

Abstract
Human macrophages express oestrogen receptors and are therefore competent to respond to the hormone present in their microenvironment, which is implicated in sexual dimorphism observed in several immune and autoimmune phenomena. An earlier study from this laboratory demonstrated 17β‐oestradiol (E2) induced apoptosis in macrophages derived from human peripheral blood monocytes and THP‐1 acute monocytic leukaemia cell line when Bcl‐2 was down‐regulated; however, the involvement of E2 receptor subtypes in the modulation of death pathways in these cells remain unknown. Using macrophages derived from THP‐1 human acute monocytic leukaemia cells as a model, we demonstrate that plasma membrane associated oestrogen receptor (ER) ‐α participate in E2 induced Bcl‐2 increase, through activation of the mitogen activated protein kinase (MAPK) pathway whereas cytosolic ER‐β transmits signals for the pro‐apoptotic event of Bax translocation. The mechanistic basis of Bax translocation comprised of ER‐β mediated increase in intracellular pH, facilitated by activation of the Na+–H+ exchanger. Intracellular alkalinization accompanied by concomitant Bcl‐2 increase and Bax migration does not cause cellular apoptosis; however, siRNA mediated down‐regulation of ER‐α during E2 exposure leads to inhibition of Bcl‐2 increase and consequently apoptosis due to the unopposed action of mitochondrial Bax. In summary, this study underscores the importance of integrative signalling modality from multiple oestrogen receptor pools in modulating oestrogen effects on human monocyte‐derived macrophage apoptotic signalling pathway, which opens new vistas to explore the use of selective oestrogen receptor modulators in apoptosis‐based therapies.
Keywords: oestrogen, macrophage, oestrogen receptor, Bcl‐2, Bax, apoptosis, alkalinization
Introduction
Macrophages derived from the differentiation of monocytes express steroid hormone receptors and are therefore sensitive to the hormones present in their microenvironment. The study of steroid hormone action on modulation of human macrophage function is of significant interest because these versatile cells are involved in the regulation of immune response and consequently are relevant to pathogenesis of many diseases. The ovarian steroid oestrogen is able to exert pleiotropic effect on macrophages, including modulation of the death pathway, for example, it exerts paradoxical effects on human U937 macrophages where cell death is induced by oestrogen [1], but the same hormone accords protection to these cells from TNF‐α induced apoptosis [2]. Similar effect is exerted on murine osteoclasts, where oestrogen exposure leads to caspase‐dependent apoptosis [3, 4]. A previous study from this laboratory demonstrated 17β‐oestradiol (E2) induced apoptosis in macrophages derived from human peripheral blood monocytes and THP‐1 acute monocytic leukaemia cell line, when Bcl‐2 was down‐regulated [5]. It is well established that the survival of a cell in response to certain apoptotic stimuli depends on the critical ratio of mitochondrial Bcl‐2 and Bax. Consequent to sensing of apoptotic stimuli, Bax, which exists as an inactive monomer in the cell cytoplasm, migrates to the mitochondria to interact with the existing mitochondrial Bcl‐2 and the resulting interaction determines the fate of the cell, higher or lower ratio of Bcl‐2/Bax being anti‐apoptotic or pro‐apoptotic [6], respectively. For the change of location of Bax, a conformational alteration occurs in the Bax protein, leading to exposure of the mitochondrial targeting sequence resulting in its translocation to the outer mitochondrial membrane. Multiple mechanisms have been proposed as instrumental in initiating Bax translocation, which include neutralization of several pro‐survival proteins by BH3 only members of the Bcl‐2 family such as Noxa and Puma [7], phosphorylation events mediated by kinases [8, 9], down‐modulation of clusterin [10], intracellular acidification [11] or alkalinization [12] and caspase‐dependent cleavage of Bax [13]. Because modulation of Bcl‐2 and Bax could form part of a strategy for manipulation of cell survival, it is important to establish the associated processes that lead to such changes.
Although it is known that E2 can influence cell death pathways in human macrophages, the involvement of oestrogen receptor‐α (ER‐α) and ‐β (ER‐β) expressed at multiple subcellular locations [14] in these cells in mediating death or survival signals is not known. It is important to understand the relative contribution of the two receptors on effects elicited by oestrogen because signalling through separate subtypes could have diverse outcome [15]. This is evident from the distinct phenotypes obtained with ER‐α and ER‐β knockout mice [16], and in addition, evidence for differential receptor activity come from studies showing overlapping but exclusive sets of downstream target genes for the two subtypes [17]. The classical model of ER action is where ligand bound ER interacts with oestrogen response elements in target genes and initiates transcription by modulating co‐repressors and co‐activators. Conversely, ERs can also interact with other transcription factors like activating protein‐1 and stimulating protein‐1 to initiate transcription [18, 19]. In addition to the above mode of actions, oestrogen may elicit effects through genomic or non‐genomic mechanisms by binding to oestrogen receptors localized on the plasma membrane of target cells [20] and activate mitogen activated protein kinase (MAPK) signalling [21] or induce intracellular Ca2+ fluxes [22]. Elicitation of a particular event in response to E2 could be dependent on the relative concentrations of the two ER subtypes, for example, U937 monocytes expressing mostly ER‐β are sensitive to oestrogen induced apoptosis; however, after differentiation to macrophages, receptor population expressed is predominantly ER‐α, as a result of which the apoptosis inducing effect of oestrogen becomes ineffective [23].
The purpose of this study was to investigate the involvement of the two ER subtypes in mediating apoptosis associated events in human macrophages using THP‐1 monocyte derived macrophages as a model system. We show that THP‐1 human macrophage survival is compromised in the presence of E2 if ER‐α but not ER‐β receptor levels are down‐regulated. This is because E2 signalling via ER‐α mediates the anti‐apoptotic event of Bcl‐2 up‐regulation, whereas ER‐β signals for the pro‐apoptotic event of Bax translocation to the mitochondria via Na+‐H+ exchanger mediated intracellular alkalinization.
Materials and methods
Cell lines and culture
THP‐1, a human acute monocytic leukaemia cell line, and MCF‐7, a human breast carcinoma cell line (ATCC, Manassas, VA, USA), were maintained in Roswell Park Memorial Institute (RPMI)‐1640 (Biological Industries, Kibbutz Beit Haemek, Israel), supplemented with 10% FCS (Biological Industries, Kibbutz Beit Haemek, Israel). Differentiation of THP‐1 monocytes to macrophages was induced by treatment with 10 ng/mL PMA for 36 hrs. Forty eight hours prior to experimentation, the cells were transferred to phenol‐red free RPMI‐1640 supplemented with 10% dextran‐coated charcoal stripped FCS to remove all extraneous sources of oestrogen.
Reagents
E2 (cyclodextrin encapsulated), E2 cojugated to BSA (E2‐BSA), E2‐BSA conjugated to FITC (E2‐BSA‐FITC), PD98,059, nigericin, amiloride and propidium iodide (PI) were obtained from Sigma‐Aldrich (St. Louis, MO, USA). ICI 182,780 was obtained from Tocris Cookson (Bristol, UK). Negative control siRNA was purchased from Ambion (Austin, TX, USA), whereas Bcl‐2, ER‐α and ER‐β siRNAs were obtained from Dharmacon (Lafayette, CO, USA). The siRNA transfection reagent Transpass R2 was purchased from New England Biolabs (Ipswich, MA, USA). All reagents for Western blotting and ECL development were from Amersham Biosciences (Piscataway, NJ, USA). SNARF (5‐(and‐6)‐carboxy SNARF®1‐AM), Sodium Green™ tetra‐acetate, secondary antimouse IgG conjugated to Alexa fluor 488 and Hoechst 33342 nuclear dye were purchased from Molecular Probes (Eugene, OR, USA). Antibodies for oestrogen receptor α/β, oestrogen receptor‐α and actin were procured from Calbiochem (Darmstadt, Germany), whereas antibodies against phospo‐ERK and whole‐ERK were from StressGen Biotechnologies (Victoria, BC, Canada). Anti‐Bcl‐2, anti‐Bax and anti‐cytochrome c antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti‐GAPDH antibody was from Ambion (Austin, TX, USA), whereas anti‐histone dimethyl lysine antibody was purchased from Upstate (VA, USA). Secondary antimouse and anti‐rabbit IgG conjugated to horseradish peroxidase were procured from Jackson Immunoresearch (Cambridgeshire, UK). The Vybrant apoptosis detection system was purchased from Promega (Madison, WI, USA). Anti‐clusterin antibody was a kind gift from Dr. C. Yan Cheng of the Population Council, NY, USA. All other chemicals used were purchased from Sigma‐Aldrich (St. Louis, MO, USA) unless mentioned otherwise.
Intracellular pH measurement
Intracellular pH measurement was performed with the long‐wavelength fluorescent pH indicator carboxy SNARF‐1 AM following manufacturer’s protocol. Briefly, the cells (106/ml) were resuspended in serum‐free RPMI‐1640 and incubated with a final concentration of 1 μM SNARF‐1 AM, diluted from a stock solution of 1 mM in DMSO for 15 min. at room temperature. Cells were washed and incubated for 20 min. at room temperature for complete de‐esterification of AM esters. In situ calibration of SNARF‐1 AM was performed with the ionophore nigericin at 10 μM concentration in a high‐K+ buffer to equilibrate intracellular pH with that of the controlled extracellular medium. Appropriate groups were subjected to different treatments, and fluorescence measurements were commenced in a spectrofluorometer (Perkin Elmer, Waltham, MA, USA), followed by kinetic analysis. The pH was calculated from the fluorescence measurements using the following formula:
where pKa of carboxy SNARF‐1 AM is 7.5. R is the ratio of fluorescent intensities (F) measured at two emission wavelengths, 580 (λ1) and 640 nm (λ2), with fixed excitation at 514 nm. The subscripts A and B represent the limiting values at the acidic and basic end‐points of the titration, respectively. Na+‐free and HCO3 −‐free buffer were prepared as described by Khaled et al.[24].
Intracellular Na+ measurement
For intracellular Na+ measurement, cells (106/mL) were labelled for 20 min. at room temperature with the cell permeable fluorescent Na+ indicator Sodium Green™ tetra‐acetate, diluted to 1 μM in RPMI‐1640 from a 5 mM stock solution made in DMSO. After washing the cells to remove excess probe, kinetic fluorescent measurements were carried out with a spectrofluorometer at an excitation of 480 nm and emission of 520 nm (BMG Fluostar Optima, BMG technologies, Offenburg, Germany). In situ Calibration was accomplished by using the indicator in solutions of precisely known free Na+ concentration in the presence of the pore forming antibiotic gramicidin (10 μM). Intracellular Na+ was calculated using the following equation:
where Kd of the dye is 5.7 mM at 37°C, F is the fluorescence of the experimental sample, F min is fluorescence in the absence of Na+ and F max is fluorescence in the presence of saturating concentrations of Na+.
siRNA transfection
THP‐1 macrophages were transfected with specific siRNAs using Transpass R2 transfection reagent as described previously [5]. Briefly, Bcl‐2 siRNA (15 pmol), ER‐α and ER‐β siRNA (100 pmol) or negative control siRNA (pre‐designed siRNA with no known target genes) at similar concentrations were added to transfection reagent TranspassR2, diluted in serum‐free medium, and incubated for 20 min. to allow the formation of transfection complexes. The formed complexes were added to 105 cells/well grown in 24‐well plates and incubated for 6 hrs, following which fresh complete medium was added. Transfection efficiency was estimated by observing Cy3 fluorescence of the negative control siRNA with a Nikon TE2000‐E fluorescence microscope using a tetramethyl rhodamine filter (530–580 nm). For all transfections, target protein knockdown was assessed 24 hrs after transfection by probing extracts of transfected cells on Western blots with anti‐Bcl‐2 and anti ER‐α/β antibody.
Subcellular fractionation
THP‐1 macrophages were allowed to swell for 10 min. in hypotonic buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris‐HCl, pH 7.5) followed by homogenization with a Dounce homogenizer (50 strokes). Immediately after cell lysis, the mitochondria were stabilized by addition of mitochondrial stabilization buffer (525 mM mannitol, 175 mM sucrose, 12.5 mM Tris‐HCl, pH 7.5; 2.5 mM EDTA, pH 7.5), and the homogenate was centrifuged at 1300 ×g for 15 min. to isolate the nuclear fraction. The post‐nuclear supernatant was further centrifuged at 17,000 ×g for 15 min. in an ultracentrifuge (Optima XL‐100K, Beckman, Fullerton, CA, USA) to isolate the mitochondria. The post‐mitochondrial supernatant was centrifuged at 100,000 ×g for 1 hr to obtain the membranous fraction as a pellet and the supernatant as the cytosol. The homogeneity of the obtained fractions was determined by Western blotting with probes specific for each fraction.
Cell viability assay
To assess cell viability, PI dye exclusion assay was performed by incubating the cells with 1 μg/ml PI for 5 min. at 37°C, followed by one wash with ice‐cold PBS. The cells were analysed under a Nikon TE2000‐E fluorescence microscope using Nikon G2A filter cube. The percentage cell death was calculated as the number of cells with PI positive nuclei as against the total number of cells. Annexin‐V‐PI staining was performed as described previously [5], and data acquisition was performed on a BD‐LSR flow cytometer equipped with a 488 nm air‐cooled argon ion laser. The acquired data was analysed using WinMDI software (Microsoft v.2.9).
Reverse transcription‐polymerase chain reaction
Total RNA was isolated using TRIzol reagent (GIBCO, CA, USA), and cDNA was synthesized as described previously [5]. The specific primers used were
ER‐α (sense) GTGGGAATGATGAAAGGTGG; ER‐α (antisense) TCCAGAGACTTCAGGGTGCT
ER‐β (sense) TGAAAAGGAAGGTTAGTGGGAACC; ER‐β (antisense) TGGTCAGGGACATCATCATGG
Actin(sense),5′‐GTGGGGCGCCCCAGGCACCA‐3′; Actin(antisense), 5′CTCCTTAATGTCACGCACGATTTC‐3′.
PCR was performed after determining the cycle number in which a linear amplification of serially diluted template could be achieved. The PCR products were then resolved on 1.5% agarose gel and visualized by ethidium bromide staining and quantitated by densitometry.
Immunocyotochemistry
Cells were fixed with 4% formaldehyde for 20 min., followed by several washes with ice‐cold PBS. Saponin (0.1%) was used for cell permeabilization, and 3% normal goat serum was used as a blocking reagent to reduce non‐specific binding. The permeabilized cells were incubated with primary antibody recognizing ER‐α/β (1 : 100) followed by incubation with secondary antimouse IgG conjugated to Alexa fluor 488 (1 : 200). For live‐cell staining, all incubations were performed at 4°C with anti‐ER‐α antibody in addition to reagents as described above. For nuclear labelling of cells, Hoechst 33342 was used. All stainings were visualized using a Nikon TE2000‐E fluorescence microscope using appropriate filter blocks, and the image acquisition was carried out with a high‐resolution Retiga Exi camera (Q‐imaging, Surrey, BC, Canada), the mask of co‐localization was created and the co‐efficient of colocalization was calculated using Image‐Pro Plus software (Media cybernetics, Silver Spring, MD, USA).
SDS‐PAGE and Western blot
Whole cell extracts were prepared by treating the cells with lysis buffer (0.125 M Tris, 4% SDS, 20% glycerol and 10% 2‐ME), and protein estimation was performed with CBX protein assay kit (G‐Biosciences, St. Louis, MO, USA). Lysates were resolved on 12% SDS‐PAGE gel, following which they were transferred onto nitrocellulose membrane as described before [5]. Non‐specific binding sites were blocked by incubating the blots in 5% non‐fat skimmed milk with 0.05% PBS‐Tween 20 for 1 hr. Primary (1: 5000) and secondary antibody (1 : 10,000) incubations were carried out for 1 hr each, and immunoreactivity was visualized by enhanced chemiluminescence using ECL reagent, as described previously [5].
Densitometry and statistical analysis
Quantitative assessment of reaction intensity in Western blots was performed using a UVP gel‐documentation instrument, and the data were analysed with the LabWorks image analysis and acquisition software (v4.0.0.8; UVP, Upland, CA, USA). At least three Western blots per experiment were quantitated to arrive at the average value of the signal. All measurements were normalized to internal loading controls. Data are expressed as mean ± standard error (SE) unless mentioned. Comparisons were made between different groups using unpaired Student’s t‐test. The values were considered to be significantly different at P < 0.05. All analysis was performed on data acquired from three or more independent experiments.
Results
Human macrophages express both oestrogen receptor‐α and β at multiple subcellular locations
E2 initiates cellular signalling pathways via interaction with its receptors expressed primarily as two subtypes – the ER‐α and ER‐β[25]– found in the nucleus, plasma membrane or cytosol, the distribution varying with different cell types [26]. Human macrophages are known to express both ER‐α and ER‐β[27, 28], however, the subcellular localization of these receptors is not known. In the current study, membrane bound E2 receptors were detected on viable differentiated THP‐1 macrophages by labelling live cells at 4°C with membrane impermeable E2‐BSA linked to FITC (E2‐BSA‐FITC), and flow cytometric analysis showed an obvious shift in fluorescence labelling intensity in these cells compared with those labelled with only BSA‐FITC used as a control (Fig. 1A). Decreased fluorescence readings obtained with cells incubated with E2‐BSA‐FITC in the presence of unconjugated E2 compared with cells exposed to only E2‐BSA‐FITC confirmed specificity of this binding (Fig. 1A).
Figure 1.
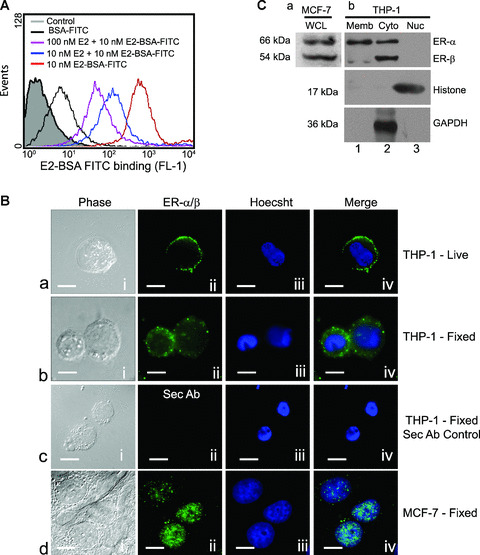
Human macrophages express oestrogen receptor α and β on the plasma membrane and cytoplasm. (A) The histogram represents flow‐cytometric analysis of live THP‐1 macrophages incubated with E2 conjugated to BSA‐FITC (red line) or co‐incubation with different concentrations of E2 (blue, 10 nM E2 and pink, 100 nM E2 lines) under similar conditions. The area shaded grey represents unlabelled cells, and the black line represents cells labelled with only BSA‐FITC. Note the distinct shift in staining of the cells treated with E2‐BSA‐FITC demonstrating recognition of cell surface localization of ERs. (B) Indirect immunofluorescence on live as well as formaldehyde‐fixed cells stained with anti‐ER‐α/β antibody. (a) THP‐1 live; (b), THP‐1 fixed; (c), secondary antibody control, (d) fixed MCF‐7 cells – (i), nomarski image; (ii), ER‐α/β staining; (iii), nuclear staining with Hoechst 33342; (iv), overlap of (ii) and (iii). The MCF‐7 cells (d, i–iv) used as positive controls show presence of nuclear receptors. The bar represents 10 μm. All data are representative of at least three independent experiments. (C) Western blots of subcellular fractions of THP‐1 cells probed with anti‐ER‐α/β showing presence of both forms in the cytoplasm (b, Cyto, lane 2), predominantly ER‐α in membrane fraction (b, Memb, lane 1) and absence of receptors in nuclear fraction (b, Nuc, lane 3). (a) MCF‐7 cell extracts show the presence of both ER‐α and ER‐β. Western blot for histone and GAPDH was performed to assess the homogeneity of the obtained nuclear and cytoplasmic fractions, respectively.
Further analysis of presence of ERs in different subcellular locations in these cells showed surface labelling of ERs on live cells (Fig. 1B, a, i–iv) with an anti‐E2 receptor antibody raised against common epitopes on ER‐α and ER‐β receptor proteins, thus corroborating the above data obtained with E2‐BSA‐FITC.
Intracellular receptors were detected by staining fixed and permeabilized cells with the same antibody that demonstrated the presence of receptors within the cytosol and the membrane but not in the nucleus (Fig. 1B, b, i–iv). Figure 1B, c, i–iv shows absence of fluorescence in secondary antibody controls. MCF‐7 cells, where E2 receptors are predominantly nuclear [29], were used as positive controls and showed distinct nuclear staining with the same antibody (Fig. 1B, d, i–iv). Cytosolic ERs dimerize and migrate to the nuclei upon ligand engagement [18]; this phenomenon was used as a control for cytosolic receptors, and Figure S1 shows that upon E2 exposure the cytosolic ERs migrate to the nuclei (E2, b and d) compared with control (Control, b and d) and E2‐BSA treatment (E2‐BSA, b and d). Measurement of the mask of colocalization clearly shows significant colocalization of ER receptor staining with nuclear staining after E2 treatment (E2, e) compared with vehicle treated controls (Control, e) and after E2‐BSA treatment (E2‐BSA, e).
To determine the subtype specific distribution of receptors, Western blots of subcellular fractions of THP‐1 cells were probed with the same antibody as above, and it recognized both receptor types in the cellular cytosolic fraction (Fig. 1C, b, lane 2) whereas the membranous fraction primarily showed reactivity for ER‐α subtype, the expression of ER‐β being low (Fig. 1C, b, lane 1). The nuclear fraction did not show any reactivity, suggesting the absence of nuclear oestrogen receptors in this particular cell type (Fig. 1C, b, lane 3). Total extract of MCF‐7 cells known to express both ER‐α and ER‐β was used as positive control (Fig. 1C, a). Collectively, the above data demonstrated the presence of ERs on the cell surface as well as in the cytosol of THP‐1 monocyte derived macrophages, nucleus being devoid of such receptors. Both ER‐α and ER‐β are present intracellularly, whereas the plasma membrane appears to be primarily populated by ER‐α.
E2 modulates Bcl‐2 and Bax via different sets of receptors in THP‐1 cells
To investigate if the receptors at different subcellular locations transmit similar or different signals for modulation of the mitochondrial apoptotic pathway, both membrane permeable and impermeable E2 were used. E2‐BSA, where E2 is conjugated to BSA through a six atom hydrocarbon tether [30] restricting its diffusion through the plasma membrane, was used to distinguish signals originating from membrane bound receptors only [20, 30].
Equivalent increase in Bcl‐2 levels was obtained with membrane permeable E2 as well as membrane impermeable E2‐BSA at 6 hrs (Fig. 2A, lanes 2 and 4). Pure E2 receptor antagonist ICI 182,780 inhibited this increase (Fig. 2A, lanes 3 and 5), confirming that in both cases, E2 receptors were involved. In contrast to its ability to modulate Bcl‐2, E2‐BSA was unable to exert any effect on subcellular localization of Bax (Fig. 2B, lanes 5 and 6), whereas unconjugated E2 was able to stimulate Bax translocation (Fig. 2B, lanes 3 and 4). The inability of E2‐BSA to induce migration of Bax from cytosol to the mitochondria unlike free E2 suggested that Bax migration was independent of membrane receptor mediated signalling.
Figure 2.
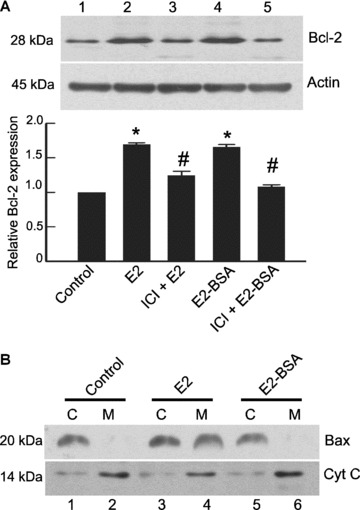
Regulation of Bax and Bcl‐2 is mediated by E2 receptors localized at different sub‐cellular locations. (A) Extracts of THP‐1 cells treated with E2 (10 nM)(lane 2) and E2‐BSA for 6 hrs (10 nM)(lane 4) show Bcl‐2 up‐regulation. Presence of ICI 182,780 (1 μM) during E2 (lane 3) and E2 BSA (lane 5) treatment inhibited Bcl‐2 up‐regulation. Same blots were stripped and reprobed for actin, which was used as a loading control. The bar graph represents densitometric measurements of Bcl‐2 expression relative to control and normalized to actin (n= 3). The error bars represent ± SEM. *P < 0.05 compared with control. #P < 0.05 compared with E2 or E2‐BSA treated group. (B) THP‐1 macrophages were exposed for 6 hrs to E2 (lanes 3 and 4) or E2 BSA (lanes 5 and 6) and subcellular fractions of mitochondria (M) and cytosol (C) were analysed for the localization of Bax. Note the absence of Bax translocation in the E2‐BSA treated groups. The blot was stripped and reprobed for cytochrome c to determine the homogeneity of mitochondrial fractions. Note the heavy presence of cytochrome c in the mitochondrial fraction. All data are representative of at least three independent experiments. Cyt C: cytochrome c.
Based on the above data, attempt was made to identify receptor subtypes involved in mediating the above responses. ER‐α and ER‐β mRNA and protein were selectively down‐regulated with siRNA to test the effect of this down‐regulation on Bcl‐2 and Bax and, eventually, cell death. Figure 3A shows the RT‐PCR for ER‐α mRNA performed with primers specific for ER‐α, where lane 2 shows decrease in ER‐α mRNA when siRNA to ER‐α was used but no decrease with ER‐β siRNA (Fig. 3A, lane 3). Figure 3B, shows the RT‐PCR for ER‐β mRNA performed with ER‐β specific primers, where lane 3 shows down‐regulation of ER‐β mRNA levels with ER‐β siRNA but not with ER‐α siRNA (lane 2). Negative control siRNA did not show any interference with RNA levels of either receptors (Fig. 3A and B, lanes 1). Status of protein levels of ER‐α and ER‐β after ER‐α siRNA (Fig. 3C, lane 3) and ER‐β siRNA (Fig. 3C, lane 2) transfection shows that protein levels of both receptors were significantly down‐regulated with respective siRNA treatment. A distinct reduction of surface receptor population was observed with siRNA for ER‐α (Fig. 3D, b, i‐iii) compared with a negative control siRNA (Fig. 3D, a, i‐iii), as visualized by immunostaining of treated and untreated cells with a specific anti‐ER‐α antibody.
Figure 3.
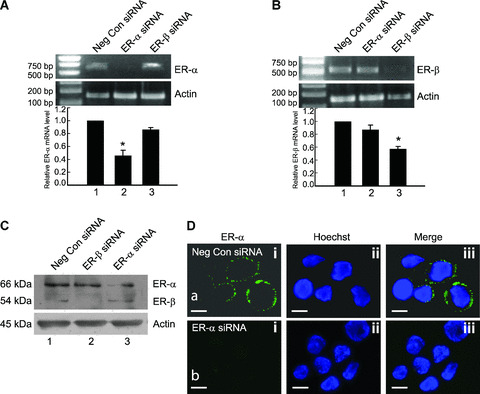
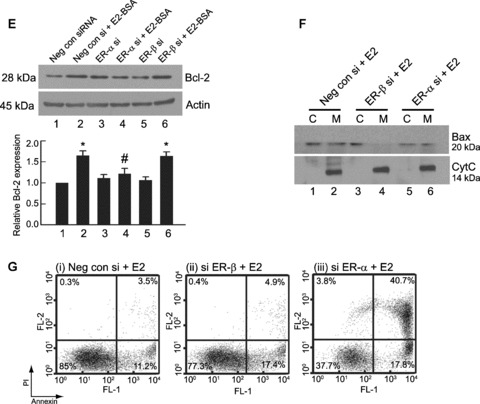
Bcl‐2 increase and Bax translocation are regulated by distinct oestrogen receptor subtypes. (A) RT‐PCR for ER‐α mRNA. Down‐regulation of ER‐α mRNA at 24 hrs of treatment by siRNA against ER‐α (lane 2) in THP‐1 cells as visible by amplification of mRNA for ER‐α by RT‐PCR. Note that there is no down‐regulation of ER‐α mRNA in cells transfected with ER‐β siRNA (lane 3). (B) RT‐PCR for ER‐β mRNA. Down‐regulation of ER‐β mRNA by siRNA against ER‐β (lane 3) at 24 hrs of treatment as visible by amplification of mRNA for ER‐β by RT‐PCR. Note that there is no down‐regulation of ER‐β mRNA (lane 2) in cells transfected with ER‐α siRNA. (C) Western blot analysis on whole cell lysates for expression of ER‐α and ER‐β in THP‐1 cells transfected with negative control siRNA (lane 1), ER‐β siRNA (lane 2) or ER‐α siRNA (lane 3), 24 hrs after transfection. Note the down‐regulation of ER‐α and ER‐β after treatment with respective siRNAs. The blot was stripped and reprobed for actin, which was used as an endogenous loading control. (D) Live cell fluorescence microscopic analysis of ER‐α expression on the plasma membrane by immunostaining with an ER‐α specific antibody in negative control siRNA transfected (a, i–iii) and ER‐α siRNA (b, i‐iii) transfected THP‐1 macrophages at 24 hrs. The blue stain (ii) is labelling with the nuclear dye Hoechst 33342. Note the loss of membrane ER‐α expression in ER‐α siRNA transfected cells (b, i‐iii). The bar represents 10 μm. (E) Analysis of Bcl‐2 expression by Western blotting in THP‐1 macrophages transfected with negative control siRNA (Neg con siRNA), ER‐α siRNA (ER‐α si) or ER‐β siRNA (ER‐β si) for 24 hrs and subsequently treated with or without 10 nM E2‐BSA for 6 hrs. Note the reduction in Bcl‐2 when ER‐α siRNA and E2‐BSA (lane 4) was used compared with ER‐β siRNA and E2‐BSA, respectively (lane 6). Western blot for actin was used as loading control. Bar graph is densitometric representation of the relative Bcl‐2 expression compared with negative control siRNA transfected cells. *P < 0.05 compared with the respective control groups. #P < 0.05 compared with negative control siRNA transfected cells treated with E2‐BSA. (F) Western blot analysis for subcellular localization of Bax was performed in cells transfected with negative control siRNA (lanes 1, 2), ER‐β siRNA (lanes 3, 4) or ER‐α siRNA (lanes 5, 6) for 24 hrs, followed by treatment with 10 nM E2 for 6 hrs. Note the decrease in Bax translocation in lanes 3 and 4. The blots were stripped and reprobed for cytochrome c, which served as a control to determine the homogeneity of the obtained mitochondrial and cytosolic fractions. All data are representative of at least three independent experiments. (G) Flow‐cytometric analysis of Annexin‐V‐PI staining in ER‐α (iii) or ER‐β (ii) knockdown cells, treated with respective siRNAs for 24 hrs, followed by exposure to 10 nM E2 for 6 hrs. Note that ER‐α down‐regulation in the presence of E2 shows high number of apoptotic cells. Cells in the lower left quadrant represent viable cells. Neg Con si: negative control siRNA; siER‐α: ER‐α siRNA; si ER‐β: ER‐β siRNA.
Next, the effects of ER‐α and ER‐β down‐regulation on Bcl‐2 and Bax modulation were examined. E2‐BSA was able to increase Bcl‐2 in the presence of negative control siRNA (Fig. 3E, lane 2) but not when the cells were transfected with ER‐α siRNA (Fig. 3E, lane 4), thus linking the involvement of ER‐α in Bcl‐2 increase. This treatment with ER‐α siRNA, therefore, creates a condition within the cell, which is pro‐apoptotic in nature because presence of E2 will induce Bax translocation, and in the absence of Bcl‐2, Bax will induce apoptosis. In contrast, the knockdown of ER‐β during E2‐BSA exposure did not affect Bcl‐2 increase (Fig. 3E, lane 6), showing the absence of any effect of membrane associated ER‐β on Bcl‐2 levels. Knockdown of ER‐β (Fig. 3F, lanes 3 and 4) but not of ER‐α (Fig. 3F, lanes 5 and 6) in the presence of E2 resulted in inhibition of Bax migration, thereby creating an anti‐apoptotic condition because Bcl‐2 will not have to counteract the effect of translocated Bax to the mitochondria. The translocation was complete in cells transfected with negative control siRNA (Fig. 3F, lanes 1 and 2). Together, the above data demonstrate a dichotomous effect of E2 on the components of the mitochondrial cell death pathway mediated through the two ER subtypes, ER‐α mediating Bcl‐2 increase and ER‐β arbitrating Bax translocation.
Arguably, the modulation of the two receptor levels leading to changes in the pro‐apoptotic Bax and anti‐apoptotic Bcl‐2 would affect cell survival. Annexin‐V/PI staining showed that ER‐β knockdown in the presence of E2 did not induce any apoptotic death (Fig. 3G, ii), whereas ER‐α knockdown resulted in about 40% late apoptotic cells and 17% early apoptotic cells at 6 hrs after E2 exposure (Fig. 3G, iii). This corroborated our findings that ER‐α but not ER‐β was involved with the survival pathway, and interference with this pathway resulted in increased cell death in the presence of E2.
Bcl‐2 modulation is mediated through ERK phosphorylation whereas Bax translocation is dependent upon intracellular alkalinization
Downstream to ER engagement by E2‐BSA and E2, inhibition of ERK phosphorylation by MEK inhibitor PD 98,059 resulted in abrogation of Bcl‐2 up‐regulation (Fig. 4A, lane 5 and 3). Furthermore, E2‐BSA was able to induce phosphorylation of ERK in as early as 10 min. (Fig. 4B, lane 4), which could be inhibited by ICI 182,780 (Fig. 4B, lane 5). Therefore, this indicated the competence of the membrane associated ERs to transmit Bcl‐2 up‐ regulation signals. Following the observation that cytosolic ER‐β was involved in Bax translocation, the mechanisms that lead to this change in subcellular localization was investigated. Clusterin and cytosolic pH statuses were checked, as these are known to mediate Bax translocation [10, 11, 12]. E2 exposure did not suppress the expression of secretory form of clusterin in THP‐1 cells (Fig. 5A), which is the pro‐survival form, thereby ruling out its involvement in E2‐induced Bax translocation as opposed to earlier reports in fibrosarcoma and prostate cancer cells, where suppression of clusterin was shown to induce Bax translocation [10]. Also, the level of pro‐apoptotic nuclear clusterin remained unaltered (Fig. 5A).
Figure 4.
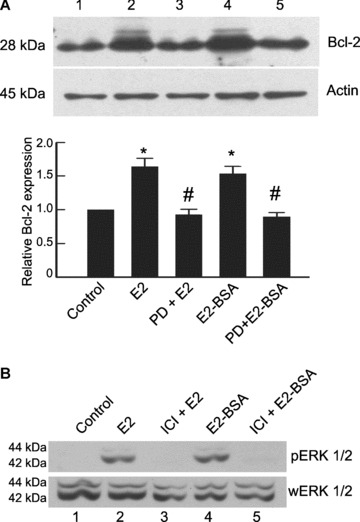
E2‐BSA regulates Bcl‐2 increase through ERK phosphorylation. (A) THP‐1 macrophages were treated with 10 nM E2 or E2‐BSA for 6 hrs with or without pre‐ incubation with the MEK inhibitor PD 98,059 (25 μM, 30 min. prior to E2 addition), and extracts were probed for Bcl‐2 levels by Western blotting. The bar graph represents densitometric measurements of Bcl‐2 expression relative to control and normalized to actin (n= 3). The error bars represent ± SEM. *P < 0.05 compared with control. #P < 0.05 compared with E2 or E2‐BSA treated group. PD, PD 98,059 (MEK inhibitor). (B) Figure shows detection of phosphorylated ERK1/2 as an early response by Western blotting in THP‐1 macrophages treated with E2 and E2‐BSA for 10 min. with or without incubation with ICI 182,780. The blot was stripped and reprobed for whole ERK1/2, which served as a loading control.
Figure 5.
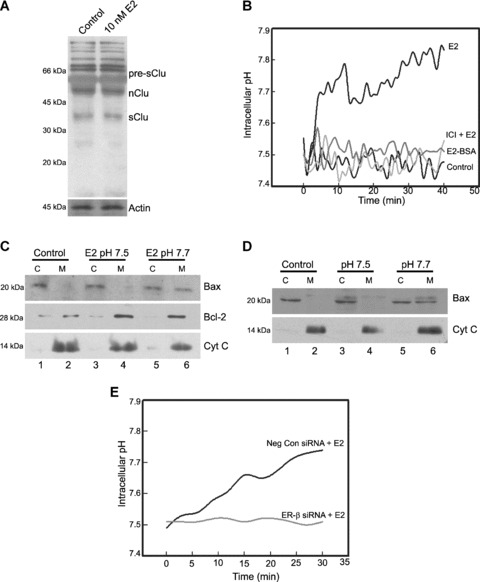
Bax translocation is dependent on E2‐induced change in intracellular pH mediated via ER‐β. (A) Western blot analysis of clusterin expression after E2 treatment indicates no change in the expression of the different isoform levels. Pre‐sClu: precursor to secretory clusterin; nClu: nuclear clusterin; sClu: secretory clusterin. (B) Intracellular pH measurement was performed with the pH sensitive dye SNARF‐1 AM. The graph represents the calculated intracellular pH of THP‐1 macrophages exposed to various drug treatments as indicated over a time‐course of 40 min. Note that although E2 increased the intracellular pH, this was prevented in the presence of antioestrogen ICI 182,780 but E2‐BSA was unable to alter intracellular pH. ICI: ICI 182,780. (C) Appropriate groups of THP‐1 macrophages were resuspended in high‐K+ buffer of pH 7.5 or 7.7. The control group was left untreated whereas the other two groups were treated with 1 μM nigericin and 10 nM E2 for 6 hrs. The cytosolic (C) and mitochondrial (M) fractions of the appropriate groups were probed for Bax and Bcl‐2 expression by immunoblotting. Western blotting for cytochrome c was performed to determine the homogeneity of the obtained fractions. Note the lack of Bax migration in cells maintained at basal pH (lanes 3 and 4). Cyt C: cytochrome c. (D) Appropriate groups of THP‐1 macrophages were resuspended in high‐K+ buffer of pH 7.5 or 7.7 and permeabilized with 1 μM nigericin to maintain the intracellular pH the same as that of the extracellular medium. Lysates of cytosolic (C) and mitochondrial (M) fractions were probed for Bax and cytochrome c by Western blotting. Note the migration of Bax at pH 7.7. Cyt C: cytochrome c. (E) Intracellular pH measurement in cells transfected with negative control siRNA and ER‐β siRNA following treatment with 10 nM E2. Note the inhibition of increase in pH with knockdown of ER‐β. Neg Con si: negative control siRNA; ER‐β si: ER‐β siRNA.
In other cellular systems like B cells and thymocytes, it has been demonstrated that Bax translocation could occur in response to a pH change [11, 12, 31]. As shown in Figure 5B, E2 treatment resulted in an increase in intracellular pH from a basal level of 7.5 to about 7.7–7.8, but this increase was inhibited by the E2 receptor antagonist ICI 182,780 (Fig. 5B). Involvement of membrane‐associated receptors was ruled out, as E2‐BSA was unable to effect any change in pH. When pH change was prevented by placing the cells in high‐K+ buffer of desired pH in the presence of nigericin, an activator of K+/H+ antiporter, E2 was not able to induce Bax translocation (Fig. 5C, lane 3 and 4). On the contrary, the translocation was complete when cells were maintained at a pH of 7.7 (Fig. 5C, lane 5 and 6), suggesting that E2‐induced intracellular alkalinization acted as a signal for Bax translocation. In contrast, E2‐induced Bcl‐2 up‐regulation was unaffected by pH alterations (Fig. 5C). To ascertain if translocation of Bax could occur whenever there was a pH change independent of other stimuli, an increase in intracellular pH was induced by nigericin treatment in high‐K+ buffer in the absence of E2, and a change of pH to 7.7 resulted in Bax translocation but not when pH was maintained at 7.5 (Fig. 5D, lanes 5, 6 and 3, 4, respectively). These data point out that an increase in intracellular pH was sufficient to induce the translocation of Bax, independent of other signalling pathways that might be activated by E2. Also, siRNA mediated down‐regulation of ER‐β prevented E2 induced pH change (Fig. 5E), providing evidence that signals generated through ER‐β was capable of altering the pH and also supports that data presented above that signals for Bax translocation is mediated through ER‐β.
Becuse intracellular pH is maintained by the co‐ordinated activity of a number of ion channels and their respective ions, the most important of which are the Na+–H+ exchangers (NHE) and the HCO3 − transporters, involvement of each of these elements in the E2‐induced increase in intracellular pH was investigated. NHE functions in the maintenance of intracellular pH by pumping out intracellular H+ for extracellular Na+, and hence its activity is indicated by both an increase in intracellular Na+ and alkalinization of the cytoplasm due to expulsion of H+ ions. When THP‐1 macrophages were suspended in a Na+‐free media, E2 was unable to induce a pH change indicating that influx of Na+ was essential for alkalinization (Fig. 6A). HCO3 − was not required for E2‐induced intracellular alkalinization because E2 was able to induce a pH change in a HCO3 − free media (Fig. 6B). A possible role for NHE in mediating cellular alkalinization was indicated by an increase in intracellular Na+ in response to E2 as observed by an increase in Sodium Green™ fluorescence (Fig. 6C). This was further confirmed when amiloride, an NHE inhibitor lowered Na+ levels and also prevented alkalinization of the cytoplasm (Fig. 6C and D). If amiloride could prevent intracellular alkalinization, ideally then amiloride should be able to prevent translocation of Bax if pH increase and Bax translocation were linked. Western blots of cytosolic and mitochondrial fraction obtained from cells treated with E2 in the presence of amiloride showed absence of Bax translocation to mitochondria (Fig. 6E, lanes 5 and 6) compared with cells treated with E2 only (Fig. 6E, lanes 3 and 4). In summary, the above data suggest that E2 signals, through ER‐β, to induce intracellular alkalinization via activation of NHE, which results in Bax translocation.
Figure 6.
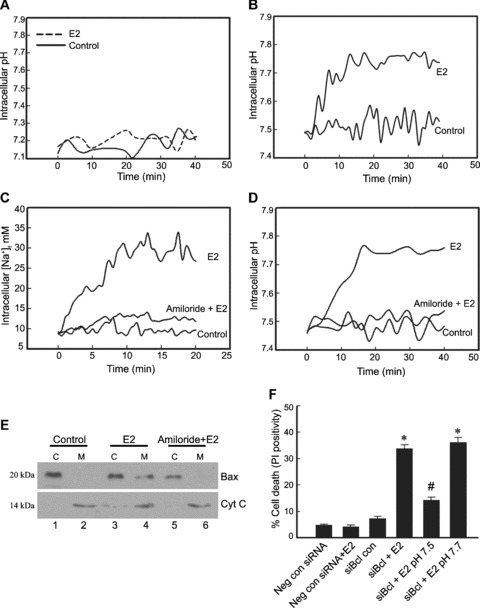
Oestrogen increases intracellular pH by activation of Na+–H+ exchanger. THP‐1 macrophages were resuspended in Na+‐free buffer (A) or HCO3 −‐free buffer (B), and intracellular pH was measured using SNARF‐1 AM dye after treatment with 10 nM E2 over a time period of 40 min. (C) Intracellular Na+ was monitored in THP‐1 macrophages using the fluorescent dye Sodium Green™ that shows a distinct increase in Na+ in response to E2. Amiloride, a Na+–H+ exchanger inhibitor, was used at a concentration of 2 μM 10 min. prior to E2 treatment, which was able to inhibit the increase in intracellular Na+ levels. (D) Intracellular pH measurement in THP‐1 macrophages exposed to E2 and pre‐incubated with or without the Na+–H+ exchanger inhibitor amiloride (2 μM) shows an abrogation of E2 induced increase in pH in the presence of amiloride. (E) Lysates of cytosolic (C) and mitochondrial (M) fractions prepared from the above groups after 6 hrs of incubation, and probed for Bax and cytochrome c on Western blots show the absence of Bax translocation to the mitochondria in the presence of amiloride (lanes 5 and 6). Cyt C: cytochrome c. (F) Cell viability. THP‐1 macrophages were transfected with negative control siRNA or Bcl‐2 siRNA, and 24 hrs post‐transfection cells were subjected to appropriate treatments with E2 as indicated. All cells were resuspended in high‐K+ buffer and appropriate groups, where intracellular pH was to be maintained at 7.5 or 7.7, were treated with 1 μM nigericin. E2 treatment was given for 6 hrs and viability was analysed by propidium iodide dye exclusion method performed with fluorescence microscopy. The bar graph represents the percentage cell death in the various treatment groups. *P < 0.05, compared with cells transfected with negative control siRNA and treated with E2. #P < 0.05 compared with Bcl‐2 siRNA transfected cells treated with E2. Neg Con si: negative control siRNA; siBcl: Bcl‐2 siRNA.
To confirm that alkalinization‐induced change in Bax translocation is the major pro‐apoptotic event induced by E2, Bcl‐2 knockdown macrophages were treated with E2, a situation that normally precipitates cell death (Fig. 6F, siBcl + E2), but when these cells are maintained at pH of 7.5, the apoptosis inducing effect of E2 was abrogated (Fig. 6F, siBcl +E2, pH 7.5).
Discussion
Insights into the role of oestrogen in macrophage survival and associated mechanisms are of great relevance because the findings would have direct bearing on the development of tumour targeting therapies [32]. Our earlier study showed that E2 was able to induce apoptosis in human macrophages when Bcl‐2 was down‐regulated [5]. The work described in this manuscript explores the involvement of E2 receptor subtypes localized in distinct subcellular compartments in the regulation of mitochondrial death pathway in human THP‐1 macrophages. We demonstrate that (i) signals for Bcl‐2 increase is primarily mediated through membrane associated ER‐α; (ii) the translocation of Bax to mitochondria is mediated via signalling through intracellular ER‐β receptors and (iii) the E2‐induced Bax translocation is dependent on intracellular alkalinization mediated through activation of Na+/H+ exchangers.
Ratio of Bcl‐2/Bax is crucial in maintaining cell viability under certain conditions, and therefore the relative involvement of the ERs in regulating this ratio was important to examine. Recognition of ER‐α binding sites on live cells by specific anti‐ER‐α antibody and knockdown of surface ER‐α by siRNA for ER‐α clearly confirmed the presence of surface localized ER‐α in THP‐1 cells, which is in agreement with a growing body of evidence showing the presence of membrane associated ERs in various cell types [33, 34]. Interestingly, the surface located ER‐α emerges as the major transducer of survival signal during E2 treatment, as demonstrated by the ability of E2‐BSA, the membrane impermeable form of E2, to up‐regulate Bcl‐2, as well as abrogation of this effect upon siRNA mediated knockdown of surface localized ER‐α resulting in cell death. Although we show that membrane associated ER‐α is sufficient to transduce the survival signal, the relative contribution of cytosolic ER‐α in the survival response could not be determined due to the non‐availability of specific inhibitors to intracellular versus the membrane receptors. The anti‐apoptotic role of ER‐α as noted in our studies is in concurrence with other reports that implicate ER‐α in mediating E2 induced protective role during H2O2 induced apoptosis in murine skeletal muscle C2C12 cells [35] or in human osteosarcoma cell line [36]. The downstream events after engagement of E2 on membrane associated ER‐α involved the activation of MAPK pathway for an induction of Bcl‐2 increase because E2‐BSA was competent to phosphorylate ERK, and MEK inhibition could inhibit Bcl‐2 up‐regulation. The functional role of surface receptors in mediating survival signals was of interest because surface receptors are amenable to selective manipulation by cell impermeable agonists, providing opportunities to exploit the surface receptors for induction or inhibition of specific cellular functions.
In contrast to E2‐induced Bcl‐2 increase, Bax translocation was independent of membrane bound receptors because E2‐BSA, which interacted with surface receptors, was unable to induce Bax migration. Because knockdown of ER‐β but not of ER‐α resulted in abrogation of Bax translocation, it indicated the importance of ER‐β in death inducing arm of the mitochondrial apoptotic pathway. This particular function has not been demonstrated in cells of monocytic origin, but mediation of pro‐apoptotic events by ER‐β is known in cells of non‐myeloid lineage like breast and colon cancer cells [37, 38]. As Bax translocation is the primary event that initiates changes pertaining to cell death, the mechanism of translocation consequent to ER‐ β mediated signalling by E2 is of importance. It is known that translocation of Bax to the mitochondria is linked to alteration in its conformation resulting in the exposure of its N‐terminal or BH‐3 domain [39, 40], which is under the control of various physiological factors, possibly of different natures [41]. Prior knowledge that a change in pH could trigger Bax movement [11, 12, 31] prompted us to focus on the possibility of E2 inducing a pH change in THP‐1 cells. Because Bax translocation could be initiated upon intracellular alkalinization in the presence or in the absence of E2, movement of Bax was likely to be facilitated by any stimulus capable of altering cellular pH. Importantly, the consequence of Bcl‐2/Bax ratio changes would affect cell survival, and a substantial decrease in cell death was observed after E2‐induced pH change was blocked at the time of Bcl‐2 knockdown, presumably due to lack of Bax translocation to the mitochondria, thus validating the observation that Bax migration to mitochondria due to pH change in the absence of concomitant Bcl‐2 up‐regulation is responsible for increased apoptosis.
Therefore, intracellular alkalinization was an important event, and this appeared to be mediated by NHE because the process was Na+‐dependent and could be inhibited by amiloride, a NHE inhibitor. A number of studies show that E2 can alter NHE functions [42] through a NHE regulatory factor (NHE‐RF), which is a primary response gene under ER control [43]. However, the rapid increase in pH in response to E2 observed in our system makes transcriptional regulation through NHE‐RF unlikely. The mechanism of NHE involvement remains unknown.
In summary, this study highlights the importance of oestrogen signalling through distinct ER subtypes in modulating the mitochondrial death pathway of human monocyte derived macrophages. The observations raise interesting possibilities of exploring the use of selective oestrogen receptor modulators specific for ER‐α or ER‐β or those which could signal exclusively through the membranous or cytoplasmic pool of receptors to manipulate death pathway in human macrophages. For example, estren, which is an oestrogen agonist signalling selectively on the membranous ER with no known transcriptional effects via the classical ER mechanism [44], could be used for generating anti‐apoptotic effects. The development and use of such agonists and antagonists could be utilized to target specific receptor population in target cells to achieve desired therapeutic effects like manipulation of death pathways in favour or against cell survival.
Supporting information
Fig. S1 Translocation of oestrogen receptors into the nucleus upon E2 treatment. Sub‐cellular distribution of ERs consequent to treatment with 10 nM E2 or E2‐BSA for 2 h is shown by immunofluorescence using an antibody, which recognizes both ER‐α and ER‐β. The panel stained green represents the ER‐α/β staining (b), the blue staining represents nuclear staining with Hoecsht 33342 nuclear dye (c). The merge of images in panel b and c is shown in panel d. The panel "coloc mask" (e) represents the area within the cell showing colocalization of estrogen receptors with the nucleus. The value within the panel "coloc mask" represents the coefficient of colocalization of ER‐α/β staining with the nuclear staining. Panel a represents phase contrast images. The bar represents 10 μm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgements
This work was supported by grants from the Department of Biotechnology, New Delhi, India. Anti‐clusterin antibody was a kind gift from Dr. C. Yan Cheng of the Population Council, NY, USA. Technical assistance of Mr. Neelaram is appreciated.
References
- 1. Carruba G, D’Agostino P, Miele M, et al . Estrogen regulates cytokine production and apoptosis in PMA‐differentiated, macrophage‐like U937 cells. J Cell Biochem . 2003; 90: 187–96. [DOI] [PubMed] [Google Scholar]
- 2. Vegeto E, Pollio G, Pellicciari C, et al . Estrogen and progesterone induction of survival of monoblastoid cells undergoing TNF‐alpha‐induced apoptosis. FASEB J . 1999; 13: 793–803. [DOI] [PubMed] [Google Scholar]
- 3. Saintier D, Khanine V, Uzan B, et al . Estradiol inhibits adhesion and promotes apoptosis in murine osteoclasts in vitro . J Steroid Biochem Mol Biol . 2006; 99: 165–73. [DOI] [PubMed] [Google Scholar]
- 4. Zecchi‐Orlandini S, Formigli L, Tani A, et al . 17beta‐estradiol induces apoptosis in the preosteoclastic FLG 29.1 cell line. Biochem Biophys Res Commun . 1999; 255: 680–5. [DOI] [PubMed] [Google Scholar]
- 5. Subramanian M, Shaha C. Up‐regulation of Bcl‐2 through ERK phosphorylation is associated with human macrophage survival in an estrogen microenvironment. J Immunol . 2007; 179: 2337–8. [DOI] [PubMed] [Google Scholar]
- 6. Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol . 2001; 2: 63–7. [DOI] [PubMed] [Google Scholar]
- 7. Adams JM, Cory S. The Bcl‐2 apoptotic switch in cancer development and therapy. Oncogene . 2007; 26: 1324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghatan S, Larner S, Kinoshita Y, et al . p38 MAP kinase mediates bax translocation in nitric oxide‐induced apoptosis in neurons. J Cell Biol . 2000; 150: 335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Linseman DA, Butts BD, Precht TA, et al . Glycogen synthase kinase‐3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci . 2004; 24: 9993–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang H, Kim JK, Edwards CA, et al . Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol . 2005; 7: 909–15. [DOI] [PubMed] [Google Scholar]
- 11. Ahmad KA, Iskandar KB, Hirpara JL, et al . Hydrogen peroxide‐mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug‐induced apoptosis of tumor cells. Cancer Res . 2004; 64: 7867–78. [DOI] [PubMed] [Google Scholar]
- 12. Khaled AR, Kim K, Hofmeister R, et al . Withdrawal of IL‐7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc Natl Acad Sci USA . 1999; 96: 14476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi WS, Lee EH, Chung CW, et al . Cleavage of Bax is mediated by caspase‐dependent or ‐independent calpain activation in dopaminergic neuronal cells: protective role of Bcl‐2. J Neurochem . 2001; 77: 1531–41. [DOI] [PubMed] [Google Scholar]
- 14. Beato M. Gene regulation by steroid hormones. Cell . 1989; 56: 335–44. [DOI] [PubMed] [Google Scholar]
- 15. Zhao C, Dahlman‐Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal . 2008; 6: e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us Endocr Rev . 1999; 20: 358–417. [DOI] [PubMed] [Google Scholar]
- 17. Kian TM, Rogatsky I, Tzagarakis‐Foster C, et al . Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors alpha and beta. Mol Biol Cell . 2004; 15: 1262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol . 2005; 19: 833–42. [DOI] [PubMed] [Google Scholar]
- 19. Kushner PJ, Agard DA, Greene GL, et al . Estrogen receptor pathways to AP‐1. J Steroid Biochem Mol Biol . 2000; 74: 311–7. [DOI] [PubMed] [Google Scholar]
- 20. Razandi M, Pedram A, Merchenthaler I, et al . Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol . 2004; 18: 2854–65. [DOI] [PubMed] [Google Scholar]
- 21. Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol . 2006; 20: 1996–2009. [DOI] [PubMed] [Google Scholar]
- 22. Stefano GB, Prevot V, Beauvillain JC, et al . Estradiol coupling to human monocyte nitric oxide release is dependent on intracellular calcium transients: evidence for an estrogen surface receptor. J Immunol . 1999; 163: 3758–63. [PubMed] [Google Scholar]
- 23. Mor G, Sapi E, Abrahams VM, et al . Interaction of the estrogen receptors with the Fas ligand promoter in human monocytes. J Immunol . 2003; 170: 114–22. [DOI] [PubMed] [Google Scholar]
- 24. Khaled AR, Moor AN, Li A, et al . Trophic factor withdrawal: p38 mitogen‐activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol . 2001; 21: 7545–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv . 2003; 3: 281–92. [DOI] [PubMed] [Google Scholar]
- 26. Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol . 2005; 19: 1951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kramer PR, Wray S. 17‐Beta‐estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J Steroid Biochem Mol Biol . 2002; 81: 203–16. [DOI] [PubMed] [Google Scholar]
- 28. Phiel KL, Henderson RA, Adelman SJ, et al . Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol Lett . 2005; 97: 107–13. [DOI] [PubMed] [Google Scholar]
- 29. Monsma FJ Jr, Katzenellenbogen BS, Miller MA, et al . Characterization of the estrogen receptor and its dynamics in MCF‐7 human breast cancer cells using a covalently attaching antiestrogen. Endocrinology 1984; 115: 143–53. [DOI] [PubMed] [Google Scholar]
- 30. Taguchi Y, Koslowski M, Bodenner DL. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA). Nucl Recept . 2004; 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belaud‐Rotureau MA, Leducq N, Macouillard Poulletier dG, et al . Early transitory rise in intracellular pH leads to Bax conformation change during ceramide‐induced apoptosis. Apoptosis . 2000; 5: 551–60. [DOI] [PubMed] [Google Scholar]
- 32. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res . 2006; 66: 605–12. [DOI] [PubMed] [Google Scholar]
- 33. Prevot V, Croix D, Rialas CM, et al . Estradiol coupling to endothelial nitric oxide stimulates gonadotropin‐releasing hormone release from rat median eminence via a membrane receptor. Endocrinology . 1999; 140: 652–9. [DOI] [PubMed] [Google Scholar]
- 34. Benten WP, Lieberherr M, Giese G, et al . Estradiol binding to cell surface raises cytosolic free calcium in T cells. FEBS Lett . 1998; 422: 349–53. [DOI] [PubMed] [Google Scholar]
- 35. Vasconsuelo A, Milanesi L, Boland R. 17Beta‐estradiol abrogates apoptosis in murine skeletal muscle cells through estrogen receptors: role of the phosphatidylinositol 3‐kinase/Akt pathway. J Endocrinol . 2008; 196: 385–97. [DOI] [PubMed] [Google Scholar]
- 36. Kallio A, Guo T, Lamminen E, et al . Estrogen and the selective estrogen receptor modulator (SERM) protection against cell death in estrogen receptor alpha and beta expressing U2OS cells. Mol Cell Endocrinol . 2008; 289: 38–48. [DOI] [PubMed] [Google Scholar]
- 37. Hodges‐Gallagher L, Valentine CD, Bader SE, et al . Estrogen receptor beta increases the efficacy of antiestrogens by effects on apoptosis and cell cycling in breast cancer cells. Breast Cancer Res Treat . 2008; 109: 241–50. [DOI] [PubMed] [Google Scholar]
- 38. Qiu Y, Waters CE, Lewis AE, et al . Oestrogen‐induced apoptosis in colonocytes expressing oestrogen receptor beta. J Endocrinol . 2002; 174: 369–77. [DOI] [PubMed] [Google Scholar]
- 39. Schinzel A, Kaufmann T, Schuler M, et al . Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol . 2004; 164: 1021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cartron PF, Oliver L, Mayat E, et al . Impact of pH on Bax alpha conformation, oligomerisation and mitochondrial integration. FEBS Lett . 2004; 578: 41–6. [DOI] [PubMed] [Google Scholar]
- 41. Tafani M, Cohn JA, Karpinich NO, et al . Regulation of intracellular pH mediates Bax activation in HeLa cells treated with staurosporine or tumor necrosis factor‐alpha. J Biol Chem . 2002; 277: 49569–76. [DOI] [PubMed] [Google Scholar]
- 42. Hillebrand U, Hausberg M, Stock C, et al . 17beta‐estradiol increases volume, apical surface and elasticity of human endothelium mediated by Na+/H+ exchange. Cardiovasc Res . 2006; 69: 916–24. [DOI] [PubMed] [Google Scholar]
- 43. Ediger TR, Kraus WL, Weinman EJ, et al . Estrogen receptor regulation of the Na+/H+ exchange regulatory factor. Endocrinology . 1999; 140: 2976–82. [DOI] [PubMed] [Google Scholar]
- 44. Kousteni S, Chen JR, Bellido T, et al . Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science . 2002; 298: 843–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Translocation of oestrogen receptors into the nucleus upon E2 treatment. Sub‐cellular distribution of ERs consequent to treatment with 10 nM E2 or E2‐BSA for 2 h is shown by immunofluorescence using an antibody, which recognizes both ER‐α and ER‐β. The panel stained green represents the ER‐α/β staining (b), the blue staining represents nuclear staining with Hoecsht 33342 nuclear dye (c). The merge of images in panel b and c is shown in panel d. The panel "coloc mask" (e) represents the area within the cell showing colocalization of estrogen receptors with the nucleus. The value within the panel "coloc mask" represents the coefficient of colocalization of ER‐α/β staining with the nuclear staining. Panel a represents phase contrast images. The bar represents 10 μm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item