality in platelet-depleted plasma.
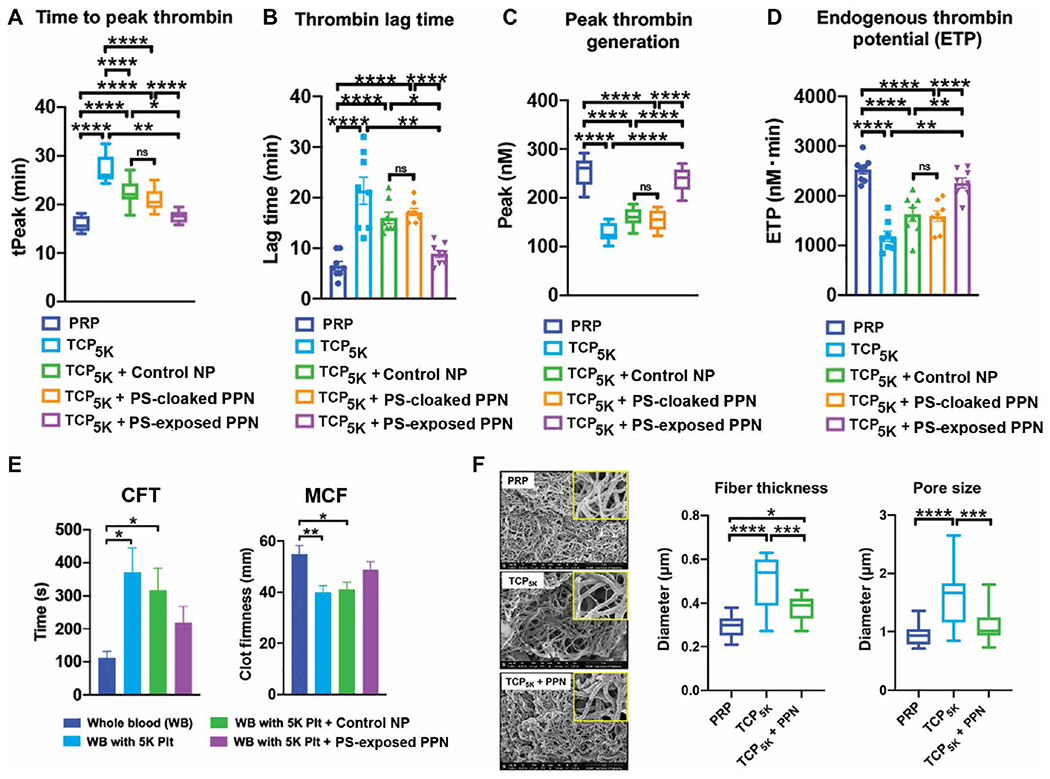
(A to D) Shown are thrombin generation studies in human thrombocytopenic plasma with ~5000 platelets/μl (TCP5K) compared to platelet-rich human plasma (PRP). Time to peak thrombin (tPeak), thrombin lag time, peak thrombin generation, and endogenous thrombin potential are shown after the addition of control nanoparticles (Control NP) or PPNs with exposed phosphatidylserine (PS-exposed) or without exposed phosphatidylserine (PS-cloaked). (E) Rotational thromboelastometry analysis of human whole blood (WB) or human whole blood with ~5000 platelets /μl (WB with 5K Plt) was conducted after addition of Control NP or PPNs with exposed phosphatidylserine (PS-exposed), and clot formation time (CFT) and maximum clot firmness (MCF) were measured. (F) Shown are representative scanning electron microscopy (SEM) images of fibrin clots generated in human TCP5K compared to PRP before and after the addition of PPNs with exposed phosphatidylserine (PS-exposed). Insets show a magnified view (×5) of the main image. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, one way analysis of variance (ANOVA) with Tukey’s multiple comparisons test.
This procoagulant effect of PS-exposed PPNs reached a maximum at about 10 to 15 mol % DSPS incorporation and then started to decrease (fig. S6, A and B). Also, adding an equivalent amount of DSPS directly (not preassembled into PPNs) to plasma did not improve thrombin generation (fig. S6, A and B). Additional studies were carried out to test the effect of PS-exposed PPNs in platelet-free plasma (fig. S7). Here, control nanoparticles or PS-cloaked PPNs could not restore thrombin generation, whereas PS-exposed PPNs partly rescued thrombin generation. When added to PRP, PS-exposed PPNs did not further increase thrombin output (fig. S8).
Given that PS-exposed PPNs could rescue thrombin generation in thrombocytopenic plasma, we next tested whether this thrombin could increase fibrin formation and stability in thrombocytopenic human whole blood after addition of PS-exposed PPNs. For these studies, viscoelastic testing of clots using rotational thromboelastometry allowed temporal assessment of specific clot characteristics (fig. S9, A and B) including clot formation time (CFT) and maximum clot firmness (MCF). Compared to healthy human whole blood (no platelet depletion, control), blood with only 5000 platelets/μl showed delayed (increased) CFT and reduced MCF. Adding PS-exposed PPNs improved both parameters to values closer to those for healthy human whole blood (Fig. 2E, fig. S9C, and tables S8 and S9). Scanning electron microscopy (SEM) analysis of fibrin formed from PRP, thrombocytopenic plasma alone, and thrombocytopenic plasma treated with PS-exposed PPNs was conducted to assess fibrin thickness and porosity (41). Addition of PS-exposed PPNs enhanced fibrin fiber morphology and density in thrombocytopenic plasma (Fig. 2F and tables S10 and S11).
PPNs increase fibrin generation and reduce clot lysis
We first used the overall hemostatic potential (OHP) assay in platelet-free plasma in the presence of tissue plasminogen activator (tPA) (42) to measure the generation and degradation kinetics of fibrin when plasma was treated with PS-cloaked PPNs. Here, the in situ generation of plasmin by tPA was expected to cleave off the PEG from the cholesterol-KTFKC-PEG2000 “cloak” to expose PS at the PPN surface. The assay results showed a balance between fibrin generation and fibrin degradation that was not affected by addition of control nanoparticles (fig. S10). However, addition of PPNs increased OHP, indicating increased fibrin generation (fig. S10). We next used an in vitro microfluidic setup with human plasma clots exposed to tPA (fig. S11) to evaluate the effect of PPNs on fibrin in a fibrinolytic environment. Addition of PS-cloaked PPNs resulted in a significant delay in fibrinolysis compared to addition of control nanoparticles that lacked a PS component or saline (P ≤ 0.05) (Fig. 3, A and B, and table S12). PPNs were found to be incorporated effectively into clots by colocalizing with activated platelets (fig. S12). The ability of PPNs to reduce clot lysis was further corroborated by the observation that the PPN-treated plasma showed decreased fibrin D-dimer values (Fig. 3C and table S13). Parallel thromboelastometry studies were carried out with tPA added to human whole blood with or without the addition of PPNs. Addition of tPA significantly reduced MCF and increased maximum lysis (ML) within 30 min, without affecting CFT, indicating increased fibrinolysis (P ≤ 0.001) (Fig. 3D). We next assessed MCF maintenance time as a measure of clot stability. Treatment with PPNs significantly improved MCF maintenance time and reduced fibrinolysis (P ≤ 0.001) (Fig. 3, E and F, and tables S14 and S15). Comparable results were observed when tranexamic acid (a direct plasmin inhibitor) was added to tPA-treated human whole blood (fig. S13). Treatment with PPNs delayed, but did not eliminate, clot lysis even when a higher tPA dose was added to human whole blood (fig. S14).
Fig. 3. PPNs enhance human plasma and whole blood clot stability under fibrinolytic conditions.
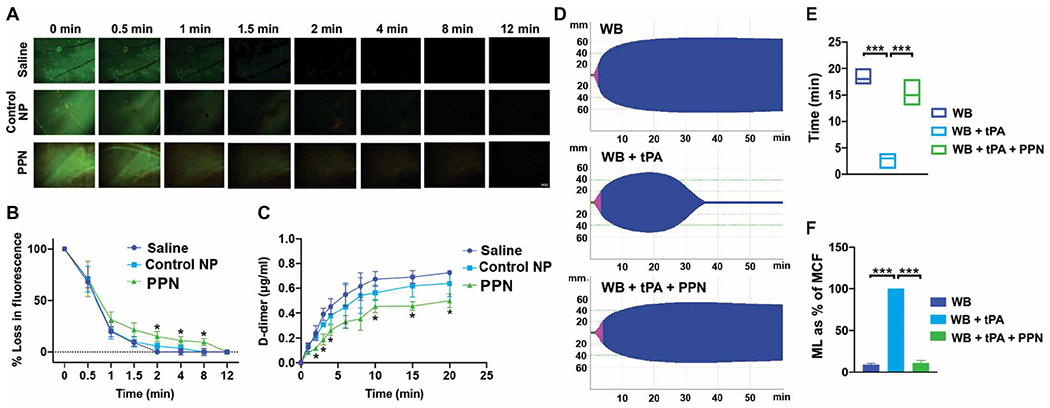
(A to C) Shown is microfluidic analysis of human plasma clots exposed to tissue plasminogen activator (tPA) to create a fibrinolytic environment. Addition of PPNs, with exposed phosphatidylserine in response to plasmin (PPN) in human plasma, delayed clot lysis as indicated by fibrin green fluorescence (A and B) and reduced D-dimers in the clot lysate (C) compared to control nanoparticles (Control NP). (D to F) Shown is rotational thromboelastometry analysis of human whole blood in the presence of tPA. Addition of PPNs, with exposed phosphatidylserine in response to plasmin (PPN), enhanced clot stability as demonstrated by the MCF maintenance time (E) and reduced maximum lysis (ML) (ML as % of MCF) (F). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001, two-way ANOVA with Tukey’s multiple comparisons for microfluidic data and one-way ANOVA with Tukey’s multiple comparisons for rotational thromboelastometry data.
PPNs reduce bleeding in thrombocytopenic mice
All in vivo studies were conducted using PS-cloaked PPNs with the rationale that injury site–localized plasmin would cleave off the PEG from the cholesterol-KTFKC-PEG2000 cloak to expose PS at the surface of PPNs. Before evaluating hemostatic efficacy of PPNs in vivo, we first assessed whether these nanoparticles carried an inherent risk of thrombosis. This was evaluated in a FeCl3-induced arterial thrombosis model in mice (43), where the time to occlusion of the carotid artery in the presence or absence of PPNs was compared. PPN administration did not accelerate time to occlusion of the carotid artery compared to saline administration (fig. S15), indicating that the PPNs were not inherently prothrombotic. The circulation half-life of PPNs in mice, evaluated by administering rhodamine B–labeled PPNs through tail-vein injection and collecting blood over 24 hours to analyze PPN-associated fluorescence, indicated a circulation half-life of ~12 hours (fig. S16).
The hemostatic efficacy of PPNs was next evaluated in the tail-transection bleeding mouse model in mice that were rendered thrombocytopenic by administration of anti-CD42b (anti-GPIb) antibody (Fig. 4A) (44, 45). For these experiments, the antibody dose (0.5 μg/kg) reduced the platelet count by about 80% at 18 to 24 hours after antibody administration (platelet count reduced from ~1500/nl to ~250/nl) (Fig. 4B and table S16). Platelet recovery started at around 36 to 48 hours after antibody administration. The extent of the platelet count reduction was within the range of clinical TCP in humans. Control nanoparticles (liposomes containing all other lipid and lipid-peptide components but not DSPS or cholesterol-KTFKC-PEG) or PPNs were administered at a dose of 2 mg/kg. Additional comparisons were done with syngeneic mouse platelets (250/nl dose).
Fig. 4. PPNs reduce tail bleeding in thrombocytopenic mice.
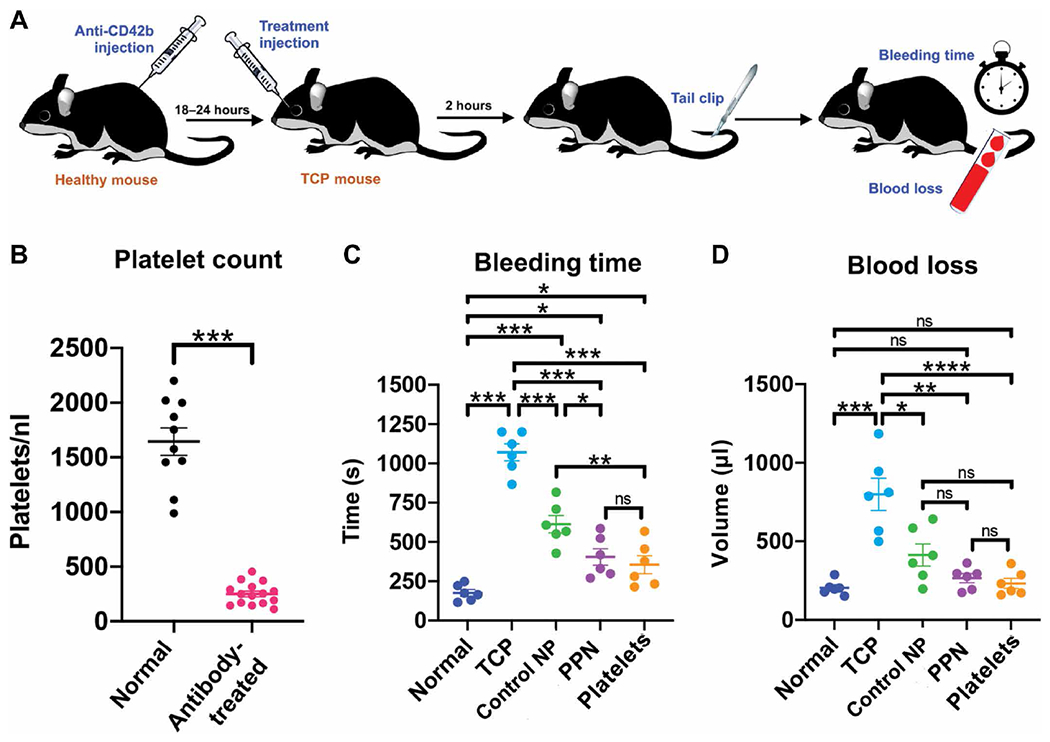
(A) In the tail transection bleeding mouse model, mice were rendered thrombocytopenic through intraperitoneal injection of anti-CD42b (anti-GPIb) antibody (0.5 μg/kg) to deplete platelets. Mice were administered by retro-orbital injection either Control NP or PPNs, ~18 to 24 hours later. Two hours after treatment, mouse tails were clipped 1 mm from the tip and bleeding time and blood loss were measured. (B) Shown are platelet counts in normal (wild-type) mice and antibody-treated thrombocytopenic mice. (C) Shown are bleeding times in normal (wild-type) mice and antibody-treated thrombocytopenic (TCP) mice before and after addition of control nanoparticles or PPNs. Also shown are bleeding times when mice were transfused with syngeneic platelets. (D) Shown is blood loss analysis using a hemoglobin assay in normal (wild-type) mice and antibody-treated thrombocytopenic (TCP) mice before and after addition of control nanoparticles or PPNs. Also shown is blood loss when mice were transfused with syngeneic platelets. n = 6 animals per group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, one-way ANOVA with Tukey’s multiple comparisons test. ns, not significant.
After transection of tails of normal control (wild-type) or thrombocytopenic mice, bleeding times were monitored (Fig. 4C, fig. S17, and table S17). Bleeding stopped at 175.67 ± 51.4 s in normal mice but at 1071 ± 130.8 s in thrombocytopenic mice (Fig. 4C). Treating the thrombocytopenic mice with retro-orbitally injected control nanoparticles 2 hours before tail transection reduced the bleeding time to 613.3 ± 134.9 s (Fig. 4C). Treatment with PPNs reduced the bleeding time even further to 404.5 ± 127.9 s (P ≤ 0.001) (Fig. 4C). When thrombocytopenic mice were treated with syngeneic platelets, the bleeding time was reduced to 355 ± 131.4 s (Fig. 4C). Blood loss analysis by spectrophotometric assessment of hemoglobin demonstrated that treatment with PPNs reduced blood loss from clipped tails in thrombocytopenic mice (Fig. 4D and table S18) in a manner comparable to treatment with syngeneic platelets.
PPNs enhance hemostasis and survival in rodent traumatic hemorrhage models
An acute liver injury model (46, 47) in Sprague-Dawley rats (Fig. 5A) was used to evaluate the ability of PPNs (2 mg/kg dose) administered by tail vein injection to treat traumatic hemorrhage. In this model, blood loss was measured at 1 hour after treatment and survival was monitored for up to 3 hours after treatment. D-dimer and plasmin-antiplasmin analyses of blood samples at early time points (0 to 30 min after injury) in this rat model confirmed high fibrinolysis (fig. S18). Treatment with control nanoparticles (liposomes containing all other lipid and lipid-peptide components of PPNs but not DSPS or cholesterol-KTFKC-PEG) was able to reduce blood loss by ~25% compared to saline treatment (Fig. 5B and table S19). Treatment with PPNs reduced blood loss still further by ~38% (P ≤ 0.05) (Fig. 5B). In a 3-hour survival analysis, injured rats treated with PPNs showed 100% survival, whereas those treated with control nanoparticles showed 80% survival and those treated with saline showed 60% survival (Fig. 5C). Representative fluorescence images showed that PPNs became incorporated efficiently into hemostatic clots (Fig. 5D). Corresponding histology analysis using Carstairs’ staining indicated that PPN-treated animals had more fibrin-rich clots (orange-red staining; Fig. 5E and fig. S19A) compared to saline-treated or control nanoparticle–treated animals. PPN administration did not affect rat vital signs such as heart rate, temperature, or SpO2 (fig. S19, B to D). An initial perturbation of SpO2 (a measure of peripheral oxygenation) was observed in all animals for 5 to 10 min after hemorrhagic injury but stabilized as soon as the hemostatic response started to reduce the hemorrhage. This stabilization occurred slightly faster in the PPN-treated animals compared to saline-treated or control nanoparticle–treated animals (fig. S19D). No systemic thrombosis was observed in other organs in the posteuthanasia analysis after the 3-hour observation period (fig. S19E). Further studies were carried out in a murine acute traumatic coagulopathy model involving liver laceration and cardiac puncture (fig. S20, A to C) in a small group of animals (n = 4 per group). We monitored whether PPN treatment (administered 30 min before injury) could improve survival over 3 days. Treatment with PPNs led to 100% survival over the 3-day period compared to 50% for the saline control group (fig. S20D).
Fig. 5. PPNs reduce blood loss and improve survival in rats with hemorrhagic liver injury.
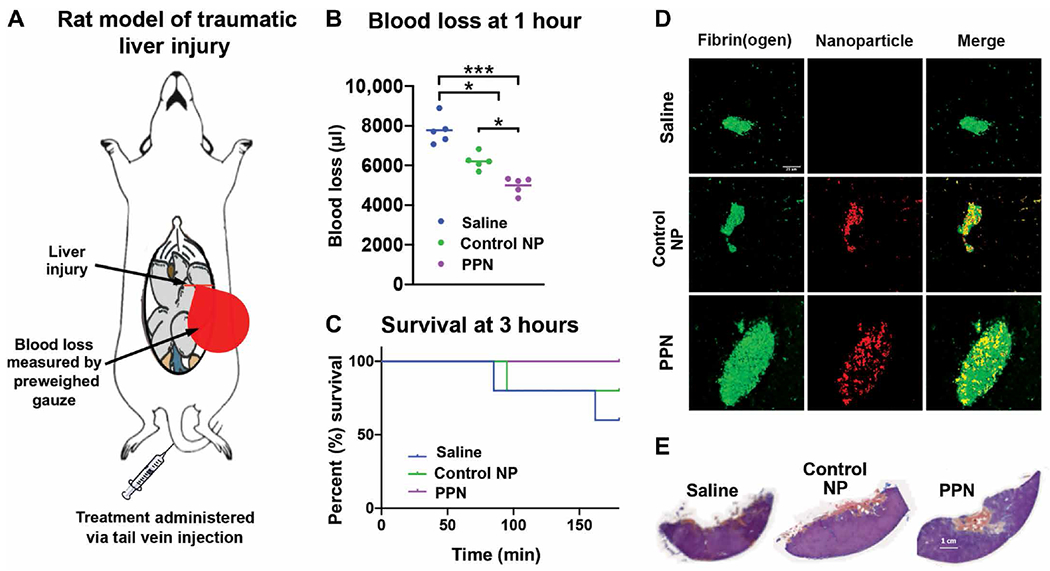
(A) In the rat liver injury hemorrhagic model, >30% of the liver was resected to cause intra-peritoneal hemorrhage and treatment after injury was administered via tail vein. (B) Shown is blood loss at 1 hour after treatment with PPNs compared to control nanoparticles (Control NP) or saline. (C) Shown is animal survival 3 hours after treatment with PPNs compared to control nanoparticles (Control NP) or saline. (D) Shown are representative immunofluorescence images of the liver injury site indicating greater fibrin fluorescence in hemostatic clots after PPN treatment compared to either control nanoparticles (Control NP) or saline. (E) Shown is Carstairs’ staining of representative hemostatic clots at the liver injury site indicating higher fibrin content (orange-red staining) in the clots of PPN-treated rats compared to Control NP– or saline-treated rats. n = 5 animals per group. *P ≤ 0.05 and ***P ≤ 0.001, one-way ANOVA with Tukey’s multiple comparisons test.
DISCUSSION
There are many challenges facing platelet transfusions including platelet availability, portability, contamination risks, and a very short shelf life. Pathogen reduction, low temperature storage, and stem cell–based in vitro platelet production (11–15, 48) are currently being studied to address these challenges. However, synthetic platelet surrogates provide another translationally feasible alternative (49–54). Most synthetic platelet surrogate designs have involved coating nanoparticles with fibrinogen or fibrinogen-derived peptides to amplify platelet aggregation. However, the hemostatic response of platelets requires critical steps including rapid adhesion to vWF and collagen at the injury site, followed by aggregation and procoagulant activity at that site (1, 2). The fibrinogen-coated (and fibrinogen-derived peptide) nanoparticles do not have both adhesion and aggregation capabilities.
Our original platelet surrogate design involved nanoparticles bearing vWF-binding, collagen-binding, and fibrinogen-mimetic peptides. These nanoparticles successfully mimicked the adhesion and aggregation function of platelets needed for primary hemostasis (32–34), but they did not exhibit the procoagulant function of amplifying thrombin and enhancing fibrin needed for efficient hemostasis. In the current study, we built on our original platelet surrogate design to develop a next-generation platelet surrogate, termed PPN, that not only enabled platelet-mimetic adhesion and aggregation functions but also incorporated PS into the nanoparticles for thrombin-amplifying procoagulant function. In this next-generation PPN design, PS is exposed at the surface of PPNs in the presence of high plasmin locally at the injury site enabling site-specific enhancement of procoagulant hemostatic activity. This design could be potentially advantageous in treating trauma-induced hyperfibrinolysis where high amounts of tPA production and plasmin generation occur at the injury site (55–60). Our in vitro studies confirmed that, under no or low plasmin conditions, the PS exposure on PPNs was minimal, whereas, at high plasmin concentrations, the PEG on PPNs was removed to enable high exposure of PS that could amplify thrombin generation even in the absence of native platelets. Circulating active plasmin is rapidly inhibited by antiplasmin and α2-macroglobulin, whereas, at the injury site, the plasmin produced from plasminogen by fibrin-localized tPA is protected from rapid inhibition (55). Therefore, we rationalized that the local plasmin concentration at an injury site would be high enough to cleave off the PEG and expose the PS on PPNs, enabling procoagulant function. We show that PPN-generated thrombin could enhance fibrin generation and preserve clot morphology and stability even under fibrinolytic conditions. Our finding that adding DSPS directly (not preassembled into PPNs) to plasma did not improve thrombin generation suggested that spatial presentation of PS in “patches” at the PPN surface mimicking the membrane surface of procoagulant platelets was important for its function (61, 62). A freeze-dried platelet product derived from group O platelets has been reported to have a PS-rich surface that amplifies hemostasis and is currently in clinical trials (63). Another recent study has shown that plasma-derived PS-positive microparticles improved clot quality in blood samples from patients with hemophilia; however, this was only demonstrated in vitro as such particles cannot be administered in vivo due to a systemic procoagulant risk (64).
Our PPNs circulated in mice in vivo without risk of thrombosis most likely because the PS of PPNs remained unexposed until the PEG cloak was removed by injury site–localized plasmin. Our in vivo rodent studies demonstrated that PPNs provided hemostatic benefit to stanch bleeding in thrombocytopenic mice at a level comparable to that for syngeneic platelets. Our in vivo studies in the rat acute liver injury model and mouse acute hemorrhagic trauma model further demonstrated the hemostatic capability of PPNs, with improved survival of PPN-treated animals compared to saline-treated or control nanoparticle–treated animals. The saline control was a no treatment control as the saline was administered at the same injection volume as the PPN solution and not at a resuscitation volume. Saline-based hypotensive resuscitation remains relevant in pre-hospital management of patients with acute hemorrhage (65) because blood product transfusion is currently not available outside a hospital setting. Also, note that, in our in vitro and in vivo studies, the control nanoparticles showed partial hemostatic benefit because these nanoparticles were our original platelet surrogate design, that is, liposomes that mimicked platelet adhesion and aggregation (32–34). The PPNs used in the current study substantially augmented hemostatic benefit compared to the original platelet surrogate particles due to the ability of exposed PS to boost procoagulant hemostatic function.
The current study further demonstrated that even in plasma completely lacking platelets (platelet-free plasma), PPNs could partly rescue thrombin generation; in the presence of only a small number of endogenous platelets, PPNs could substantially amplify thrombin kinetics. This could be of particular importance in treating trauma-induced or drug-induced platelet dysfunction where native platelets may become hyporesponsive (66, 67). A recent study has shown that, in certain platelet dysfunction settings, transfusion of native platelets may not provide optimal hemostatic output (68). In such scenarios, one can envision hemostatic benefit from PPNs as this synthetic platelet surrogate is not expected to be influenced by inhibitory signals that affect native platelets. Also, given that PPNs are liposomes, opportunities exist for the delivery of adjunctive agents (e.g., platelet agonists, antifibrinolytics, and coagulation factors) as payloads. We and others have recently demonstrated this possibility by delivering agents like tranexamic acid and adenosine diphosphate packaged in clot-targeted liposomes (47, 69). In the future, platelet surrogates such as PPNs could be potentially combined with other blood surrogates such as red blood cell mimics to create biosynthetic whole blood (70, 71).
Our study has a number of limitations. First, we note that PPNs do not perform all of the functions of native platelets (72, 73), although certain platelet functions may not be necessary for hemostasis. Second, for all of our in vivo studies, we used a single bolus dose of PPNs. The effect of recurrent or escalating doses will need to be studied further to establish a therapeutic window and maximum tolerated dose for treating uncontrolled bleeding. Third, the life span of PPNs in the mouse circulation was about 1 day, whereas the life span of native mouse platelets is ~5 days, for native human platelets is ~9 days, and for room temperature–stored transfused human platelets is 3 to 4 days in humans. However, in certain hemostatic applications (e.g., treating hemorrhagic trauma), rapid function to reduce bleeding may be more important than platelet number recovery or platelet-equivalent circulation time, and PPNs may address this need. Fourth, a risk factor of PPNs that has not been extensively evaluated is the potential for systemic thrombosis at higher doses or in certain unique disease settings. Whereas all of our in vivo studies with PPNs at a single therapeutic dose did not show a systemic thrombotic effect, primed platelets or high concentrations of uninhibited active plasmin in the circulation may pose a systemic thrombotic risk that may be aggravated by PPNs. Several recent clinical studies have emphasized that there is hypofunction of circulating platelets in severe trauma even if there is no reduction in platelet count and have suggested that this may be due to an initial hyperactivation followed by exhaustion of platelets after injury (74–76). In parallel, some studies have also indicated that fibrinolytic processes may become systemically activated in trauma to protect against thrombosis in noninjured tissue, although it remains unclear whether this results in high amounts of uninhibited active plasmin in the circulation (77). Therefore, future studies will need to be directed at investigating whether such maladaptive platelet and coagulation responses can aggravate off-target thrombotic effects when PPNs are introduced into the circulation at high doses. Last, the current study has not extensively investigated whether PPNs present dose-dependent immunological risks, including complement activation and effects on innate or adaptive immune processes of myeloid or lymphoid origin. Several liposome-based nanoparticle formulations have been translated successfully into clinical applications (78, 79). In other research studies, small peptides have been shown to elicit reduced immunogenicity (80). Therefore, peptide-decorated liposomal designs for platelet surrogates may have reduced immunogenic risks. However, escalating or recurrent dosing of such nanoparticles may elicit unique immune responses, and this will be an important avenue for future investigations.
In conclusion, we have designed and evaluated procoagulant platelet-mimetic nanoparticles that mimic not only the adhesive and aggregation properties of platelets but also the procoagulant hemostatic function of platelets. Translational advancement of PPN technology for the transfusion management of bleeding warrants further investigation.
MATERIALS AND METHODS
Study design
In this study, we manufactured and evaluated procoagulant platelet-mimetic nanoparticles (PPNs) that mimic platelet adhesion, aggregation, and hemostatic functions at the vascular injury site. We evaluated these PPNs in vitro and in vivo for hemostatic properties and ability to staunch bleeding and improve survival in rodent models of traumatic hemorrhage. PPNs that contained cholesterol-KTFKC-PEG2000 were designed to expose PS at the PPN surface in response to plasmin. Control nanoparticles lacked cholesterol-KTFKC-PEG2000. For all in vitro and in vivo studies, the nanoparticle:platelet ratio was maintained at about 100:1 (see Supplementary Materials and Methods and figs. S21 and S22 for theoretical calculations and rationale). In vitro studies included the thrombin generation assay, rotational thromboelastometry, SEM, OHP assay, and microfluidic analysis.
In vivo studies were carried out in accordance with protocols approved by Case Western Reserve University Institutional Animal Care and Use Committee (IACUC) (Protocol 2016-0012) and following standard International Society on Thrombosis and Haemostasis guidelines. These studies involved a mouse model of TCP and rat and mouse models of traumatic hemorrhage. Treatment of antibody-induced thrombocytopenic mice with PPNs or control nanoparticles was randomized, and tail bleeding time and blood loss were measured. In the rat liver injury traumatic hemorrhage model, treatment with PPNs compared to control nanoparticles or saline was randomized, and blood loss was measured at 1 hour after treatment and survival was calculated at 3 hours after treatment. The mouse traumatic hemorrhage model was used as an additional group to assess long-term survival at 3 days after injury, with PPNs or control nanoparticles administered 30 min before injury.
PPN and control nanoparticle manufacture and characterization
Liposome nanoparticles were manufactured using the thin-film rehydration and extrusion technique. For control liposomes, DSPC, cholesterol, DSPE-PEG2000-VBP, DSPE-PEG2000-CBP, DSPE-PEG2000-FMP, and DSPE-mPEG1000 were homogeneously mixed at 0.625, 0.3, 0.0125, 0.0125, 0.025, and 0.025 mol fractions, respectively, in 1:1 chloroform:methanol. Solvent was removed via rotary evaporation, and the thin lipid film was rehydrated with 0.9% NaCl at a concentration of 1 × 105 mol lipid/ml. This lipid suspension was subjected to 10 freeze per thaw cycles and subsequent extrusion through 200-nm pore diameter polycarbonate membrane using a pneumatic extruder (Northern Lipids, Burnaby, Canada) to create “VBP + CBP + FMP”–decorated control nanoparticles that mimic the adhesion and aggregation functions of platelets. PPNs were manufactured using the same fabrication protocol as above, with the following membrane component mole fractions: DSPC (0.55), cholesterol (0.3), cholesterol-KTFKC-PEG2000 (0.15), DSPS (0.1), DSPE-PEG2000-VBP (0.0125), DSPE-PEG2000-CBP (0.0125), and DSPE-PEG2000-FMP (0.025). The DSPS incorporation allowed for the PS to be presented on the PPN surface and the cholesterol-KTFKC-PEG2000 incorporation allowed for the cloaking of this PS with the cloak amenable to be removed via plasmin-triggered cleavage of the KTFKC sequence. DLS, cryo-TEM, and AFM were used to characterize PPN size distribution. Incorporation of PS into PPN membrane was confirmed using annexin V staining. For this, PPNs were modified with DSPE-PEG2000-biotin (0.01 mol fraction), incubated on avidin-coated glass slides, washed, and stained with Alexa Fluor 647–conjugated annexin V. To evaluate concentration-dependent plasmin-triggered exposure of PS on the PPN surface, DSPE-PEG2000-biotin–incorporated PPNs containing cholesterol-KTFKC-PEG2000 were first incubated on avidin-coated glass slides, and then the immobilized PPNs were incubated with (or without) plasmin (1, 10, 50, 100, and 200 nM) and subsequently stained with annexin V. To confirm FVa and FXa immobilization on PPN, similarly biotinylated control nanoparticles or PPNs were immobilized on avidin-coated glass slide, incubated with platelet-poor plasma (PPP) for an hour, then washed, incubated with Factor Va and Factor Xa antibodies for an hour each, and then further incubated with Alexa Fluor 488–conjugated secondary antibody to FVa and FXa.
Thrombin generation and ROTEM assays in plasma and SEM imaging of fibrin clots
PRP was obtained by centrifuging human whole blood (WB) at 1500g for 15 min at room temperature. The PRP was further centrifuged (13,000g for 5 min) to obtain PPP. The platelet counts in PRP and PPP were monitored using a Multisizer 3 Coulter Counter (Beckman Coulter, Brea, CA). The PRP platelet count was maintained at 150,000/μl (lower limit of normal human platelet count), and the PRP was adjusted with PPP to create thrombocytopenic plasma with platelet counts of 20,000/μl (TCP20K) and 5000/μl (TCP5K). For thrombin generation studies, a thrombin-sensitive fluorescent substrate rhodamine 110 bis-(p-tosyl-l-glycyl-l-prolyl-l-arginine amide) and TF were added to 100 μl of recalcified normal or thrombocytopenic plasma to study the effects of control nanoparticle versus PS-cloaked PPN versus PS-exposed PPN (at dose of 100 particles per platelet) on thrombin generation kinetics. The effect of control nanoparticles and PPN treatment on clot characteristics in platelet-depleted whole blood was evaluated using Rotational Thromboelastometry (ROTEM). For this, normal or thrombocytopenic plasma was combined with initially separated hematocrit to form reconstituted whole blood with normal or depleted platelet count and subjected to ROTEM (EXTEM) assays with nanoparticles added in it. CFT and MCF were measured. In addition, separate studies were done to assess fibrin morphology of clots formed in calcified thrombocytopenic plasma in the presence of PPN using SEM. For these studies, the fibrin clots were fixed with 3% glutaraldehyde, washed, fixed with 2% osmium tetroxide, washed again, dehydrated with increasing concentrations of ethanol (35, 50, 70, 95, and 100%), critical point–dried using hexamethyldisilazane, mounted, and sputter-coated. The dried clots were imaged with a Helios NanoLab 650 SEM at the Swagelok Center for Surface Analysis of Materials. Fiber thickness and pore size in the images obtained were analyzed using ImageJ.
Measurement of PPN effects on fibrin generation and clot stability under fibrinolytic conditions
The effect of control nanoparticles versus PPNs on fibrin kinetics under a lytic environment was studied using the OHP assay (42). Two types of buffers were prepared before the assay, one for the overall coagulation potential (OCP) and the other for OHP. The OCP buffer contained CaCl2 (40 mM) and thrombin (0.5 IU/ml) in tris buffer [66 mM tris and 130 mM NaCl (pH 7)]. For OHP assay, tPA (660 ng/ml) was added to the above-described OCP. In a 96-well plate, 60 μl of plasma was added to each well and mixed with the respective buffers for OCP or OHP. Absorbance values at 405 nm were recorded over time for a 40-min period to construct fibrin generation versus degradation curves. The effect of control nanoparticles versus PPNs on clot characteristics was further evaluated in ROTEM under tPA-induced lytic conditions. For this, tPA (25 μg/ml) was added to whole blood and the effects on ROTEM parameters were evaluated in the presence of SP or PPN (nanoparticle dose of 100 particles per platelet). Specifically, two parameters were assessed: (i) MCF maintenance time, defined as the time for which at least 80% MCF was maintained post reaching MCF and (ii) ML as a % of MCF at 60 min. In a separate experimental setup, PRP was spiked with Alexa Fluor 647–labeled fibrinogen, 50-μl volume of this plasma was placed on collagen-coated glass slide, and fluorescent clots were formed by adding thrombin (100 nM). The clots on the glass slide were vacuum-sealed within parallel plate microfluidic chamber and exposed to flow of plasma containing tPA (25 μg/ml) plus control nanoparticles or PPNs. The flow was maintained at a shear stress of 5 dyne cm−2, and the clot was imaged under inverted fluorescence microscope to monitor “loss” of fibrin fluorescence under flow due to the fibrinolytic action of tPA-generated plasmin. Correspondingly, the fibrin lysis product was collected and analyzed by D-dimer ELISA assay to quantify the extent of fibrinolysis.
In vivo evaluation of PPNs in a thrombocytopenic mouse model
Wild-type C57/BL6J mice were injected intraperitoneally with anti-CD42b (anti-GPIbα) antibody (0.5 μg/g), and 18 to 24 hours later, platelet counts were measured on retro-orbitally drawn blood using a HEMAVET 950 (Drew Scientific, Miami Lakes, FL). After confirming significant TCP at this time point, additional batches of mice (3 male + 3 female = 6 per group) were administered with the antibody, and 18 to 24 hours after antibody administration, control nanoparticles or PPNs (nanoparticle dose of 2 mg/kg) were injected in these TCP mice retro-orbitally. Two hours after nanoparticle administration, the tails of these mice were transected 1 mm from the tip with a sharp surgical blade and immersed in warm (37°C) saline. The time needed for bleeding to stop (bleeding time) was recorded, and the collected blood was analyzed for hemoglobin (marker for blood loss) via the sodium lauryl sulfate method using UV spectrometry at 535 nm. In separate experiments, syngeneic platelets were isolated from mice by collecting whole blood and centrifuging to get PRP, which was further centrifuged to obtain platelet pellet. This platelet pellet was resuspended in Tyrode’s buffer containing PGI2 to prevent clumping. Additional mice were injected intraperitoneally with anti-CD42b antibody (0.5 μg/g) as before, and 18 to 24 hours later, platelet counts were measured on retro-orbitally drawn blood to confirm TCP as before. Tail bleeding assessment was carried out on three normal mice and three TCP mice to confirm that the bleeding time and blood loss in these mice are statistically similar to the normal and TCP mice used for the positive and negative control groups in the nanoparticle treatment studies. After confirming this, additional six TCP mice (three male and three female) were administered retro-orbitally with syngeneic platelet dose at 250 platelets/nl. Thirty minutes after syngeneic platelet administration, the tail was clipped as before, and tail bleeding time and blood loss were measured. Because the positive (normal mice) and negative control (TCP mice) data for these studies were statistically similar to the normal and TCP mice in the nanoparticle treatment studies, the syngeneic platelet treatment group data were pooled and statistically compared with the nanoparticle treatment data.
In vivo evaluation of PPNs in a rat liver injury traumatic hemorrhage model
A rat liver injury model (Fig. 5A) was used with >30% liver resection that causes severe intraperitoneal hemorrhage. SAS Sprague-Dawley rats (~300 g) were acclimated to laboratory space for 48 hours. Rats were anesthetized with isoflurane and maintained on 100% oxygen with a non-rebreather mask, and ophthalmic ointment was applied. To monitor vitals, a rectal thermometer, pulse oximeter, and blood pressure cuff were placed. A tail vein catheter was placed to enable treatment (saline or control nanoparticle or PPN) administration. The abdomen of the rats was shaved, and subcutaneous buprenorphine and intramuscular injection of lidocaine at the incision site was administered. The peritoneal cavity was opened, and the liver was exposed. The cavity was packed with preweighed absorbent gauze, and the liver was cut sharply 1.5 cm from the superior vena cava, removing both median and lateral lobes. The abdomen was immediately closed. Rats were then administered with 0.5 ml of saline or 0.5 ml of control nanoparticles or PPNs at 2 mg/kg dose via the tail vein catheter at 0.25 ml/min. These studies were powered to blood loss at 60 min after treatment (n = 5 per treatment group) because, in traumatic injury, the first hour after injury is considered to be the most critical for controlling hemorrhage and improving survival (termed the “golden hour”). The vitals were monitored every 1 min for the first 30 min and every 5 min for the next 150 min. One hour after injury, the gauze was removed from the abdomen and weighed to determine blood loss (assuming blood density of 1 g/ml), the abdomen was sutured close, and the wound area was injected with lidocaine. Three hours after treatment, rats were euthanized with an overdose of pentobarbital. After euthanasia, clearance organs (heart, lungs, liver, spleen, and kidney) were harvested for histology and immunostaining. For this, organs were fixed in formalin and processed, and slides were made for Carstairs’ and immunostaining. Sections of the liver injury site were deparaffinized and rehydrated by washing with xylene and then ethanol. Antigen retrieval was performed by tris-EDTA at 60°C overnight. Slides were washed, blocked in 10% serum with 1% bovine serum albumin in tris-buffered saline for 2 hours, incubated with antifibrin(ogen) antibody, followed by incubation with the Alexa Fluor 488–labeled secondary antibody, washed, protected with coverslips, and imaged using Olympus FV1000 fluorescence confocal microscope. SP or PPN (red RhB) and fibrin(ogen) (green fluorescein isothiocyanate) fluorescence were imaged for the same field of view.
In vivo evaluation of PPNs in a mouse traumatic hemorrhage model
Eight- to 12-week-old male C57 BL/6 mice were anesthetized with 2% isoflurane, injected with either PPN or control nanoparticles (2 mg/kg) via femoral vein and allowed to recover for 30 min. After 30 min, 25% of circulating blood volume was removed by blind cardiac puncture followed by midline laparotomy, resection of a standardized piece of the left liver lobe, and skin closure. Mice were placed in their cages with ample food and water and observed for 3 days. Mortality, if occurred, was noted. All procedures were performed using sterile technique.
Statistical analysis
For analysis of annexin V fluorescence and FVa/FXa antibody fluorescence on immobilized control nanoparticles versus PPNs, a two-tailed t test was used. For analysis of clot fluorescence in the microfluidic setup under fibrinolytic conditions, a two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used. All other in vitro data were analyzed using a one-way ANOVA with Tukey’s multiple comparisons test. Blood loss was assessed with a log-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test. For blood loss analysis in the thrombocytopenic mouse bleeding model, we considered 50% reduction in bleeding time (seconds) between the saline (control) group versus the PPN-treated group with 0.9 (90%) power and 95% confidence; the power analysis estimated that six animals per group were needed. For blood loss analysis in the rat liver injury traumatic hemorrhage model, we considered 33% reduction in blood loss (volume) between the saline group versus the PPN-treated group with 0.9 (90%) power and 95% confidence; the power analysis estimated that five animals per group were needed. Studies were powered to measure blood loss because to study survival with 90% power and 95% confidence would have required 21 animals per group. P < 0.05 was considered statistically significant.
Supplementary Material
Funding:
This work was supported by NIH grants R01 HL121212 to A.S.G.; R01 HL098217 to M.N.; R15 HL145573 to W.L.; R35 GM119526 to M.D.N.; F32 HL149207 to J.A.; VA grant A2611-R, A-3077, and IK6RX003077 to J.C.; and VA grant I01RX003420-01A1 to A.S. Y.K. was supported by a NIH NIBIB training grant T32EB004314 (principal investigator: J.C.) in the Department of Biomedical Engineering at Case Western Reserve University. The University of Pittsburgh holds a Physician-Scientist Institutional Award from the Burroughs Welcome Fund 1018885 that supported J.A. The work at Case Western Reserve University made use of biomedical engineering research facilities funded by National Center for Research Resources Grant Number C06 RR12463-01 (PI: K. Kutina).
Footnotes
Competing interests: A.S.G. is a cofounder of Haima Therapeutics LLC, which is focused on development of platelet-inspired hemostat technologies. A.S.G. and M.D.N. serve on the scientific advisory board of Haima. A.S.G. is coinventor on U.S. patent 9107845 entitled “Synthetic platelets” that is licensed to Haima. U.D.S.S. and A.S.G. are coinventors on a provisional patent application no. 63/147411 entitled “Bioresponsive nanoparticles for targeted amplification of coagulation and apoptosis signals,” filed by Case Western Reserve University. The other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. PPNs can be made available through a materials transfer agreement with Case Western Reserve University.
REFERENCES AND NOTES
- 1.Hoffman M, Monroe DM III, A cell-based model of hemostasis. Thromb. Haemost 85, 958–965 (2001). [PubMed] [Google Scholar]
- 2.Versteeg HH, Heemskerk JWM, Levi M, Reitsma PH, New fundamentals in hemostasis. Physiol. Rev 93, 327–358 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Etchill EW, Myers SP, Raval JS, Hassoune A, Sen Gupta A, Neal MD, Platelet transfusion in critical care and surgery: Evidence-based review of contemporary practice and future directions. Shock 47, 537–549 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Mhaskar R, Grossman BJ, Kaufman RM, Tobian AA, Kleinman S, Gernsheimer T, Tinmouth AT, Djubegovic B; AABB Platelet Transfusion Guidelines Panel, Platelet transfusion: A systematic review of the clinical evidence. Transfusion 55, 1116–1127 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Cohen MJ, Kutcher M, Redick B, Nelson M, Call M, Knudson MM, Schreiber MA, Bulger EM, Muskat P, Alarcon LH, Myers JG, Rahbar MH, Brasel KJ, Phelan HA, del Junco DJ, Fox EE, Wade CE, Holcomb JB, Cotton BA, Matijevic N; PROMMTT Study Group, Clinical and mechanistic drivers of acute traumatic coagulopathy. J. Trauma Acute Care Surg 75, S40–S47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, del Junco DJ, Brasel KJ, Bulger EM, Callcut RA, Cohen MJ, Cotton BA, Fabian TC, Inaba K, Kerby JD, Muskat P, O’Keeffe T, Rizoli S, Robinson BR, Scalea TM, Schreiber MA, Stein DM, Weinberg JA, Callum JL, Hess JR, Matijevic N, Miller CN, Pittet JF, Hoyt DB, Pearson GD, Leroux B, van Belle G; PROPPR Study Group, Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 313, 471–482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardenas JC, Zhang X, Fox EE, Cotton BA, Hess JR, Schreiber MA, Wade CE, Holcomb JB; PROPPR Study Group, Platelet transfusions improve hemostasis and survival in a substudy of the prospective, randomized PROPPR trial. Blood Adv. 2, 1696–1704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinella PC, Dunne J, Beilman GJ, O’Connell RJ, Borgman MA, Cap AP, Rentas F, Constant challenges and evolution of US military transfusion medicine and blood operations in combat. Transfusion 52, 1146–1153 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Lambert MP, Sullivan SK, Fuentes R, French DL, Poncz M, Challenges and promises for the development of donor-independent platelet transfusions. Blood 121, 3319–3324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humbrecht C, Kientz D, Gachet C, Platelet transfusion: Current challenges. Transfus. Clin. Biol 25, 151–164 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Magron A, Laugier J, Provost P, Boilard E, Pathogen reduction technologies: The pros and cons for platelet transfusion. Platelets 29, 2–8 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Reddoch-Cardenas KM, Bynum JA, Meledeo MA, Nair PM, Wu X, Darlington DN, Ramasubramanian AK, Cap AP, Cold-stored platelets: A product with function optimized for hemorrhage control. Transfus. Apher. Sci 58, 16–22 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Cap AP, Platelet storage: A license to chill. Transfusion 56, 13–16 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Milford EM, Reade MC, Comprehensive review of platelet storage methods for use in the treatment of active hemorrhage. Transfusion 56, S140–S148 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Apelseth TO, Cap AP, Spinella PC, Hervig T, Strandenes G, Cold stored platelets in treatment of bleeding. ISBT Sci. Ser 12, 488–495 (2017). [Google Scholar]
- 16.Stubbs JR, Tran SA, Emery RL, Hammel SA, Haugen AL, Zielinski MD, Zietlow SP, Jenkins D, Cold platelets for trauma-associated bleeding: Regulatory approval, accreditation approval, and practice implementation- just the “tip of the iceberg”. Transfusion 57, 2836–2844 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Hegde S, Akbar H, Zheng Y, Cancelas JA, Towards increasing shelf life and haemostatic potency of stored platelet concentrates. Curr. Opin. Hematol 25, 500–508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancelas JA, Future of platelet formulations with improved clotting profile: A short review on human safety and efficacy data. Transfusion 59, 1467–1473 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Hoffmeister KM, Felbinger TW, Falet H, Denis CV, Bergmeier W, Mayadas TN, von Andrian UH, Wagner DD, Stossel TP, Hartwig JH, The clearance mechanism of chilled blood platelets. Cell 112, 87–97 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Josefsson EC, Hartwig JH, Hoffmeister KM, Platelet storage temperature – how low can we go? Transfus. Med. Hemother 34, 253–261 (2007). [Google Scholar]
- 21.Getz TM, Physiology of cold-stored platelets. Transfus. Apher. Sci 58, 12–15 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Roberts N, James S, Delaney M, Fitzmaurice C, The global need and availability of blood products: A modelling study. Lancet Haematol. 6, e606–e615 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Modery-Pawlowski CL, Tian LL, Pan V, McCrae KR, Mitragotri S, Sen Gupta A, Approaches to synthetic platelet analogs. Biomaterials 34, 526–541 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Sen Gupta A, Bio-inspired nanomedicine strategies for artificial blood components. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 9, e1464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Girish A, Sekhon U, Sen Gupta A, Bioinspired artificial platelets for transfusion applications in traumatic hemorrhage. Transfusion 60, 229–231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lentz BR, Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res 42, 423–438 (2003). [DOI] [PubMed] [Google Scholar]
- 27.van Meer G, Voelker DR, Feigenson GW, Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol 9, 112–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harper MT, Poole AW, Chloride channels are necessary for full platelet phosphatidylserine exposure and procoagulant activity. Cell Death Dis. 4, e969 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Witt SM, Verdoold R, Cosemans JMEM, Heemskirk JWM, Insights into platelet-based control of coagulation. Thromb. Res 133, S139–S148 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Obydennyy SI, Sveshnikova AN, Ataullakhanov FI, Panteleev MA, Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost 14, 1867–1881 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Monroe DM, Hoffman M, Roberts HR, Platelets and thrombin generation. Arterioscler. Thromb. Vasc. Biol 22, 1381–1389 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Shukla M, Sekhon UDS, Betapudi V, Li W, Hickman DA, Pawlowski CL, Dyer MR, Neal MD, McCrae KR, Sen Gupta A, In vitro characterization of SynthoPlate™ (synthetic platelet) technology and its in vivo evaluation in severely thrombocytopenic mice. J. Thromb. Haemost 15, 375–387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dyer MR, Hickman D, Luc N, Haldeman S, Loughran P, Pawlowski C, Sen Gupta A, Neal MD, Intravenous administration of synthetic platelets (SynthoPlate) in a mouse liver injury model of uncontrolled hemorrhage improves hemostasis. J. Trauma Acute Care Surg 84, 917–923 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hickman DA, Pawlowski CL, Shevitz A, Luc NF, Kim A, Girish A, Marks J, Ganjoo S, Huang S, Niedoba E, Sekhon UDS, Sun M, Dyer M, Neal MD, Kashyap VS, Sen Gupta A, Intravenous synthetic platelet (SynthoPlate) nanoconstructs reduce bleeding and improve ‘golden hour’ survival in a porcine model of traumatic arterial hemorrhage. Sci. Rep 8, 3118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Thin-film hydration followed by extrusion method for liposome preparation, in Liposomes. Methods in Molecular Biology, D’Souza G, Ed. (Humana Press, 2016), vol. 1522, pp. 17–22. [DOI] [PubMed] [Google Scholar]
- 36.Ravikumar M, Modery CL, Wong T, Sen Gupta A, Peptide-decorated liposomes promote arrest and aggregation of activated platelets under flow on vascular injury relevant protein surfaces in vitro. Biomacromolecules 13, 1495–1502 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA, Synthesis of positional-scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nat. Biotechnol 18, 187–193 (2000). [DOI] [PubMed] [Google Scholar]
- 38.van Engeland M, Nieland LJ, Ramaekers FC, Schuttle B, Reutelingsperger CP, Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 31, 1–9 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Beguin S, Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol. Haemost. Thromb 33, 4–15 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Szlam F, Levy JH, Tanaka KA, Tissue plasminogen activator and thrombin generation measurements using the Thrombinoscope. Blood Coagul. Fibrinolysis 17, 603–604 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Wolberg AS, Campbell RA, Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci 38, 15–23 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curnow JL, Morel-Kopp MC, Roddie C, Aboud M, Ward CM, Reduced fibrinolysis and increased fibrin generation can be detected in hypercoagulable patients using the overall hemostatic potential assay. J. Thromb. Haemost 5, 528–534 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Li W, Nieman M, Sen Gupta A, Ferric chloride-induced murine thrombosis models. J. Vis. Exp 115, 54479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H, Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood 96, 2520–2527 (2000). [PubMed] [Google Scholar]
- 45.Greene TK, Schiviz A, Hoellriegl W, Poncz M, Muchitsch EM; Animal Models Subcommittee of the Scientific and Standardization Committee of the ISTH, Towards a standardization of the murine tail bleeding model. J. Thromb. Haemost 8, 2820–2822 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Weber B, Lackner I, Haffner-Luntzer M, Palmer A, Pressmar J, Scharfetter-Kochanek K, Knoll B, Shrezenemeier H, Relja B, Kalbitz M, Modeling trauma in rats: Similarities to humans and potential pitfalls to consider. J. Transl. Med 17, 305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Girish A, Hickman DA, Banerjee A, Luc N, Ma Y, Miyazawa K, Sekhon UDS, Sun M, Huang S, Sen Gupta A, Trauma-targeted delivery of Tranexamic Acid improves hemostasis and survival in rat liver hemorrhage model. J. Thromb. Haemost 17, 1632–1644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strassel C, Gacjhet C, Lanza F, On the way to in vitro platelet production. Front. Med 5, 239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coller BS, Springer KT, Beer JH, Mohandas N, Scudder LE, Norton KJ, West SM, Thromboerythrocytes: In vitro studies of a potential autologous semi-artificial alternative to platelet transfusions. J. Clin. Invest 89, 546–555 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levi M, Friedrich PW, Middleton S, de Groot PG, Wu YP, Harris R, Biemond BJ, Hiejnen HFG, Levin J, Ten Cate JW, Fibrinogen-coated albumin microcapsules reduce bleeding in severely thrombocytopenic rabbits. Nat. Med 5, 107–111 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Yen RCK, Ho TWC, Blajchman MA, A new haemostatic agent: Thrombospheres shorten the bleeding time in thrombocytopenic rabbits. Thromb. Haemost 73, 986 (1995). [Google Scholar]
- 52.Bertram JP, Williams CA, Robinson R, Segal SS, Flynn NT, Lavik EB, Intravenous hemostat: Nanotechnology to halt bleeding. Sci. Trans. Med 1, 11–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gkikas M, Peponis T, Mear T, Hong C, Avery RK, Roussakis E, Yoo H-J, Parakh A, Patino M, Sahani DV, Watkins MT, Oklu R, Evans CL, Albadawi H, Velmahos G, Olsen BD, Systemically administered hemostatic nanoparticles for identification and treatment of internal bleeding. ACS Biomat. Sci. Eng 5, 2563–2576 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Okamura Y, Fujie T, Nogawa M, Maruyama H, Handa M, Ikeda Y, Takeoka S, Haemostatic effects of polymerized albumin particles carrying fibrinogen γ-chain dodecapeptide as platelet substitutes in severely thrombocytopenic rabbits. Transfus. Med 18, 158–166 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Chapin JC, Hajjar KA, Fibrinolysis and the control of blood coagulation. Blood Rev. 29, 17–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schöchl H, Voelckel W, Maegele M, Solomon C, Trauma-associated hyperfibrinolysis. Hamostaseologie 32, 22–27 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Taylor JR III, Fox EE, Holcomb JB, Rizoli S, Inaba K, Schreiber MA, Brasel K, Scalea TM, Wade CE, Bulger E, Cotton BA; PROPPR Study Group, The hyperfibrinolytic phenotype is the most lethal and resource intense presentation of fibrinolysis in massive transfusion patients. J. Trauma Acute Care Surg 84, 25–30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cardenas JC, Matijevic N, Baer LA, Holcomb JB, Cotton BA, Wade CE, Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock 41,514–521 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, Mitra S, Ghasabyan A, Chin TL, Sauaia A, Banerjee A, Silliman CC, Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg 80, 16–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu X, Darlington DN, Cap AP, Procoagulant and fibrinolytic activity after polytrauma in rat. Am. J. Physiol. Regul. Integr. Comp. Physiol 310, R323–R329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Agbani EO, Williams CM, Hers I, Poole AW, Membrane ballooning in aggregated platelets is synchronised and mediates a surge in microvesiculation. Sci. Rep 7, 2770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reddy EC, Rand ML, Procoagulant phosphatidylserine-exposing platelets in vitro and in vivo. Front. Cardiovasc. Med 7, 15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bynum JA, Meledeo MA, Peltier GC, McIntosh CS, Taylor AS, Montgomery RK, Reddoch-Kardenas KM, Getz TM, Fitzpatrick MJ, Cap AP, Evaluation of a lyophilized platelet-derived hemostatic product. Transfusion 59, 1490–1498 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Zhong Y, Pruner I, Antovic A, Taxiarchis A, Vila ZP, Soutari N, Mobarrez F, Chaireti R, Weidengren J, Piguet J, Antovic JP, Phosphatidylserine positive microparticles improve hemostasis in in-vitro hemophilia A plasma models. Sci. Rep 10, 7871 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Owattanapanitch N, Chittawatanarat K, Benyakorn T, Sirikun J, Risks and benefits of hypotensive resuscitation in patients with traumatic hemorrhagic shock: A meta-analysis. Scand. J. Trauma Resusc. Emerg. Med 26, 107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ, Characterization of platelet dysfunction after trauma. J. Trauma Acute Care Surg 73, 13–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramsey MT, Fabian TC, Shahan CP, Sharpe JP, Mabry SE, Weinberg JA, Croce MA, Jennings LK, A prospective study of platelet function in trauma patients. J. Trauma Acute Care Surg 80, 726–732 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Lee RH, Piatt R, Dhenge A, Lozano ML, Palma-Barqueros V, Rivera J, Bergmeier W, Impaired hemostatic activity of healthy transfused platelets in inherited and acquired platelet disorders: Mechanisms and implications. Sci. Transl. Med 11, eaay0203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hagisawa K, Kinoshita M, Takikawa M, Takeoka S, Saitoh D, Seki S, Sakai H, Combination therapy using fibrinogen γ-chain peptide-coated, ADP-encapsulated liposomes and hemoglobin vesicles for trauma-induced massive hemorrhage in thrombocytopenic rabbits. Transfusion 59, 3186–3196 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Shaffer L, News feature: Making and storing blood to save lives. Proc. Natl. Acad. Sci. U.S.A 117, 7542–7545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cap AP, Cannon JW, Reade MC, Synthetic blood and blood products for combat casualty care and beyond. J. Trauma Acute Care Surg 91, S26–S32 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC; 2009 Platelet Colloquium Participants, Platelet functions beyond hemostasis. J. Thromb. Haemost 7, 1759–1766 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Ware J, Corken A, Khetpal R, Platelet function beyond hemostasis and thrombosis. Curr. Opin. Hematol 20, 451–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kornblith LZ, Moore HB, Cohen MJ, Trauma induced coagulopathy: The past, present, and future. J. Thromb. Haemost 17, 852–862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Starr NE, Matthay ZA, Fields AT, Nunez-Garcia B, Callcut RA, Cohen MJ, Kornblith LZ, Identification of injury and shock driven effects on ex vivo platelet aggregometry: A cautionary tale of phenotyping. J. Trauma Acute Care Surg 89, 20–28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, Schöchl H, Hunt BJ, Sauaia A, Trauma-induced coagulopathy. Nat. Rev. Dis. Primers 7, 30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Banerjee A, Silliman CC, Moore EE, Dziesiatskowa M, Kelher M, Sauaia A, Jones K, Chapman MP, Gonzalez E, Moore HB, D’Alessandro A, Peltz E, Huebner BE, Einerson P, Chandler J, Ghasabayan A, Hansen K, Systemic hyperfibrinolysis after trauma: A pilot study of targeted proteomic analysis of superposed mechanisms in patient plasma. J. Trauma Acute Care Surg 84, 929–938 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Torchilin VP, Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Disc 4, 145–160 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Crommelin DJA, van Hoogevest P, Storm G, The role of liposomes in clinical nanomedicine development. What now? Now what? J. Control. Rel 318, 256–263 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Van Regenmortel MH, Antigenicity and immunogenicity of synthetic peptides. Biologicals 29, 209–213 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.