

Abstract
Regulation of the profibrotic and angiogenesis modulating cytokine connective tissue growth factor (CTGF) occurs primarily at the transcriptional level. Therefore, we hypothesized that histone deacetylating enzymes (HDAC), which modulate the accessibility of transcriptionally active promoter regions, might play a role in the regulation of CTGF gene expression. We analyzed microvascular endothelial cells, which showed immunoreactivity for acetylated histone in kidney sections, and compared them with renal tubular epithelial cells. Treatment of cultured endothelial cells with different HDAC inhibitors up‐regulated CTGF mRNA and protein. Pre‐treatment with HDAC inhibitors facilitated induction of CTGF by transforming growth factor–beta (TGF‐β) or lysophosphatidic acid. Transcription factors of the FoxO family were involved in the up‐regulation of CTGF as shown at protein level and by reporter gene analyses. In tubular epithelial cells, up‐regulation of CTGF was only observed when these cells were cultured as subconfluent cells. Dense cells, which are more likely to resemble tubular cells in vivo, showed no up‐regulation upon treatment with HDAC inhibitors and were protected against CTGF induction by TGF‐β. Taken together, our data indicate that the effect of HDAC inhibitors on CTGF expression is largely cell dependent in non‐tumour cells. Different cell type–specific transcription factors seem to determine whether CTGF expression is reduced or increased in cells exposed to HDAC inhibitors.
Keywords: connective tissue growth factor, trichostatin A, histone deacetylase, renal, epithelial cells, endothelial cells, FoxO proteins
Introduction
Post‐translational modifications of histone proteins affect chromatin organization, among them acetylation and deacetylation of the N‐terminal tails of the core histones by histone acetyltransferases (HATs) and histone deacetylases (HDACs) [1]. Acetylation of histones switches to a locally relaxed chromatin structure and recruitment of transcription factors, which often results in transcriptional activation. HATs and HDACs are embedded in multi‐meric protein–DNA complexes, thereby interacting with different regulatory proteins. Because deregulated HDAC activity is associated with different pathological conditions, inhibition of HDACs seems to be a promising therapeutic goal [2, 3].
The beneficial effects of histone deacetylase inhibitors (HDIs) in clinical trials of solid malignancies are not only attributed to their direct repression of tumour cell growth but also to their anti‐angiogenic activity [4, 5]. Up‐regulation of tumour suppressor p53 and VHL by HDIs resulted in reduced activity of hypoxia inducible factor–1α (HIF‐1α) and diminished secretion of vascular endothelial growth factor (VEGF), one of the major angiogenic factors [6]. These results were confirmed in several cancer cell lines. The angiostatic effect of HDIs is also related to direct effects on endothelial cells of tumour neovessels, interfering with DNA synthesis, migration and tube formation in vitro and in vivo[4]. HDIs interfere with VEGF signalling in endothelial cells, complementing the reduced synthesis of VEGF in cancer cells. Up‐regulation of VEGF receptors, VEGFR1, VEGFR2 and neuropilin‐1 during in vitro angiogenesis was prevented by HDIs [7].
HDIs are not only considered as additive treatment of tumours but are considered as potentially useful in neurodegenerative diseases like spinal muscular atrophy [8] or ischaemic injury of the brain [9], in metabolic disorders like diabetes mellitus [10], in inflammatory [11] or autoimmune diseases [12]. There are indications that HDIs might interfere with tissue remodelling as observed in cardiac hypertrophy [13, 14]. Thus far, there is limited information about the effects of HDIs in the kidney in vivo and in renal cells in vitro. Yoshikawa et al. reported that HDIs might also interfere with transforming growth factor (TGF)‐β–induced myofibroblastic transformation of renal tubular epithelial cells by up‐regulation of BMP‐7 and Id2 mRNA [15]. These data suggested that HDIs might also affect the expression of connective tissue growth factor (CTGF), a down‐stream mediator of TGF‐β related to renal fibrosis.
CTGF is a multi‐functional, matricellular protein that has been implicated in wound healing and scar formation and, when synthesized in excess, in the development of fibrosis. CTGF is an immediate early response gene that is potently induced by a variety of stimuli of tissue remodelling and neovascularization, including TGF‐β[16], bioactive lipids [17] or VEGF [18]. The role of CTGF in angiogenesis is under debate ([19] and citations therein). By forming a complex with VEGF‐165, CTGF inhibited VEGF‐induced angiogenesis [20]. Reduced microvessel density was detected within human tumours overexpressing CTGF [21], whereas earlier studies suggested that tumour cell‐derived CTGF facilitated angiogenic microcapillary formation [22]. In several in vivo and in vitro models, CTGF by itself was shown to promote endothelial cell adhesion, migration and tube formation [23]. The functional role of CTGF thus seems to be dependent on the cellular context, especially on the presence of other growth factors such as VEGF, which may serve as binding partners.
CTGF expression is primarily regulated at the transcriptional level, integrating multiple signalling pathways [24]. Besides soluble mediators, mechanical forces generated by blood flow control CTGF gene expression in endothelial cells [25]. Alterations of the microtubular network and the actin cytoskeleton are involved in the transcriptional regulation of CTGF gene expression in microvascular endothelial cells [26]. To the best of our knowledge, regulation of CTGF expression by alterations of the chromatin structure has not been addressed so far. Gene expression profiling of HDI‐treated cancer cells provided conflicting data. Most array data did not suggest CTGF to be regulated by HDACs, whereas variable up‐regulation was observed in certain hepatoma cell lines [27]. Gray et al. reported strong up‐regulation of CTGF in renal cell carcinoma cells and Hep3B cells upon stimulation with different types of HDIs [28]. Furthermore, epigenetic silencing by hypermethylation of CTGF was correlated with a loss of function in ovarian cancer cells [29]. Apart from microarray data, CTGF regulation by HDAC inhibitors in non‐tumour cells has not been investigated yet.
Our previous work concentrated on the regulation of CTGF in different renal cells, including endothelial cells and tubular epithelial cells (e.g.[26, 30]). As outlined earlier, regulation of CTGF was likely to occur in the context of HDIs interfering with TGF‐β–mediated tubular epithelial‐to‐mesenchymal transition, but could also be envisaged related to HDI–mediated alterations of gene expression in endothelial cells. Therefore, it seemed warranted to investigate the regulation of CTGF by HDIs in different renal cell types and to address the molecular mechanisms, which might play a role in CTGF regulation.
Materials and methods
Materials
Mithramycin, valproic acid and sodium butyrate were obtained from Sigma (Taufkirchen, Germany). Trichostatin A from Streptomyces platensis was from Serva (Heidelberg, Germany). Suberoylanilide hydroxamic acid (SAHA) was kindly provided by D. P. Zlotos (Pharmaceutical Institute, University of Wuerzburg, Germany). Lysophosphatidic acid (LPA) was purchased from Biomol (Hamburg, Germany) and human recombinant TGF‐β1 was obtained from tebu‐bio (Offenbach, Germany). Appropriate solvent controls were used whenever cells were treated with substances, which had to be dissolved in DMSO. Protease inhibitor cocktail (Complete™) was obtained from Roche Diagnostics (Mannheim, Germany). Fetal calf serum (FCS) was from Gibco‐BRL (Eggenstein, Germany) and Accutase™ from PAA Laboratories (Pasching, Austria).
Cell culture
The murine glomerular microvascular endothelial cell line (glEND.2) was kindly provided by R. Hallmann (Erlangen, Germany). Cells were characterized by positive staining for typical endothelial cell markers MECA‐32, CD‐31 and the lack of staining for mesangial cell markers such as α‐smooth muscle actin, α8‐integrin, as well as epithelial cell markers such as WT‐1 (Wilms tumour gene 1) and cytokeratin [31]. The cells were cultured as described [26]. Human umbilical vein endothelial cells (HUVECs) were isolated from freshly delivered umbilical cords and grown on 0.1% gelatine‐coated dishes as described [17]. After detachment with Accutase™ and expanding the cells at a 1:2 ratio, nearly confluent HUVEC cells were used the next day for the experiments. A human tubular epithelial cell line (HKC‐8) was kindly provided by J. Racusen and cultured as described [32]. If not indicated otherwise, confluent cells were used for the experiments.
Western blot analysis
Western blot analyses were performed with cellular lysates by standard procedures as described [17]. Proteins were separated by 10% and 12% SDS‐PAGE and transferred onto polyvinylidene fluoride (PVDF) membrane (Pall, Biosupport Division, Dreieich, Germany). Immunoreactive proteins were visualized by the enhanced chemiluminescence detection system (ECL‐Plus, Amersham Biosciences, Freiburg, Germany). The immunoreactive bands were quantified using the luminescent image analyzer (LAS‐1000 Image Analyzer, Fujifilm, Berlin, Germany) and AIDA 4.15 image analyzer software (Raytest, Berlin, Germany). To correct for equal loading and blotting, all blots were re‐detected with antibodies directed against tubulin or vinculin. For quantification purposes, the ratio of the specific protein band and a control protein was calculated.
The following antibodies were used: goat polyclonal anti‐CTGF (SC‐14939), rabbit polyclonal anti‐vinculin (SC‐5573), donkey anti‐goat IgG (SC‐2020) conjugated to horseradish‐peroxidase (Santa Cruz Biotechnology, Heidelberg, Germany), mouse monoclonal anti‐tubulin antibody E7, developed by Klymkowsky (Developmental Studies Hybridoma Bank, University of Iowa, IA); peroxidase‐conjugated sheep anti‐mouse IgG and donkey anti‐rabbit IgG secondary antibodies (Amersham Biosciences); rabbit polyclonal anti‐histone H3 and anti‐acetyl histone H3 (Upstate, Charlottesville, VA).
Histone isolation
Preparation of nuclear histones was performed by acidic extraction. In brief, cells were rinsed twice with ice‐cold PBS, scraped into 500 μl ice‐cold lysis buffer containing 10 mM PIPES (pH 6.8), 1 mM EGTA, 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% Triton X‐100 and protease inhibitor cocktail complete™. The cellular samples were incubated on ice for 10 min. and briefly mixed by vortexing. After centrifugation at 600 ×g for 10 min. at 4°C, the supernatant was removed and the nuclear pellet was mixed with 1 M sulfuric acid, incubated for 1 hr at 4°C and centrifuged at 12,000 ×g for 10 min. The extracted histones were precipitated by acetone (final concentration 90%) over night at −20°C. After centrifugation at 12,000 ×g for 10 min., the precipitated protein was dried for 30 min., resuspended in water and used for SDS‐PAGE.
RNA isolation and real‐time RT‐PCR
Total RNA was prepared from cultured endothelial cells using TriFast™ reagent from Peqlab (Erlangen, Germany). Ten nanograms of RNA were reverse transcribed with TaqMan reverse transcription reagents (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. cDNA was amplified using Power SYBR® MM reaction buffer including AmpliTaq® Gold DNA polymerase, dNTPs and the fluorescent dye SYBR Green, 200 nM of specific primers (100 nM each). The PCR reactions were carried out using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems) using the following thermal cycling profile: 2 min. 50°C, 40 cycles with 95°C, 15 sec. and 60°C, 1 min. Melting curve analysis at the end of the PCR monitored specificity of the amplified products. Primers used in real‐time RT‐PCR were designed to span exon–exon junctions to avoid detection of genomic DNA using Primer Express® software (Applied Biosystems). The following primers were used: murine CTGF (NM_010217.2) forward primer: 5′‐GCC CTA GCT GCC TAC CGA CT‐3′, reverse primer: 5′‐CAT AGT TGG GTC TGG GCC AA‐3′; 18S rRNA forward primer: 5′‐TTG ATT AAG TCC CTG CCC TTT GT‐3′, reverse primer: 5′‐CGA TCC GAG GGC CTC ACT A‐3′. Quantification of mRNA expression was carried out with respect to 18S rRNA as reference as described by PE Applied Biosystems (Perkin Elmer, Foster City, CA).
Reporter gene assays
The following constructs were used for transient transfection. The CTGF promoter construct (−418 to +42) cloned into pEGFP‐1 was kindly provided by R. Goldschmeding, Utrecht, The Netherlands (human CTGF, AF316367. It was subcloned into unique HindIII and EcoRI sites of pSEAP2‐basic (Clontech, Paolo Alto, CA). The putative Sp1/Sp3 binding sites 5′ and 3′ of the TATA‐box in the pCTGF‐418 promoter construct were mutagenized by introducing point mutations using the following primer pairs: for the 5′ mutation forward 5′‐GCC GCG AAA T CC CGG AGC GTA TAA AAG CCT CGG‐3′ and reverse 5′‐CCG GGA TTT CGC GGC TCG CCA ATG AGC TG‐3′, and for the 3′ mutation forward 5′‐CTC ATG CCG CAA TCG CCC AAA CTC ACA CAA‐3′ and reverse 5′‐CG A TT G CGG C AT GAG GCT TTT ATA CGC TCG GG‐3′. Mutations were verified by sequencing of the vector DNA.
Endothelial cells (glEND.2) were seeded at 70% confluence the day before transfection of the reporter constructs with jetPEI™ (Qbiogene, Heidelberg, Germany) according to the manufacturer’s instructions. Four hours after transfection, the cells were stimulated with TSA as indicated. Secreted alkaline phosphatase (SEAP) activity in the supernatant was detected with CSPD as chemiluminescent substrate (Phospho‐Light™ Reporter Gene Assay Kit, Tropix, Bedford, U.K.). Cells were cotransfected with a CMV promoter‐driven vector encoding β‐galactosidase to control for differences in transfection efficiency. Galactosidase activity was determined in cellular homogenates.
siRNA transfection
To down‐regulate FoxO1 (forkhead protein class O) or FoxO3a expression, endothelial cells (glEND.2) were transfected with FoxO1 (NM_019739.2) siRNA (sense 5′‐GCG‐GGC‐UGG‐AAG‐AAU‐UCA‐A‐3′, 50 nM), FoxO3a (NM_019740.2) siRNA (sense 5′‐GCU‐CUU‐GGU‐GGA‐UCA‐UCA‐A‐3′, 50 nM) or luciferase siRNA (100 nM) 3 hrs after seeding using HiPerFect (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s instructions. siRNAs were designed and synthesized by Eurogentec, Seraing, Belgium. Experiments were performed 24 hrs after transfection. The efficiency of the siRNA treatment was confirmed by Western blot analyses of FoxO1 and FoxO3a. FoxO3a was reduced by 73.5 ± 11.8%, means ± SD, n= 6. FoxO1 seemed to be down‐regulated to a similar extent, but expression in glEND.2 cells was too low to allow quantification.
Immunocytochemistry
glEND.2 cells were fixed with 3.5% paraformaldehyde in Dulbecco’s PBS (140 mM NaCl, 2.68 mM KCl, 1.47 mM KH2PO4 and 8.1 mM Na2HPO4) for 10 min. and afterwards permeabilized by 0.2% Triton X‐100 in Dulbecco’s PBS for 10 min. Cells were incubated with primary antibodies against CTGF (1:500, SC‐14939), FoxO3a (#7508, Cellular Signaling) and flag‐tag (Sigma‐Aldrich, Munich, Germany) overnight, and with secondary Alexa Fluor‐labelled antibodies (1:500, Invitrogen, Eugene, OR) for 45 min. After mounting, slides were viewed by using a Nikon fluorescent microscope (Nikon Eclipse 80i, Nikon, Duesseldorf, Germany). Digital images were recorded using MetaVue software (Universal Imaging, Downington, PA).
To analyze the effect of FoxO3a, cells were transfected 16 hrs before fixation with the triple mutant of FoxO3a (FLAG‐FoxO3a‐TM), kindly provided by J. Behrens, University of Erlangen‐Nuremberg [33]. For quantification purposes, about 100 cells in six randomly chosen fields were counted.
Immunohistochemistry
All aspects of care and handling of the animals were in accordance with the institutional guidelines. Part of the kidneys of 6‐week‐old male NMRI mice was fixed in methyl‐Carnoy solution (60% methanol, 30% chloroform and 10% glacial acetic acid). Tissues were dehydrated by bathing in increasing concentrations of methanol or isopropanol, respectively. After embedding in paraffin, 3‐μm sections were cut with a Leitz SM 2000 R microtome (Leica Instruments, Nussloch, Germany) and processed for immunohistochemistry. The following antibodies were used: goat anti‐CTGF (1:500, SC‐14939), rat anti‐Meca32 (undiluted hybridoma supernatant, Developmental Studies Hybridoma Bank); mouse anti‐acetylated histone H3 (1:500, ab1191, Abcam, Cambridge, U.K.). Alexa fluor‐labelled secondary antibodies were obtained from Invitrogen. After mounting, slides were viewed using a Zeiss or Nikon fluorescent microscope and digital images were recorded using MetaVue or Spot software (Diganostic Instruments, Sterling Heights, MI).
Statistical analysis
Data are presented as means ± S.D. To compare multiple measurements, ANOVA with Tukey Kramer multiple comparison test or Dunnett post hoc test was used (PRISM software, Graph Pad, San Diego, CA). The paired Student’s t‐test was used to compare two conditions. A P value < 0.05 was considered significant.
Results
Expression of acetylated histone H3 and CTGF in mouse kidneys
To get an insight into the histone acetylation status of the kidney, tissue sections of mice were stained with an antibody directed against acetylated histone H3. Acetylated histone was primarily localized to interstitial cells with only low staining detectable in tubular epithelial cells (Fig. 1, upper panel). Double staining with the endothelial cell marker Meca32 showed partial colocalization of acetylated histone with endothelial cells indicating that endothelial cells as well as additional interstitial cells were transcriptionally active.
Figure 1.
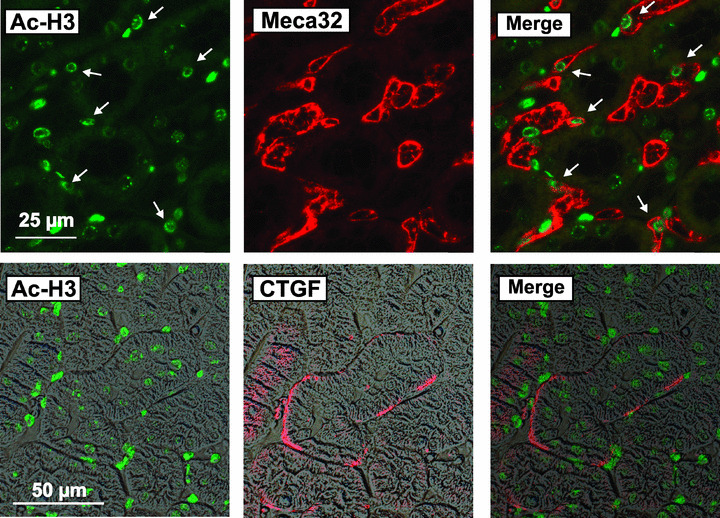
Detection of CTGF and acetylated histone H3 in mouse kidney sections. Sections of mouse kidneys were double‐stained for acetylated histone H3 (Ac‐H3) and the endothelial cell marker Meca32 (upper panels) or Ac‐H3 and CTGF (lower panels). Images taken with polarization filters were used as background in the lower panels to show the localization of CTGF staining. Data are representative of eight individual kidneys. Arrows indicate cells positive for Ac‐H3 and Meca32.
CTGF expression was weak in renal tissue of untreated mice. Immunoreactivity was detectable at the basolateral membrane of individual tubules (Fig. 1, lower panel). CTGF is a secreted protein and bound to the extracellular matrix. Therefore, it was interesting to note that active interstitial cells, that is, cells with a strong expression of acetylated histone H3, were often found in areas of CTGF expression (Fig. 1, lower panel). This suggested that CTGF was not derived exclusively from epithelial cells but might also be secreted by neighbouring interstitial cells.
To get a better insight into the regulatory mechanisms of CTGF expression in terms of HDAC activity, we investigated CTGF regulation in different types of endothelial cells and in the established human tubular proximal epithelial cell line HKC‐8.
Regulation of CTGF protein and mRNA in endothelial cells by the HDAC inhibitor trichostatin A (TSA)
Because microvascular endothelial cells are the target of angiogenesis and vascular remodelling, we analyzed the regulation of CTGF gene expression by the HDI trichostatin A (TSA) in a renal microvascular endothelial cell line, glEND.2. Treatment with TSA (330 or 660 nM corresponding to 100 and 200 ng/ml) induced CTGF protein expression in a time‐ and concentration‐dependent manner as determined by Western blot analysis of cell‐associated protein (Fig. 2A). A significant four‐ to fivefold up‐regulation of CTGF was obtained with 330 nM TSA after 6 hrs. Lower concentrations (e.g. 65 nM) were sufficient to up‐regulate CTGF after 24 hrs (Fig. 2B). CTGF protein was barely detectable in cell culture supernatants and was not significantly altered upon treatment with TSA. Corresponding to the increase in CTGF protein, up‐regulation of CTGF mRNA was detected (Fig. 2C). The increase of CTGF expression was also observed in primary cultures of HUVEC, although the induction was less pronounced (about threefold after 24 hrs; Fig. 2D).
Figure 2.
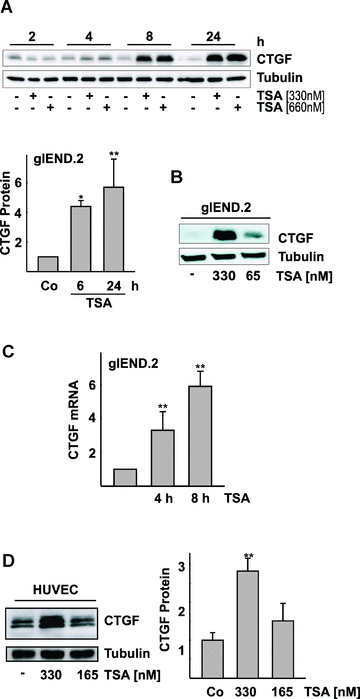
Time‐ and concentration‐dependent induction of CTGF protein and mRNA in endothelial cells by the HDAC inhibitor TSA. (A) Microvascular renal endothelial cells, glEND.2, were treated with of TSA (330 and 660 nM) for the times indicated. CTGF was detected by Western blot analysis. The graph summarizes the data of three (6 hrs) and five (24 hrs) experiments (stimulation with 330 nM TSA). Expression of CTGF in control cells was set to 1 at both time points. *P < 0.05, **P < 0.01 compared with control cells. (B) glEND.2 cells were treated with 330 and 65 nM TSA for 24 hrs. CTGF was detected by Western blotting. (C) glEND. 2 cells were stimulated with TSA (330 nM) for 4 and 8 hrs. CTGF mRNA expression was analyzed by real‐time RT‐PCR. The mRNA expression of control cells was set to 1. Data are means ± S.D. of four experiments. P < 0.05 compared with control cells. (D) HUVEC were stimulated with TSA (165 or 330 nM) overnight. CTGF was detected by Western blot analysis. The graph summarizes n= 3 experiments, **P < 0.01 compared with control‐stimulated cells.
Induction of CTGF in endothelial cells by chemically diverse HDI
Induction of CTGF expression was not only restricted to TSA but also observed with other HDAC inhibitors (Fig. 3A). Compared to TSA, higher concentrations of SAHA (0.5–10 μM) were needed in accordance with the lower potency of this compound [34]. Valproic acid was less effective, whereas sodium butyrate, which does not inhibit HDAC‐6, a tubulin deacetylase [35], clearly induced CTGF expression. Induction of CTGF by TSA and sodium butyrate correlated with a comparable increase in histone acetylation (Fig. 3B). These results indicated that the induction of CTGF protein was not dependent on the chemical class of HDAC inhibitor, and occurred independently of tubulin acetylation.
Figure 3.
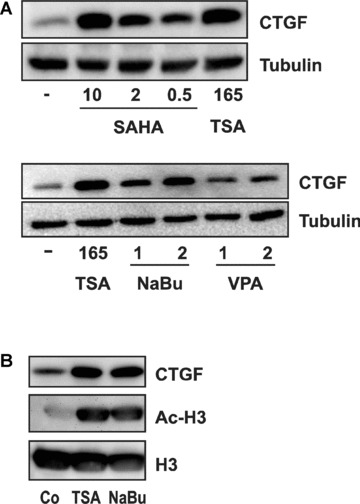
Induction of CTGF by chemically different HDIs. (A) glEND.2 cells were stimulated for 24 hrs with different HDAC inhibitors: TSA (165 nM), SAHA (0.5, 2 and 10 μM), sodium butyrate (NaBu, 1 and 2 mM) and valproic acid (VPA, 1 and 2 mM). Cellular extracts were separated by SDS PAGE and CTGF protein was detected by Western blotting. (B) glEND.2 cells were stimulated with TSA (165 nM) or sodium butyrate (NaBu, 2 mM) for 6 hrs. Histone H3 and acetylated histone H3 (Ac‐H3) were detected in nuclear extracts, whereas CTGF was detected in the cytosolic fraction.
Cooperative effect of receptor‐mediated induction of CTGF and TSA
To address the interaction of TSA with known stimuli of CTGF expression, glEND.2 cells were pre‐incubated with TSA overnight and then stimulated with TGF‐β or LPA. Pre‐treatment with TSA facilitated the up‐regulation of CTGF protein by these stimuli, which induce CTGF via Smads and RhoA, respectively (Fig. 4).
Figure 4.
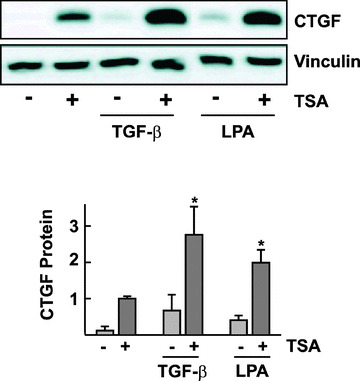
Facilitation of TGF‐β‐ and LPA‐mediated CTGF protein expression by TSA. glEND.2 cells were pre‐incubated for 20 hrs with TSA (330 nM) and then stimulated with TGF‐β (TGF‐β, 5 ng/ml, 5 hrs) or LPA (LPA, 10 μM, 2 hrs). CTGF protein from cellular homogenates was analyzed by Western blotting. The graph summarizes the results of n= 4 experiments; *P < 0.05 ANOVA with Tukey’s post‐test, TSA plus TGF‐beta or TSA plus LPA compared with any stimulus alone,
Molecular mechanisms involved in TSA‐mediated up‐regulation of CTGF in endothelial cells
Specificity of HDIs is obtained by interaction with transcription factors. This became obvious when promoter constructs of different length were compared, both of which were stimulated significantly compared with non‐stimulated controls (P < 0.05, n= 3). Stimulation of a construct that consisted of 59 bp of the CTGF promoter was significantly less than stimulation of the core promoter of 418 bp (Fig 5A). Sp1/Sp3 transcription factors seem to be common interaction partners of HDACs ([36] and citations therein). Therefore, we analyzed mithramycin, which intercalates into GC‐rich sequences and thus interferes with Sp1/Sp3 binding to the DNA. Mithramycin reduced the basal and the TSA‐stimulated CTGF expression of glEND.2 cells within 4 hrs (Fig. 5B). However, more detailed analyses did not confirm a specific role for Sp1/Sp3. Transfection of glEND.2 cells with dominant negative Sp1 and Sp3 cDNAs did not reduce CTGF promoter activity (data not shown). Furthermore, the 418 bp CTGF core promoter construct was activated by TSA even when the Sp1/Sp3 sites located 5′and 3′ to the TATA box were mutated (Fig. 5C). Taken together, these data suggested that the effect of mithramycin was not mediated by the Sp1/Sp3 sites that are essential for CTGF expression in scleroderma fibroblasts [37], not excluding interaction with Sp1/Sp3 transcription factors bound elsewhere in the gene. In additional studies, we compared the core promoter construct to a promoter construct where the Smad binding site and the so‐called basal control element BCE‐1 were mutated [38]. However, TSA‐mediated activation of the promoter was not affected by these mutations (data not shown).
Figure 5.
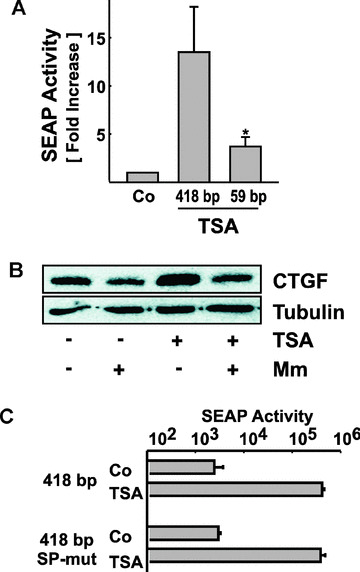
Regulation of CTGF promoter activity by TSA. (A) The 418 bp CTGF core promoter (418 bp) or a shortened promoter (59 bp) were transiently transfected into glEND.2 cells. SEAP activities were analyzed after treatment with 33 nM TSA for 24 hrs. In each experiment, the activity of the pCTGF‐418 core promoter in cells treated with solvent was set to 1. Data are means ± S.D. of four independent experiments performed in duplicate. *P < 0.05, paired Student’s t‐test, stimulation of 418 bp versus 59 bp. (B) glEND.2 cells were pre‐incubated for 30 min. with mithramycin (Mm, 10 μM) and then stimulated with TSA (330 nM, 4 hrs). CTGF protein was detected in cellular homogenates by immunoblotting. (C) glEND.2 cells were transfected with the 418 bp CTGF core promoter or with a construct with mutated proximal Sp1/Sp3 sites. Transfected cells were treated with or without TSA (33 nM) for 24 hrs. The graph shows a representative experiment performed with triplicate incubations (means ± S.D.).
FoxO transcription factors as regulators of CTGF expression
To investigate the effect of more endothelial cell‐specific transcription factors, we turned to FoxOs (forkhead protein class O), which have been shown to activate CTGF in endothelial cells but not in thymocytes [39]. FoxO proteins are negatively regulated by phosphatidylinositol‐3 kinase (PI3K)–AKT signalling [40]. When this pathway was inhibited in glEND.2 cells by the PI3K inhibitor LY294002 (10 μM), FoxO3a was translocated to the nucleus, whereas control cells showed cytosolic staining of FoxO3a (Fig. 6A, upper panel). Only few of the control cells showed CTGF immunoreactivity. Inhibition of PI3K – AKT signalling strongly induced CTGF synthesis. As a secreted protein CTGF was detected most prominently in the Golgi. To provide further evidence for a role of FoxO proteins in CTGF regulation, a constitutively active form of FoxO3a, the triple mutant FLAG‐FoxO3a‐TM [33] was transiently transfected into glEND.2 cells. The cells are poorly transfectable with a maximal transfection efficiency of about 20% (Fig. 6A, lower panel). As an active form of FoxO3a was transfected, it localized predominantly to the nucleus. Sixteen hours after transfection, about 20% of all cells showed strong staining for CTGF (18%± 5% in six randomly chosen sections), of which 54%± 8% were positive for FoxO3a. Colocalization of FoxO3a and CTGF was significant compared with CTGF expression in the whole cell population (P < 0.01). These results showed directly that the transcription factor FoxO3a activates CTGF expression.
Figure 6.
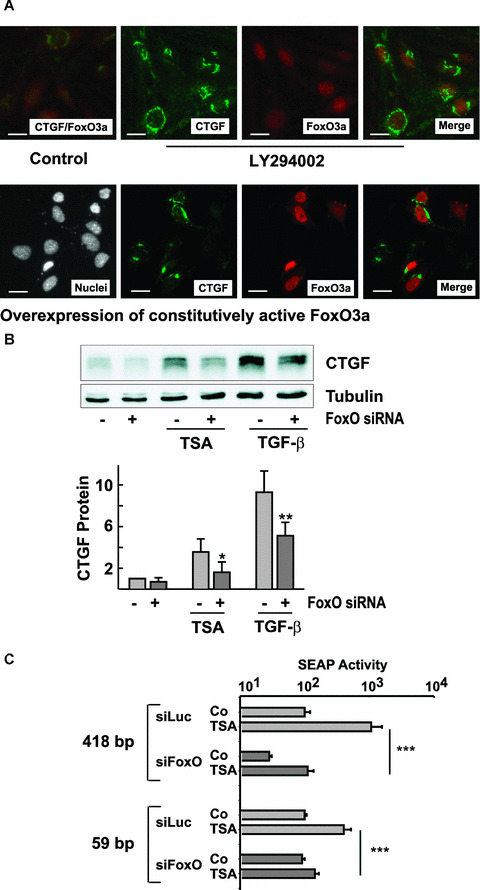
Role for FoxO proteins in CTGF induction. (A) Upper panel: glEND.2 cells were treated with the PI3K inhibitor LY294002 (10 μM) for 2 hrs. Lower panel: glEND.2 cells were transfected with a flag‐tagged construct coding for constitutively active FoxO3a (FLAG‐FoxO3a‐TM) for 16 hrs. FoxO3a, flag‐tag (representing FoxO3a) and CTGF were detected by immunocytochemistry. Nuclei were stained with Hoechst 33258. To show co‐expression of CTGF and FoxO3a, images were merged using Spot software. (B) glEND.2 cells were incubated with FoxO1/3a siRNA (50 ng/ml each) or with luciferase siRNA (100 ng/ml) overnight and then stimulated with TSA (165 nM) or TGF‐β (5 ng/ml) for 6 hrs. The graph summarizes data of n= 5 experiments. *P < 0.05, **P < 0.01 paired Student’s t‐test, cells treated with FoxO siRNA versus cells incubated with luciferase siRNA. (C) glEND2 cells were transfected with siRNA against FoxO1/3a or against luciferase. The next day the cells were transfected with the promoter constructs as indicated. Stimulation with TSA (33 nM) was overnight. Data are means ± S.D. of two experiments with triplicate transfections. SEAP activity in luciferase‐treated control cells was set to 100. ***P < 0.001, ANOVA with Tukey Kramer multiple comparison test.
To analyze the role of FoxO proteins in TSA‐induced expression of CTGF, endothelial cells were transfected with siRNA directed against FoxO1 and FoxO3a, the main FoxO transcription factors in endothelial cells, which have been shown to partially compensate each other [41]. Up‐regulation of CTGF by TSA was significantly reduced in FoxO1/3a siRNA‐treated cells compared with cells treated with siRNA directed against luciferase (Fig. 6B). Furthermore, TGF‐β–mediated induction of CTGF was also partially reduced when FoxO1/3 proteins were down‐regulated (42%± 20%, n= 5, P < 0.05 compared with cells treated with siRNA against luciferase). The involvement of FoxO proteins in the induction of CTGF expression was confirmed by promoter analyses (Fig. 6C). Activation of the core promoter was significantly inhibited when FoxO proteins were down‐regulated by siRNA. As also shown in Fig. 5A, the shortened promoter was much less regulated by TSA, but still sensitive to FoxO siRNA. These data indicated that FoxO proteins are involved in the up‐regulation of CTGF by HDAC inhibitors in endothelial cells.
Regulation of CTGF expression in human tubular epithelial cells
In contrast to endothelial cells, CTGF protein levels were not increased by TSA in confluent human proximal tubular epithelial cells (cell line HKC‐8), but rather decreased upon prolonged incubation (Fig. 7A). Histone acetylation was observed upon treatment with TSA (100 ng/ml) or SAHA (10 μM) for 6 hrs, indicating that the missing up‐regulation of CTGF was not due to a poor response of the epithelial cells to treatment with HDIs (Fig. 7B). When subconfluent cells were analyzed, an increase of CTGF was observed after prolonged incubation (Fig. 7C). A comparable cell density‐dependent regulation of CTGF was also detected in another well‐established human proximal tubular cell line (HK‐2, data no shown). Inhibition of PI3K–AKT signalling with the PI3K inhibitor LY294002 rapidly down‐regulated AKT activity as shown by the reduced expression of phosphorylated AKT (Fig. 7D). In contrast to the up‐regulation of CTGF observed in endothelial cells, inhibition of PI3K–AKT signalling by LY294002 inhibited TSA‐mediated up‐regulation of CTGF in subconfluent HKC‐8 cells (Fig. 7C). These results indicated that FoxO proteins, which are activated upon inhibition of PI3K/AKT signalling, were not involved in TSA‐ mediated up‐regulation of CTGF in subconfluent epithelial cells.
Figure 7.
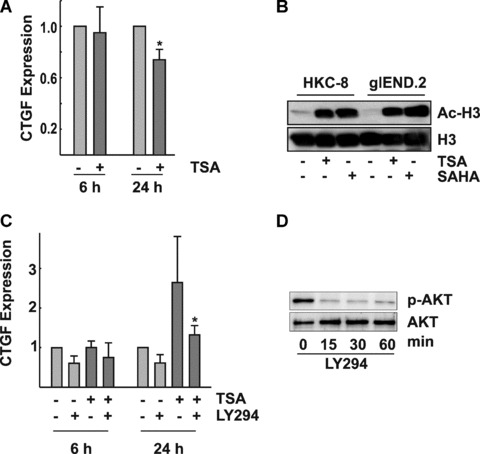
Cell density–dependent modulation of CTGF expression by TSA in epithelial cells. (A) Confluent HKC‐8 cells were incubated with TSA (330 nM) for the times indicated. CTGF expression was detected in cellular homogenates by Western blotting. The graph summarizes data of n= 4 experiments, CTGF expression in control cells was set to 1 at each time point; *P < 0.05. (B) HKC‐8 and for comparison glEND.2 cells were treated with SAHA (10 μM) or TSA (330 nM) for 6 hrs. Immunoreactive‐acetylated histone H3 (Ac‐H3) was detected in nuclear extracts. Re‐detection of total histone H3 served as loading control. (C) HKC‐8 cells were seeded at low density (20,000 cells/cm2) and were stimulated the next day with TSA (330 nM) in the presence or absence of 10 μM LY294002 for the times indicated. Expression of CTGF in control cells was set to 1 at each time point. Data are means ± S.D. of n= 3 experiments. *P < 0.05 compared with cells stimulated with TSA. (D) HKC‐8 cells were treated with 10 μM LY294002 for the times indicated. Expression of phospho‐AKT (p‐AKT) and AKT was detected by Western blotting.
TGF‐β is one of the most important mediators of mesenchymal transition of epithelial cells and up‐regulates CTGF in these cells. A threefold up‐regulation of CTGF protein was observed, when HKC‐8 cells were stimulated for 6 hrs (Fig. 4). In contrast to endothelial cells, where pre‐incubation with TSA facilitated TGF‐β–mediated induction of CTGF, pre‐incubation with TSA abrogated induction of CTGF in epithelial cells (Fig. 8).
Figure 8.
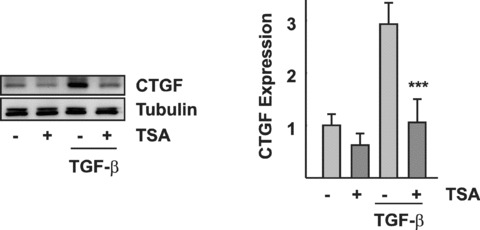
Inhibition of TGF‐β–mediated induction of CTGF in epithelial cells. Confluent HKC‐8 cells were pre‐incubated with TSA (330 nM) overnight and then stimulated with TGF‐β (5 ng/ml) for 6 hrs. CTGF was detected by Western blot analysis. The graph summarizes the results of n= 3 experiments. ***P < 0.001 compared with TGF‐β–stimulated cells.
Discussion
The effects of HDIs have been intensively studied in tumour cells, whereas much less is known about other tissues. In the present study, we provide evidence for different basal activity of the HAT/HDAC system in renal endothelial and epithelial cells in situ and differential regulation of gene expression in renal endothelial and epithelial cells treated with HDIs.
There was a striking difference in the expression of acetylated histones in different cell types of the kidney, especially tubular cells, which hardly expressed acetylated histone H3, whereas interstitial cells, among them endothelial cells, showed strong immunoreactivity. This indicated that even in control conditions certain types of renal cells are characterized by a high transcriptional activity. Although this study focussed on the molecular mechanisms of HDI‐mediated regulation of CTGF expression, it will be interesting to extend the in situ studies to gain insight into alterations of the transcriptional activity of the different cell types in the kidney, which is expected to occur when HDIs are used for the treatment of tumours or other diseases.
CTGF is a secreted matricellular protein, which remains attached to the extracellular matrix. Accordingly, CTGF immunoreactivity was detected primarily as a focussed line between interstitial cells and epithelial cells. Interstitial cells with increased levels of acetylated histone H3 were frequently detected in the areas of CTGF deposition. This raised the possibility that these cells might contribute to the secreted CTGF protein. The role of interstitial cells, endothelial cells and possibly fibroblasts or dendritic cells, in renal CTGF expression has not yet been investigated in detail and needs further consideration.
In terms of molecular regulation of CTGF expression, marked differences were detectable between epithelial cells and endothelial cells, CTGF being up‐regulated in endothelial cells upon HDAC inhibition, whereas no effect or down‐regulation was observed in confluent epithelial cells. It was interesting to note that prolonged incubation of subconfluent epithelial cells led to a variable degree of CTGF up‐regulation. The long time lag between stimulation and up‐regulation suggested an indirect effect, which may be related to rather non‐specific toxic effects of HDIs, which have been reported recently in rat renal proximal tubular cells [42]. Subconfluent epithelial cells do not represent epithelial cell as found in healthy kidneys but may rather represent epithelial cells in injured tubuli. In confluent epithelial cells, we did not observe any signs of cytotoxicity upon treatment with HDIs.
Furthermore, pre‐treatment of epithelial cells with TSA prevented TGF‐β–mediated up‐regulation of CTGF in epithelial cells. These data are in accordance with previous findings of Yoshikawa et al., who observed reduced expression of collagen type I under comparable conditions [15]. Furthermore, they observed up‐regulation of BMP‐7, which is a functional antagonist of CTGF [43, 44] and may also play a role in the observed down‐regulation of CTGF, not excluding other factors. Whether, based on these results, HDIs can be regarded as tubular protective agents needs some caution. Considering the different effects of TSA in confluent and subconfluent epithelial cells, the latter being more likely to be found in injured kidneys, an in vivo effect of HDIs cannot be easily predicted based on the present data.
CTGF was clearly up‐regulated when endothelial cells were incubated with various types of HDIs. Different chemical types of HDI regulate overlapping sets of genes [28, 45], based on common alterations of histone acetylation. In our studies, we detected quantitative but no qualitative differences in the regulation of CTGF expression when different HDIs were used. This was especially notable related to sodium butyrate and valproate, which inhibit all HDACs with the exception of HDAC6 and HDAC10 [36, 46]. In addition to histones, HDAC6 acetylates tubulin and thus affects the organization of the cytoskeleton [35, 47]. The expression of CTGF is strongly dependent on changes of the cytoskeleton [24]. This raised the possibility that modulation of tubulin might be relevant for HDI‐mediated induction of CTGF. However, sodium butyrate proved to be an efficient inducer of CTGF arguing against a major role of HDAC6/tubulin modification in CTGF regulation.
Transient transfection experiments were used to get an insight into transcription factors possibly involved in TSA‐mediated up‐regulation of CTGF. Involvement of multiple transcription factors was suggested by the higher activation of the core promoter compared with the shortened promoter. Sp1/Sp3 transcription factors seem to be common interaction partners of HDACs ([48] and citations therein) and different models have been suggested to contribute to the multiple interactions between Sp1/Sp3 and HDACs [49]. Our analyses, however, did not support a major role for Sp1/Sp3 as regulators in TSA‐mediated activation of the core promoter of CTGF in endothelial cells. Zhang et al. demonstrated a link between PI3K and SP1, which was essential for TSA‐activated gene expression of the luteinizing hormone receptor [49]. In fibroblasts, up‐regulation of CTGF was inhibited when the cells were treated with inhibitors against PI3K, mTOR, Sp1 or Smad3 [50]. Whether a comparable link is involved in the PI3K‐dependent up‐regulation of CTGF in subconfluent epithelial cells remains to be investigated.
Most interestingly, we observed a role for transcription factors of the FoxO family, FoxO1 and FoxO3a in CTGF regulation in endothelial cells. These transcription factors belong to a group of multi‐functional proteins involved in various cellular functions such as differentiation, proliferation or metabolism [40, 51] and exert cell type‐specific functions in endothelial cells [39]. FoxO3a is negatively regulated by PI3K–AKT signalling, and accordingly we observed nuclear localization of FoxO3a when this pathway was inhibited. Concomitantly, up‐regulation of CTGF was detectable. The link between FoxO3a and CTGF was confirmed by overexpression of an active mutant of FoxO3a. Targeting FoxO1 and FoxO3a, the most abundant FoxO proteins in endothelial cells, by siRNA we observed interference with TSA‐mediated up‐regulation of CTGF at the protein as well as the promoter level. FoxO binding sites are found in the core promoter of CTGF [40, 42], and our data suggest that FoxOs may interact with the basic transcription complex activated by TSA. Rather unexpectedly, FoxO siRNA also affected the activation of a shortened promoted construct, which did not contain FoxO binding sites as deduced from its sequence. In a recent report, it was shown that FoxO1, and possibly other FoxO proteins as well, are able to decondense linker chromatin [52]. It remains to be determined how FoxOs interact with HDAC inhibitors in the activation of the short promoter construct, which was far less pronounced than the activation of the core promoter.
In contrast to endothelial cells, there was no evidence for an up‐regulation of CTGF by FoxO proteins in epithelial cells. Inhibition of PI3K/AKT signalling did not induce CTGF expression, but inhibited the late up‐regulation of CTGF by TSA, which was observed in subconfluent cells. FoxO proteins may thus contribute to the differential regulation of CTGF by HDIs.
Cell type–specific regulation of CTGF was most obvious when cells were treated with TSA and other (patho)physiologically relevant stimuli, for example, TGF‐β. In epithelial cells, TSA impaired TGF‐β–induced up‐regulation of CTGF, whereas increased CTGF expression was observed in endothelial cells. Interactions between HDI signalling and TGF‐β signalling have been observed in several cellular systems resulting in both interference with or support of gene regulation. Modulation of TGF‐β receptor expression has been described in several tumour cell lines [53, 54] and may also play a role in non‐tumour cells. RhoA‐Rho kinase signalling is activated by LPA, and in an earlier study we provided evidence for G‐actin and serum response factor as downstream regulators in RhoA‐mediated expression of CTGF [26]. Pre‐treatment of glEND.2 cells with TSA facilitated LPA‐induced CTGF expression, suggesting interaction of the activated basal transcription complex with multiple transcription factors, including serum response factor. In summary, our data indicate that HDAC inhibition per se is not sufficient to induce CTGF expression, but is functional only in the context of other transcription factors, stimulatory and/or inhibitory.
CTGF is a matricellular protein and its biological effects are largely regulated by the interaction with other extracellular proteins, matrix proteins as well as growth factors. It is thus no contradiction that CTGF on its own or overexpressed in cells and tissues has been described as a pro‐angiogenic factor (summarized in [23]), whereas it is anti‐angiogenic when VEGF is present [20, 55]. In tumours treated with HDIs, endothelial‐derived CTGF may thus contribute to the anti‐angiogenic properties of HDIs observed in vivo, similarly as suggested for semaphorin III, a competitor of VEGF‐165 binding to neuropilin‐1, which was induced by HDIs in endothelial cells [7]. CTGF may thus represent an additional target for the interruption of the angiogenic loop by HDIs in solid tumour therapy.
The role of HDI‐modulated CTGF expression in the kidney needs further investigation. Thus far, no in vivo data have been obtained to appreciate the contribution of individual cell types to CTGF synthesis in healthy or diseased kidneys. Most notably, epigenetic regulation of CTGF in non‐endothelial interstitial cells has not yet been addressed. These cells showed a very high immunoreactivity of acetylated histones and are thus expected to be highly active cells.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft SFB 423‐B3 and the Interdisciplinary Center for Clinical Research (IZKF) at the University Hospital of the University of Erlangen‐Nuremberg, project D5. C.K. received a fellowship from the IZKF, Erlangen. The excellent technical assistance of M. Rehm and A. Ebenau is highly appreciated.
References
- 1. Eberharter A, Ferreira R, Becker P. Dynamic chromatin: concerted nucleosome remodelling and acetylation. Biol Chem . 2005; 386: 745–51. [DOI] [PubMed] [Google Scholar]
- 2. Drummond DC, Noble CO, Kirpotin DB, et al . Clinical development of histone deacetylase inhibitors as anticancer agents. Ann Rev Pharmacol Toxicol . 2005; 45: 495–528. [DOI] [PubMed] [Google Scholar]
- 3. Schneider‐Stock R, Ocker M. Epigenetic therapy in cancer: molecular background and clinical development of histone deacetylase and DNA methyltransferase inhibitors. IDrugs . 2007; 10: 557–61. [PubMed] [Google Scholar]
- 4. Hellebrekers DMEI, Griffioen AW, vam Engeland M. Dual targeting of epigenetic therapy in cancer. Biochim Biophys Acta – Rev Cancer . 2007; 1775: 76–91. [DOI] [PubMed] [Google Scholar]
- 5. Qian DZ, Kato Y, Shabbeer S, et al . Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006; 634–42. [DOI] [PubMed] [Google Scholar]
- 6. Kim MS, Kwon HJ, Lee YM, et al . Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med . 2001; 7: 437. [DOI] [PubMed] [Google Scholar]
- 7. Deroanne CF, Bonjean K, Servotte S, et al . Histone deacetylases inhibitors as anti‐angiogenic agents altering vascular endothelial growth factor signaling. Oncogene . 2002; 21: 427–36. [DOI] [PubMed] [Google Scholar]
- 8. Hahnen E, Eyupoglu IY, Brichta L, et al . In vitro and ex vivo evaluation of second‐generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem . 2006; 98: 193–202. [DOI] [PubMed] [Google Scholar]
- 9. Faraco G, Pancani T, Formentini L, et al . Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol . 2006; 70: 1876–84. [DOI] [PubMed] [Google Scholar]
- 10. Gray SG, De Meyts P. Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes Metab Res Rev . 2005; 21: 416–33. [DOI] [PubMed] [Google Scholar]
- 11. Bhavsar P, Ahmad T, Adcock IM. The role of histone deacetylases in asthma and allergic diseases. J Allergy Clin Immunol . 2008; 121: 580–4. [DOI] [PubMed] [Google Scholar]
- 12. Gray SG, Dangoud, F . Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics . 2006; 1: 67–75 [DOI] [PubMed] [Google Scholar]
- 13. Kee HJ, Sohn IS, Nam KI, et al . Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation . 2006; 113: 51–9. [DOI] [PubMed] [Google Scholar]
- 14. Lee TM, Lin MS, Chang NC. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol . 2007; 293: H968–77. [DOI] [PubMed] [Google Scholar]
- 15. Yoshikawa M, Hishikawa K, Marumo T, et al . Inhibition of histone deacetylase activity suppresses epithelial‐to‐mesenchymal transition induced by TGF‐beta1 in human renal epithelial cells. J Am Soc Nephrol . 2007; 18: 58–65. [DOI] [PubMed] [Google Scholar]
- 16. Grotendorst GR. Connective tissue growth factor: a mediator of TGF‐beta action on fibroblasts. Cytokine Growth Factor Rev . 1997; 8: 171–9. [DOI] [PubMed] [Google Scholar]
- 17. Muehlich S, Schneider N, Hinkmann F, et al . Induction of connective tissue growth factor (CTGF) in human endothelial cells by lysophosphatidic acid, sphingosine‐1‐phosphate, and platelets. Atherosclerosis . 2004; 175: 261–8. [DOI] [PubMed] [Google Scholar]
- 18. Suzuma K, Naruse K, Suzuma I, et al . Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, flt1, and phosphatidylinositol 3‐kinase‐ akt‐dependent pathways in retinal vascular cells. J Biol Chem . 2000; 275: 40725–31. [DOI] [PubMed] [Google Scholar]
- 19. Kuiper EJ, Roestenberg P, Ehlken C, et al . Angiogenesis is not impaired in connective tissue growth factor (CTGF) knock‐out mice. J Histochem Cytochem . 2007; 55: 1139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Inoki I, Shiomi T, Hashimoto G, et al . Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF‐induced angiogenesis. FASEB J . 2002; 16: 219–21. [DOI] [PubMed] [Google Scholar]
- 21. Chang CC, Lin MT, Lin BR, et al . Effect of connective tissue growth factor on hypoxia‐inducible factor 1alpha degradation and tumor angiogenesis. J Natl Cancer Inst . 2006; 98: 984–95. [DOI] [PubMed] [Google Scholar]
- 22. Shimo T, Nakanishi T, Nishida T, et al . Involvement of CTGF, a hypertrophic chondrocyte‐specific gene product, in tumor angiogenesis. Oncology . 2001; 61: 315–22. [DOI] [PubMed] [Google Scholar]
- 23. Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine‐rich 61 (CYR61). Angiogenesis . 2002; 5: 153–65. [DOI] [PubMed] [Google Scholar]
- 24. Chaqour B, Goppelt‐Struebe M. Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J . 2006; 273: 3639–49. [DOI] [PubMed] [Google Scholar]
- 25. Cicha I, Goppelt‐Struebe M, Muehlich S, et al . Pharmacological inhibition of RhoA signaling prevents connective tissue growth factor induction in endothelial cells exposed to non‐uniform shear stress. Atherosclerosis . 2008; 196: 136–45. [DOI] [PubMed] [Google Scholar]
- 26. Muehlich S, Cicha I, Garlichs CD, et al . Actin‐dependent regulation of connective tissue growth factor (CTGF). Am J Physiol Cell Physiol . 2007; 292: 1732–8. [DOI] [PubMed] [Google Scholar]
- 27. Chiba T, Yokosuka O, Fukai K, et al . Cell growth inhibition and gene expression induced by the histone deacetylase inhibitor, trichostatin A, on human hepatoma cells. Oncology . 2004; 66: 481–91. [DOI] [PubMed] [Google Scholar]
- 28. Gray SG, Qian CN, Furge K, et al . Microarray profiling of the effects of histone deacetylase inhibitors on gene expression in cancer cell lines. Int J Oncol . 2004; 24: 773–95. [DOI] [PubMed] [Google Scholar]
- 29. Kikuchi R, Tsuda H, Kanai Y, et al . Promoter hypermethylation contributes to frequent inactivation of a putative conditional tumor suppressor gene connective tissue growth factor in ovarian cancer. Cancer Res . 2007; 67: 7095–105. [DOI] [PubMed] [Google Scholar]
- 30. Kroening S, Solomovitch S, Sachs M, et al . Regulation of connective tissue growth factor (CTGF) by hepatocyte growth factor in human tubular epithelial cells. Nephrol Dial Transplant . 2008. epub. [DOI] [PubMed] [Google Scholar]
- 31. Li ZD, Bork JP, Krueger B, et al . VEGF induces proliferation, migration, and TGF‐beta1 expression in mouse glomerular endothelial cells via mitogen‐activated protein kinase and phosphatidylinositol 3‐kinase. Biochem Biophys Res Commun . 2005; 334: 1049–60. [DOI] [PubMed] [Google Scholar]
- 32. Racusen LC, Monteil C, Sgrignoli A, et al . Cell lines with extended in vitro growth potential from human renal proximal tubule: characterization, response to inducers, and comparison with established cell lines. J Lab Clin Med . 1997; 129: 318–29. [DOI] [PubMed] [Google Scholar]
- 33. Dehner M, Hadjihannas M, Weiske J, et al . Wnt signaling inhibits Forkhead box O3a‐induced transcription and apoptosis through up‐regulation of serum‐ and glucocorticoid‐inducible kinase 1. J Biol Chem . 2008; 283: 19201–10. [DOI] [PubMed] [Google Scholar]
- 34. Wegener D, Hildmann C, Schwienhorst A. Recent progress in the development of assays suited for histone deacetylase inhibitor screening. Mol Genet Metab . 2003; 80: 138–47. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y, Li N, Caron C, et al . HDAC‐6 interacts with and deacetylates tubulin and microtubules in vivo . EMBO J . 2003; 22: 1168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guardiola AR, Yao TP. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J Biol Chem . 2002; 277: 3350–6. [DOI] [PubMed] [Google Scholar]
- 37. Holmes A, Abraham DJ, Chen Y, et al . Constitutive connective tissue growth factor expression in scleroderma fibroblasts is dependent on Sp1. J Biol Chem . 2003; 278: 41728–33. [DOI] [PubMed] [Google Scholar]
- 38. Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ . 1996; 7: 469–80. [PubMed] [Google Scholar]
- 39. Paik JH, Kollipara R, Chu G, et al . FoxOs are lineage‐restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell . 2007; 128: 309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci . 2007; 120: 2479–87. [DOI] [PubMed] [Google Scholar]
- 41. Gomis RR, Alarcon C, He W, et al . A FoxO‐Smad synexpression group in human keratinocytes. Proc Natl Acad Sci USA . 2006; 103: 12747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dong G, Wang L, Wang CY, et al . Induction of apoptosis in renal tubular cells by histone deacetylase inhibitors, a family of anticancer agents. J Pharmacol Exp Ther . 2008; 325: 978–84. [DOI] [PubMed] [Google Scholar]
- 43. Nguyen TQ, Goldschmeding R. Bone morphogenetic protein‐7 and connective tissue growth factor: novel targets for treatment of renal fibrosis Pharm Res . 2008; 25: 2416–26. [DOI] [PubMed] [Google Scholar]
- 44. Wang S, Hirschberg R. BMP7 antagonizes TGF‐beta‐dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol . 2003; 284: F1006–13. [DOI] [PubMed] [Google Scholar]
- 45. Peart MJ, Smyth GK, van Laar RK, et al . Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA . 2005; 102: 3697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gurvich N, Tsygankova OM, Meinkoth JL, et al . Histone deacetylase is a target of valproic acid‐mediated cellular differentiation. Cancer Res . 2004; 64: 1079–86. [DOI] [PubMed] [Google Scholar]
- 47. Hubbert C, Guardiola A, Shao R, et al . HDAC6 is a microtubule‐associated deacetylase. Nature . 2002; 417: 455–8. [DOI] [PubMed] [Google Scholar]
- 48. Gan Y, Shen YH, Utama B, et al . Dual effects of histone deacetylase inhibition by trichostatin A on endothelial nitric oxide synthase expression in endothelial cells. Biochem Biophys Res Commun . 2006; 340: 29–34. [DOI] [PubMed] [Google Scholar]
- 49. Zhang Y, Liao M, Dufau ML. Phosphatidylinositol 3‐kinase/protein kinase Czeta‐induced phosphorylation of Sp1 and p107 repressor release have a critical role in histone deacetylase inhibitor‐mediated derepression [corrected] of transcription of the luteinizing hormone receptor gene. Mol Cell Biol . 2006; 26: 6748–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khoo YT, Ong CT, Mukhopadhyay A, et al . Upregulation of secretory connective tissue growth factor (CTGF) in keratinocyte‐fibroblast coculture contributes to keloid pathogenesis. J Cell Physiol . 2006; 208: 336–43. [DOI] [PubMed] [Google Scholar]
- 51. van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol . 2007; 8: 440–50. [DOI] [PubMed] [Google Scholar]
- 52. Hatta M, Cirillo LA. Chromatin opening and stable perturbation of core histone: DNA contacts by FoxO1. J Biol Chem . 2007; 282: 35583–93. [DOI] [PubMed] [Google Scholar]
- 53. Ammanamanchi S, Brattain MG. Restoration of transforming growth factor‐beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J Biol Chem . 2004; 279: 32620–5. [DOI] [PubMed] [Google Scholar]
- 54. Moody TW, Nakagawa T, Kang Y, et al . Bombesin/gastrin‐releasing peptide receptor antagonists increase the ability of histone deacetylase inhibitors to reduce lung cancer proliferation. J Mol Neurosci . 2006; 28: 231–8. [DOI] [PubMed] [Google Scholar]
- 55. Hashimoto G, Inoki I, Fujii Y, et al . Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem . 2002; 277: 36288–95. [DOI] [PubMed] [Google Scholar]