

Abstract
Background
Many people with cancer experience moderate to severe pain that requires treatment with strong opioids, such as oxycodone and morphine. Strong opioids are, however, not effective for pain in all people, neither are they well tolerated by all people. The aim of this review was to assess whether oxycodone is associated with better pain relief and tolerability than other analgesic options for adults with cancer pain. This is an updated Cochrane review previously published in 2017.
Objectives
To assess the effectiveness and tolerability of oxycodone by any route of administration for pain in adults with cancer.
Search methods
For this update, we searched the Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library, MEDLINE and MEDLINE In‐Process (Ovid), Embase (Ovid), Science Citation Index, Conference Proceedings Citation Index ‐ Science (ISI Web of Science), BIOSIS (ISI), and PsycINFO (Ovid) to November 2021. We also searched four trial registries, checked the bibliographic references of relevant studies, and contacted the authors of the included studies. We applied no language, date, or publication status restrictions.
Selection criteria
We included randomised controlled trials (parallel‐group or cross‐over) comparing oxycodone (any formulation or route of administration) with placebo or an active drug (including oxycodone) for cancer background pain in adults by examining pain intensity/relief, adverse events, quality of life, and participant preference.
Data collection and analysis
Two review authors independently sifted the search, extracted data and assessed the included studies using standard Cochrane methodology. We meta‐analysed pain intensity data using the generic inverse variance method, and pain relief and adverse events using the Mantel‐Haenszel method, or summarised these data narratively along with the quality of life and participant preference data. We assessed the overall certainty of the evidence using GRADE.
Main results
For this update, we identified 19 new studies (1836 participants) for inclusion. In total, we included 42 studies which enrolled/randomised 4485 participants, with 3945 of these analysed for efficacy and 4176 for safety. The studies examined a number of different drug comparisons.
Controlled‐release (CR; typically taken every 12 hours) oxycodone versus immediate‐release (IR; taken every 4‐6 hours) oxycodone
Pooled analysis of three of the four studies comparing CR oxycodone to IR oxycodone suggest that there is little to no difference between CR and IR oxycodone in pain intensity (standardised mean difference (SMD) 0.12, 95% confidence interval (CI) ‐0.1 to 0.34; n = 319; very low‐certainty evidence). The evidence is very uncertain about the effect on adverse events, including constipation (RR 0.71, 95% CI 0.45 to 1.13), drowsiness/somnolence (RR 1.03, 95% CI 0.69 to 1.54), nausea (RR 0.85, 95% CI 0.56 to 1.28), and vomiting (RR 0.66, 95% CI 0.38 to 1.15) (very low‐certainty evidence). There were no data available for quality of life or participant preference, however, three studies suggested that treatment acceptability may be similar between groups (low‐certainty evidence).
CR oxycodone versus CR morphine
The majority of the 24 studies comparing CR oxycodone to CR morphine reported either pain intensity (continuous variable), pain relief (dichotomous variable), or both. Pooled analysis indicated that pain intensity may be lower (better) after treatment with CR morphine than CR oxycodone (SMD 0.14, 95% CI 0.01 to 0.27; n = 882 in 7 studies; low‐certainty evidence). This SMD is equivalent to a difference of 0.27 points on the Brief Pain Inventory scale (0‐10 numerical rating scale), which is not clinically significant. Pooled analyses also suggested that there may be little to no difference in the proportion of participants achieving complete or significant pain relief (RR 1.02, 95% CI 0.95 to 1.10; n = 1249 in 13 studies; low‐certainty evidence).
The RR for constipation (RR 0.75, 95% CI 0.66 to 0.86) may be lower after treatment with CR oxycodone than after CR morphine. Pooled analyses showed that, for most of the adverse events, the CIs were wide, including no effect as well as potential benefit and harm: drowsiness/somnolence (RR 0.88, 95% CI 0.74 to 1.05), nausea (RR 0.93, 95% CI 0.77 to 1.12), and vomiting (RR 0.81, 95% CI 0.63 to 1.04) (low or very low‐certainty evidence). No data were available for quality of life. The evidence is very uncertain about the treatment effects on treatment acceptability and participant preference.
Other comparisons
The remaining studies either compared oxycodone in various formulations or compared oxycodone to different alternative opioids. None found any clear superiority or inferiority of oxycodone for cancer pain, neither as an analgesic agent nor in terms of adverse event rates and treatment acceptability. The certainty of this evidence base was limited by the high or unclear risk of bias of the studies and by imprecision due to low or very low event rates or participant numbers for many outcomes.
Authors' conclusions
The conclusions have not changed since the previous version of this review (in 2017). We found low‐certainty evidence that there may be little to no difference in pain intensity, pain relief and adverse events between oxycodone and other strong opioids including morphine, commonly considered the gold standard strong opioid. Although we identified a benefit for pain relief in favour of CR morphine over CR oxycodone, this was not clinically significant and did not persist following sensitivity analysis and so we do not consider this important. However, we found that constipation and hallucinations occurred less often with CR oxycodone than with CR morphine; but the certainty of this evidence was either very low or the finding did not persist following sensitivity analysis, so these findings should be treated with utmost caution. Our conclusions are consistent with other reviews and suggest that, while the reliability of the evidence base is low, given the absence of important differences within this analysis, it seems unlikely that larger head‐to‐head studies of oxycodone versus morphine are justified, although well‐designed trials comparing oxycodone to other strong analgesics may well be useful. For clinical purposes, oxycodone or morphine can be used as first‐line oral opioids for relief of cancer pain in adults.
Keywords: Adult; Humans; Analgesics, Opioid; Analgesics, Opioid/adverse effects; Cancer Pain; Cancer Pain/drug therapy; Constipation; Constipation/chemically induced; Morphine; Morphine/adverse effects; Nausea; Nausea/chemically induced; Nausea/drug therapy; Neoplasms; Neoplasms/complications; Neoplasms/drug therapy; Oxycodone; Oxycodone/adverse effects; Pain; Pain/drug therapy; Pain/etiology; Quality of Life; Reproducibility of Results; Sleepiness; Vomiting; Vomiting/chemically induced
Plain language summary
Oxycodone for cancer‐related pain in adults
Background
Many people with cancer experience moderate to severe pain that requires treatment with strong painkillers that are classified as opioids.
Oxycodone and morphine are examples of these opioids that are used for the relief of cancer pain. However, strong painkillers are not effective for pain in all people, neither are they well tolerated by all people. The aim of this review was to assess whether oxycodone is associated with better pain relief and fewer side effects than other strong painkillers for adults with cancer pain.
Study characteristics
For this update, in November 2021, we found 19 additional relevant studies. In total, we included 42 studies with 4485 participants. These studies compared the painkilling ability (benefit) and side effects (harms) of different types of oxycodone to each other or to other strong painkillers.
Key results
In general, the studies showed no difference between oxycodone taken every 4‐6 (immediate‐release) or every 12 (controlled‐release) hours. In general, the studies also showed no difference between oxycodone and other strong pain killers such as morphine.
All the strong painkillers examined in the studies also have a number of unwanted effects, such as vomiting, constipation, and drowsiness. Overall, these do not differ between oxycodone and the other strong painkillers. Hallucinations (where people experience imaginary things, e.g. hearing voices) are much less common as a side effect of strong painkillers, and we found that they were less likely with oxycodone than with morphine.
Overall, we found that the current evidence is comprised of studies that contained small numbers of people, of which many (12.2%) did not finish the studies. However, since there was very little difference between oxycodone and morphine, more research in this area is unlikely to be undertaken. This is partly because recruitment and retention of participants in this context is challenging. Studies looking at oxycodone compared to other strong painkillers may be useful.
Certainty of the evidence
We rated the certainty of the evidence from studies using four levels: very low, low, moderate, or high. Very low‐certainty evidence means that we are very uncertain about the results. High‐certainty evidence means that we are very confident in the results. Overall, the certainty of the evidence in this review was rated low or very low, meaning that we are not sure about the results because of problems with study quality and small size.
Summary of findings
Summary of findings 1. CR oxycodone compared with IR oxycodone for cancer‐related pain in adults.
| CR oxycodone compared with IR oxycodone for cancer‐related pain in adults | ||||||
|
Patient or population: adults with cancer‐related pain Settings: in‐ or outpatients Intervention: CR oxycodone Comparison: IR oxycodone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| IR oxycodone | CR oxycodone | |||||
| Pain intensity (mean across treatment or at end of treatment; length of treatment varied across trials; various pain intensity scales; SMD) |
The mean pain intensity in the CR oxycodone group was 0.12 standard deviations higher (0.1 lower to 0.34 higher) than in the IR oxycodone group | SMD 0.12 (‐0.1 to 0.24) | 319 (3 studies) | ⊕⊕⊝⊝ very lowa,b | ‐ | |
| Constipation (Event rate during treatment, length of treatment varied across trials) |
224 per 1000 | 159 per 1000 (101 to 253) | RR 0.71 (0.45 to 1.13) | 317 (3 studies) | ⊕⊝⊝⊝ very lowc,d | ‐ |
| Drowsiness/somnolence (Event rate during treatment, length of treatment varied across trials) |
224 per 1000 | 230 per 1000 (154 to 344) | RR 1.03 (0.69 to 1.54) | 317 (3 studies) | ⊕⊝⊝⊝ very lowc,d | ‐ |
| Nausea (Event rate during treatment, length of treatment varied across trials) |
242 per 1000 | 206 per 1000 (136 to 310) | RR 0.85 (0.56 to 1.28) | 317 (3 studies) | ⊕⊝⊝⊝ very lowc,d | ‐ |
| Vomiting (Event rate during treatment, length of treatment varied across trials) |
174 per 1000 | 115 per 1000 (66 to 200) | RR 0.66 (0.38 to 1.15) | 317 (3 studies) | ⊕⊝⊝⊝ very lowc,d | ‐ |
| Quality of life | No data available, but there appeared to be no difference in treatment acceptability between the treatment groups (measured on various scales, not pooled); 578 participants (3 studies); quality of the evidence low.a | |||||
| Participant preference | No data available | ‐ | ‐ | ‐ | ‐ | |
| *The assumed risk is reported as the observed risk in the control group across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CR: controlled‐release; IR: immediate‐release; RR: risk ratio; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a Downgraded twice for very serious limitations to study quality due to risk of bias (attrition bias and under‐reporting of the domain of selection bias)
b Downgraded once for imprecision due to low event rates c Downgraded twice for imprecision due to very low event rates d Downgraded twice for very serious limitations to study quality due to risk of bias (performance/detection bias, and inadequate titration and under‐reporting of the domains of selection, performance, detection and attrition bias, and whether the participants were adequately titrated)
Summary of findings 2. CR oxycodone compared with CR morphine for cancer‐related pain in adults.
| CR oxycodone compared with CR morphine for cancer‐related pain in adults | ||||||
|
Patient or population: adults with cancer‐related pain Settings: in‐ or outpatients Intervention: CR oxycodone Comparison: CR morphine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| CR morphine | CR oxycodone | |||||
| Pain intensity (various pain intensity scales; SMD) |
The mean pain intensity in the CR oxycodone group was 0.14 standard deviations higher (0.01 lower to 0.27 higher) than in the CR morphine group | SMD 0.14 (0.01 to 0.27) |
882 (7 studies) | ⊕⊕⊝⊝ lowa | This estimate did not include the data from the Chinese language studies (n = 2) as sensitivity analyses indicated they were not comparable. Converting the SMD as a difference in Brief Pain Inventory scores (0 to 10 numerical rating scale from no pain to worst pain imaginable) between the treatments gave an estimated difference of 0.27 between the treatments, which was not clinically significant. |
|
| Constipation ‐ all available data (Event rate during treatment, length of treatment varied across trials) |
322 per 1000 | 241 per 1000 (212 to 277) | RR 0.75 (0.66 to 0.86) | 1894 (18 studies) | ⊕⊕⊝⊝ lowb | This estimate did include the data from the Chinese language studies. |
| Constipation ‐ only English language studies (Event rate during treatment, length of treatment varied across trials) |
355 per 1000 | 348 per 1000 (291 to 412) | RR 0.98 (0.82 to 1.16) | 797 (5 studies) | ⊕⊝⊝⊝ very lowb,c | This estimate did not include the data from the Chinese language studies (n = 13) as sensitivity analyses indicated they were not comparable. |
| Drowsiness/somnolence (Event rate during treatment, length of treatment varied across trials) |
228 per 1000 | 201 per 1000 (169 to 239) | RR 0.88 (0.74 to 1.05) | 1486 (15 studies) | ⊕⊕⊝⊝ lowb | This estimate did include the data from the Chinese language studies as sensitivity analyses indicated they were comparable. |
| Nausea (Event rate during treatment, length of treatment varied across trials) |
231 per 1000 | 215 per 1000 (178 to 259) | RR 0.93 (0.77 to 1.12) | 1388 (13 studies) | ⊕⊕⊝⊝ lowb | This estimate did include the data from the Chinese language studies as sensitivity analyses indicated they were comparable. |
| Vomiting (Event rate during treatment, length of treatment varied across trials) |
157 per 1000 | 127 per 1000 (99 to 163) | RR 0.81 (0.63 to 1.04) | 1388 (13 studies) | ⊕⊝⊝⊝ very lowb,c | This estimate did include the data from the Chinese language studies as sensitivity analyses indicated they were comparable |
| Quality of life | No data available, but CR oxycodone appeared to be associated with similar or lower treatment acceptability than CR morphine (measured on various scales, not pooled); 149 participants (3 studies); quality of the evidence verylowd,e | ‐ | ||||
| Participant preference (end of treatment in a cross‐over trial with each phase lasting seven days) | 8/23 participants preferred CR oxycodone while 11/23 participants preferred treatment with CR morphine. | ‐ | 23 (1 study) |
⊕⊝⊝⊝ very lowd,e | ‐ | |
| *The assumed risk is reported as the observed risk in the control group across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CR: controlled‐release; RR: risk ratio; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a Downgraded twice for very serious limitations to study quality due to risk of bias (performance/detection bias and attrition bias and under‐reporting of the domain of selection bias) b Downgraded twice for very serious limitations to study quality due to risk of bias (performance/detection bias and/or attrition bias and under‐reporting) c Downgraded once for imprecision due to low event rates/participant numbers d Downgraded twice for imprecision due to very low event rates/participant numbers e Downgraded twice for very serious limitations to study quality due to risk of bias (attrition bias and under‐reporting of the domain of selection bias)
Background
This review is an update of a previously published review in the Cochrane Database of Systematic Reviews, 2017, Issue 8 on oxycodone for cancer‐related pain.
Description of the condition
Pain from cancer can be caused by direct invasion of a tumour into soft tissue or bone and is often a presenting symptom at the time of diagnosis of cancer. One European survey published in 2009 found that, of 5000 people with cancer (including 617 community‐based National Health Service (NHS) patients in the UK), 72% experienced pain (77% of UK patients) which was of moderate to severe intensity in 90% of this group (Breivik 2009). This is consistent with a systematic review that demonstrated cancer pain prevalence of up to 75% in advanced disease, and that at least 30% of people with cancer are undertreated (Greco 2014). Recent research has also shown that less than half of all people with cancer that die are prescribed a strong opioid, and that median treatment duration is only nine weeks before death (Ziegler 2016). Pain in people with cancer may also be caused by cancer treatments and by comorbid conditions. In this review, we define cancer pain as pain arising as a direct consequence of the cancer, and not from other aetiologies.
Description of the intervention
Oxycodone is a strong opioid analgesic indicated for the treatment of moderate to severe chronic pain, including cancer pain. It is available orally as immediate‐release (IR) solution and tablets (for four‐hourly dosing) and as sustained (controlled (CR)) release tablets (for 12‐hourly dosing). It is also available as a parenteral injection. In some countries, oxycodone is available as a compound with paracetamol (acetaminophen) or ibuprofen.
How the intervention might work
Oxycodone works primarily as an agonist of mu‐opioid receptors in the spinal cord and brain. It has some activity at kappa‐opioid receptors (which are also involved in nociception or analgesia) though the importance of this mechanism in the overall analgesic effect of oxycodone is unclear. Despite animal studies suggesting differences in pharmacodynamics, these have not been demonstrated in clinical studies to date. Therefore, the shared mechanism of action to other strong opioids (i.e. agonist activity at mu‐opioid receptors) means that clinical benefits and adverse effects are likely to be similar. However, important differences exist in the pharmacokinetics of strong opioids (e.g. morphine undergoes second‐phase elimination via glucuronidation, while oxycodone undergoes extensive first‐phase metabolism via CYP2D6 and CYP3A4 pathways) so clinical equivalence cannot be inferred (Gudin 2012; Leppert 2010).
Why it is important to do this review
The World Health Organization (WHO) published the Method for Cancer Pain Relief (WHO analgesic ladder) in 1986 (WHO 1986), which advocates a stepwise approach to analgesia for cancer pain and revolutionised the use of oral opioids. It recommended that morphine be used first‐line for moderate to severe cancer pain. Over 30 years on, WHO guidelines continue to support opioids for moderate to severe cancer pain management (WHO 2018). Observational studies have suggested that this approach results in pain control for 73% of people (Bennett 2008) with a mean reduction in pain intensity of 65% (Ventafridda 1987).
Many people with cancer experience moderate to severe pain that requires treatment with strong analgesics. Oxycodone and morphine are examples of strong opioids that are used for the relief of cancer pain. However, strong opioids are not effective for pain in all people, neither are they well tolerated by all people. Guidance by the European Association for Palliative Care on the use of opioids in cancer pain suggests that oxycodone could be used as first‐line treatment of moderate to severe cancer pain as an alternative to morphine (Caraceni 2012). The aim of this review is to assess whether oxycodone is associated with better pain relief and tolerability than other analgesic options for people with cancer pain. The protocol for this review was updated from Reid 2010.
Objectives
To assess the effectiveness and tolerability of oxycodone by any route of administration for pain in adults with cancer.
Methods
Criteria for considering studies for this review
Types of studies
Randomised trials are the best design to minimise bias when evaluating the effectiveness of an intervention. We included randomised controlled trials (RCTs), with parallel‐group or cross‐over design, comparing oxycodone (any formulation and any route of administration) with placebo or an active drug (including oxycodone) for cancer background pain. We did not examine studies on breakthrough pain.
Types of participants
Adults (aged ≥ 18 years) with cancer pain. We did not restrict inclusion by cancer type or body region affected, but rather included adults with pain from any type of cancer in any body region.
Types of interventions
Oxycodone (any dose/frequency, formulation, and route of administration) versus oxycodone (any other dose/frequency, formulation, and route of administration)
Oxycodone (any dose/frequency, formulation, and route of administration) versus other active drug (any dose/frequency, formulation, and route of administration)
Oxycodone (any dose/frequency, formulation, and route of administration) versus placebo
Types of outcome measures
Primary outcomes
Pain was the primary outcome. This is often reported as pain intensity (typically reported as a continuous measure, measured on a visual assessment scale (VAS) or numerical rating scale (NRS)) or pain relief (typically reported as a categorical measure), which we have treated as measures of essentially the same thing, albeit from opposite starting points (i.e. effectiveness is demonstrated by pain intensity going down and by pain relief going up). The majority of the included studies reported pain intensity, with much fewer studies reporting pain relief apart from the newly added Chinese‐language studies (see also Subgroup analysis and investigation of heterogeneity; Sensitivity analysis; Excluded studies). We therefore meta‐analysed pain intensity and treated this as our primary outcome measure (see also Measures of treatment effect; Data synthesis), but we also meta‐analysed pain relief, where possible.
Both these outcomes had to be participant‐reported and could be reported in any transparent manner (e.g. by using numerical or verbal rating scales). We did not consider these outcomes when reported by physicians, nurses, or carers. If possible, we aimed to distinguish between nociceptive and neuropathic pain, but the data were not presented in a manner that made this possible.
Secondary outcomes
Adverse events (e.g. constipation, nausea, vomiting, drowsiness, confusion, respiratory depression).
Quality of life (or treatment acceptability as a proxy).
Participant preference.
We considered all these outcomes as they were reported in the included studies.
Search methods for identification of studies
We applied no language, date, or publication status (published in full, published as abstract, unpublished) restrictions to the search.
Electronic searches
For this update, we identified relevant trials by searching the following databases:
Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library (Issue 11 of 12, 2021);
MEDLINE and MEDLINE In‐Process (Ovid) (Nov 2016 to 24 November 2021);
Embase (Ovid) (Nov 2016 to 2021 4 November);
Science Citation Index (Web of Science) (2016 to 29 November 2021);
Conference Proceedings Citation Index ‐ Science (Web of Science) (2016 to 29 November 2021);
BIOSIS (Web of Science) (2016 to 30 November 2021);
PsycINFO (EBSCO) (2016 to November 2021).
We applied the Cochrane highly sensitive search strategy for identifying RCTs to this search (Lefebvre 2021). When these searches were run for the original review in March 2014, PubMed was also searched. We did not search PubMed for the subsequent updates as it did not yield any records that were not found by the other databases in the original review. The search strategies used can be found in Appendix 1.
Searching other resources
For this update, we ran searches on 30 November 2021, on Clinicaltrials.gov, EU Clinical Trials Register, WHO International Clinical Trials Registry Platform (ICTRP), and UK Clinical Trials Gateway (UKCTG), but not metaRegister of Controlled Trials (mRCT) as this is no longer available, but rather signposted to EU Clinical Trials Register and UK Clinical Trials Gateway (UKCTG). For both the previous review and the updates, we checked the bibliographic references of relevant identified studies to find additional trials not identified by the electronic searches and contacted authors of the included studies to ask if they knew of any other relevant studies.
Data collection and analysis
Selection of studies
Two of three review authors (MSH, NB, AJP) assessed the titles and abstracts of all the studies identified by the search for potential inclusion. We independently considered the full records of all potentially relevant studies for inclusion by applying the selection criteria outlined in the Criteria for considering studies for this review section. We resolved any disagreements by discussion. We did not restrict the inclusion criteria by date, language, or publication status (published in full, published as abstract, unpublished).
Data extraction and management
Using a standardised, piloted data extraction form, two review authors (MSH, JSH) extracted data pertaining to study design, participant details (including age, cancer characteristics, previous analgesic medication, and setting), interventions (including details about titration), and outcomes. We resolved any disagreements by discussion. If there were studies for which only a subgroup of the participants met the inclusion criteria for the current review, we would have only extracted data on this subgroup provided randomisation had not been broken; however, no such studies were identified for inclusion.
Assessment of risk of bias in included studies
Two review authors (MSH and JSH or NB or YC) independently assessed the methodological quality of each of the included studies by using the risk of bias assessment method outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). For each study, we assessed the risk of bias for the following domains: selection bias (study level; two items; random sequence generation and allocation concealment), performance bias (outcome level; two items; blinding of participants and blinding of treating personnel), detection bias (outcome level; one item; blinding of outcome assessment), attrition bias (outcome level; one item; incomplete outcome data), and reporting bias (study level; one item; selective reporting). For the question on allocation concealment, in the absence of explicit explanation about allocation concealment, we accepted 'central randomisation' as sufficient indication of adequate allocation concealment and consequently rated such studies as having low risk of bias for this item. We also included an item that assessed the adequacy of titration (with judgements made based on any available relevant information, including design features, inclusion criteria, and interim pain assessments) and another item that captured whether data were available for both time periods in cross‐over trials; we also listed under 'Other bias' any other biases identified, including carry‐over effects for cross‐over trials. Each of the risk of bias items required a 'low risk,' 'high risk,' or 'unclear risk' response. We also documented the reasons for each response in accordance with Higgins 2011, and resolved any disagreements on the risk of bias ratings through discussion. For the item assessing whether data were available for both time periods in cross‐over trials, we inputted 'unclear' and 'not applicable' as the rating and reason for parallel‐group trials. Finally, we also extracted and reported whether a study was free from commercial funding.
Measures of treatment effect
For pain intensity, we extracted the means and standard deviations (SDs) and we used these to estimate the standardised mean difference (SMD) between the treatments along with the 95% confidence interval (CI), as the outcome was not measured on the same scale across studies. For this update, the majority of the 18 new studies did not report pain intensity as a continuous measure, but rather pain relief as a categorical measure. In order to be able to meta‐analyse the pain data from these studies, we therefore extracted the event rates in each of these pain relief categories also to calculate and analyse them as risk ratios (RRs) with 95% CIs. For adverse events, we extracted event rates to calculate RR with 95% CIs as the summary estimates (see also Data synthesis).
Unit of analysis issues
The participant was the unit of analysis but, in a number of cases, the pain intensity data reported as a continuous measure in the included cross‐over trials could not otherwise be incorporated into the analyses (see Dealing with missing data), so we included them as if the design had been parallel‐group. Higgins 2011 (in Chapter 16) pointed out that this approach, while giving rise to unit of analysis errors, is nevertheless conservative as it results in an underweighting of the data. However, in order to assess the impact of this strategy, we also performed sensitivity analyses when we included cross‐over trial data in this manner by excluding the cross‐over trials from the meta‐analyses. We did not include dichotomous pain relief or adverse event data from cross‐over trials in this manner, but rather reported them per study. Where a study with more than two intervention arms was included, only data from intervention and control groups that met the eligibility criteria were extracted. We included studies with more than two eligible intervention arms in separate comparisons to avoid the double‐counting of participants.
Dealing with missing data
In cases where data were missing, we contacted the authors to request the missing data. This strategy did not result in any additional data. We limited imputation of missing data to the imputation of missing SDs, either by calculating the SD if enough information was available or by using SDs from similar samples or studies, both according to the methods outlined by Higgins 2011. We only imputed SDs for pain intensity for Lux 2014, which were not reported for the subgroup of participants with malignant pain, by using the reported SDs for the whole sample of participants with either malignant (n = 31) or non‐malignant pain (n = 15), and for Yu 2014, for the primary outcome of the study "mean pain at its worst in the past 24 hours", by using the standard deviations for the same outcome measured at baseline in the full analysis set. We recorded the dropout/missing data rates in the risk of bias tables under the items on attrition bias and in the 'Participants' section of the Characteristics of included studies table, and we addressed the potential effect of the missing data on the results in the Discussion. It was not possible to assess the impact of missing data in sensitivity analyses due to the low number of included studies within each comparison that were not subject to attrition bias. In all cases, we aimed to perform intention‐to‐treat (ITT) analyses.
Assessment of heterogeneity
We quantified heterogeneity by using the I2 statistic. We considered I2 values above 50% to represent substantial heterogeneity in line with Higgins 2011, and we planned to assess potential sources of heterogeneity through subgroup analyses as outlined in Subgroup analysis and investigation of heterogeneity. See also Data synthesis.
Assessment of reporting biases
In addition to implementing the comprehensive search strategy outlined in the Search methods for identification of studies section, the risk of outcome reporting bias was illustrated in the risk of bias summary figures that we constructed for each study and each type of assessed bias.
Data synthesis
We entered the data extracted from the included studies into Review Manager 5 (RevMan 2014), which was used for data synthesis. We analysed pain intensity using the generic inverse variance method in accordance with Higgins 2011. However, given the limitations of this analysis strategy as outlined in the Unit of analysis issues section, we also considered the results of the individual studies. We meta‐analysed the pain relief and adverse events data by using the Mantel‐Haenszel method; however, as this method is not suitable for cross‐over trial data, we only included the data from parallel‐group trials in these analyses. In addition, we have also presented all reported adverse events from the included studies in tables. As we have assumed that there is a single common intervention effect which we are aiming to estimate, we used a fixed‐effect model in all analyses. However, we did not pool the data if the I2 statistic was above 50%, although we note that in some instances the pooled estimate of analyses where the I2 statistic exceeds 50% will appear on the presented figures or analyses due to the way we have presented the results (in comparison‐based subgroups for each outcome) as Review Manager does not allow for the selective presentation of pooled estimates within only a subset of subgroups. In such cases, we have clearly stated in the text that the pooled estimates should be disregarded. We reported the pain intensity and pain relief data for the included studies that could not be meta‐analysed narratively, along with any other outcome data that could not be meta‐analysed (such as quality of life data).
Subgroup analysis and investigation of heterogeneity
Different aspects of the trials are likely to contribute heterogeneity to the proposed main analyses. If there were sufficient data, we therefore planned to perform subgroup analyses based on doses, titration, formulations (e.g. IR, sustained‐release), routes of administration (e.g. oral, rectal), length of the trials, and populations (e.g. opioid‐naive participants). We grouped the studies by formulation and route of administration, but as there were insufficient data, we were unable to perform any further subgroup analyses. As outlined and discussed in detail in the Excluded studies section, for this update, we performed additional sensitivity analyses that assessed the impact of including Chinese language studies.
Sensitivity analysis
If sufficient data had been available, we planned to examine the robustness of the meta‐analyses by conducting sensitivity analyses using different components of the risk of bias assessment, particularly those relating to whether allocation concealment and blinding were adequate. We also planned to conduct further sensitivity analyses to examine the impact of missing data on the results if a large proportion of the studies were at an 'unknown' or 'high risk' of attrition bias and, finally, we planned to use sensitivity analyses to examine whether publication status and trial size influenced the results. Unfortunately, we were unable to perform any such sensitivity analyses due to the low number of studies within each comparison. As already outlined in Unit of analysis issues, we performed sensitivity analyses when we included cross‐over trial data in analyses as if their designs were parallel‐group by excluding the cross‐over trials from the meta‐analyses and, as outlined in Excluded studies, we also performed additional sensitivity analyses to assess the impact of the inclusion of Chinese language studies in the analyses of the CR oxycodone versus CR morphine comparison.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE system to rank the certainty of the evidence using the GRADEprofiler Guideline Development Tool software (GRADEpro GDT 2015), and the guidelines provided in Chapter 12.2 of the CochraneHandbook for Systematic Reviews of Interventions (Higgins 2011).
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning grade of evidence:
high: we are very confident that the true effect lies close to that of the estimate of the effect;
moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different;
low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect;
very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
We decreased the grade rating by one (‐1) or two (‐2) if we identified:
serious (‐1) or very serious (‐2) limitations to study quality based on a qualitative assessment of the extent the body of evidence was at risk of bias;
important inconsistency (‐1) if the I2 was above 50%. We did not downgrade twice for this domain;
some (‐1) or major (‐2) uncertainty about directness based on a qualitative assessment of the extent the body of evidence met the inclusion criteria;
imprecise (‐1) or very imprecise data (‐2) based on number of participants/events. For dichotomous outcomes, we downgraded once or twice if the total number of events was below 300 or 150, respectively, and for continuous outcomes we downgraded once or twice if the total number of participants was below 400 or 200, respectively;
high probability of reporting bias (‐1) based on a qualitative assessment of the likelihood of reporting bias. We did not downgrade twice for this domain.
'Summary of findings' tables
We included two summary of findings tables to present the main findings in a transparent and simple tabular format. In particular, we included key information concerning the certainty of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes pain intensity, adverse events (constipation, drowsiness/somnolence, nausea, and vomiting), quality of life (or treatment acceptability as a proxy) and participant preference.
See Schmidt‐Hansen 2013 for the published protocol for this review.
Results
Description of studies
Results of the search
The updated searches of the electronic databases retrieved 817 records (see Electronic searches). Our searches of the trials registers identified 46 further studies. Our screening of the reference lists of the included publications revealed one additional potentially relevant study, and the search identified a systematic review of Chinese studies (Zhou 2020) with 16 potentially relevant studies. We therefore had a total of 880 records.
Once duplicates had been removed, we had 616 records. We excluded 564 records based on titles and abstracts. We obtained the full text of the remaining 52 records. We excluded 14 studies for this update. We identified 10 new ongoing studies for this update and there were three studies we could not classify so they are listed under 'studies awaiting classification'. Two of the records were additional references for Corli 2016.
From the search, we included a total of 19 new studies reported in 23 references for this update (Cao 2015; Gao 2012; Inoue 2017; Inoue 2018; Lee 2017; Li 2013; Liu 2021; Nosek 2017; Ren 2012; Song 2015; Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Zhang 2011; Zhang 2016a), of which one had originally been included under 'ongoing studies'. For a further description of our screening process, see the study flow diagram (Figure 1).
1.
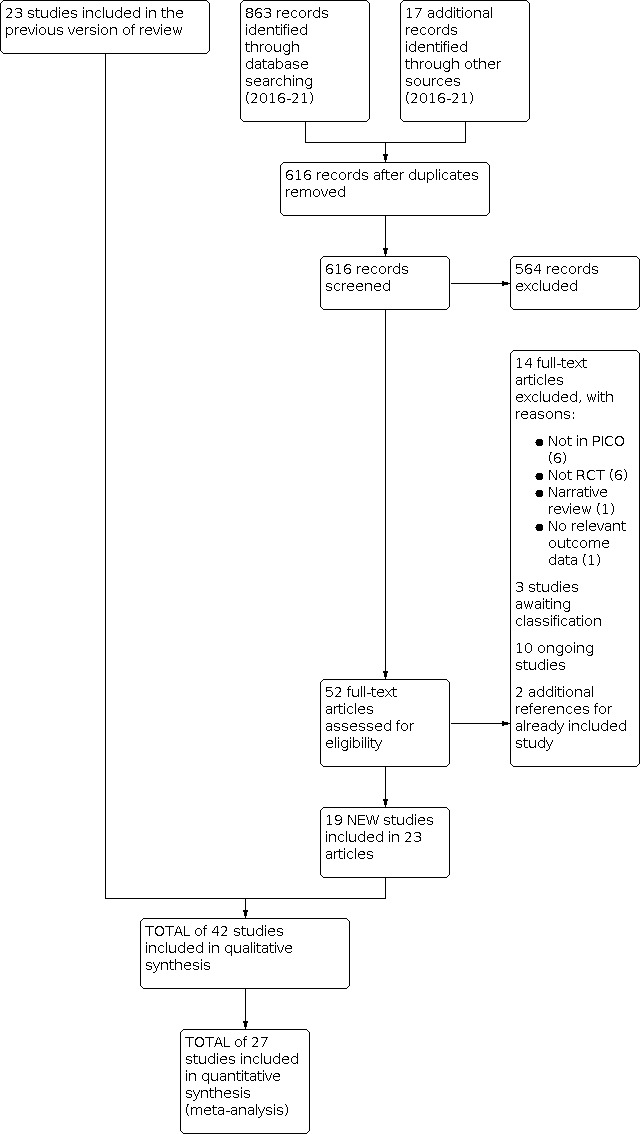
Study flow diagram
In total, we included 42 studies (see Characteristics of included studies) and excluded 77 studies (see Characteristics of excluded studies) in this review. There are nine studies awaiting classification (see Characteristics of studies awaiting classification), and 18 ongoing studies (see Characteristics of ongoing studies).
Included studies
The 19 new studies included an additional 1836 randomised participants, such that the 42 included studies enrolled/randomised 4485 participants (2347 men, 1869 women; for the remaining 268 participants, gender was not specified) with 3945 of these analysed for efficacy and 4176 for safety. The reported mean/median ages of the participant populations in the studies ranged from 45 years to 75.3 years. Eleven of the studies were cross‐over trials (Beaver 1978a; Beaver 1978b; Bruera 1998; Gabrail 2004; Hagen 1997; Heiskanen 1997; Kalso 1990; Lauretti 2003; Leow 1995; Lux 2014; Stambaugh 2001), and the remainder were parallel‐group trials, with eight of the studies conducted in the USA (Beaver 1978a; Beaver 1978b; Gabrail 2004; Kaplan 1998; Mucci‐LoRusso 1998; Parris 1998; Salzman 1999; Stambaugh 2001); two in Canada (Bruera 1998; Hagen 1997); two in Finland (Heiskanen 1997; Kalso 1990); 18 in China (Cao 2015; Gao 2012; Li 2013; Liu 2021; Ren 2012; Song 2015; Su 2015; Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Yu 2014; Zhang 2011; Zhang 2014; Zhang 2016a); three in Italy (Corli 2016; Mercadante 2010; Zecca 2016), two in Japan (Inoue 2017; Inoue 2018); and one each in Germany/Poland/Switzerland (Lux 2014), Australia (Leow 1995), Brazil (Lauretti 2003), Poland (Nosek 2017), Korea (Lee 2017), the UK (Riley 2015), and Japan/Korea (Imanaka 2013). The length of the trials ranged from single‐dose treatment to one year, and the studies reported the following comparisons:
CR oxycodone versus IR oxycodone (Kaplan 1998; Parris 1998; Salzman 1999; Stambaugh 2001);
CR oxycodone versus extended‐release (ER) oxycodone (Lux 2014);
CR oxycodone versus CR morphine (Bruera 1998; Cao 2015; Corli 2016; Gao 2012; Heiskanen 1997; Lauretti 2003; Li 2013; Mercadante 2010; Mucci‐LoRusso 1998; Nosek 2017; Ren 2012; Riley 2015; Song 2015; Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Zecca 2016; Zhang 2011; Zhang 2014; Zhang 2016a), with two of these studies including a further two arms of transdermal (TD) buprenorphine and TD fentanyl (Corli 2016; Nosek 2017); and two of the studies comparing two different brands of slow‐release morphine to CR oxycodone (Zhang 2014; Zhang 2016a);
CR oxycodone versus CR hydromorphone (Hagen 1997);
CR oxycodone versus ER hydromorphone (Inoue 2017; Yu 2014);
CR oxycodone versus ER oxymorphone (Gabrail 2004);
CR oxycodone versus ER tapentadol (Imanaka 2013);
CR oxycodone versus TD fentanyl (Corli 2016; Nosek 2017; Su 2015);
CR oxycodone versus TD buprenorphine (Corli 2016; Nosek 2017);
CR oxycodone versus oral ibuprofen (Liu 2021)
IR oxycodone versus IR hydromorphone (Inoue 2018);
intravenous (IV) oxycodone versus rectal oxycodone (Leow 1995);
IV oxycodone versus IV morphine (Lee 2017);
IV oxycodone followed by IR oxycodone versus IV morphine followed by IR morphine (Kalso 1990);
intramuscular (IM) oxycodone versus oral oxycodone (Beaver 1978a);
IM oxycodone versus IM morphine versus IM codeine (Beaver 1978b).
See also Characteristics of included studies table for further details about the studies.
Excluded studies
For this update, we excluded 14 studies. Of the records identified by the original and the update search, altogether we excluded 77 studies. A number of the studies identified in the searches compared oxycodone in combination with another drug (e.g. naloxone or acetaminophen) against oxycodone alone, another active drug or placebo. Such studies were not included as they would not answer our primary question, which concerned the effectiveness of oxycodone for cancer pain. The majority of the 77 studies were excluded because they did not include the population or comparison of interest (42 studies), while others were excluded because they were systematic (11) or narrative reviews (seven), not RCTs/RCT‐based analyses (13), letters to the editor (two), case reports (one) or did not report relevant outcome data (one). See also Characteristics of excluded studies table.
Moreover, we also explored the possibility of extending our searches to cover Chinese databases. These exploratory searches in the four main Chinese databases (China Network Knowledge Infrastructure (CNKI), Chinese Scientific Journals Database (VIP), Wanfang data and SinoMed) identified 2087 de‐duplicated records, and after full‐text screening, we found over 200 potentially eligible studies that on balance we decided against including. This decision was not taken lightly but ultimately arrived at for two reasons: 1) At that point, we had already undertaken data extraction and risk of bias assessment of the 14 new eligible studies (Cao 2015; Gao 2012; Li 2013; Ren 2012; Song 2015; Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Zhang 2011; Zhang 2016a) identified from the systematic review of Chinese studies (Zhou 2020), all published in Chinese and, during this work, it became apparent that all of these studies were compromised by extensive methodological under‐reporting which is illustrated by the fact that we were only able to arrive at a risk of bias assessment (low or high) in 11 out of 140 possible ratings, leaving the remaining 129 ratings as unclear (Figure 2). We had no reason to expect that this would be different for Chinese studies identified through a systematic search of Chinese databases, an assertion which is also supported by Tong 2018 and Zhang 2016b. This, in turn, would have implications for the conclusions we would be able to arrive at based on these data, and there was a risk that this extensive uncertainty would over‐shadow any conclusions we would otherwise be able to arrive at with some degree of certainty, especially given the large number of potentially relevant studies. 2) The second reason for not pursuing the search of Chinese databases concerns the understanding or employment of the term "randomised" in the context of patient allocation in Chinese studies published in Chinese journals, with a number of studies finding that a large proportion of Chinese studies described as randomised are in fact not (Tong 2018; Wu 2009a; Zhou 2019). Although the inclusion of Chinese studies identified through searches of Chinese databases would have served to limit the geographical/Western world bias of our included studies and thereby would have increased the applicability of our results, the large extent of uncertainty surrounding the methodological quality of such studies coupled with the real possibility that many of them would indeed not be randomised studies (and would therefore put the 'systematic' aspect of our review at risk due to the potential inadvertent inclusion of observational non‐randomised studies, which is an exclusion criterion of this review) meant that on balance and, in accordance with our protocol, we did not pursue the search of Chinese databases further. We note that all these concerns of course also apply to the majority of the new studies found for this update as 14 of the 18 studies were from Chinese journals, however, they were identified as part of the search strategy agreed in our protocol and were therefore included. They all examined the same comparison (CR oxycodone versus CR morphine) and we performed additional sensitivity analyses to assess the impact of their inclusion.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Studies awaiting classification
Nine studies are awaiting classification because they are either ongoing but of unclear relevance, or published in such a way that not enough information is available to ascertain whether they meet the inclusion criteria (e.g. as an abstract only). In some cases, we have attempted to contact the authors, but have not successfully obtained a response. We await further information, including study completion and publication, before we can ascertain their relevance to the current review and classify them accordingly. See also Characteristics of studies awaiting classification table.
Ongoing studies
Eighteen studies are ongoing. These studies examine some new drug comparisons involving oxycodone compared to those reported in this review, and some comparisons that have already been included. Upon their completion and publication, we hope to be able to include data from all of them in future updates of this review. See also Characteristics of ongoing studies table.
Risk of bias in included studies
See Figure 2 and Figure 3 for summaries of the risk of bias judgements made for the included studies.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
Allocation
Only seven studies were considered to be at low risk of bias for both generation of randomisation sequence and allocation concealment (Corli 2016; Heiskanen 1997; Imanaka 2013; Inoue 2018; Lux 2014; Yu 2014; Zecca 2016), while a further seven studies were considered at low risk of bias for randomisation sequence but at unclear risk of bias for allocation concealment (Cao 2015; Liu 2021; Riley 2015; Song 2015; Sun 2013; Xie 2018; Zhang 2011), and one study was at high risk of inadequate allocation concealment while at unclear risk for randomisation sequence generation (Lee 2017). The remainder of the studies did not report enough information for us to assess whether the methods employed to generate the randomisation sequence and to ensure allocation concealment were adequate. In nine studies, we could make a judgement that the treatment groups were comparable at baseline (Corli 2016; Imanaka 2013; Inoue 2017; Kaplan 1998; Liu 2021; Riley 2015; Salzman 1999; Su 2015; Zhang 2014). Three further studies reported that the groups were comparable at baseline apart from there being more participants with bone metastasis and an Eastern Cooperative Oncology Group (ECOG) performance status of 3 in the hydromorphone group than in the oxycodone group (Yu 2014), more participants with a Karnofsky Performance Status (from 0 to 100) ≤ 70 in the CR oxycodone group than in the CR morphine group (Zecca 2016), or more people with pancreatic and gastric cancers in the oxycodone group and more gastric, lung, and colorectal cancers in the morphine group (Lee 2017). In the remaining studies, it was unclear whether the participant selection methods employed had resulted in comparable, balanced groups at the start of the study.
Blinding
The problem of under‐reporting was also an issue when assigning risk of bias estimates to the items assessing performance and detection bias, that is, blinding. In only one instance was it directly and unequivocally reported who was blinded; for the most part so we had to infer, on the basis of supplementary information, whether we were reasonably certain that blinding had been adequately executed for a given individual (that is, participant, treating personnel, outcome assessors, or a combination of these, where not the participants themselves).
On this basis, the risk of performance bias was considered to be low for the primary outcome of pain in 13 of the studies (Beaver 1978a; Beaver 1978b; Bruera 1998; Hagen 1997; Heiskanen 1997; Imanaka 2013; Kaplan 1998; Lauretti 2003; Lux 2014; Mucci‐LoRusso 1998; Parris 1998; Stambaugh 2001; Yu 2014), high in nine of the studies (Corli 2016; Lee 2017; Leow 1995; Liu 2021; Mercadante 2010; Nosek 2017; Riley 2015; Salzman 1999; Zecca 2016) that were all described as open‐label (with the exceptions of Liu 2021 and Nosek 2017), and unclear in the remaining studies. For adverse events, the risk of performance bias was low in 11 studies (Beaver 1978a; Beaver 1978b; Bruera 1998; Hagen 1997; Imanaka 2013; Kaplan 1998; Lauretti 2003; Lux 2014; Mucci‐LoRusso 1998; Stambaugh 2001; Yu 2014), high in the same nine studies as was the case for pain (Corli 2016; Lee 2017; Leow 1995; Liu 2021; Mercadante 2010; Nosek 2017; Riley 2015; Salzman 1999; Zecca 2016), and unclear in the remaining studies. The pattern of judgements was identical for detection bias, for both outcomes. This was the case for the primary outcome of pain because, according to our criteria, this outcome had to be participant‐reported. It was, therefore, at risk of detection bias to the same extent that it was at risk of performance bias, since both depend on participant blinding. As is also evident from the bias judgements (see Characteristics of included studies), when a study was described as double‐blind but did not describe who was blinded, additional information in the studies generally led us to the conclusion that at least the participants seemed to be blinded, although we did not feel able to gauge with sufficient confidence who else might have been blinded. Given that it was not always clear who assessed the adverse events, this accounts for the similar judgements for performance and detection bias for this outcome.
Incomplete outcome data
Overall, the data from 88% of the total number of enrolled/randomised participants were analysed for pain and 93.1% for adverse events, which indicates that attrition bias was a substantial problem in this dataset especially for pain, with only 12 studies considered at low risk (Corli 2016; Inoue 2017; Inoue 2018; Kalso 1990; Kaplan 1998; Lee 2017; Leow 1995; Liu 2021; Parris 1998; Su 2015; Zecca 2016; Zhang 2011), and 14 studies considered at high risk (Bruera 1998; Gabrail 2004; Hagen 1997; Heiskanen 1997; Imanaka 2013; Lux 2014; Mercadante 2010; Mucci‐LoRusso 1998; Nosek 2017; Riley 2015; Salzman 1999; Stambaugh 2001; Yu 2009; Yu 2014), while the rest of the studies were at unclear risk for the primary outcome of pain. For adverse events, the risk of attrition bias was slightly less, with 16 studies considered at low risk (Corli 2016; Imanaka 2013; Inoue 2017; Inoue 2018; Kalso 1990; Kaplan 1998; Lee 2017; Leow 1995; Liu 2021; Mucci‐LoRusso 1998; Parris 1998; Salzman 1999; Su 2015; Yu 2014; Zecca 2016; Zhang 2011), eight studies considered at high risk (Bruera 1998; Hagen 1997; Heiskanen 1997; Mercadante 2010; Nosek 2017; Riley 2015; Stambaugh 2001; Yu 2009), and the remainder at unclear risk.
Selective reporting
We considered 21 of the included studies to be at low risk of selective reporting bias, whereas six of the studies either did not report adverse events ± pain or did not report them in a manner so they could be scrutinised for (and potentially included in) an evidence synthesis (Beaver 1978a; Beaver 1978b; Bruera 1998; Lauretti 2003; Lux 2014; Nosek 2017); these studies were therefore judged as being at high risk. One study only reported four adverse events in a transparent manner and was therefore considered at unclear risk of reporting bias (Hagen 1997). All of the 14 newly included Chinese studies were considered at unclear risk of selective reporting bias due to lack of information reported (see also Excluded studies).
Other potential sources of bias
Adequate titration
One study examined titration as its main objective (Salzman 1999). In the other 40 studies, the participants appeared to be adequately titrated in the majority of the studies (Bruera 1998; Corli 2016; Gabrail 2004; Hagen 1997; Heiskanen 1997; Imanaka 2013; Inoue 2017; Inoue 2018; Kalso 1990; Lauretti 2003; Lux 2014; Mucci‐LoRusso 1998; Nosek 2017; Parris 1998; Riley 2015; Stambaugh 2001; Su 2015; Yu 2009; Yu 2014; Zecca 2016; Zhang 2011; Zhang 2014), although this was not the case in one study (Kaplan 1998) and unclear in the remaining studies.
Availability of data from both time periods of cross‐over trials
For all 11 cross‐over trials, data were available for all cross‐over phases.
Other bias
Twenty‐three of the included studies were considered at low risk of any other biases (e.g. carry‐over effects in the cross‐over trials) with the remainder being judged to be at unclear risk of other bias due to the limited manner in which the trials were reported (Beaver 1978a; Beaver 1978b; Cao 2015; Gao 2012; Li 2013; Liu 2021; Ren 2012; Song 2015; Su 2015;Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Yu 2014; Zhang 2011; Zhang 2014; Zhang 2016a).
Study funding
Eighteen of the included studies had received commercial funding or had authors who were employees of the drug manufacturers, or both (Beaver 1978a; Beaver 1978b; Gabrail 2004; Hagen 1997; Heiskanen 1997; Imanaka 2013; Inoue 2017; Inoue 2018; Kaplan 1998; Lee 2017; Leow 1995; Lux 2014; Mucci‐LoRusso 1998; Parris 1998; Salzman 1999; Stambaugh 2001; Yu 2014; Zecca 2016). Only seven studies were considered free from the potential influence of commercial funding (Corli 2016; Kalso 1990; Li 2013; Liu 2021; Nosek 2017; Riley 2015; Zhang 2011), with the rest of the studies having unclear status.
Effects of interventions
Analysis 1.1 and Figure 4 shows the pain intensity scores for each of the listed treatment groups, subgrouped according to overall treatment comparisons. We felt that presenting the pain intensity data this way, for the studies where it was possible, gave a comprehensive overview of the pain intensity data for the majority of the included studies, although the actual analyses should be treated with some caution as outlined in the Unit of analysis issues section. The inclusion of, in particular, the Chinese studies for this update, presented challenges in terms of analysing the pain data comprehensively as almost all of these studies only reported pain relief in categorical terms, and not pain intensity as a continuous measure. We therefore included an additional meta‐analysis of the pain data of the proportions of participants who achieved "complete" and/or "significant" pain relief (Analysis 1.2), which is illustrated in Figure 5 . Where possible, we have captured the exact definitions used by the individual studies in the Characteristics of included studies, but these were not always fully reported in the studies.
1.1. Analysis.

Comparison 1: Pain, Outcome 1: Pain intensity and pain relief (continuous)
4.

Forest plot of comparison: 1 Pain, outcome: 1.1 Pain intensity and pain relief (continuous)
1.2. Analysis.

Comparison 1: Pain, Outcome 2: Complete and/or significant pain relief (categorical)
5.

Forest plot of comparison: 1 Pain, outcome: 1.2 Complete and/or significant pain relief (categorical)
Controlled‐release oxycodone versus immediate‐release oxycodone
Four studies compared CR oxycodone to IR oxycodone (Kaplan 1998; Parris 1998; Salzman 1999; Stambaugh 2001).
Pain intensity and pain relief
Pooled analysis of three of the four studies suggests that there is little to no difference in pain intensity after treatment with either CR or IR oxycodone (SMD 0.12, 95% CI ‐0.1 to 0.34; participants = 319; studies = 3; I2 = 38%; Figure 4), which was also in line with the finding that none of the included studies reported that pain intensity differed between the treatment groups. Salzman 1999 could not be included in the pooled analysis due to the titration design of the study, so was instead summarised narratively below. Sensitivity analysis excluding the cross‐over trial (Stambaugh 2001) did not change the overall results although heterogeneity increased substantially (SMD 0.16, 95% CI ‐0.08 to 0.41; participants = 259; studies = 2; I2 = 62%). Kaplan 1998 analysed 156 participants for efficacy evaluation rather than 160 participants; however, it was unclear from which groups these participants were missing. In the meta‐analyses, we removed two from each group, and sensitivity analyses showed none of the other possible options made any difference to the conclusions.
Salzman 1999 examined, in a parallel‐group trial lasting up to 21 days, whether CR oxycodone could be used as readily as IR oxycodone for titration to stable pain control and found that 22/24 and 19/24 participants in the CR and IR groups, respectively, achieved stable pain control within a mean time of 1.6 days (SE = 0.4) and 1.7 days (SE = 0.6), respectively.
We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from attrition bias and under‐reporting) and by one level due to imprecision (arising from low participant numbers). See Table 1.
Adverse events
The evidence is very uncertain about the effect of CR oxycodone compared with IR oxycodone on adverse events, including asthenia (RR 0.58, 95% CI 0.20 to 1.68; participants = 208; studies = 2; I2 = 30%; Analysis 2.5), confusion (RR 0.78, 95% CI 0.20 to 3.02; participants = 157; studies = 2; I2 = 25%; Analysis 2.6), constipation (RR 0.71, 95% CI 0.45 to 1.13; participants = 317; studies = 3; I2 = 38%; Analysis 2.7; Figure 6), dizziness/lightheadedness (RR 0.74, 95% CI 0.40 to 1.37; participants = 317; studies = 3; I2 = 15%; Analysis 2.9), drowsiness/somnolence (RR 1.03, 95% CI 0.69 to 1.54; participants = 317; studies = 3; I2 = 0%; Analysis 2.10; Figure 7), dry mouth (RR 1.14, 95% CI 0.48 to 2.75; participants = 317; studies = 3; I2 = 0%; Analysis 2.11), insomnia (RR 1.04, 95% CI 0.31 to 3.53; participants = 269; studies = 2; I2 = 35%; Analysis 2.16), nausea (RR 0.85, 95% CI 0.56 to 1.28; participants = 317; studies = 3; I2 = 0%; Analysis 2.18; Figure 8), nervousness (RR 0.57, 95% CI 0.20 to 1.64; participants = 208; studies = 2; I2 = 0%; Analysis 2.20), pruritus (RR 1.46, 95% CI 0.65 to 3.25; participants = 317; studies = 3; I2 = 33%; Analysis 2.21), vomiting (RR 0.66, 95% CI 0.38 to 1.15; participants = 317; studies = 3; I2 = 18%; Analysis 2.23; Figure 9), and discontinuation due to adverse events (RR 0.60, 95% CI 0.29 to 1.22; participants = 317; studies = 3; I2 = 0%; Analysis 2.24). The I2 statistic was 55% for sweating, so the pooled results reported in Analysis 2.22 should be disregarded. The results were also very inconsistent for headache (I2 statistic was 61%) and it was unclear whether there were any differences between the interventions (Analysis 2.15). Parris 1998 analysed 109 participants for safety evaluation; however, it was unclear which group had 55 and which had 54 participants. In the meta‐analyses of adverse events, we allocated 54 participants to the CR oxycodone group and 55 to the IR oxycodone group. Sensitivity analyses showed that allocating 55 participants to the CR oxycodone group and 54 participants to the IR oxycodone group made no difference to the conclusions.
2.5. Analysis.

Comparison 2: Adverse events, Outcome 5: Asthenia
2.6. Analysis.

Comparison 2: Adverse events, Outcome 6: Confusion
2.7. Analysis.

Comparison 2: Adverse events, Outcome 7: Constipation
6.

Forest plot of comparison: 2 Adverse events, outcome: 2.7 Constipation
2.9. Analysis.

Comparison 2: Adverse events, Outcome 9: Dizziness/lightheadedness
2.10. Analysis.

Comparison 2: Adverse events, Outcome 10: Drowsiness/somnolence
7.

Forest plot of comparison: 2 Adverse events, outcome: 2.10 Drowsiness/somnolence
2.11. Analysis.

Comparison 2: Adverse events, Outcome 11: Dry mouth
2.16. Analysis.

Comparison 2: Adverse events, Outcome 16: Insomnia
2.18. Analysis.

Comparison 2: Adverse events, Outcome 18: Nausea
8.

Forest plot of comparison: 2 Adverse events, outcome: 2.18 Nausea
2.20. Analysis.

Comparison 2: Adverse events, Outcome 20: Nervousness
2.21. Analysis.

Comparison 2: Adverse events, Outcome 21: Pruritus
2.23. Analysis.

Comparison 2: Adverse events, Outcome 23: Vomiting
9.

Forest plot of comparison: 2 Adverse events, outcome: 2.23 Vomiting
2.24. Analysis.

Comparison 2: Adverse events, Outcome 24: Discontinuation due to adverse events
2.22. Analysis.

Comparison 2: Adverse events, Outcome 22: Sweating
In a parallel‐group trial lasting five days, Parris 1998 reported that all the adverse events observed during the study resolved. Stambaugh 2001 conducted a cross‐over study with a duration of three to seven days per phase, and stated that: "The study showed similar incidences and numbers of reports of individual adverse events considered related to the IR and CR drug" (page 505), but did not report any formal statistical comparisons of the adverse event rates between the study groups. Table 3 contains all the adverse events reported by the included studies comparing CR oxycodone and IR oxycodone.
1. Controlled‐release (CR) oxycodone versus immediate‐release (IR) oxycodone: adverse events.
| Comparison | CR oxycodone versus IR oxycodone | |||||||
| Study | Kaplan 1998 | Parris 1998a | Salzman 1999 | Stambaugh 2001 | ||||
| Treatment | CR | IR | CR | IR | CR | IR | CR | IR |
| Any adverse events | ‐ | ‐ | 38/54‐55 | 38/54‐55 | ‐ | ‐ | 10/30 | 10/30 |
| Total adverse events | 109 | 186 | 138 | 142 | ‐ | ‐ | ‐ | ‐ |
| Abdominal pain | ‐ | ‐ | 3/54‐55 | 1/54‐55 | ‐ | ‐ | ‐ | ‐ |
| Anxiety | 0/78 | 4/82 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Asthenia | 3/78 | 8/82 | ‐ | ‐ | 2/24 | 1/24 | 2/30 | 2/30 |
| Confusion | ‐ | ‐ | 0/54‐55 | 2/54‐55 | 3/24 | 2/24 | ‐ | ‐ |
| Constipation | 9/78 | 17/82 | 12/54‐55 | 10/54‐55 | 4/24 | 9/24 | 1/30 | 1/30 |
| Dizziness, lightheadedness | 5/78 | 11/82 | 8/54‐55 | 10/54‐55 | 2/24 | 0/24 | 3/30 | 3/30 |
| Drowsiness, somnolence | 14/78 | 17/82 | 13/54‐55 | 12/54‐55 | 9/24 | 7/24 | 3/30 | 2/30 |
| Dry mouth | 3/78 | 5/82 | 4/54‐55 | 3/54‐55 | 3/24 | 1/24 | 1/30 | 1/30 |
| Headache | 0/78 | 6/82 | 7/54‐55 | 3/54‐55 | 1/24 | 1/24 | ‐ | ‐ |
| Insomnia | 2/78 | 4/82 | 3/54‐55 | 1/54‐55 | ‐ | ‐ | ‐ | ‐ |
| Nausea | 14/78 | 21/82 | 11/54‐55 | 13/54‐55 | 7/24 | 5/24 | 4/30 | 3/30 |
| Nervousness | 3/78 | 5/82 | ‐ | ‐ | 2/24 | 4/24 | 0/30 | 1/30 |
| Postural hypotension | ‐ | ‐ | ‐ | ‐ | 5/24 | 4/24 | ‐ | ‐ |
| Pruritus | 2/78 | 4/82 | 7/54‐55 | 5/54‐55 | 4/24 | 0/24 | 1/30 | 2/30 |
| Sweating | 4/78 | 3/82 | 1/54‐55 | 5/54‐55 | ‐ | ‐ | 2/30 | 1/30 |
| Vomiting | 8/78 | 14/82 | 5/54‐55 | 11/54‐55 | 5/24 | 3/24 | 2/30 | 0/30 |
| Discontinuation due to adverse events | 6/78 | 10/82 | 4/54‐55 | 7/54‐55 | 1/24 | 2/24 | ‐ | ‐ |
a Total number of participants for safety evaluation = 109. Not clear which group had 55 and 54 participants, respectively.
‐: not reported CR: controlled‐release IR: immediate‐release
We judged the certainty of evidence for adverse events to be very low in all cases. We downgraded the certainty of the evidence by two levels due to imprecision (arising from very low event rates); and we downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (performance/detection bias, and inadequate titration and under‐reporting of the domains of selection, performance, detection and attrition bias, and whether the participants were adequately titrated).
Quality of life
There were no data for quality of life, but three studies reported treatment acceptability and their results showed that there may be little to no difference in treatment acceptability between CR and IR oxycodone. In particular, Kaplan 1998 reported in a parallel‐group study lasting six days that there was no difference in treatment acceptability between the study groups (mean at study end 3.2, SE 0.1, in both groups), and Parris 1998 found no differences in acceptability of treatment between the study groups at any time point. In Stambaugh 2001, 30/30 and 29/30 participants rated IR and CR oxycodone, respectively, as of 'fair', 'good' or 'excellent' acceptability during the last 24 hours of the treatment phases, with 24/30 and 22/30 participants rating the drugs 'good' or 'excellent', respectively. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from attrition bias and under‐reporting) and by one level due to imprecision (arising from low participant numbers).
Participant preference
None of the studies reported data for participant preference.
See also Table 1.
Controlled‐release oxycodone versus extended‐release oxycodone
One study compared CR oxycodone to ER oxycodone (Lux 2014).
Pain intensity and pain relief
Lux 2014 suggests there may be little to no difference in pain intensity between CR and ER oxycodone in a cross‐over trial with each of the two phases lasting 10 days (Figure 4). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by one level for serious limitations to study quality due to risk of bias (arising from attrition bias) and by two levels due to imprecision (arising from very low participant numbers).
Adverse events
Lux 2014 also included participants with non‐malignant pain and only reported adverse events for the whole sample. Therefore, we have not reported results for adverse events.
Quality of life
The study did not report quality of life.
Participant preference
The study did not report participant preference.
Controlled‐release oxycodone versus controlled‐release morphine
Twenty‐four studies compared CR oxycodone to CR morphine (Bruera 1998; Cao 2015; Corli 2016; Gao 2012; Heiskanen 1997; Lauretti 2003; Li 2013; Mercadante 2010; Mucci‐LoRusso 1998; Nosek 2017; Ren 2012 ; Riley 2015; Song 2015, Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Zecca 2016; Zhang 2011; Zhang 2014; Zhang 2016a).
Pain intensity and pain relief
Pain intensity: Fourteen studies could not be included in the pooled analysis due to the design of the study (Lauretti 2003) or because pain intensity was not reported as a continuous variable (Gao 2012; Li 2013; Nosek 2017; Ren 2012; Song 2015; Sun 2013; Tu 2015; Wang 2008; Ye 2012; Yu 2009; Zhang 2011; Zhang 2014; Zhang 2016a), and the results of these studies are therefore summarised separately below. Pooled analysis of Bruera 1998; Cao 2015; Corli 2016; Heiskanen 1997, Mercadante 2010; Mucci‐LoRusso 1998; Riley 2015; Xie 2018; Yu 2007; and Zecca 2016 showed that the pain intensity scores may be lower after treatment with CR oxycodone than with CR morphine (SMD ‐0.18, 95% CI ‐0.30 to ‐0.06; n = 1137; studies = 10; I2 = 96%; Analysis 1.1; Figure 4), however, the I2 indicated excessive heterogeneity and inspection of Figure 4 indicated two clear outlying studies (Cao 2015; Xie 2018) which were among the studies added from the group of Chinese language studies. Sensitivity analyses omitting the Chinese language studies (Cao 2015; Xie 2018, and Yu 2007) indicated that the pain intensity scores may be lower after treatment with CR morphine than with CR oxycodone (SMD 0.14, 95% CI 0.01 to 0.27; n = 882; studies = 7; I2 = 7%). Due to the concerns outlined in Excluded studies, we consider the latter analysis our main analysis. Using the SD of the baseline average pain score of the full sample (200 participants; SD 1.94) in Riley 2015 to express this SMD as a difference in Brief Pain Inventory scores (0 to 10 numerical rating scale from no pain to worst pain imaginable) between the treatments gave an estimated difference of 0.27 between the treatments, which was not clinically significant. Moreover, sensitivity analysis excluding the two cross‐over trials (Bruera 1998, Heiskanen 1997) provided wider confidence intervals, which included no effect between CR oxycodone and CR morphine (SMD 0.12, 95% CI ‐0.02 to 0.26; n = 782; studies = 5; I2 = 24%).
Pain relief: Pooled analysis of those studies that reported the number of participants who experienced complete or significant pain relief (Corli 2016; Gao 2012; Li 2013; Ren 2012; Song 2015; Sun 2013; Tu 2015; Wang 2008; Ye 2012; Yu 2007; Yu 2009; Zhang 2011; Zhang 2016a) showed that there may be little to no difference in the proportions of participants achieving complete or significant pain relief between CR oxycodone and CR morphine (RR 1.02, 95% CI 0.95 to 1.10; n = 1249; studies = 13; I2 = 0%; Analysis 1.2; Figure 5). This was regardless of whether the CR oxycodone data were compared to the CR morphine sulfate or CR morphine hydrochloride data in Zhang 2016a.
Additional results reported by the individual studies: Gao 2012, in a parallel‐group trial of unclear duration, did not report pain intensity but rather that 20, 9 and 1 participants in the CR oxycodone group experienced complete, partial and no pain relief, respectively. The corresponding numbers after treatment with CR morphine were 18, 9 and 1.
In a four‐arm parallel‐group trial lasting 28 days, Corli 2016 compared CR oxycodone with CR morphine (and also included a TD fentanyl and a TD buprenorphine arm), and found that there may be little to no difference between CR oxycodone and CR morphine in terms of requirement for additional opioids (CR oxycodone: 33/125 participants; CR morphine: 36/122 participants; P = 0.59), the opioid escalation index > 5% (CR oxycodone: 24/125 participants; CR morphine: 13/122 participants; P = 0.06), or premature discontinuation for pain‐related reasons (CR oxycodone: 19/125 participants; CR morphine: 33/122 participants; P = 0.051); however, the proportion of participants requiring adjuvant drugs may be higher in the CR oxycodone group (CR oxycodone: 102/125 participants; CR morphine: 84/122 participants; P = 0.02), whereas the proportion of participants requiring switches (CR oxycodone: 15/125 participants; CR morphine: 27/122 participants; P = 0.03) may be lower in the CR oxycodone group compared to the CR morphine group.
Lauretti 2003 conducted a two‐phase (each lasting 14 days) cross‐over study to examine IR morphine consumption (which was the main outcome) during treatment with CR oxycodone and CR morphine, keeping the ratio of CR oxycodone and CR morphine constant (1:1.8). IR morphine was used as rescue medication and the participants were allowed to take as much as necessary to keep the visual analogue scale (VAS) pain score below 4. The participants consumed 38% more IR morphine during treatment with CR morphine than with CR oxycodone. Lauretti 2003 concluded that the results indicated that CR oxycodone combined with IR morphine was associated with superior analgesia and lower, or similar, rates of adverse events (see 'Adverse events' below) than a combination of CR and IR morphine.
Li 2013 conducted a parallel‐group trial lasting three days, and did not report pain intensity, but rather that 27, 13, and 2 of the 42 participants in the CR oxycodone group experienced complete, partial and mild pain relief, respectively. The corresponding numbers after treatment with CR morphine were 21, 17 and 2 out of 40 participants. Li 2013 also found that the mean onset to pain relief may be faster after CR oxycodone (mean (SD) = 44 (12.71) minutes) than after CR morphine (mean (SD) = 85 (12.96) minutes) treatment.
Mucci‐LoRusso 1998 conducted a parallel‐group trial lasting up to 12 days and found that 40/48 and 42/52 participants achieved stable pain control after receiving CR oxycodone and CR morphine, respectively, within a median of 2 days for both groups (ranges were 1 to 10 and 1 to 9 days, respectively).
Nosek 2017, in a four‐arm trial lasting 28 days comparing CR oxycodone to TD fentanyl, TD buprenorphine and CR morphine, could not be included in the analysis as the authors did not report the actual data for this outcome. Rather, they reported the pain data overall for all analysed participants, which they analysed using four treatment‐by‐time analyses of variance (ANOVA) which yielded significant interactions for all four pain variables analysed (pain at its worst; pain at its least; pain on average; pain right now). However, the authors did then not go on to perform (and present) simple main effects analyses in order to ascertain what drug and time differences underlaid these interactions. Two emails sent by our team to the authors have failed to elicit a response from the authors so, at this point, we cannot further examine these data.
In a parallel‐group trial of 14 days' duration, Ren 2012 found that out of the 40 participants in the CR oxycodone group 38, 1 and 1 achieved significant, moderate and mild pain relief, respectively. For the 40 participants in the CR morphine group, the numbers were 37, 2 and 1, respectively. Ren 2012 also found that there may be little to no difference in quality of life between the CR oxycodone (mean (SD) = 71 (8)) and CR morphine (mean (SD) = 69 (7)) groups.
In an open‐label, parallel‐group trial of one‐year duration, Riley 2015 compared CR oxycodone to CR morphine and found that 67% and 62% of the participants achieved a response to first‐line oxycodone and morphine, respectively, and their inferential analyses indicated that there may be little to no difference between these proportions. Moreover, in the participants who achieved a response to their assigned first‐line treatment, there may be little to no difference between the treatments in the five pain indices studied (that is, 'worst pain', 'least pain', 'average pain', 'pain right now', and 'percentage relief').
Song 2015 conducted a one‐month long parallel‐group trial and found that 23, 20 and 12 of the 55 participants in the CR oxycodone group experienced markedly effective, effective and ineffective pain relief, respectively. For the 55 participants who received CR morphine, the corresponding numbers were 22, 18 and 15. The mean (SD) onset to pain relief was 1.27 (0.45) hours in the CR oxycodone group and 1.59 (0.61) hours after treatment with CR morphine. Song 2015 also found that there may be little to no difference in quality of life between the CR oxycodone (mean (SD) = 37.25 (8.14)) and CR morphine (mean (SD) = 36.98 (7.59)) groups.
In a parallel‐group trial of 30 days' duration, Sun 2013 reported that, of the 102 participants in the CR oxycodone group, 81 achieved an NRS pain score of 0‐3, 20 achieved a score of 4‐6 and 1 had a score of 7‐10. In the CR oxycodone group, the corresponding numbers were 77, 24, and 1, respectively, out of a total of 102 participants. After CR oxycodone, the onset of pain relief was < 1 hour for 45 participants and > 1 hour for 57 participants. It was < 1 hour for 14 participants and > 1 hour for 88 participants in the CR morphine group.
In a parallel‐group trial by Tu 2015, which lasted > 14 days, 18, 11, 9 and 5 participants (of a total of 43 participants), respectively, achieved significant, moderate, mild and no pain relief after CR oxycodone treatment, whereas 19, 10, 8 and 6 (of a total of 43 participants), respectively, achieved significant, moderate, mild and no pain relief after CR morphine treatment.
Wang 2008 conducted a parallel‐group trial that lasted ≥ 14 days, however, the pain outcomes appear to have only been reported at 1, 2, 4, 8 and 12 hours. We have reported here those outcomes reported for 12 hours, where 9, 12, 6 and 3 (out of 30) participants, respectively, achieved complete, significant, moderate and mild pain relief in the CR oxycodone group. In the CR morphine group 10, 12, 6 and 2 (out of 30) participants, respectively, achieved complete, significant, moderate and mild pain relief. The mean (range) onset to pain relief was 43 (22‐65) minutes in the CR oxycodone group and 82 (58‐102) minutes in the CR oxycodone group. The mean (duration of?) analgesia time was 12.2 (range 8.5‐14.5) hours in the CR oxycodone group and 12.5 (range 9.5‐15.5) hours in the CR oxycodone group.
Ye 2012 in a parallel‐group trial of seven days' duration reported that 27, 11, 3 and 1 (of 42) participants, respectively, experienced complete, partial, mild and no pain relief after treatment with CR oxycodone; and that 24, 7, 6 and 4 (of 41) participants, respectively, experienced complete, partial, mild and no pain relief after treatment with CR morphine.
In a parallel‐group trial lasting five days, Yu 2007 found that on the fifth day, 6, 7 and 2 (of 15) participants experienced complete, significant and moderate pain relief, respectively, in the CR oxycodone group and 5, 7 and 3 (of 15) participants in the CR morphine group experienced complete, significant and moderate pain relief, respectively.
Yu 2009 conducted a 18‐day parallel‐group trial and found that 0, 23, 7, and 2 (of 32) participants in the CR oxycodone group achieved complete, significant, moderate and mild pain relief. The corresponding numbers were 0, 22, 6 and 2 (of 30) participants in the CR morphine group. The number of break‐through pain events were 22 and 15 in the CR oxycodone and CR morphine groups, respectively. The mean amount of rescue medication used may be lower in the CR oxycodone group (mean 23.43 (SD or SE [study did not report which] 30.23) mg) than in the CR morphine group (mean 40.33 (SD/SE 34.39) mg).
Zecca 2016, in a parallel‐group trial lasting two weeks, reported that there may be little to no difference in opioid dose escalation between treatments (CR oxycodone 8.3%; CR morphine 6.5%).
In a 4‐day long parallel‐group trial, Zhang 2011 found that 3, 21, 7 and 4 (of 35) participants, respectively, experienced complete, significant, moderate and' mild or no' pain relief in the CR oxycodone group. In the CR morphine group, the corresponding numbers of participants were 3, 19, 8 and 2 of a total of 32 participants.
Zhang 2014 conducted a three‐arm parallel‐group trial of unknown duration comparing CR oxycodone to CR morphine and CR MS Contin, and found that there may be little to no difference in pain relief rates (i.e. participants experiencing at least moderate pain relief) between the three groups (CR oxycodone 53/57 participants; CR morphine 51/57 participants; CR MS Contin 52/57 participants).
Zhang 2016a, in a three‐arm parallel‐group trial lasting one month, found that the number of participants who achieved complete, significant, moderate, mild, or no pain relief (of 40 participants in each of the 3 arms) were 9, 23, 4, 4 and 0, respectively, in the CR oxycodone group; 7, 25, 5, 3 and 0, respectively, in the CR morphine sulfate group; and 10, 24, 2, 4 and 0, respectively, in the morphine hydrochloride group.
We judged the certainty of evidence for this outcome to be low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance/detection bias, and attrition bias). See Table 2.
Adverse events
Meta‐analyses: Pooled analyses showed that for most of the adverse events the CIs were wide, including no effect as well as potential benefit and harm, for the comparison between CR oxycodone and CR morphine including anorexia/appetite loss (RR 1.20, 95% CI 0.36 to 3.94; participants = 263; studies = 3; I2 = 0%; Analysis 2.4), confusion (RR 1.01, 95% CI 0.78 to 1.31; participants = 584; studies = 3; I2 = 16%; Analysis 2.6), dizziness/lightheadedness (RR 0.87, 95% CI 0.58 to 1.31; participants = 941; studies = 11; I2 = 0%; Analysis 2.9), drowsiness/somnolence (RR 0.88, 95% CI 0.74 to 1.05; participants = 1486; studies = 15; I2 = 0%; Analysis 2.10; Figure 7), dry mouth (RR 0.97, 95% CI 0.78 to 1.22; participants = 888; studies = 5; I2 = 34%; Analysis 2.11), dysuria/uroschesis (RR 0.64, 95% CI 0.38 to 1.07; participants = 887; studies = 7; I2 = 0%; Analysis 2.12), nausea (RR 0.93, 95% CI 0.77 to 1.12; participants = 1388; studies = 13; I2 = 0%; Analysis 2.18; Figure 8), vomiting (RR 0.81, 95% CI 0.63 to 1.04; participants = 1388; studies = 13; I2 = 0%; Analysis 2.23; Figure 9), nausea and vomiting (RR 0.77, 95% CI 0.56 to 1.06; participants = 637; studies = 6; I2 = 37%; Analysis 2.19), pruritus (RR 0.76, 95% CI 0.51 to 1.14; participants = 1108; studies = 8; I2 = 0%; Analysis 2.21), sweating (RR 4.52, 95% CI 0.54 to 37.94; participants = 220; studies = 2; I2 = 0%; Analysis 2.22), and discontinuation due to adverse events (RR 0.79, 95% CI 0.36 to 1.73; participants = 618; studies = 7; I2 = 9%; Analysis 2.24). However, the RRs for constipation (RR 0.75, 95% CI 0.66 to 0.86; participants = 1894; studies = 18; I2 = 38%; Analysis 2.7; Figure 6), hallucinations (RR 0.52, 95% CI 0.28 to 0.97; participants = 696; studies = 4; I2 = 0%; Analysis 2.14; Figure 10) and insomnia and lethargy (RR 0.48, 95% CI 0.26 to 0.90; participants = 314; studies = 2; I2 = 0%; Analysis 2.17) may be lower after treatment with CR oxycodone than after CR morphine. The I2 statistic was 83% for the outcome 'any adverse events' so the pooled results reported for this outcome (Analysis 2.1) should be disregarded.
2.4. Analysis.

Comparison 2: Adverse events, Outcome 4: Appetite loss/anorexia
2.12. Analysis.

Comparison 2: Adverse events, Outcome 12: Dysuria/uroschesis
2.19. Analysis.

Comparison 2: Adverse events, Outcome 19: Nausea & vomiting
2.14. Analysis.

Comparison 2: Adverse events, Outcome 14: Hallucinations
10.

Forest plot of comparison: 2 Adverse events, outcome: 2.14 Hallucinations
2.17. Analysis.

Comparison 2: Adverse events, Outcome 17: Insomnia & lethargy
2.1. Analysis.

Comparison 2: Adverse events, Outcome 1: Any adverse events
Zhang 2014 compared CR oxycodone to both CR morphine and CR MS Contin. In the meta‐analyses of adverse events, we included CR morphine as the comparison group. Sensitivity analyses substituting the CR morphine data with the CR MS Contin data showed that whether the comparison group was CR morphine or CR MS Contin made no difference to the conclusions.
Zhang 2016a compared treatment with CR oxycodone, CR morphine sulfate and CR morphine hydrochloride and, in the analyses described above, we have used the CR morphine sulfate data. Sensitivity analyses showed that using the CR morphine hydrochloride data instead made no difference to the conclusions.
Sensitivity analysis excluding the Chinese language studies made no difference to the overall results, with the exception of constipation, which indicated that there may be little to no difference between CR oxycodone and CR morphine in constipation (RR 0.98, 95% CI 0.82 to 1.16; participants = 797; studies = 5; I2 = 33%). For the outcome 'any adverse events', the I2 was reduced, but still too high to pool the results (53%).
Additional results reported by the individual studies: The evidence was very uncertain about the effect of CR oxycodone versus CR morphine on adverse events. Bruera 1998 reported that: "There were no statistically significant differences by treatment in mean severity for any of the elicited adverse events or in the frequency of reporting of unelicited events" (page 3225), but presented only data on sedation and nausea VAS ratings. Corli 2016 found that there may be little to no difference between the two treatment groups in the incidence of gastralgia and breathlessness, whether they were 'any degree' or 'severe.' Severe but not 'any degree' muscle spasm myoclonus may, however, occur more often in the CR morphine group than in the CR oxycodone group. Heiskanen 1997 conducted a cross‐over trial lasting three to six days per phase and found that vomiting may be more common during morphine treatment while constipation may be more common during oxycodone treatment; and that for the remaining adverse events reported there may be little to no difference between the drugs. In a parallel‐group trial lasting four weeks (with an extension of another four weeks), Mercadante 2010 found that there may be little to no difference in the reported adverse events between the groups. Lauretti 2003 found that there may be higher rates of nausea and vomiting in the CR and IR morphine group compared to the group who had a combination of CR oxycodone and IR morphine. Nosek 2017 reported only the bowel function index data split by treatment group, which may not differ between the groups. Similarly, the number of participants discontinuing treatment due to adverse events was very low and may not differ between the groups. From the ANOVAs reported by the authors, there was an indication (by non‐significant main effects of treatment and non‐significant interactions between treatment and time) that there may be little to no difference between the groups in terms of fatigue, insomnia, drowsiness, nausea, vomiting, constipation, (loss of) appetite, dyspnoea, depression and anxiety. Riley 2015 reported that there may be little to no difference in adverse event reaction scores between oxycodone and morphine, either in first‐line responders or non‐responders. The adverse event data from the remaining studies (Cao 2015; Gao 2012; Li 2013; Ren 2012; Song 2015; Sun 2013; Tu 2015; Wang 2008; Xie 2018; Ye 2012; Yu 2007; Yu 2009; Yu 2014; Zhang 2011; Zhang 2016a) have all been included in the meta‐analyses reported above with the following exceptions: mental disorders (Song 2015), dizziness and fatigue (Wang 2008), abdominal distension (Xie 2018) and insanity and 'other adverse events' (Sun 2013). They can all be found in Table 4 and Table 5 which contain all the adverse events reported by the included studies comparing CR oxycodone and CR morphine.
2. Controlled‐release (CR) oxycodone versus CR morphine (English‐language studies): adverse events.
| Comparison | CR oxycodone versus CR morphine | ||||||||||||||||||||
| Study | Bruera 1998 | Corli 2016 | Heiskanen 1997 | Lauretti 2003 | Mercadante 2010a | Mucci‐LoRusso 1998 | Nosek 2017 | Riley 2014 | Zhang 2014 | Zecca 2016 | |||||||||||
| Treatment | Oxy | Mor | Oxy | Mor | Oxy | Mor | Oxy | Mor | Oxy | Mor | Oxy | Mor | Oxy | Mor | Oxy | Mor | Oxy | Mor | MS Contin | Oxy | Mor |
| Any adverse events | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 40/48 | 39/52 | ‐ | ‐ | ‐ | ‐ | 25/57 | 34/57 | 31/57 | 77/81 | 79/94 |
| Severe/moderate adverse events | ‐ | ‐ | 48.8% | 58.9% | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Abnormal dreams | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/81 | 1/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anorexia, appetite loss | ‐ | ‐ | ‐ | ‐ | 0/27 | 1/27 | 14/22 | 13/22 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Chills | ‐ | ‐ | ‐ | ‐ | 1/27 | 0/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Confusion ‐ serious |
‐ | ‐ | 55/129 12/129 |
59/129 20/129 |
‐ | ‐ | ‐ | ‐ | 0.37 (0.49) | 0.25 (0.44) |
‐ | ‐ | ‐ | ‐ | 7/81 3/81 |
2/72 0/72 |
‐ | ‐ | ‐ | 12/85 | 12/88 |
| Constipation ‐ serious |
‐ | ‐ | 75/129 40/129 |
82/129 50/129 |
18/27 | 14/27 | 4/22 | 5/22 | 0.63 (0.68) | 0.7 (0.92) | 10/48 | 10/52 | 1.91 (1.15)b | 2.14 (1.01)b | 18/81 2/81 |
24/72 5/72 |
6/57 | 3/57 | 5/57 | 30/85 | 22/87 |
| Decreased mobility | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 2/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Depression | ‐ | ‐ | ‐ | ‐ | 1/27 | 0/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Diarrhoea | ‐ | ‐ | ‐ | ‐ | 2/27 | 2/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Dizziness, lightheadedness | ‐ | ‐ | ‐ | ‐ | 6/27 | 6/27 | ‐ | ‐ | ‐ | ‐ | 4/48 | 7/52 | ‐ | ‐ | 3/81 | 2/72 | 2/57 | 3/57 | 4/57 | ‐ | ‐ |
| Double vision | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 1/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Drowsiness, somnolence ‐ serious (with/without hallucinations) |
‐ | ‐ | 74/129 34/129 |
79/129 38/129 |
‐ | ‐ | 7/22 | 11/22 | 0.37 (0.6) | 0.35 (0.59) | 7/48 | 10/52 | ‐ | ‐ | 12/81 1/81 |
13/72 0/72 |
1/57 | 1/57 | 1/57 | 27/85 | 31/88 |
| Drunken feeling | ‐ | ‐ | ‐ | ‐ | 1/27 | 1/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Dry mouth ‐ severe |
‐ | ‐ | 66/129 27/129 |
66/129 31/129 |
12/27 | 15/27 | 3/22 | 2/22 | 0.63 (0.68) | 0.6 (0.68) | 1/48 | 7/52 | ‐ | ‐ | 3/81 | 2/72 | ‐ | ‐ | ‐ | 19/85 | 14/88 |
| Dyspnoea ‐severe |
‐ | ‐ | 12/129 3/129 |
17/129 4/129 |
2/27 | 2/27 | 0/22 | 0/22 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Dysuria ‐ severe |
‐ | ‐ | 17/129 4/129 |
22/129 2/129 |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/57 | 2/57 | 1/57 | ‐ | ‐ |
| Extrasystoles | ‐ | ‐ | ‐ | ‐ | 1/27 | 0/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Faecal incontinence | ‐ | ‐ | ‐ | ‐ | 1/27 | 1/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Fall | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 3/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Feeling abnormal | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 1/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Flatus | ‐ | ‐ | ‐ | ‐ | 0/27 | 1/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Gastralgia ‐ severe |
‐ | ‐ | 21/129 6/129 |
24/129 3/129 |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Hallucinations ‐ severe |
‐ | ‐ | 8/129 1/129 |
17/129 6/129 |
‐ | ‐ | 0/22 | 0/22 | ‐ | ‐ | 0/48 | 2/52 | ‐ | ‐ | 3/81 | 4/72 | ‐ | ‐ | ‐ | 3/91 | 4/94 |
| Hollow feeling | ‐ | ‐ | ‐ | ‐ | 1/27 | 0/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Lethargy | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Memory impairment | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 1/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Muscle twitches | ‐ | ‐ | ‐ | ‐ | 1/27 | 1/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 2/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Muscle spasm myoclonus ‐ severe |
‐ | ‐ | 23/129 0/129 |
14/129 6/129 |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Nausea ‐ serious (with vomiting)/severe |
12.3 | 13.9 | 63/129 22/129 |
64/129 19/129 |
14/27 | 16/27 | 1/22 | 8/22 | 0.84 (0.9) | 0.6 (0.75) | 6/48 | 8/52 | ‐ | ‐ | 10/81 1/81 |
6/72 0/72 |
11/57 | 15/57 | 14/57 | 18/85 | 13/88 |
| Nightmares | ‐ | ‐ | ‐ | ‐ | 0/27 | 3/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | |
| Pain | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 1/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Paresthesia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Pruritus ‐ severe |
‐ | ‐ | 16/129 2/129 |
24/129 3/129 |
10/27 | 7/27 | 1/22 | 1/22 | ‐ | ‐ | 4/48 | 5/52 | ‐ | ‐ | 3/81 | 2/72 | ‐ | ‐ | ‐ | 6/85 | 5/88 |
| Sedation | 21.4 | 25 | ‐ | ‐ | 16/27 | 18/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Sensation of empty head | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/22 | 0/11 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Slow speech | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Sweating, hyperhidrosis | ‐ | ‐ | ‐ | ‐ | 12/27 | 9/27 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Serious toxicity secondary to infection | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Urinary hesitation | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/81 | 1/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Visual impairment | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/81 | 0/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Vomiting ‐ severe |
‐ | ‐ | 29/129 12/129 |
35/129 12/129 |
5/27 | 10/27 | 0/22 | 7/22 | ‐ | ‐ | 6/48 | 5/52 | ‐ | ‐ | 9/81 | 4/72 | 5/57 | 10/57 | 6/57 | 11/85 | 10/88 |
| Discontinuation due to adverse events | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/48 | 6/52 | 0/16 | 1/14 | ‐ | ‐ | 0/57 | 0/57 | 0/57 | 6/91 | 3/94 |
| Unexpected serious adverse events | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2/81 | 7/72 | ‐ | ‐ | ‐ | ‐ | ‐ |
a Mean (SD) ratings (out of 3) experienced during week 4
bBowel function index mean (standard deviation) (range 0‐10, with constipation indicated if score = 3 or above) at 28 days
‐: not reported CR: controlled‐release Mor: morphine Oxy: oxycodone
3. Controlled‐release (CR) oxycodone versus CR morphine (Chinese‐language studies): adverse events.
| Comparison | CR oxycodone versus CR morphine | ||||||||||||||||||||
| Study |
Treat‐ ment |
Adverse event (AE) | |||||||||||||||||||
|
Any AE |
Se‐ ve‐ re AE |
Ab‐ domi‐ nal dis‐ ten‐ sion |
Ano‐ rexia/ appe‐ tite loss |
Con‐ sti‐ pa‐ tion |
Dizzi‐ ness |
Dizzi‐ ness & fa‐ tigue |
Drow‐ si‐ ness |
Drow‐ siness & fa‐ tigue |
Dry mouth |
Dys‐ uria/ uro‐ sche‐ sis |
Insa‐ nity/ men‐ tal disor‐ ders |
In‐ som‐ nia & le‐ thar‐ gy |
Nau‐ sea |
Nau‐ sea & vo‐ mi‐ ting |
Pru‐ ritis |
Swea‐ ting |
Res‐ pira‐ tory de‐ pres‐ sion |
Vo‐ mi‐ ting |
Dis‐ con‐ tinu‐ ation due to AE |
||
| Cao 2015 | Oxy | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/65 | ‐ | ‐ | ‐ | ‐ | 1/65 | ‐ |
| Mor | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 11/65 | ‐ | ‐ | ‐ | ‐ | 7/65 | ‐ | |
| Gao 2012 | Oxy | ‐ | ‐ | ‐ | ‐ | 2/30 | 3/30 | ‐ | 1/30 | ‐ | ‐ | ‐ | ‐ | ‐ | 4/30 | ‐ | ‐ | ‐ | ‐ | 2/30 | ‐ |
| Mor | ‐ | ‐ | ‐ | ‐ | 5/28 | 2/28 | ‐ | 2/28 | ‐ | ‐ | ‐ | ‐ | ‐ | 3/28 | ‐ | ‐ | ‐ | ‐ | 3/28 | ‐ | |
| Li 2013 | Oxy | 16/42 | ‐ | ‐ | ‐ | 9/42 | ‐ | ‐ | 4/42 | ‐ | ‐ | 0/42 | ‐ | ‐ | 3/42 | ‐ | ‐ | ‐ | ‐ | 0/42 | ‐ |
| Mor | 24/40 | ‐ | ‐ | ‐ | 12/40 | ‐ | ‐ | 5/40 | ‐ | ‐ | 1/40 | ‐ | ‐ | 4/40 | ‐ | ‐ | ‐ | ‐ | 2/40 | ‐ | |
| Ren 2012 | Oxy | ‐ | 0/40 | ‐ | 2/40 | 12/40 | 7/40 | ‐ | 5/40 | ‐ | ‐ | ‐ | ‐ | ‐ | 9/40 | ‐ | 2/40 | ‐ | ‐ | 13/40 | ‐ |
| Mor | ‐ | 0/40 | ‐ | 1/40 | 22/40 | 6/40 | ‐ | 6/40 | ‐ | ‐ | ‐ | ‐ | ‐ | 8/40 | ‐ | 2/40 | ‐ | ‐ | 14/40 | ‐ | |
| Song 2015 | Oxy | ‐ | ‐ | ‐ | ‐ | 5/55 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/55 | 3/55 | ‐ | 6/55 | 2/55 | ‐ | ‐ | ‐ | ‐ |
| Mor | ‐ | ‐ | ‐ | ‐ | 13/55 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 7/55 | 10/55 | ‐ | 15/55 | 4/55 | ‐ | ‐ | ‐ | ‐ | |
| Sun 2013a | Oxy | 64/102 | ‐ | ‐ | ‐ | 19/102 | ‐ | ‐ | ‐ | ‐ | 1/102 | 0/102 | 2/102 | 10/102 | ‐ | 25/102 | 3/102 | ‐ | ‐ | ‐ | ‐ |
| Mor | 99/102 | ‐ | ‐ | ‐ | 36/102 | ‐ | ‐ | ‐ | ‐ | 4/102 | 3/102 | 6/102 | 17/102 | ‐ | 21/102 | 5/102 | ‐ | ‐ | ‐ | ‐ | |
| Tu 2015 | Oxy | ‐ | ‐ | ‐ | ‐ | 5/43 | 3/43 | ‐ | ‐ | 2/43 | ‐ | 1/43 | ‐ | ‐ | ‐ | 5/43 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Mor | ‐ | ‐ | ‐ | ‐ | 12/43 | 3/43 | ‐ | ‐ | 3/43 | ‐ | 1/43 | ‐ | ‐ | ‐ | 4/43 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Wang 2008 | Oxy | ‐ | ‐ | ‐ | ‐ | 9/30 | ‐ | 1/30 | 2/30 | ‐ | ‐ | 1/30 | ‐ | ‐ | 4/30 | ‐ | ‐ | ‐ | 0/30 | 3/30 | 0/30 |
| Mor | ‐ | ‐ | ‐ | ‐ | 18/30 | ‐ | 2/30 | 4/30 | ‐ | ‐ | 2/30 | ‐ | ‐ | 5/30 | ‐ | ‐ | ‐ | 0/30 | 4/30 | 0/30 | |
| Xie 2018 | Oxy | 3/48 | ‐ | 0/48 | ‐ | 1/48 | 1/48 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/48 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Mor | 10/47 | ‐ | 2/47 | ‐ | 2/47 | 3/47 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/47 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Ye 2012 | Oxy | 18/42 | ‐ | ‐ | ‐ | 3/42 | 2/42 | ‐ | 2/42 | ‐ | ‐ | 1/42 | ‐ | ‐ | 7/42 | ‐ | ‐ | ‐ | ‐ | 3/42 | ‐ |
| Mor | 28/41 | ‐ | ‐ | ‐ | 5/41 | 2/41 | ‐ | 3/41 | ‐ | ‐ | 1/41 | ‐ | ‐ | 10/41 | ‐ | ‐ | ‐ | ‐ | 7/41 | ‐ | |
| Yu 2007 | Oxy | ‐ | ‐ | ‐ | 2/15 | 5/15 | 5/15 | ‐ | 2/15 | ‐ | ‐ | ‐ | ‐ | ‐ | 1/15 | ‐ | 2/15 | ‐ | ‐ | 2/15 | ‐ |
| Mor | ‐ | ‐ | ‐ | 3/15 | 7/15 | 4/15 | ‐ | 2/15 | ‐ | ‐ | ‐ | ‐ | ‐ | 2/15 | ‐ | 3/15 | ‐ | ‐ | 3/15 | ‐ | |
| Yu 2009 | Oxy | ‐ | ‐ | ‐ | ‐ | 7/32 | 8/32 | ‐ | 5/32 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 10/32 | ‐ | ‐ | ‐ | ‐ | 1/32 |
| Mor | ‐ | ‐ | ‐ | ‐ | 15/30 | 7/30 | ‐ | 4/30 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 18/30 | ‐ | ‐ | ‐ | ‐ | 3/30 | |
| Zhang 2011 | Oxy | ‐ | ‐ | ‐ | ‐ | 15/35 | ‐ | ‐ | 3/35 | ‐ | ‐ | ‐ | ‐ | ‐ | 10/35 | ‐ | ‐ | 2/35 | ‐ | 4/35 | 0/35 |
| Mor | ‐ | ‐ | ‐ | ‐ | 13/32 | ‐ | ‐ | 1/32 | ‐ | ‐ | ‐ | ‐ | ‐ | 10/32 | ‐ | ‐ | 0/32 | ‐ | 4/32 | 0/32 | |
| Zhang 2016a | Oxy | 6/40 | ‐ | ‐ | ‐ | 0/40 | 1/40 | ‐ | 2/40 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/40 | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Mor sulfate |
14/40 | ‐ | ‐ | ‐ | 2/40 | 5/40 | ‐ | 4/40 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/40 | ‐ | ‐ | ‐ | ‐ | ‐ | |
|
Mor hydro‐ chloride |
13/40 | ‐ | ‐ | ‐ | 1/40 | 4/40 | ‐ | 5/40 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 3/40 | ‐ | ‐ | ‐ | ‐ | ||
aSun 2013 also reported that 4/102 participants in the CR oxycodone group and 8/102 participants in the CR morphine group experienced "Other adverse events".
‐: not reported AE: adverse event CR: controlled‐release Mor: morphine Oxy: oxycodone
We judged the certainty of evidence for this outcome to be low or very low in all cases. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance/detection bias, attrition bias, or a combination of these), and we downgraded the certainty of the evidence by no, one or two levels due to imprecision (arising from low or very low event rates).
Quality of life
The evidence was very uncertain about the effect of CR oxycodone versus CR morphine on quality of life. Mucci‐LoRusso 1998 reported no clinically significant changes in quality of life for either treatment group, but did not show results or analyses. The authors also found there may be little to no difference in treatment acceptability between the treatment groups with 74% and 77% of the CR oxycodone and CR morphine participants, respectively, rating the acceptability of treatment as good to excellent and the mean acceptability ratings at the study end being 4 (SE = 0.1) in the CR oxycodone and 3.9 (SE = 0.1) in the CR morphine participants. Heiskanen 1997 found that the mean daily acceptability of treatment ratings may be higher for morphine (3.49/5; SE = 0.12) than for oxycodone (3.19/5; SE = 0.11), but Lauretti 2003 also found that there may be little to no difference in treatment acceptance between treatment with CR and IR morphine and treatment with the combination of CR oxycodone and IR morphine. Again, Nosek 2017 did not report the quality of life results by treatment group or analyse any significant interactions fully, which means that it is impossible to know what the results were. We are, therefore, limited to reporting what the authors reported, which was that there were no significant differences between the groups in emotional functioning or overall quality of life, however, there was a significant interaction between treatment and time for "well‐being" (not further analysed) and a significant main effect for physical functioning, which the authors reported as "the most beneficial effect was obtained for morphine" (page 3217). Finally, the Karnofsky Performance Scale score, which was reported by treatment, may not differ between the (four) treatment groups (CR oxycodone mean = 65, SD = 11.55; CR morphine mean = 66.67, SD = 9.85).
We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and performance, detection and attrition bias), and we downgraded one level for imprecision (arising from low participant numbers).
Participant preference
The evidence was very uncertain about the effect of CR oxycodone versus CR morphine on participant preference. In a cross‐over trial with each phase lasting seven days, Bruera 1998 reported that 8/23 participants preferred CR oxycodone treatment while 11/23 participants preferred treatment with CR morphine. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and attrition bias), and we downgraded the certainty of the evidence by two levels due to imprecision (arising from very low participant numbers).
See also Table 2.
Controlled‐release oxycodone versus controlled‐release hydromorphone
One study compared CR oxycodone to CR hydromorphone (Hagen 1997).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus CR hydromorphone on pain intensity. In a cross‐over trial lasting seven days per phase, Hagen 1997 found that there may be little to no difference in pain intensity between treatment with CR oxycodone and CR hydromorphone (Figure 4). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and attrition bias), and two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus CR hydromorphone on adverse events. Hagen 1997 observed that there may be little to no difference in the frequency of adverse events between treatment groups with the exception of drowsiness, which may occur more frequently during treatment with oxycodone (see Table 6). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and attrition bias), and two levels for imprecision (arising from very low participant/event numbers).
4. Other oral oxycodone comparisons: adverse events.
| Comparison | CR oxycodone versus CR hydromorphone | CR oxycodone versus ER hydromorphone | CR oxycodone versus ER oxymorphone | CR oxycodone versus ER tapentadol | CR oxycodone versus oral ibuprofen | IR oxycodone versus IR hydromorphone | ||||||||
| Study | Hagen 1997 | Inoue 2017 | Yu 2014 | Gabrail 2004 | Imanaka 2013 | Liu 2021 | Inoue 2018 | |||||||
| Treatment | Oxy | Hyd | Oxy | Hyd | Oxy | Hyd | Oxy | Oxymo | Oxy | Tap | Oxy | Ibu | Oxy | Hyd |
| Any adverse events | ‐ | ‐ | 77/92 | 71/88 | 117/126 | 111/128 | ‐ | ‐ | 155/172 | 147/168 | ‐ | ‐ | 65/84 | 73/88 |
| Total adverse events | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Severe adverse events | ‐ | ‐ | 10/92 | 7/88 | 30/126 | 30/128 | ‐ | ‐ | ‐ | ‐ | 0/34 | 0/32 | ‐ | ‐ |
| Serious adverse events (including death) | ‐ | ‐ | 14/92 | 11/88 | 18/126 | 11/128 | ‐ | ‐ | ‐ | ‐ | 0/34 | 0/32 | 8/84 | 7/88 |
| Serious adverse events (including death), considered related to study drugs | ‐ | ‐ | 6/92 | 4/88 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/34 | 0/32 | 3/84 | 4/88 |
| Abdominal discomfort | ‐ | ‐ | ‐ | ‐ | 7/126 | 4/128 | ‐ | ‐ | ‐ | ‐ | 5/34 | 10/32 | ‐ | ‐ |
| Abdominal distension | ‐ | ‐ | ‐ | ‐ | 7/126 | 7/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anaemia | ‐ | ‐ | ‐ | ‐ | 14/126 | 14/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anorexia, appetite loss | ‐ | ‐ | 3/92 | 7/88 | 21/126 | 20/128 | ‐ | ‐ | 24/172 | 23/168 | ‐ | ‐ | ‐ | ‐ |
| Asthenia | ‐ | ‐ | ‐ | ‐ | 9/126 | 11/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Bone marrow failure | ‐ | ‐ | ‐ | ‐ | 9/126 | 9/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Chest discomfort | ‐ | ‐ | ‐ | ‐ | 6/126 | 9/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Confusion | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Constipation | ‐ | ‐ | 14/92 | 11/88 | 45/126 | 43/128 | 19/41 | 21/43 | 64/172 | 51/168 | 9/34 | 3/32 | 19/84 | 21/88 |
| Delirium | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 6/172 | 10/168 | ‐ | ‐ | 5/84 | 2/88 |
| Diarrhoea | ‐ | ‐ | 17/92 | 16/88 | 9/126 | 12/128 | ‐ | ‐ | 19/172 | 11/168 | ‐ | ‐ | 9/84 | 12/88 |
| Dizziness or lightheadedness | ‐ | ‐ | 5/92 | 6/88 | 22/126 | 21/128 | 9/41 | 7/43 | ‐ | ‐ | 10/34 | 4/32 | ‐ | ‐ |
| Drowsiness, somnolence | 28/31 | 19/31 | 18/92 | 23/88 | ‐ | ‐ | ‐ | ‐ | 36/172 | 29/168 | 3/34 | 0/32 | 21/84 | 23/88 |
| Fever | ‐ | ‐ | 5/92 | 7/88 | 27/126 | 24/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Hallucinations | 0/31 | 2/31 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Hypoproteinaemia | ‐ | ‐ | ‐ | ‐ | 5/126 | 9/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Insomnia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 11/172 | 9/168 | ‐ | ‐ | ‐ | ‐ |
| Malaise | ‐ | ‐ | 6/92 | 3/88 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Nausea | 15 (3)a | 13 (3)a | 21/92 | 36/88 | 45/126 | 43/128 | 15/41 | 17/43 | 61/172 | 48/168 | 3/34 | 0/32 | 14/84 | 14/88 |
| Neutrophil count decreased | ‐ | ‐ | ‐ | ‐ | 5/126 | 7/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Oedema, peripheral | ‐ | ‐ | ‐ | ‐ | 6/126 | 11/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Platelet count decreased | ‐ | ‐ | ‐ | ‐ | 7/126 | 8/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Pruritus | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 8/41 | 13/43 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Rash | ‐ | ‐ | ‐ | ‐ | 4/126 | 7/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Respiratory depression | ‐ | ‐ | 0/92 | 0/88 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Sedation | 24 (4)a | 18 (3)a | ‐ | ‐ | ‐ | ‐ | 13/41 | 18/43 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Sweating or hyperhidrosis | ‐ | ‐ | ‐ | ‐ | 8/126 | 3/128 | 9/41 | 12/43 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Transaminases elevated | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2/34 | 7/32 | ‐ | ‐ |
| Urinary retention | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Urinary tract infection | ‐ | ‐ | ‐ | ‐ | 7/126 | 4/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Urination difficulty | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1/34 | 0/32 | ‐ | ‐ |
| Vomiting | ‐ | ‐ | 16/92 | 32/88 | 47/126 | 43/128 | 7/41 | 5/43 | 41/172 | 42/168 | ‐ | ‐ | 15/84 | 17/88 |
| White blood cell count decreased | ‐ | ‐ | ‐ | ‐ | 17/126 | 13/128 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Discontinuation due to adverse events | ‐ | ‐ | 14/92 | 10/88 | 18/126 | 19/128 | ‐ | ‐ | 29/172 | 22/168 | ‐ | ‐ | 8/84 | 6/88 |
| Deaths due to study drug treatment | ‐ | ‐ | ‐ | ‐ | 0/126 | 0/128 | ‐ | ‐ | ‐ | ‐ | 0/34 | 0/32 | ‐ | ‐ |
aMean (standard error) visual analogue scale across all days
‐: not reported CR: controlled‐release ER: extended‐release Hyd: hydromorphone Ibu: Ibuprofen IR: Immediate‐release IV: intravenous Mor: morphine Oxy: oxycodone Oxymo: oxymorphone Tap: tapentadol
Quality of life
The study did not report quality of life.
Participant preference
The evidence was very uncertain about the effect of CR oxycodone versus CR hydromorphone on participant preference. Hagen 1997 found that 25.8% of participants had no treatment preference, with approximately half of the remaining participants preferring oxycodone (35.5%) while the other half preferred hydromorphone (38.7%). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and attrition bias), and two levels for imprecision (arising from very low participant numbers).
Controlled‐release oxycodone versus extended‐release hydromorphone
Two studies compared CR oxycodone to ER hydromorphone (Inoue 2017; Yu 2014).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus ER hydromorphone on pain intensity and pain relief. Pooled analysis including both the parallel‐group Inoue 2017 trial of seven days' duration and the 28‐day parallel‐group trial by Yu 2014 showed that there may be little to no difference in pain intensity between the two treatment groups (Analysis 1.1; Figure 4). Yu 2014 also found that there may be little to no difference in pain intensity or pain relief between treatment with CR oxycodone and ER hydromorphone on any of the following additional pain measures: mean pain on average (CR oxycodone 3.3; ER hydromorphone 2.9), mean pain at its least in past 24 hours (CR oxycodone 1.9; ER hydromorphone 1.6), mean pain 'right now' (CR oxycodone 2.8; ER hydromorphone 2.7), mean pain relief in past 24 hours (CR oxycodone 62.2%; ER hydromorphone 64.5%), and the number of rescue medication doses taken during the overall maintenance phase (CR oxycodone 29.3; ER hydromorphone 24.2). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of selection, performance and detection bias, and attrition bias), and one level for imprecision (arising from low participant numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus ER hydromorphone on adverse events. Pooled analysis of the event rates showed that there may be little to no difference between the treatment groups for any adverse event (RR 1.06, 95% CI 0.98 to 1.14; participants = 434; studies = 2; I2 = 0%; Analysis 2.1), severe adverse events (RR 1.08, 95% CI 0.73 to 1.62; participants = 434; studies = 2; I2 = 0%; Analysis 2.2), serious adverse events including death (RR 1.44, 95% CI 0.86 to 2.39; participants = 434; studies = 2; I2 = 0%; Analysis 2.3), appetite loss/anorexia (RR 0.89, 95% CI 0.54 to 1.49; participants = 434; studies = 2; I2 = 42%; Analysis 2.4), constipation (RR 1.10, 95% CI 0.80 to 1.49; participants = 434; studies = 2; I2 = 0%; Analysis 2.7; Figure 6), diarrhoea (RR 0.91, 95% CI 0.55 to 1.49; participants = 434; studies = 2; I2 = 0%; Analysis 2.8), dizziness (RR 1.00, 95% CI 0.61 to 1.64; participants = 434; studies = 2; I2 = 0%; Analysis 2.9), fever (RR 1.04, 95% CI 0.66 to 1.62; participants = 434; studies = 2; I2 = 0%; Analysis 2.13) and discontinuation of treatment due to adverse events (RR 1.09, 95% CI 0.69 to 1.75; participants = 434; studies = 2; I2 = 0%; Analysis 2.24). For both nausea and vomiting, the I2 statistics were very high (80% and 86%, respectively), so the pooled results in Figure 8 (Analysis 2.18) and Figure 9 (Analysis 2.23), respectively, for this treatment subgroup should be ignored. For both nausea and vomiting, Inoue 2017 reported that the rates may be lower in the CR oxycodone group compared to the ER hydromorphone group, whereas Yu 2014 found that there may be little to no difference between the interventions. See also Table 6 for other adverse events that were reported only by one or the other of the studies. We judged the certainty of evidence for this outcome to be moderate (for 'any adverse event') or very low (for all other reported adverse events), depending on the event rates of the individual adverse events. We downgraded the certainty of evidence by one level for risk of bias (due to under‐reporting of selection, performance and detection bias) and by none or two levels for imprecision (arising from very low event rates).
2.2. Analysis.

Comparison 2: Adverse events, Outcome 2: Severe adverse events
2.3. Analysis.

Comparison 2: Adverse events, Outcome 3: Serious adverse events, including death
2.8. Analysis.

Comparison 2: Adverse events, Outcome 8: Diarrhoea
2.13. Analysis.

Comparison 2: Adverse events, Outcome 13: Fever
Quality of life
The studies did not report quality of life.
Participant preference
The studies did not report participant preference.
Controlled‐release oxycodone versus extended‐release oxymorphone
One study compared CR oxycodone to ER oxymorphone (Gabrail 2004).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus ER oxymorphone on pain intensity. Gabrail 2004, in a cross‐over trial with each phase lasting seven to 10 days, found that there may be little to no difference in mean 24‐hour average daily pain intensity ratings between the two treatments (Analysis 1.1; Figure 4). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus ER oxymorphone on adverse events. Gabrail 2004 reported that there may be little to no difference in adverse event rates between the drugs (see Table 6), and no participants withdrew due to abnormal laboratory values, insufficient analgesia, or loss to follow‐up. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection, performance, detection, and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Quality of life
The evidence was very uncertain about the effect of CR oxycodone versus ER oxymorphone on quality of life. Gabrail 2004 reported that there may be little to no difference in quality of life (general activity, mood, walking ability, normal work, relationships with others, sleep, and enjoyment of life) between the drugs. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection, performance, detection, and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Participant preference
The evidence was very uncertain about the effect of CR oxycodone versus ER oxymorphone on participant preference. The study reported no data for participant preference, but Gabrail 2004 found that 78.3% of participants rated oxycodone as 'excellent,' 'very good,' or 'good' with 86.4% of the participants giving oxymorphone such ratings. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection, performance, and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Controlled‐release oxycodone versus extended‐release tapentadol
One study compared CR oxycodone to ER tapentadol (Imanaka 2013).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus ER tapentadol on pain intensity. Imanaka 2013, in a parallel‐group trial of four weeks' duration, found that there may be little to no difference in pain intensity between the study groups (see Figure 4) with 82/139 CR oxycodone participants and 80/126 ER tapentadol participants reporting ≥ 30% improvement in pain intensity during the last three days of treatment, and 59/139 CR oxycodone participants and ER 63/126 tapentadol participants reporting ≥ 50% improvement in pain intensity during the last three days of treatment (Figure 5). We judged the certainty of evidence for this outcome to be low. We downgraded the certainty of evidence by one level for serious limitations to study quality due to risk of bias (arising from attrition bias), and by one level for imprecision (arising from low participant numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus ER tapentadol on adverse events. Inspection of Table 6 suggested that there may be little to no difference in adverse event rates between the treatment groups, but Imanaka 2013 did not present any formal statistical analyses of this apparent equality. We judged the certainty of evidence for this outcome to be high (for 'any adverse events') or low (all other reported adverse events), depending on the event rates of the individual adverse events. We downgraded the certainty of evidence by no or two levels for imprecision (arising from very low event rates).
Quality of life
The study did not report quality of life.
Participant preference
The study did not report participant preference.
Controlled‐release oxycodone versus transdermal fentanyl
Three studies compared CR oxycodone to TD fentanyl (Corli 2016; Nosek 2017; Su 2015).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus TD fentanyl on pain intensity and pain relief. Pooled analysis including Corli 2016 and Su 2015 showed that there may be little to no difference in pain intensity scores after treatment with CR oxycodone or TD fentanyl (SMD 0.02, 95% CI ‐0.19 to 0.24; participants = 329; studies = 2; I2 = 0%; Analysis 1.1; Figure 4). This was also the case when pain relief was analysed as a dichotomous measure (RR 0.98, 95% CI 0.85 to 1.14; participants = 329; studies = 2; I2 = 0%; Analysis 1.2; Figure 5). For Nosek 2017, the situation was the same for this comparison as for the comparison between CR oxycodone and CR morphine reported above.
Corli 2016 compared CR oxycodone with TD fentanyl in a four‐arm trial of 28 days' duration, which also included a TD buprenorphine and a CR morphine group, and found that there may be little to no difference between CR oxycodone and TD fentanyl treatment in terms of the requirement for additional opioids (CR oxycodone 33/125 participants; TD fentanyl 46/124 participants), premature discontinuations for pain‐related reasons (CR oxycodone 19/125 participants; TD fentanyl 18/124 participants), proportion of participants requiring adjuvant drugs (CR oxycodone 102/125 participants; TD fentanyl 100/124 participants), or proportion of participants requiring switches (CR oxycodone 15/125 participants; TD fentanyl 16/124 participants). However, a higher proportion of participants in the CR fentanyl group may have required a mean increase in the opioid daily dose > 5% according to the opioid escalation index compared to the oxycodone group (CR oxycodone 24/125 participants; TD fentanyl 45/124 participants). In a parallel‐group trial lasting two weeks, Su 2015 found that there may be little to no difference between groups treated with CR oxycodone or TD fentanyl in pain relief (CR oxycodone 90.48%; TD fentanyl 92.11%) or pain intensity measured by response categories (CR oxycodone: 15 complete response, 21 partial response, 2 minor response, 4 no response; TD fentanyl: 13 complete response, 20 partial response, 2 minor response, 3 no response).
We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance bias, detection bias, attrition bias and selective reporting bias), and by one level for imprecision (arising from low participant/event numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus TD fentanyl on adverse events. Pooled analysis of the event rates for constipation and dysuria revealed that the I2 statistic was 83% for constipation, so the pooled results shown in Figure 6 (Analysis 2.7) for this treatment subgroup should be ignored, and that there may be little to no difference between treatment groups for dysuria (RR 1.15, 95% CI 0.62 to 2.16; participants = 336; studies = 2; I2 = 0%; Analysis 2.12).
Corli 2016 found that there may be little to no difference between the two treatment groups in the incidence of the following adverse events, whether they were 'any degree' or 'severe': drowsiness, confusion, nausea, vomiting, constipation, dry mouth, hallucinations, gastralgia, muscle spasm myoclonus, breathlessness, and itching. For Nosek 2017, the situation was the same for this comparison as for the comparison between CR oxycodone and CR morphine reported above. Su 2015 reported that the rates of nausea and vomiting and constipation may be significantly higher in the CR oxycodone group than in the TD fentanyl group, and that there may be little to no difference in the rates of dizziness and lethargy between the groups. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance bias, detection bias, attrition bias and selective reporting bias), and by one or two levels (depending on the event rates of the individual adverse events) for imprecision (arising from low or very low event rates). See Table 7.
5. Controlled‐release (CR) oxycodone versus transdermal fentanyl or buprenorphine.
| Comparison | CR oxycodone versus TD fentanyl | CR oxycodone versus TD buprenorphine | ||||||||
| Study | Corli 2016 | Nosek 2017 | Su 2015 | Corli 2016 | Nosek 2017 | |||||
| Treatment | Oxy | Fen | Oxy | Fen | Oxy | Fen | Oxy | Bup | Oxy | Bup |
| Severe/moderate adverse events | 48.8% | 50.4% | ‐ | ‐ | 48.8% | 60% | ||||
| Breathlessness | 12/129 | 22/127 | ‐ | ‐ | 12/129 | 30/130 | ||||
| Breathlessness, severe | 3/129 | 5/127 | ‐ | ‐ | 3/129 | 6/130 | ||||
| Confusion | 55/129 | 46/127 | ‐ | ‐ | 55/129 | 61/130 | ||||
| Confusion, severe | 12/129 | 8/127 | ‐ | ‐ | 12/129 | 12/130 | ||||
| Constipation | 75/129 | 77/127 | 1.91 (1.15)a | 2.77 (1.45)a | 13/42 | 3/38 | 75/129 | 87/130 | 1.91 (1.15)a | 1.76 (2.06)a |
| Constipation, severe | 40/129 | 36/127 | ‐ | ‐ | 40/129 | 39/130 | ||||
| Dizziness | ‐ | ‐ | 4/42 | 3/38 | ‐ | ‐ | ||||
| Drowsiness | 74/129 | 70/127 | ‐ | ‐ | 74/129 | 81/130 | ||||
| Drowsiness, severe | 34/129 | 26/127 | ‐ | ‐ | 34/129 | 40/130 | ||||
| Dry mouth | 66/129 | 67/127 | ‐ | ‐ | 66/129 | 73/130 | ||||
| Dry mouth, severe | 27/129 | 29/127 | ‐ | ‐ | 27/129 | 30/130 | ||||
| Dysuria | 17/129 | 13/127 | 2/42 | 3/38 | 17/129 | 16/130 | ||||
| Dysuria, severe | 4/129 | 4/127 | ‐ | ‐ | 4/129 | 4/130 | ||||
| Gastralgia | 21/129 | 26/127 | ‐ | ‐ | 21/129 | 21/130 | ||||
| Gastralgia, severe | 6/129 | 4/127 | ‐ | ‐ | 6/129 | 1/130 | ||||
| Hallucinations | 8/129 | 3/127 | ‐ | ‐ | 8/129 | 8/130 | ||||
| Hallucinations, severe | 1/129 | 0/127 | ‐ | ‐ | 1/129 | 2/130 | ||||
| Itching | 16/129 | 14/127 | ‐ | ‐ | 16/129 | 21/130 | ||||
| Itching, severe | 2/129 | 3/127 | ‐ | ‐ | 2/129 | 1/130 | ||||
| Lethargy | ‐ | ‐ | 3/42 | 2/38 | ‐ | ‐ | ||||
| Muscle spasm/myoclonus | 23/129 | 15/127 | ‐ | ‐ | 23/129 | 24/130 | ||||
| Muscle spasm/myoclonus, severe | 0/129 | 3/127 | ‐ | ‐ | 0/129 | 1/130 | ||||
| Nausea and vomiting | ‐ | ‐ | 11/42 | 2/38 | ‐ | ‐ | ||||
| Nausea | 63/129 | 57/127 | ‐ | ‐ | 63/129 | 59/130 | ||||
| Nausea, severe | 22/129 | 16/127 | ‐ | ‐ | 22/129 | 18/130 | ||||
| Vomiting | 29/129 | 29/127 | ‐ | ‐ | 29/129 | 30/130 | ||||
| Vomiting, severe | 12/129 | 10/127 | ‐ | ‐ | 12/129 | 5/130 | ||||
| Discontinuation due to adverse events | 0/16 | 1/15 | 0/16 | 1/17 | ||||||
aBowel function index mean (standard deviation) (range 0‐10, with constipation indicated if score = 3 or above) at 28 days
‐: not reported Bup: buprenorphine CR: controlled‐release Fen: fentanyl Oxy: oxycodone TD: transdermal
Quality of life
The evidence was very uncertain about the effect of CR oxycodone versus TD fentanyl on quality of life. For Nosek 2017, the situation was the same for this comparison as for the comparison between CR oxycodone and CR morphine reported above, with the exception that the Karnofsky Performance Scale mean score was 58 and the SD was 13.98 for the TD fentanyl group.
Su 2015 found that there may be little to no difference in quality of life as measured by the Karnofsky Performance Status (from 0 to 100) (CR oxycodone mean 75.79; TD fentanyl mean 74.05). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from selection, performance, detection, attrition and selective reporting bias), and by two levels for imprecision (arising from very low participant numbers).
Participant preference
None of the studies reported participant preference.
Controlled‐release oxycodone versus transdermal buprenorphine
Two studies compared CR oxycodone to TD buprenorphine (Corli 2016; Nosek 2017).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus TD buprenorphine on pain intensity and pain relief. Corli 2016 compared CR oxycodone with TD buprenorphine in a four‐arm trial of 28 days' duration, which also included a TD fentanyl and a CR morphine group, and found that there may be little to no difference between CR oxycodone and TD buprenorphine groups in terms of pain intensity (Analysis 1.1; Figure 4) or pain relief (Analysis 1.2; Figure 5). Corli 2016 found that there may be little to no difference between the groups in terms of requirement for additional opioids (CR oxycodone 33/125 participants; TD buprenorphine 48/127 participants), opioid escalation index > 5% (CR oxycodone 24/125 participants; TD buprenorphine 18/127 participants), premature discontinuations for pain‐related reasons (CR oxycodone 19/125 participants; TD buprenorphine 26/127 participants), proportion of participants requiring adjuvant drugs (CR oxycodone 102/125 participants; TD buprenorphine 100/127 participants), or proportion of participants requiring switches (CR oxycodone 15/125 participants; TD buprenorphine 21/127 participants). For Nosek 2017, the situation was the same for this comparison as for the comparison between CR oxycodone and CR morphine reported above. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from performance bias, detection bias, attrition bias and selective reporting bias), and by one level for imprecision (arising from low participant numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus TD buprenorphine on adverse events. Corli 2016 found that there may be little to no difference between the two treatment groups in the incidence of the following adverse events, whether they were 'any degree' or 'severe': drowsiness, confusion, nausea, vomiting, constipation, dry mouth, hallucinations, gastralgia, dysuria, muscle spasm myoclonus, and itching. 'Any degree,' but not severe, breathlessness may occur more often in the TD buprenorphine group than in the CR oxycodone group. For Nosek 2017, the situation was the same for this comparison as for the comparison between CR oxycodone and CR morphine reported above. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from performance bias, detection bias, attrition bias and selective reporting bias), and by one or two levels (depending on the event rates for the individual adverse events) for imprecision (arising from low or very low event rates). See Table 7.
Quality of life
The evidence was very uncertain about the effect of CR oxycodone versus TD buprenorphine on quality of life. Corli 2016 did not report quality of life. For Nosek 2017, the situation was the same for this comparison as for the comparison between CR oxycodone and CR morphine reported above with the exception that the Karnofsky Performance Scale mean score was 64.67 and the SD was 10.6 for the TD buprenorphine group. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from performance bias, detection bias, attrition bias and selective reporting bias), and by two levels for imprecision (arising from very low participant numbers).
Participant preference
Neither study reported participant preference.
Controlled‐release oxycodone versus oral ibuprofen
One study compared CR oxycodone to oral ibuprofen (Liu 2021).
Pain intensity and pain relief
The evidence was very uncertain about the effect of CR oxycodone versus oral ibuprofen on pain relief. Liu 2021, in a parallel‐group trial of seven days' duration, found that the rates of achieving complete or significant pain relief may be higher after treatment with CR oxycodone than with oral ibuprofen (Analysis 1.2; Figure 5). One of the 32 participants in the ibuprofen group achieved no pain relief whereas this was the case for none of the 34 participants in the oxycodone group. The remaining participants in both groups achieved partial pain relief. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from uncertainty about allocation concealment and performance and detection bias), and by two levels for imprecision (arising from low participant/event numbers).
Adverse events
The evidence was very uncertain about the effect of CR oxycodone versus oral ibuprofen on adverse events. Inspection of Table 6 suggested that there may be little to no difference in adverse event rates between the treatment groups, which was also confirmed by the analyses reported by Liu 2021. All of the adverse events were grade I or II. No grade III or above adverse events occurred. We judged the certainty of evidence for this outcome to be very low, We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from uncertainty about allocation concealment and performance and detection bias), and by two levels for imprecision (arising from low participant/event numbers).
Quality of life
The evidence was very uncertain about the effect of CR oxycodone versus oral ibuprofen on quality of life. Liu 2021 assessed quality of life using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire‐Core15_Palliative (EORTC QLQ‐C15‐PAL) scale and the Edmonton Symptom Assessment System (ESAS) and found that there may be little to no difference in quality of life between the treatment groups (mean (SD) EORTC QLQ‐C15‐PAL scores after treatment = 20 (2.67) in the oxycodone group and 21.09 (2.37) in the ibuprofen group; mean (SD) ESAS scores after treatment = 18.82 (5.59) in the oxycodone group and 20.19 (4.3) in the ibuprofen group). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from uncertainty about allocation concealment and performance and detection bias), and by two levels for imprecision (arising from low participant/event numbers).
Participant preference
The study did not report participant preference.
Immediate‐release oxycodone versus immediate‐release hydromorphone
One study compared IR oxycodone to IR hydromorphone (Inoue 2018).
Pain intensity and pain relief
The evidence was very uncertain about the effect of IR oxycodone versus IR hydromorphone on pain intensity. Inoue 2018, in a parallel‐group trial of five days' duration, found that there may be little to no difference in pain intensity between the study groups (Analysis 1.1; Figure 4). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by one level for serious limitations to study quality due to risk of bias (arising from under‐reporting of performance and detection bias), and by two levels for imprecision (arising from low participant numbers).
Adverse events
The evidence was very uncertain about the effect of IR oxycodone versus IR hydromorphone on adverse events. Inspection of Table 6 suggested that there may be little to no difference in adverse event rates between the treatment groups, which was also confirmed by the analyses reported by Inoue 2018. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by one level for serious limitations to study quality due to risk of bias (arising from under‐reporting of performance and detection bias), and by two levels for imprecision (arising from low participant/event numbers).
Quality of life
The study did not report quality of life.
Participant preference
The study did not report participant preference.
Intravenous oxycodone versus rectal oxycodone
One study compared IV oxycodone to rectal oxycodone (Leow 1995).
Pain intensity and pain relief
The evidence was very uncertain about the effect of IV oxycodone versus rectal oxycodone on pain relief. Leow 1995 conducted a single‐dose cross‐over study in 12 participants, with each phase lasting 24 hours, and found that while IV oxycodone may be associated with faster onset of pain relief relative to rectal oxycodone, rectal oxycodone may be associated with a longer duration of pain relief compared to IV oxycodone. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting and performance bias and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of IV oxycodone versus rectal oxycodone on adverse events. Leow 1995 found that there may be little to no difference in the adverse event profiles for the two study arms (see Table 8). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance bias and detection bias), and by two levels for imprecision (arising from very low participant numbers).
6. Intravenous (IV) oxycodone comparisons: adverse events.
| Comparison | IV oxycodone versus IV morphine | IV oxycodone versus rectal oxycodone | IV oxycodone followed by IR oxycodone versus IV morphine followed by IR morphine | |||||
| Study | Lee 2017 | Leow 1995a | Kalso 1990b | |||||
| Treatment | Oxy | Mor | IV | Rectal | IV oxy | IR oxy | IV mor | IR mor |
| Any adverse events | 29/34 | 26/32 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Total adverse events | ‐ | ‐ | 82 | 94 | ‐ | ‐ | ‐ | ‐ |
| Severe adverse events | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Serious adverse events | 3/34 | 2/32 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Unexpected adverse events | 9/34 | 16/32 | ||||||
| Adverse drug reaction | 14/34 | 11/32 | ||||||
| Abdominal discomfort | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Abdominal distension | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anaemia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anorexia, appetite loss | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Asthenia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Bone marrow failure | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Chest discomfort | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Confusion | ‐ | ‐ | ‐ | ‐ | 0/19 | 1/19 | 0/19 | 1/19 |
| Constipation | 13/34 | 6/32 | ‐ | ‐ | 6/19 | 6/19 | 8/19 | 8/19 |
| Delirium | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Diarrhoea | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Dizziness or lightheadedness | ‐ | ‐ | 0.54 (0.74) | 0.71 (0.9) | ‐ | ‐ | ‐ | ‐ |
| Drowsiness, somnolence | ‐ | ‐ | 0.68 (0.81) | 0.79 (0.93) | 7/19 | 4/19 | 4/19 | 5/19 |
| Dyspnoea (serious) | 0/34 | 1/32 | ||||||
| Fever (serious) | 1/34 | 0/32 | ||||||
| Gastrointestinal disorders | 22/34 | 16/32 | ||||||
| General disorders and administration site | 6/34 | 6/32 | ||||||
| Hallucinations | ‐ | ‐ | ‐ | ‐ | 0/19 | 0/19 | 2/19 | 3/19 |
| Hypoproteinaemia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Insomnia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Nausea | 10/34 | 8/32 | 0.02 (0.15) | 0.12 (0.45) | 7/19 | 7/19 | 7/19 | 12/19 |
| Neutrophil count decreased/neutropenia | 1/34 | 1/32 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Nervous system disorders | 7/34 | 5/32 | ||||||
| Oedema, peripheral | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Platelet count decreased | 1/34 | 0/32 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Pruritus | ‐ | ‐ | 0.05 (0.21) | 0.05 (0.21) | 3/19 | 1/19 | 3/19 | 2/19 |
| Pyrexia | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Rash | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Sedation | ‐ | ‐ | ‐ | ‐ | 12/19 | 13/19 | 12/19 | 14/19 |
| Skin and subcutaneous tissue disorders | 5/34 | 4/32 | ||||||
| Sweating or hyperhidrosis | ‐ | ‐ | 0.04 (0.19) | 0.07 (0.3) | 4/19 | 2/19 | 1/19 | 1/19 |
| Urinary retention | ‐ | ‐ | ‐ | ‐ | 1/19 | 1/19 | 2/19 | 0/19 |
| Urinary tract infection | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Vomiting | ‐ | ‐ | 0.01 (0.11) | 0.01 (0.11) | ‐ | ‐ | ‐ | |
| White blood cell count decreased | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Other disorders | 15/34 | 16/32 | ||||||
| Discontinuation due to adverse events | 2/34 | 0/32 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Deaths due to study drug treatment | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
aMean (standard deviation) ratings (out of 3) experienced during the 24 hours of drug administration, apart from the total number of adverse events which is read from the authors' Figure 3
bThe measure is the sum of positive responses after each study period: moderate = 1, severe = 2.
‐: not reported CR: controlled‐release ER: extended‐release Hyd: hydromorphone IV: intravenous Mor: morphine Oxy: oxycodone Oxymo: oxymorphone Tap: tapentadol
Quality of life
The study did not report quality of life.
Participant preference
The study did not report participant preference.
Intravenous oxycodone versus intravenous morphine
One study compared IV oxycodone to IV morphine (Lee 2017).
Pain intensity and pain relief
The evidence was very uncertain about the effect of IV oxycodone versus IV morphine on pain intensity and pain relief. Lee 2017 compared IV oxycodone with IV morphine in a 5‐day parallel‐group trial in 66 participants, and found that there may be little to no difference in pain intensity between the two treatments (Analysis 1.1; Figure 4). There may also be little to no difference in pain relief with 78.8% and 75% of participants experiencing a reduction in pain of at least 30% from baseline to end of treatment with oxycodone and morphine, respectively, while for 63.6% and 62.5% of participants in the oxycodone and morphine groups, respectively, the reduction in pain was at least 50% (Analysis 1.2; Figure 5). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance bias and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of IV oxycodone versus IV morphine on adverse events. Lee 2017 found that there may be little to no difference in the adverse event profiles for the two study arms (see Table 8) although they did report "There was a difference in the unexpected AEs with more (29) events affecting 16 patients (50.0%) in the morphine group compared to 12 events in the oxycodone group affecting 9 patients (26.5%) (P value 0.049)" (page 7). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance bias and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Quality of life
The study did not report quality of life.
Participant preference
The evidence was very uncertain about the effect of IV oxycodone versus IV morphine on participant preference. The study did not report participant preference, but it did report treatment satisfaction, both as assessed by the participants and by the investigator. Nineteen of the 32 participants in the oxycodone group and 18 of the 31 participants in the morphine group rated their overall analgesic treatment satisfaction regarding pain as "very much improved" or much "improved", with 12 participants in each group indicating minimal improvement and one in each group indicating "no change". When this outcome was assessed by the investigator, the corresponding numbers were 26 and 24 in the oxycodone and morphine groups, respectively, rated as "very much improved" or "much improved" and five and six participants in the oxycodone and morphine groups, respectively, experiencing "minimal improvement", and again one participant in each group experiencing "no change". We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting, performance bias and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Intravenous oxycodone followed by immediate‐release oxycodone versus intravenous morphine followed by immediate‐release morphine
One study compared IV oxycodone followed by IR oxycodone to IV morphine followed by IR morphine (Kalso 1990).
Pain intensity and pain relief
There evidence was very uncertain about the effect of IV oxycodone followed by IR oxycodone versus IV morphine followed by IR morphine on pain intensity and pain relief. In a cross‐over study comparing IV oxycodone titration (two days) followed by IR oxycodone titration (two days) with IV morphine titration (two days) followed by IR morphine titration (two days) in 19 analysed participants, Kalso 1990 found that the participants achieved equal analgesia with both drugs, but around 30% more IV oxycodone was needed compared to IV morphine and around 25% less IR oxycodone was needed than IR morphine to achieve this. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection, performance, and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of IV oxycodone followed by IR oxycodone versus IV morphine followed by IR morphine on adverse events. Kalso 1990 found that nausea may be more common with oral morphine treatment compared to the other three treatment modalities (see also Table 8). We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection, performance, and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Quality of life
The study did not report quality of life.
Participant preference
The evidence was very uncertain about the effect of IV oxycodone followed by IR oxycodone versus IV morphine followed by IR morphine on participant preference. Kalso 1990 reported that 10 participants expressed no treatment preference while five participants preferred oxycodone while another five participants preferred treatment with morphine. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection, performance, and detection bias), and by two levels for imprecision (arising from very low participant numbers).
Intramuscular oxycodone versus oral oxycodone
One study compared IM oxycodone to oral oxycodone (Beaver 1978a).
Pain intensity and pain relief
The evidence was very uncertain about the effect of IM oxycodone versus oral oxycodone on pain intensity and pain relief. In a single‐dose, cross‐over study, Beaver 1978a compared 5 mg and 15 mg IM oxycodone to 10 mg and 30 mg oral oxycodone in 17 participants, of whom 13 completed at least one cross‐over round of the study medications. Beaver 1978a reported that oral oxycodone may be 0.57 (95% CI 0.22 to 1.84) times as potent as IM oxycodone for pain relief and 0.78 (95% CI 0.3 to 8.82) times as potent for change in pain intensity. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of IM oxycodone versus oral oxycodone on adverse events. Beaver 1978a reported that the adverse effects for both oral and IM oxycodone, although infrequent, were related to dose, but otherwise provided no further details on the observed adverse effects. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Quality of life
The study did not report quality of life.
Participant preference
The study did not report participant preference.
Intramuscular oxycodone versus intramuscular morphine versus intramuscular codeine
Two studies (reported in one publication) compared IM oxycodone, IM morphine and IM codeine (Beaver 1978b).
Pain intensity and pain relief
The evidence was very uncertain about the effect of IM oxycodone versus IM morphine versus IM codeine on pain intensity and pain relief. In another single‐dose, cross‐over study Beaver 1978b compared 7.5 mg, 15 mg and 30 mg IM oxycodone to 8 mg, 16 mg and 32 mg IM morphine in 34 participants, of whom 28 completed at least one round of the study drugs. In this study, IM oxycodone was found to be 0.74 (95% CI 0.36 to 1.2) times as potent as IM morphine for pain relief and 0.68 (95% CI 0.32 to 1.07) times as potent as IM morphine for change in pain intensity. In a further study of similar design, Beaver 1978b compared 7.5 mg, 15 mg and 30 mg IM oxycodone to 90 mg and 180 mg IM codeine and to 16 mg IM morphine in 30 participants, of whom 26 completed at least one cross‐over round of the study medications. Beaver 1978b reported that IM oxycodone may be 10.72 (95% CI not reported) times as potent as IM codeine for pain relief and 8.44 (95% CI 2.13 to 44.69) times as potent as IM codeine for change in pain intensity. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Adverse events
The evidence was very uncertain about the effect of IM oxycodone versus IM morphine versus IM codeine on adverse events. Beaver 1978b noted that, in both studies, side effects typical of narcotic analgesics were observed, although not in sufficient numbers to allow meaningful analysis, and they reported no further details on adverse events. We judged the certainty of evidence for this outcome to be very low. We downgraded the certainty of evidence by two levels for very serious limitations to study quality due to risk of bias (arising from under‐reporting of the domains of selection and attrition bias), and by two levels for imprecision (arising from very low participant numbers).
Quality of life
The studies did not report quality of life.
Participant preference
The studies did not report participant preference.
Discussion
For the current update, we identified 19 additional studies for inclusion, which allowed us to perform further meta‐analyses, both on pain relief data and on more adverse event data. We were also able to examine three new treatment comparisons with oxycodone, and further assess the robustness of our primary pain intensity meta‐analyses by performing new sensitivity analyses. The main new issue in the context of this review that was identified in this update was the inclusion of 14 Chinese language studies and we have discussed the issues that arise in this context in detail already (see Excluded studies). Despite the addition of a further 19 studies in this update, the updated results and conclusions remain the same. This is because the only potentially new results were driven by the Chinese language studies and not corroborated by sensitivity analyses (e.g. for CR oxycodone superiority over CR morphine in terms of pain intensity and constipation).
Summary of main results
Overall, we included 42 studies which enrolled/randomised 4485 participants, with 3945 of these analysed for efficacy and 4176 for safety. The studies examined a number of different drug comparisons.
Four studies compared CR oxycodone to IR oxycodone, and pooled analysis of three of these studies showed that there may be little to no difference in pain intensity after treatment with either CR or IR oxycodone (SMD 0.1, 95% CI ‐0.06 to 0.26). This is also in line with the finding that none of the included studies reported that the pain intensity differed between the treatment groups. Pooled analyses of the adverse event data from three of the studies found that there may be little to no difference between the treatments in RRs for any of the adverse events, and the fourth study, which could not be included in the pooled analyses, reported no differences in adverse events either. Three of the four studies also found that there may be little to no difference in treatment acceptability between the comparisons. We noted that IR oxycodone was given every six hours rather than every four hours in these studies. This might have biased the efficacy data in favour of CR oxycodone; however, the adverse effect data suggest that giving IR oxycodone every four hours (more frequently) would have resulted in greater adverse effects, which would have mitigated advantages in efficacy.
Twenty‐four studies compared CR oxycodone to CR morphine and pooled analysis suggested that pain intensity may be lower (better) after treatment with CR morphine than CR oxycodone (SMD 0.14, 95% CI 0.01 to 0.27). However, this result was not corroborated by a sensitivity analysis that excluded the two cross‐over trials included in the overall analysis (SMD 0.12, 95% CI ‐0.02 to 0.26). Further pooled analysis showed that there may be little to no difference in the proportions of participants achieving complete or significant pain relief between CR oxycodone and CR morphine (RR 1.02, 95% CI 0.95 to 1.10). No data were available for quality of life. The evidence is very uncertain about the treatment effects on treatment acceptability and participant preference. Pooled analyses showed that, for most of the adverse events, the CIs were wide, including no effect as well as potential benefit and harm, for the comparison between CR oxycodone and CR morphine. Participants treated with CR morphine may be at about double the risk of experiencing hallucinations and insomnia and lethargy compared to participants treated with CR oxycodone, and participants treated with CR oxycodone may be at 25% lower risk of experiencing constipation. However, the latter result did not remain significant in sensitivity analyses, excluding the Chinese language studies. These findings also contrast somewhat with those reported in Lauretti 2003, which was different in design to the other studies and examined IR morphine consumption during treatment with CR oxycodone and CR morphine while keeping the ratio of CR oxycodone and CR morphine constant. Lauretti 2003 found that the participants consumed 38% more IR morphine during treatment with CR morphine than with CR oxycodone, and that CR and IR morphine may be associated with more nausea and vomiting and a similar acceptance to the study drugs compared to the combination of CR oxycodone and IR morphine, and therefore concluded that CR oxycodone combined with IR morphine is associated with superior analgesia and lower or similar rates of adverse events than a combination of CR and IR morphine.
Three studies compared CR oxycodone to TD fentanyl and pooled analysis of two of them found that there may be little to no difference in pain intensity (SMD 0.02, 95% CI ‐0.19 to 0.24) or complete/significant pain relief rates (RR 0.98, 95% CI 0.85 to 1.14) after treatment with CR oxycodone and TD fentanyl. One of the studies also found that quality of life may not differ between the treatments, but there was some disagreement between the study results in terms of adverse events with one of the studies finding that the rates of nausea and vomiting, and constipation may be higher in the CR oxycodone group than in the TD fentanyl group, whereas the other study reported that there may be little to no difference in these (and other) adverse event rates between the treatment groups.
Two studies compared CR oxycodone to TD buprenorphine but one of the studies did not report pain data in a manner where it could be included and assessed. The study that did contribute data suggested that there may be little to no difference between the treatments in terms of pain intensity, complete/significant pain relief rates and adverse event rates with the exception of 'any degree', but not severe, breathlessness, which may occur more often in the TD buprenorphine group than in the CR oxycodone group
Two studies compared CR oxycodone to ER hydromorphone. Pooled analysis showed that there may be little to no difference between the treatment groups in pain intensity (SMD 0.04, 95% CI ‐0.21 to 0.28) or in adverse event rates. The data for nausea and vomiting could not be pooled due to excessive between‐study heterogeneity and, in line with this, for both nausea and vomiting, Inoue 2017 found that the rates of nausea and vomiting may be lower in the CR oxycodone group compared to the ER hydromorphone group, whereas Yu 2014 found that there may be little to no difference in them between the interventions.
The remaining studies all compared either oxycodone in different formulations or oxycodone to different alternative opioids and none of them found any clear superiority or inferiority of oxycodone for cancer pain, neither as an analgesic agent nor in terms of adverse event rates or treatment acceptability.
Overall completeness and applicability of evidence
Although the findings of this review are applicable to the population and comparisons defined for this review, that is, adults with cancer who need treatment with strong opioids for cancer pain, they should be taken in the context that this review found 42 studies that were eligible for inclusion and these studies reported on 16 different comparisons involving oxycodone and included only 4485 participants. Moreover, for some of the outcomes (participant preference and quality of life) there were extremely few data available. To somewhat mitigate this shortfall, we reported treatment acceptability as a proxy. However, that does not change the fact that the evidence base for the effectiveness and tolerability of oxycodone (relative or absolute) for pain in adults with cancer was very limited and it did not allow us to examine the effectiveness and tolerability of oxycodone in detail through participant or treatment subgroup analyses. The current evidence base would therefore benefit from more well‐designed, large RCTs.
Quality of the evidence
The certainty of the evidence for all the outcomes was low or very low, meaning we have little confidence in the effect estimate and the true effect may be substantially different from the estimate of the effect. This is due to imprecision (low participant numbers) in some cases, and serious or very serious study limitations in all cases. In general, the assessment of the quality of the included studies was limited by a great extent of under‐reporting in the studies, especially for Chinese language studies in general. For the other studies, the participant selection items (random sequence generation and allocation concealment) were also severely under‐reported, while blinding appeared to be reasonably well undertaken overall, both in terms of treatment performance and outcome assessment. However, as is not unusual for pain research, the results were substantially compromised by attrition, with data missing from 12% of the enrolled/randomised participants for efficacy, and from just under 7% for safety. These are substantial proportions and, while it did not appear to be selective attrition, the results must be interpreted with caution, especially as they are likely to be under‐estimates given the amount of under‐reporting associated with the Chinese language studies in particular, which meant we could not be sure we had captured study dropouts/the extent of missing data fully.
Potential biases in the review process
We undertook the review according to the methods specified in our protocol, which were all in line with the recommendations of Cochrane as outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and included a thorough search strategy designed to maximise the chances of identifying all relevant studies. Contacting authors resulted in no additional studies being identified, that is, the review therefore only contains data from published studies, some of which have not reported all the outcome data despite having apparently collected these data. The review may therefore be at some risk of publication bias, although publication bias is usually associated with positive results, and the majority of the included studies did not find differences between their treatment groups in terms of efficacy and safety. Although our meta‐analyses of pain intensity included data from cross‐over studies that were analysed as if they were parallel‐group studies, which as outlined in Unit of analysis issues results in unit of analysis error (although, in turn, this leads to an under‐weighting rather than over‐weighting of the data), the results were also subjected to sensitivity analyses that excluded the cross‐over trials, and that needed to confirm the results in order for them to be accepted as true results. In this section, we return again to the issue of whether or not we should have included Chinese databases in our search strategy to increase the applicability of our results and decrease the geographical bias associated with mainly including studies conducted in the West and published in English. As outlined already in the Excluded studies section, we would have liked to include studies from searches of Chinese databases if we could have been sufficiently confident in the integrity of the data and the correct use of the term 'random' in the context of patient allocation and if the study methods and data were reported in full. Unfortunately, this is not currently the case, but we will keep this option open in future updates of this review.
Agreements and disagreements with other studies or reviews
King 2011 conducted a systematic review without meta‐analysis that also included observational studies and concluded that, "There is no evidence from the included trials of a significant difference in analgesia or adverse effects between oxycodone and morphine or hydromorphone" (page 454). Caraceni 2011 reached a similar conclusion in their systematic review without meta‐analysis. Bekkering 2011 and Reid 2006 both included meta‐analyses in their systematic reviews and they also concluded that the effectiveness of oxycodone and morphine did not differ, although the inclusion criteria employed by Bekkering 2011 differed from ours, with Bekkering 2011 excluding cross‐over trials and including trials of chronic non‐malignant pain, whereas the publication of Reid 2006 before the trial of Mercadante 2010 precluded its inclusion. That said, the conclusions of all these reviews are all in agreement with those that we have reached in this review dealing with the same comparisons as the aforementioned reviews. In a more recent meta‐analysis that included seven trials that were also included in this review and purported to compare the analgesic effect of oxycodone and morphine on patients with moderate and advanced cancer pain, Guo 2018 concluded that "The results of this meta‐analysis demonstrate clinical non‐inferiority of morphine compared with oxycodone in alleviating cancer pain, with respect to [having] achieved a comparable clinical response whether morphine or oxycodone was used as first‐line opioid in the treatment of cancer‐related pain" (page 5). While this conclusion is also in general agreement with our review, it cannot actually be made based on the data Guo 2018 has analysed and presented because the seven included trials, which were all analysed together and treated as if they examined the same drug comparison (i.e. oxycodone versus morphine), were in fact a mix of comparisons with some of them comparing CR oxycodone with CR morphine (Corli 2016; Mercadante 2010; Riley 2015; Zecca 2016) while others compared CR oxycodone to ER hydromorphone (Inoue 2017; Yu 2014) or to ER oxymorphone (Gabrail 2004). It is unclear to us why these seven trials have been analysed together and the results presented by Guo 2018 are therefore not comparable to any of the results in this review.
Authors' conclusions
Implications for practice.
Since the last version of this review, none of the new relevant studies have provided additional information to change the conclusions.
For adults with cancer pain
We found low‐certainty evidence that there may be little to no difference between oxycodone and other strong opioids in pain intensity, pain relief and adverse events for adults with cancer. Although we identified a clinically insignificant benefit on pain relief in favour of controlled‐release (CR) morphine compared to CR oxycodone, this did not persist following sensitivity analysis excluding cross‐over trials and so we do not consider this important. We did find that the frequency of hallucinations may be increased after treatment with CR morphine (7.8%) compared to CR oxycodone (4%).
For clinicians
We found low‐certainty evidence that there may be little to no difference in pain intensity, cancer pain relief and adverse events between oxycodone and other strong opioids including morphine, which is commonly considered the gold standard strong opioid. Although we identified a clinically insignificant benefit on pain relief in favour of CR morphine compared to CR oxycodone, this did not persist following sensitivity analysis and so we do not consider this important. We found that the risk of hallucinations may be increased with CR morphine but we also found a numerically higher frequency of myoclonus (another excitatory opioid adverse effect) with CR oxycodone and we did not find any differences in reported drowsiness or confusion. The interpretation of increased relative risk of hallucinations should therefore be treated with caution given the low certainty of evidence. We also found low‐certainty evidence that there may be little to no difference in pain intensity, cancer pain relief and adverse events between CR and IR oxycodone, which suggests there is no benefit of OR over IR oxycodone. However, evidence on patient preference was lacking in these studies.
For policy makers
The findings of this review are consistent with current international guidance on using oxycodone or morphine as first‐line opioids for adults with cancer‐related pain.
For funders of the intervention
We did not undertake cost‐effectiveness analysis.
Implications for research.
General
We found that the current evidence base is comprised of studies that contained small numbers of participants in which there was a significant (> 12%) dropout rate. For example, the direct comparison meta‐analysis between CR oxycodone and CR morphine was based on fewer than 450 cancer participants in each treatment group; this was a very small evidence base. However, given the absence of important differences within this analysis, it seems unlikely that larger head‐to‐head studies of oxycodone versus morphine will be undertaken. In part, this is because recruitment and retention of participants is challenging in this context. Well‐designed randomised controlled trials comparing oxycodone to other strong analgesics may well be useful.
Design
Future randomised controlled trials assessing the effectiveness and tolerability of oxycodone for pain in adults with cancer need to be adequately powered, well‐designed, protocol‐driven and fully reported following the most up‐to‐date CONSORT (Schulz 2010) trial reporting guidelines, including adequate reporting of participant baseline characteristics and co‐interventions.
Measurement (endpoints)
For future cancer pain studies, developing a single outcome that combines good pain control (no more than mild on a verbal rating scale) with acceptable adverse effects (perhaps no more than mild severity on any adverse event) would enable a clearer comparison between any analgesics used in this context.
What's new
| Date | Event | Description |
|---|---|---|
| 7 June 2021 | New search has been performed | This review has been updated to include the results of a new search on 30 November 2021. |
| 30 March 2021 | New citation required but conclusions have not changed | An update search run on 30 November 2021 identified 616 new records of which 19 new eligible studies have been included in this review. The studies added a further 1836 participants to the review and examined three new comparisons, namely immediate‐release (IR) oxycodone versus IR hydromorphone, controlled‐release (CR) oxycodone versus oral ibuprofen and intravenous (IV) oxycodone versus IV morphine, and also contained data to include in the original CR oxycodone versus CR morphine comparison and add to other comparisons (CR oxycodone versus transdermal (TD) fentanyl, TD buprenorphine and extended‐release hydromorphone). The additional studies allowed us to meta‐analyse the rates of complete or significant pain relief, and perform further meta‐analyses of the adverse event data and further sensitivity analyses of both the pain and safety data. The conclusions of the updated review remain the same as that of the previous version of the review. Previous readers of the review may enjoy re‐reading this update. |
History
Protocol first published: Issue 4, 2002 Review first published: Issue 2, 2015
| Date | Event | Description |
|---|---|---|
| 12 March 2019 | Review declared as stable | See Published notes. |
| 25 June 2018 | Amended | Updated Other published versions of this review |
| 25 October 2017 | Amended | Minor correction to Discussion |
| 30 January 2017 | New citation required but conclusions have not changed | An update search run on 29 November 2016 identified 267 new records of which 6 new eligible studies (Corli 2016; Lux 2014; Su 2015; Yu 2014; Zecca 2016; Zhang 2014), have been included in this review. The studies added a further 1258 participants to the review and examined four new comparisons with oxycodone, namely transdermal (TD) fentanyl (2 studies), TD buprenorphine, extended‐release (ER) oxycodone, and ER hydromorphone, and also contained data to include in the original controlled‐release (CR) oxycodone versus CR morphine comparison. The additional studies allowed us to meta‐analyse the adverse event data (which were summarised narratively and in tables in the original review), to examine the robustness of our primary outcome pain intensity meta‐analyses in sensitivity analyses, and to include an additional meta‐analysis of pain intensity for the comparison of CR oxycodone and TD fentanyl. None of the meta‐analyses of pain intensity and adverse events were significant with two exceptions: 1) Pain intensity was statistically significantly, but not clinically significantly, higher after treatment with CR oxycodone compared to CR morphine. Sensitivity analysis of this result however did not find a statistically significant difference in pain intensity between the two treatments, and 2) the risk ratio of experiencing hallucinations was significantly higher after treatment with CR morphine than CR oxycodone. Otherwise, the conclusions of the updated review remain the same as that of the original review. Previous readers of the review may enjoy re‐reading this update. |
| 18 January 2017 | New search has been performed | An update search run on 29 November 2016 revealed 6 new eligible studies. These have now been added. |
| 22 February 2013 | New citation required and major changes | This protocol has been significantly updated by new authors. See Published notes. |
| 11 February 2010 | New citation required and major changes | This protocol was originally published in Issue 4, 2002. As the authors were unable to commit time to the completion of the full review it was then withdrawn in January 2009. The original authors are now able to work on completing the full review and plan to do so by the end of 2010. |
| 13 January 2009 | New citation required and major changes | Withdrawn: the review group was unable to maintain contact with the contact author. New authors are being sought to take over this protocol, please contact the PaPaS Review Group if you are interested in working on this review title. |
| 22 September 2008 | Amended | Converted to new review format. |
Notes
A full update of this review has been undertaken. This is the second full update of the review.
Acknowledgements
We would like to thank Yuhong Yuan (Department of Medicine, Division of Gastroenterology McMaster University, Canada) and Yu‐Tian Xiao (School of Clinical Medicine, Fudan University, China) for help with the translation of Chinese papers, Xiaofeng Du (Liaoning University of Traditional Chinese Medicine, China) for peer reviewing the search strategy for Chinese databases, Mei Wang (Liaoning University of Traditional Chinese Medicine, China) for downloading Chinese full texts, Wencong Cao (Guangzhou University of Chinese Medicine) and Ziwei Zhang (Beijing University of Chinese Medicine) for screening of Chinese abstracts and full texts and Joanne Abbott at the Cochrane Pain, Palliative and Supportive Care Review Group (PaPaS) for her assistance in devising and executing the search strategy.
Cochrane Review Group funding acknowledgement: this project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to Cochrane Pain, Palliative and Supportive Care Review Groups (PaPaS). The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health and Social care.
The Cochrane Pain, Palliative and Supportive Care Review Group (PaPaS) supported the authors in the development of this review. The following people conducted the editorial process for this article: Sign‐off Editor (final editorial decision): Dr Neil O'Connell, PaPaS Co‐ordinating Editor, and Reader at Brunel University London Managing Editor (provided editorial guidance to authors, edited the article): Anna Erskine (Oxford University Hospitals (OUH) NHS Foundation Trust, Oxford, UK)
Assistant Managing Editor (selected peer reviewers, collated peer‐reviewer comments, conducted editorial checks and supported editorial team): Kerry Harding (Oxford University Hospitals (OUH) NHS Foundation Trust, Oxford, UK)
Contact Editor (editorial guidance): McKenzie Ferguson, Southern Illinois University Edwardsville, USA Information Specialist (searching support): Joanne Abbott (Oxford University Hospitals (OUH) NHS Foundation Trust, Oxford, UK) Copy‐editing (initial copy‐edit and final proofread): [NAME], Copy‐edit Group
OR
Copy‐editing (initial copy‐edit): Anne Lethaby, Copy‐edit Group Copy‐editing (final proofread): [NAME], Copy‐edit Group
Peer‐reviewers (provided comments and recommended an editorial decision): Dr Christina Abdel Shaheed Academic Fellow, School of Public Health University of Sydney, and Sydney Musculoskeletal Health (clinical review), Dulce Estêvão, University of Algarve, School of Health, Faro, Portugal (consumer review), Bruno Saragiotto, Masters and Doctoral Programs in Physical Therapy, Universidade Cidade de São Paulo, Brazil (clinical review),
Appendices
Appendix 1. Search strategies
Update search run in November 2021
CENTRAL (Cochrane Library)
#1 MeSH descriptor: [Oxycodone] explode all trees
#2 (ox?codon* or oxycontin or oxycodeinon or oxycone or oxycdn or ox?conum or oxydose or oxyfast or oxygesic or oxynorm or oxynormoro or oxyrapid):ti,ab,kw (Word variations have been searched)
#3 (dazidox or dihydrohydroxycodeinone or dihydrone or dinarkon):ti,ab,kw (Word variations have been searched)
#4 (endocet or endocodone or endone or eu?odal or eubine):ti,ab,kw (Word variations have been searched)
#5 ("m oxy" or oxecta or oxydihydrocodeinonum or pancodine or pavinal or percocet or percolone or proladone):ti,ab,kw (Word variations have been searched)
#6 (remoxy or roxicet or rox?codone or roxilox):ti,ab,kw (Word variations have been searched)
#7 (supeudol or thecodinum or theocodin or tylox):ti,ab,kw (Word variations have been searched)
#8 #1 or #2 or #3 or #4 or #5 or #6 or #7
#9 MeSH descriptor: [Neoplasms] explode all trees
#10 (cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk?emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*):ti,ab,kw (Word variations have been searched)
#11 #9 or #10
#12 #8 and #11
MEDLINE and MEDLINE In‐Process (OVID)
1 Oxycodone/
2 (ox?codon$ or oxycontin or oxycodeinon or oxycone or oxycdn or ox?conum or oxydose or oxyfast or oxygesic or oxynorm or oxynormoro or oxyrapid).tw.
3 (dazidox or dihydrohydroxycodeinone or dihydrone or dinarkon).tw.
4 (endocet or endocodone or endone or eu?odal or eubine).tw.
5 ("m oxy" or oxecta or oxydihydrocodeinonum or pancodine or pavinal or percocet or percolone or proladone).tw.
6 (remoxy or roxicet or rox?codone or roxilox).tw.
7 (supeudol or thecodinum or theocodin or tylox).tw.
8 or/1‐7
9 exp Neoplasms/
10 (cancer$ or neoplas$ or tumo$ or carcinoma$ or hodgkin$ or nonhodgkin$ or adenocarcinoma$ or leuk?emia$1 or metasta$ or malignan$ or lymphoma$ or sarcoma$ or melanoma$ or myeloma$ or oncolog$).tw.
11 or/9‐10
12 8 and 11
13 randomized controlled trial.pt.
14 controlled clinical trial.pt.
15 randomized.ab.
16 placebo.ab.
17 drug therapy.fs.
18 randomly.ab.
19 trial.ab.
20 or/13‐19
21 exp animals/ not humans.sh.
22 20 not 21
23 12 and 22
Embase (OVID)
1. Oxycodone/
2. (ox?codon$ or oxycontin or oxycodeinon or oxycone or oxycdn or ox?conum or oxydose or oxyfast or oxygesic or oxynorm or oxynormoro or oxyrapid).tw.
3. (dazidox or dihydrohydroxycodeinone or dihydrone or dinarkon).tw.
4. (endocet or endocodone or endone or eu?odal or eubine).tw.
5. ("m oxy" or oxecta or oxydihydrocodeinonum or pancodine or pavinal or percocet or percolone or proladone).tw.
6. (remoxy or roxicet or rox?codone or roxilox).tw.
7. (supeudol or thecodinum or theocodin or tylox).tw.
8. or/1‐7
9. exp Neoplasms/
10. (cancer$ or neoplas$ or tumo$ or carcinoma$ or hodgkin$ or nonhodgkin$ or adenocarcinoma$ or leuk?emia$1 or metasta$ or malignan$ or lymphoma$ or sarcoma$ or melanoma$ or myeloma$ or oncolog$).tw.
11. or/9‐10
12. 8 and 11
13. random$.tw.
14. factorial$.tw.
15. crossover$.tw.
16. cross over$.tw.
17. cross‐over$.tw.
18. placebo$.tw.
19. (doubl$ adj blind$).tw.
20. (singl$ adj blind$).tw.
21. assign$.tw.
22. allocat$.tw.
23. volunteer$.tw.
24. Crossover Procedure/
25. double‐blind procedure.tw.
26. Randomized Controlled Trial/
27. Single Blind Procedure/
28. or/13‐27
29. (animal/ or nonhuman/) not human/
30. 28 not 29
31. 12 and 30
Web of Science (ISI) SCI & CPCI‐S
#22 #21 AND #9
#21 #20 OR #17 OR #16 OR #15 OR #14 OR #11 OR #10
#20 #19 AND #18
#19 TS=random* OR TI=random*
#18 TS=(allocate* OR assign*) OR TI=(allocate* OR assign*)
#17 TS=crossover* OR TI=crossover*
#16 TS=(mask* OR blind*) OR TI=(mask* OR blind*)
#15 TS=(singl* OR Doubl* OR Tripl* OR Trebl*) OR TI=(singl* OR Doubl* OR Tripl* OR Trebl*)
#14 #13 AND #12
#13 TS=trial* OR TI=trial*
#12 TI=clin* OR TS=clin*
#11 TI=randomi* OR TS=randomi*
#10 TS=Randomized clinical trial* OR TI=Randomized clinical trial*
#9 #8 AND #7
#8 Topic=((cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk?emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*))
#7 #6 OR #5 OR #4 OR #3 OR #2 OR #1
#6 Topic=((supeudol or thecodinum or theocodin or tylox))
#5 Topic=((remoxy or roxicet or rox?codone or roxilox))
#4 Topic=(("m oxy" or oxecta or oxydihydrocodeinonum or pancodine or pavinal or percocet or percolone or proladone))
#3 Topic=((endocet or endocodone or endone or eu?odal or eubine))
#2 Topic=((dazidox or dihydrohydroxycodeinone or dihydrone or dinarkon))
#1 Topic=((ox?codon* or oxycontin or oxycodeinon or oxycone or oxycdn or ox?conum or oxydose or oxyfast or oxygesic or oxynorm or oxynormoro or oxyrapid))
BIOSIS (ISI)
#21 #20 AND #19 AND #12
#20 Topic=(((cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk?emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*)))
#19 #18 OR #17 OR #16 OR #15 OR #14 OR #13
#18 Topic=(((supeudol or thecodinum or theocodin or tylox)))
#17 Topic=(((remoxy or roxicet or rox?codone or roxilox)))
#16 Topic=((("m oxy" or oxecta or oxydihydrocodeinonum or pancodine or pavinal or percocet or percolone or proladone)))
#15 Topic=(((endocet or endocodone or endone or eu?odal or eubine)))
#14 Topic=(((dazidox or dihydrohydroxycodeinone or dihydrone or dinarkon)))
#13 Topic=(((ox?codon* or oxycontin or oxycodeinon or oxycone or oxycdn or ox?conum or oxydose or oxyfast or oxygesic or oxynorm or oxynormoro or oxyrapid)))
#12 #11 OR #8 OR #7 OR #6 OR #5 OR #2 OR #1
#11 #10 AND #9
#10 DS=random* OR TS=random* OR TI=random*
#9 DS=(allocate* OR assign*) OR TS=(allocate* OR assign*) OR TI=(allocate* OR assign*)
#8 DS=crossover* OR TS=crossover* OR TI=crossover*
#7 DS=(mask* OR blind*) OR TS=(mask* OR blind*) OR TI=(mask* OR blind*)
#6 DS=(singl* OR Doubl* OR Tripl* OR Trebl*) OR TS=(singl* OR Doubl* OR Tripl* OR Trebl*) OR TI=(singl* OR Doubl* OR Tripl* OR Trebl*)
#5 #4 AND #3
#4 DS=trial* OR TS=trial* OR TI=trial*
#3 DS=clin* OR TI=clin* OR TS=clin*
#2 DS=randomi* OR TI=randomi* OR TS=randomi*
#1 MQ=Randomized clinical trial* OR DS=Randomized clinical trial* OR TS=Randomized clinical trial* OR TI=Randomized clinical trial*
PsycINFO (EBSCO)
S20 S10 AND S19
S19 S12 OR S13 OR S14 OR S15 OR S16 OR S17 OR S18
S18 (singl* OR doubl* OR trebl* OR tripl*) N3 (blind* OR mask*)
S17 clinical N3 trial* OR research N3 design OR evaluat* N3 stud* OR prospectiv* N3 stud*
S16 placebo* OR random* OR "comparative stud*"
S15 DE "Followup Studies"
S14 DE "Placebo"
S13 DE "Treatment Outcomes" OR DE "Psychotherapeutic Outcomes" OR DE "Side Effects (Treatment)" OR DE "Treatment Compliance" OR DE "Treatment Duration" OR DE "Treatment Refusal" OR DE "Treatment Termination" OR DE "Treatment Withholding"
S12 DE "Treatment Effectiveness Evaluation"
S11 S7 AND S10
S10 S8 OR S9
S9 (cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk#emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*)
S8 DE "Neoplasms" OR DE "Benign Neoplasms" OR DE "Breast Neoplasms" OR DE "Endocrine Neoplasms" OR DE "Leukemias" OR DE "Melanoma" OR DE "Metastasis" OR DE "Nervous System Neoplasms" OR DE "Terminal Cancer"
S7 S1 OR S2 OR S3 OR S4 OR S5 OR S6
S6 (supeudol or thecodinum or theocodin or tylox)
S5 (remoxy or roxicet or rox#codone or roxilox)
S4 ("m oxy" or oxecta or oxydihydrocodeinonum or pancodine or pavinal or percocet or percolone or proladone)
S3 (endocet or endocodone or endone or eu#odal or eubine).
S2 (dazidox or dihydrohydroxycodeinone or dihydrone or dinarkon)
S1 (ox#codon* or oxycontin or oxycodeinon or oxycone or oxycdn or ox#conum or oxydose or oxyfast or oxygesic or oxynorm or oxynormoro or oxyrapid)
Data and analyses
Comparison 1. Pain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Pain intensity and pain relief (continuous) | 23 | Std. Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 Controlled‐release (CR) oxycodone vs immediate‐release (IR) oxycodone | 3 | 319 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.10, 0.34] |
| 1.1.2 CR oxycodone vs extended‐release (ER) oxycodone | 1 | 62 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.08 [‐0.42, 0.57] |
| 1.1.3 CR oxycodone vs CR morphine | 10 | 1137 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.30, ‐0.06] |
| 1.1.4 CR oxycodone vs CR hydromorphone | 1 | 62 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.13 [‐0.63, 0.37] |
| 1.1.5 CR oxycodone vs ER hydromorphone | 2 | 259 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.04 [‐0.21, 0.28] |
| 1.1.6 CR oxycodone vs ER oxymorphone | 1 | 74 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.23 [‐0.23, 0.69] |
| 1.1.7 CR oxycodone vs ER tapentadol | 1 | 265 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.06 [‐0.30, 0.18] |
| 1.1.8 CR oxycodone vs transdermal (TD) fentanyl | 2 | 329 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.19, 0.24] |
| 1.1.9 CR oxycodone vs TD buprenorphine | 1 | 252 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.15, 0.35] |
| 1.1.10 IR oxycodone vs IR morphine | 1 | 38 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.15 [‐0.79, 0.49] |
| 1.1.11 IR oxycodone vs IR hydromorphone | 1 | 172 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.15 [‐0.15, 0.45] |
| 1.1.12 IV oxycodone vs IV morphine | 1 | 65 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.17 [‐0.66, 0.31] |
| 1.2 Complete and/or significant pain relief (categorical) | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 CR oxycodone vs CR morphine | 13 | 1249 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.95, 1.10] |
| 1.2.2 CR oxycodone vs ER tapentadol | 1 | 265 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.65, 1.10] |
| 1.2.3 CR oxycodone vs oral ibuprofen | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [1.24, 3.07] |
| 1.2.4 CR oxycodone vs transdermal (TD) fentanyl | 2 | 329 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.85, 1.14] |
| 1.2.5 CR oxycodone vs TD buprenorphine | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.82, 1.09] |
| 1.2.6 IV oxycodone vs IV morphine | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.70, 1.48] |
Comparison 2. Adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Any adverse events | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1.1 Controlled‐release (CR) oxycodone vs CR morphine | 8 | 943 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.70, 0.85] |
| 2.1.2 CR oxycodone vs extended‐release (ER) hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.98, 1.14] |
| 2.2 Severe adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.2.1 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.73, 1.62] |
| 2.3 Serious adverse events, including death | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.3.1 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.86, 2.39] |
| 2.4 Appetite loss/anorexia | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.4.1 CR oxycodone vs CR morphine | 3 | 263 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.36, 3.94] |
| 2.4.2 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.54, 1.49] |
| 2.5 Asthenia | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.5.1 CR oxycodone vs immediate‐release (IR) oxycodone | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.20, 1.68] |
| 2.6 Confusion | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.6.1 CR oxycodone vs IR oxycodone | 2 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.20, 3.02] |
| 2.6.2 CR oxycodone vs CR morphine | 3 | 584 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.78, 1.31] |
| 2.7 Constipation | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.7.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.45, 1.13] |
| 2.7.2 CR oxycodone vs CR morphine | 18 | 1894 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.66, 0.86] |
| 2.7.3 CR oxycodone vs transdermal (TD) fentanyl | 2 | 336 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.88, 1.32] |
| 2.7.4 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.80, 1.49] |
| 2.8 Diarrhoea | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.8.1 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.55, 1.49] |
| 2.9 Dizziness/lightheadedness | 16 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.9.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.40, 1.37] |
| 2.9.2 CR oxycodone vs CR morphine | 11 | 941 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.58, 1.31] |
| 2.9.3 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.61, 1.64] |
| 2.10 Drowsiness/somnolence | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.10.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.69, 1.54] |
| 2.10.2 CR oxycodone vs CR morphine | 15 | 1486 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.74, 1.05] |
| 2.11 Dry mouth | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.11.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.48, 2.75] |
| 2.11.2 CR oxycodone vs CR morphine | 5 | 888 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.78, 1.22] |
| 2.12 Dysuria/uroschesis | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.12.1 CR oxycodone vs CR morphine | 7 | 887 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.38, 1.07] |
| 2.12.2 CR oxycodone vs TD fentanyl | 2 | 336 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.62, 2.16] |
| 2.13 Fever | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.13.1 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.66, 1.62] |
| 2.14 Hallucinations | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.14.1 CR oxycodone vs CR morphine | 4 | 696 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.28, 0.97] |
| 2.15 Headache | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.15.1 CR oxycodone vs IR oxycodone | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.16 Insomnia | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.16.1 CR oxycodone vs IR oxycodone | 2 | 269 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.31, 3.53] |
| 2.17 Insomnia & lethargy | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.17.1 CR oxycodone vs CR morphine | 2 | 314 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.26, 0.90] |
| 2.18 Nausea | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.18.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.56, 1.28] |
| 2.18.2 CR oxycodone vs CR morphine | 13 | 1388 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.77, 1.12] |
| 2.18.3 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.63, 1.08] |
| 2.19 Nausea & vomiting | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.19.1 CR oxycodone vs CR morphine | 6 | 637 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.56, 1.06] |
| 2.20 Nervousness | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.20.1 CR oxycodone vs IR oxycodone | 2 | 208 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.20, 1.64] |
| 2.21 Pruritus | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.21.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.65, 3.25] |
| 2.21.2 CR oxycodone vs CR morphine | 8 | 1108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.51, 1.14] |
| 2.22 Sweating | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.22.1 CR oxycodone vs IR oxycodone | 2 | 269 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.22, 1.93] |
| 2.22.2 CR oxycodone vs CR morphine | 2 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.52 [0.54, 37.94] |
| 2.23 Vomiting | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.23.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.38, 1.15] |
| 2.23.2 CR oxycodone vs CR morphine | 13 | 1388 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.63, 1.04] |
| 2.23.3 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.63, 1.10] |
| 2.24 Discontinuation due to adverse events | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.24.1 CR oxycodone vs IR oxycodone | 3 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.29, 1.22] |
| 2.24.2 CR oxycodone vs CR morphine | 7 | 618 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.36, 1.73] |
| 2.24.3 CR oxycodone vs ER hydromorphone | 2 | 434 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.69, 1.75] |
2.15. Analysis.

Comparison 2: Adverse events, Outcome 15: Headache
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beaver 1978a.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: USA |
|
| Participants |
Participants: 17 participants entered, "13 patients completed at least one round" (see 'Interventions' below) and were analysed for efficacy ("The 4 patients who failed to complete a single round did so for reasons extraneous to the drugs under study"); 5 men/8 women, mean (range) age 51 (23‐68) years. "One of these patients appeared twice in the study, and 5 completed a second round, yielding 19 cross‐over comparisons." Inclusion criteria: "The subjects were patients with a variety of malignant tumours on the wards of James Ewing Hospital. Each patient was first examined to ascertain the nature and location of his or her pain, the extent of disease, prior experience with narcotics and analgesic drugs and ability to communicate meaningful information about pain. At this time, the patient was also told how the studies were to be conducted and that, while all test medications might appear the same, they would actually include a number of different drugs, some probably more effective than others in relieving pain. Many of the patients had some prior experience with oral or parenteral narcotics, and several had a history of sufficient recent narcotic use to warrant the assumption that they possessed some tolerance to narcotics. Patients were placed on a routine analgesic other than those included in the study during non‐study hours, and, insofar as was possible, concomitant administration of psychoactive drugs was avoided." Exclusion criteria: see 'Inclusion criteria.' No other information provided |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone + oral placebo ‐ Dose/dosing: 5 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: assessed by participant "hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' Comparison arm 1 ‐ Drug: oxycodone + oral placebo ‐ Dose or dosing: 15 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: assessed by participant "hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' Comparison arm 2 ‐ Drug: oxycodone + intramuscular placebo ‐ Dose or dosing: 10 mg ‐ Formulation: Immediate‐release? ‐ Route of administration: oral ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: assessed by participant "hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' Comparison arm 3 ‐ Drug: oxycodone + intramuscular placebo ‐ Dose or dosing: 30 mg ‐ Formulation: Immediate‐release? ‐ Route of administration: oral ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: assessed by participant "hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' ‐ For cross‐over trials, cross‐over schedule: "Treatments were assigned to patients according to a series of randomly chosen Latin squares, and each study medication was administered on a separate day. Each patient received a low and a high dose of both the "standard" and the "test drug", chosen at equilog intervals. Unless a patient completed all doses of the crossover comparison or "round," his data were excluded from the relative potency analysis. After completing the first round, some patients were able to repeat the course, allowing for comparison of replicate rounds within the same individual." |
|
| Outcomes | ‐ Pain intensity: assessed by participant "hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication);" using a 4‐point categorical scale (0 = none, 1 = slight, 2 = moderate, 3 = severe) ‐ Pain relief: assessed by participant hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hours had elapsed since administration of the study medication); using a 5‐point categorical scale (0 = none, 1 = slight, 2 = moderate, 3 = lots, 4 = complete) "Patients who were remedicated before 6 hr [hours] elapsed after administration of a study medication were assigned scores of zero (0) for change in pain intensity and pain relief for the remaining observation points of the 6‐hr observation period." ‐ Adverse effects: "The observer also recorded apparent and volunteered side‐effects, but leading questions were avoided." |
|
| Notes | ‐ Study free of commercial funding? No: "This work was supported by grants awarded by the Committee on Problems of Drug Dependence, National Academy of Sciences, National Research Council, from funds contributed by a group of interested pharmaceutical manufacturers, and by National Cancer Institute Grant CA‐08748." ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Treatments were assigned to patients according to a series of randomly chosen Latin squares." No further information reported |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "Neither the patient nor the observer was aware of the identity of the medications, which were physically indistinguishable and identified only by a numerical code on individual dosage envelopes. To maintain double‐blind conditions, both capsules and an injection, one of which was a dummy, were administered each time a patient was given a study medication." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Participant reported. See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | Participant reported. See cell above |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | ITT analyses not undertaken: "Unless a patient completed all doses of the crossover comparison or "round," his data were excluded from the relative potency analysis". Data from 13/17 participants reported. "One of these patients appeared twice in the study, and 5 completed a second round, yielding 19 crossover comparisons." |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Data from 13/17 participants reported. "One of these patients appeared twice in the study, and 5 completed a second round, yielding 19 crossover comparisons." |
| Selective reporting (reporting bias) | High risk | No side effects or adverse events reported in detail: "The side‐effects are both intramuscular and oral oxycodone were dose‐related and qualitatively similar to those noted in the codeine study." (which were also not reported in any detail at all: "While a dose‐response regression was generally evident, side‐effects did not occur with sufficient frequency to allow meaningful analysis.") |
| Were the participants adequately titrated? | Unclear risk | Not enough information provided |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available from all the cross‐over periods. |
| Other bias | Unclear risk | It was unclear if this study was subject to a high risk of other biases. |
Beaver 1978b.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: USA |
|
| Participants | "The patient population and method of evaluating analgesic efficacy were similar to those employed in the oral/parenteral analgesic relative potency assays of codeine and oxycodone described in the previous paper" (Beaver 1978a). Paper contained 2 studies: 'Intramuscular morphine and oxycodone' and 'intramuscular codeine, oxycodone and morphine' 'Intramuscular morphine and oxycodone' Participants: 34 participants entered, "28 patients completed at least one round" (see 'Interventions' below) and were analysed for efficacy ("All of the patients who failed to complete a single round did so for reasons extraneous to the drugs under study"); 14 men/14 women, mean (range) age 46 (23‐68) years. "Of the 28 patients participating in the study, 4 appeared twice in a single series, and 2 appeared in each of two series. Twenty‐four patients completed a second round, yielding a total of 58 crossover comparisons." 'Intramuscular codeine, oxycodone and morphine' Participants: 30 participants entered, "26 completed at least one round" (see 'Interventions' below) and were analysed for efficacy ("The 4 who failed to complete a single round did so for reasons extraneous to the drugs under study"); 14 men/12 women, mean (range) age 45 (23‐80) years. "Series I was carried out in 11 patients, one of whom appeared twice in the series and 10 of whom completed a second round, yielding 22 cross‐over comparisons of 90 and 180 mg of codeine, 7.5 and 15 mg of oxycodone and 16 mg of morphine. Series II consisted of 27 cross‐over comparisons in 16 patients of 90 and 180 mg codeine, 15 and 30 mg of oxycodone, and 16 mg of morphine. One patient appeared in both Series I and Series II." Inclusion criteria: see above Exclusion criteria: see above |
|
| Interventions |
'Intramuscular morphine and oxycodone': "This assay consisted of three series, each comparing two doses of morphine sulfate (the standard) with two doses of oxycodone hydrochloride (the test drug) by intramuscular injection." "The distribution of patients and doses in the various series is presented in table 1. In general, the more obviously tolerant patients were given series II treatments, which consisted of double the dosage in series I." Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose or dosing: series I: 7.5 mg and 15 mg; series II and III: 15 mg and 30 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: "Assessed by patient hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' Comparison arm ‐ Drug: morphine sulphate ‐ Dose or dosing: series I and III: 8 mg and 16 mg; series II: 16 mg and 32 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: "Assessed by patient hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' 'Intramuscular codeine, oxycodone and morphine': "This assay consisted of two series, each comparing 90 and 180 mg codeine phosphate (the standard) with two doses of oxycodone hydrochloride (the test drug) and a single 16 mg dose of morphine sulfate." Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose or dosing: series I: 7.5 mg and 15 mg; series II: 15 mg and 30 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: "Assessed by patient hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' Comparison arm 1 ‐ Drug: morphine sulphate ‐ Dose or dosing: series I and II: 16 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration. ‐ Rescue medication: "Assessed by patient hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' Comparison arm 2 ‐ Drug: codeine phosphate ‐ Dose/dosing: series I and II: 90 mg and 180 mg ‐ Formulation: Intramuscular ‐ Route of administration: Intramuscular ‐ Length of treatment: appeared to be single dose ‐ Titration schedule: no titration ‐ Rescue medication: "Assessed by patient hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication)." No further information reported ‐ Other medication: see 'Rescue medication' and 'Inclusion criteria' ‐ For cross‐over trials, cross‐over schedule: "Treatments were assigned to patients according to a series of randomly chosen Latin squares, and each study medication was administered on a separate day. Each patient received a low and a high dose of both the "standard" and the "test drug," chosen at equilog intervals. Unless a patient completed all doses of the cross‐over comparison or "round," his data were excluded from the relative potency analysis. After completing the first round, some patients were able to repeat the course, allowing for comparison of replicate rounds within the same individual." |
|
| Outcomes | ‐ Pain intensity: assessed by participant "hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr [hours] had elapsed since administration of the study medication);" using a 4‐point categorical scale (0 = none, 1 = slight, 2 = moderate, 3 = severe) ‐ Pain relief: assessed by participant hourly for 6 hours after administration of the study medication or until pain returned to the premedication level and a routine analgesic was administered (if at least 3 hr had elapsed since administration of the study medication); using a 5‐point categorical scale (0 = none, 1 = slight, 2 = moderate, 3 = lots, 4 = complete) "Patients who were remedicated before 6 hr elapsed after administration of a study medication were assigned scores of zero (0) for change in pain intensity and pain relief for the remaining observation points of the 6‐hr observation period." ‐ Adverse effects: "The observer also recorded apparent and volunteered side‐effects, but leading questions were avoided." |
|
| Notes | ‐ Study free of commercial funding? No: "This work was supported in part by grants awarded by the Committee on Problems of Drug Dependence, National Academy of Sciences, National Research Council, from funds contributed by a group of interested pharmaceutical manufacturers, and by National Cancer Institute Grant CA‐08748." ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Treatments were assigned to patients according to a series of randomly chosen Latin squares". No further information reported |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "Neither the patient nor the observer was aware of the identity of the medications, which were physically indistinguishable and identified only by a numerical code on individual dosage envelopes. To maintain double‐blind conditions, both capsules and an injection, one of which was a dummy, were administered each time a patient was given a study medication." From Beaver 1978a |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Participant reported. See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | Participant reported. See cell above |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | ITT analyses not undertaken: "Unless a patient completed all doses of the crossover comparison or “round,” his data were excluded from the relative potency analysis." Data included from 28/34 and 26/30 participants in the two studies, respectively |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Data included from 28/34 and 26/30 participants in the two studies, respectively |
| Selective reporting (reporting bias) | High risk | No side effects or adverse events reported in detail: Study 1: "While side‐effects observed after both morphine and oxycodone were typical of the narcotic analgesics, they did not occur with sufficient frequency to allow a meaningful comparison of the side‐effect liability of the two drugs. Noteworthy was the virtual absence of side‐effects in patients in series II, an observation consistent with these patients' substantial tolerance to narcotics." Study 2: "Side‐effects were qualitatively similar to those noted in the oxycodone‐morphine comparison, but they did not occur with sufficient frequency to allow a meaningful comparison among treatments." |
| Were the participants adequately titrated? | Unclear risk | Not enough information provided |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available from all the cross‐over periods. |
| Other bias | Unclear risk | It was unclear if this study was subject to a high risk of other biases. |
Bruera 1998.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: Canada |
|
| Participants |
Participants: 32 participants entered, 23 participants analysed for efficacy and VAS variables (5 participants dropped out during the CR morphine phase: 3 in phase 1, 2 in phase 2; 1 due to lack of pain control and adverse event, 1 due to protocol violation, 3 due to adverse events; 4 participants dropped out during the CR oxycodone phase: 2 in phase 1, 2 in phase 2; 1 due to lack of pain control, 2 due to adverse events, 1 was lost to follow‐up); 13 women, 10 men; age not reported; cancer type: lung (7), breast (7), prostate (1), other (8): cancer stage not reported; type of pain not reported; setting: palliative care programme; previous analgesic medication: IR morphine (8), CR morphine (10), IR oxycodone ± acetaminophen (11), CR hydromorphone (1), CR codeine (1), IR codeine + acetaminophen (1); duration of opioid use: 6.6 (± 10) months; duration of chronic pain: 8 (± 13) months Inclusion criteria: "The study included 32 patients from the Palliative Care Program at the Cross Cancer Institute and Grey Nuns Hospital in Edmonton, Canada. All patients were ≥ 18 years of age, gave written informed consent, had pain due to cancer, and were receiving treatment with an oral opioid analgesic at study entry. Life expectancy for all patients was estimated by the treating physician to be at least 4 months." Exclusion criteria: use of active anticancer therapy, with the exception of hormones, within 2 weeks of study entry; physical or mental inability to answer questions and comply with the treatment protocol; history of intolerance of oxycodone or any related compound; impaired renal or hepatic function; significantly impaired ventilatory function (clinically present dyspnoea at rest); current use of an investigational drug; pregnancy or lactation; unwillingness or inability to co‐operate or give written, informed consent; and inability to take oral medications |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone + placebo morphine ‐ Dose and dosing: mean dose 46.5 (± 57) mg every 12 hours ‐ Formulation: Controlled‐release (CR) ‐ Route of administration: oral ‐ Length of treatment: 7 days ‐ Titration schedule: "≥ 3 day prestudy history of stable analgesia (defined as a daily rescue opioid consumption ≤ 20% of the scheduled daily opioid dose)." ‐ Rescue medication: Immediate‐release oxycodone hydrochloride, at doses of ca 10% of daily scheduled opioid dose. Mean daily number of doses 2.3 (± 2.3) ‐ Other medication: no other analgesic agents. All other prestudy medications were maintained with no changes allowed later than 72 hours before randomisation. Comparison arm ‐ Drug: morphine + placebo oxycodone ‐ Dose and dosing: mean dose = 72.6 (± 102) mg every 12 hours ‐ Formulation: Controlled‐release (CR) ‐ Route of administration: oral ‐ Length of treatment: 7 days ‐ Titration schedule: "≥ 3 day prestudy history of stable analgesia (defined as a daily rescue opioid consumption ≤ 20% of the scheduled daily opioid dose)." ‐ Rescue medication: Immediate‐release (IR) morphine, at doses of ca 10% of daily scheduled opioid dose. Mean daily number of doses 1.7 (± 2.1) ‐ Other medication: no other analgesic agents; all other prestudy medications were maintained with no changes allowed later than 72 hours before randomisation. "Patients who had been receiving narcotic analgesics other than morphine or single‐entity oxycodone before the start of the study were transferred to an equianalgesic oral dose of controlled‐release oxycodone or controlled‐release morphine at the start of phase 1. The initial dose of controlled‐release oxycodone was determined busing a 1:1.5 conversion ratio between controlled‐release oxycodone and controlled‐release morphine." ‐ For cross‐over trials, cross‐over schedule: on day 8 participants crossed over to receive the alternative drug and placebo at a dose equivalent to that received at the start of phase 1. During both study phases, blind‐labelled dose adjustments were permitted if participants required more than 3 rescue analgesic doses over 24 hours. |
|
| Outcomes | ‐ Pain intensity: assessed by participant 4 times per day before dosing and at the end of each phase; 100 mm VAS (0 = no pain to 100 = worst possible pain) and 5‐point categorical scale (0 = no pain to 4 = worst possible pain) ‐ Overall effectiveness of the study medication: assessed by participant on days 8 and 15; verbal rating scale (0 = not effective to 3 = highly effective) ‐ Nausea and sedation: days 8 and 15; 100 mm VAS (0 = no nausea or sedation to 100 extreme nausea or sedation) ‐ Adverse events: recorded by participants; checklist (nausea, vomiting, constipation, dry mouth, drowsiness, dizziness, difficulty concentrating, fatigue, poor sleep, vivid dreams, hallucinations, headache, agitation, twitching, itching, sweating (from 0 = none to 4 = intolerable) and non‐directed adverse events questionnaire ‐ Treatment preference: assessed by participants and investigators at the end of study |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomised. No further information provided |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "Blinding was maintained by the double‐dummy technique using matching placebos of controlled release oxycodone and controlled‐release morphine. The immediate‐release oxycodone and morphine formulations were also blinded." Trial labelled as 'double‐blind' |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | "Blinding was maintained by the double‐dummy technique using matching placebos of controlled release oxycodone and controlled‐release morphine. The immediate‐release oxycodone and morphine formulations were also blinded." Trial labelled as 'double‐blind' |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above. Outcome was participant‐reported. |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above. Outcome was participant‐reported. |
| Incomplete outcome data (attrition bias) Pain | High risk | The analyses restricted to the 23/32 participants who completed both study phases |
| Incomplete outcome data (attrition bias) Adverse events | High risk | The only safety data analyses that were reported analysed 23/32 participants. |
| Selective reporting (reporting bias) | High risk | The majority of the adverse events were not reported beyond the sentence "There were no statistically significant differences by treatment in mean severity for any of the elicited adverse events or in frequency of reporting of unelicited events." |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes. Data only analysed if available from both time periods |
| Other bias | Low risk | The authors reported that "There was no evidence of period or sequence (carry‐over) effect." No other biases were identified. |
Cao 2015.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2012‐2014 Country: China |
|
| Participants |
Participants: 130 participants randomised ‐ Oxycodone: 65 participants, 39 men, 26 women; no further information reported ‐ Morphine: 65 participants, 41 men, 24 women; no further information reported Inclusion criteria: Elderly patients admitted to the authors' hospital with cancer and neuropathic pain and a numerical rating scale score ≥ 4 Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 10 mg, but not reported how often medication was given ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 4 weeks ‐ Titration schedule: Dose increased by 25% if pain was not relieved after 24 hours, then personnel observed for 24 hours, and increased the dose by 25% every 24 hours until the pain was controlled ‐ Rescue medication: Not reported ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine ‐ Dose and dosing: Starting dose = 10 mg, but not reported how often medication was given ‐ Formulation: Controlled‐release (CR) ‐ Route of administration: oral ‐ Length of treatment: 4 weeks ‐ Titration schedule: Dose increased by 25% if pain was not relieved after 24 hours, then personnel observed for 24 hours, and increased the dose by 25% every 24 hours until the pain was controlled ‐ Rescue medication: Not reported ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain intensity: assessed before treatment and 1, 2, 3 and 4 weeks after treatment on numerical rating scale ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses undertaken: data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Corli 2016.
| Study characteristics | ||
| Methods |
Design: randomised, parallel‐group, open‐label, superiority, phase IV clinical trial Year: 2011‐2014 Country: Italy |
|
| Participants |
Participants: of 520 randomised participants, 515 included in safety analyses and 498 included in ITT analyses ‐ Morphine: 122 participants, 67 men, 55 women; mean (SD) age 67.5 (± 11.7) years; cancer type: lung/pleura (34), colon/rectum (11), breast (17), prostate (6), pancreas (14), genitourinary (10), oesophagus/stomach/duodenum (3), head/neck (9), gynaecological (11), myeloma (1), sarcoma (0), other (6); tumour metastatic: yes (101); ongoing cancer therapy (53); previous administration of weak opioids for the background pain (88); type of pain: only nociceptive (98), only neuropathic (0), nociceptive and neuropathic (23), insufficient information to classify (1), missing (0) ‐ Oxycodone: 125 participants, 72 men, 53 women; mean (SD) age 66.9 (± 11.1) years; cancer type: lung/pleura (31), colon/rectum (14), breast (16), prostate (13), pancreas (8), genitourinary (8), oesophagus/stomach/duodenum (8), head/neck (8), gynaecological (6), myeloma (4), sarcoma (2), other (7); tumour metastatic: yes (112); ongoing cancer therapy (48); previous administration of weak opioids for the background pain (81); type of pain: only nociceptive (106), only neuropathic (0), nociceptive and neuropathic (18), insufficient information to classify (1), missing (0) ‐ Buprenorphine: 127 participants, 68 men, 59 women; mean (SD) age 65.2 (± 13.5) years; cancer type: lung/pleura (39), colon/rectum (15), breast (22), prostate (4), pancreas (13), genitourinary (3), oesophagus/stomach/duodenum (1), head/neck (13), gynaecological (8), myeloma (2), sarcoma (3), other (4); tumour metastatic: yes (107); ongoing cancer therapy (48); previous administration of weak opioids for the background pain (89); type of pain: only nociceptive (102), only neuropathic (1), nociceptive and neuropathic (23), insufficient information to classify (1), missing (0) ‐ Fentanyl: 124 participants, 70 men, 54 women; mean (SD) age 68 (± 10.6) years; cancer type: lung/pleura (37), colon/rectum (17), breast (10), prostate (6), pancreas (4), genitourinary (10), oesophagus/stomach/duodenum (6), head/neck (12), gynaecological (9), myeloma (5), sarcoma (3), other (5); tumour metastatic: yes (104); ongoing cancer therapy (42); previous administration of weak opioids for the background pain (85); type of pain: only nociceptive (106), only neuropathic (0), nociceptive and neuropathic (15), insufficient information to classify (2), missing (1) Inclusion criteria: Participants with diagnostic (histological or cytological) evidence of locally advanced or metastatic solid tumour; with persistent moderate to severe cancer pain (average pain intensity ≥ 4, measured on a 0‐10 numerical rating scale (NRS) and related to the last 24 hours); requiring for the first time an analgesic treatment with third‐step WHO opioids; aged > 18 years Exclusion criteria: Participants with a diagnosis of primary brain tumour or leukaemia, or undergoing concurrent radiotherapy, first‐line chemotherapy started 7 days before randomisation, or on any nonpharmacological analgesic treatment or with preexisting renal failure |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: "The initial dose of opioid was based on the recommendations of the European Association for Palliative care." "The suggested initial daily dose was 60 mg of oral M[orphine] for participant with previous WHO step‐II treatment or half of this (30 mg/day) for opioid‐naïve participants. The doses of the other opioids were regulated on the basis of the oral M equivalent dose ratio." Protocol lists 40 mg/24 hours. Mean (± SD) initial dose (as oral morphine equivalent daily dose) 44.6 (± 16) mg/day. Mean (± SD) final dose (as oral morphine equivalent daily dose) 71.1 (± 60.8) mg/day ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 28 days ‐ Titration schedule: "During the follow‐up [active treatment phase], any adjustments necessary for better control of pain was allowed for clinical and ethical reasons. Physicians could change the dose, add another opioid or adjuvant drug or change the opioid (switch). In case of constantly unsatisfactory analgesia or severe toxicity, the opioid could be discontinued." ‐ Rescue medication: not reported ‐ Other medication: see 'Titration schedule' Comparison arm 1 ‐ Drug: morphine ‐ Dose and dosing: "The initial dose of opioid was based on the recommendations of the European Association for Palliative care." The suggested initial daily dose was 60 mg/24 hours for participants who had previously received WHO step‐II opioids and 30 mg/24 hours for opioid‐naive participants. Mean (± SD) initial dose 45.7 (± 16.2) mg/day. Mean (± SD) final dose 58.9 (± 38.6) mg/day ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 28 days ‐ Titration schedule: "During the follow‐up [active treatment phase], any adjustments necessary for better control of pain was allowed for clinical and ethical reasons. Physicians could change the dose, add another opioid or adjuvant drug or change the opioid (switch). In case of constantly unsatisfactory analgesia or severe toxicity, the opioid could be discontinued." ‐ Rescue medication: not reported ‐ Other medication: see 'Titration schedule' Comparison arm 2 ‐ Drug: buprenorphine ‐ Dose and dosing: "The initial dose of opioid was based on the recommendations of the European Association for Palliative care" "The suggested initial daily dose was 60 mg of oral M[orphine] for participants with previous WHO step‐II treatment or half of this (30 mg/day) for opioid‐naïve participants. The doses of the other opioids were regulated on the basis of the oral M equivalent dose ratio." Protocol listed 35 µg/hour. Mean (SD) initial dose (as oral morphine equivalent daily dose) 53.7 (12.5) mg/day. Mean (± SD) final dose (as oral morphine equivalent daily dose) 80.1 (± 40.4) mg/day ‐ Formulation: patch ‐ Route of administration: Transdermal ‐ Length of treatment: 28 days ‐ Titration schedule: "During the follow‐up [active treatment phase], any adjustments necessary for better control of pain was allowed for clinical and ethical reasons. Physicians could change the dose, add another opioid or adjuvant drug or change the opioid (switch). In case of constantly unsatisfactory analgesia or severe toxicity, the opioid could be discontinued." ‐ Rescue medication: not reported ‐ Other medication: see 'Titration schedule' Comparison arm 3 ‐ Drug: fentanyl ‐ Dose and dosing: "The initial dose of opioid was based on the recommendations of the European Association for Palliative care" The suggested initial daily dose was 60 mg of oral M[orphine] for participants with previous WHO step‐II treatment or half of this (30 mg/day) for opioid‐naïve participants. The doses of the other opioids were regulated on the basis of the oral M equivalent dose ratio." Protocol listed 25 µg/hour. Mean (± SD) initial dose (as oral morphine equivalent daily dose) 53.4 (± 14.2) mg/day. Mean (± SD) final dose (as oral morphine equivalent daily dose) 111.4 (± 74.9) mg/day ‐ Formulation: patch ‐ Route of administration: Transdermal ‐ Length of treatment: 28 days ‐ Titration schedule: "During the follow‐up [active treatment phase], any adjustments necessary for better control of pain was allowed for clinical and ethical reasons. Physicians could change the dose, add another opioid or adjuvant drug or change the opioid (switch). In case of constantly unsatisfactory analgesia or severe toxicity, the opioid could be discontinued." ‐ Rescue medication: not reported ‐ Other medication: see 'Titration schedule' |
|
| Outcomes | ‐ Pain intensity (average and worst) measured on a 0 to 10 NRS ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Study particularly funded by Grünenthal‐Italia, but funder "had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript." ‐ Groups comparable at baseline? No inferential statistics reported. Treatment groups appeared to be reasonably comparable at baseline. Protocol links: http://www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2010‐021017‐23‐IT; https://clinicaltrials.gov/show/NCT01809106 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation |
| Allocation concealment (selection bias) | Low risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | High risk | Open‐label |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) Pain | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | Open‐label |
| Incomplete outcome data (attrition bias) Pain | Low risk | ITT analyses undertaken: 498/520 randomised participants were included in the analyses. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | ITT analyses not reported to have been undertaken: The study stated that "Only patients who started on opioid were included in the safety analysis, which considered patients in the arm of the treatment they actually received" (page 1108), but according to their Figure 1 (page 1109), the analyses appeared to be ITT‐based. 515/520 randomised participants were included in the safety analyses. |
| Selective reporting (reporting bias) | Low risk | All the obvious outcomes appeared to be reported. |
| Were the participants adequately titrated? | Low risk | The participants appeared to be adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to other bias. |
Gabrail 2004.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: USA |
|
| Participants |
Participants: 58 participants screened, 47 participants titrated, 45 participants randomised, and 44 participants received ≥ 1 dose of study medication and had ≥ 1 pain intensity evaluation after treatment and were therefore analysed for safety (1/45 never received any double‐blind study medication and was excluded from all analyses). 37/45 randomised participants completed the first double‐blind phase and ≥ 5 days of the second phase and were analysed for efficacy (2/45 participants had insufficient visits or assessments to be included in the efficacy population); 5/45 randomised participants discontinued the drug during the double‐blind treatment periods: 2 participants withdrew due to adverse events unrelated to the study drug, 2 participants withdrew consent and 1 participant due to protocol violation. 0 participants discontinued the study due to insufficient analgesia or loss to follow‐up. 21 safety and 18 efficacy participants received extended‐release oxymorphone followed by controlled‐release oxycodone and 23 safety and 19 efficacy participants received controlled‐release oxycodone followed by extended‐release oxymorphone. 21 men, 23 women, mean age (range) 59.3 (26‐81) years; 80% had severe untreated pain and 20% had moderate untreated pain. Previous anticancer therapy included surgery (68%), chemotherapy (82%), radiotherapy (50%), and/or immunotherapy (2.3%). Inclusion criteria: men and women aged ≥ 18 years with moderate to severe pain secondary to cancer who required long‐term outpatient treatment with an opioid analgesic. People hospitalised for reasons unrelated to cancer were also eligible. Exclusion criteria: allergy or sensitivity to morphine, extended‐release oxymorphone, controlled‐release oxycodone or their components; requirement for a concurrent opioid analgesic other than the study medication; contraindication to opioid therapy; pregnancy; lactation; plan for pregnancy; uncontrolled emesis; inability to take adequate oral food and hydration; levels of hepatic enzymes (gamma‐glutamyl transpeptidase, alanine aminotransferase, and aspartate aminotransferase) ≥ 3 times the upper limit of the normal range; receipt of radiotherapy or therapeutic radionuclides within the previous 2 weeks preceding study enrolment |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: mean daily dose 91.9 mg (any dose adjustments were made during the first 3 days of the double‐blind phase; dosage remained fixed thereafter) ‐ Formulation: Controlled‐release (CR) ‐ Route of administration: oral ‐ Length of treatment: 7‐10 days, take medication at 8 a.m. and 8 p.m. ‐ Titration schedule: "During the open‐label titration/stabilization phase, patients received either oxymorphone immediate‐release (IR) or oxycodone CR to determine a stable dosage, defined as a fixed dosage that provided adequate analgesia for at least 2 consecutive days, required no more than 2 doses of rescue medication/day, and produced tolerable AEs [adverse events]." ‐ Rescue medication: tablets of 15 mg oral morphine sulphate (IR) every 4‐6 hours as needed. Participants requiring > 2 doses/day after the first 3 days of double‐blind treatment were discontinued. Mean daily dose (range) = 12.6 (0‐75) mg ‐ Other medication: not reported Comparison arm ‐ Drug: oxymorphone ‐ Dose and dosing: mean daily dose 45.9 mg (any dose adjustments were made during the first 3 days of the double‐blind phase; dosage remained fixed thereafter) ‐ Formulation: Extended‐release, take medication at 8 a.m. and 8 p.m. ‐ Route of administration: oral ‐ Length of treatment: 7‐10 days ‐ Titration schedule: "During the open‐label titration/stabilization phase, patients received either oxymorphone immediate‐release (IR) or oxycodone CR to determine a stable dosage, defined as a fixed dosage that provided adequate analgesia for at least 2 consecutive days, required no more than 2 doses of rescue medication/day, and produced tolerable AEs [adverse events]." ‐ Rescue medication: tablets of 15 mg oral morphine sulphate (IR) every 4‐6 hours as needed. Participants requiring > 2 doses/day after the first 3 days of double‐blind treatment were discontinued. Mean daily dose (range) 16.6 (0‐90) mg ‐ Other medication: not reported ‐ For cross‐over trials, cross‐over schedule: "Following the first double‐blind treatment period, patients crossed over to the alternative double‐blind treatment (oxymorphone ER or oxycodone CR) for an additional 7‐10 days." |
|
| Outcomes | ‐ Pain intensity: Assessed by daily diary recording by the participants of all study drugs taken (including supplemental pain medication) and their 24‐hour pain intensity, using an 11‐point numerical scale (0 = no pain to 10 = worst possible pain) and the Brief Pain Inventory ‐ Quality of life: assessed by the Brief Pain Inventory to assess the interference of pain with 7 domains of quality of life (general activity, mood, walking ability, normal work, relations with other people, sleep, and enjoyment of life). Appeared to be rated by the participants during the study visits that marked the end of each double‐blind treatment phase ‐ Global assessment of current pain medication, rated by participants and physicians independently following each double‐blind phase. Physicians were asked "Please rate the subject's current pain medication used for treating their cancer pain". ‐ Karnofsky performance status: assessed by physicians at each visit ‐ Safety analysis: assessed by physical examination, vital signs, clinical laboratory tests (serum chemistry profile, complete blood count, urinalysis), electrocardiograms, and the monitoring of adverse events (which were rated by the investigators as mild, moderate, severe intensity, and as unlikely, possibly, probably related to study medication) |
|
| Notes | ‐ Study free of commercial funding? No, the study was funded by Endo Pharmaceuticals Inc., Chadds Ford, PA and Penwest Pharmaceuticals Co., Danbury, CT. ‐ Groups comparable at baseline? The authors reported that there were no significant differences in the demographic or baseline characteristics of the treatment groups, but did not report these characteristics split by treatment group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomised. No further information provided |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | The study was described as "double‐blind". No further information reported, so it was unclear who was blinded and whether it was adequately executed |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | 37/45 randomised participants were analysed for efficacy. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | 41 to 43 of 45 randomised participants were analysed for efficacy. |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes were reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available for both study periods for 40/45 participants. |
| Other bias | Low risk | The authors reported that "There were no sequence effects observed during the study; comparable pain scores and other efficacy measures were obtained irrespective of the order in which patients received the study medication." No other potential biases were identified. |
Gao 2012.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: Not reported Country: China |
|
| Participants |
Participants: 58 participants randomised ‐ Oxycodone: 30 participants, no further information reported ‐ Morphine: 28 participants, no further information reported Inclusion criteria: Patients with advanced pathologically diagnosed malignant tumour, severe tumour‐associated pain (NRS > 6), WHO pain degree classified as severe, All patients had not used other analgesic drugs 6 hours before administration, and the effect of radiotherapy and chemotherapy on analgesic effect could be excluded. Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: Not reported ‐ Titration schedule: Not reported ‐ Rescue medication: Not reported ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 20 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: Not reported ‐ Titration schedule: Not reported ‐ Rescue medication: Not reported ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain relief: assessed according to the WHO grading of pain and by NRS, with 0 as painless, 1‐3 as mild, 4‐6 as moderate, 7‐9 as severe, and 10 as extremely severe. The efficacy was evaluated according to the degree of pain relief and the NRS value before and after treatment. The analgesic effect was divided into complete relief, partial relief and no relief. Complete relief + partial relief = total relief ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation This study was only partially dual‐extracted as the translation software did not perform very well in the translation of this study to allow the second non‐Chinese speaking author to fully extract and appraise this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients reported to be randomised, but no information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses were undertaken: Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Hagen 1997.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: Canada |
|
| Participants |
Participants: 44 participants enrolled, 31 participants completed the study. Reasons for withdrawal included adverse events (8), inadequate pain control (3), intercurrent illness (1), and voluntary withdrawal (1). "Failure to complete both phases of the study did not appear to be related to toxicity of one of the study drugs over another." The analysis of all efficacy outcome variables, including VAS and categorical pain intensity, sedation VAS and nausea VAS were restricted to participants completing both study phases. Spontaneously reported safety variables were analysed for all enrolled participants. 13 men/18 women, mean age (± SE) = 56 (± 3) years. Primary tumour: breast (7), colorectal (5), lung (1), urological/prostate (5), CNS (4), unknown primary (2), other (7). Type of pain: bone (61%), soft tissue (29%), visceral (23%), neuropathic (45%). Pain described as "lancinating" (16%): steady pain (61%), incident pain with or without steady pain (52%) Inclusion criteria: Patients with chronic cancer pain and stable analgesic requirements Exclusion criteria: Known hypersensitivity to opioid analgesics, intolerance of oxycodone or hydromorphone, presence of a medical or surgical condition likely to interfere with drug absorption in the gastrointestinal tract, concurrent use of other opioid analgesics during the study period, presence of intractable nausea and vomiting, and patients who had undergone or were expected to undergo therapeutic procedures likely to influence their pain during the study period |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: mean daily initial dose = 120 ± 22 mg, mean final dose = 124 ± 22 mg (blind‐label dose changes were permitted, and in case of a dose change, the rescue analgesic dose was modified accordingly) ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 7 days, 12‐hourly dosing of active drug ‐ Titration schedule: "Patients with 3 days of stable analgesic requirements on a prestudy opioid were randomized to controlled‐release oxycodone or controlled‐release hydromorphone. Stable analgesia was defined as 2 or fewer rescue doses of opioid analgesic per 24‐hour period, calculated over 3 or more days." ‐ Rescue medication: incident and non‐incident breakthrough pain was treated with immediate‐release oxycodone at a dosage of approximately 10% of the daily scheduled dose. Mean daily frequency of rescue use (SD) = 1.4 ± 0.3 mg ‐ Other medication: no other opioids were permitted. Non‐opioid analgesics, such as corticosteroids, antidepressants, anticonvulsants, bisphosphonates, and psychostimulants, that had been part of the participant's therapy were continued at the same dose level throughout the study. Comparison arm ‐ Drug: hydromorphone ‐ Dose/dosing: mean daily initial dose 24 (± 4) mg, mean (± SD) final dose 30 (± 6) mg (blind‐label dose changes were permitted, and in case of a dose change, the rescue analgesic dose was modified accordingly) ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 7 days, 12‐hourly dosing of active drug ‐ Titration schedule: "Patients with 3 days of stable analgesic requirements on a prestudy opioid were randomized to controlled‐release oxycodone or controlled‐release hydromorphone. Stable analgesia was defined as 2 or fewer rescue doses of opioid analgesic per 24‐hour period, calculated over 3 or more days." ‐ Rescue medication: Incident and nonincident breakthrough pain was treated with immediate‐release hydromorphone at a dosage of approximately 10% of the daily scheduled dose. Mean daily frequency of rescue use (SD) = 1.6 ± 0.3 mg ‐ Other medication: no other opioids were permitted. Non‐opioid analgesics, such as corticosteroids, antidepressants, anticonvulsants, bisphosphonates, and psychostimulants, that had been part of the participant's therapy were continued at the same dose level throughout the study. ‐ For cross‐over trials, cross‐over schedule: "At the end of Phase I, patients were crossed over to the alternative treatment in Phase II without an intervening washout period." |
|
| Outcomes | ‐ Pain intensity: Assessed by participants 4 times daily (8.00, 12.000, 16.00, and 20.00) on a 100 mm visual analogue scale (going from no pain to excruciating pain) and on a 5‐point categorical scale (0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = excruciating) ‐ Nausea and sedation: Assessed by participants 4 times daily (8.00, 12.000, 16.00, and 20.00) on a 100 mm visual analogue scale (going from no nausea or sedation to severe nausea or extreme sedation) ‐ Spontaneously reported; investigator‐observed and elicited adverse events were recorded at the end of each phase. ‐ Participant and investigator treatment preferences were recorded at the end of both phases. |
|
| Notes | ‐ Study free of commercial funding? No information reported, but the second author (Najib Babul) is an employee of Purdue Frederick, which is the manufacturer of the controlled‐release oxycodone study drug used in the study. ‐ Groups comparable at baseline? No information provided about initial group allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "Blinding was maintained by the double‐dummy technique, which involved matching placebos. In the active treatment phases, patients received either active controlled‐release oxycodone and placebos matching controlled‐release hydromorphone or active controlled‐release hydromorphone and placebos matching controlled‐release oxycodone." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | The analyses were restricted to 31/44 participants. |
| Incomplete outcome data (attrition bias) Adverse events | High risk | The analyses were restricted to 31/44 participants, or not reported in a manner that allowed them to be included in any meta‐analysis. |
| Selective reporting (reporting bias) | Unclear risk | The adverse event reporting was restricted to 4 adverse events in a manner that did not allow them to be included in the meta‐analysis. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available for both study periods for 31/44 participants. |
| Other bias | Low risk | The authors reported that analysis of treatment sequence revealed no significant carry‐over effects. |
Heiskanen 1997.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: Finland |
|
| Participants |
Participants: 45 participants enrolled, and 27/45 participants were evaluated for efficacy and safety. Reasons for withdrawal included adverse events (all were nausea/vomiting; 7), unstable pain control at the end of titration (5), non‐compliance (3), sudden deterioration unrelated to the study (1), and a technical error (1); 1 participant was withdrawn due to suspected incomplete absorption of controlled‐release oxycodone. 16 men/11 women, mean age (range) 60 (39‐76) years. Primary tumour: breast (2), rectum (5), lung (4), prostate (6), kidney (1), pancreas (4), unknown primary (2), and other (3). Former analgesics: morphine alone or in combination with other analgesic (20), oxycodone alone or in combination with other analgesic (5). 12 participants were randomised to titration with CR oxycodone and 15 participants with CR morphine. Inclusion criteria: Patients with chronic cancer pain requiring opioid analgesics, who were co‐operative, and able to take oral medication and keep a simple diary Exclusion criteria: Patients receiving radiation therapy or other cancer treatment that could affect their pain |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone + morphine‐matched placebo ‐ Dose and dosing: mean daily initial dose 123 mg at the end of titration ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 3‐6 days, unclear how many doses per day ‐ Titration schedule: in opioid‐naive participants, the open‐label titration phase (of 21‐day maximum duration) was started with a total daily dose of 40 mg oxycodone. Dose titration was continued until effective pain relief with acceptable adverse effects was achieved for ≥ 48 hours. The controlled‐release dose was titrated upwards if pain continued at the moderate to severe level or if > 2 doses of escape analgesic were used in a 24‐hour period. The controlled‐release dose was titrated downwards in case of unacceptable opioid adverse effects which were not manageable with appropriate treatment. ‐ Rescue medication: oxycodone hydrochloride solution in a dose of approximately 1/6 to 1/8 of the daily dose of controlled‐release oxycodone; mean total amount per participant during the last 3 days of the titration phase = 79 mg. Mean daily number of doses (SE) during double‐blind phase 1.26 (± 0.22) mg ‐ Other medication: Nonsteroidal anti‐inflammatory analgesics, if used by the participant before the study, were continued at the same dose. Comparison arm ‐ Drug: morphine + oxycodone‐matched placebo ‐ Dose and dosing: mean daily initial dose 180 mg at the end of titration ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 3‐6 days, unclear how many doses per day ‐ Titration schedule: in opioid‐naive participants the open‐label titration phase (of 21‐day maximum duration) was started with a total daily dose of 40 mg oxycodone. Dose titration was continued until effective pain relief with acceptable adverse effects was achieved for ≥ 48 hours. The controlled‐release dose was titrated upwards if pain continued at the moderate to severe level or if > 2 doses of escape analgesic were used in a 24‐hour period. The controlled‐release dose was titrated downwards in case of unacceptable opioid adverse effects which were not manageable with appropriate treatment. ‐ Rescue medication: morphine hydrochloride solution in a dose of approximately 1/6 to 1/8 of the daily dose of controlled‐release morphine; mean total amount per participant during the last 3 days of the titration phase = 74 mg. Mean daily number of doses (SE) during double‐blind phase = 0.79 ± 0.18 mg ‐ Other medication: Nonsteroidal anti‐inflammatory analgesics, if used by the participant before the study, were continued at the same dose. ‐ For cross‐over trials, cross‐over schedule: after 3‐6 days of dosing, the participant visited the Pain Relief Unit for an end of phase visit. A similar 3‐ to 6‐day period was then completed in a cross‐over fashion using the other opioid. |
|
| Outcomes | ‐ Pain intensity: assessed by participants 4 times daily (morning, noon, evening, and bedtime) on a 4‐point verbal rating scale (none, slight, moderate, severe) ‐ Acceptability of therapy: assessed by participants twice daily, considering pain intensity and adverse effects during the previous 12‐hour period on a 5‐point verbal rating scale (very poor, poor, fair, good, excellent) ‐ Adverse experiences: recorded by participant in diary along with each dose of scheduled and escape study medication, concomitant medications, and intercurrent illnesses ‐ At each double‐blind phase ends, a Modified Specific Drug Effect Questionnaire was completed by the participants and a trained research nurse or investigator. |
|
| Notes | ‐ Study free of commercial funding? No, the study was funded by Purdue Frederick, which is the manufacturer of the controlled‐release oxycodone study drug used in the study, and the Academy of Finland. ‐ Groups comparable at baseline? No information provided about initial group allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer generated randomisation for the open‐label titration phase and again for the double‐blind phase was performed by the Purdue Frederick Company and a list of randomisation codes was kept by the hospital pharmacist." |
| Allocation concealment (selection bias) | Low risk | See cell above. No further details reported. Probably adequate |
| Blinding of participants and personnel (performance bias) Pain | Low risk | A double‐blind placebo controlled design was used. It is unclear who was blinded, but it appeared that at least the participants were. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Participant‐reported. See also cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | The analyses were restricted to 27/45 participants. |
| Incomplete outcome data (attrition bias) Adverse events | High risk | The analyses were restricted to 27/45 participants. |
| Selective reporting (reporting bias) | Low risk | All expected main outcomes appeared to be reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available for both study periods for 27/45 participants. |
| Other bias | Low risk | It was unclear whether there were any carry‐over effects, but there probably were none. |
Imanaka 2013.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group multicentre, non‐inferiority trial Year: 25 August 2010 to 16 August 2012 Country: Japan, Korea |
|
| Participants |
Participants: 343 participants enrolled, and 340/343 participants received ≥ 1 dose of study drug (172 oxycodone, 168 tapentadol); 236/343 participants completed treatment (123 oxycodone, 113 tapentadol), and 231/343 participants completed the study (121 oxycodone, 110 tapentadol). Reasons for withdrawal included adverse events (14 oxycodone, 12 tapentadol), progressive disease (15 oxycodone, 11 tapentadol), withdrawal of consent (8 oxycodone, 8 tapentadol), physician decision (1 oxycodone, 8 tapentadol), protocol violation (5 oxycodone, 5 tapentadol), lack of efficacy (1 oxycodone, 4 tapentadol), non‐compliance (4 oxycodone, 1 tapentadol), death (1 oxycodone, 0 tapentadol), other (0 oxycodone, 6 tapentadol). Oxycodone: 172, 100 men, 72 women, mean age (± SD) 64.9 (± 11.41) years, 110 Japanese and 62 Korean. Primary tumour: gastrointestinal (65); respiratory or mediastinal (46); > 92% participants had metastatic cancer. Former analgesics: not reported Tapentadol: 168, 90 men, 78 women, mean age (± SD) 65.5 (11.21) years, 111 Japanese and 57 Korean. Primary tumour: gastrointestinal (70); respiratory or mediastinal (53); > 92% participants had metastatic cancer. Former analgesics: not reported Inclusion criteria: Patients aged ≥ 20 years with a diagnosis of any type of cancer, experiencing chronic malignant tumour‐related pain, with an average pain intensity score over the past 24 hours ≥ 4 on an 11‐point numerical rating scale (NRS) (0 = ‘no pain’ to 10 = ‘pain as bad as you can imagine’) on the day of randomisation, who "had not taken opioid analgesics (except for codeine phosphate (≤ 60 mg/day), or dihydrocodeine phosphate (≤ 30 mg/day) as antitussives) within 28 days before screening. Patients must have been dissatisfied with the pain relief achieved on their current analgesic treatment for cancer pain and must have had pain requiring treatment with an opioid analgesic (based on the investigator’s assessment)." Exclusion criteria: "an uncontrolled or clinically significant arrhythmia; a history of or current disease that could result in increased intracranial pressure, disturbance of consciousness, lethargy, or respiratory problems; any disease for which opioids are contraindicated; a history of surgery intended for the cure of the primary disease or for the treatment of cancer pain within 28 days before screening or during the study; radiation therapy within 7 days before screening; or a psychiatric disorder or concurrent symptoms with accompanying pain that could interfere with efficacy and safety evaluations. Patients were also excluded if they had any of the following laboratory values at screening: white blood cell count ≤ 3000/mL, platelet count ≤ 10 x 104/uL, haemoglobin ≤ 9.5 g/dL, corrected total serum calcium level > 12.5 mg/dL, alanine aminotransferase or aspartate aminotransferase ≥ 3 times the upper limit of normal, total bilirubin ≥ 1.5 times the upper limit of normal, or creatinine ≥ 2 mg/dL. The following medications were prohibited: opioid analgesics (including codeine phosphate and dihydrocodeine phosphate as antitussives), except morphine IR 5 mg as rescue medication); opioid antagonists (e.g. naloxone, levallorphan), except for the treatment of respiratory depression; anti‐parkinsonian drugs; neuroleptics (including antipsychotics, except for prochlorperazine); monoamine oxidase inhibitors; serotonin norepinephrine reuptake inhibitors; noradrenergic and specific serotonergic antidepressants; radiotherapy; nerve block; stimulation analgesia; other investigational drugs. The following drugs were prohibited on an as‐needed basis as newly started treatment (but could be continued at the same regimen if started before study entry): Selective serotonin reuptake inhibitors; tricyclic or tetracyclic antidepressants; anti‐anxiety agents (e.g. benzodiazepines); hypnotics (e.g. benzodiazepines, non‐benzodiazepine hypnotics, barbiturates); anticonvulsants; central muscle relaxants; bisphosphonates; corticosteroids; anti‐arrhythmics; non‐opioid analgesics (nonsteroidal anti‐inflammatory drugs (e.g. cyclo‐oxygenase II inhibitors)); pyrazolone antipyretic agents (e.g. sulpyrine) and analine antipyretic agents (e.g. acetaminophen); neurotropin; pregabalin. The following were permitted as needed during the study: topical corticosteroids; lidocaine (as a local anaesthetic); acetaminophen (≤ 1.5 g/day (Japan) or ≤ 4 g/day (Korea) for fever reduction); supportive therapy for chemotherapy; stable doses of very short‐acting, non‐benzodiazepine hypnotic drugs (for insomnia); medications for nausea, vomiting, and constipation; and rescue medication (as described below). Chemotherapy could be continued at the same dose or chemotherapy doses could be reduced, discontinued, or restarted (if deemed necessary by the investigator); however, if a patient’s chemotherapy was considered by the investigator to be interfering with efficacy or safety evaluations of the study drug, that patient was excluded from the study." |
|
| Interventions |
Oxycodone arm ‐ Drug: Oxycodone HCI ‐ Dose/dosing: 5 to 40 mg bid. The median of the mean total daily dose = 13.8 mg. The median modal (or most frequently used) total daily dose = 10 mg ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: "4 week double‐blind treatment period (including titration and maintenance periods), and a 1 week post‐treatment period." Median duration of treatment 28 days ‐ Titration schedule: "Study treatment was initiated with twice daily doses of oxycodone HCl [hydrochlorine] CR 5 mg. During the titration period, doses of study treatment could be increased if necessary to achieve adequate pain control to a maximum of oxycodone HCl CR 40 mg bid [twice daily] after a patient had received the same dose at least four consecutive times. Dose escalations could begin on Day 3 of the titration period. Although not required for dose escalation, the following criteria were evaluated in patients who needed a dose escalation (based on the investigators assessment): 24 hour pain intensity score (11‐point NRS) of at least 4 on the previous evaluation and rescue medication used for breakthrough pain at least three times per day. Doses could be decreased during the study as needed for safety reasons to the minimum doses of oxycodone HCl CR (5 mg bid). Study drug doses were titrated to each patient’s optimal dose, balancing efficacy and tolerability, until sufficient analgesia was attained. Patients with a pain intensity score of no more than 3 who did not take rescue medication more than twice a day while taking stable doses of study drug (six consecutive identical doses) over a consecutive 3 day period were considered eligible to formally enter the maintenance period; patients who did not meet these criteria were permitted to continue in the double‐blind treatment period while continuing to titrate their dose. During the maintenance period, patients continued taking the optimal dose of study drug determined during the titration period. Dose adjustments were permitted during the maintenance period except during the last 3 days. Dose levels during the last 3 days of the maintenance period were to be kept stable." ‐ Rescue medication: "Oral morphine IR 5 mg was permitted throughout the study (except during the screening period) as rescue medication for breakthrough pain, with no limit on the number and timing of doses per day." The mean (SD) of the average number of morphine IR doses taken per day = 1.4 (0.43); mean (SD) of the average total daily dose = 6.7 (2.15) mg morphine IR ‐ Other medication: See the inclusion and exclusion criteria in cell above Comparison arm ‐ Drug: tapentadol ‐ Dose and dosing: 25 to 200 mg bid. The median of the mean total daily dose = 64.5 mg. The median modal (or most frequently used) total daily dose = 50 mg ‐ Formulation: Extended‐release ‐ Route of administration: oral ‐ Length of treatment: "4 week double‐blind treatment period (including titration and maintenance periods), and a 1 week post‐treatment period." Median duration of treatment 28 days ‐ Titration schedule: "Study treatment was initiated with twice daily doses of tapentadol ER 25 mg. During the titration period, doses of study treatment could be increased if necessary to achieve adequate pain control to a maximum of tapentadol ER 200 mg bid [twice daily] after a patient had received the same dose at least four consecutive times. Dose escalations could begin on Day 3 of the titration period. Although not required for dose escalation, the following criteria were evaluated in patients who needed a dose escalation (based on the investigators assessment): 24 hour pain intensity score (11 point NRS) of at least 4 on the previous evaluation and rescue medication used for breakthrough pain at least three times per day. Doses could be decreased during the study as needed for safety reasons to the minimum doses of tapentadol ER (25 mg bid). Study drug doses were titrated to each patient’s optimal dose, balancing efficacy and tolerability, until sufficient analgesia was attained. Patients with a pain intensity score of no more than 3 who did not take rescue medication more than twice a day while taking stable doses of study drug (six consecutive identical doses) over a consecutive 3 day period were considered eligible to formally enter the maintenance period; patients who did not meet these criteria were permitted to continue in the double‐blind treatment period while continuing to titrate their dose. During the maintenance period, patients continued taking the optimal dose of study drug determined during the titration period. Dose adjustments were permitted during the maintenance period except during the last 3 days. Dose levels during the last 3 days of the maintenance period were to be kept stable." ‐ Rescue medication: "Oral morphine IR 5 mg was permitted throughout the study (except during the screening period) as rescue medication for breakthrough pain, with no limit on the number and timing of doses per day." The mean (SD) of the average number of morphine IR doses taken per day = 1.4 (0.46); mean (SD) of the average total daily dose = 7 (2.3) mg morphine IR ‐ Other medication: See the inclusion and exclusion criteria in cell above |
|
| Outcomes | ‐ Pain intensity: Assessed by participants once daily (evening on an 11‐point numerical rating scale from 0 (no pain) to 10 (= pain as bad as you can imagine). Primary efficacy endpoint was the mean change in average pain intensity from baseline to the last 3 days of study. ‐ Patient global impression of change: questionnaire completed at weeks 1, 2, 3 of double‐blind treatment and at the end of study or early withdrawal. Participants rated their overall condition on a scale from 1 (very much improved) to 7 (very much worse) by completing the following statement, "Since the start of this treatment, my cancer‐related pain overall is..." ‐ Adverse events: monitored and coded using the Medical Dictionary for Regulatory Activities. Each instance of disease progression was considered an adverse event and included in the analysis of treatment‐emergent adverse events. |
|
| Notes | ‐ Study free of commercial funding? No, the study was funded by Janssen Research and Development. ‐ Groups comparable at baseline? The groups appeared to be comparable at baseline. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patient assignments to study treatment were based on a computer‐generated randomization schedule prepared by the sponsor prior to the study; randomization was balanced using randomly permuted blocks and stratified by study site. An Interactive Voice Response System (IVRS) assigned each patient a unique treatment code, which determined that patient’s treatment assignment." |
| Allocation concealment (selection bias) | Low risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "The blind was not broken until all patients completed the study and the database was finalized, except in case of emergency." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | Per‐protocol analyses including 139/172 oxycodone participants and 126/168 tapentadol participants |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The safety population included all randomised participants who received at least one dose of study drug, that is 340/343 randomised participants (172 oxycodone participants and 168 tapentadol participants). |
| Selective reporting (reporting bias) | Low risk | All main expected outcomes were reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Inoue 2017.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group multicentre, non‐inferiority trial Year: 2014 to 2015 Country: Japan |
|
| Participants |
Participants: 184 participants enrolled, 181 randomised (93 to oxycodone and 88 to hydromorphone) of whom all received study drugs and 147 participants completed the study (75 oxycodone, 72 hydromorphone). 34 participants discontinued the study (18 oxycodone, 16 hydromorphone). Reasons for discontinuation included adverse events (14 oxycodone, 10 hydromorphone). Oxycodone: 92 (1 participant excluded due to major protocol deviation), 61 men, 31 women, mean age (± SD) 68.4 (± 9.17) years. Primary tumour: lung (25); breast (6), gastrointestinal (39), hepatic‐biliary‐pancreatic (15), urogenital (6); other (1). Former analgesics: not reported Hydromorphone: 86 (2 participants excluded as they had no evaluable data after study treatment; reason for this not reported), 47 men, 39 women, mean age (± SD) 70.1 (± 10.19) years. Primary tumour: lung (31); breast (6), gastrointestinal (26), hepatic‐biliary‐pancreatic (12), urogenital (10); other (1). Former analgesics: not reported Inclusion criteria: Cancer patients aged ≥ 20 years, receiving non‐opioid analgesics for cancer pain, with an average pain intensity score in the past 24 hours ≥ 35 mm on VAS and an Eastern Cooperative Oncology Group performance status ≤ 3, who had not used opioid analgesics within 14 days of enrolment, and eligible for treatment with potent opioid analgesics Exclusion criteria: Patients with symptoms for which oxycodone or morphine were contraindicated or relatively contraindicated; receipt of a monoamine oxidase inhibitor within 2 weeks of enrolment; opioid analgesics; narcotic antagonists; participation in another clinical trial within 4 weeks of enrolment; serious hepatic, renal, or respiratory disorder of Common Terminology Criteria for Adverse Events grade 3; starting new treatment with/changing the dosing regimen of systemic non‐opioid analgesics during treatment; adjuvant analgesics for pain relief during treatment; bisphosphonates or anti‐RANKL antibody preparations during treatment; radiotherapy during treatment; nerve block during treatment; percutaneous vertebroplasty during treatment; surgery during treatment; receipt of any new cancer chemotherapy or immunotherapy for the first time during treatment |
|
| Interventions |
Oxycodone arm ‐ Drug: Oxycodone hydrochloride + or placebo hydromorphone extended‐release tablets ‐ Dose/dosing: Twice daily dosing of active drug. See also "Titration schedule" ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 7 days ‐ Titration schedule: Intial starting dose of 10 mg/day; every 24 hours up to a maximum of 5 times and 80 mg/day; the dose could be increased if necessary due to insufficient analgesic efficacy. 65/92 participants completed or discontinued the study with no dose increase from the initial dose. ‐ Rescue medication: Oral morphine hydrochloride (5 mg when coupled with oxycodone daily doses of 10‐30 mg, 10 mg when coupled with oxycodone daily dose of 40 mg, 15 mg when coupled with oxycodone daily dose of 60 mg, and 20 mg when coupled with oxycodone daily dose of 80 mg). The mean number of doses per day ≤ 1 on all evaluation days ‐ Other medication: Magnesium oxide at 2 g/day and prochlorperazine maleate at 15 mg/day were administered to ensure balanced evaluation of constipation and nausea/vomiting. See the also the exclusion criteria in cell above Comparison arm ‐ Drug: Hydromorphone + or placebo oxycodone extended‐release tablets ‐ Dose/dosing: Once daily dosing of active drug. See also "Titration schedule" ‐ Formulation: Extended‐release ‐ Route of administration: oral ‐ Length of treatment: 7 days ‐ Titration schedule: Intial starting dose of 4 mg/day, every 24 hours up to a maximum of 5 times and 24 mg/day; the dose could be increased if necessary due to insufficient analgesic efficacy. 67/86 participants completed or discontinued the study with no dose increase from the initial dose. ‐ Rescue medication: Oral morphine hydrochloride (5 mg when coupled with hydromorphone daily doses of 4‐8 mg, 10 mg when coupled with hydromorphone daily dose of 12 mg, 15 mg when coupled with hydromorphone daily dose of 18 mg, and 20 mg when coupled with hydromorphone daily dose of 24 mg). The mean number of doses per day ≤ 1 on all evaluation days ‐ Other medication: Magnesium oxide at 2 g/day and prochlorperazine maleate at 15 mg/day were administered to ensure balanced evaluation of constipation and nausea/vomiting. See the also the exclusion criteria in cell above |
|
| Outcomes | ‐ Pain intensity: Average pain severity for the past 24 hours assessed by participants once daily (using a VAS on which ratings ≥ 35 mm) indicates moderate–severe pain that interferes with functioning. Full range not reported. Primary efficacy endpoint was the mean change in average pain intensity from baseline to completion or discontinuation of treatment. ‐ Sleep quality: Assessed by patients at baseline and on each evaluation day (days 1, 8/discontinuation). Participants rated their sleep on a scale from 0 (very unsatisfactory/did not sleep at all) to 3 (satisfactory). ‐ Adverse events: monitored and coded using the Medical Dictionary for Regulatory Activities. Severity of adverse events graded as mild, moderate, or severe |
|
| Notes | ‐ Study free of commercial funding? No, the study was funded by Daiichi Sankyo Co Ltd which also manufactured ER hydromorphone (the second largest pharmaceutical company in Japan according to Wikipedia on 1 Dec 2020). Three authors employed by this company (Daiichi Sanko) ‐ Groups comparable at baseline? The groups appeared to be comparable at baseline. Clinical trial number: JapicCTI‐142666 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Subjects were randomized at a ratio of 1:1". No further information provided |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | "A double‐dummy method was used for blinding, and each randomized subject received either hydromorphone extended‐release tablets plus placebo oxycodone hydrochloride extended‐release tablets or placebo hydromorphone extended‐release tablets plus oxycodone hydrochloride extended‐release tablets orally for 7 days (once‐daily dosing for hydromorphone and twice‐daily dosing for oxycodone)." No further information provided about who exactly was blinded or how blinding was undertaken in terms of how the drugs looked, and daily dosing which differed between oxycodone and hydromorphone |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | Low risk | 1 patient excluded due to major protocol deviation and 2 further patients excluded as they had no evaluable data (not reported why this was the case). Data included for 92/93 in the oxycodone group and for 86/88 in the hydromorphone group |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | 1 patient excluded due to major protocol deviation and 2 further patients excluded as they had no evaluable data (not reported why this was the case). Data included for 92/93 in the oxycodone group and for 86/88 in the hydromorphone group |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes were reported. |
| Were the participants adequately titrated? | Low risk | Yes, they appeared to be. Use of rescue medication very low (mean number of doses was less than 1 dose/day for both groups) |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | No other biases identified |
Inoue 2018.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group multicentre, non‐inferiority trial Year: 2013 to 2014 Country: Japan |
|
| Participants |
Participants: 183 participants enrolled, 181 randomised (89 to oxycodone and 92 to hydromorphone) of whom 179 received study drugs and 160 participants completed the study (75 oxycodone, 85 hydromorphone). 21 participants discontinued the study (14 oxycodone, 7 hydromorphone). Reasons for discontinuation included adverse events (8 oxycodone, 6 hydromorphone). Oxycodone: 89 (5 participants excluded due to major protocol deviation), 62 men, 22 women, mean age (± SD) 66.8 (± 10.14) years. Primary tumour: lung (33), gastrointestinal (26), hepatic‐biliary‐pancreatic (9), urogenital (9); other (7). Former analgesics: not reported Hydromorphone: 92 (4 participants excluded due to major protocol deviation), 54 men, 34 women, mean age (± SD) 67.7 (± 10.29) years. Primary tumour: Head/neck (2), lung (30), breast (4), gastrointestinal (24), hepatic‐biliary‐pancreatic (13), urogenital (11); other (4). Former analgesics: not reported Inclusion criteria: Cancer patients aged ≥ 20 years, receiving non‐opioid analgesics for cancer pain, with an average pain intensity score in the past 24 hours ≥ 35 mm on VAS and an Eastern Cooperative Oncology Group performance status ≤ 3, who had not used opioid analgesics within 14 days of registration, and eligible for treatment with potent opioid analgesics Exclusion criteria: Patients with symptoms for which oxycodone or morphine are contraindicated or relatively contraindicated; receipt of a monoamine oxidase inhibitor within 2 weeks of registration; participation in another clinical trial within 4 weeks of enrolment; serious hepatic, renal, or respiratory disorder of Common Terminology Criteria for Adverse Events grade 3; starting new treatment with/changing the dosing regimen of systemic non‐opioid analgesics during treatment; adjuvant analgesics for pain relief during treatment; bisphosphonates or anti‐RANKL antibody preparations during treatment; new initiation of radiotherapy, nerve block, percutaneous vertebroplasty, surgery, or cancer chemotherapy or immunotherapy |
|
| Interventions |
Oxycodone arm ‐ Drug: Oxycodone hydrochloride + or placebo hydromorphone immediate‐release tablets ‐ Dose/dosing: Four times daily dosing. See also "Titration schedule" ‐ Formulation: Immediate‐release powder ‐ Route of administration: oral ‐ Length of treatment: 5 days ‐ Titration schedule: Intial starting dose of 10 mg/day, every 24 hours up to a maximum of 60 mg/day in increments of 10 (day 1 to 2) or 20 (day 2‐4) mg/day. 60/84 participants completed or discontinued the study with no dose increase from the initial dose. ‐ Rescue medication: Oral morphine hydrochloride (5 mg when coupled with oxycodone daily doses of 10‐20 mg, 10 mg when coupled with oxycodone daily dose of 40 mg, and 15 mg when coupled with oxycodone daily dose of 60 mg). The mean number of doses per day ≤ 1 on all evaluation days ‐ Other medication: Magnesium oxide at 2 g/day and prochlorperazine maleate at 15 mg/day were administered to ensure balanced evaluation of constipation and nausea/vomiting. See the also the exclusion criteria in cell above Comparison arm ‐ Drug: Hydromorphone + or placebo oxycodone immediate‐release (not specified that it was powder) ‐ Dose/dosing: Four times daily dosing. See also "Titration schedule" ‐ Formulation: Immediate‐release tablet ‐ Route of administration: oral ‐ Length of treatment: 5 days ‐ Titration schedule: Intial starting dose of 4 mg/day, every 24 hours up to a maximum of 16 mg/day in increments of 4 mg/day. 66/88 participants completed or discontinued the study with no dose increase from the initial dose. ‐ Rescue medication: Oral morphine hydrochloride (5 mg when coupled with hydromorphone daily doses of 4‐8 mg, 10 mg when coupled with hydromorphone daily dose of 12 mg, and 15 mg when coupled with hydromorphone daily dose of 16 mg). The mean number of doses per day ≤ 1 on all evaluation days ‐ Other medication: Magnesium oxide at 2 g/day and prochlorperazine maleate at 15 mg/day were administered to ensure balanced evaluation of constipation and nausea/vomiting. See the also the exclusion criteria in cell above |
|
| Outcomes | ‐ Pain intensity: Average pain severity for the past 24 hours assessed by participants once daily (using a 0‐100 mm VAS). Primary efficacy endpoint was the mean change in average pain intensity from baseline to completion or discontinuation of treatment. ‐ Sleep quality: Assessed by patients at baseline and on each day (days 1‐6/discontinuation). Participants rated their sleep on a scale from 0 (very unsatisfactory/did not sleep at all) to 3 (satisfactory). ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? No, the study was funded by Daiichi Sankyo Co Ltd which also manufactured ER hydromorphone (the second largest pharmaceutical company in Japan according to Wikipedia on 1 Dec 2020). Three authors employed by this company (Daiichi Sanko) ‐ Groups comparable at baseline? Possible differences in proportion of males (61.4% in the hydromorphone group and 73.8% in the oxycodone group) and proportion of patients with ECOG PS of 0 (33% in the hydromorphone group and 20.2% in the oxycodone group). The authors stated that all other characteristics were similar between the two groups. Clinical trial number: JapicCTI‐132338 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The computer‐generated block random allocation sequence was provided by Bell Medical Solutions Inc. (Tokyo, Japan) and was stratified according to history of opioid usage". |
| Allocation concealment (selection bias) | Low risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | "A double‐dummy method was employed for blinding, and patients received either hydromorphone plus placebo or a placebo tablet plus oxycodone hydrochloride powder orally four times daily for 5 days." No further information provided about who exactly was blinded or how blinding was undertaken in terms of how the drugs looked |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data analysed for 84/89 patients in the oxycodone group and 88/92 in the hydromorphone group |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data analysed for 84/89 patients in the oxycodone group and 88/92 in the hydromorphone group |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes were reported. |
| Were the participants adequately titrated? | Low risk | Yes, they appeared to be. Use of rescue medication very low (mean number of doses was less than 1 dose/day for both groups) |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | No other biases identified |
Kalso 1990.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: Finland |
|
| Participants |
Participants: 20 participants entered, 19 participants analysed (1 participant excluded as her morphine dose had to be considerably reduced due to adverse effects); 11 women, 9 men; median age (range): 56 (20‐75) years; cancer type: pancreatic (3), breast (5), prostate (1), gastric (1), rectal (2), other (8): cancer stage: metastatic; type of pain: visceral (6), nerve (7), bone (5), bone‐fracture (1), bone‐nerve (1), soft tissue (1); setting: not reported, tertiary?; previous analgesic medication: buprenorphine (7), oxycodone (1), dextropropoxyphene (1), aspirin + codeine (1), ibuprofen + buprenorphine (2), indomethacin + buprenorphine (1), dextropropoxyphene + buprenorphine (1), diclofenac + buprenorphine (1), indomethacin + codeine (2), naproxen + dextropropoxyphene (1), noramidopyrin + pitofenone (1), ketoprofen + dextropropoxyphene (1) Inclusion criteria: "Twenty patients, 11 women and nine men, who had metastasised cancer and severe pain and who required a change from weaker narcotic analgesic agents (codeine, dextropropoxyphene, buprenorphine) to morphine, participated in the study." Exclusion criteria: none reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: previous opioid treatment was discontinued 12‐24 hours before commencing the study, and during this time 1 mg/kg meperidine was given intramuscularly when requested. The participants titrated themselves free from pain in 48 hours using a patient‐controlled analgesia (PCA) device. The concentration of both morphine hydrochloride and oxycodone hydrochloride was 10 mg/mL.This treatment was continued for another 48 hours with the use of the same drug, which was now taken orally. The oral dose was calculated from the IV consumption during the previous 24 hours. The daily oral dose was calculated in mL by assuming that the bioavailability of morphine was either 44% (first 10 participants, group 1) or 33% (last 10 participants, group 2) and that the bioavailability of oxycodone hydrochloride was 66% (group 1) and 50% (group 2). To overcome the differences in bioavailabilities of the two drugs, the concentrations of the oral solutions were 2.7 mg/mL for oxycodone hydrochloride and 4 mg/mL for morphine. The dosing interval was 4 hours and the dose was increased by 1 mL at a time if the participant was not pain‐free during the 4‐hour period. If the participant was pain‐free, but too sedated, the dose was decreased by 1 mL. PCA device: The bolus dose was 3 mg, which was given over a period of 60 seconds, followed by a tail dose of 2 mg over 1 hour. The lockout time, during which the participant was unable to initiate another dose, was 15 minutes. If the participant was not free from pain with this regimen, the tail dose was increased by 2 mg at a time. ‐ Formulation: Immediate‐release (oral) ‐ Route of administration: IV (2 days) then oral (2 days) ‐ Length of treatment: 4 days ‐ Titration schedule: see 'Dose and dosing' ‐ Rescue medication: see 'Dose and dosing.' No further information was reported. ‐ Other medication: any pre‐existing treatment with nonsteroidal anti‐inflammatory drugs was continued. Comparison arm: Same as oxycodone arm, just replacing oxycodone with morphine ‐ For cross‐over trials, cross‐over schedule: "The same protocol was then repeated with the other drug for another 96 hours." |
|
| Outcomes | ‐ Pain severity: Assessed by participant at study start and every 4 hours from 8 a.m. to 8 p.m.; VAS from 0 to 10 ‐ Adverse effects: determined by questioning (have you had nausea, constipation, drowsiness, sedation symptoms, hallucinations, or any other symptoms you would connect with the analgesic?) scored according to grade (moderate = 1, severe = 2); registered on the second day of each study period ‐ Sleep quality, registered on the second day of each study period ‐ Participant preference or acceptability with reason The last 24 hours of each of the four study stages were considered as the steady state and the drug consumption, and the ratings from the VAS during this period were used for the statistical calculations. |
|
| Notes | ‐ Study free of commercial funding? Yes. Supported by the Paolo (non‐profit) Foundation, Helsinki, Finland ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomised. No further information provided |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | The study was described as "double‐blind". No further information reported, so it was unclear who was blinded and whether it was adequately executed |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | Low risk | The data from 1/20 participants were excluded. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The data from 1/20 participants were excluded. |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes were reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available for both study periods for 19/20 participants. |
| Other bias | Low risk | The authors reported that "The order in which the drugs were given (either as the first or the second study drug) had no effect on the drug consumption." No other potential biases were identified. |
Kaplan 1998.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group trial Year: not reported Country: USA |
|
| Participants |
Participants: 180 participants enrolled (108 before protocol amendment allowing dose titration before randomisation and 72 after the amendment; 16/72 participants discontinued before randomisation due to lack of acceptable pain control (6), intercurrent illness (4), adverse event (2), death (1), withdrawal of consent (1), other (2). 164 participants were randomised (controlled‐release oxycodone: N = 81; immediate‐release oxycodone: 83); 156 were included in efficacy analyses (4 participants did not receive the study medication, 3 participants did not complete the efficacy ratings, and 1 participant may have received unblinded treatment). All 160 participants who received at least 1 dose of study medication were included in the safety analyses (of adverse events). 74% of participants were white; mean (SE) age = 59 (1) years; 58% were male; most participants were receiving oral morphine at study entry; cancer type: gastrointestinal (22%), lung (21%), prostate (17%), breast (10%), gynaecological (10%): predominant pain sites were bone and viscera, with an additional 15 participants (9 in controlled‐release oxycodone group and 6 in the immediate‐release oxycodone group) reporting neuropathic pain. Inclusion criteria: "Male and female patients with cancer‐related pain were enrolled at 17 centers. The study received institutional review board approval at each center and all patients gave written informed consent. At the time of enrollment, patients were being treated with a strong single‐entity opioid or 10 or more tablets per day of a fixed‐dose opioid/nonopioid analgesic; were receiving a stable opioid dose; and had stable coexistent disease. Under the original protocol, patients were excluded if they were receiving any other analgesics (opioid or nonopioid) or if they were to receive radiotherapy immediately before enrollment or during the study period. After the study had begun, these exclusion criteria were eliminated by an amendment to facilitate enrollment into the study, which had been slow." Exclusion criteria: See above |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose/dosing: Oxycodone tablets (10 mg) every 12 hours (8 a.m. and 8 p.m.) and placebo tablets every 2 p.m. and bedtime. Mean daily dose (range) = 114 (20 to 400) mg ‐ Formulation: Controlled‐release ‐ Route of administration: Oral ‐ Length of treatment: 6 days ‐ Titration schedule: The original study design did not allow dose titration or use of rescue medication for breakthrough/incident pain. Participants whose pain was not effectively controlled at the initial oxycodone dose calculated from previous opioid use were discontinued from the study. However, an interim analysis conducted to determine whether dose adjustments were required showed that dropout rates were too high for relevant conclusions. This suggested that the initial conversion dose estimate was not adequate for a subgroup of participants, and the protocol was amended to include open‐label titration with immediate‐release oxycodone before the participants were randomised to double‐blind treatment, as well as the use of immediate‐release oxycodone 5 mg tablets as rescue medication throughout the trial. Supplemental doses could be taken no more frequently than every 4 hours at no more than approximately 1/6 of the daily dose of study medication. No further information was reported. ‐ Rescue medication: See 'Titration schedule' above. Mean number of rescue medication doses per day = 0.6 ‐ Other medication: See 'Inclusion criteria' above. No further information reported Comparison arm ‐ Drug: Oxycodone ‐ Dose and dosing: Oxycodone tablets (5 mg) every 6 hours (8 p.m., bedtime (≥ 3 hours after 8 p.m., but not after 2 a.m.), 8 a.m. and 2 p.m.). Mean daily dose (range) = 127 (40 to 640) mg ‐ Formulation: Immediate‐release ‐ Route of administration: oral ‐ Length of treatment: 6 days ‐ Titration schedule: The original study design did not allow dose titration or use of rescue medication for breakthrough/incident pain. Participants whose pain was not effectively controlled at the initial oxycodone dose calculated from previous opioid use were discontinued from the study. However, an interim analysis conducted to determine whether dose adjustments were required showed that dropout rates were too high for relevant conclusions. This suggested that the initial conversion dose estimate was not adequate for a subgroup of participants, and the protocol was amended to include open‐label titration with immediate‐release oxycodone before the participants were randomised to double‐blind treatment, as well as the use of immediate‐release oxycodone 5 mg tablets as rescue medication throughout the trial. Supplemental doses could be taken no more frequently than every 4 hours at no more than approximately 1/6 of the daily dose of study medication. No further information was reported. ‐ Rescue medication: see 'Titration schedule.' Mean number of rescue medication doses per day 1 ‐ Other medication: see 'Inclusion criteria.' No further information reported |
|
| Outcomes | ‐ Pain intensity: Assessed by participant at study start and 4 times daily at 8 a.m., 2 p.m., 8 p.m. and bedtime; categorical verbal scale from 0 (= none, 1 = slight, 2 = moderate) to 3 (= severe) ‐ Acceptability of treatment: Assessed by participant at study start and twice daily at 8.a.m. and 8 p.m.; categorical verbal scale from 1 (= very poor; 2 = poor, 3 = fair, 4 = good) to 5 (= excellent) ‐ Adverse events: Those spontaneously reported by participants or observed by investigators were recorded, and their severity and relationship to study drug (none, possible, probable, definite) were assessed by each investigator. |
|
| Notes | ‐ Study free of commercial funding? No, some or 1 of the authors (including the corresponding author) were or was employee(s) of Purdue Pharma Ltd, the manufacturer of the study drugs. ‐ Groups comparable at baseline? The authors reported, "There were no significant differences in the primary pain site, prestudy opioid, or cancer diagnosis between the two treatment groups." No other information reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The authors stated that the participants were randomised, but give no further details. |
| Allocation concealment (selection bias) | Unclear risk | The authors stated that the participants were randomised, but give no further details. |
| Blinding of participants and personnel (performance bias) Pain | Low risk | Double‐blind placebo‐controlled study. To maintain the blind, all doses of the study medication were encapsulated in green size #00 lactose‐filled capsules. |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | Low risk | A total of 156/164 participants were included in the efficacy analyses. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | A total of 160/164 participants were included in the safety analyses. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes were reported. |
| Were the participants adequately titrated? | High risk | No before amendment; unclear after amendment |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Lauretti 2003.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: Brazil |
|
| Participants |
Participants: 22/26 enrolled participants evaluated (withdrawals due to death unrelated to the study (1), uncontrollable nausea/vomiting (1), and unstable pain control requiring spinal drugs (2)); mean/median (?) (SD/interquartile range?) age 59 (19) years; 15 men, 7 women; cancer type: oropharynx (9), lung (3), prostate gland (2), colon (4), gastric (2), ovary (2); pain types were somatic and visceral; adjuvant therapy: radiation (1), chemotherapy (6), radiation/chemotherapy (4), none (11) Inclusion criteria: "26 patients with chronic cancer pain of the visceral and somatic type..... Before enrolling in this actual study, patients received 3‐4 mg/kg‐1 tramadol, plus nonsteroidal anti‐inflammatory drugs: however they still complained of pain VAS ≥ 4 cm." Exclusion criteria: none listed |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose/dosing: The optimum dosage was calculated on a daily basis, and the consumption ratio of oxycodone to morphine was set at 1:1.8. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 14 days ‐ Titration schedule: The study started with an open‐label, randomised titration phase to achieve stable pain control for 7 days. Participants only used immediate‐release morphine and had free access to it to keep pain VAS < 4. ‐ Rescue medication: At any point, participants were allowed to use immediate‐release morphine (10 mg tablets) as needed to keep pain VAS ≤ 4. ‐ Other medication: as part of the protocol, all participants were taking oral 25 mg amitriptyline at bedtime. Comparison arm ‐ Drug: morphine ‐ Dose/dosing: The optimum dosage was calculated on a daily basis, and the consumption ratio of oxycodone to morphine was set at 1:1.8. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 14 days ‐ Titration schedule: The study started with an open‐label, randomised titration phase to achieve stable pain control for 7 days. Participants only used immediate‐release morphine and had free access to it to keep pain VAS < 4. ‐ Rescue medication: at any point, participants were allowed to use immediate‐release morphine (10 mg tablets) as needed to keep pain VAS ≤ 4. ‐ Other medication: as part of the protocol, all participants were taking oral 25 mg amitriptyline at bedtime. ‐ For cross‐over trials, cross‐over schedule: "After stable pain relief was achieved [during titration phase], this was followed by a double‐blind, cross‐over phase in two periods, 14 days each...... and no period of washout was allowed for ethical reasons". |
|
| Outcomes | ‐ Pain intensity: assessed by participants; 10‐cm VAS from 0 (= no pain at all) to 10 (= worst possible pain) ‐ Participant satisfaction: assessed by participant ‐ Adverse events: assessed by participant (possibly using a 10‐cm VAS similar to pain intensity, but data not reported that way) ‐ Number of rescue morphine tablets: Assessed by participant It also appeared that an investigator recorded these data on a weekly basis. |
|
| Notes | ‐ Study free of commercial funding? No information reported ‐ Groups comparable at baseline? Unclear, no information reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Low risk | The participants were blinded, but it was unclear whether the investigator administering the drugs was. |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | The participants were blinded, but it was unclear whether the investigator administering the drugs was. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | The participants and outcome assessor were blinded. |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | The participants and outcome assessor were blinded. |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Data from 22/26 participants included |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Data from 22/26 participants included |
| Selective reporting (reporting bias) | High risk | All obvious outcomes were reported, although not in the most useful manner (e.g. no collapsing across study phase weeks, that is, mean final weekly dose of CR oxycodone and morphine were reported for 4 weeks, not 2 weeks). |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available from both time periods, although not reported by arm (see two cells above). |
| Other bias | Low risk | It was unclear whether there were any carry‐over effects, but there probably were none. |
Lee 2017.
| Study characteristics | ||
| Methods |
Design: randomised, open‐label, parallel‐group trial Year: 2015 to 2016 Country: Korea |
|
| Participants |
Participants: 68 participants screened, 66 randomised (34 to oxycodone and 32 to morphine) of whom 65 received study drugs and 57 participants completed the study (28 oxycodone, 29 morphine). 8 participants discontinued the study (5 oxycodone, 3 morphine). Reasons for discontinuation included adverse events (1 oxycodone, 0 morphine; although under study results the authors reported that 2 patients in the oxycodone group discontinued due to adverse events). Oxycodone: n = 33, 21 men, 12 women, mean age (± SD) 66.6 (± 9.1) years. Primary tumour: pancreatic (8), gastric (7), gall bladder/biliary tract (3), lung (2), liver (2), breast (2), colorectal (1); other (8). Cancer stage I (0), II (2), III (4), IV (27). Former analgesics: not reported, but prior medication excluding anticancer therapy was: 32 Morphine: n = 32, 22 men, 10 women, mean age (± SD) 64.1 (± 13) years. Primary tumour: pancreatic (3), gastric (6), lung (5), colorectal (5); other (13). Cancer stage I (0), II (0), III (2), IV (29), unknown (1). Former analgesics: not reported, but prior medication excluding anticancer therapy was: 31 Inclusion criteria: Cancer patients aged ≥ 19 years, with moderate‐to‐severe pain (NRS ≥ 4) over the past 7 days, who were either hospitalised or scheduled for hospitalisation and not planned for discharge during the study period Exclusion criteria: Receipt of at least one of the following opioid analgesic doses: oral morphine dose 195 mg/day, oral oxycodone dose 130 mg/day, or patch fentanyl dose 75 g/hour for cancer pain prior to screening; medical history of hypersensitivity to oxycodone or morphine or other opioid analgesics; clinically significant respiratory disorder or severe respiratory dysfunction; current treatment with monoamine oxidase inhibitors; moderate‐to‐severe hepatic impairment (ALT or AST > 3.0 upper limit of normal (ULN), total bilirubin > 1.5 × ULN); respiratory depression or hypotension; current receipt of anticancer therapy that may affect pain control measurement, at the discretion of the investigator, or scheduled for radiotherapy during the study period; clinically significant cardiovascular or renal dysfunction; or pregnancy |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone (Oxynorm (R), EP[NOS]) ‐ Dose/dosing: At baseline, pain was stabilised with IV bolus injection of oxycodone "at a dose determined by the investigator based on the dose of the previous analgesics. Where the patient was using a strong opioid for the first time, 2 mg of oxycodone..... was initially administered by IV bolus injection to stabilize the pain." "All medication doses were diluted in 0.9% normal saline. The dose administered was adjusted at the investigator’s discretion according to the subject’s pain intensity". Mean (SD) cumulative dose at the end of treatment = 226.8 (110.4) mg. ‐ Formulation: Intravenous, in 10 mg/1 ml or 20 mg/2 ml ampoules. ‐ Route of administration: Intravenous ‐ Length of treatment: 5 days. ‐ Titration schedule: See "Dose/dosing" ‐ Rescue medication: Not reported. ‐ Other medication: Prophylocatic laxatives and antiemetics. Comparison arm ‐ Drug: morphine (BC Morphine sulfate hydrate injection (R)) ‐ Dose/dosing: At baseline, pain was stabilized with IV bolus injection of morphine "at a dose determined by the investigator based on the dose of the previous analgesics. Where the patient was using a strong opioid for the first time, 2 mg of ..... morphine was initially administered by IV bolus injection to stabilize the pain." "All medication doses were diluted in 0.9% normal saline. The dose administered was adjusted at the investigator’s discretion according to the subject’s pain intensity". Mean (SD) cumulative dose at the end of treatment = 226.6 (135.1) mg ‐ Formulation: Intravenous, in 5 mg/5 mL or 30 mg/2 mL ampoules ‐ Route of administration: Intravenous ‐ Length of treatment: 5 days ‐ Titration schedule: See "Dose/dosing" ‐ Rescue medication: Not reported ‐ Other medication: Prophylocatic laxatives and antiemetics |
|
| Outcomes | ‐ Pain intensity: assessed by participants; 0‐10 point NRS from 0 (= no pain at all) to 10 (= worst pain) ‐ Participant satisfaction: assessed by participant on days 3 and 5; 7‐point Patient Global Impression of Change scale from 1 (very much improved) to 7 (very much worse), and assessed by investigators on days 3 and 5; 7‐point Clinical Global Impression of Change scale from 1 (very much improved) to 7 (very much worse) ‐ Adverse events: assessed by participant |
|
| Notes | ‐ Study free of commercial funding? No, study supported by Mundi‐pharma Korea, which manufactures oxycodone ‐ Groups comparable at baseline? No, pancreatic and gastric cancers were more common in the oxycodone group while gastric, lung, and colorectal cancers were more common in the morphine group. ‐ Trial registration number: NCT02660229. Please note this record also included some results, however the adverse events data did not always directly seem to correspond with those reported in the full‐text publication and we therefore only included the data where there did not seem to be any conflict between the two reports and/or we knew the full definition of the adverse events. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details reported about randomisation sequence generation |
| Allocation concealment (selection bias) | High risk | “investigator administered the pertinent drug (oxycodone or morphine) following the order in the randomization list when patients were enrolled and was not blinded to the treatment allocation.” High risk because investigators enrolling participants could possibly foresee assignments and thus introduce selection bias |
| Blinding of participants and personnel (performance bias) Pain | High risk | Open‐label study |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | High risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data from 65/66 participants analysed |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data from 66/66 participants analysed |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes were reported. |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | NA |
| Other bias | Low risk | No other biases identified |
Leow 1995.
| Study characteristics | ||
| Methods |
Design: randomised, open‐label, single‐dose, cross‐over trial Year: not reported Country: Australia |
|
| Participants |
Participants: 12 participants entered; 5 women, 7 men; mean age (± SD): 68.8 (± 12.6) years; cancer type: cervical (2), breast (1), prostate (1), bowel (1), anal (1), endometrial (1), renal (1), lung/bronchial (2), skeletal or thoracic‐vertebral metastases (2); all inpatients; all receiving oral nutrition; none hypovolaemic; all opioid‐naive apart from 1 participant who was receiving paracetamol + dextropropoxyphene. 2 participants had compromised renal function, and 5 participants had impaired liver function to varying degree. Inclusion criteria: inpatients with moderate to severe cancer pain Exclusion criteria: known hypersensitivity to oxycodone or other opioid analgesics or a history of drug dependence, or both |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: single dose of IV oxycodone hydrochloride in a concentration of 5 mg/mL, equivalent to 4.5 mg/mL oxycodone base. The mean (SD) IV oxycodone dose administered was 0.11 (0.02) mg/kg (range 5.4 to 9 mg), which a previous study by the authors had shown to produce satisfactory analgesia in participants with moderate to severe cancer. Participants with impaired liver function received the lower doses of IV oxycodone. The IV oxycodone dose was administered into a forearm vein. The rate of injection (0.5 to 5 min) was titrated by the anaesthetist. ‐ Formulation: IV ‐ Route of administration: IV ‐ Length of treatment: 24 hours, 1 dose ‐ Titration schedule: See 'Dose and dosing' section ‐ Rescue medication: oral paracetamol (up to 1 g every 4 hours) or Di‐Gesic (up to 2 tablets every 4 hours) were available as rescue medication on participant request. 9 participants asked for supplementary analgesics after 4 hours post‐dosing. ‐ Other medication: "Medications that had been taken routinely by patients before the study, were permitted." Comparison arm ‐ Drug: oxycodone ‐ Dose and dosing: single dose of 30 mg oxycodone base in a rectal suppository ‐ Formulation: suppository ‐ Route of administration: rectal ‐ Length of treatment: 24 hours, 1 dose ‐ Titration schedule: see 'Dose and dosing' section ‐ Rescue medication: oral paracetamol (up to 1 g every 4 hours) or digesic (up to 2 tablets every 4 hours) were available as rescue medication on participant request. 9 participants asked for supplementary analgesics after 6‐8 hours post‐dosing. ‐ Other medication: "Medications that had been taken routinely by patients before the study, were permitted." ‐ For cross‐over trials, cross‐over schedule: "Patients were randomly assigned to begin treatment with either a single dose of... The second treatment was administered 24 h [hours] after the first dose." |
|
| Outcomes | ‐ Pain intensity: Assessed by participant at study start at 0.5, 1, 2, 4, 8, 12 and 24 hours post‐dosing; 10‐cm VAS with delimiters 'no pain' and 'worst pain imaginable' ‐ Adverse effects: assessed by questioning the participant at study start at 0.5, 1, 2, 4, 8, 12, and 24 hours post‐dosing; participants were asked to report any adverse effects, but were specifically asked whether they experienced nausea, vomiting, pruritus, lightheadedness, or drowsiness, using a 4‐point verbal rating scale going from 0 to 3 (none = 0, mild = 1, moderate = 2, severe = 3) |
|
| Notes | ‐ Study free of commercial funding? No. Supported by the Boots Company (Australia; manufacturer of the rectal suppository study drug), Pty Ltd, the University of Queensland Cancer Research Fund, and the Queensland Cancer Fund ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned to begin treatment with..." No further information reported |
| Allocation concealment (selection bias) | Unclear risk | "Patients were randomly assigned to begin treatment with..." No further information reported |
| Blinding of participants and personnel (performance bias) Pain | High risk | The study was open‐label. |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | The study was open‐label. |
| Blinding of outcome assessment (detection bias) Pain | High risk | The study was open‐label. |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | The study was open‐label. |
| Incomplete outcome data (attrition bias) Pain | Low risk | All data appeared to be included. It was not possible to confirm if based on the presented data, but no information to the contrary was reported. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | All data appeared to be included. It was not possible to confirm if based on the presented data, but no information to the contrary was reported. |
| Selective reporting (reporting bias) | Low risk | All expected outcomes seemed to be reported. |
| Were the participants adequately titrated? | Unclear risk | Not enough information reported |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available from both time periods. |
| Other bias | Low risk | "An absence of carryover effects (P > 0.05) between Treatments 1 and 2 was confirmed using the Grizzle analysis for cross‐over designs". |
Li 2013.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2010‐2013 Country: China |
|
| Participants |
Participants: 82 participants randomised; 48 men, 34 women, aged 32‐80 years; cancer types were lung (38), pancreatic (11), liver (6), breast (8), prostate (7), colorectal (5), gastric (3); no further information reported and not reported by intervention group ‐ Oxycodone: 42 participants ‐ Morphine: 40 participants Inclusion criteria: Patients with severe pain (numerical rating scale score > 7) from advanced cancer (verified by histopathology and imaging) Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: Starting dose = 10 mg every 12 hours up to 60 mg every 12 hours. Mean (SD) maintenance dose = 28.4 (4.2) mg/12 hours ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 3 days ‐ Titration schedule: Dose adjusted according to degree of pain relief assessed 3 hours after administration such that if NRS 0‐3, no dose change; if NRS 4‐6 dose, increase of 50%; and if NRS 7‐10, then dose increased to 100% (see also "Dose and dosing"; no further information reported) ‐ Rescue medication: Not reported ‐ Other medication: No other analgesics were taken 4 hours before oxycodone. No further information reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 20 mg every 12 hours up to 120 mg every 12 hours. Mean (SD) maintenance dose = 70.2 (4.5) mg/12 hours ‐ Formulation: Sustained‐release ‐ Route of administration: oral ‐ Length of treatment: 3 days ‐ Titration schedule: Dose adjusted according to degree of pain relief assessed 3 hours after administration such that if NRS 0‐3, no dose change; if NRS 4‐6, dose increase of 50%; and if NRS 7‐10, then dose increased to 100% (see also "Dose and dosing"; no further information reported) ‐ Rescue medication: Not reported ‐ Other medication: No other analgesics were taken 4 hours before oxycodone. No further information reported |
|
| Outcomes | ‐ Pain relief: assessed at 3 days according to the WHO criteria (complete response/painless [CR], partial response/pain was significantly reduced relative to before treatment and sleep was not disturbed [PR], mild relief/pain was slightly relieved compared to before treatment [MR], no response/treatment, gave no pain relief [NR]. Response rate = (CR number + PR number)/total number of cases ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Yes, government‐funded ‐ Groups comparable at baseline? Unclear; although the data were not presented, the authors reported that the groups did not differ at baseline, but unclear whether the authors only examined age, sex and tumour type |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients reported to be randomised, but no information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses were undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Liu 2021.
| Study characteristics | ||
| Methods | Design: randomised controlled trial Year: 2016‐2018 Country: China |
|
| Participants | Oxycodone: N = 34 (34 entered, 34 analysed), 17 males and 17 females; mean (SD) age 63.94 (2.25) years; stage III/IV: 3/31; cancer types: lung (19), breast (2) digestive tract (9), other (4); cause of pain disease‐related/treatment‐related 31/3; pain location: chest (11), abdomen (10), back (5), limbs (5), bone (3); nature of pain: Dull (18), soreness (10), compression‐like (4), other (2); mean (SD) duration of pain 26.03 (32.793) days(?) Ibuprofen: N = 32 (33 entered, 32 analysed; 1 patient excluded due to acute pulmonary embolism), 17 males and 15 females; mean (SD) age 62.88 (2.27) years; stage III/IV: 3/29; cancer types: lung (21), breast (1) digestive tract (6), other (4); cause of pain disease‐related/treatment‐related 28/4; pain location: chest (11), abdomen (6), back (5), limbs (7), bone (3); nature of pain: Dull (17), soreness (9), compression‐like (2), other (4); mean (SD) duration of pain 26.44 (33.625) days(?) Inclusion criteria: Opioid‐naive patients aged 18 or above, with somatic or visceral pain < 4 on NRS from clinically or pathologically confirmed malignant tumours and expected survival > 3 months suitable for oral medication and who "could be excluded from the effect of anti‐neoplastic therapy (e.g. radiotherapy, chemotherapy, targeted therapy) on analgesia" (page 3413) Exclusion: Inability to take oral medication, treatment with strong opioids prior to study start, neuropathic pain, inability "to perform and cooperate with the follow‐up visits (page 3413), contraindication to opioids, including hypoxic respiratory depression, head injury, paralytic intestinal obstruction, acute abdomen, delayed gastric emptying, COPD, pulmonary heart disease, acute or severe bronchial asthma, hypercapnia, allergy to oxycodone, moderate to severe liver dysfunction, severe renal dysfunction (creatinine clearance < 10 mL/min), chronic constipation, and taking MAOIs or discontinuing them for < 2 weeks), unsuitable for the current study "according to the judgment of the investigators, for any reasons other than the inclusion and exclusion criteria, including allergy to ibuprofen, aspirin or other nonsteroidal anti‐inflammatory drugs, a history of peptic ulcer, gastrointestinal bleeding or perforation, taking nonsteroidal inflammatory drugs before this study and acute pulmonary embolism" (page 3413) |
|
| Interventions |
Oxycodone arm: Drug: Oxycodone hydrochloride Dose/dosing: 10 mg every 12 hours as starting dose Formulation: controlled‐release Route of administration: oral Length of treatment: 7 days Titration schedule: "the dose was adjusted according to the pain status of the patient" (page 3413) Rescue medication: not reported Other medication: not reported Comparison arm: Drug: Ibuprofen Dose/dosing: 300 mg orally twice daily Formulation: not applicable Route of administration: Oral Length of treatment: 7 days Titration schedule: not reported Rescue medication: not reported Other medication: not reported |
|
| Outcomes |
|
|
| Notes | Study free of commercial funding? Yes. Funded by “Dalian Medical Science Research Program Project Funding” (page 3418) Groups comparable at baseline? Yes ITT analyses undertaken? Yes, although 1 randomised patient excluded due to pulmonary embolism |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Allocation by random number table |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | High risk | Study described as single‐blind and that it was the patients who were blinded. However, no details reported |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | High risk | See cell above. Personnel involved in outcome reporting |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data from 66/67 patients analysed |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data from 66/67 patients analysed |
| Selective reporting (reporting bias) | Low risk | Main outcomes appeared to be reported. |
| Were the participants adequately titrated? | Unclear risk | No information provided |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | Limited details reported, so not possible to assess |
Lux 2014.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, non‐inferiority cross‐over trial Year: 2010‐2012 Country: Germany, Poland, Switzerland |
|
| Participants |
Participants: 85 participants entered into titration/stabilisation phase, 71 participants randomised and 68 took at least 1 dose of the study drug and comprised the safety dataset. The full analysis set comprised the 60 participants with at least 1 measurement of the primary efficacy parameter. 56 participants completed the trial of whom 46 qualified for the per‐protocol dataset (23 in each sequence); of the 60 participants in the full analysis set, 40 had malignant pain of the following type: lung (7), breast (6), cervix (5), prostate (5), colon/rectum/anus (4), oropharynx (3), skin (2), lymphoma (2), and other (6). 32 women, 36 men. Mean age (SD) of the 68 participants in the safety dataset was 60.8 (10) years. Inclusion criteria: white men and women, ≥ 18 years of age with chronic cancer pain (in the protocol) or non‐cancer pain (not in the protocol) and predominantly non‐neuropathic pain, requiring at least 40 mg oxycodone per day (or equivalent), ECOG (Eastern Cooperative Oncology Group) performance status < 3, life expectancy of at least 3 months, adequate analgesia (mean 'current' pain intensity per day ≤ 40 mm on VAS) prior to randomisation for at least 3 consecutive days and stable analgesic requirements prior to randomisation for at least 3 days (stable maintenance dose of oxycodone; requirement of ≥ 40 mg oxycodone per day; ≤ 2 doses of rescue medication per day) Exclusion criteria: hypersensitivity to oxycodone or any of the excipients of the study drugs, requirement of > 120 mg oxycodone per day (or equivalent), surgery within 1 month prior to study start and/or anticipated or scheduled surgical intervention during the study, intravenous chemotherapy and/or radiotherapy for pain alleviation and/or neural blockade within 2 weeks prior to study start, significant hepatic impairment or severe renal impairment (creatinine clearance < 30 mL/min), and pregnancy or lactation However, only outcome data pertaining to the participants with malignant pain alone were included in this review. |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone (oxygesic/oxycontin) ‐ Dose and dosing: twice‐daily administration of oxycodone at 8.00 and 20.00 hours. No dose adjustment of oxycodone was allowed during the double‐blind treatment phase after the titration/stabilisation phase. The total daily dose was determined during the titration/stabilisation phase and fixed throughout the study. The mean daily dose was not reported. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 10 days per cross‐over phase (data only analysed for days 6‐10) ‐ Titration schedule: after a screening phase (maximum 14 days), a titration/ stabilisation period followed (maximum 14 days) where the participants were switched to oxycodone and the dose was adjusted for sufficient pain relief, i.e. daily mean current pain ≤ 40 mm on VAS. ‐ Rescue medication: 10 mg immediate‐release morphine sulfate (≤ 2 doses of rescue medication per day) ‐ Other medication: not reported Comparison arm ‐ Drug: oxycodone + placebo ‐ Dose and dosing: once‐daily administration of oxycodone at 8.00 hours and placebo at 20.00 hours. No dose adjustment of oxycodone was allowed during the double‐blind treatment phase after the titration/stabilisation phase. Total daily dose was determined during the titration/stabilisation phase and fixed throughout the study. Mean daily dose was not reported. ‐ Formulation: Extended‐release ‐ Route of administration: oral ‐ Length of treatment: 10 days per cross‐over phase (data only analysed for days 6‐10) ‐ Titration schedule: after a screening phase (maximum 14 days), a titration/stabilisation period followed (maximum 14 days) where the participants were switched to oxycodone and the dose was adjusted for sufficient pain relief, i.e. daily mean current pain ≤ 40 mm on VAS. ‐ Rescue medication: 10 mg immediate‐release morphine sulfate (≤ 2 doses of rescue medication per day) ‐ Other medication: not reported ‐ For cross‐over trials, cross‐over schedule: "After 10 days of treatment with the first study medication of the respective sequence, patients were directly switched to the second medication without wash‐out." "During the double‐blind phase of the study, for each study medication the current pain and recalled pain scores obtained from days 6 to 10 were employed for statistical evaluations. Days 1 to 5 of each period were regarded as an active 5 day run‐in phase in order to avoid any potential carry‐over effects between the different study periods." |
|
| Outcomes |
Primary outcome measures: ‐ Overall 'current' pain intensity (PI) on 0 to 100 mm VAS (mean 'current' PI of the last 5 days of each treatment period). Pain intensity (PI) was assessed five times daily, i.e. at 08:00 h (before study drug intake), 11:00 h, 14:00 h, 17:00 h, and 20:00 h (before study drug intake; allowed deviation ± 20 min for all assessments) on a 0 to 100 mm VAS ('current' pain). PI assessment at 08:00 h and 20:00 h also comprised ratings of PI over the past 12 hours ('recalled' pain during day‐ and night‐time). From the PI scores, the mean 'current' PI over all time points of the last 5 treatment days of period 1 and period 2 (= overall mean 'current' PI) will be calculated for each participant as the primary efficacy endpoint. Secondary outcome measures: ‐ mean 'current' pain intensity (PI) per day ‐ mean 'current' PI per time point ‐ mean 'recalled' PI over the past 12 hours at 08:00 h ‐ mean 'recalled' PI over the past 12 hours at 20:00 h ‐ overall effectiveness on 4‐point categorical scale (CAT; 0 = not effective, 3 = highly effective) by participant and investigator (assessed at the end of each treatment period) ‐ daily dose of rescue medication for each of the last 5 days of period 1 and 2 ‐ mean daily dose of rescue medication over the last 5 treatment days of period 1 and 2 ‐ total amount of rescue medication over the last 5 treatment days of period 1 and 2 ‐ Adverse events and serious adverse events recorded using the Medical Dictionary for Regulatory Activities (MedDRA). ‐ Nausea and sedation assessed on 0 to 100 mm VAS at 08:00 h and 20:00 h by participants However, only the primary outcome data was reported on a per‐protocol basis for participants with malignant pain alone. These are therefore the only study results included in this review. |
|
| Notes | ‐ Study free of commercial funding? No. Study was funded by study sponsor Develco Pharma Schweiz AG. ‐ Groups comparable at baseline? Unclear. The groups comprising the safety dataset were comparable at baseline in terms of age, sex, weight, height, and body mass index, but this was not the analysis set used for the present review (i.e. included both benign and malignant pain). ‐ Trial registration details: DRKS00000577 (https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00000577) and www.clinicaltrialsregister.eu/ctr-search/trial/2010-020402-15/results |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The treatment sequence (test–reference; reference–test) was assigned by randomization code and central randomization procedure." |
| Allocation concealment (selection bias) | Low risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "OTD, OOD and a dummy were blinded using the same type of over‐encapsulation. The patients received the same number of encapsulated tablets in both periods of the double‐blind treatment phase in the morning (OTD or OOD) and in the evening (OTD or dummy)." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | The only data available for the participants with malignant pain only was per‐protocol. Outcome data only available for 31/40 participants |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Adverse events only reported for the 68 participants in the safety dataset of whom 40 had malignant pain. These data were therefore not included in this review. |
| Selective reporting (reporting bias) | High risk | Adverse events were not reported separately for the participants with malignant pain and could therefore not be extracted for the purposes of this review. |
| Were the participants adequately titrated? | Low risk | Adequate titration was an inclusion criterion. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available for both time periods collapsed across the time periods. |
| Other bias | Low risk | The study appeared to be free of other bias. |
Mercadante 2010.
| Study characteristics | ||
| Methods |
Design: randomised, parallel‐group trial Year: not reported Country: Italy |
|
| Participants |
Participants: 60 participants randomised; 46/60 participants completed baseline evaluation (21 participants in group oxycodone and 25 participants in group morphine, 14/60 participants did not complete baseline evaluation as they were lost to follow‐up); 27 females, 19 males; mean age (SD): 63.2 (9.48) years. 19 oxycodone and 20 morphine participants completed 4 weeks of study participation and 7 and 10 participants, respectively, completed 8 weeks of study participation. Inclusion criteria: Patients with pancreatic cancer with local disease, presenting with abdominal pain at an intensity ≥ 4/10 numerical rating scale from 0 to 10, and no longer responsive no nonopioid analgesics Exclusion criteria: Distant and bone metastases, or prevalent somatic pain due to evident peritoneal involvement, changes in chemotherapy regimen, hepatic or renal failure, cognitive failure, lack of cooperation, aged < 18 or > 80 years, and a Karnofsky performance status < 50 |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: starting dose of 20 mg/day, according to an approximate morphine to oxycodone ratio of 1.5:1. For participants requiring an increase in the dose for increasing pain (> 4/10 or > 3 breakthrough pain medications per day) during the study period, opioid doses were increased according to clinical needs. Mean dose (SD) at week 1: 23.8 (8) mg/day; mean dose (SD) at week 2: 25.5 (8) mg/day; mean dose (SD) at week 3: 27.9 (9) mg/day; mean dose (SD) at week 4: 33.1 (14) mg/day; mean dose (SD) at week 8: 45.7 (24) mg/day ‐ Formulation: Sustained‐release ‐ Route of administration: oral ‐ Length of treatment: 4 weeks (with a study extension up to 8 weeks) ‐ Titration schedule: "Patients were recruited and followed during admission to the palliative care unit, as outpatients and at home. Physicians provided frequent call contacts to adjust the opioid dose at any time." See also 'Dose and dosing' section. No further information provided ‐ Rescue medication: oral morphine in doses of 1/6 of the daily dose was provided (starting at 5 mg). ‐ Other medication: "Adjuvants and symptomatic drugs were prescribed as indicated by the clinical situation." Comparison arm ‐ Drug: morphine ‐ Dose and dosing: starting dose of 30 mg/day, according to an approximate morphine:oxycodone ratio of 1.5:1. For participants requiring an increase in the dose for increasing pain (> 4/10 or > 3 breakthrough pain medications per day) during the study period, opioid doses were increased according to the clinical needs. Mean dose (SD) at week 1: 35 (9) mg/day; mean dose (SD) at week 2: 36.2 (14) mg/day; mean dose (SD) at week 3: 41 (19) mg/day; mean dose (SD) at week 4: 42.6 (21) mg/day; mean dose (SD) at week 8: 60 (46) mg/day ‐ Formulation: Sustained‐release ‐ Route of administration: oral ‐ Length of treatment: 4 weeks (with study extension up to 8 weeks) ‐ Titration schedule: "Patients were recruited and followed during admission to the palliative care unit, as outpatients and at home. Physicians provided frequent call contacts to adjust the opioid dose at any time." See also 'Dose and dosing' section. No further information provided ‐ Rescue medication: oral morphine in doses of 1/6 of the daily dose was provided (starting at 5 mg). ‐ Other medication: "Adjuvants and symptomatic drugs were prescribed as indicated by the clinical situation." |
|
| Outcomes | ‐ Pain intensity (average in the last 24 hours): Assessed by participant, using a numerical rating scale from 0 to 10 ‐ Opioid‐related symptoms (including nausea and vomiting, drowsiness and confusion): Assessed by participant, using a categorical scale from 0 (= absent, 1 = slight, 2 = moderate) to 3 (= severe) ‐ Constipation: Assessed by participant, using a categorical scale from 0 (= 1 passage, 1 to 2 days; 1 = 1 passage, 3 to 4 days; 2 = 1 passage, 4 days) to 3 (= only by enema) |
|
| Notes | ‐ Study free of commercial funding? Unclear. No details reported ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomized by a computer system in 2 groups." No further information reported |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | High risk | Unblinded study |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) Pain | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) Pain | High risk | From baseline to study end at 4 weeks, 11/30 oxycodone participants and 10/30 morphine participants dropped out of the study and only the data from participants who completed the study phases were reported/analysed by week (0, 1, 2, 3, 4, and 8). |
| Incomplete outcome data (attrition bias) Adverse events | High risk | From baseline to study end at 4 weeks, 11/30 oxycodone participants and 10/30 morphine participants dropped out of the study and only the data from participants who completed the study phases were reported/analysed by week (0, 1, 2, 3, 4, and 8). |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to be reported. |
| Were the participants adequately titrated? | Unclear risk | Not enough information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Mucci‐LoRusso 1998.
| Study characteristics | ||
| Methods |
Design: randomised, parallel‐group trial Year: not reported Country: USA |
|
| Participants |
Participants: 101 participants randomised; 100/101 participants received ≥ one dose of study medication; N = 48 in oxycodone group and 52 in the morphine group, 55% participants were male, mean (range) age = 59 (30 to 83) years; bone and viscera were most common pain sites; nerve pain was the primary pain type in 10/48 oxycodone and 9/52 morphine participants; most common pre‐study pain medication was fixed‐dose oxycodone‐acetaminophen combination (22 participants in each group), followed by single‐entity morphine (13 oxycodone and 17 morphine participants). Most participants were receiving > 1 pain medication pre‐study and all but 3 participants (all in the oxycodone group) were receiving opioids prior to enrolment, the mean (range) oral oxycodone equivalent of the pre‐study dose = 64 (14 to 280) mg in the oxycodone group and 70 (14‐235) mg in the morphine group. 7 oxycodone and 9 morphine participants discontinued the study before achieving stable pain control due to adverse experiences (2 oxycodone and 6 morphine participants), intercurrent illness (3 oxycodone participants), ineffective treatment (1 oxycodone and 1 morphine participant), participant request (1 oxycodone and 1 morphine participant), and protocol violation (1 morphine participant). An additional 4 participants dropped out of the study after achieving stable pain control due to adverse experience (1 oxycodone participant), protocol violation (1 oxycodone participant), intercurrent illness (1 morphine participant) and worsening of pre‐existing condition (1 morphine participant). Inclusion criteria: Patients who required around‐the‐clock treatment with opioid analgesics for chronic cancer‐related pain with the equivalent of 30 to 340 mg of oral oxycodone daily. Patients whose pain was not controlled by maximum recommended doses of non‐opioid analgesics were also eligible if they would require ≥ 30 mg. Exclusion criteria: "a history of sensitivity to oxycodone or morphine, any contra‐indication for opioid therapy (such as paralytic ileus or severe pulmonary disease) or severely compromised organ function that could obscure efficacy or adversely affect safety. Patients whose pain control was so fragile they could not switch opioids were also excluded." |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: multiples of 20 mg tablets, every 12 hours (8 a.m. and 8 p.m.). Starting dose calculated from participants' prestudy daily opioid dose and could be adjusted based on the investigator's judgement. Dose was titrated until stable pain control was achieved. Pain control was considered stable when, over a 48‐hour period, the 'every 12 hours' dose was unchanged, ≤ 2 supplemental analgesic doses were taken per day, the dosing regimen for any non‐opioids or adjuvants was unchanged, and the participant reported that pain control was acceptable and any adverse effects were tolerable. Participants who could not be stabilised within 10 days were discontinued. Mean final daily doses of every 12 hour (range): 101 (40‐360) mg ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: up to 12 days ‐ Titration schedule: see 'Dose and dosing' section ‐ Rescue medication: Immediate‐release oxycodone in multiples of 2 × 5 mg tablets. Each supplemental medication dose was 1/4 to 1/3 of every 12 hours scheduled dose. Participants were instructed to take a supplemental dose as needed for breakthrough pain, but not more frequently than once every 2‐4 hours or 1 hour before activity associated with incident pain. Median dose use on the next to last study day (during stable pain control) 1 (range 0‐4) and median dose use on last study day (during stable pain control) 1 (range 0‐3) ‐ Other medication: "Non‐opioid analgesics and adjuvant medications were allowed during the study provided they had been given on a regular basis (not as needed) before the study." Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: multiples of 30 mg tablets, every 12 hours (8 a.m. and 8 p.m.). Starting dose was calculated from the participants' prestudy daily opioid dose and could be adjusted based on the investigator's judgement. Dose was titrated until stable pain control was achieved. Pain control was considered stable when, over a 48‐hour period, the q12h dose (= dose every 12 hours) was unchanged, ≤ 2 supplemental analgesic doses were taken per day, the dosing regimen for any non‐opioids or adjuvants was unchanged, and the participant reported that pain control was acceptable and any adverse effects were tolerable. Participants who could not be stabilised within 10 days were discontinued. Mean final daily doses every 12 hours (range): 140 (60‐300) mg ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: ≤ 12 days ‐ Titration schedule: see 'Dose and dosing' section ‐ Rescue medication: Immediate‐release morphine in multiples of 15 mg tablets. Each supplemental medication dose was 1/4 to 1/3 of every 12 h scheduled dose. Participants were instructed to take a supplemental dose as needed for breakthrough pain, but not more frequently than once every 2 to 4 hours or 1 hour before activity associated with incident pain. Median dose use on the next to last study day (during stable pain control) = 1 (range 0 to 3) and median dose use on last study day (during stable pain control) = 1 (range 0 to 3) ‐ Other medication: "Non‐opioid analgesics and adjuvant medications were allowed during the study provided they had been given on a regular basis (not as needed) before the study." |
|
| Outcomes | ‐ Pain intensity (average since previous evaluation): Assessed by participant at baseline and before every q12h dose, using a categorical scale from 0 (= none) (1 = slight, 2 = moderate) to 3 (= severe). Also assessed after ≥ 48 hours of stable pain control using the categorical scale and a 100 mm VAS scale from 0 (= no pain) to 100 (worst possible pain) ‐ Adverse experiences and drug effects: Assessed by participant in a daily diary, and after ≥ 48 hours of stable pain control by using the Specific Drug Effect Questionnaire 100 mm VAS scale (?) from 0 (= not at all) to 100 (an awful lot); also assessed by observers after ≥ 48 hours of stable pain control by using the Specific Drug Effect Questionnaire 100 mm VAS scale (?) from 0 (= not at all) to 100 (extremely) ‐ Drowsiness and nausea: Assessed by participant after ≥ 48 hours of stable pain control (?), using a categorical scale from 0 (= none, 1 = slight, 2 = moderate) to 3 (= severe) and a 100‐mm VAS scale from 0 (= none) to 100 (worst possible) ‐ Acceptability of therapy: Assessed by participant at baseline and study end, using a categorical scale from 1 (= very poor, 2 = poor, 3 = fair, 4 = good) to 5 (= excellent) ‐ Quality of life: Assessed by participant at baseline and study end, using the Functional Assessment of Cancer Therapy‐General (FACT‐G), a 28‐item questionnaire consisting of 5 subscales measuring different aspects of quality of life: Physical, social/family, relationship with physician, emotional and functional |
|
| Notes | ‐ Study free of commercial funding? No. The authors were either "financially compensated for their efforts" or employees of the study drug manufacturer. ‐ Groups comparable at baseline? Unclear. No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Block randomization was used to ensure that all centers had a comparable number of patients in each treatment group." No further information reported |
| Allocation concealment (selection bias) | Unclear risk | No further information reported than that in the cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "The double‐dummy technique was used to blind the study medications." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | "The double‐dummy technique was used to blind the study medications." |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Participant recorded. See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | A total of 79/101 participants who achieved stable pain control and had simultaneous pharmacokinetic‐pharmacodynamic assessments were analysed for efficacy (39 oxycodone, 40 morphine). |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | A total of 100/101 participants were analysed for safety. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes were reported. |
| Were the participants adequately titrated? | Low risk | The participants were adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Nosek 2017.
| Study characteristics | ||
| Methods |
Design: randomised, parallel‐group, multicentre clinical trial Year: 2013 to 2015 Country: Poland |
|
| Participants |
Participants: of 62 randomised participants, 53 completed the trial. Sex and cancer type not presented by treatment, but only overall: 30 men, 32 women; cancer type: lung (14), colon (10), stomach (3), oesophagus (1), pancreas (1), breast (8), prostate (7), bladder (3), kidney (3), uterus (4), other isolated cases (7), unknown primary (8). Use of non‐opioid analgesics/step‐2 opioids,and adjuvant analgesics: Tramadol (57), paracetamol (52), ketoprofen (30), metamizol (10), diclofenac (3), dexamethasone (3), meloxicam (2) and ibuprofen (2). ‐ Morphine: 14 participants (2 discontinued treatment due to death (1) or adverse effects (1)), mean (SD) age 62 (± 13.4) years; type of pain (participants may experience more than 1 type): visceral (11), bone (8), neuropathic (8), superficial somatic (0) ‐ Oxycodone: 16 participants (0 discontinued treatment), mean (SD) age 72.3 (± 13.2) years; type of pain (participants may experience more than 1 type): visceral (10), bone (10), neuropathic (8), superficial somatic (2) ‐ Buprenorphine: 17 participants (2 discontinued treatment due to death (1) or adverse effects (1)), mean (SD) age 70 (± 13.4) years; type of pain (participants may experience more than 1 type): visceral (11), bone (11), neuropathic (7), superficial somatic (5) ‐ Fentanyl: 15 participants (5 discontinued treatment due to death (3), adverse effects (1), or withdrawal of consent (1)), mean (SD) age 70.7 (± 10.9) years; type of pain (participants may experience more than 1 type): visceral (8), bone (8), neuropathic (7), superficial somatic (1) Inclusion criteria: Participants aged > 18 years with cancer with severe pain (scores of 6‐10 on a 0‐10 numerical rating scale) treated in the Home Hospice or Palliative Care Outpatient Clinic, who had failed to respond to step‐1 WHO analgesic ladder drugs and/or weak opioids such as tramadol, codeine, dihydrocodeine and had an expected survival time ≥ 40 days and no renal or liver dysfunction or cognitive disorders according to the Mini–Mental State Examination Exclusion criteria: Previous treatment with strong opioids; symptoms of respiratory insufficiency; disorders of consciousness; central nervous system primary neoplasm or brain metastases; inability to receive medications by oral/transdermal route; clinically significant liver dysfunction (bilirubin or transaminase level twice exceeding the norm) and/or renal dysfunction (creatinine level above the norm or estimated glomerular filtration rate [eGFR] < 60); and patients undergoing chemotherapy |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: See 'Titration schedule' below. Initial dose 10 mg/day. Mean (± SD) final dose 31.64 (± 13.7) mg/day ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 28 days ‐ Titration schedule: Gradual increase in dose to reach NRS pain intensity ≤ 4 and acceptable adverse effects according to following schedule administered every 12 hours: 2 × 5, 2 × 10, 2 × 15, 2 × 20, 2 × 30, 2 × 45, 2 × 60, 2 × 80, 2 × 100, 2 × 120 mg ‐ Rescue medication: Immediate‐related morphine "administered by an oral route, titrated to satisfactory analgesic effect". Mean (SD) total dose 338.13 (± 273.94) mg ‐ Other medication: The authors stated: "The patients could take adjuvant analgesics in bone pain and neuropathic pain." but provided no further details. Lactulose in doses of 10 mL administered twice daily to prevent constipation, but no antiemetics were used as prophylaxis Comparison arm 1 ‐ Drug: morphine ‐ Dose and dosing: See 'Titration schedule' below. Initial dose 20 mg/day. Mean (± SD) final dose 56.66 (± 11.54) mg/day ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 28 days ‐ Titration schedule: Gradual increase in dose to reach NRS pain intensity ≤ 4 and acceptable adverse effects according to following schedule administered every 12 hours: 2 × 10, 2 × 20, 2 × 30, 2 × 40, 2 × 60, 2 × 90, 2 × 120, 2 × 150, 2 × 180, 2 × 200 mg ‐ Rescue medication: Immediate‐related morphine "administered by an oral route, titrated to satisfactory analgesic effect". Mean (SD) total dose 260 (± 327.04) mg ‐ Other medication: The authors stated: "The patients could take adjuvant analgesics in bone pain and neuropathic pain." but provided no further details. Lactulose in doses of 10 mL administered twice daily to prevent constipation, but no antiemetics were used as prophylaxis Comparison arm 2 ‐ Drug: buprenorphine ‐ Dose and dosing: See 'Titration schedule' below. Initial dose 35 μg/hour. Mean (± SD) final dose 63 (± 24.57) μg/hour ‐ Formulation: patch ‐ Route of administration: Transdermal ‐ Length of treatment: 28 days ‐ Titration schedule: Gradual increase in dose to reach NRS pain intensity ≤ 4 and acceptable adverse effects according to following schedule administered every 60‐84 hours: 35, 52.5, 70, 105, 140, 175, and 210 μg/hour ‐ Rescue medication: Immediate‐related morphine "administered by an oral route, titrated to satisfactory analgesic effect". Mean (SD) total dose 457.65 (± 479.47) mg ‐ Other medication: The authors stated: "The patients could take adjuvant analgesics in bone pain and neuropathic pain." but provided no further details. Lactulose in doses of 10 mL administered twice daily to prevent constipation, but no antiemetics were used as prophylaxis Comparison arm 3 ‐ Drug: fentanyl ‐ Dose and dosing: See 'Titration schedule' below. Initial dose 25 μg/hour. Mean (± SD) final dose 45 (± 25.82) μg/hour ‐ Formulation: patch ‐ Route of administration: Transdermal ‐ Length of treatment: 28 days ‐ Titration schedule: Gradual increase in dose to reach NRS pain intensity ≤ 4 and acceptable adverse effects according to following schedule administered every 48‐72 hours: 25, 37.5, 50, 75, 100, 125, and 150 μg/hour ‐ Rescue medication: Immediate‐related morphine "administered by an oral route, titrated to satisfactory analgesic effect". Mean (SD) total dose 390.67 (± 377.48) mg ‐ Other medication: The authors stated: "The patients could take adjuvant analgesics in bone pain and neuropathic pain." but provided no further details. Lactulose in doses of 10 mL administered twice daily to prevent constipation, but no antiemetics were used as prophylaxis |
|
| Outcomes | ‐ Pain intensity (average, least, worst and right now): Assessed by patient on Days 1, 3, 6, 9, 13, 16, 19, 22, 25 and 28; Brief Pain Inventory‐Short Form (BPI‐SF) (11‐point scale (range 0–10), with a lower score corresponding to a smaller intensity of pain) ‐ Adverse events: Edmonton Symptom Assessment System (ESAS) on Days 1, 3, 6, 9, 13, 16, 19, 22, 25 and 28 ‐ Bowel movements: Bowel Function Index (BFI) (scale 1‐10; normal values 0‐2.99, constipation 3‐10), on days 1, 7, 14, 21 and 28 ‐ Quality of life |
|
| Notes | ‐ Study free of commercial funding? Yes ‐ Groups comparable at baseline? Inferential statistics suggested no difference in age and sex between the groups, but they may not have been powerful enough to detect difference. Mean age of the morphine group seemed a lot lower than those of the other groups. Other characteristics not reported by treatment group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Allocation described as randomised. No further details reported |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | High risk | Open‐label |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | High risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | Data from 53 of 62 randomised patients were analysed. Unfortunately, the results were not presented split by treatment group, only overall along with ANOVA analyses, which means this outcome could not be included in the review. |
| Incomplete outcome data (attrition bias) Adverse events | High risk | Unclear if adverse events were reported for all randomised patients. Unfortunately, the results were not presented split by treatment group, apart from for bowel function index, which means that was the only adverse event other than withdrawal due to adverse events that could be included. |
| Selective reporting (reporting bias) | High risk | Most of the outcomes could not be included due to the manner in which they were reported. See the 2 cells above |
| Were the participants adequately titrated? | Low risk | The patients appeared to be adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | No other biases identified |
Parris 1998.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group trial Year: not reported Country: USA |
|
| Participants |
Participants: 111 participants randomised; 103/111 participants received ≥ one dose of study medication; N = 52 in controlled‐release group and 51 in the immediate‐release group, 50% participants were female, average (mean?) (range) age = 57 (31 to 80) years; bone (45%) and viscera (28%) were most common pain sites; most common cancer diagnoses were breast, gastrointestinal, lung, and gynaecological. 66/111 participants completed the 5‐day study period (33 in each group). Pre‐study analgesics: Oxycodone and acetaminophen (71%), most lower‐dose participants received a total daily pre‐study oxycodone dosage of 30 to 45 mg with 2.0 to 2.9 g of acetaminophen; higher‐dose participants received a daily oxycodone dosage of 50 to 60 mg with 3.2 to 3.9 g of acetaminophen; other prior opioids included codeine and acetaminophen (17%), hydrocodone and acetaminophen (10%), propoxyphene napsylate and acetaminophen (2%), and transdermal fentanyl (1%) (protocol violation). A total of 19 controlled‐release and 18 immediate‐release participants discontinued the study due to adverse events (4 controlled‐release and 7 immediate‐release participants), unrelated illness (1 in each group), ineffective treatment (10 controlled‐release and 4 immediate‐release participants), protocol violation (4 controlled‐release and 5 immediate‐release participants), and other (1 immediate‐release participant). Inclusion criteria: "The study included adult patients recruited from 15 centers in the United States who were receiving 6 to 12 tablets or capsules per day of fixed‐combination analgesics for cancer‐related pain. Patients were of either gender and had stable coexistent disease." Exclusion criteria: "Patients were excluded if their pain was not already acceptably controlled; if they had surgery or radiotherapy within 10 days prior to study or anticipated these procedures during study; of they had compromised function of a major organ system; or of they were receiving nonopioid analgesics (before the protocol was amended). Of course, concomitant non‐analgesic therapies were allowed during the study. To encourage participation and to lower the discontinuation rate, the protocol was modified during the study to include patients undergoing or recently given radiotherapy and those receiving stable doses of nonopioid analgesics or analgesic adjuvants. In addition, patients receiving ten or more tablets or capsules of fixed‐combination analgesics were no longer permitted to enter the study, but could be enrolled in a companion study intended for patients with greater opioid requirements." |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: 30 mg, every 12 hours, total daily dosage 60 mg. Mean daily dosage 60 mg (see 'Titration schedule') ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 5 days ‐ Titration schedule: participants needing titration of analgesic or supplemental medication were required to discontinue from the study. ‐ Rescue medication: see 'Titration schedule' ‐ Other medication: see 'Titration schedule.' "Of course, concomitant non‐analgesic therapies were allowed during the study." Comparison arm ‐ Drug: oxycodone ‐ Dose and dosing: 15 mg, 4 times daily, total daily dose 60 mg, mean daily dose 60 mg. See 'Titration schedule' ‐ Formulation: Immediate‐release ‐ Route of administration: oral ‐ Length of treatment: 5 days ‐ Titration schedule: participants needing titration of analgesic or supplemental medication were required to discontinue from the study. ‐ Rescue medication: see 'Titration schedule' ‐ Other medication: see 'Titration schedule.' "Of course, concomitant non‐analgesic therapies were allowed during the study." |
|
| Outcomes | ‐ Pain intensity: Assessed by participant at baseline and 4 times daily, that is, morning (overnight pain rating), midday (morning pain rating), evening (afternoon pain rating), and bedtime (evening pain rating), using a categorical scale from 0 (= none) (1 = slight, 2 = moderate) to 3 (= severe) ‐ Acceptability of current therapy: Assessed by participant at baseline and 2 times daily, that is, for both day and night, using a categorical scale from 1 (= very poor) (2 = poor, 3 = fair, 4 = moderate) to 5 (= excellent) ‐ Adverse experiences: "Observers contacted patients by telephone daily throughout the 5‐day study period and recorded information about adverse events and changes in the patients' condition." |
|
| Notes | ‐ Study free of commercial funding? No. The study was sponsored by the drug manufacturers (The Purdue Frederick Company and Purdue Pharma L.P.) and some of the authors were employees of the study drug manufacturer. ‐ Groups comparable at baseline? No participant details reported by initial treatment allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported beyond that "This was a randomized, double‐blind, parallel‐group study" |
| Allocation concealment (selection bias) | Unclear risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "This was a randomized, double‐blind, parallel‐group study".... "using a double‐dummy technique". No further information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. We assumed that the participants were blinded, but it was unclear whether the personnel administering the study medication or the personnel assessing some of the outcomes, or both, were also blinded. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | This outcome was participant‐assessed. See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. We assumed that the participants were blinded, but it was unclear whether the personnel administering the study medication and/or the personnel assessing some of the outcomes were also blinded. |
| Incomplete outcome data (attrition bias) Pain | Low risk | A total of 103/111 participants who took ≥ 1 study drug dose constituted the ITT population (52 controlled‐release, 51 immediate‐release), 8/111 participants were excluded for administrative reasons, which were not further specified. The pain data appeared to include these 103 remaining participants. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | A total of 109/111 participants were analysed for safety. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to be reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated because otherwise they were discontinued. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Ren 2012.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2009‐2012 Country: China |
|
| Participants |
Participants: 80 participants randomised ‐ Oxycodone: 40 participants, 21 men, 19 women; aged 39‐75 years, mean (SD) age 55.2 (8.9) years; NRS pain score 4‐6 (17) or 7‐10 (23); type of pain: bone (12), neuralgia (13), visceral (10), soft tissue (5); no further information reported ‐ Morphine: 40 participants, 20 men, 20 women; aged 41‐77 years, mean (SD) age 54.5 (7.9) years; NRS pain score 4‐6 (18) or 7‐10 (22); type of pain: bone (11), neuralgia (14), visceral (9), soft tissue (6); no further information reported Inclusion criteria: Patients ≥ 18 years with moderate‐severe cancer pain admitted to the authors' hospital, with efficacy evaluation during the study period, no anti‐tumour treatment (e.g. radiotherapy, chemotherapy), numerical rating scale pain score > 4, no communication difficulties, no serious heart, liver and kidney dysfunction, no hypoxic respiratory depression, no chronic obstruction respiratory diseases who have expected survival ≥ 3 months Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 14 days ‐ Titration schedule: If the pain score of patients decreases ≤ 4 points at 24 hours, the dosage should be increased. The dosage should be increased by 25% to 50% each time, depending on the patient's condition, and the number of times of administration should not be changed until satisfactory pain relief is achieved. ‐ Rescue medication: If patients experience poor short‐term results and sudden exacerbations of pain, they can be treated with short‐acting morphine injections. No further information ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 30 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 14 days ‐ Titration schedule: If the pain score of patients decreases ≤ 4 points at 24 hours, the dosage should be increased. The dosage should be increased by 25% to 50% each time, depending on the patient's condition, and the number of times of administration should not be changed until satisfactory pain relief is achieved. ‐ Rescue medication: If patients experience poor short‐term results and sudden exacerbations of pain, they can be treated with short‐acting morphine injections. No further information ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain relief: assessed using a NRS with a drop in scores of 0 indicating no relief, 1‐2 or 1 indicating mild relief, 3‐4 or 2‐4 or 2‐3 or 3 indicating moderate relief, and 4 or 5 or more indicating significant relief ‐ Quality of life assessed using the Kaspersky Performance Status (percentage system with higher percentages indicating better outcome) ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information reported ‐ Groups comparable at baseline? Unclear, although the authors did report that the groups were comparable in terms of gender, age, type of pain and baseline NRS score. It was unclear whether any other characteristics were examined. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients reported to be randomised, but no information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear if ITT analyses were undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Riley 2015.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group trial (with cross‐over to other arm for non‐responders to first‐line opioid) Year: 2006‐2011 Country: UK |
|
| Participants |
Participants: 200 participants randomised; 198/200 participants received ≥ 1 dose of study medication; N = 100 in the oxycodone group and 98 in the morphine group; 198 were included in the intention‐to‐treat analyses: ‐ Oxycodone: N = 100; 38 males and 62 females, mean (SD) age = 58.9 (13.2) years; cancer diagnosis: breast (18), lower gastrointestinal (16), upper gastrointestinal (2), pancreas and hepatobiliary (4), sarcoma (8), lung (13), gynaecological (9), urinary tract (3), prostate (8), haematological (7), malignant melanoma (6), head and neck (3), other (3); concomitant opioid medications before randomisation: As required morphine (40), as required oxycodone (3), codeine (45), tramadol (45), dihydrocodeine (5), dextropropoxyphene (1), buprenorphine (3). A total of 20/100 participants who received first‐line oxycodone withdrew from the trial for drug (16) or trial (4) reasons ‐ Morphine: N = 100; 50 males and 50 females, mean (SD) age = 59.2 (11.6) years; cancer diagnosis: breast (14), lower gastrointestinal (11), upper gastrointestinal (10), pancreas and hepatobiliary (10), sarcoma (11), lung (5), gynaecological (7), urinary tract (12), prostate (2), haematological (6), malignant melanoma (4), head and neck (2), other (6); concomitant opioid medications before randomisation: as required morphine (51), as required oxycodone (1), codeine (47), tramadol (47), dihydrocodeine (3), dextropropoxyphene (0), buprenorphine (0). 13/98 participants who received first‐line oxycodone withdrew from the trial for drug (10) or trial (3) reasons. Inclusion criteria: "Inpatients and outpatients were identified and recruited at a tertiary referral cancer center by the specialist palliative care team. Patients were eligible if they needed to begin a regular oral strong opioid for cancer‐related pain and were strong opioid‐naive, that is, had not taken a regular strong opioid within the previous month. The use of an "as required" strong opioid was permitted (less than six doses in 24 hours). Patients were recruited before, or within 24 hours, of starting a regular strong opioid." Exclusion criteria: renal impairment (serum creatinine ≥ 1.5 times the upper limit of normal), requirement of parenteral opioids, previous poor response to either morphine or oxycodone, and pregnancy |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: see 'Titration schedule.' No further information reported ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 1 year ‐ Titration schedule: "Patients were initially titrated on immediate‐release preparations, administered at four‐hourly intervals with additional as required doses available for breakthrough pain .... the starting dose was determined by the treating physician on an individual patient basis and titrated accordingly.... until adequate pain control was achieved or intolerable side effects were reported by the patient. At this stage, patients were converted to the comparable modified‐release preparations. Nonresponders to the first opioid were switched to the alternative opioid. As this was not a stable analgesic setting, the ratio of oral morphine:oxycodone (2:1).... Doses were retitrated according to response." ‐ Rescue medication: see 'Titration schedule' ‐ Other medication: "adjuvant medications (laxatives, antiemetics, co‐analgesics) were either started or continued where indicated." Comparison arm ‐ Drug: morphine ‐ Dose and dosing: see 'Titration schedule.' No further information reported ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 1 year ‐ Titration schedule: "Patients were initially titrated on immediate‐release preparations, administered at four‐hourly intervals with additional as required doses available for breakthrough pain .... the starting dose was determined by the treating physician on an individual patient basis and titrated accordingly.... until adequate pain control was achieved or intolerable side effects were reported by the patient. At this stage, patients were converted to the comparable modified‐release preparations. Nonresponders to the first opioid were switched to the alternative opioid. As this was not a stable analgesic setting, the ratio of oral morphine:oxycodone (2:1).... Doses were retitrated according to response." ‐ Rescue medication: see 'Titration schedule' ‐ Other medication: "Adjuvant medications (laxatives, antiemetics, co‐analgesics) were either started or continued where indicated." |
|
| Outcomes | ‐ Pain intensity: Assessed by participant at baseline and daily during titration in addition to the following times: (1) when the participant was clinically stabilised on first‐line opioid, (2) if the participant did not respond to first‐line opioid and required switching to alternative opioid, (3) when participant was clinically stabilised on second‐line opioid, (4) if the participant's analgesic requirement increased by 200% of their initial stable opioid dose, and (5) if the participant did not respond to second‐line opioid or fitted the criteria to exit the study, using an 11‐point numerical rating scale (the Brief Pain Inventory) with five pain modalities from 0 (= no pain) to 10 (= worst pain imaginable) ‐ Adverse experiences: Assessed by participant at baseline and daily during titration in addition to the following times: (1) when the participant was clinically stabilised on first‐line opioid, (2) if the participant did not respond to first‐line opioid and required switching to alternative opioid, (3) when participant was clinically stabilised on second‐line opioid, (4) if the participant's analgesic requirement increased by 200% of their initial stable opioid dose, and (5) if the participant did not respond to second‐line opioid or fitted the criteria to exit the study, using an 11‐point numerical rating scale from 0 (= no symptom) to 10 (= worst symptom severity imaginable) for nausea, vomiting, constipation, diarrhoea, drowsiness, confusion or disorientation or hallucinations, bad dreams and other notable symptoms. During assessments, participants were also asked to report any new adverse events. ‐ Responding participants (primary outcome): Defined as participants who responded clinically to morphine and oxycodone when used as the first‐line strong opioid in cancer‐related pain, that is, opioid nonresponse was classified as inadequate analgesia despite dose escalation or intolerable adverse effects, or both, and adequacy of pain control and tolerability of adverse effects were defined by participants' subjective assessment, regardless of score. |
|
| Notes | ‐ Study free of commercial funding? "This study was funded by the Palliative Care Research Fund from the Royal Marsden Hospital, St. Joseph's Hospice, the Asmarley Trust, and an unrestricted educational grant from Napp Pharmaceuticals. None of the funding bodies had any role in the design and conduct of the study, the collection, management, analysis, or interpretation of the data, and the preparation, review, and approval of the manuscript, or in the decision to submit for publication. The authors reported no conflicts of interest. The study also was supported by the National Institute for Health Research Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield National Health Service Foundation Trust and Imperial College London." ‐ Groups comparable at baseline? Yes, the groups seemed to be comparable at baseline. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomized to either morphine or oxycodone in a 1:1 ratio via computer‐generated random permuted blocks." |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | High risk | "This independent study was an open‐label one because of safety, logistical, and financial considerations." |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | High risk | Participant‐assessed. See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | Data only available for 80/100 participants in the oxycodone group and 85/100 in the morphine group for the meta‐analyses |
| Incomplete outcome data (attrition bias) Adverse events | High risk | Adverse events reported for 153/198 participants |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes were reported. |
| Were the participants adequately titrated? | Low risk | Yes, the participants appeared to be adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Salzman 1999.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group trial Year: not reported Country: USA |
|
| Participants |
Participants: 50 participants randomised; 48/50 participants received ≥ 1 dose of study medication; N = 24 in each group. 35/50 participants completed the titration period, 3 participants discontinued the study due to adverse events, 8 due to ineffective treatment or intercurrent illnesses, and 2 due to other reasons Controlled‐release group: 8 males and 16 females, mean (range) age = 60 (25 to 77) years; participants taking pre‐study opioids: Yes: N = 23, No: N = 1 Immediate‐release group:13 males and 11 females, mean (range) age = 61 (39 to 91) years; participants taking pre‐study opioids: Yes: N = 22, No: N = 2 Inclusion criteria: Patients aged ≥ 18 years with stable cancer pain not adequately controlled by prior analgesic therapy with or without opioids. Among patients who were receiving nonopioid analgesic therapy, the dosing regimen was stabilised ≥ 1 week before the initiation of study medication and remained stable for the duration of the studies. Exclusion criteria: "Patients excluded from the studies included individuals with an allergy or contraindication to opioid therapy; patients with a history of substance abuse; patients receiving an opioid analgesic that could not be discontinued; cancer patients prescribed oral oxycodone at a total dose of more than 400 mg/day" |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: starting dose for opioid‐naive participants 20 mg/day, and for non‐opioid‐naive participants starting dose was based on prior 3 days of analgesic therapy; every 12 hours at 8 a.m. and 8 p.m. (± 1 hour each time). Mean final daily dose (SE) = 104 (20) mg ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: Up to 21 days ‐ Titration schedule: "The starting dose was titrated upward in each study to a limit of 400 mg/day..... Among those who required titration, the dose was increased until the patients rated their level of pain at an intensity of no greater than "slight" (1.5) on the CAT scale. The dose could be adjusted every 24 to 48 hours if necessary. Criteria for stable pain control were said to be met if pain was stabilized at 1.5 or below for 48 hours while patients were taking no more than two doses per day of supplemental analgesic." ‐ Rescue medication: "Supplemental analgesic was permitted as needed for control of breakthrough or incident pain and was provided in doses of 5 mg IR oxycodone (1 tablet) for patients titrated to 20 to 40 mg/day and 10 mg IR oxycodone (2 X 5 mg tablets) for patients titrated to 60 to 80 mg/day. For patients receiving doses greater than 80 mg/day, the supplemental analgesic dose was approximately 1/6 of the patient's total daily oxycodone dose rounded to the nearest 5 mg. Rescue medication was taken no more than once every 4 hours." ‐ Other medication: "All other opioid analgesics were prohibited. Besides nonopioid analgesic medications (discussed above), other medications necessary for patients' welfare were administered under the supervision of the investigator/physician." Comparison arm ‐ Drug: oxycodone ‐ Dose and dosing: starting dose for opioid‐naive participants 20 mg/day, and for non‐opioid‐naive participants starting dose was based on prior 3 days of analgesic therapy; 4 times daily at 8 a.m., 2 p.m., 8 p.m., and bedtime (± 1 hour each time). Bedtime dose was to be taken ≥ 3 hours after the 8 p.m. dose. Mean final daily dose (SE) = 113 (24) mg ‐ Formulation: Immediate‐release ‐ Route of administration: oral ‐ Length of treatment: Up to 21 days ‐ Titration schedule: "The starting dose was titrated upward in each study to a limit of 400 mg/day..... Among those who required titration, the dose was increased until the patients rated their level of pain at an intensity of no greater than "slight" (1.5) on the CAT scale. The dose could be adjusted every 24 to 48 hours if necessary. Criteria for stable pain control were said to be met if pain was stabilized at 1.5 or below for 48 hours while patients were taking no more than two doses per day of supplemental analgesic." ‐ Rescue medication: "Supplemental analgesic was permitted as needed for control of breakthrough or incident pain and was provided in doses of 5 mg IR oxycodone (1 tablet) for patients titrated to 20 to 40 mg/day and 10 mg IR oxycodone (2 × 5 mg tablets) for patients titrated to 60 to 80 mg/day. For patients receiving doses greater than 80 mg/day, the supplemental analgesic dose was approximately 1/6 of the patient's total daily oxycodone dose rounded to the nearest 5 mg. Rescue medication was taken no more than once every 4 hours." ‐ Other medication: "All other opioid analgesics were prohibited. Besides nonopioid analgesic medications... other medications necessary for patients' welfare were administered under the supervision of the investigator/physician." |
|
| Outcomes | ‐ Pain intensity: Assessed by participant in daily diary, using a categorical scale from 0 (= none) (1 = slight, 2 = moderate) to 3 (= severe). Also assessed at the clinic visit at the end of the titration period ‐ Adverse events: Assessed by participant in daily diary, using a categorical scale from 0 (= none) (1 = slight, 2 = moderate) to 3 (= severe). Also assessed at the clinic visit at the end of the titration period ‐ Time to stable pain control was recorded as zero for participants meeting the criteria for success in the first 48 hours (i.e. no titration was needed)." |
|
| Notes | ‐ Study free of commercial funding? No. The study was sponsored by the drug manufacturer (Purdue Pharma L.P.) and some of the authors were employees of the study drug manufacturer. ‐ Groups comparable at baseline? The groups appeared to be comparable at baseline. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | High risk | The study was open‐label. |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | The study was open‐label. |
| Blinding of outcome assessment (detection bias) Pain | High risk | The study was open‐label. |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | The study was open‐label. |
| Incomplete outcome data (attrition bias) Pain | High risk | Data reported for 35/50 participants |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data reported for 48/50 participants |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to be reported. |
| Were the participants adequately titrated? | Unclear risk | Not applicable. This study was a titration study. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Song 2015.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2012‐2014 Country: China |
|
| Participants |
Participants: 110 participants randomised ‐ Oxycodone: 55 participants; 31 men, 24 women, aged 56‐85 years, mean (SD) = 69.82 (9.39) years; cancer types were lung (16), pancreatic (3), liver (5), breast (7), oesophagus (3), cervix (8), gastric (13); mean (SD) NRS pain score = 7.21 (1.23), range 4‐10 points; no further information reported ‐ Morphine: 55 participants; 32 men, 23 women, aged 55‐87 years, mean (SD) = 68.85 (9.97) years; cancer types were lung (18), pancreatic (2), liver (4), breast (8), oesophagus (2), cervix (9), gastric (12); mean (SD) NRS pain score = 7.22 (1.35), range 4‐10 points; no further information reported Inclusion criteria: Patients admitted to hospital with severe pain from clinical stage III‐IV cancer (verified by pathology and imaging) who were sane (without mental disease) and able to communicate Exclusion criteria: Liver and kidney dysfunction, history of opioid abuse |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 1 month ‐ Titration schedule: Every 24 hours, pain was assessed and the drug dosage adjusted accordingly. If the pain was poorly controlled, the dose was increased by 30%~50%, but the number of administrations was still once every 12 hours until the NRS score was controlled at 0 to 3 points. ‐ Rescue medication: In case of sudden onset of pain, immediate‐release morphine tablets were administered. If the sudden pain treatment reached 2 times or more, the dosage had to be increased. ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 20 mg every 12 hours ‐ Formulation: Sustained‐release ‐ Route of administration: oral ‐ Length of treatment: 1 month ‐ Titration schedule: Every 24 hours pain was assessed and the drug dosage adjusted accordingly. If the pain was poorly controlled, the dose was increased by 30%~50%, but the number of administrations was still once every 12 hours until the NRS score was controlled at 0 to 3 points. ‐ Rescue medication: In case of sudden onset of pain, immediate‐release morphine tablets were administered. If the sudden pain treatment reached 2 times or more, the dosage had to be increased. ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain relief: assessed on numerical rating scale from 0 (painless)‐10. Effective pain relief if reduction ≥ 75%. Ineffective: no relief of pain after medication or < 25% ‐ Quality of life; assessed on 0‐60 scale (with higher scores indicating better outcome) for patients with malignant tumours ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear ‐ Groups comparable at baseline? Yes, at least for age and gender, but otherwise unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses were undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed. |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Stambaugh 2001.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, cross‐over trial Year: not reported Country: USA |
|
| Participants |
Participants: 40 participants entered; 30/40 participants completed both of the double‐blind periods with 100% compliance; 9 participants discontinued the study during the titration phase due to adverse events (2), lack of efficacy (4), intercurrent illness (1), and 'other' reasons (2), and 1 participant discontinued the study during the double‐blind phase due to weakness secondary to progressive disease. 10 males and 20 females, mean (range) age = 60 (34 to 83) years; primary pain site was bone (27), viscera (1), and other (2). All participants were receiving therapy that included opioids pre‐study. Inclusion criteria: Patients aged > 18 years with moderate or severe cancer‐related pain who did not require > 240 mg/day oral oxycodone equivalent for pain relief who were able to take oral medication and practiced a medically acceptable method of birth control if female with childbearing potential Exclusion criteria: Primary tumour or metastatic disease in the brain, received chemotherapy within 3 days of study entry, drug abuse, severe cognitive impairment, compromised hepatic or renal function, radiotherapy to the pain site, or hypersensitivity to oxycodone |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone + placebo ‐ Dose and dosing: The total 24‐hour oxycodone dose was equal to the stable daily dose obtained at the end of the titration phase. Drug administration 4 times daily consisting of oxycodone interspersed with placebo, resulting in q12h dosing of oxycodone. Mean final daily dose was not reported. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: Up to 35 days, consisting of a titration period of 2‐21 days, followed by 2 double‐blind cross‐over periods each lasting 3‐7 days ‐ Titration schedule: open‐label with immediate‐release oxycodone, starting dose was comparable to that calculated, based on the past 3 days of analgesia therapy. "The subjects completed the titration phase at home while monitored on a daily basis by telephone by the research monitor. Recommendations regarding changes in medication were used to minimize oxycodone use while providing adequate analgesia. More than 2 rescue medication doses per 24‐hour period or a moderate or severe global pain score indicated inadequate pain control. Participants whose pain was inadequately controlled after 21 days or who required more than 240 mg or less than 20 of oxycodone daily were discontinued from the study". ‐ Rescue medication: Immediate‐release oxycodone in 5 mg tablets ‐ Other medication: "Concurrent, stable therapy with acetaminophen, NSAIDs, or analgesic adjuvants and co‐analgesics were allowed. Opioids other than the study medication were prohibited. All medically necessary but noninvestigational medications were permitted." Comparison arm ‐ Drug: oxycodone ‐ Dose and dosing: The total 24‐hour oxycodone dose was equal to the stable daily dose obtained at the end of the titration phase. Drug administration 4 times daily, qid dosing of oxycodone. Mean final daily dose was not reported. ‐ Formulation: Immediate‐release ‐ Route of administration: oral ‐ Length of treatment: Up to 35 days, consisting of a titration period of 2‐21 days, followed by 2 double‐blind cross‐over periods each lasting 3‐7 days ‐ Titration schedule: open‐label with immediate‐release oxycodone, starting dose was comparable to that calculated, based on the past 3 days of analgesia therapy. "The subjects completed the titration phase at home while monitored on a daily basis by telephone by the research monitor. Recommendations regarding changes in medication were used to minimize oxycodone use while providing adequate analgesia. More than 2 rescue medication doses per 24‐hour period or a moderate or severe global pain score indicated inadequate pain control. Participants whose pain was inadequately controlled after 21 days or who required more than 240 mg or less than 20 of oxycodone daily were discontinued from the study". Stable pain control for 48 hours to 10 days was required before entry into the double‐blind phase. ‐ Rescue medication: Immediate‐release oxycodone in 5 mg tablets ‐ Other medication: "Concurrent, stable therapy with acetaminophen, NSAIDs, or analgesic adjuvants and co‐analgesics were allowed. Opioids other than the study medication were prohibited. All medically necessary but noninvestigational medications were permitted." ‐ For cross‐over trials, cross‐over schedule: "After successful completion of period 1, patients were crossed over into the double‐blind period 2 without a washout." The procedures for this period were identical to those in period 1. |
|
| Outcomes | ‐ Pain intensity or pain relief: Assessed by participant in daily diary, using an 11‐point scale from 0 (= no pain or no relief) to 10 (= severe pain or complete relief) ‐ Acceptability of treatment: Assessed by participant in daily diary, using a 5‐point scale from 1 (= very poor) (2 = poor, 3 = fair, 4 = good) to 5 (= excellent) ‐ Adverse events: Spontaneously reported by participant in daily telephone contact |
|
| Notes | ‐ Study free of commercial funding? No. The study was sponsored by the drug manufacturer (Purdue Frederick Company) and one of the authors was employed by the study drug manufacturer. ‐ Groups comparable at baseline? No details reported about initial group allocation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "The double‐blind periods were blinded by using three tablets identical in appearance: 5 mg IR oxycodone, 10 mg CR oxycodone, and placebo." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Participant‐reported outcome. See also cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | Data from 30/40 participants analysed |
| Incomplete outcome data (attrition bias) Adverse events | High risk | See cell above |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to be reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. Pain intensity dropped from 6 (SD = 2.2) at the beginning of titration to 2.7 at the completion of the titration phase. |
| For cross‐over trials: are data available for both time periods? | Low risk | Yes, data were available for both study periods for 30/40 participants. |
| Other bias | Low risk | The study did not appear to be subject to high risk of other biases. |
Su 2015.
| Study characteristics | ||
| Methods |
Design: randomised, parallel‐group trial Year: 2011‐2014 Country: China |
|
| Participants |
Participants: 80 participants selected; cancer types were lung (N = 22), breast (N = 20), gastric (N = 18), colon (N = 9), prostate (N = 6) and oesophageal (N = 5). Pain was assessed on a numerical rating scale (NRS) going from 0 (no pain), 1‐3 (mild pain), 4‐6 (moderate pain), 7‐9 (severe pain), to 10 (very severe pain). All participants enrolled were NRS ≥ 4 points. The participants were randomly allocated to 2 treatment groups: ‐Oxycodone: N = 42; 25 males/17 females; mean age = 55.48 (SD = 11.54; range = 29‐76) years; mean Karnofsky score (KPS) = 55.14 (SD = 5.25); mean NRS = 6.93 (SD = 1.73), N = 16 with NRS of 4‐6 points, and N = 26 with NRS of 7‐9 points. Pain types were chest pain (N = 18), abdominal pain (N = 7), ostealgia (N = 11), and shoulder and back pain (N = 6). ‐Fentanyl: N = 38; 22 males/16 females; mean age = 54.89 (SD = 11.07; range 32‐83) years, mean KPS score = 56.05 (SD = 5.77); mean NRS = 7.16 (SD = 1.64), N = 14 with NRS of 4‐6 points, and N = 24 with NRS of 7‐9 points. Pain types were chest pain (N = 16), abdominal pain (N = 8), ostealgia (N = 10), and shoulder and back pain (N = 4). Inclusion criteria: Patients with a diagnosis of malignant tumours with moderate‐severe pain, and no radiotherapy for the pain; no respiratory, cardiovascular and cerebrovascular dysfunction, and no obstruction and serious liver and kidney dysfunction; and no history of psychosis and opioids drug abuse history Exclusion criteria: not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone (oxycontin) ‐ Dose and dosing: 10 mg every 12 hours as initial dose for morphine‐naive participants, while participants who had previously received morphine were given oxycontin according to used dosage (total daily dose of morphine × 0.5 and then divided into 2 equal doses for 12 hourly treatment). The drug should be taken as whole tablets, but not grinded pieces. Mean final daily dose was not reported. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 2 weeks ‐ Titration schedule: effectiveness and adverse events were evaluated 15 minutes after the drugs were administered. If NRS increased or did not change, dose increased by 50‐100%. If NRS decreased to 4‐6, the same dose was administered after 15 min. If NRS decreased to 0‐3, the basic treatment for alleviating pain was continued, and the same dose was administered if needed. On the second day, the needed dose was calculated according to the total amount of analgesics used during the first day, in an attempt to maintain the NRS at 0‐3. ‐ Rescue medication: IV short‐acting morphine was used as rescue medication for breakthrough pain. The initial dose for those who had not used morphine before was 2‐5 mg. For those who had used morphine before, the total amount of analgesics needed in the first 24 hours was calculated and converted to the equivalent dose of morphine for injection, of which 10‐20% was the initial dose. ‐ Other medication: not reported Comparison arm ‐ Drug: fentanyl (durogesic) ‐ Dose and dosing: 25 μg/hour replaced after 72 hours as initial dose for morphine‐naive participants, while participants who had previously received morphine, received a dose 0.5 × morphine dose. Mean final daily dose was not reported. ‐ Formulation: patch ‐ Route of administration: Transdermal ‐ Length of treatment: 2 weeks ‐ Titration schedule: effectiveness and adverse events were evaluated 15 minutes after the drugs were administered. If NRS increased or did not change, the dose increased by 50‐100%. If NRS decreased to 4‐6, the same dose was administered after 15 minutes. If NRS decreased to 0‐3, the basic treatment for alleviating pain was continued, and the same dose was administered if needed. On the second day, the needed dose was calculated according to the total amount of analgesics used during the first day, in an attempt to maintain the NRS at 0‐3. ‐ Rescue medication: IV short‐acting morphine was used as rescue medication for breakthrough pain. The initial dose for those who had not used morphine before was 2‐5 mg. For those who had used morphine before, the total amount of analgesics needed in the first 24 hours was calculated and converted to the equivalent dose of morphine for injection, of which 10‐20% was the initial dose. ‐ Other medication: not reported |
|
| Outcomes | ‐ Pain relief: Categorised as complete remission (CR; pain disappeared), partial remission (PR; medication significantly reduced pain), mild remission (MR, pain after treatment reduced, but sleep still affected?), and invalid (NR; no pain relief after medication). Pain relief rate (%) = (CR + PR + MR)/total number of cases × 100% ‐ Quality of life: Assessed using Karnofsky (KPS) score, with participants' daily life, mental status, and appetite evaluated before and after treatment. "Interpersonal communication and other aspects of the situation" were also assessed before and after treatment. ‐ Adverse events: "Sleep, dizziness, nausea and vomiting, constipation, dysuria and other adverse reactions" observed using WHO standards |
|
| Notes | ‐ Study free of commercial funding? Unclear; no information appeared to be reported. ‐ Groups comparable at baseline? The treatment groups did not differ statistically significantly in terms of sex, age, KPS, type of pain, and degree of pain (P > 0.05). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Low risk | The data from all the participants were reported. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The data from all the participants were reported. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to have been reported. |
| Were the participants adequately titrated? | Low risk | The participants appeared to be adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Sun 2013.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2008‐2012 Country: China |
|
| Participants |
Participants: 204 participants randomised ‐ Oxycodone: 102 participants, 56 men, 44 women [56 + 44 = 100, not 102, but this is what was reported in paper], aged median (range) = 65 (42‐90) years; no further information reported ‐ Morphine: 102 participants, 54 men, 48 women, aged median (range) = 67 (37‐93) years; no further information reported Inclusion criteria: Patients with advanced malignant tumours diagnosed by physical and imaging studies with severe pain (NRS > 7), not sufficiently treated by weak opioids, with expected survival time > 1 month, able to take oral medication Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 15 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 30 days ‐ Titration schedule: Pain assessment carried out every 48 hours, dose adjustment accordingly with dose increase if the pain was not well controlled by 30‐50%, strictly in accordance with the number of administrations every 12 hours until the NRS 0‐3 points. If the patient had adverse reactions, symptomatic treatment should be given. ‐ Rescue medication: If breakthrough pain occurred tylenol (oxycodone 5 mg, paracetamol 325 mg). If episodes ≥ 2 per day, oxycodone dose increased. ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 30 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 30 days ‐ Titration schedule: Pain assessment carried out every 48 hours, dose adjustment accordingly with dose increase if the pain was not well controlled by 30‐50%, strictly in accordance with the number of administrations every 12 hours until the NRS 0‐3 points. If the patient had adverse reactions, symptomatic treatment should be given. ‐ Rescue medication: If breakthrough pain occurred tylenol (oxycodone 5 mg, paracetamol 325 mg). If episodes ≥ 2 per day, morphine dose increased. ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain relief/intensity: assessed using NRS from 0 (no pain), through 1‐3 (mild pain), 4‐6 (moderate pain), 7‐9 (severe pain) to 10 (extreme pain) ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Unclear, although the authors did report that the groups were comparable in terms of gender, age, type of disease and type of pain. It was unclear whether any other characteristics were examined. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses were undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Tu 2015.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2012‐2013 Country: China |
|
| Participants |
Participants: 86 participants randomised ‐ Oxycodone: 43 participants, 22 men, 21 women, mean (SD) age = 55.73 (6.14) years; pain severity moderate (23) or severe (20); no further information reported ‐ Morphine: 43 participants, 30 men, 13 women, mean (SD) age = 55.13 (6.08) years; pain severity moderate (21) or severe (22); no further information reported Inclusion criteria: Patients with advanced cancer with moderate to severe cancer pain Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: > 14 days ‐ Titration schedule: The dosage and duration of the medication were adjusted according to the patient's pain assessment until it was painless or virtually painless. If there are adverse reactions, timely symptomatic treatment should be given. ‐ Rescue medication: In the event of an outbreak of pain, IR morphine administered immediately ‐ Other medication: Other adjuvant drugs used according to the patient's condition. Ondansetron or metoclopramide for nausea and vomiting; lactulose oral solution or phenyl tablets for constipation, or enema for serious constipation, urethral catheterisation for dysuria patients Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 30 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: > 14 days ‐ Titration schedule: Within 24 hours, the dose was gradually increased by 50% to 100% with the aim of a pain score at 72 hours of 0/3 points? ‐ Rescue medication: In the event of an outbreak of pain, IR morphine administered immediately ‐ Other medication: Other adjuvant drugs used according to the patient's condition. Ondansetron or metoclopramide for nausea and vomiting; lactulose oral solution or phenyl tablets for constipation, or enema for serious constipation, urethral catheterisation for dysuria patients |
|
| Outcomes | ‐ Pain relief/intensity: assessed using VAS from 0 (no pain), through 1‐3 (mild pain), 4‐6 (moderate pain) to 7‐10 (severe pain). Pain relief degree = (pre‐score – post‐score)/pre‐score > 75% = significant relief, 74‐50% = moderate relief, 49‐25% = mild relief, < 25% = no relief) ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Authors stated the groups were comparable, but did not present many characteristics. This study was only partially dual‐extracted as the translation software could not be fully employed for the translation of this study to allow the second non‐Chinese speaking author to fully extract and appraise this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients reported to be randomised, but no information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses were undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Wang 2008.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: Not reported Country: China |
|
| Participants |
Participants: 60 participants randomised ‐ Oxycodone: 30 participants, 22 men, 8 women, mean (range) age = 59.8 (26‐78) years; cancer type: lung (14), colorectal (7), gastric (5), breast (2), ovarian (1), and cervical (1); type of pain: visceral (17), bone (6), soft tissue infiltration (5), neuropathic (2); previous analgesics: none (23), morphine sulfate CR (4), non‐steroidal drugs or tramadol (3); pain severity moderate (3) or severe (27) ‐ Morphine: 30 participants, 21 men, 9 women, mean (range) age = 57.7 (32‐75) years; cancer type: lung (12), colorectal (6), gastric (6), breast (4), ovarian (1), and oesophageal (1); type of pain: visceral (19), bone (5), soft tissue infiltration (4), neuropathic (2); previous analgesics: not reported; pain severity moderate (2) or severe (28) Inclusion criteria: Patients with pathologically confirmed cancer (except pancreatic and liver cancer) with moderate to severe cancer pain (NRS scores 4‐10) Exclusion criteria: No serious heart, liver or kidney dysfunction; no other analgesics 4 hours before study drug administration |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: ≥ 14 days ‐ Titration schedule: Dose titrated once every 24 hours, adjusted according to the degree of pain relief. If the pain score reduction < 4 points in 24 hours, the dose increased the following day. Each dose was increased by 25%‐50%, without increasing the number of doses until satisfactory pain relief. ‐ Rescue medication: In the event of an outbreak of pain, a short‐acting morphine injection added ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 30 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: ≥ 14 days ‐ Titration schedule: Dose titrated once every 24 hours, adjusted according to the degree of pain relief. If the pain score < 4 points in 24 hours, the dose increased the following day. Each dose was increased by 25%‐50%, without increasing the number of doses until satisfactory pain relief. ‐ Rescue medication: In the event of an outbreak of pain, a short‐acting morphine injection added ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain relief/intensity: assessed by physician using NRS from 0 (no pain), through 1‐3 (mild pain), 4‐6 (moderate pain) to 7‐10 (severe pain); 0 degrees is no relief; 1 degree is mild relief (relief by 1/4); 2 degrees is moderate relief (relief by 1/2); 3 degrees is significant relief (relief by > 3/4); 4 degrees is complete relief (pain is gone) [note: no data on control group for some outcomes] ‐ Quality of life using Karnofsky Performance Score ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Unclear, although the authors did report that the groups were comparable in terms of gender, age, type of disease, and type and degree of pain. It was unclear whether any other characteristics were examined. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients reported to be randomised, but no information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Xie 2018.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2016‐2017 Country: China |
|
| Participants |
Participants: 95 participants randomised ‐ Oxycodone: 48 participants, 27 men, 21 women, mean (SD/SE?; range) age = 75.3 (0.7; 65‐82) years; mean (SD/SE; range) length of illness = 2.3 (0.4; 6 months‐4 years) years; no other information reported ‐ Morphine: 47 participants, 27 men, 20 women, mean (SD/SE?; range) age = 75.3 (0.8; 64‐83) years; mean (SD/SE; range) length of illness = 2.5 (0.3; 8 months‐4 years) years; no other information reported Inclusion criteria: Elderly patients with cancer; no other information reported Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: Not reported ‐ Titration schedule: The entire drug dose was adjusted so that the VAS score was less than 3 points. ‐ Rescue medication: Morphine, not otherwise specified ‐ Other medication: According to the patient’s depression and convulsions, supplementary auxiliary drug treatment appeared to be available. Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 20 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: Not reported ‐ Titration schedule: The entire drug dose was adjusted so that the VAS score was less than 3 points. ‐ Rescue medication: Morphine, not otherwise specified ‐ Other medication: According to the patient’s depression and convulsions, supplementary auxiliary drug treatment appeared to be available. |
|
| Outcomes | ‐ Pain relief/intensity: assessed by on 0 (no pain) to 10 (severe pain)‐point scale ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? The authors reported that the groups were comparable, but very few characteristics reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Ye 2012.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: Not reported Country: China |
|
| Participants |
Participants: 83 participants randomised ‐ Oxycodone: 42 participants, 25 men, 17 women, mean (SD) age = 58.5 (not reported) years; cancer type: lung (14), colorectal (8), gastric (6), breast (5), oesophageal (2), liver (5), nasopharyngeal (1) and pancreatic (1); no other information reported ‐ Morphine: 41 participants, 22 men, 19 women, mean (SD) age = 59.7 (not reported) years; cancer type: lung (15), colorectal (7), gastric (4), breast (4), oesophageal (3), liver (6), and nasopharyngeal (2); no other information reported Inclusion criteria: Patients aged 18‐72 years with pathologically confirmed cancer (except pancreatic and liver cancer) with moderate to severe cancer pain (NRS scores 6‐10), and expected survival > 2 months Exclusion criteria: No severe disease of gastrointestinal tract. One week before treatment no morphine‐type analgesics used; no other analgesics or adjuvant drugs during the treatment. No radiotherapy or chemotherapy was given within 3 weeks before treatment and during treatment. |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: Starting dose = 10 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 7 days ‐ Titration schedule: After 24 hours, the daily dose could be adjusted according to the degree of pain. The doses started at 10 mg/12 hours, but could go up through 20 mg, 30 mg, 40 mg, 50 mg to 60 mg. ‐ Rescue medication: Not reported ‐ Other medication: No other analgesics or adjuvant drugs during the treatment. No radiotherapy or chemotherapy was given within 3 weeks before treatment and during treatment. Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 30 mg every 12 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 7 days ‐ Titration schedule: After 24 hours, the daily dose could be adjusted according to the degree of pain. The doses started at 30 mg/12 hours, but could go up through 60 mg, 90 mg, 120 mg, 150 mg to 160 mg. ‐ Rescue medication: Not reported ‐ Other medication: No other analgesics or adjuvant drugs during the treatment. No radiotherapy or chemotherapy was given within 3 weeks before treatment and during treatment. |
|
| Outcomes | ‐ Pain relief/intensity: assessed by physician using NRS from 0 (no pain), through 1‐3 (mild pain), 4‐6 (moderate pain) to 7‐10 (severe pain); classified to complete remission/relief (no pain; 0), partial remission/relief (pain significantly reduced, sleep undisturbed, and part of being able to live normally; 1‐3), mild remission/relief (pain slightly reduced but not satisfactory; 4‐5); no pain remission/relief (> 6 points) ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Appeared comparable in terms of gender, age and type of cancer, but no other characteristics reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients reported to be randomised, but no information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Yu 2007.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: Not reported Country: China |
|
| Participants |
Participants: 30 participants randomised ‐ Oxycodone: 15 participants, 7 men, 8 women, median age 52.7 years; no further information reported ‐ Morphine: 15 participants, 9 men, 6 women, median age 53.73 years; no further information reported Inclusion criteria: Inpatient and outpatient cancer patients, with expected survival > 2 months, and meeting one of the following criteria: (1) In the past 4 weeks, the pain intensity after treatment with morphine (daily dose 20‐60 mg) was ≤ 4, while the pain intensity increased ≥ 5 after opioid therapy was stopped; (2) In the past 4 weeks, the pain intensity was ≥ 4 (that is, the pain intensity was not controlled satisfactorily due to insufficient dose) after continuous use of 20‐40 mg morphine for more than 7 days, and the pain intensity further increased ≥ 5 after withdrawal of the drug; (3) Never used morphine and other analgesic drugs, and the pain intensity was ≥ 5 Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: Starting dose = 10 mg every 12 hours, with dose adjustment every 24 hours up to a maximum of 40 mg ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 5 days ‐ Titration schedule: See "Dose and dosing". No further information reported ‐ Rescue medication: Not reported ‐ Other medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 30 mg every 12 hours, with dose adjustment every 24 hours up to a maximum of 90 mg ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 5 days ‐ Titration schedule: See "Dose and dosing". No further information reported ‐ Rescue medication: Not reported ‐ Other medication: Not reported |
|
| Outcomes | ‐ Pain relief/intensity: assessed using on scale from 0 (no pain), through 1‐4 (mild pain), 5‐7 (moderate pain), 8‐9 (severe pain) to 10 (also severe pain), assessed by medical staff at baseline and on days 1‐5 ‐ Pain response: assessed using the WHO analgesic efficacy classification standard: 0 (no pain relief), 1 (mild pain relief; approximately 1/4 pain reduction); 2 (moderate pain relief; pain is reduced by about 1/2), 3 (obvious pain relief; pain is reduced by about 3/4), 4 (complete pain relief: no pain); assessed by medical staff after at least 5 days ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Unclear. The groups appeared comparable in terms of age and gender, but no further characteristics reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study described as randomised. No further information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Yu 2009.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2008‐2009 Country: China |
|
| Participants |
Participants: 72 participants randomised, 62 of whom completed the study. Reason for withdrawals in the oxycodone group were intolerant side effects (1), non‐visceral pain (2) and financial reasons (1); and in the morphine group were intolerant side effects (3), non‐visceral pain (1), financial reasons (1) and death (1). ‐ Oxycodone: 36 participants, 21 men, 15 women, average (SD?) [NOS] age 62.4 (11.3) years; VAS score 4‐6 (13) or 7‐10 (23); cancer type: gastric (6), cardiac (1), liver (11), gallbladder cholangiocarcinoma (2), pancreatic (9), colon (2) rectal (5); no further information reported split by group ‐ Morphine: 36 participants, 23 men, 13 women, average (SD?) [NOS] age 61.8 (11.7) years; VAS score 4‐6 (12) or 7‐10 (24); cancer type: gastric (10), cardiac (3), liver (13), gallbladder cholangiocarcinoma (2), pancreatic (3), colon (2) rectal (3); no further information reported split by group Inclusion criteria: Patients with pathologically (histology or cytology) confirmed abdominal cancer tumours admitted to the authors' hospital with moderate or severe visceral cancer pain (VAS > 4) not satisfactorily relieved by step 1 and 2 analgesics who could stop chemotherapy and radiotherapy and other analgesics, and were able to take medications orally and provide informed consent Exclusion criteria: Severe heart, lung, liver, and kidney disease or central nervous system and biliary diseases; history of drug abuse; breathing inhibition or head injury, paralytic intestinal obstruction, acute abdomen, delayed emptying or history of severe constipation; mental confusion who could not self‐evaluate; new type of pain emerging during the study |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: Starting dose not reported. See 'Titration schedule' ‐ Formulation: Sustained‐release/controlled‐release (after titration with IR morphine) ‐ Route of administration: oral ‐ Length of treatment: 18 days (including 3 days of titration) ‐ Titration schedule: 3‐day titration phase with titration with immediate‐release morphine with a dose continually evaluated and adjusted to effective analgesic effect (VAS ≤ 3). The dose then converted to controlled‐release equivalent dosing of oxycodone hydrochloride at a ratio of 1.5:1 for dosing every 12 hours. See also 'Rescue medication' ‐ Rescue medication: Immediate‐release morphine for breakthrough pain. The daily dose should not exceed 1/2 of the total dose. When VAS > 3, the dose was increased from the previous dose by 25%‐50%, with the aim of keeping VAS ≤ 3. ‐ Other medication: If necessary, metoclopramide, senna, etc. to relieve opioids‐induced adverse reactions Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose not reported. See 'Titration schedule' ‐ Formulation: Sustained‐release/controlled‐release (after titration with IR morphine) ‐ Route of administration: oral ‐ Length of treatment: 18 days (including 3 days of titration) ‐ Titration schedule: 3‐day titration phase with titration with immediate‐release morphine with a dose continually evaluated and adjusted to effective analgesic effect (VAS ≤ 3). The dose then converted to controlled‐release equivalent dosing of morphine sulfate at a ratio of 1:1 for dosing every 12 hours. See also 'Rescue medication' ‐ Rescue medication: Immediate‐release morphine for breakthrough pain. The daily dose should not exceed 1/2 of the total dose. When VAS > 3, the dose was increased from the previous dose by 25%‐50%, with the aim of keeping VAS ≤ 3. ‐ Other medication: If necessary, metoclopramide, senna, etc. to relieve opioids‐induced adverse reactions |
|
| Outcomes | Pain relief: Assessed using VAS according to the following criteria: No relief (decrease in VAS < 1/4), mild relief (decrease in VAS of around 1/4), moderate relief (decrease in VAS by 1/2), obvious relief (decrease in VAS of 3/4), complete relief (VAS = 0) ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Unclear, although the groups appeared comparable in terms of the reported characteristics (see 'Participants' row above), it was unclear whether they differed on other characteristics. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study described as randomised. No further information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | High risk | Data included for 32/36 and 30/36 oxycodone and morphine patients, respectively. Dropouts were explained. |
| Incomplete outcome data (attrition bias) Adverse events | High risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Low risk | Yes probably |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Yu 2014.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, non‐inferiority parallel‐group trial Year: 2009‐2011 (from ClinicalTrials.gov) Country: China |
|
| Participants |
Participants: Of 260 randomised participants, 137 completed the maintenance (active treatment) phase of the study (70 in the hydromorphone group and 67 in the oxycodone group), and these 137 participants presumably comprised the per‐protocol dataset, although the criteria for the per‐protocol set were also that they needed to have completed all efficacy evaluations with good compliance. The full analysis set comprised all randomised participants with at least one dose of medication administered during the titration phase and one measurement of efficacy. The safety dataset comprised all randomised participants with at least one dose of medication administered during the titration phase and one measurement of safety. ‐ Hydromorphone: N = 125, 82 males / 43 females; mean/median (SD, range) age = 53.5/54 (10.86; 22‐70) years; cancer type: breast (N = 8), lung (N = 38), bone (N = 0), oral cavity (N = 1), gastrointestinal (N = 46), genitourinary (N = 13), lymphoma (N = 0), leukaemia (N = 0), other (N = 17), not known (N = 2); tumour metastatic: yes (N = 118), no (N = 7); concomitant cancer therapy (N = 75); administration of strong opioids (N = 104), weak opioids (N = 21) ‐ Oxycodone: N = 123, 80 males / 43 females; mean/median (SD, range) age = 52.7/55 (10.75; 18‐68) years; cancer type: breast (N = 7), lung (N = 34), bone (N = 0), oral cavity (N = 0), gastrointestinal (N = 46), genitourinary (N = 17), lymphoma (N = 0), leukaemia (N = 0), other (N = 16), not known (N = 3); tumour metastatic: yes (N = 112), no (N = 11); concomitant cancer therapy (N = 72); administration of strong opioids (N = 105), weak opioids (N = 18) Inclusion criteria: Patients aged 18 to 70 years; currently receiving strong oral or transdermal (through the skin) opioid analgesics with inadequate control of moderate to severe cancer pain or currently receiving weak opioids for cancer pain and were eligible according to the study protocol to receive treatment with a strong opioid analgesic; who required or were expected to require between 40 mg and 184 mg of oral morphine or morphine equivalents every 24 hours and who were reasonably expected to achieve a stable dose of opioid study medication during the study; with a life expectancy of 12 weeks or longer Exclusion criteria: Patients with pure neuropathic pain, pain of unknown origin, acute pain, or only pain on movement; requiring other opioid analgesics (apart from immediate‐release morphine hydrochloride as rescue medication for breakthrough pain), with any significant central nervous system (CNS) disorder or any disorder that predisposed the patient to respiratory depression or any condition wherein the risks of treatment with study drug might outweigh the potential benefits; and women of childbearing potential who were pregnant or lactating |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: twice daily administration of oxycodone morning and evening at 12‐hourly intervals. The mean (SD) (daily?) dose of study medication in the overall maintenance phase 38.5 (20.94) mg oxycodone CR ‐ Formulation: Controlled‐release provided in 10, 20 and 40 mg over‐encapsulated tablets ‐ Route of administration: oral ‐ Length of treatment: 28 days ‐ Titration schedule: Up to 8 days during which participants were converted from their prior opioids to their morphine equivalents (morphine to oxycodone CR, 2:1) and titrated to adequate effect (as determined by pain assessments and supplementary analgesic requirements). Dosage adjustments were made no more frequently than every 2 days, both upwards and downwards. Maximum total daily dose allowed was 80 mg oxycodone. For breakthrough pain episodes observed within a 2‐day period, rescue medication (IR morphine hydrochloride tablets, 5 mg or 10 mg) was administered once every 4 hours as needed. The participants had to achieve a stable dose providing pain control (maximum use of rescue medication allowed was 3 times per day on average) at least in the last 2 days the titration phase (2‐8 days) and this dose was continued for 28 consecutive days. In the 28‐day active treatment phase "upward and downward dose titrations were not to exceed a total daily dose of " 80 mg oxycodone CR. ‐ Rescue medication: see "'Titration schedule.' "A single dose of rescue medication was approximately 15% of the corresponding total daily dose of study medication." ‐ Other medication: other opioids (than study drugs and rescue medication) not allowed during the study. The following therapies were not allowed during the study or within 2 weeks before study entry: monoamine oxidase inhibitors, neuroablative procedures, therapy with isotopes, anaesthetic procedures including acupuncture, or surgical procedures relevant to cancer pain. Fentanyl patches were not allowed during the study or within 5 days of study entry. Adjuvant medications (e.g. paracetamol, NSAIDs, anxiolytics, antidepressants, antiarrhythmic drugs, hormone therapy, anticonvulsants, corticosteroids, and neuroleptics) were allowed if the participant, at study start, was on a stable dose, which was to be maintained. Comparison arm ‐ Drug: hydromorphone + placebo ‐ Dose and dosing: twice‐daily administration of hydromorphone and placebo morning and evening at 12‐hourly intervals (i.e. once daily dosing of active drug). The mean (SD) (daily?) dose of study medication in the overall maintenance phase 16 (8.51) mg hydromorphone ER ‐ Formulation: Extended‐release provided in 8 and 16 mg over‐encapsulated tablets ‐ Route of administration: oral ‐ Length of treatment: 28 days ‐ Titration schedule: Up to 8 days during which participants were converted from their prior opioids to their morphine equivalents (morphine to hydromorphone ER, 5:1) and titrated to adequate effect (as determined by pain assessments and supplementary analgesic requirements). Dosage adjustments were made no more frequently than every 2 days, both upwards and downwards. Maximum total daily dose allowed was 32 mg hydromorphone. For breakthrough pain episodes observed within a 2‐day period, rescue medication (IR morphine hydrochloride tablets, 5 mg or 10 mg) was administered once every 4 hours as needed. The participants had to achieve a stable dose providing pain control (maximum use of rescue medication allowed was 3 times per day on average) at least in the last 2 days the titration phase (2‐8 days) and this dose was continued for 28 consecutive days. In the 28‐day active treatment phase "upward and downward dose titrations were not to exceed a total daily dose of " 32 mg oxycodone CR. ‐ Rescue medication: see 'Titration schedule.' "A single dose of rescue medication was approximately 15% of the corresponding total daily dose of study medication." ‐ Other medication: other opioids (than study drugs and rescue medication) not allowed during the study. The following therapies were not allowed during the study or within 2 weeks before study entry: monoamine oxidase inhibitors, neuroablative procedures, therapy with isotopes, anaesthetic procedures including acupuncture, or surgical procedures relevant to cancer pain. Fentanyl patches were not allowed during the study or within 5 days of study entry. Adjuvant medications (e.g. paracetamol, nonsteroidal anti‐inflammatory drugs, anxiolytics, antidepressants, antiarrhythmic drugs, hormone therapy, anticonvulsants, corticosteroids, and neuroleptics) were allowed if the participant, at study start, was on a stable dose, which was to be maintained. |
|
| Outcomes | ‐ Participant assessment of "pain at its worst in the last 24 hours," included as an item in the Brief Pain Inventory (BPI) Short Form (0 = no pain and 10 = pain as bad as you can imagine). "Endpoint was defined as the last recorded BPI score of worst pain, just before taking the morning dose of study drug." ‐ "Pain at its least in the past 24 hours", "average pain," "pain right now;" all assessed in the same way as "pain at its worst in the past 24 hours" ‐ Number of breakthrough pain medication doses taken, recorded by participants in diaries ‐ Number of participants with treatment‐emergent adverse events, serious adverse events, and adverse events leading to discontinuation from the study |
|
| Notes | ‐ Study free of commercial funding? No. Some of the authors were from Janssen Pharmaceutical Company. ‐ Groups comparable at baseline? "The baseline characteristics were comparable between the 2 treatment groups, except in the hydromorphone ER group, in which a higher percentage of participants with bone metastasis (hydromorphone ER: 52.0%; oxycodone CR: 37.4%) and Eastern Cooperative Oncology Group performance status of 3 (hydromorphone ER: 25.6%; oxycodone CR: 18.7%)." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation by an online dynamic minimisation allocation programme with stratification for centre, concomitant cancer therapy, and administration of opioids during the last 14 days before study entry |
| Allocation concealment (selection bias) | Low risk | See cell above |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "The interactive web based response system designated a unique patient number and treatment code, which dictated the treatment assignment for each patient. The blind was broken only if specific emergency treatment dictated knowing the treatment status." All study drugs, including placebo, provided in over‐encapsulated tablets |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above |
| Incomplete outcome data (attrition bias) Pain | High risk | Only data from 81/260 randomised participants were analysed. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The data from 254/260 randomised participants were analysed. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to have been reported. |
| Were the participants adequately titrated? | Low risk | The participants appeared to be adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The results did not appear to be subject to other bias. |
Zecca 2016.
| Study characteristics | ||
| Methods |
Design: randomised, open‐label, superiority parallel‐group trial Year: 2006‐2007 Country: Italy |
|
| Participants |
Participants: 187 participants randomised to oxycodone or morphine: ‐ Oxycodone: N = 92, 60 males and 32 females, mean (SD) age = 62.1 (12.5) years; cancer types were GI tract (digestive tract, liver, pancreas; N = 23), urogenital system (N = 16), breast (N = 10), lung (N = 20), sarcoma (N = 9), head and neck (N = 3), other (N = 10) and unknown (N = 1); anatomical pain site(s) were lower back (N = 25), abdomen (N = 21), lower limb (N = 3), thorax (N = 11); 81 participants had metastatic disease and 11 had localised disease; and 36 participants were receiving ongoing chemotherapy while 56 participants were not. ‐ Morphine: N = 95, 56 males and 39 females, mean (SD) age = 61.8 (11.5) years; cancer types were GI tract (digestive tract, liver, pancreas; N = 23), urogenital system (N = 15), breast (N = 16), lung (N = 16), sarcoma (N = 1), head and neck (N = 5), other (N = 17) and unknown (N = 2); anatomical pain site(s) were lower back (N = 11), abdomen (N = 20), lower limb (N = 7), thorax (N = 25); 80 participants had metastatic disease and 15 had localised disease; and 39 participants were receiving ongoing chemotherapy while 56 participants were not. Inclusion criteria: Patients aged 18 years or above; previous 24 hours average pain intensity score of at least 5 on a 0‐10 numerical rating scale; Karnofsky performance status score of at least 40; > 1 month expected survival; and minimum expected follow‐up of two weeks at the study centre Exclusion criteria: treatment with WHO step‐III opioids within 30 days of study entry; severe renal impairment; severe hepatic failure; dyspnoea or severe chronic obstructive pulmonary disease; inability to take oral medications; history of psychiatric illness; cerebral metastasis; cognitive impairment; medical history of intolerance to morphine or oxycodone; pregnancy; or breastfeeding |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone ‐ Dose and dosing: every 12 hours. "The initial daily dose of the study medication was decided by the treating physician, based on patient characteristics, pain intensity, and previous analgesic dosage, according to usual clinical practice." "and the dose was titrated to effect. Dose adjustments during follow‐up were encouraged if patients required more than two rescue analgesic doses over 24 hours." Mean(?; 95% CI) oral morphine equivalent daily dose at baseline = 36.6 (33.6 to 39.6) mg; mean(?; 95% CI) oral morphine equivalent daily dose at study end = 69.8 (58.3 to 81.4) mg ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 14 days ‐ Titration schedule: see "Dose and dosing" ‐ Rescue medication: "Oral immediate release or parenteral morphine could be prescribed as supplemental analgesics for breakthrough pain." ‐ Other medication: "Administration of hormonotherapy, chemotherapy, analgesic adjuvants (steroids, anticonvulsants, and antidepressants) was permitted only if started before study entry and had to be kept unchanged during the study period." "Use of antiemetics and laxatives was permitted for treatment of adverse effects as required." Comparison arm ‐ Drug: morphine ‐ Dose and dosing: every 12 hours. "The initial daily dose of the study medication was decided by the treating physician, based on patient characteristics, pain intensity, and previous analgesic dosage, according to usual clinical practice." "and the dose was titrated to effect. Dose adjustments during follow‐up were encouraged if patients required more than two rescue analgesic doses over 24 hours." Mean(?; 95% CI) oral morphine equivalent daily dose at baseline = 30.9 (27.7 to 34.1) mg; mean(?; 95% CI) oral morphine equivalent daily dose at study end = 53.9 (44.4 to 63.4) mg ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 14 days ‐ Titration schedule: see 'Dose and dosing' ‐ Rescue medication: "Oral immediate release or parenteral morphine could be prescribed as supplemental analgesics for breakthrough pain." ‐ Other medication: "Administration of hormonotherapy, chemotherapy, analgesic adjuvants (steroids, anticonvulsants, and antidepressants) was permitted only if started before study entry and had to be kept unchanged during the study period." "Use of antiemetics and laxatives was permitted for treatment of adverse effects as required." |
|
| Outcomes | ‐ Participant assessment of "average pain intensity in the previous 24 hours" from 0 (no pain) to 10 (worst possible pain), numerical rating scale at baseline, 7 days and 14 days ‐ Participant assessment of "average intensity of adverse events in the previous week" from 0 (no symptom) to 10 (worst possible symptom), numerical rating scale at baseline, 7 days and 14 days of the following adverse events: nausea, vomiting, confusion, constipation, somnolence, dry mouth, itching, and hallucinations (rated as present/absent). Other than hallucinations, an adverse event was considered to occur when there was a worsening, relative to baseline of ≥ 2 points. |
|
| Notes | ‐ Study free of commercial funding? No. Study supported by Mundipharma Pharmaceuticals; Floriani Foundation, Milan (del.CDA 22/11/12) and by Associazione Italiana per la Ricerca sul Cancro (IG15314). The authors stated that the funders had no part in study design, data analysis and interpretation, nor in writing the report. ‐ Groups comparable at baseline? Yes apart from Kanofsky Performance Status: Higher number of participants in the oxycodone group (N = 46/52%) with a performance status of 70 or below compared to morphine (N = 36/39%) Study terminated early due to slow accrual Trial registration: https://www.clinicaltrialsregister.eu/ctr‐search/search?query=2006‐003151‐21) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation to treatment group in a 1:1 ratio via a computer‐generated block randomisation with a block size of 4 and stratification by age (< 70 v 70 years or above) and ongoing (administered from 15 days before to 15 days after randomisation) vs not ongoing chemotherapy |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation by a trial office. Randomisation sequence concealed until interventions assignment |
| Blinding of participants and personnel (performance bias) Pain | High risk | Open‐label study |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) Pain | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data included from 85/92 participants in the oxycodone arm and 88/95 participants in the morphine arm |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data from 185 of 187 randomised participants analysed |
| Selective reporting (reporting bias) | Low risk | The obvious outcomes were reported. |
| Were the participants adequately titrated? | Low risk | The participants appeared to be adequately titrated. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Low risk | The study did not appear to be subject to other bias. |
Zhang 2011.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: Not reported Country: China |
|
| Participants |
Participants: 67 participants randomised with the following cancer types: colorectal (16), lung (11), gastric (11), breast (8), pancreatic (6), malignant lymphoma (5), bile duct (2), prostate (2), oesophageal (1), liver (1), cervical (1), malignant melanoma (1), thyroid (1), multiple myeloma (1) ‐ Oxycodone: 35 participants, 21 men, 14 women, average (SD?) [NOS] age 52.7 (9.6) years; no further information reported split by group ‐ Morphine: 32 participants, 18 men, 14 women, average (SD?) [NOS] age 53.5 (10.5) years; no further information reported split by group Inclusion criteria: Opioid‐naive patients with moderate pain from cancer Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: Starting dose = 5 mg every 12 hours(?) ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 4 days ‐ Titration schedule: If necessary, the dose was adjusted every 12 hours: If the NRS score still 4‐6, the dose gradually increased from 50% to 100%. Once the pain decreased to < 3, current dose maintained. No further information reported ‐ Rescue medication: Not reported ‐ Other medication: Phenolphthalein tablets, lactulose oral solution, and oral metoclopramide appeared to be allowed. No further details reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: Starting dose = 10 mg every 12 hours(?) ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 4 days ‐ Titration schedule: If necessary, the dose was adjusted every 12 hours: If the NRS score still 4‐6, the dose gradually increased from 50% to 100%. Once the pain decreased to < 3, current dose maintained. No further information reported ‐ Rescue medication: Not reported ‐ Other medication: Phenolphthalein tablets, lactulose oral solution, and oral metoclopramide appeared to be allowed. No further details reported |
|
| Outcomes | ‐ Pain relief/intensity: assessed using an NRS scale from 0 (no pain), through 1‐3 (mild pain), 4‐6 (moderate pain), 7‐9 (severe pain) to 10 (also severe pain), assessed at baseline(?), and at 0.5, 1, 1.5, 2, 4, 8, 12, 24, 48, 36, 72, 96 hours ‐ Adverse events, including sleep quality |
|
| Notes | ‐ Study free of commercial funding? Yes, government‐funded (Fund Project: Supported by Shanghai Key Discipline Construction Project (B905)) ‐ Groups comparable at baseline? The authors stated that the groups were comparable in terms of age, gender and tumour stage, but no further characteristics reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Low risk | Unclear whether ITT analyses were undertaken. Data appeared to be included and analysed for all patients, and study reported that there were no dropouts. Quote: “None of the patients withdrew from the study due to intolerance.” |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Low risk | A total of 46 patients were successfully titrated (NRS ≤ 3), 24 in oxycodone group and 22 in morphine group. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
Zhang 2014.
| Study characteristics | ||
| Methods |
Design: randomised, double‐blind, parallel‐group trial Year: 2013‐2014 Country: China |
|
| Participants |
Participants: 171 participants selected; 94 males and 77 females, median (range) age = 62 (39 to 74) years; cancer types were lung (N = 48), pancreatic (N = 13), liver (N = 30), gastric (N = 15), nasopharynx (N = 21), colorectal (N = 25) and bone metastases (N = 19); 49 participants had moderate pain and 122 had severe pain. The participants were randomly allocated to 3 treatment groups with N = 57 in each. Inclusion criteria: Not explicitly given, but "All patients had moderate or severe stable pain, the KPS score > 50, expected survival > 3 months, no obvious respiratory depression or airway obstruction, normal liver function and blood routine examination, no drug allergy history, no use of other analgesics at 5 h before treatment." Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone (supplied by Mundipharma (China) Pharmaceutical Co. Ltd.) ‐ Dose and dosing: 10 mg every 12 hours as initial dose. Mean final daily dose was not reported. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: not reported ‐ Titration schedule: "The above‐mentioned drugs must be swallowed wholly, not partially or trituration. If the patients cannot take the drugs, the same dose of rectal administration was considered. The dose was evaluated once every 48 h and regulated according to the degrees of pain relief. The dose was added and each dose was increased by 50%~100% due to poor control of disease but the administration frequency was not changed until the cancer pain was relived satisfactorily." ‐ Rescue medication: "During the treatment, if [the] unsound short‐term effect or sudden aggravated pain, a short‐acting morphine injection was given." ‐ Other medication: not reported Comparison arm 1 ‐ Drug: morphine (supplied by Taiji Group, Southwest Pharmaceutical Co Ltd) ‐ Dose and dosing: 30 mg every 12 hours as initial dose. Mean final daily dose was not reported. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: not reported ‐ Titration schedule: "The above‐mentioned drugs must be swallowed wholly, not partially or trituration. If the patients cannot take the drugs, the same dose of rectal administration was considered. The dose was evaluated once every 48 h and regulated according to the degrees of pain relief. The dose was added and each dose was increased by 50%~100% due to poor control of disease but the administration frequency was not changed until the cancer pain was relived satisfactorily." ‐ Rescue medication: "During the treatment, if [the] unsound short‐term effect or sudden aggravated pain, a short‐acting morphine injection was given." ‐ Other medication: not reported Comparison arm 2 ‐ Drug: MS Contin (supplied by Mundipharma (China) Pharmaceutical Co Ltd) ‐ Dose and dosing: 30 mg every 12 hours as initial dose. Mean final daily dose was not reported. ‐ Formulation: Controlled‐release ‐ Route of administration: oral ‐ Length of treatment: not reported ‐ Titration schedule: "The above‐mentioned drugs must be swallowed wholly, not partially or trituration. If the patients cannot take the drugs, the same dose of rectal administration was considered. The dose was evaluated once every 48 h and regulated according to the degrees of pain relief. The dose was added and each dose was increased by 50%~100% due to poor control of disease but the administration frequency was not changed until the cancer pain was relived satisfactorily." ‐ Rescue medication: "During the treatment, if [the] unsound short‐term effect or sudden aggravated pain, a short‐acting morphine injection was given." ‐ Other medication: not reported |
|
| Outcomes | ‐ Pain: Classified according to World Health Organisation (WHO) from level 0 (painless), through level 1 (mild pain, no need to use drugs), level 2 moderate pain, influences sleep, analgesics needed) to level 3 (severe pain, strong impact on sleep, analgesics needed) ‐ Pain relief: Measured on degree scale from 0 (non‐remission pain), through Ⅰ degree (mild pain relief, pain which is reduced by 1/4), Ⅱ degrees (moderate pain relief, pain reduced by 1/2), Ⅲ degrees (obvious pain relief, pain reduced by 3/4), and Ⅳ degrees (complete pain relief, pain disappears). "The pain relief rate refers to the rate of moderate or above pain, that is, the pain relief rate = patients of [The] Ⅱ degrees and [the] above/the total selected patients." (page 8798) ‐ Adverse events: Nausea, vomiting, constipation, dizziness, dysuria and somnolence. No further details reported |
|
| Notes | ‐ Study free of commercial funding? Unclear, the study drugs were provided by Taili Group Southwest Pharmaceuticals Co. Ltd. (morphine) and Mundipharma (China) Pharmaceuticals Co. Ltd. ‐ Groups comparable at baseline? No differences were found between the 3 groups in mean age, gender, disease categories, KPS score, pain types, and degrees (P > 0.05). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear how many were entered into the trial |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Unclear how many were entered into the trial |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appeared to be reported. |
| Were the participants adequately titrated? | Low risk | The participants were probably adequately titrated. About 90% of patients in each of the three groups achieved at least moderate pain relief. |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | It cannot be evaluated whether this study is at risk of other bias due to the sparse reporting of study details. |
Zhang 2016a.
| Study characteristics | ||
| Methods |
Design: randomised, parallel trial Year: 2014‐2015 Country: China |
|
| Participants |
Participants: 120 participants randomised, 62 men, 58 women, median (range) age 57 (28‐83) years; cancer type: gastric (19), pancreatic (2), colorectal (23), oesophageal (6), breast (20), liver (10), head and neck (12), and lung cancer (28); no further information reported, and none split by group ‐ Oxycodone: 40 participants ‐ Morphine sulfate: 40 participants ‐ Morphine hydrochloride: 40 participants Inclusion criteria: Patients with cancer pain Exclusion criteria: Not reported |
|
| Interventions |
Oxycodone arm ‐ Drug: oxycodone hydrochloride ‐ Dose and dosing: 10 mg every 10 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 1 month ‐ Titration schedule: Not reported ‐ Rescue medication: Not reported Comparison arm ‐ Drug: morphine sulfate ‐ Dose and dosing: 30 mg every 10 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 1 month ‐ Titration schedule: Not reported ‐ Rescue medication: Not reported Comparison arm ‐ Drug: morphine hydrochloride ‐ Dose and dosing: 30 mg every 10 hours ‐ Formulation: Sustained‐release/controlled‐release ‐ Route of administration: oral ‐ Length of treatment: 1 month ‐ Titration schedule: Not reported ‐ Rescue medication: Not reported |
|
| Outcomes | Pain intensity: Assessed using NRS from 0 (no pain) through 1‐4 (mild pain), 5‐6 (moderate pain), 7‐9 (severe pain) to 10 (extreme pain) Pain relief: NRS score decreased: to 0 (complete relief), by 75% (significant relief), by 50% (moderate relief), by 25% (mild relief), no decrease or increase (no relief) ‐ Adverse events |
|
| Notes | ‐ Study free of commercial funding? Unclear. No information provided ‐ Groups comparable at baseline? Unclear. The authors reported that they were comparable, but presented no characteristics split by group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study described as randomised. No further information reported |
| Allocation concealment (selection bias) | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Unclear whether ITT analyses were undertaken. Data appeared to be included and analysed for all patients, but study did not report whether there were any dropouts. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | Very limited reporting so could not be assessed |
| Were the participants adequately titrated? | Unclear risk | No information reported |
| For cross‐over trials: are data available for both time periods? | Unclear risk | Not applicable |
| Other bias | Unclear risk | No information reported |
AE: Adverse events a.m.: Ante meridiem ANOVA: Analysis of variance bid: Bis in die (twice a day) BFI: Bowel Function Index BPI‐SF: Brief Pain Inventory‐Short Form ca: Circa CAT: Categorial scale cm: centimetres CNS: central nervous system CR: controlled‐release CTCAE: Common Terminology Criteria for Adverse Events ECOG: Eastern Cooperative Oncology Group; eGFR: Estimated glomerular filtration rate EORTC QLQ‐C15‐PAL: European Organization for Research and Treatment of Cancer Quality of Life Questionnaire‐Core15_Palliative ER: extended‐release ESAS: Edmonton Symptom Assessment System FACT‐G: Functional Assessment of Cancer Therapy‐General g: grams g/dL: grams per decilitre GI: Gastrointestinal h or hr: hour(s) HCI: Hydrochloride IM: intramuscular IR: Immediate‐release ITT: intention‐to‐treat IV: intravenous IVRS: Interactive Voice Response System kg: kilograms KPS: Karnofsky Performance Status M: morphine MAOI: Monoamine oxidase inhibitors max: maximum MedDRA: Medical Dictionary for Regulatory Activities mg: milligrams mg/dL: milligrams per decilitre min: minute(s) mL: millilitres mm: millimetres MR: Mild relief N: number of participants NA: Not applicable NOS: Not otherwise reported NR: No response NRS: numerical rating scale NSAID: non‐steroidal anti‐inflammatory drug OOD: oxycodone once daily OTD: oxycodone twice daily PCA: patient‐controlled analgesia PI: pain intensity p.m.: Post meridiem PR: Partial response PS: Performance status q12h: Every 12 hours qid: Four times per 24 hours RANKL: Receptor activator of nuclear factor kappa‐Β ligand SD: standard deviation SE: standard error SR: sustained‐release TD: transdermal uL: microlitres vs: versus VAS: visual analogue scale WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ahmedzai 2012 | Comparison not in PICO: Oxycodone‐naloxone versus oxycodone |
| Awerbuch 2011 | Not cancer pain |
| Bekkering 2011 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Bell 2006 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Borchgrevink 2004 | Narrative review |
| Caraceni 2011 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Carroll 2011 | Comparison not in PICO: Patients received a variety of sustained‐release opioids + immediate‐release morphine or oxycodone versus placebo |
| Chary 1994 | Intervention not in PICO |
| Chen 2009 | Comparison not in PICO: Oxycodone‐acetominophen versus morphine |
| De Conno 1991 | Not RCT |
| Dunlop 2013 | Comparison not in PICO: Oxycodone‐naloxone versus oxycodone |
| Fallon 2011 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Gao 2020 | Comparison did not appear to be in PICO: The authors referred to the synergistic effect of oxycodone and rosuvastatin in the title and last paragraph of the introduction, but did not actually say anywhere explicitly that they were comparing oxycodone + rosuvastatin versus oxycodone alone; they also reported as an outcome the effective dose of oxycodone in each group and did not report anywhere methods for converting rosuvastatin to "effective oxycodone dose" so we have assumed the participants received oxycodone in both groups as how else could the authors report the effective oxy dose. |
| Garassino 2010 | Comparison not in PICO: Fixed‐dose oxycodone and increasing dose of pregabalin versus increasing dose of oxycodone and fixed‐dose pregabalin |
| Garassino 2011 | Comparison not in PICO: Fixed‐dose oxycodone and increasing dose of pregabalin versus increasing dose of oxycodone and fixed‐dose pregabalin |
| Garassino 2013 | Comparison not in PICO: Fixed‐dose oxycodone and increasing dose of pregabalin versus increasing dose of oxycodone and fixed‐dose pregabalin |
| George 2003 | Narrative review |
| Guo 2017 | Not RCT |
| Hanks 2002 | Narrative review |
| Hongmei 2013 | Not RCT |
| Huang 2015 | Did not appear to be RCT “According to the different analgesic drugs, they were divided into a study group and a control group with 34 cases each” ([translated], page 2022) |
| Igarashi 2015 | Letter to the editor |
| Katz 2008 | Letter to the editor |
| Kim 2015 | Comparison not in PICO: Transdermal fentanyl versus transdermal fentanyl + oral oxycodone |
| King 2011 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Koyyalagunta 2012 | Systematic review.Checked for relevant, previously unidentified studies for current review |
| Kummer 2011 | Particpants were healthy individuals, not people with cancer |
| LeBon 2009 | Systematic review.Checked for relevant, previously unidentified studies for current review |
| Leppert 2011 | Comparison not in PICO: Oxycodone‐naxolone versus oxycodone |
| Li 2008 | Comparison not in PICO: Oxycodone‐acetominophen versus oxycodone |
| Li 2010 | Comparison not in PICO: Oxycodone + gabapentin versus oxycodone |
| Liang 2021 | Participants randomised to titration with CR oxycodone or (IR?) morphine tablets for hours 0‐12 (with 10 mg morphine tablets available for breakthrough pain in both groups), then at 12 hours both intervention groups converted to CR oxycodone based on the previous 12 hours' consumption of CR oxycodone + morphine or morphine, respectively. The interventions in both randomised groups therefore were identical from hours 12 to 72 (study end), and the only outcome presented at 12 hours was number of breakthrough pain episodes in each group as well as mean NRS score without the associated SD or SE. All other outcome data presented at 24 hours or later. Data pertaining to the differential effect of oxycodone and morphine, respectively, therefore not available |
| Lin 2013 | Did not appear to be RCT. Only reference to allocation is as in abstract: "were divided into 40 cases in the observation group and 38 cases in the control group". No mention of anything to do with random in whole paper |
| Ma 2016 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Marineo 2012 | Not randomised enrolment |
| Meng 2008 | Published completely in Chinese. Translator confirmed that the study was not an RCT, but rather a retrospective review of cancer patient charts. |
| Moertel 1974 | Comparison not in PICO: Aspirin versus aspirin + codeine versus aspirin + oxycodone versus aspirin + pentazocine hydrochloride |
| Moksnes 2012 | Comparison not in PICO: Study compared two switching strategies from morphine/oxycodone to methadone |
| Mosley 2018 | Comparison not in PICO: Genotype‐guided or conventional pain management strategy |
| Nadstawek 2008 | Comparison not in PICO: Comparing different doses of naloxone in patients on oxycodone |
| Nalamachu 2013 | Not RCT |
| NCT01859715 | Population not in PICO: "Patients with pain and/or nausea are enrolled in the Emergency Department (ED). They are given either oxycodone, hydrocodone, or ondansetron at the discretion of the Emergency Department (ED) provider or the triage nurse by triage protocol. Detailed prescription, over the counter, herbal, supplement, and illicit drug ingestion histories are taken from the patient or their health care proxy. Serial visual analogue scales are captured prior to study drug administration then between 30 and 90 minutes following drug administration." "Subjects given either oxycodone 5 mg or hydrocodone/acetaminophen 5 mg/500 mg by ED provider decision or by triage nurse randomization." Unclear whether it was an RCT |
| NCT01885182 | Comparison not in PICO: Oxycodone‐naxolone versus oxycodone |
| Nunez Olarte 2008 | Narrative review |
| Oosten 2015 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Pan 2019 | Comparison not in PICO: Oxycodone group received treatment with 10 mg CR oxycodone and IR morphine as needed. If this was not effective, they got more IR morphine (not oxycodone) versus IR morphine + more IR morphine if needed in the comparison group. |
| Pang 2009 | Comparison not in PICO: fixed doses of oxycodone‐acetominophen versus background doses of oxycodone‐acetominophen plus additional dose for breakout pain versus controlled‐release oxycodone plus oxycodone‐acetominophen for breakout pain |
| Passik 2014 | Comparison/population not in PICO (N = 2 with cancer) |
| Reid 2006 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Rentz 2009 | Not RCT‐based analyses |
| Riley 2008 | Narrative review |
| Shi 2008 | Comparison not in PICO: Oxycodone‐acetominophen versus morphine |
| Shi 2018 | Comparison not in PICO: During titration, oxycodone group received treatment with 10 mg CR oxycodone and, if this was not effective, they got IR morphine (not oxycodone) versus IR morphine + more IR morphine if needed in the comparison group. After titration, all patients appeared to go on to receive CR oxycodone (with some uncertainty about doses/their calculations). |
| Sima 2010a | Comparison not in PICO: Oxycodone + aceteminophen versus placebo |
| Sima 2010b | Comparison not in PICO: Oxycodone + aceteminophen versus placebo |
| Sima 2012 | Comparison not in PICO: Oxycodone + paracetamol versus placebo |
| Stambaugh 1980a | Comparison not in PICO: Oxycodone + aspirin + caffeine + phenaticin (Percodan) versus zomepirac versus placebo |
| Stambaugh 1980b | Comparison not in PICO: Oxycodone‐acetominophen (tylox) versus oxycodone‐aspirin (percodan) |
| Stambaugh 1981 | Comparison not in PICO: Oxycodone + aspirin + caffeine + phenaticin (percodan) versus zomepirac versus placebo |
| Stambaugh 1985 | Comparison not in PICO: Ibuprofen vs. placebo |
| Stambaugh 1987 | Comparison not in PICO: Xorphanol versus oxycodone‐acetominophen versus placebo |
| Stambaugh 1990 | Comparison not in PICO: Flurbiprofen versus oxycodone‐acetominophen versus placebo |
| Stambaugh 1991 | Narrative review |
| Taeron 2002 | Narrative review |
| Tanaka 2017 | Not RCT |
| Wallace 2013 | Case report |
| Wang 2012 | Systematic review. Checked for relevant, previously unidentified studies for current review |
| Watanabe 2008 | N = 1 received oxycodone |
| Watanabe 2020 | Not RCT |
| Wei 2016 | Comparison not in PICO: Oxycodone versus acupoint catgut embedding |
| Wu 2009 | Comparison not in PICO: Oxycodone‐acetominophen versus tramadol |
| Wu 2015 | Comparison not in PICO: Morphine versus oxycodone + morphine |
| Xiong 2008 | Comparison not in PICO: Oxycodone‐acetominophen versus morphine |
| Xu 2008 | Not RCT |
| Yoshimoto 2018 | Both trials were single‐arm non‐randomised studies |
| Zhu 2019 | Exact opioid regimen used in the comparison groups unclear, but comparison did not appear to be in PICO: Oxycodone group received treatment with 10 mg CR oxycodone (if opioid‐naive) or with a dose calculated based on previous step‐II or III opioid use and IR morphine as needed. If this was not effective, they appeared to get more IR morphine (not oxycodone) versus IR morphine + more IR morphine if needed in the comparison group. We note that this was not clear from the paper as the results also included reporting of a "Comparison of consumption of oxycodone hydrochloride among the four groups [naive and tolerant oxycodone and morphine] during the maintenance period (W1 and W2), *P < 0.001, naive‐IR group vs. naive‐CR group. (Figure 3, page 7323) |
| Zou 2009 | Comparison not in PICO: Oxycodone + acetaminophen versus increased dose of existing opioid treatment |
CR: Controlled‐release ED: Emergency department IR: Immediate‐release NRS: Numerical rating scale PICO: P = participant, problem, or population; I = intervention; C = comparison, control, or comparator; O = outcome RCT: randomised controlled trial SD: Standard deviation SE: Standard error vs.: Versus
Characteristics of studies awaiting classification [ordered by study ID]
Aurilio 2009.
| Methods | Randomised parallel‐group controlled trial |
| Participants | 40 patients (13 men and 27 women), affected by severe chronic pain (mean NRS 8) |
| Interventions | Prolonged‐release (PR) oxycodone 10 mg/morning and 20 mg/evening versus PR oxycodone 20 mg twice a day Trial duration was 28 days with 5 visits, once a week. |
| Outcomes | Pain intensity measured by NRS Nausea, vomiting, somnolence, stypsis and itching Use of rescue medication (immediate‐release oral morphine 10 mg) |
| Notes | Emailed authors to ask for clarification re population on 23 May 2013 |
JapicCTI‐090789/090/091.
| Methods | JapicCTI‐090789: An open‐label study of intravenous (i.v.) S‐811717 (oxycodone hydrochloride solution for injection) in patients with cancer pain JapicCTI‐090790: An extension study of S‐811717 (oxycodone hydrochloride solution for injection) in patients with cancer pain JapicCTI‐090791: An open‐label study of subcutaneous injection (s.c.) S‐811717 (oxycodone hydrochloride solution for injection) in patients with cancer pain |
| Participants | Inpatients with pain associated with various cancers aged ≥ 20 years |
| Interventions | S‐811717 |
| Outcomes | ‐ To evaluate the efficacy and safety of S‐811717 in patients with pain caused by various cancers ‐ To determine the pharmacokinetics of S‐811717 and its metabolites. No other information available |
| Notes | Location: Japan Sponsors, collaborators, investigators: Shionogi & Co, Ltd., Research and Development No other information available |
JapicCTI‐111388.
| Methods | Described as "Multicenter Open Study" "To evaluate the safety and efficacy of TK‐641 when switching from existing opioid to TK‐641 for patients who have controlled cancer pain" |
| Participants |
Inclusion criteria: "1) Cancer patients who can be hospitalized for clinical trial 2) Patients who are 20‐74 years old at the time of informed consent 3) Patients who have been received cancer pain therapy with opioid etc." Exclusion criteria: "1) Patients who have any complications or history of cardiac conduction disturbance 2) Patients who have any complications or history of abnormality of respiratory function 3) Patients with any organic encephalopathies etc." |
| Interventions | Investigational material(s) described as "TK‐641", "Oxycodone Hydrochloride Hydrate" and "opium alkaloids preparations" |
| Outcomes | Primary outcome: "Efficacy and Safety" |
| Notes | Location: Japan Primary sponsor: TEIKOKU SEIYAKU CO., LTD. Clinical development department, rinsho@teiyaku.co.jp Study described as completed |
NCT00378937.
| Methods | An open, randomized, parallel group study in patients with cancer pain, to compare a two‐step analgesic ladder (non‐opioid to oxycodone) with conventional management using a three‐step approach |
| Participants | Disease characteristics: ‐ Diagnosis of cancer ‐ Requires regular step‐2 analgesia for the management of cancer‐related pain Patient characteristics: ‐ Aged ≥ 18 years ‐ Not pregnant or nursing ‐ Fertile patients must use effective contraception ‐ Must be able to take oral medication Must be willing and able to complete a daily patient assessment booklet (PAB) ‐ No history of the following conditions: Depression, personality disorders that may lead to self‐harm, admission to the hospital for psychiatric reasons, any other psychological disorder that, in the opinion of the investigator, would preclude study treatment ‐ Not at risk of additional CNS depressant effects due to study drugs ‐ No known history of alcohol or drug abuse or, in the opinion of the investigator, tendency towards drug abuse or addiction ‐ No current abuse of alcohol or drugs ‐ No known sensitivity to oxycodone hydrochloride or other opioids ‐ No history of a specific or allergic reaction to study drugs ‐ No contraindications as a result of adverse drug reaction or drug interactions of oxycodone or other opioid drugs ‐ No other condition that, in the opinion of the investigator, would make the patient unsuitable for study participation Prior concurrent therapy: ‐ More than 30 days since prior and no concurrent chemotherapy or radiotherapy ‐ At least 2 weeks since prior regular (i.e. 4 times per day) step‐2 analgesics ‐ More than 3 months since prior regular use of opioids, defined as having a regular prescription of an opioid medication ‐ Not planning to undergo cancer‐related surgery ‐ No other concurrent opioid‐based medication other than oxycodone hydrochloride capsules as escape medication (arm II) ‐ No concurrent participation in another clinical trial involving a new chemical entity |
| Interventions |
Arm 1: Participants receive an analgesic regimen, according to their level of pain, for up to 18 weeks ‐ Step 1: Participants in mild pain receive oral acetaminophen 4 times daily ‐ Step 2: Participants in mild‐to‐moderate pain receive oral codeine or oral dextropropoxyphene hydrochloride 4 times daily and oral acetaminophen 4 times daily. ‐ Step 3: Participants in moderate‐to‐severe pain receive oral morphine or oral oxycodone hydrochloride 6 times daily (every 4 hours) with or without a non‐opioid analgesic Participants may also receive an adjuvant drug (i.e. for side effects or for primary indication other than pain management that is analgesic in selected circumstances) versus Arm 2: Participants receive oral oxycodone hydrochloride twice daily for up to 18 weeks. Participants may receive a different opioid analgesic or analgesia or adjuvant medication as in arm I, if needed Participants in both arms may also receive additional medication for breakthrough pain. Participants complete a participant‐assessment booklet (PAB) daily which includes a Box‐Scale (BS)‐11 rating for average pain; questions regarding contact (e.g. telephone or visit) with healthcare professionals on that day; and information regarding the number of times escape medication is used. Quality of life and levels of cancer pain are assessed using the short form of the Brief Pain Inventory (BPI). After completion of study treatment, participants are followed at 4 weeks. |
| Outcomes |
Primary Outcome Measures: ‐ Percentage of time in assessment periods 1 and 2 (i.e. first 4 weeks) with a BS‐11 pain score of ≤ 4 (i.e. mild pain) Secondary Outcome Measures: ‐ Percentage of time in assessment periods 3 and 4 with a BS‐11 pain score of ≤ 4 ‐ Mean BS‐11 pain scores ‐ Time to reach stable pain control ‐ Mean escape medication use ‐ Quality of sleep ‐ Global assessment of pain relief with study drugs ‐ Mean pain intensity, pain interference, and pain relief scores as measured by the BPI ‐ Overall number of phone calls, home visits by a nurse, home visits by a doctor, and unscheduled visits to a healthcare provider, related to pain control or analgesic medication during study treatment |
| Notes | Location: US Sponsors and collaborators: University Hospitals Bristol NHS Trust Study chair: Geoff Hanks, University Hospitals Bristol NHS Trust Target enrolment: N = 30 Study dates: ? Other study ID numbers: CDR0000507650, CRUK‐ON/2003/1772, EU‐20640, EUDRACT‐2004‐004235‐66, NAPP‐CRUK‐ON/2003/1772 |
NCT00726830.
| Methods | Randomised, parallel‐group, open‐label controlled trial: A randomized comparison of oral methadone as a "first‐switch" opioid versus opioid switching between sustained‐release morphine and oxycodone for Oncology‐Hematology outpatients with pain management problems: the "Simply Rotate" study |
| Participants | Disease characteristics: ‐ Receiving ongoing care in the outpatient medical oncology setting ‐ Self‐reported pain (of any cause) for which long‐acting strong opioids (morphine or oxycodone) have been prescribed or administered oral morphine‐equivalent daily dose (MEDD) of existing opioid regimen (long‐acting or immediate‐release) 40 to 300 mg/day ‐ Worst pain score on a scale of 0 (no pain) to 10 (worst pain) of ≥ 5 for ≥ 1 week duration based on verbal self‐report or ≥ 1 persistently bothersome symptom attributed to an opioid side effect (e.g. fatigue, confusion, depressed level of consciousness, memory loss, personality change, anorexia, constipation, dehydration, nausea, vomiting, weight loss, pruritus, urticaria, impotence, reduced libido, and urinary retention or hesitancy), or both Patient characteristics: ‐ Aged ≥ 18 years ‐ None of the following conditions that could predispose the patient to prolonged QT interval‐associated tachycardia: serum potassium < 3.0 mg/dL; cocaine abuse within the past 3 months; family history of sudden death; advanced heart failure (ejection fraction < 40% or New York Heart Association (NYHA) class III or IV heart disease, or both ‐ No known or suspected cognitive impairment that could interfere with adherence to the medication plan or self‐report of symptoms and side effects ‐ Not pregnant or nursing ‐ Fertile patients must use effective contraception Prior concurrent therapy: ‐ See 'Disease characteristics' ‐ More than 4 weeks since prior radiotherapy or surgery for local control of cancer or pain palliation ‐ More than 60 days since prior use of the same long‐acting opioid (i.e. the new long‐acting opioid) that patient is switching to on the study ‐ More than 12 weeks since prior methadone therapy ‐ More than 3 days since prior and no concurrent transdermal fentanyl, oxymorphone, or buprenorphine ‐ Concurrent systemic anticancer therapy or bisphosphonates allowed provided therapy was initiated ≥ 4 weeks ago ‐ Concurrent tricyclic antidepressants, nonsteroidal anti‐inflammatory drugs (NSAIDs), anticonvulsants, or other adjuvant analgesics or psychostimulants allowed provided therapy was initiated ≥ 2 weeks ago; dose expected to remain stable until after the first week of opioid rotation on study ‐ No concurrent methadone maintenance therapy for opioid addiction ‐ No concurrent intrathecal infusion of analgesics ‐ No concurrent antiarrhythmic medications (e.g. amiodarone or quinidine) |
| Interventions | Opioid rotation to oral methadone (participants are switched from their current opioid medication (oxycodone or morphine) to methadone Participants receive oral methadone 2 to 3 times daily for 4 weeks) versus Opioid rotation to another long‐acting strong opioid (participants currently receiving oxycodone are switched to sustained‐release (SR) morphine. Participants currently receiving morphine are switched to SR oxycodone. Participants receive either oral SR morphine or oxycodone 2 to 3 times daily for 4 weeks) |
| Outcomes |
Primary outcome measures: ‐ Number of participants with at least a 3‐point reduction in pain score on the M.D. Anderson Symptom Inventory (MDASI) (time frame: 28 days) ‐ MDASI questionnaire completed on days 8, 15, and 22 after enrolment. The 'primary success' is defined as a 3‐point reduction in pain score on the MDASI. Scores from baseline and from four weeks later compared using the MDASI average pain intensity on a scale of 0 (no pain) to 10 (worst pain) Secondary outcome measures: ‐ Number of participants with 30% reduction in total summary score for the Individual Composite Drug Toxicity Score Items (time frame: 28 days) (designated as safety issue) |
| Notes | Location: US Sponsors and collaborators: M.D. Anderson Cancer Institute, National Cancer Institute Principal investigators: Michael J Fisch, MD, Anderson Cancer Center; James D Bearden, CCOP ‐ Upstate Carolina Target enrolment: N = ? Study dates: March 2009 to October 2010 Other study ID numbers: 2007‐0791, MDA‐2007‐0791, CDR0000598283 |
NCT01493635.
| Methods | Randomised, parallel‐group, open‐label controlled trial: An international, multicentre, open randomised parallel group trial comparing a two step approach for cancer pain relief with the standard three step approach of the WHO analgesic ladder in patients with cancer pain requiring step‐2 analgesia |
| Participants |
Inclusion Criteria: ‐ 18 years of age and over ‐ Patient has a cancer diagnosis (based on radiological, histological, cytological, or operative evidence). Those with haematological malignancies are eligible ‐ Cancer related pain ‐ which in the opinion of the clinician is caused by the presence of tumour or metastases ‐ Average pain score > 4, on a numerical rating scale from 0 to 10, requiring step‐2 analgesia (weak opioid) ‐ Patient is able to comply with trial procedures Exclusion criteria: ‐ Patients who have received radiotherapy in the previous 6 weeks or are planned to receive radiotherapy during the trial period where in either case, it is expected to affect pain during the trial period ‐ Pain due to surgery in the preceding 4 weeks ‐ Life expectancy less than two months (based on clinical impression) ‐ Patients with psychotic disorders or cognitive impairment ‐ Patients who have received regular doses (scheduled doses ‐ not as required dosing) of weak or strong opioids in the preceding two weeks ‐ Patients using immediate‐release opioids > 2 doses/24 hours, in the previous 24 hours |
| Interventions | Standard 3‐Step approach (participants will be managed according to the standard 3‐Step approach of the WHO analgesic ladder (Step‐1 ‐ step‐2 ‐ Step‐3)) versus 2‐Step approach (participants managed according to the WHO analgesic ladder bypassing Step‐2, i.e. participants will move from Step‐1 of the WHO analgesic ladder to Step‐3) Drugs to be used: oral morphine, oral oxycodone, oral tramadol, codeine |
| Outcomes |
Primary outcome measures: ‐ Time to achieving stable pain control, where stable pain control is defined as the first day of three consecutive days with average pain score less than or equal to 3 using scores from the Patient Diary and participant assessments. (Time frame: up to 20 days) Secondary outcome measures: ‐ Mean of daily average pain scores from the Patient Diary ‐ Mean of daily worst pain scores from the Patient Diary ‐ Percentage of days with average pain score ≥ 6 from the Patient Diary ‐ Percentage of days with worst pain score ≥ 6 from the Patient Diary ‐ Pain intensity, pain relief, and pain interference scores at day 10 and 20 from the Brief Pain Inventory ‐ Patient distress score at day 10 and 20 from the NCCN Distress Thermometer |
| Notes | Location: UK, Norway, Australia, Italy, Germany, Uganda, Spain Sponsors and collaborators: University of Edinburgh, NHS Lothian, Mundipharma (UK), St Olavs Hospital (Norway) Principal investigators: Marie Fallon, University of Edinburgh Target enrolment: N = 450 Study dates: March 2012 to December 2014 Other study ID numbers: 2012‐001578‐26, 11/SS/0079 |
NCT03439904.
| Methods | Randomised, parallel‐group, open‐label controlled trial: "This study is a prospective multicenter randomized controlled study to investigate the impact of pharmaceutical care on cancer pain treatment for opioid‐tolerant outpatients treated with sustained released morphine, oxycodone, and transdermal fentanyl." |
| Participants |
Inclusion Criteria: ‐ aged ≥ 18 years ‐ histologically confirmed solid tumour ‐ chronic cancer pain ‐ opioid‐tolerant ‐ expected overall survival ≥ 3 months ‐ Karnofsky performance score ≥ 50 ‐ willing and able to comply with the protocol Exclusion Criteria: Unclear as the record lists the same criteria as the inclusion criteria |
| Interventions | Group 1: "Patients will receive individualized pharmaceutical care in addition to usual medical care". "Patients receive pharmaceutical care including individualized evaluation and intervention of adherence, efficacy and safety in cancer pain treatment." Group 2: "Patients will receive usual medical care". |
| Outcomes |
Primary outcome measures: ‐ Change in medication adherence (using the Morisky Scale from baseline to 1 months, which has 4 questions and a total score range of 0‐4 with lower scores indicating higher adherence) (time frame: 1 month) Secondary outcome measures: ‐ Change in pain score (using a NRS ranging from 0‐10 with lower scores indicating less pain) (time frame: 1 month) ‐ Change in quality of life (using the EuroQol‐ 5 Dimension (EQ‐5D) with its 5 domains and VAS) (time frame: 1 month) ‐ Change in patients' knowledge of cancer pain and analgesics for cancer pain patients (using an investigator designed questionnaire with 16 items and score ranging from 0‐16 with higher scores indicating better knowledge) (time frame: 1 month) ‐ Incidence of adverse events (using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Event (CTCAE) Version 4.0) (time frame: 1 month) |
| Notes | Location: China Sponsors and collaborators: Zhejiang Cancer Hospital Principal investigators/responsible party: Ping Huang, Chief of Pharmacy, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, China, 310022. Tel: +86‐571‐88122118. Email: NCT03439904,%20ECCOPG‐003,%20Individualized%20Pharmaceutical‐care%20in%20Outpatients%20With%20Cancer%20Pain" type="EXTERNAL">huangping1841@zjcc.org.cn Target enrolment: N = 200 Study dates: 30 June 2018 to 30 June 2019 Other study ID numbers: ECCOPG‐003 |
Song 2009.
| Methods | It was unclear whether this was a retrospective study or a randomised controlled trial. Authors emailed on 14 January 2014 for clarification Design: 'Randomized', parallel‐group Year: 2006 to 2008 Country: China |
| Participants |
Participants: ‐ Oxycodone (commercial name Tai Lening): N = 42, 42 analysed, M/F = unclear, median (range) age = 55 (28 to 83) years. Primary tumours were: lung cancer (12) , breast cancer (5), liver cancer (6), gastric cancer (4), nasopharyngeal carcinoma (3), colorectal cancer (3), oesophageal cancer (3), lymphoma (2), osteosarcoma (2), chordoma (1), pancreatic cancer (1). ‐ Morphine sulfate controlled‐release (MS contin): N = 45, 45 analysed, 27 males and 18 females; median (range) age = 53 (30 to 76) years. Primary tumours were: Lung cancer (14), breast cancer (6), liver cancer (6); gastric cancer (6), oesophageal cancer (3), pancreatic cancer (2), nasopharyngeal (2), colorectal cancer (2), non‐Hodgkin's lymphoma (2), ovarian cancer (2). Inclusion criteria: "87 patients who were diagnosed with malignant tumour based on histopathology and cytology, with moderate to severe cancer pain and who did not respond to non‐steroidal anti‐inflammatory drugs and weak opioid analgesics" Exclusion criteria: Not reported |
| Interventions |
Oxycodone arm ‐ Drug: Oxycodone + 1 tablet (each containing oxycodone 5 mg, acetaminophen 325 mg) ‐ Dose and dosing: every 6 h (2 oxycodone tables has equal titration dose with oral morphine 30 to 40 mg) ‐ Formulation: Controlled‐release ‐ Route of administration: Oral ‐ Length of treatment: 5 days ‐ Titration schedule: Not clear but seemed they have same dose increased as the contin group ‐ Rescue medication: During the treatment, if participants have short term unsatisfactory treatment efficacy or have sudden intensified pain, then short‐acting morphine injection was administrated. The participants were considered treatment failure if the pain relief was not relieved until the observation period had ended or the limit dose was reached. ‐ Other medication: Unclear Comparison arm ‐ Drug: Morphine sulfate (MS contin) ‐ Dose and dosing: 20 mg/day as the first dose ‐ Formulation: Controlled‐release ‐ Route of administration: Oral ‐ Length of treatment: 5 days ‐ Titration schedule: MS Contin group with 20 mg/day as the first dose, if the pain could be relieved, then continued using the same dose as maintenance treatment. If the pain was not relieved after 24 hr, then increased the dose until a satisfactory pain relief, or till reached the maximum dose (the maximum dose = 270 mg/day) ‐ Rescue medication: During the treatment, if participants have short‐term unsatisfactory treatment efficacy or have sudden intensified pain, then short‐acting morphine injection was administrated. The participants were considered treatment failure if the pain relief was not relieved until the observation period had ended or the limit dose was reached. ‐ Other medication: Unclear For both groups, if the participants had intolerable adverse reactions when increasing the dose, the drugs could be discontinued at any time, then the participants were observed for 30 days and then treatment efficacy was evaluated. The participant was also considered as treatment failure if the treatment had to be stopped due to intolerable adverse events. |
| Outcomes | ‐ Pain Intensity (PI) and pain relief: the WHO linear Visual Analog Scale VAS was used; the degree of pain was graded using by dividing a line into 10 segments: 0 = no pain, 1 to 3 as mild, 4 to 7 as moderate, severe pain as 8 to 9, 10 = extreme pain. Complete remission (CR): completely no pain after treatment, with a pain score of 0 on a 0 to 10 VAS. Partial remission (PR): pain reduced significantly, there was no sleep disturbance, have normal daily life, the pain reduced 4 or more grades in the segments.(note: the authors did not say scores lower than 4, they said reduced 4 or more, CR can be translated as complete relief). Mild remission (NC): certain degree of pain relief, but require enhanced pain control, participants had sleep disturbances, VAS score reduced 1 to 3 grades in the 0 to 10 VAS line. (note, NC normally means no changes). Treatment failure (PD): no pain relief compared to baseline. (note ‐ PD normally means progression of disease). The authors considered participants who were CR or PR as “treatment was effective”. ‐ Adverse reactions. Participants were observed for all kinds of adverse reactions: constipation, nausea, vomiting, dizziness, drowsiness, skin rash or itching, abdominal discomfort etc. |
| Notes | Study free of commercial funding? Unclear Were the participants adequately titrated? Unclear, possibly? Groups comparable at baseline? Unclear, probably if properly randomised ITT analyses undertaken? Yes |
Zhang 2016.
| Methods |
Design: Randomised, parallel‐group (patient allocation described as random, but no further information included) Year: 2014 to 2015 Country: China |
| Participants |
Participants: ‐ Oxycodone rectal: N = 44 (no further details reported) ‐ Oxycodone oral: N = 44 (no further details reported) Inclusion criteria: Patients with severe cancer pain (no further details reported) Exclusion criteria: Not reported |
| Interventions |
Oxycodone arm ‐ Drug: Oxycodone (oxycontin) ‐ Dose and dosing: Not reported ‐ Formulation: Not reported ‐ Route of administration: Rectal ‐ Length of treatment: Not reported ‐ Titration schedule: Not reported. ‐ Rescue medication: Not reported ‐ Other medication: Not reported Comparison arm ‐ Drug: Oxycodone (oxycontin) ‐ Dose and dosing: Not reported ‐ Formulation: Not reported ‐ Route of administration: Oral ‐ Length of treatment: Not reported ‐ Titration schedule: Not reported ‐ Rescue medication: Not reported ‐ Other medication: Not reported |
| Outcomes | ‐ "efficacy" (no further details reported) ‐ "adverse effects" (no further details reported |
| Notes | Study free of commercial funding? Unclear Were the participants adequately titrated? Unclear Groups comparable at baseline? Unclear ITT analyses undertaken? Unclear The study was published as an abstract only and the results were reported in the following way "The effect of the treatment group [rectal] was significantly better than that of the control group [oral]. In addition, [and] the adverse effects was less than that of the control group." |
BPI: Brief Pain Inventory BS(‐11): Box Scale‐11 CNS: Central nervous system CR: Controlled‐release CTCAE: Common Terminology Criteria for Adverse Event EQ‐5D: EuroQol‐ 5 Dimension ITT: Intention‐to‐treat MDASI: M.D. Anderson Symptom Inventory MEDD: Morphine‐equivalent daily dose M/F: Male/female NC: No changes NCCN: National Comprehensive Cancer Network NCI: National Cancer Institute NHS: National Health Service NRS: numerical rating scale NSAID: Non‐steroidal anti‐inflammatory drug NYHA: New York Heart Association PAB: Participant‐assessment booklet PD: Progression of disease PI: Pain intensity PR: prolonged‐release QT: This is a measure of the Q and T waves. SR: sustained‐release VAS: visual analogue scale WHO: World Health Organization
Characteristics of ongoing studies [ordered by study ID]
2008‐002273‐12.
| Study name | Long term opioid administration in oncologic chronic pain: open label, prospective study on efficacy, safety and pharmacogenetic factors |
| Methods | Randomised, parallel‐group, open controlled trial |
| Participants |
Inclusion criteria: ‐ age > 18 years ‐ oncologic, chronic, neuropathic or nociceptive peripheral pain Exclusion criteria: ‐ abuse history ‐ opioid analgesic use history ‐ opioid allergies |
| Interventions |
Morphine (oral solution) versus morphine (oral tablet) versus oxycodone (oral tablet) versus fentanyl (transdermal patch) versus buprenorphine (transdermal patch) versus hydromorphone (prolonged‐release oral tablet) |
| Outcomes | Pain reduction at least 40% in VAS scale |
| Starting date | Not reported |
| Contact information | Location: Italy Sponsors: Ospedale Policlinico S. Matteo Principal investigators: Not reported |
| Notes | Target enrolment: N = 320 Study completion date: ? but of 3‐year duration Other study ID numbers: None reported, but is it the same as NCT00916890 below? |
2009‐013118‐28.
| Study name | Bukkaalinen fentanyyli syöpäpotilaiden toimenpidekivun hoidossa ("The buccal fentanyl in cancer pain management measure") |
| Methods | Randomised, cross‐over (open or blind?) controlled trial |
| Participants |
Inclusion criteria: ‐ cancer metastatic to the bone ‐ beginning radiotherapy to bone metastases (?) Exclusion criteria: ‐ severe hepatic, renal or cardiac dysfunction ‐ uncontrolled or rapidly increasing pain ‐ dry mouth ‐ oral mucositis or stomatitis ‐ pregnancy or breastfeeding ‐ impaired cognitive performance ‐ increased intracranial pressure ‐ drug abuse or history of drug use within the previous 5 years, or of use of CYP3A4 inhibition drug(s?) (translated from Finnish) |
| Interventions |
Fentanyl (buccal) versus oxycodone (oral) (oxynorm) |
| Outcomes | Pain relief and speed of effect for fentanyl compared to oxycodone, radiation therapy‐related acute, short‐term pain relief (translated from Finnish), side effects |
| Starting date | Not reported |
| Contact information | Location: Finland Sponsors: Tarja Heiskanen Principal investigators: Not reported |
| Notes | Target enrolment: N = ? Study completion date: ? Other study ID numbers: EUCTR2009‐013118‐28‐FI |
ChiCTR1800014268.
| Study name | Effect and efficacy of hydrochloride oxycodone controlled‐release tablets with dose titration at 12h for cancer pain |
| Methods | Randomised parallel‐group controlled trial |
| Participants |
Inclusion criteria: ‐ Aged ≥ 18 years ‐ Cancer (clinical diagnosis and/or pathological diagnosis) ‐ Mean pain intensity (NRS) during the past 24 hours ≥ 4 points ‐ "The patients who are intolerable to opioids (including the patients without a long‐term use of opioids as the daily basic medications); therefore, there is no manifestation of obvious tolerance either. Defined by FDA, patients who are considered opioid‐tolerant are those who have been taking at least 60 mg of morphine daily, or at least 30 mg of oral oxycodone daily for a week or longer)" ‐ Patients who can orally take drugs and are judged by the investigator to be suitable to use the oxycodone sustained‐release tablet simplified titration ‐ "The patients can rule out the influence of anti‐tumor treatment (radiotherapy, chemotherapy, and targeted therapy etc.) on the analgesic effects during the analgesic drug dose adjustment" ‐ Using release morphine analgesia to relive breakthrough pain at least 1 time within first 12h ‐ Cognitively able to understand the guidance of medication regimen ‐ Informed consent Exclusion criteria: ‐ Pregnancy or lactation ‐ Known allergy to any other component in oxycodone ‐ Pain unrelated to the tumour or the pain with unclear causes (e.g. osteoarthritis‐induced pain, low back pain) ‐ Emergency treatment due to tumour pain ‐ Complicated tumour pain or intractable tumour pain ‐ Refractory constipation ‐ Monoamine oxidase inhibitors (MAOIs) or this type of drugs within 2 weeks ‐ Potential gastrointestinal diseases and/or the risk of surgical treatment, which may induce gastrointestinal stenosis, blind loop or gastrointestinal tract obstruction ‐ Unstable concomitant diseases or important organ dysfunction ‐ Infection, abscess or fever symptoms ‐ Liver and renal functional abnormalities (e.g. creatinine value is not less than 2 times of the upper limit of normal values or ALT or AST is not less than 2.5 times of the upper limit of normal values (as for the patients with hepatic metastasis, it is not less than 5 times of the upper limit of normal values)) ‐ Anti‐epileptic or antiarrhythmic drugs ‐ Contraindications to oxycodone or morphine, adverse drug reactions (ADR) and drug interactions as stated in the package insert or investigators brochure ‐ History of drug or alcohol abuse ‐ Participation in another compound clinical trial study within 1 month before the study ‐ Potential change of the combined medications (excluding those used to treat the adverse reactions of opioids) during the study ‐ Too unsuitable to participate in this study due to any reason other than the inclusion criteria and exclusion criteria according to the judgement of the investigator |
| Interventions | Oxycodone: Hydrochloride oxycodone CR with dose titration at 12 h Oxycodone: Hydrochloride oxycodone CR with dose titration at 24 h |
| Outcomes |
Primary outcomes: ‐ Proportion of patients with pain remission 24 h, 48 h and 72 h after the titration Secondary outcomes: ‐ Quality of life ‐ Safety ‐ Rescue analgesic medications within first 24 h,48 h and 72 h ‐ Satisfaction of treatment (investigator‐ and patient‐evaluated) |
| Starting date | 1 January 2018 |
| Contact information | Location: China Sponsors, collaborators, investigators: Zhang Yiping and Song Zhengbo, Zhejiang Cancer Hospital, 1 Banshan Road East, Gongshu District, Hangzhou, Zhejiang, China. Tel: +86 13750881678 and +86 13857153345. Email: lmy19841002@163.com and songzb@zjcc.org.cn Principal investigator: Not reported |
| Notes | Target enrolment: 206 Study completion date: "2018‐12‐01" or "2018‐12‐09" Other study ID numbers: None reported |
ChiCTR1800017461.
| Study name | Efficacy and safety of different titration regimens for oxycodone hydrochloride sustained‐release tablet in the treatment of cancer pain |
| Methods | Randomised parallel‐group controlled trial |
| Participants |
Inclusion criteria: ‐ Age ≥ 18 years ‐ Pathologically or cytologically diagnosed tumours ‐ Inpatient with cancer pain ‐ Pain scores (NRS) on admission ≥ 4 points ‐ Conscious, with a certain level of education, able to understand doctor's guidance on dosing regimen ‐ Informed consent Exclusion criteria: ‐ Pregnancy or lactation ‐ Allergy to oxycodone or any other ingredients in the study drug ‐ Presence of non‐cancer pain or unexplained pain such as osteoarthritis pain, low back pain ‐ Acute cancer pain ‐ Intractable constipation ‐ Monoamine oxidase inhibitors (MAOIs) or this type of drugs within the past 2 weeks ‐ Potential risks for gastrointestinal disorders and/or surgical treatment, possibly leading to gastrointestinal stenosis, blind loop or gastrointestinal obstruction ‐ Unstable comorbidities; or existence of vital organ dysfunction ‐ Ongoing infection, abscess or fever symptoms ‐ Liver and kidney dysfunction, e.g. creatinine ≥ 2 X ULN or ALT or AST ≥ 2.5 X ULN (which can be relaxed to ≥ 5 X ULN in patients with liver metastases) or liver function Child Class C ‐ Antiepileptic or arrhythmic drugs ‐ Oxycodone or morphine contraindications, adverse drug reactions (ADRs) and drug interactions as described in product package insert or investigators brochure ‐ History of drug or alcohol abuse ‐ Participation in clinical trial of another compound within 1 month before this study ‐ Patients who may change their drug combination (except for treatment of adverse opioid reactions) during the study period |
| Interventions | Oxycodone: Hydrochloride oxycodone CR?, assess and adjust the dose every 12 hours Oxycodone: Hydrochloride oxycodone CR?, assess and adjust the dose every 24 hours |
| Outcomes |
Primary outcomes: ‐ Pain relief rate at 24 h (NRS scores at 24 h decreased to ≤ 3 points) Secondary outcomes: ‐ Pain relief rate at 48 h and 72 h ‐ Dosage of oxycodone hydrochloride sustained‐release tablet at 24 h, 48 h and 72 h ‐ Number of breakthrough pain (episodes?) ‐ Quality of life (measured by EORTC QLQ‐30) |
| Starting date | "2018‐08‐01" |
| Contact information | Location: China Sponsors, collaborators, investigators: Yong Liu, Xuzhou Central Hospital, 199 Jiefang Road South, Xuzhou, Jiangsu, China. Tel: +86 15295641568. Email: liuyong20180724@163.com Principal investigator: Not reported |
| Notes | Target enrolment: 128 Study completion date: "2020‐08‐01" or "2020‐02‐01" Other study ID numbers: None reported |
ChiCTR1900022566.
| Study name | Comparison of oxycodone versus morphine in the treatment of patients with severe cancer pain: a randomized controlled trial |
| Methods | Randomised parallel‐group controlled trial |
| Participants | Inclusion criteria: 1. Aged 18 to 75 years 2. Patients with pathological or cytological diagnosis of tumour 3. Pain intensity (NRS) score for severe pain 4. Be conscious; be able to understand the doctor's medication plan guidance 5. Patients with opioid intolerance Exclusion criteria 1. Pregnancy or lactation 2. Pain crisis or other oncology emergency 3. Contraindications to strong opioids 4. Patients with severe liver and kidney function (ALT/AST/creatinine/urea nitrogen is 3 times higher than the upper limit of normal) 5. History of drug or alcohol abuse |
| Interventions | 30 mg oxycodone initial titration treatment versus Immediate‐release morphine titration |
| Outcomes | Pain measured every 24 hours for 7 days on numerical rating scale |
| Starting date | "2019‐04‐18" |
| Contact information | Location: China Sponsors, collaborators, investigators: Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Hankou, Wuhan, Hubei, China Principal investigator: Mei Qi (tel: +86 15871708675; email: borismq@hotmail.com) and/or Hu Guangyuan (tel: +86 13098834328; email: huguangyuan2018@sohu.com) |
| Notes | Target enrolment: 252 Study completion date: "2020‐04‐30" Other study ID numbers: None reported |
ChiCTR2000037845.
| Study name | A multicenter randomized controlled clinical trial of hydromorphone injection PCA versus oxycodone sustained release tablets in the treatment of moderate and severe cancer pain |
| Methods | Randomised, parallel‐group, controlled trial |
| Participants |
Inclusion criteria: Consenting patients with cancer pain, aged ≥ 18 years, NRS cancer pain score ≥ 4 in the past 24 hours; who did not receive radiotherapy during the observation period; patients who needed chemotherapy, long‐term hormone therapy, targeted therapy or bisphosphonate therapy who had received stable anti‐tumour therapy before randomisation; able to complete the survey form either alone or with their nursing staff; without mental illness who can correctly understand and cooperate with the medication guidance of medical staff; no history of allergy to narcotic drugs or opiate addiction; ECOG‐PS ≤ 2; who had not participated in any drug trial within 1 month before the trial Exclusion criteria: Patients with non‐cancer or postoperative pain, paralytic intestinal obstruction, uncontrolled brain metastases, allergic diseases and allergic constitution those with similar drug structure in the study [sic], abnormal and clinically significant laboratory results, such as creatinine >= 2 times of the upper limit of normal value, ALT or AST >= 2.5 times of the upper limit of normal value (liver metastasis or primary liver cancer >= 5 times of the upper limit of normal value), or child C grade of liver function; pregnant, lactating or planning pregnancy within one month of trial (including male participants), opioid addition, cognitive impairment or severe skin oedema, or poor peripheral and subcutaneous circulation, or where it is not suitable to implant subcutaneous analgesia pump |
| Interventions | Oxycodone oral titration versus subcutaneous PCA titration of hydromorphone |
| Outcomes |
Primary outcomes ‐ Time to successful titration Secondary outcomes ‐ Number of episodes of breakthrough pain within 24 and 48 hours ‐ Average pain scores at 24 hours and 48 hours ‐ Number of patients with successful titration at 24 hours and 48 hours ‐ Quality of Life assessed with the Chinese version of the Edmonton Symptom Assessment System |
| Starting date | 6 September 2020 |
| Contact information | Location: China Sponsors, collaborators, investigators: Xiaoguang Xiao (Tel: +86 15629387266; email: 1425128151@qq.com) and Yuan Chen (Tel: +86 27‐83663407; email: chenyuan008@163.com), Tongji Hospital Affiliated to Tongji Medical College, Huazhong University of Science and Technology; 1095 Jiefang Avenue, Wuhan, Hubei, China Principal investigator: Not reported https://trialsearch.who.int/Trial2.aspx?TrialID=ChiCTR2000037845; http://www.chictr.org.cn/showproj.aspx?proj=60437 |
| Notes | Target enrolment: 105 in each group Study completion date: 31 December 2021 Other study ID numbers: None reported |
ChiCTR2100042972.
| Study name | Comparative study of sufentanil self‐controlled analgesic pump and oxycodone hydrochloride controlled‐release tablets in the treatment of moderate and severe cancer pain |
| Methods | Randomised clinical trial |
| Participants |
Inclusion criteria: ‐ Tissue or pathology diagnosed as cancer ‐ Aged 18‐80 years ‐ ECOG score ≤ 2 ‐ Expected survival period ≥ 2 months ‐ Cancer‐related pain of 4‐10 on NRS ‐ opioid‐naive ‐ normal cognitive ability to record the relief of cancer pain ‐ no intestinal obstruction, dysphagia and other special circumstances where oral analgesics cannot be taken Exclusion criteria: Patients with mental system disease, severe liver and kidney dysfunction, multiple organ failure and cachexia, etc. or during pregnancy and lactation |
| Interventions | Oxycodone hydrochloride controlled‐release versus sufentanil self‐controlled analgesic pump |
| Outcomes |
Primary outcome ‐ Pain relief Secondary outcomes ‐ Number of breakthrough pain episodes ‐ Quality of life ‐ Adverse events |
| Starting date | 1 March 2019 |
| Contact information | Location: China Sponsors, investigator: Xiao Yan (Tel: +86 13669808150; email: xiaoyan199409@163.com) and Yao Weirong (tel: +86 13907002901; email: 13907002901@126.com) Jiangxi Provincial People's Hospital, 152 Aiguo Road, Donghu District, Nanchang, Jiangxi, China Principal investigator: Not reported http://www.chictr.org.cn/showproj.aspx?proj=64416 |
| Notes | Target enrolment: 26 in each group Study completion date: 31 August 2020 Other study ID numbers: None reported |
Elsayem 2010.
| Study name | No study name reported beyond 'TPS324' which could be the abstract number |
| Methods | Randomised, parallel‐group, open‐label trial |
| Participants | 300 cancer patients in the outpatient community setting with inadequate pain control and/or intolerable opioid‐related side effects and prescribed either sustained‐release morphine or oxycodone, with an oral morphine equivalent daily dose between 40 mg and 300 mg |
| Interventions | Rotation to either oral methadone or oral sustained‐release morphine or oxycodone with the new opioid dose determined using study‐specific equianalgesic tables Patients also receive immediate‐release opioids for breakthrough pain and supportive measures for side effects, and patients have their opioids titrated according to study protocol. |
| Outcomes | Pain intensity/relief and adverse events measured at enrolment and then weekly for a total of 4 weeks using validated tools that include: M. D. Anderson Symptom Inventory (MDASI), Composite Drug Toxicity Score, and Revised Edmonton Staging System (rESS) for Cancer Pain |
| Starting date | 2010? |
| Contact information | |
| Notes | The authors noted no significant financial relationships to disclose, but study was NCI‐funded. |
IRCT20201202049575N1.
| Study name | Comparison of the effect of morphine and oxycodone in relieving pain in patients with bone metastasis pain |
| Methods | Randomised clinical trial, with parallel groups, double‐blind, phase 3 on 32 patients, block method and online software www.sealedenvelope.com were used for randomisation. |
| Participants |
Inclusion criteria: Patients aged 18‐60 years with malignancy whose bone metastasis has been confirmed by bone scan and are candidates for analgesia. Pain intensity (VAS) < 6 Exclusion criteria: Patients with history of diabetes and kidney failure, bone fracture hypotension, bradypnoea |
| Interventions | Two 5 mg oxycodone tablets (Shafa, Iran) orally and simultaneously, and only once at the beginning of the intervention and an injectable placebo of 5 mL, once at the beginning of the intervention versus Morphine sulfate (Darupakhsh, Iran) 5 mg intravenously once at the beginning of the intervention and two placebo tablets orally once at the beginning of the intervention |
| Outcomes |
Primary outcomes ‐ Pain (measured on VAS before the intervention, at 30 mins, 2 and 6 hours after the intervention) Secondary outcomes: None reported |
| Starting date | 21 December 2020 |
| Contact information | Location: Iran Sponsors: Alireza Kamali, Arak University of Medical Sciences, Payambar‐e‐azam Complex, Basij Sq., Sardasht, Arak, Markazi 3848176341; Tel: +98 86 3417 3639; email: research@arakmu.ac.ir Recruitment: Mahdi Farahani, Valiasr hospital, Valiasr sq., Arak, Markazi 3814957558; tel: +98 86 3417 3505, +98 86 3222 2003 ; email: m.mahdi.f.13732324@gmail.com, pr_valieasr@arakmu.ac.ir Principal investigator: Not reported https://trialsearch.who.int/Trial2.aspx?TrialID=IRCT20201202049575N1http://en.irct.ir/trial/52795 |
| Notes | Target enrolment: 16 in each group Study completion date: 20 January 2021 Other study ID numbers: None reported |
Matsouka 2017.
| Study name | Selection of opioids for cancer‐related pain using a biomarker: a randomized, multi‐institutional, open‐label trial (RELIEF study) |
| Methods | Randomised (parallel‐group), multi‐institutional, open‐label controlled trial |
| Participants |
Inclusion criteria: ‐ Patients with advanced malignant tumours ‐ Non‐daily use of opioids ‐ Cancer pain targeted for daily treatment with opioids, NSAIDs or acetaminophen ‐ NRS ≥ 3 (average over 24 h) ‐ Opioid treatment‐naive within 30 h ‐ No chemotherapy, radiotherapy, or bisphosphonate administration newly started within 2 weeks ‐ Written informed consent Exclusion criteria: ‐ Patients with chronic renal failure (glomerular filtration rate, 30 mL/min) ‐ Patients with severe hepatic or respiratory failure ‐ Patients deemed ineligible for the study by the study coordinator or a collaborative investigator (e.g. neuropathic pain or predominant spontaneous pain only, and history of opioid/drug abuse or alcoholism) |
| Interventions | All patients will be genotyped for SNPs with a Taqman SNP Genotyping Assay (Life Technologies) into a GG group and a non‐GG group based on the COMT rs4680 SNP. Each group will be randomised to morphine or oxycodone treatment:
Morphine: IR morphine 5 mg (Tmax about 1 hour), doses subject to titration: Dose titration to decrease pain by ≥ 33% on the NRS pain scale and to reduce NRS to ≤ 3, then conversion to CR morphine Oxycodone: IR oxycodone 2.5 mg (Tmax about 2 hours), doses subject to titration: Dose titration to decrease pain by ≥ 33% on the NRS pain scale and to reduce NRS to ≤ 3, then conversion to CR oxycodone |
| Outcomes |
Primary outcomes ‐ "proportion of subjects requiring high‐dose opioids calculated from use of the immediate‐release preparation on day 0 in a parallel group comparison" (page 2‐3) Secondary outcomes ‐ Anxiety and depression (measured by the Hospital Anxiety and Depression Scale) ‐ Quality of life (measured by the European Organization for Research and Treatment of Cancer QLQ‐C15‐PAL for score) ‐ Pain (measured by the Short‐Form McGill Pain Questionnaire 2 score for pain characterisation; and the Pain Catastrophizing Scale) ‐ Adverse events (e.g. constipation, somnolence, nausea, pruritus, ischuria; measured by the Common Terminology Criteria for Adverse Events version 4.0) |
| Starting date | November 2014 |
| Contact information | Location: Japan Sponsors, collaborators, investigators: K Nakagawa and H Matsuoka, 377‐2, Ohno‐higashi, Kinki University, Faculty of Medicine Department of Medical Oncology; Osakasayama City, Osaka, 589‐8511, Japan. Tel: 072‐366‐0221. Email: nakagawa@med.kindai.ac.jp and matsuoka_h@med.kindai.ac.jp |
| Notes | Target enrolment: N = 140 Study completion date: Not reported Other study ID numbers: UMIN000015579 |
NCT00916890.
| Study name | Chronic administration of opioids in cancer chronic pain: an open prospective study on efficacy, safety and pharmacogenetic factors influence |
| Methods | Randomised (parallel‐group), single‐blind (outcome‐assessor) controlled trial |
| Participants |
Inclusion criteria: ‐ Adult oncologic patients (≥ 18 years old) ‐ Chronic peripheral neuropathic or nociceptive pain, or both ‐ Written informed consent Exclusion criteria: ‐ Paediatric patients ‐ Mentally impaired patients ‐ Substance abuse disorder ‐ Opioid allergy ‐ History of opioids use or addiction ‐ Severe immunodeficiency, severe renal impairment, severe liver disease ‐ Cachectic state ‐ HIV‐positive patients |
| Interventions |
Morphine (after a titration phase with fast‐release oral morphine, once the optimal dosage (no side effects and less than two rescue doses per day) is reached, an equipotent dose of oral sustained‐release morphine will be randomly assigned to a participant) versus oxycodone (after a titration phase with fast‐release oral morphine, once the optimal dosage (no side effects and less than two rescue doses per day) is reached, an equipotent dose of oral extended‐release oxycodone will be randomly assigned to a participant) versus fentanyl (after a titration phase with fast‐release oral morphine, once the optimal dosage (no side effects and less than two rescue doses per day) is reached, an equipotent dosage of transdermal fentanyl will be randomly assigned to a participant) versus buprenorphine (after a titration phase with fast‐release oral morphine, once the optimal dosage (no side effects and less than two rescue doses per day) is reached, an equipotent dosage of transdermal buprenorphine will be randomly assigned to a participant) |
| Outcomes |
Primary outcome measures: ‐ To identify the drug with the best clinical‐pharmacological safety‐efficacy profile among the four opioids: oral extended‐release morphine, oral extended‐release oxycodone, transdermal fentanyl and transdermal buprenorphine (time frame: 15 days after randomisation (reduction of at least 40% of median daily pain, on a NRS)) "We will define a treatment effective if it will produce a mean reduction of NRS values at least of 40% [than] of basal values. Among all effective treatments, we will identify the best as the one that will have a reduction of NRS to a value of 4 or less in 90% of participants compared to the 70% of the others treatments. To evaluate pharmacological safety the plasma concentrations of the drugs and their metabolites will be measured. We will branch participants population in 3 groups to evaluate the correlation between clinical‐pharmacological response and genetics (responder, partially and not responder)." Secondary outcome measures: ‐ Pharmacokinetic of opioids and of their metabolites during long‐term administration; correlation between specific genotypes and clinical response or the clinical/pharmacological susceptibility to side effects on administration of a specific opioid (time frame: 6 months (each participant will be followed for 6 months after enrolment with clinical and pharmacological evaluations once a month [for] [and if] inefficacy, tolerance or side effects)) ‐ Comparison of plasma levels of opioids and of their metabolites in 'responder' participants (clinical effectiveness without side effects), 'partial responder' participants (clinical effectiveness without side effects but taking not more than 2 rescue doses per day), and in 'non‐responder' participants (3 groups: clinical inefficacy, side effects, tolerance or opioid induced hyperalgesia). Evaluation of the correlation between the polymorphisms studied and clinical response; the frequency of allelic variants of interest will be compared in 'responder', 'partial responder' and 'non‐responder'. |
| Starting date | February 2009 |
| Contact information | Location: Italy Sponsors, collaborators, investigators: IRCCS Policlinico S. Matteo, University of Pavia, Italy Principal investigator: Massimo Allegri, IRCCS Foundation Policlinico "San Matteo", Pavia, Italy; e‐mail: NCT00916890, PT‐SM‐1‐Op‐Cancer, Prospective Study About Clinical and Pharmacogenetic Safety of Opioid Use for Chronic Pain" type="EXTERNAL">m.allegri@smatteo.pv.it, Tel: 00390382502627 |
| Notes | Target enrolment: N = 320 Study completion date: December 2015 Other study ID numbers: PT‐SM‐1‐Op‐Cancer |
NCT01165281.
| Study name | A randomized, double‐blind, active controlled, optimal dose titration, multicenter study to evaluate the safety and efficacy of oral JNS024 extended‐release (ER) in Japanese and Korean subjects with moderate to severe chronic malignant tumor related cancer pain |
| Methods | Randomised (parallel‐group), double‐blind (participant, caregiver, investigator) controlled trial |
| Participants |
Inclusion criteria: ‐ Aged ≥ 20 years ‐ Documented clinical diagnosis of any type of cancer ‐ Diagnosis of chronic malignant tumour‐related cancer pain with an average score for pain intensity in the past 24 hours of ≥ 4 on the 11‐point numerical rating scale (NRS) on the day of randomisation (day ‐1) ‐ Have not received treatment with opioid analgesics within 28 days before screening (note: codeine phosphate (≤ 60 mg/d) or dihydrocodeine phosphate (≤ 30 mg/d) for antitussive use are allowed) ‐ Dissatisfied with pain relief by the current treatment and for whom the investigator or designee judges that treatment with opioid analgesics is required Exclusion criteria: ‐ Have complicated with uncontrolled or clinically significant arrhythmia ‐ Have previous or concurrent presence of any disease which may develop increased intracranial pressure, disturbance of consciousness, lethargy, or respiratory problems such as traumatic encephalopathy with cerebral contusion, intracranial haematoma, disturbance of consciousness, brain tumour, cerebral infarction, transient ischaemic attack, epilepsy or convulsive diseases ‐ Have history of alcohol or drug abuse ‐ Have any disease for which opioids are contraindicated such as serious respiratory depression of serious chronic obstructive pulmonary disease, bronchial asthma attack, cardiac failure secondary to chronic pulmonary disease, paralytic ileus, status epileptics, tetanus, strychnine poisoning, acute alcohol poisoning, hypersensitivity to opium alkaloid, haemorrhagic colitis, or bacterial diarrhoea |
| Interventions |
R331333 ((referred to as JNS024 ER or CG5503) one 25 mg to 200 mg capsules twice daily for 4 weeks) versus Oxycodone CR (one 5 mg to 40 mg capsules twice daily for 4 weeks) |
| Outcomes |
Primary outcome measures: ‐ The average pain intensity score using an 11‐point numerical rating scale (NRS) (time frame: change from baseline to the last 3 days of study drug administration) Secondary outcome measures: ‐ The Patient Global Impression of Change (PGIC) (time frame: at the end of the 4‐week double‐blind treatment phase) ‐ The duration of rescue medication (time frame: during the 4‐week double‐blind treatment phase) ‐ The concentration of JNS024 in blood samples from participants (time frame: protocol‐specified time points during weeks 1, 2, and 4) ‐ The proportion of participants responding to treatment, including at least 30% and 50%, based on the per cent change from baseline using an 11‐point numerical rating score (NRS) (time frame: at week 4 of the double‐blind treatment phase on an 11‐point NRS) ‐ Adverse events and findings from clinical laboratory tests, physical examinations, vital signs measurements, and ECG measurements reported (time frame: from time of screening (days ‐7 to ‐1) to post‐treatment (week 5) or time of early termination from study) |
| Starting date | August 2012 |
| Contact information | Location: Japan, Republic of Korea Sponsors, collaborators, investigators, study director: Janssen Research & Development, L.L.C. Clinical Trial (no other contact information reported) |
| Notes | Target enrolment: N = 343 Study completion date: August 2012 Other study ID numbers: CR017188, JNS024ER‐KAJ‐C02 |
NCT01675622.
| Study name | A comparative study of immediate‐release oxycodone capsules versus immediate‐release morphine tablets for the treatment of Chinese patients with cancer pain |
| Methods | Randomised, parallel‐group, double (triple?)‐blind (participant, caregiver, investigator, outcome assessor) controlled trial |
| Participants |
Inclusion criteria: ‐ Patients of either sex aged 18 to 80 years inclusive, with cancers of all types ‐ Patients with moderate to severe cancer pain, whose pain intensity NRS ≥ 4 ‐ Patients who can understand and are able to complete NRS and BPI assessment ‐ Patients who have given written informed consent to participate in the study Exclusion criteria: ‐ Patients who are pregnant, or lactating ‐ Patients who are unable to manage their pain effectively with opioids ‐ Patient who need ≥ 120 mg morphine or equivalent for treatment of pain at time of study entry ‐ Patients who are receiving chemotherapy, or still under the responsive period of chemotherapy (patients who are at the interval period of chemotherapy can be enrolled into study. That is to say, patients who completed chemotherapy for more than 2 weeks can be enrolled, or patients who have completed chemotherapy for at least one week could be enrolled at the discretion of the investigator) ‐ Patients who have received radiotherapy for bony metastasis, patients receiving radiotherapy within the 4‐week period before study entry (patient receiving radiotherapy for area other than pain area can be enrolled), or patients who were scheduled to receive radiotherapy for pain area during study period ‐ Patients are receiving or should receive anticonvulsive drugs or antidepressant drugs considered by investigator for the treatment of neuropathy pain ‐ Patients are receiving or should receive any analgesic other than study medicine, including NSAIDs ‐ Patients with other unstable disease, or with dysfunction of important organ ‐ Patients with an ongoing infection, abscess or fever ‐ Patient with serious abnormal liver or renal function (ALT, AST, creatinine, urea nitrogen) which is higher than 3 times upper limit ‐ Paralytic or mechanical ileus ‐ Persistent asthma, chronic obstructive diseases, and cor pulmonary ‐ Intracranial neoplasms, and intracranial hypertension with central respiratory depression risk ‐ Monoamine oxidase inhibitors (MAOIs) or same type drugs have been administered in last 2 weeks ‐ Patients who are currently taking active treatment for epilepsy or arrhythmias ‐ Patients with known sensitivity or record of specific or allergic reaction to oxycodone or morphine ‐ Patients excluded by the contraindications, adverse drug reaction (ADRs) and drug interactions of oxycodone or morphine as detailed in the data sheet, summary of product characteristics or investigator's brochure ‐ Patients with a history of drug or alcohol abuse ‐ Patients who participated in another clinical research study involving a new chemical entity within one month prior to study entry ‐ Patients whose concomitant medication is likely to be changed within the study period, with the exception of treatment for opioid side effects ‐ Patients who, in the opinion of the investigator, are unsuitable to participate in the study for any other reason not mentioned in the inclusion and exclusion criteria |
| Interventions |
Oxycodone (5 mg, l0 mg and 20 mg capsules every 6 h, 5 to 8 days) versus morphine (tablets 10 mg and 20 mg, oral every 4 to 6 hours) |
| Outcomes |
Primary outcome measures: ‐ NRS (Numerical Rating Scale) score (time frame: 5 to 8 days). To compare the average for decrease of NRS score after double‐blind treatment between the two treatment groups ‐ The average dose of study medicine used during double‐blind treatment period (time frame: 5 to 8 days). To compare the average dose of study medicine used during double‐blind treatment period between the two treatment groups Secondary outcome measures: ‐ BPI (Brief pain inventory) (time frame: 19 to 22 days). To compare BPI score at baseline, after completion of double‐blind treatment and open‐label treatment to baseline between the two treatment groups ‐ Times and frequency of breakthrough pain and the total dose of rescue medicine for breakthrough pain (time frame: 19 to 22 days). To compare the times and frequency of breakthrough pain and the total dose of rescue medicine for breakthrough pain during double‐blind phase between the two treatment groups ‐ Participant assessments of satisfaction for pain management (time frame: 19 to 22 days). To compare participant assessments of satisfaction for pain management between the two treatment groups at the end of double‐blind treatment and the open‐label treatment period ‐ Average time for titration (time frame: 1 to 3 days). To compare the average time for titration between the two treatment groups |
| Starting date | December 2010 |
| Contact information | Location: China Sponsors, collaborators: Mundipharma Principal investigator: Shiying Yu, Wuhan Tong Ji Hospital |
| Notes | Target enrolment: N = 240 Study completion date: July 2012 Other study ID numbers: OXYC10‐CN‐303 |
NCT02084355.
| Study name | Efficacy and safety of opioid rotation compared with opioid dose escalation in patients with moderate to severe cancer pain ‐ open label, randomized, prospective study |
| Methods | Open‐label, randomised, prospective study |
| Participants |
Inclusion criteria: ‐ age > 18 years ‐ patients who are being treated with one of strong opioids including oral oxycodone, oral hydromorphone, or fentanyl patch with range from 60 mg to 200 mg of oral morphine equivalent daily dose (MEDD) ‐ moderate to severe cancer pain (numeric rating scale more than 3) at screening ‐ patients without uncontrolled adverse effects associated with currently applied opioid Exclusion criteria: ‐ previous opioid rotation ‐ unable to take oral medication ‐ life expectancy less than a month ‐ newly started chemotherapy or radiotherapy within past 2 weeks of screening ‐ serum aspartate aminotransferase, alanine aminotransferase, or alkaline phosphatase > 2.5 times upper normal limit ‐ serum total bilirubin or creatinine > 1.5 times of upper normal limit |
| Interventions |
Opioid rotation: Participants who are randomised to opioid rotation are treated with strong opioid other than currently used strong opioid (reduce the dose by 25% to 50% to allow for incomplete cross‐tolerance between different opioids): Oral oxycodone: convert to oral hydromorphone or fentanyl patch Oral hydromorphone: convert to oral oxycodone or fentanyl patch Fentanyl patch: convert to oral oxycodone or oral hydromorphone versus opioid dose escalation: Participants who are randomised to opioid dose escalation will be treated for cancer pain by escalation dose of same strong opioid: Oral oxycodone: maintain oral oxycodone and titrate the dose Oral hydromorphone: maintain oral hydromorphone and titrate the dose Fentanyl patch: maintain fentanyl patch and titrate the dose |
| Outcomes |
Primary outcome measures: The rate of successful pain control defined as a 30% or 2‐point reduction in the numeric rating scale (time frame: 18 months) (designated as safety issue: yes) |
| Starting date | April 2014 |
| Contact information | Location: Republic of Korea Sponsors, collaborators: Gyeongsang National University Hospital Principal investigator/contact: Se‐Il Go, M.D., tel@ +82 55 750 9454 ext 9454, e‐mail: gose1@hanmail.net |
| Notes | Target enrolment: N = 136 Study completion date: January 2016 Other study ID numbers: GNUH‐2013‐07‐014 |
NCT03024515.
| Study name | A pilot randomized open‐labelled study comparing a structured titration method of immediate‐ and sustained‐release oxycodone versus opioids titration of investigators' choice in advanced cancer patients in Hong Kong |
| Methods | Randomised, parallel‐group, open‐label controlled trial |
| Participants |
Inclusion Criteria: ‐ opioid‐naïve adults with moderate to severe cancer pain ‐ previous treatment with NSAIDs or weak opioids and currently with poor pain control, intention to be treated with strong opioids ‐ patients who need long‐term administration of hormone or targeting therapy or bisphosphonates therapy: "treatments will maintain from 3 days prior to randomization to end of the study as much as possible". ‐ patients who need radiotherapy or chemotherapy: "these therapies should be conducted during maintaining phase and completed as assuring as possible before last follow‐up." Exclusion Criteria: ‐ pure neuropathic pain or unexplained pain, pain that only occurs during moving; acute pain ‐ patients for whom oral administration is not applicable ‐ any disease that may lead to respiration inhibition ‐ monoamine oxidase inhibitor (MAOI) one week before randomisation ‐ abnormal results, with obvious clinical significance, from lab testing, such as the creatinine is ≥ 2‐fold of upper limit of normal value, or ALT or AST is ≥ 2‐fold of upper limit of normal value, or liver function is Child C grade ‐ potential gastrointestinal diseases or the risk of surgical operation, which may lead to gastrointestinal stenosis, blind loop or gastrointestinal obstruction ‐ prior exposure to prolonged‐release oxycodone tablets or other strong opioids drugs before study |
| Interventions |
"Structured titration method with predefined titration steps with the use of oxycodone immediate‐release and oxycodone sustained‐release preparations" versus "Standard Practice Arm with opioids titration of physicians' choice" |
| Outcomes |
Primary outcome measures: Time to stabilisation of pain control during titration phase Secondary outcome measures: Time of analgesic onset, number of breakthrough medications required during the titration phase, quality of life (EORTC QLQ‐C15‐PAL), a descriptive assessment of opioids prescription practice amongst practicing oncologists in Hong Kong, safety profile and adverse events, descriptive summary of the use of opioids and titration by practicing clinicians |
| Starting date | 23 August 2018 |
| Contact information | Location: Hong Kong Sponsors, collaborators: Chinese University of Hong Kong Principal investigator/contact: Herbert Loong and Jane Koh, Department of Clinical Oncology, Prince of Wales Hospital, Hong Kong, emails: h_loong@clo.cuhk.edu.hk and jane@clo.cuhk.edu.hk |
| Notes | Target enrolment: N = 60 Study completion date: Estimated to be December 2021 Other study ID numbers: None reported |
NCT03176199.
| Study name | A study to compare the titration efficacy and safety of control‐released oxycodone and immediate‐released oxycodone in patients with moderate to severe cancer pain |
| Methods | Randomised, parallel‐group, open‐label controlled trial |
| Participants |
Inclusion Criteria: ‐ Cancer patients aged ≥ 20 years ‐ Patients with background cancer pain ≥ NRS 4 during previous 24 hours, or patients who receive ≥ 3 times/day for breakthrough pain medication management ‐ ECOG ≤ 2 ‐ Opioid‐naive patients who have not received any strong opioid for at least one month prior to the index treatments, currently with poor pain control and intended for treatment with strong opioids for pain relief (the FDA identifies opioid‐naive as patients who have not been receiving the following treatment for ≥ 1 week: 1) ≥ 60 mg of morphine daily, 2) ≥ 25 mcg transdermal fentanyl/hour, 3) ≥ 8 mg of oral hydromorphone daily, or 4) an equianalgesic dose of another opioid) ‐ No radiotherapy within 7 days prior to randomisation and during study ‐ Patients needing chemotherapy, long‐term administration of hormone, targeted therapy, or bisphosphonates therapy should undergo a stable anti‐tumour therapy prior to randomisation ‐ Patients or his/her caregivers able to fill out the diary and questionnaire forms Exclusion Criteria: ‐ Non‐cancer pain or unexplained pain ‐ Postop pain ‐ Ineligible for oral administration ‐ Severe constipation defined by CTCAE grade 3 and above ‐ Disease that may easily lead to respiratory depression ‐ Monoamine oxidase inhibitor (MAOI) one week before randomisation ‐ Abnormal lab results, with obvious clinical significance, such as creatinine ≥ 2 times upper limit of normal value, or ALT or AST ≥ 2.5 times upper limit of normal value (≥ 5 times, to the patients with liver metastasis or primary liver cancer), or liver function of Child C grade ‐ Potential risk for surgical operation, which may lead to gastrointestinal stenosis, blind loop or gastrointestinal obstruction; or patient unable to effectively absorb oral medication through gastrointestinal tract ‐ Drug or alcohol abuse ‐ Moderate to severe psychiatric problems ‐ Hypersensitivity to oxycodone ‐ Pregnancy or lactation ‐ Clinically unstable or have a life expectancy < 3 months making completion of the trial unlikely |
| Interventions | Oxycodone 1: CR oxycodone every 12 hours (oxycontin), initial daily dose is 20 mg + immediate‐released oxycodone for PRN Oxycodone 2: IR oxycodone every 6 hours (oxynorm), initial daily dose is 20mg + immediate‐released oxycodone for PRN |
| Outcomes |
Primary outcomes: ‐ "The change from baseline of NRS pain score and the daily number of breakthrough pain" episodes Secondary outcomes: ‐ Proprortion of patients in each titration cycle ‐ Proportion of patients who switched/discontinued therapy due to serious adverse events or lack of pain control ‐ Total opioid taken within 24 hrs daily from baseline to day 14 ‐ "Mean daily NRS score of patients from baseline to day 14" ‐ "The total daily rescue dose taken (immediate‐released oxycodone capsule) for treatment of breakthrough pain among patients from baseline to day 14" ‐ Rate of adverse events and physical examination status ‐ "To evaluate the change from baseline in questionnaire [time frame: up to 14 days]" |
| Starting date | September 2016 |
| Contact information | Location: Taiwan Investigaros, sponsors, collaborators: Chih‐Jen Hung, MSc; Taichung Veterans General Hospital. Taiwan; Taiwan Mundipharma Pharmaceuticals Ltd. |
| Notes | Target enrolment: N = 30 Study completion date: December 2018 Other study ID numbers: OXY15‐TW‐401 |
NCT04808531.
| Study name | NanaBis™ an oro‐buccal administered delta9‐tetrahydrocannabinol (d9‐THC) & cannabidiol (CBD) medicine for the management of bone pain from metastatic cancers |
| Methods | Randomised parallel‐group clinical trial |
| Participants |
Inclusion criteria: Participants aged 18‐70 years with metastatic bone pain from a cancer diagnosis as the only major cause of pain, pathologically confirmed (blood, imaging) metastatic bone cancer, meeting the International Classification of Diseases‐10 (ICD‐10) codes for pain management criteria (i.e. bone cancer pain). During the screening period, the participant is on stable opioid pain management and pain severity (NPRS) ≤ 8 with a maximum variation of ± 1, pain Detect score > 18, and willing and able to provide informed consent and follow study procedures Exclusion criteria: History of epilepsy or recurrent seizures; moderate to severe medical conditions such as severe hepatic; cardiovascular or renal impairment; or psychiatric disorders (i.e. unstable schizophrenia, recent drug‐induced psychosis, severe mood disorders), assessed at the medical screen; substance abuse disorder such as nicotine or alcohol, or other illicit or prescription drug dependence (e.g. opioid dependence), or methadone or buprenorphine treatment for opioid dependence; pregnancy, lactation or planning to become pregnant; identified concerns by the nursing/medical team relevant to the safe storage of medications (i.e., NanaBis™ or standard medical therapy) and participant who may not be available for follow‐up (i.e. planned or expected travel or other) |
| Interventions | Oxycodone + placebo: Spray placebo + oxycodone CR spray. Placebo is a nanoparticle water‐soluble solution without cannabinoids containing a small amount of hemp seed oil (for fragrance purposes only) as defined by Australian ODC (https://www.odc.gov.au/hemp‐products). One dose is equivalent to 2 actuations of the pump delivering 280 µL volume. Oxycodone controlled‐release (CR) used as a comparator will be oxycontin tablets 10 mg‐70 mg po bd. versus NanaBis™ + tablet placebo: NanaBis™ is a nanoparticle water‐soluble equimolar solution of d9‐THC and CBD. One dose is equivalent to 2 actuations of the pump delivering 280 µL volume containing 2.5 mg d9‐THC and 2.5 mg CBD. The dose administered will be 2‐3 doses per 4 hours unless asleep. versus Double placebo arm: spray placebo + tablet placebo spray. Placebo is a nanoparticle water‐soluble solution without cannabinoids containing a small amount of hemp seed oil (for fragrance purposes only) as defined by Australian Office of Drug Control (ODC) (https://www.odc.gov.au/hemp‐products). One dose is equivalent to 2 actuations of the pump delivering 280 µL volume. Tablet placebo will be identical to the oxycontin tablets. |
| Outcomes |
Primary outcome ‐ Responder rate Secondary outcomes ‐ Health‐related quality of life ‐ pain DETECT score ‐ Adverse events ‐ Extension request rate (of NanaBis treatment) |
| Starting date | May 2021 |
| Contact information | Location: USA, Australia Sponsors, investigator: Dr. Michael Lyon (Tel: +1 604 777 5500; email: doctorlyon@me.com), Prof. Luis Vitetta (Tel: +61 8188 0311 ext 106email: luis_vitetta@medlab.co), Medlab Clinical; George Clinical Pty Ltd; WriteSource Medical Pty Ltd Principal investigator: Not reported https://clinicaltrials.gov/show/NCT04808531 |
| Notes | Target enrolment: 360 Study completion date: January 2024 Other study ID numbers: MDC‐NB‐P3‐01 |
UMIN000011756.
| Study name | Randomized study of fentanyl citrate versus oxycodone hydrochloride hydrate in patients with unresectable advanced pancreatic cancer (FRONTIER) |
| Methods | Randomised, single‐arm (?), phase III, open trial |
| Participants |
Inclusion criteria: ‐ Aged 20 to < 100 years ‐ unresectable advanced pancreatic cancer ‐ ≥ 15 to 25 mg oxycodone hydrochloride hydrate per day required for cancer pain Exclusion criteria: ‐ Serious liver, kidney, cardiac disorders ‐ pulmonary impairment ‐ nervous system and psychic disorders |
| Interventions | Oxycodone hydrochloride hydrate: 10 mg every 12 hours, versus Transdermal fentanyl citrate: 1 mg once a day |
| Outcomes |
Primary outcome measures: The rates of gastrointestinal disorders events in four weeks Secondary outcome measures: Quality of life, rates of opioid rotation, pain score, time until stable pain control, overall survival time, adverse events |
| Starting date | 27 March 2014 |
| Contact information | Location: Japan Sponsors, collaborators: National Cancer Center Hospital East; Welfare labor science research cost (MHLW(Japan)) Principal investigator/contact: Minori Odanaka, Clinical Trial Support Office, National Cancer Center Hospital, Tsukiji 5‐1‐1, Chuo‐ku, Tokyo 104‐0045, Japan Tel: +81‐3‐3547‐5201, e‐mails: minochant23@yahoo.co.jp; modanaka@ncc.go.jp |
| Notes | Target enrolment: N = 80 Study completion date: Not reported Other study ID numbers: None reported |
ADR: Adverse drug reaction ALT: alanine transaminase AST: aspartate aminotransferase bd: Bis die (twice daily) BPI: Brief Pain Inventory cbd: Cannabidiol CR: controlled‐release CTCAE: Common Terminology Criteria for Adverse Events ECG: electrocardiogram ECOG(‐PS): Eastern Cooperative Oncology Group EORTC QLQ‐C15‐PAL: European Organization for Research and Treatment of Cancer Quality of Life Questionnaire‐Core15_Palliative EORTC QLQ‐30: European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 FDA: Food and drug administration FRONTIER: Randomised study of fentanyl citrate versus oxycodone hydrochloride hydrate in patients with unresectable advanced pancreatic cancer (FRONTIER) GG: GG is a genotype h: Hour HIV: Human immunodeficiency virus ICD‐10: International Classification of Diseases 10th Revision IR: Immediate‐release MAOI: Monoamine oxidase inhibitors MDASI: M. D. Anderson Symptom Inventory NPRS: Numerical pain rating scale NRS: numerical rating scale NSAID: Non‐steroidal anti‐inflammatory drug ODC: Office of Drug Control PCA: Patient‐controlled analgesia PGIC: Patient Global Impression of Change po: Oral administration PRN: Pro re nata (as needed) QLQ‐CIS‐PAL: Cancer Quality of Life Questionnaire‐Core15_Palliative RELIEF: Selection of opioids for cancer‐related pain using a biomarker: a randomized, multi‐institutional, open‐label trial (RELIEF study) rESS: revised Edmonton Staging System SNP: Single‐nucleotide polymorphism thc: Tetrahydrocannabinol ULN: Upper limit of normal VAS: visual analogue scale
Differences between protocol and review
2015: In the risk of bias assessments, we also included an item that captured whether data were available for both time periods in cross‐over trials, in order to make explicit this potential source of bias. We reported treatment acceptability as a proxy for quality of life as this outcome was rarely reported.
2021: We performed sensitivity analyses to assess the impact of the inclusion of Chinese trials published in Chinese journals for the comparison of CR oxycodone versus CR morphine. Please see Excluded studies for a detailed rationale for these additional analyses.
Contributions of authors
For the original review
MSH and MIB conceived and designed the review and wrote the protocol.
SA devised and undertook the search strategy.
MSH, NB, and JSH screened the search results and performed the data extraction and risk of bias assessment of the included studies.
MSH devised and performed the analysis strategy, and wrote the first draft of the full review.
MIB interpreted the results and wrote the 'Authors conclusions' section.
All the authors approved the final version of the review.
For the 2015 update
SA undertook the search strategy.
MSH and NB screened the search results and performed the data extraction and risk of bias assessment of the included studies.
MSH devised and performed the analysis strategy, and wrote the first draft of the updated review.
MIB interpreted the results and wrote the 'Authors conclusions' section.
All the authors approved the final version of the review.
For the 2021 update
SA undertook the search strategy. YC conducted the exploratory searches of the Chinese databases.
MSH, AJP, JSH, YC and NB screened the search results and performed the data extraction and risk of bias assessment of the included studies.
MSH devised and performed the analysis strategy, and wrote the first draft of the updated review.
MIB and AJP interpreted the results and wrote the 'Authors conclusions' section.
All the authors approved the final version of the review.
Sources of support
Internal sources
No sources of support provided
External sources
No sources of support provided
Declarations of interest
MSH: none known.
MIB: none known.
SA: none known.
NB: none known.
JSH: none known. JSH is a Network Associate Editor for the Cochrane MOSS Network. PaPaS CRG is a member of the MOSS Network. JSH had no involvement in the editorial process and/or management of the peer review of this manuscript.
AJP: none known. AJP is a Palliative Care Specialty Trainee (ST5), Leeds Teaching Hospitals NHS Foundation Trust.
YC: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Beaver 1978a {published data only}
- Beaver WT, Wallenstein SL, Rogers A, Houde RW. Analgesic studies of codeine and oxycodone in patients with cancer. I. Comparisons of oral with intramuscular codeine and of oral with intramuscular oxycodone. Journal of Pharmacology and Experimental Therapeutics 1978;207(1):92-100. [PubMed] [Google Scholar]
Beaver 1978b {published data only}
- Beaver WT, Wallenstein SL, Rogers A, Houde RW. Analgesic studies of codeine and oxycodone in patients with cancer. II. Comparisons of intramuscular oxycodone with intramuscular morphine and codeine. Journal of Pharmacology and Experimental Therapeutics 1978;207(1):101-8. [PubMed] [Google Scholar]
Bruera 1998 {published data only}
- Bruera E, Belzile M, Pituskin E, Fainsinger R, Darke A, Harsanyi Z, et al. Randomized, double-blind, cross-over trial comparing safety and efficacy of oral controlled-release oxycodone with controlled-release morphine in patients with cancer pain. Journal of Clinical Oncology 1998;16(10):3222-9. [DOI] [PubMed] [Google Scholar]
- Comerford T, Bruera E. Efficacy of controlled-release oxycodone. Journal of Clinical Oncology 1999;17:738. [PubMed] [Google Scholar]
Cao 2015 {published data only}
- Cao Q, Wang LX. The analysis of the two different opioid efficacy in the elderly cancer patients and its safety. Journal of Practical Medical Techniques 2015;22(4):360-1. [Google Scholar]
Corli 2016 {published data only}
- Corli O, Floriani I, Roberto A, Montanari M, Galli F, Greco MT, et al, CERP Study of Pain Group. Are strong opioids equally effective and safe in the treatment of chronic cancer pain? A multicenter randomized phase IV 'real life' trial on the variability of response to opioids. Annals of Oncology 2016;27:1107-15. [DOI] [PubMed] [Google Scholar]
- Corli O, Santucci C, Corsi N, Radrezza S, Galli F, Bosetti C. The burden of opioid adverse events and the influence on cancer patients’ symptomatology. Journal of Pain and Symptom Management 2019;57(5):899-908. [DOI] [PubMed] [Google Scholar]
- NCT01809106. RCT comparing the analgesic efficacy of 4 therapeutic strategies based on 4 different major opioids (fentanyl, oxycodone, buprenorphine vs morphine) in cancer patients with moderate/severe pain, at the moment of starting 3rd step of WHO analgesic ladder. clinicaltrials.gov/show/NCT01809106 (date first received 12 March 2013).
Gabrail 2004 {published data only}
- Gabrail NY, Dvergsten C, Ahdieh H. Establishing the dosage equivalency of oxymorphone extended release and oxycodone controlled release in patients with cancer pain: a randomized controlled study. Current Medical Research and Opinion 2004;20(6):911-8. [DOI] [PubMed] [Google Scholar]
- Gabrail NY, Dvergsten C, Ma T, Frailey A, Ahdieh H. Oxymorphone extended-release (ER) provides safe, effective, and rapid analgesia during opioid radiation: results of a randomized, double-blind, crossover, comparative study with oxycodone controlled-release (CR) [abstract]. Proceedings of the American Society of Clinical Oncology 2003:737. [Google Scholar]
Gao 2012 {published data only}
- Gao W, Gu A, Zhu M, Yao L. Oxycodone hydrochloride sustained-release sustained tablets and morphine sulfate sustained-release tablets for advanced malignancy severe pain. Journal of Basic and Clinical Oncology 2012;25(6):524-6. [Google Scholar]
Hagen 1997 {published data only}
- Hagen NA, Babul N. Comparative clinical efficacy and safety of a novel controlled-release oxycodone formulation and controlled-release hydromorphone in the treatment of cancer pain. Cancer 1997;79(7):1428-37. [PubMed] [Google Scholar]
Heiskanen 1997 {published data only}
- Heiskanen T, Kalso E. Controlled-release oxycodone and morphine in cancer related pain. Pain 1997;73(1):37-45. [DOI] [PubMed] [Google Scholar]
- Heiskanen TE, Ruismaki PM, Seppala TA, Kalso EA. Morphine or oxycodone in cancer pain? Acta Oncologica 2000;39(8):941-7. [DOI] [PubMed] [Google Scholar]
Imanaka 2013 {published data only}
- Imanaka K, Tominaga Y, Etropolski M, Van Hove I, Ohsaka M, Wanibe M, et al. Efficacy and safety of oral tapentadol extended release in Japanese and Korean patients with moderate to severe, chronic malignant tumor-related pain. Current Medical Research and Opinion 2013;29:1399-409. [DOI] [PubMed] [Google Scholar]
Inoue 2017 {published data only}
- Inoue S, Saito Y, Tsuneto S, Aruga E, Ide A, Kakurai Y. A randomized, double-blind study of hydromorphone hydrochloride extended-release tablets versus oxycodone hydrochloride extended-release tablets for cancer pain: efficacy and safety in Japanese cancer patients (EXHEAL: a Phase III study of EXtended-release HydromorphonE for cAncer pain reLief). Journal of Pain Research 2017;10:1953-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Inoue 2018 {published data only}
- Inoue S, Saito Y, Tsuneto S, Aruga E, Takahashi H, Uemori M. A randomized, double-blind, non-inferiority study of hydromorphone hydrochloride immediate-release tablets versus oxycodone hydrochloride immediate-release powder for cancer pain: efficacy and safety in Japanese cancer patients. Japanese Journal of Cinical Oncology 2018;48(6):542-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kalso 1990 {published data only}
- Kalso E, Vainio A, Mattila MJ, Rosenberg PH, Seppala T. Morphine and oxycodone in the management of cancer pain: plasma levels determined by chemical and radioreceptor assays. Pharmacology & Toxicology 1990;67(4):322-8. [DOI] [PubMed] [Google Scholar]
- Kalso E, Vainio A. Morphine and oxycodone hydrochloride in the management of cancer pain. Clinical Pharmacology and Therapeutics 1990;47(5):639-46. [DOI] [PubMed] [Google Scholar]
Kaplan 1998 {published data only}
- Kaplan R, Parris WC, Citron ML, Zhukovsky D, Reder RF, Buckley BJ, et al. Comparison of controlled-release and immediate-release oxycodone tablets in patients with cancer pain. Journal of Clinical Oncology 1998;16(10):3230-7. [DOI] [PubMed] [Google Scholar]
Lauretti 2003 {published data only}
- Lauretti GR, Oliveira GM, Pereira NL. Comparison of sustained-release morphine with sustained-release oxycodone in advanced cancer patients. British Journal of Cancer 2003;89(11):2027-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2017 {published data only}
- Lee KH, Kang JH, Oh HS, Choi MK, Shim BY, Eum YJ, et al. Intravenous oxycodone versus intravenous morphine in cancer pain: a randomized, open-label, parallel-group, active-control study. Pain Research & Management 2017;2017:1-11. [DOI: 10.1155/2017/9741729] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT02660229. An interventional study for patients with cancer pain to evaluate the efficacy and safety of OxyNorm® compared to morphine sulfate through IV continuous infusion. clinicaltrials.gov/show/NCT02660229 (date received 21 January 2016).
- Oh HS, Lee KH, Kang JH, Choi MK, Shim BY, Eum YJ, at al. Efficacy and safety of oxynorm compared to morphine sulfate administering through iv continuous infusion in moderate-severe cancer-related pain.. Supportive Care in Cancer 2017;25 (2 Supplement 1):S90. [Google Scholar]
Leow 1995 {published data only}
- Leow KP, Cramond T, Smith MT. Pharmacokinetics and pharmacodynamics of oxycodone when given intravenously and rectally to adult patients with cancer pain. Anesthesia and Analgesia 1995;80(2):296-302. [DOI] [PubMed] [Google Scholar]
Li 2013 {published data only}
- Li XQ, Lv J, Jin Z. Clinical observation of oxycodone hydrochloride controlled release tablets and morphine sulfate sustained release tablets in the treatment of severe pain in advanced cancer. Journal of Chinese Practical Diagnosis and Therapy 2013;27(12):1216-7. [Google Scholar]
Liu 2021 {published data only}
- Liu MZ, Ma J, Li JD, Sun J, Zhou H, Guan S, et al. A comparison of the clinical effectiveness between low-dose strong opioids and non-steroidal anti-inflammatory drugs in the treatment of mild cancer pain: a randomized trial. Journal of Pain Research 2021;14:3411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lux 2014 {published data only}
- Lux EA, Janecki M, Maritz MA. Clinical evaluation of the first oxycodone once daily prolonged release tablet in moderate to severe chronic pain: a randomized, double-blind, multicenter, cross-over, non-inferiority study to investigate efficacy and safety in comparison with an established oxycodone twice daily prolonged release tablet. Current Medical Research and Opinion 2014;30(11):2365-75. [DOI] [PubMed] [Google Scholar]
Mercadante 2010 {published data only}
- Mercadante S, Tirelli W, David F, Arcara C, Fulfaro F, Casuccio A, et al. Morphine versus oxycodone in pancreatic cancer pain: a randomized controlled study. Clinical Journal of Pain 2010;26(9):794-7. [DOI] [PubMed] [Google Scholar]
Mucci‐LoRusso 1998 {published data only}
- Mucci-LoRusso P, Berman BS, Silberstein PT, Citron ML, Bressler L, Weinstein SM, et al. Controlled-release oxycodone compared with controlled-release morphine in the treatment of cancer pain: a randomized, double-blind, parallel-group study. European Journal of Pain 1998;2(3):239-49. [DOI] [PubMed] [Google Scholar]
Nosek 2017 {published data only}
- Leppert W, Nosek K. Comparison of the quality of life of cancer patients with pain treated with oral controlled-release morphine and oxycodone and transdermal buprenorphine and fentanyl. Current Pharmaceutical Design 2019;25(30):3216-24. [DOI] [PubMed] [Google Scholar]
- Nosek K, Leppert W, Nosek H, Wordliczek J, Onichimowski D. A comparison of oral controlled-release morphine and oxycodone with transdermal formulations of buprenorphine and fentanyl in the treatment of severe pain in cancer patients. Drug Design Development and Therapy 2017;11:2409-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parris 1998 {published data only}
- Parris WC, Johnson BW Jr, Croghan MK, Moore MR, Khojasteh A, Reder RF, et al. The use of controlled-release oxycodone for the treatment of chronic cancer pain: a randomized, double-blind study. Journal of Pain and Symptom Management 1998;16(4):205-11. [DOI] [PubMed] [Google Scholar]
Ren 2012 {published data only}
- Ren LP. The pain relief and the quality of life of patients with moderate to severe cancer pain after taking controlled-release tablets of oxycodone hydrochloride and sustained-release tablets of morphine sulfate.. Word Latest Medicine Information 2012;12(9):12-5. [Google Scholar]
Riley 2015 {published data only}
- Riley J, Branford R, Droney J, Gretton S, Sato H, Kennett A, et al. Morphine or oxycodone for cancer-related pain? A randomised, open-label, controlled trial. Journal of Pain and Symptom Management 2015;49(2):161-72. [DOI] [PubMed] [Google Scholar]
- Riley J, Branford R, Droney J, Gretton S, Sato H, Thick M, et al. A randomised controlled trial of oral morphine versus oral oxycodone for the treatment of pain in cancer patients. Palliative Medicine 2012;26(4):386. [Google Scholar]
Salzman 1999 {published data only}
- Salzman RT, Roberts MS, Wild J, Fabian C, Reder RF, Goldenheim PD. Can a controlled-release oral dose form of oxycodone be used as readily as an immediate-release form for the purpose of titrating to stable pain control? Journal of Pain and Symptom Management 1999;18(4):271-9. [DOI] [PubMed] [Google Scholar]
Song 2015 {published data only}
- Song YH. Effectiveness evaluation of oxycodone hydrochloride sustained release tablets and morphine sulfate sustained release tablets for severe cancer pain. China PracMed 2015;10(11):150-1. [Google Scholar]
Stambaugh 2001 {published data only}
- Stambaugh JE, Reder RF, Stambaugh M. Double-blind, randomized, two-period crossover efficacy and pharmacokinetic comparison of immediate-release oxycodone (IR) and controlled-release oxycodone (CR) in cancer patients with pain. Clinical Pharmacology and Therapeutics 1997;61(2):197. [Google Scholar]
- Stambaugh JE, Reder RF, Stambaugh MD, Stambaugh H, Davis M. Double-blind, randomized comparison of the analgesic and pharmacokinetic profiles of controlled- and immediate-release oral oxycodone in cancer pain patients. Journal of Clinical Pharmacology 2001;41(5):500-6. [DOI] [PubMed] [Google Scholar]
Su 2015 {published data only}
- Su J, Zhu Y, Wu W, Luo Y. Observation of curative effects and adverse effects of oxycodone hydrochloride controlled-release tablets and fentanyl transdermal patches on the treatment of moderate or severe cancer pain. Anti-Tumor Pharmacy 2015;5(6):444-8. [Google Scholar]
Sun 2013 {published data only}
- Sun YY, Chen XY, Wang KS. Curative effect analysis of oxycontin and MS contin in the treatment of severe cancerous pain. Cancer Research and Clinic 2013;25(1):36-40. [Google Scholar]
Tu 2015 {published data only}
- Tu XS, Hu LX. Oxycodone hydrochloride sustained release tablets compared with morphine sulfate sustained release tablets in the treatment of advanced moderate to severe cancer pain. Journal of China Prescription Drug 2015;13(10):88-9. [Google Scholar]
Wang 2008 {published data only}
- Wang P, Liu Y, Wang T, Cheng BZ. The observation of curative effect of oxycodone hydrochloride controlled release tablets in management of moderate and severe cancer pain. Modern Oncology 2008;16(11):1983–5. [Google Scholar]
Xie 2018 {published data only}
- Xie XM, Luo LJ, Zhang HL. Clinical efficacy of oxycodone hydrochloride in the treatment of elderly patients with cancer pain. Chinese Community Doctors 2018;34(24):65-7. [Google Scholar]
Ye 2012 {published data only}
- Ye YM, Xiao XX. Analysis of efficacy and adverse reactions of oxycodone hydrochloride controlled release tablet and morphine sulfate sustained-release tablets in the treatment of 83 cases of patients with severe cancer pain. Medical Frontier 2012;2(8):69-70. [Google Scholar]
Yu 2007 {published data only}
- Yu XH, Wu DG, Yang JF, Gao M. Curative effect of oxycodone hydrochloride controlled release tablets in the treatment of cancer pain. Chinese Journal of Hospital Pharmacy 2007;27(9):1277-8. [Google Scholar]
Yu 2009 {published data only}
- Yu H, Liang LS, Wang JF, Liu XD, Zhang GZ, Liu EY. Comparison of the effect of oxycodone and morphine controlled-release tablets in the treatment of visceral cancer pain. Chinese Journal of Current Advances in General Surgery 2009;12(9):769-811. [Google Scholar]
Yu 2014 {published data only}
- Yu S, Shen W, Yu L, Hou Y, Han J, Richards HM. Safety and efficacy of once-daily hydromorphone extended-release versus twice-daily oxycodone hydrochloride controlled-release in Chinese patients with cancer pain: a phase 3, randomized, double-blind, multicenter study. Journal of Pain 2014;15(8):835-44. [DOI] [PubMed] [Google Scholar]
Zecca 2016 {published data only}
- Zecca E, Brunelli C, Bracchi P, Biancofiore G, De Sangro C, Bortolussi R, et al. Comparison of the tolerability profile of controlled-release oral morphine and oxycodone for cancer pain treatment. An open-label randomized controlled trial. Journal of Pain and Symptom Management 2016;52(6):783-94. [DOI] [PubMed] [Google Scholar]
Zhang 2011 {published data only}
- Zhang YY, Han T, Wang Y, Liu L, Wang N, Wang YJ. Effect of the titration of oxycodone controlled-release tablets on moderate pain. Journal of Medical Research 2011;40(4):52–4. [Google Scholar]
Zhang 2014 {published data only}
- Zhang W-Z, Yu W-J, Zhao X-L, He B-X. Pharmacoeconomics evaluation of morphone, MS contin and oxycodone in the treatment of cancer pain. Asian Pacific Journal of Cancer Prevention 2014;15(20):8797-800. [DOI] [PubMed] [Google Scholar]
Zhang 2016a {published data only}
- Zhang XQ. Analgesic effect analysis of three opioids for patients with advanced cancer pain. Medical Frontier 2016;6(6):164-5. [Google Scholar]
References to studies excluded from this review
Ahmedzai 2012 {published data only}
- Ahmedzai SH, Friedemann N, Bar-Sela G, Bjorn B, Leyendecker P, Hopp M. A randomized, double-blind, active-controlled, double-dummy, parallel-group study to determine the safety and efficacy of oxycodone/naloxone prolonged-release tablets in patients with moderate/severe, chronic cancer pain. Palliative Medicine 2012;26(1):50-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Awerbuch 2011 {published data only}
- Awerbuch MS. Should opioids be used for chronic non-cancer pain? Medical Journal of Australia 2011;195(5):264-5. [DOI] [PubMed] [Google Scholar]
Bekkering 2011 {published data only}
- Bekkering GE, Soares-Weiser K, Reid K, Kessels AG, Dahan A, Treede RD, et al. Can morphine still be considered to be the standard for treating chronic pain? A systematic review including pair-wise and network meta-analyses. Current Medical Research and Opinion 2011;27:1477-91. [DOI] [PubMed] [Google Scholar]
Bell 2006 {published data only}
- Bell RF, Wisloff T, Eccleston C, Kalso E. Controlled clinical trials in cancer pain. How controlled should they be? A qualitative systematic review. British Journal of Cancer 2006;94(11):1559-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Borchgrevink 2004 {published data only}
- Borchgrevink PC, Klepstad P, Kongsgaard UE, Kaasa S. Use of opioids against severe cancer pain [Bruk av opioider mot sterke kreftrelaterte smerter]. Tidsskrift for Den Norske Laegeforening 2004;124(3):337-8. [PubMed] [Google Scholar]
Caraceni 2011 {published data only}
- Caraceni A, Pigni A, Brunelli C. Is oral morphine still the first choice opioid for moderate to severe cancer pain? A systematic review within the European Palliative Care Research Collaborative guidelines project. Palliative Medicine 2011;25:402-9. [DOI] [PubMed] [Google Scholar]
Carroll 2011 {published data only}
- Carroll EMA, Kamboj SK, Conroy L, Tookman A, Williams AC, Jones L, et al. Facial affect processing in patients receiving opioid treatment in palliative care: preferential processing of threat in pain catastrophizers. Journal of Pain and Symptom Management 2011;41(6):975-85. [DOI] [PubMed] [Google Scholar]
Chary 1994 {published data only}
- Chary S, Goughnour BR, Moulin DE, Thorpe WR, Harsanyi Z, Darke AC. The dose-response relationship of controlled-release codeine (Codeine Contin) in chronic cancer pain. Journal of Pain and Symptom Management 1994;9(6):363-71. [DOI] [PubMed] [Google Scholar]
Chen 2009 {published data only}
- Chen HP, Jiang JJ, Li JX. Efficacy and safety of oxycodone-acetaminophen for moderate and severe visceral pain in advanced cancer patients. Chinese Journal of New Drugs 2009;18(10):920-2. [Google Scholar]
De Conno 1991 {published data only}
- De Conno F, Ripamonti C, Sbanotto A, Barletta L, Zecca E, Martini C, et al. A clinical study on the use of codeine, oxycodone, dextropropoxyphene, buprenorphine, and pentazocine in cancer pain. Journal of Pain and Symptom Management 1991;6(7):423-7. [DOI] [PubMed] [Google Scholar]
Dunlop 2013 {published data only}
- Dunlop W, Neufeld K. Quality of life benefits and cost impact of prolonged release oxycodone/naloxone versus prolonged release oxycodone in patients with moderate to severe pain and opioid-induced constipation despite the use of 2 laxatives: a UK cost utility analysis. Value in Health 2013;16(7):A384. [DOI] [PubMed] [Google Scholar]
Fallon 2011 {published data only}
- Fallon MT, Laird BJA. A systematic review of combination step III opioid therapy in cancer pain: an EPCRC opioid guideline project. Palliative Medicine 2011;25(5):597-603. [DOI] [PubMed] [Google Scholar]
Gao 2020 {published data only}
- Gao Y, Zhang B, Li X, Wang D, Sui Y, Li G, et al. Synergistic effect of rosuvastatin with oxycodone hydrochloride sustained-release tablets in the treatment of cancer pain in patients with severe breast cancer and its effect on inflammatory factors. Acta Medica Mediterranea 2020;36(4):2353-7. [Google Scholar]
Garassino 2010 {published data only}
- Garassino MC, Febbraro A, Iorno V, Carbone A, Spagnoletti I, Isa L, et al. Randomized phase ii trial (NCT00637975) evaluating activity and toxicity of fixed dose of oxycodone and increasing dose of pregabalin versus increasing dose of oxycodone and fixed dose of pregabalin for the treatment of oncological neuropathic pain (neuropain-01). Annals of Oncology 2010;21:viii363. [Google Scholar]
Garassino 2011 {published data only}
- Garassino MC, Bianchi A, Febbraro A, Spagnoletti I, Iorno V, Bramati A, et al. Final results of a randomized phase II trial (NCT00637975) evaluating activity and toxicity of fixed-dose oxycodone and increasing dose of pregabalin versus increasing dose of oxycodone and fixed-dose pregabalin for the treatment of oncologic neuropathic pain (NEUROPAIN-01). Journal of Clinical Oncology (ASCO Annual Meeting) 2011;29(15 Suppl 1):9028. [Google Scholar]
Garassino 2013 {published data only}
- Garassino MC, Piva S, Verde N, Spagnoletti I, Iorno V, Carbone C, et al. Randomised phase II trial (NCT00637975) evaluating activity and toxicity of two different escalating strategies for pregabalin and oxycodone combination therapy for neuropathic pain in cancer patients. PlOS One 2013;8(4):e59981. [DOI] [PMC free article] [PubMed] [Google Scholar]
George 2003 {published data only}
- George B, Douard MC, Dubreuil ML. Oxycodone: alternative therapy in the management of chronic cancer pain [L'oxycodone: alternative therapeutique dans la prise en charge de la douleur chronique d'origine cancereuse]. Cahiers d'Anesthesiologie 2003;51:HS7-12. [Google Scholar]
Guo 2017 {published data only}
- Guo J, Lu J, Liu B, Xing L, Xiao J, Wang Z. Effect of hydrochloride oxycodone controlled-release tablets with dose titration for moderate or severe cancer pain [中重度癌痛盐酸羟考酮缓释片治疗 剂量调整临床研究]. Chinese Journal of Cancer Prevention Treatment 2017;24(18):1319-22. [Google Scholar]
Hanks 2002 {published data only}
- Hanks G. Morphine and other opioids in the treatment of cancer pain? International Journal of Cancer 2002;13(Suppl):5. [Google Scholar]
Hongmei 2013 {published data only}
- Hongmei X, Jianwen S. Compound matrine injection in liver carcinoma pain: a clinical study. Journal of Gastroenterology and Hepatology 2013;28:790. [Google Scholar]
Huang 2015 {published data only}
- Huang MQ, Li CF, Kong FL, Gan XQ, Chu XF. Clinical efficacy of oxycodone hydrochloride sustained release tablets in the treatment of pain in advanced cancer. Anhui Medical and Pharmaceutical Journal 2015;19(10):2022-3. [Google Scholar]
Igarashi 2015 {published data only}
- Igarashi T, Abe K, Miura T, Kinoshita H. Oxycodone frequently induced nausea and vomiting in oxycodone-naive patients with hepatic dysfunction. Journal of Palliative Medicine 2015;18(5):399. [DOI] [PubMed] [Google Scholar]
Katz 2008 {published data only}
- Katz MH, Kotabe S. Decreasing use of controlled-release oxycodone - response. Medical Care 2008;46(9):1002. [DOI] [PubMed] [Google Scholar]
Kim 2015 {published data only}
- Kim HJ, Kim YS, Park SH. Opioid rotation versus combination for cancer patients with chronic uncontrolled pain: a randomized study. BMC Palliative Care 2015;14:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
King 2011 {published data only}
- King SJ, Reid C, Forbes K, Hanks G. A systematic review of oxycodone in the management of cancer pain. Palliative Medicine 2011;25:454-70. [DOI] [PubMed] [Google Scholar]
Koyyalagunta 2012 {published data only}
- Koyyalagunta D, Bruera E, Solanki DR, Nouri KH, Burton AW, Toro MP, et al. A systematic review of randomized trials on the effectiveness of opioids for cancer pain. Pain Physician 2012;15(3 Suppl):ES39-58. [PubMed] [Google Scholar]
Kummer 2011 {published data only}
- Kummer O, Hammann F, Moser C, Schaller O, Drewe J, Krahenbuhl S. Effect of the inhibition of CYP3A4 or CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. European Journal of Clinical Pharmacology 2011;67(1):63-71. [DOI] [PubMed] [Google Scholar]
LeBon 2009 {published data only}
- LeBon B, Zeppetella G, Higginson IJ. Effectiveness of topical administration of opioids in palliative care: a systematic review. Journal of Pain and Symptom Management 2009;37(5):913-7. [DOI] [PubMed] [Google Scholar]
Leppert 2011 {published data only}
- Leppert W, Ahmedzai SH, Uhl R, Kremers W, Hopp M. Long-term efficacy and safety of the fixed combination oxycodone and naloxone prolonged release (PR) in patients with chronic cancer pain. European Journal of Pain Supplements 2011;5(1):172. [Google Scholar]
Li 2008 {published data only}
- Li RC, He BS. Therapeutic outcome of Tylox for cancer pain in malignant tumor patients with bone metastasis. Chinese Journal of New Drugs 2008;17(18):1619-21. [Google Scholar]
Li 2010 {published data only}
- Li XM, Liu DQ, Wu HY, Yang C, Yang L. Controlled-release oxycodone alone or combined with gabapentin for management of malignant neuropathic pain. Chinese Journal of Cancer Research 2010;22(1):80-6. [Google Scholar]
Liang 2021 {published data only}
- Liang J, Chen L, Yang S, Zhang H, Li L, Chen Z, et al. A 12-hour rapid titration method for cancer pain: a randomized, controlled, open-label study. Annals of Palliative Medicine 2021;10(1):88-96. [DOI] [PubMed] [Google Scholar]
- The feasibility study of sustained-release opioid used for quick analgesia. www.who.int/trialsearch/Trial2.aspx?TrialID=ChiCTR-TIR-17012925 (date first received 17 October 2017).
Lin 2013 {published data only}
- Lin XH, Xu JP, Mai YZ. The efficacy and safety observation of oxycodone retard tablets in the treatment of moderately severe pain. China Medicine and Pharmacy 2013;3(9):78-9. [Google Scholar]
Ma 2016 {published data only}
- Ma H, Liu Y, Huang L, Zeng XT, Jin SH, Yue GJ, et al. The adverse events of oxycodone in cancer-related pain: a systematic review and meta-analysis of randomized controlled trials. Medicine (Hagerstown) 2016;95(15):e3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marineo 2012 {published data only}
- Marineo G, Iorno V, Gandini C, Moschini V, Smith TJ. Scrambler therapy may relieve chronic neuropathic pain more effectively than guideline-based drug management: results of a pilot, randomized, controlled trial. Journal of Pain and Symptom Management 2012;43(1):87-95. [DOI] [PubMed] [Google Scholar]
Meng 2008 {published data only}
- Meng XL, Tang YL, Zhang JH. Effectiveness observation on oxycodone hydrochloride controlled-release tablets for cancer pain [盐酸羟考酮控释片用于癌性疼痛疗效观察]. Modern Journal of Integrated Traditional Chinese and Western Medicine 2008;17(2):199-200. [Google Scholar]
Moertel 1974 {published data only}
- Moertel CG, Ahmann DL, Taylor WF, Schwartau N. Relief of pain by oral medications. A controlled evaluation of analgesic combinations. Journal of the American Medical Association 1974;229(1):55-9. [PubMed] [Google Scholar]
Moksnes 2012 {published data only}
- Moksnes K, Kaasa S, Paulsen Ã, Rosland JH, Spigset O, Dale O. Serum concentrations of opioids when comparing two switching strategies to methadone for cancer pain. European Journal of Clinical Pharmacology 2012;8:1147-56. [DOI] [PubMed] [Google Scholar]
Mosley 2018 {published data only}
- Mosley SA, Hicks JK, Portman DG, Donovan KA, Gopalan P, Schmit J, et al. Design and rational for the precision medicine guided treatment for cancer pain pragmatic clinical trial. Contemporary Clinical Trials 2018;68:7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nadstawek 2008 {published data only}
- Nadstawek J, Leyendecker P, Hopp M, Ruckes C, Wirz S, Fleischer W, et al. Patient assessment of a novel therapeutic approach for the treatment of severe, chronic pain. International Journal of Clinical Practice 2008;62(8):1159-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nalamachu 2013 {published data only}
- Nalamachu S, Rauck R, Dillaha L, Parikh N. Lack of correlation between the dose of fentanyl sublingual spray for breakthrough cancer pain and the dose of around-the-clock opioid for persistent pain. Journal of Pain 2013;1:S74. [DOI] [PubMed] [Google Scholar]
NCT01859715 {published data only}
- NCT01859715. Emergency department (ED) drug interaction in emergency department patients. clinicaltrials.gov/ct2/show/results/NCT01859715 Date first received: 1 May 2013.
NCT01885182 {published data only}
- NCT01885182. Targin cancer pain. clinicaltrials.gov/ct2/show/NCT01885182 Date first received: 19 June 2013.
Nunez Olarte 2008 {published data only}
- Nunez Olarte JM. Oxycodone and the challenge of neuropathic cancer pain: a review. Oncology 2008;74:83-90. [DOI] [PubMed] [Google Scholar]
Oosten 2015 {published data only}
- Oosten AW, Oldenmenger WH, Mathijssen RH, Van der Rijt CC. A systematic review of prospective studies reporting adverse events of commonly used opioids for cancer-related pain: a call for the use of standardized outcome measures. Journal of Pain 2015;16(10):935-46. [DOI] [PubMed] [Google Scholar]
Pan 2019 {published data only}
- Pan H, Shen P, Shu Q, Lu L, Qian S, Zhou Y, et al. Efficacy and safety of sustained-release oxycodone compared with immediate-release morphine for pain titration in cancer patients: a multicenter, open-label, randomized controlled trial (SOCIAL). Medicine 2019;98(24):e15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pang 2009 {published data only}
- Pang J, Jie BJ, Li YP. Efficacy of oxycodone-acetominophen by different administrations in controlling moderate to severe cancer pain and break-out pain. Chinese Journal of New Drugs 2009;18(18):1767-70, 1776. [Google Scholar]
Passik 2014 {published data only}
- Passik SD, Narayana A, Yang R. Aberrant drug-related behavior observed during a 12-week open-label extension period of a study involving patients taking chronic opioid therapy for persistent pain and fentanyl buccal tablet or traditional short-acting opioid for breakthrough pain. Pain Medicine 2014;15(8):1365-72. [DOI] [PubMed] [Google Scholar]
Reid 2006 {published data only}
- Reid CM, Martin RM, Sterne JAC, Davies AN, Hanks GW. Oxycodone for cancer-related pain: meta-analysis of randomized controlled trials. Archives of Internal Medicine 2006;166:837-43. [DOI] [PubMed] [Google Scholar]
Rentz 2009 {published data only}
- Rentz AM, Yu R, Muller-Lissner S, Leyendecker P. Validation of the Bowel Function Index to detect clinically meaningful changes in opioid-induced constipation. Journal of Medical Economics 2009;12(4):371-83. [DOI] [PubMed] [Google Scholar]
Riley 2008 {published data only}
- Riley J, Eisenberg E, Müller-Schwefe G, Drewes AM, Arendt-Nielsen L. Oxycodone: a review of its use in the management of pain. Current Medical Research and Opinion 2008;24(1):175-92. [DOI] [PubMed] [Google Scholar]
Shi 2008 {published data only}
- Shi L, Xiong HH, Yang L, Yu SY. Analgesic effect of oxycodone-acetaminophen tablets in patients with cervical cancer. Chinese Journal of New Drugs 2008;17:1884-5. [Google Scholar]
Shi 2018 {published data only}
- Shi Z, Lou G, Gu C, Song Z, Shao L, Zhang Y. Efficacy and safety of titration with controlled-release oxycodone versus immediate-release morphine in patients with moderate cancer pain. International Journal of Clinical and Experimental Medicine 2018;11(3):2595-602. [Google Scholar]
Sima 2010a {published data only}
- Sima L, Wu X, Fang W, Li F. Oxycodone/acetaminophen addition to constant opioids in metastatic bone pain: a randomized, double-blinded, controlled, multicenter trial. Journal of Clinical Oncology 2010;28(15 Suppl 1):e19691. [Google Scholar]
Sima 2010b {published data only}
- Sima L, Wu X, Fang W. Oxycodone/acetaminophen combination tablet in patients with metastatic bone pain: a randomized, double-blinded, placebo-controlled trial. Annals of Oncology 2010;21:378. [Google Scholar]
Sima 2012 {published data only}
- Sima L, Fang WX, Wu XM, Li F. Efficacy of oxycodone/paracetamol for patients with bone-cancer pain: a multicenter, randomized, double-blinded, placebo-controlled trial. Journal of Clinical Pharmacy and Therapeutics 2012;37(1):27-31. [DOI] [PubMed] [Google Scholar]
Stambaugh 1980a {published data only}
- Stambaugh JEJ, Tejada F, Trudnowski RJ. Double-blind comparisons of zomepirac and oxycodone with aspirin phenacetin caffeine in cancer pain. Journal of Clinical Pharmacology 1980;20(4):261-70. [DOI] [PubMed] [Google Scholar]
Stambaugh 1980b {published data only}
- Stambaugh JEJ. Analgesic equivalence of Tylox® and Percodan®: double-blind crossover study of patients with pain from malignancy. Current Therapeutic Research, Clinical and Experimental 1980;27(2):302-8. [Google Scholar]
Stambaugh 1981 {published data only}
- Stambaugh JE Jr, Sarajian C. Analgesic efficacy of zomepirac sodium in patients with pain due to cancer. Journal of Clinical Pharmacology 1981;21:501-7. [DOI] [PubMed] [Google Scholar]
Stambaugh 1985 {published data only}
- Stambaugh JEJ, McAdams J. A double-blind comparison of ibuprofen vs. placebo in reducing oxycodone-acetaminophen dosage in chronic cancer pain. Clinical Research 1985;33:458A. [Google Scholar]
Stambaugh 1987 {published data only}
- Stambaugh JE, McAdams J. Comparison of the efficacy and safety of oral xorphanol, acetaminophen/oxycodone and placebo in chronic cancer pain. International Journal of Clinical Pharmacology and Therapeutics 1987;41:229. [Google Scholar]
Stambaugh 1990 {published data only}
- Stambaugh J, Drew J, Johnson J. Comparison of flurbiprofen (Ansaid), oxycodone APAP and placebo in cancer pain, after single and repeat oral doses. Clinical Pharmacology and Therapeutics 1990;47:187. [Google Scholar]
Stambaugh 1991 {published data only}
- Stambaugh JE. Multidose analgesic studies in chronic pain models. Advances in Pain Research and Therapy 1991;18:151-63. [Google Scholar]
Taeron 2002 {published data only}
- Taeron C. Extended release oxycodone. An oral opioid analgesic for cancer patients with chronic pain. Revue De L'Infirmiere 2002;85:47-9. [PubMed] [Google Scholar]
Tanaka 2017 {published data only}
- Tanaka R, Ishikawa H, Sato T, Shino M, Matsumoto T, Mori K, et al. Incidence of delirium among patients having cancer injected with different opioids for the first time. American Journal of Hospice & Palliative Medicine 2017;34(6):572-6. [DOI] [PubMed] [Google Scholar]
Wallace 2013 {published data only}
- Wallace E, Twomey M, Victory R, O'Reilly M. Intravesical baclofen, bupivacaine, and oxycodone for the relief of bladder spasm. Journal of Palliative Care 2013;29(1):49-51. [PubMed] [Google Scholar]
Wang 2012 {published data only}
- Wang YM, Liu ZW, Liu JL, Zhang L. Efficacy and tolerability of oxycodone in moderate-severe cancer-related pain: a meta-analysis of randomized controlled trials. Experimental and Therapeutic Medicine 2012;4(2):249-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Watanabe 2008 {published data only}
- Watanabe S, Pereira J, Tarumi Y, Hanson J, Bruera E. A randomized double-blind crossover comparison of continuous and intermittent subcutaneous administration of opioid for cancer pain. Journal of Palliative Medicine 2008;11(4):570-4. [DOI] [PubMed] [Google Scholar]
Watanabe 2020 {published data only}
- Wanatabe K. Analysis of symptoms relieved in addition to pain after administration of oxycodone or morphine to patients with advanced cancer living at home. Gan to Kagaku Ryoho [Japanese Journal of Cancer & Chemotherapy] 2020;47(5):797-800.. [PubMed] [Google Scholar]
Wei 2016 {published data only}
- Wei YG, Zhou CP. The curative effect observation of acupoint catgut embedding to treat lung cancer pain. Journal of Preventive Medicine of Chinese People's Liberation Army [jie fang jun yu fang yi xue za zhi] 2016;34(Suppl 1):297-8. [Google Scholar]
Wu 2009 {published data only}
- Wu H, Ouyang QC, Liu LP, Wang PH. Safety and efficacy of oxycodone/acetaminophen in treatment of moderate and advanced cancer pain. Chinese Journal of Cancer Prevention and Treatment 2009;18:1432-4. [Google Scholar]
Wu 2015 {published data only}
- Wu H, Chen Y, Shen Q, Cao X, Lu L. Application of controlled-release oxycodone and oral immediate-release morphine in the titration treatment of cancer pain [羟考酮缓释片联合吗啡片在癌痛滴定治疗中的应用]. Anti-Tumor Pharmacy 2015;2:112-5. [Google Scholar]
Xiong 2008 {published data only}
- Xiong J, Liang C, Wu X, Zhao YX, Fu L, Zhou YF. Efficacy and safety of oxycodone-acetaminophen tablet in 142 patients with different types of moderate and advanced cancer pain. Chinese Journal of New Drugs 2008;21:1877-9, 1900. [Google Scholar]
Xu 2008 {published data only}
- Xu LH, Quan SC, Wu LB. Treatment beginning with minute dose oxycontin with advanced stage cancer pain. Modern Journal of Integrated Traditional Chinese and Western Medicine 2008;6:859-60. [Google Scholar]
Yoshimoto 2018 {published data only}
- Yoshimoto T, Ryu E, Tomiyasu S, Hojo M, Kokubun H, Matoba M. Efficacy and safety of oxycodone injection for relieving cancer pain: a study in Japan consisting of two open trials for intravenous and subcutaneous administration. Biological & Pharmaceutical Bulletin 2018;41(6):850-7. [DOI] [PubMed] [Google Scholar]
Zhu 2019 {published data only}
- Zhu M, Liang X, Gu Z, Zhang L, Xie G, Yu S, et al. Multicenter trial of titration of morphine versus oxycodone for cancer pain. International Journal of Clinical and Experimental Medicine 2019;12(6):7317-26. [Google Scholar]
Zou 2009 {published data only}
- Zou J, Wang Q, Liu L. Efficacy of oxycodone-acetaminophen as an adjuvant of opioid analgesics for cancer pain. Chinese Journal of New Drugs 2009;18(10):923-4. [Google Scholar]
References to studies awaiting assessment
Aurilio 2009 {published data only}
- Aurilio C, Sansone P, Pace MC, Passavanti MB, Romano SV, Pota V. Evaluation of efficacy and safety of prolonged-release oxycodone at different dosages for the treatment of severe chronic pain. Pain Practice 2009;9:103. [Google Scholar]
JapicCTI‐090789/090/091 {unpublished data only}
- JapicCTI-090789/090/091. An open-label study of intravenous (i.v.) S-811717 (oxycodone hydrochloride solution for injection) in patients with cancer pain. clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-090789 (date first received 30 June 2009).
JapicCTI‐111388 {unpublished data only}
- CTI-111388. TK-641 phase 3 study. clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-111388 (date first received 14 January 2011).
NCT00378937 {unpublished data only}
- NCT00378937. Oxycodone or standard pain therapy in treating patients with cancer pain. clinicaltrials.gov/ct2/show/NCT00378937?=NCT00378937&draw=2&rank1 (date first received 21 September 2006).
NCT00726830 {unpublished data only}
- NCT00726830. Methadone, morphine, or oxycodone in treating pain in patients with cancer. clinicaltrials.gov/ct2/show/NCT00726830?term=NCT00726830&draw=2&rank1 (date first received 1 August 2008).
NCT01493635 {unpublished data only}
- NCT01493635. Two step versus the standard three step approach of the WHO analgesic ladder for cancer pain relief. clinicaltrials.gov/ct2/show/NCT01493635?term=NCT01493635&draw=2&rank1 (date first received 16 December 2011).
NCT03439904 {published data only}
- NCT03439904. Individualized pharmaceutical-care in outpatients with cancer pain. clinicaltrials.gov/show/NCT03439904 (date first received 20 February 2018).
Song 2009 {published data only}
- Song JF, Zhao YL, Zhou HM, Wang HJ, Li KY. Efficacies of oxycodone and morphine sulfate sustained-release tablets in treating moderate and severe cancer pain. Chinese Journal of New Drugs 2009;18(4):337-9. [Google Scholar]
Zhang 2016 {published data only}
- Zhang Y. Efficacy of oxycotin in the treatment of severe cancer pain by rectal administration. Cancer Nursing 2016;39(6 (Suppl 1)):S73. [Google Scholar]
References to ongoing studies
2008‐002273‐12 {unpublished data only}
- 2008-002273-12. Long term opioid administration in oncologic chronic pain: open label, prospective study on efficacy, safety and pharmacogenetic factors. clinicaltrialsregister.eu/ctr-search/search?query=2008-002273-12 (date first received 23 June 2008).
2009‐013118‐28 {unpublished data only}
- 2009-013118-28. The buccal fentanyl in cancer pain management measure [Bukkaalinen fentanyyli syöpäpotilaiden toimenpidekivun hoidossa]. clinicaltrialsregister.eu/ctr-search/search?query=2009-013118-28 (date first received 8 June 2009).
ChiCTR1800014268 {unpublished data only}
- ChiCTR1800014268. Effecty [sic] and efficacy of hydrochloride oxycodone controlled-release tablets with dose titration at 12h for cancer pain. who.int/trialsearch/Trial2.aspx?TrialID=ChiCTR1800014268 (date first received 2 January 2018).
ChiCTR1800017461 {unpublished data only}
- ChiCTR1800017461. Efficacy and safety of different titration regimens for oxycodone hydrochloride sustained-release tablet in the treatment of cancer pain. who.int/trialsearch/Trial2.aspx?TrialID=ChiCTR1800017461 (date first received 31 July 2018).
ChiCTR1900022566 {unpublished data only}
- ChiCTR1900022566. Comparison of oxycodone versus morphine in the treatment of patients with severe cancer pain: a randomized controlled trial. who.int/trialsearch/Trial2.aspx?TrialID=ChiCTR1900022566 (date first received 16 April 2019).
ChiCTR2000037845 {published data only}
- ChiCTR2000037845. A multicenter randomized controlled clinical trial of hydromorphone injection PCA versus oxycodone sustained release tablets in the treatment of moderate and severe cancer pain. chictr.org.cn/showprojen.aspx?proj=60437 (date first received 3 September 2020).
ChiCTR2100042972 {published data only}
- ChiCTR2100042972. Comparative study of sufentanil self-controlled analgesic pump and oxycodone hydrochloride controlled-release tablets in the treatment of moderate and severe cancer pain. chictr.org.cn/showprojen.aspx?proj=64416 (date first received 2 February 2021).
Elsayem 2010 {unpublished data only}
- Elsayem AF, Bain KT, Palmer JL, Bearden J, Fisch M. A randomized comparison of oral methadone as a "first-switch" opioid versus opioid switching between sustained-release morphine and oxycodone for oncology outpatients with pain management problems. Journal of Clinical Oncology 2010;28(15 Suppl 1):TPS324. [Google Scholar]
IRCT20201202049575N1 {published data only}
- IRCT20201202049575N1. Comparison of the effect of morphine and oxycodone in relieving pain in patients with bone metastasis pain. google.com/search?q=IRCT20201202049575N1&rlz=1C1GCEB_enGB999GB1000&oq=IRCT20201202049575N1&aqs=chrome..69i57j0i546l2.827j0j15&sourceid=chrome&ie=UTF-8 (date first received 9 December 2020).
Matsouka 2017 {published data only}
- Matsuoka H, Tsurutani J, Chiba Y, Fujita Y, Terashima M, Yoshida T, et al. Selection of opioids for cancer-related pain using a biomarker: a randomized, multi-institutional, open-label trial (RELIEF study). BMC Cancer 2017;17:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00916890 {unpublished data only}
- NCT00916890. Chronic administration of opioids in cancer chronic pain: an open prospective study on efficacy, safety and pharmacogenetic factors influence. clinicaltrials.gov/ct2/show/NCT00916890?term=NCT00916890&draw=2&rank1 (date first received 10 June 2009).
NCT01165281 {unpublished data only}
- NCT01165281. A safety and efficacy study of JNS024 Extended Release (ER) in Japanese and Korean patients with chronic malignant tumor-related cancer pain. clinicaltrials.gov/ct2/show/NCT01165281?term=NCT01165281&draw=2&rank1 (date first received 19 July 2010).
NCT01675622 {unpublished data only}
- NCT01675622. Immediate-release oxycodone capsules study in cancer pain. clinicaltrials.gov/ct2/show/NCT01675622?term=NCT01675622&draw=2&rank1 (date first received 30 August 2012).
NCT02084355 {unpublished data only}
- NCT02084355. Study of opioid rotation versus opioid escalation in patients with moderate to severe cancer pain. clinicaltrials.gov/ct2/show/NCT02084355 (date first received 5 March 2014).
NCT03024515 {unpublished data only}
- NCT03024515. A pilot randomized open-labelled study comparing a structured titration method of immediate- and sustained-release oxycodone versus opioids titration of investigators' choice in advanced cancer patients in Hong Kong. clinicaltrials.gov/ct2/show/NCT03024515?term=NCT03024515&draw=2&rank1 (date first received 19 January 2017). [Google Scholar]
NCT03176199 {unpublished data only}
- NCT03176199. A study to compare the titration efficacy and safety of control-released oxycodone and immediate-released oxycodone in patients with moderate to severe cancer pain. clinicaltrials.gov/ct2/show/NCT03176199 (date first received 5 June 2017).
NCT04808531 {published data only}
- NCT04808531. NanaBis™ an oro-buccal administered delta9-tetrahydrocannabinol (d9-THC) & cannabidiol (CBD) medicine for the management of bone pain from metastatic cancers. clinicaltrials.gov/ct2/show/NCT04808531?term=NCT04808531&draw=2&rank1 (date first received 22 March 2021).
UMIN000011756 {unpublished data only}
- UMIN000011756. Randomized study of fentanyl citrate versus oxycodone hydrochloride hydrate in patients with unresectable advanced pancreatic cancer. upload.umin.ac.jp/cgi-open-bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000010996&language=J (date first registered 13 September 2013).
Additional references
Bennett 2008
- Bennett MI. What evidence do we have that the WHO analgesic ladder is effective in cancer pain? In: McQuay HJ, Moore R, Kalso E, editors(s). Systematic Reviews in Pain Research; Methodology Refined. Seattle (WA): IASP Press, 2008:303-13. [Google Scholar]
Breivik 2009
- Breivik H, Cherny N, Collett B, De Conno F, Filbet M, Foubert AJ, et al. Cancer-related pain: a pan-European survey of prevalence, treatment, and patient attitudes. Annals of Oncology 2009;20:1420-33. [DOI] [PubMed] [Google Scholar]
Caraceni 2012
- Caraceni A, Hanks GW, Kaasa S, Bennett MI, Brunelli C, Cherny N, et al. Use of opioid analgesics in the treatment of cancer pain: evidence based recommendations from the EAPC. Lancet Oncology 2012;13:e58-68. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- GRADEpro Guideline Development Tool [Software]. McMaster University (developed by Evidence Prime, Inc.), 2015. Available from www.gradepro.org.
Greco 2014
- Greco MT, Roberto A, Corli O, Deandrea S, Bandieri E, Cavuto S, et al. Quality of cancer pain management: an update of a systematic review of under-treatment of patients with cancer. Journal of Clinical Oncology 2014;32(36):4149-54. [DOI] [PubMed] [Google Scholar]
Gudin 2012
- Gudin J. Opioid therapies and cytochrome P450 interactions. Journal of Pain and Symptom Management 2012;44:S4-14. [DOI] [PubMed] [Google Scholar]
Guo 2018
- Guo KK, Deng CQ, Lu GJ, Zhao GL. Comparison of analgesic effect of oxycodone and morphine on patients with moderate and advanced cancer pain: a meta-analysis. BMC Anesthesiology 2018;18:132. [DOI: 10.1186/s12871-018-0583-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1.
Lefebvre 2021
- LeLefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M-I, et al. Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Leppert 2010
- Leppert W. Role of oxycodone and oxycodone/naloxone in cancer pain management. Pharmacological Report 2010;62:578-91. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials.. Annals of Internal Medicine 2010;152(11):726-32. [DOI] [PubMed] [Google Scholar]
Tong 2018
- Tong Z, Li F, Ogawa Y, Watanabe N, Furukawa TA. Quality of randomized controlled trials of new generation antidepressants and antipsychotics identified in the China National Knowledge Infrastructure (CNKI): a literature and telephone interview study. BMC Medical Research Methodology 2018;18:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ventafridda 1987
- Ventafridda V, Tamburini M, Caraceni A, De Conno F, Naldi F. A validation study of the WHO method for cancer pain relief. Cancer 1987;59:851-6. [DOI] [PubMed] [Google Scholar]
WHO 1986
- World Health Organization (WHO). Cancer pain relief. apps.who.int/iris/bitstream/handle/10665/43944/9241561009_eng.pdf?sequence=1&isAllowed=y (accessed prior to 23 Feb 2022).
WHO 2018
- World Health Organization (WHO). WHO Guidelines for the pharmacological and radiotherapeutic management of cancer pain in adults and adolescents. www.who.int/publications/i/item/9789241550390 (accessed prior to 23 Feb 2022). [PubMed]
Wu 2009a
- Wu T, Li Y, Bian Z, Liu G, Moher D. Randomized trials published in some Chinese journals: how many are randomized? Trials 2009;10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2016b
- Zhang J, Chen X, Zhu Q, Cui J, Cao L, Su J. Methodological reporting quality of randomized controlled trials: a survey of seven core journals of orthopaedics from mainland China over 5 years following the CONSORT statement. Orthopaedics & Traumatology: Surgery & Research 2016;102:933-8. [DOI] [PubMed] [Google Scholar]
Zhou 2019
- Zhou Q, Yang N, Yang K, Estill J, Chen Y. Pharmacologicaltreatments for generalised anxiety disorder: Comment. Lancet 2019;394:1229. [DOI] [PubMed] [Google Scholar]
Zhou 2020
- Zhou JX, Wang Y, Jiang G. Oxycodone versus morphine for cancer pain titration: a systematic review and pharmacoeconomic evaluation. PLOS One 2020;15(4):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ziegler 2016
- Ziegler LE, Mulvey MR, Blenkinsopp A, Petty D, Bennett MI. Opioid prescribing for cancer patients in the last year of life: a longitudinal population cohort study. Pain 2016;157(11):2445-51. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Reid 2010
- Reid CM, Davies AN, Hanks GW, Martin RM, Sterne JAC. Oxycodone for cancer-related pain. Cochrane Database of Systematic Reviews 2010, Issue 3. Art. No: CD003870. [DOI: 10.1002/14651858.CD003870.pub3] [DOI] [Google Scholar]
Schmidt‐Hansen 2013
- Schmidt‐Hansen M, Bennett MI, Arnold S. Oxycodone for cancer‐related pain. Cochrane Database of Systematic Reviews 2013, Issue 3. Art. No: CD003870. [DOI: 10.1002/14651858.CD003870.pub4] [DOI] [PubMed] [Google Scholar]
Schmidt‐Hansen 2015
- Schmidt-Hansen M, Bennett MI, Hilgart J. Oxycodone for cancer pain in adult patients. JAMA 2015;314:1282-3. [DOI] [PubMed] [Google Scholar]
Schmidt‐Hansen 2018
- Schmidt-Hansen M, Bennett MI, Arnold S, Bromham N, Hilgart JS. Efficacy, tolerability and acceptability of oxycodone for cancer-related pain in adults: an updated Cochrane systematic review. BMJ Supportive & Palliative Care 2018;8:117-28. [DOI] [PubMed] [Google Scholar]