

Abstract
The temporal nature of chromatin structural changes underpinning pathologic transcription are poorly understood. We measured chromatin accessibility and DNA methylation to study the contribution of chromatin remodeling at different stages of cardiac hypertrophy and failure. ATAC-seq and reduced representation bisulfite sequencing were performed in cardiac myocytes after transverse aortic constriction (TAC) or depletion of the chromatin structural protein CTCF. Early compensation to pressure overload showed changes in chromatin accessibility and DNA methylation preferentially localized to intergenic and intronic regions. Most methylation and accessibility changes observed in enhancers and promoters at the late phase (3 weeks after TAC) were established at an earlier time point (3 days after TAC), before heart failure manifests. Enhancers were paired with genes based on chromatin conformation capture data: while enhancer accessibility generally correlated with changes in gene expression, this feature, nor DNA methylation, was alone sufficient to predict transcription of all enhancer interacting genes. Enrichment of transcription factors and active histone marks at these regions suggests that enhancer activity coordinates with other epigenetic factors to determine gene transcription. In support of this hypothesis, ChIP-qPCR demonstrated increased enhancer and promoter occupancy of GATA4 and NKX2.5 at Itga9 and Nppa, respectively, concomitant with increased transcription of these genes in the diseased heart. Lastly, we demonstrate that accessibility and DNA methylation are imperfect predictors of chromatin structure at the scale of A/B compartmentalization—rather, accessibility, DNA methylation, transcription factors and other histone marks work within these domains to determine gene expression. These studies establish that chromatin reorganization during early compensation after pathologic stimuli is maintained into the later decompensatory phases of heart failure. The findings reveal the rules for how local chromatin features govern gene expression in the context of global genomic structure and identify chromatin remodeling events for therapeutic targeting in disease.
Keywords: CTCF, Chromatin accessibility, DNA methylation, Enhancers, Heart failure
1. Introduction
In developing human cardiac myocytes, chromatin compartments (as defined by genome wide chromatin conformation capture, Hi-C) are established prior to DNA methylation patterns and, in keeping with these regions’ richness in genes versus noncoding DNA, A compartments are sites of DNA methylation dynamism [1,2]. The compartments themselves change little with disease [3,4], although the relationship with DNA methylation and general chromatin accessibility has not been explored in the diseased mammalian heart. Multiple studies have demonstrated that genetic or pharmacologic manipulation of chromatin modifying enzymes can derail normal development [5]—which itself is accompanied by orderly transitions in histone modifications as part of lineage commitment [6,7]—whereas cardiac disease phenotypes are likewise induced by targeted perturbation of histone modifying enzymes [8–10] and cardiovascular pathology is accompanied by genome-wide changes in histone modifications [11]. How the totality of chromatin marks is integrated to control local accessibility and global structure, and the rules for how this integration leads to the right transcriptome at the right time, are unknown.
It is a reasonable and convenient but perhaps unhelpful—at least in terms of gene expression and chromatin remodeling—simplification to think of disease as a recapitulation of development. It is true that expression of many genes in the diseased adult heart mimics that in the fetus [12], but this is not true for all gene expression changes, notably noncoding RNAs [13]. In the case of DNA methylation, disease-associated changes coordinate with other, protein level (i.e. histone modifications) chromatin modifications to target a subset of genes that drive cardiac pathology [14,15]—but these genes are different than the ones involved in development and the protein level changes in histone marks are more widespread than the changes in DNA methylation [14,15]. Missing from many of the prior analyses, however, is a chromatin level molecular readout for the actions of these histone and DNA methylation changes, that is, a direct measurement of chromatin accessibility. In the present study, we aimed to connect the different levels of chromatin regulation—DNA methylation, histone modifications and transcription factor localization—with chromatin structure using a readout not only of gene expression, but of chromatin accessibility, as measured by ATAC-sequencing [16]. The model system we employed is the mouse model of pressure overload, well suited to address the involvement of chromatin remodeling in disease because it recapitulates aspects of cardiac remodeling and heart failure. In addition, it is ideal for the study of temporal features of chromatin structure and transcription because pressure overload is a progressive pathology with clearly distinguishable stages of compensation and decompensation, each phenotypically resolvable by echocardiography and transcriptionally distinct. We also examine myocyte chromatin in the context of cell type specific knockout of the chromatin structural protein CTCF, which has been demonstrated to be crucial for maintaining chromatin architecture in the heart [3,4]. With this approach we ask the questions: What is the relationship between gene expression and chromatin accessibility? How do DNA methylation, histone modifications and transcription factors together influence chromatin accessibility and does this always lead to changes in transcript abundance? And, are distinct rules employed for regulation of genes, promoters, enhancers and other noncoding regions?
2. Material and methods
2.1. Transverse aortic constriction and echocardiographic measurements
All animal surgery, echocardiography and euthanasia procedures were approved by the UCLA Animal Research Committee in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Transverse aortic constriction was performed as described [3,17]. Briefly, C57BL/6J (8 weeks) animals were anesthetized, intubated, and ventilated with 2% isoflurane in 98% O2, 2% CO2 during surgery. After shaving hair, the chest was entered from the left side via the third intercostal space, the aorta identified at the T8 region and a venous vascular clamp (Fine Science Tools), which is outfitted with a band of silastic tubing at its distal edge, was placed around the vessel. The internal diameter of the resulting modified clamp was that of a 27-gauge needle. The chest was then closed using 6–0 prolene sutures, and negative pressure in the thorax was returned via removal of air by a PE 50 chest tube attached to a syringe. Mouse chest was then closed, and the animals extubated. Post-operatively, animals were administered analgesics (buprenorphine, subcutaneously, 0.05–0.1 mg/kg) if they exhibit signs of distress (vocalizing, failure to groom, immobility) until the animal improves or is euthanized. After the TAC procedure, mice were monitored by echocardiography under anesthesia (2% isoflurane in 98% O2, 2% CO2 using an anesthesia chamber for initial sedation and a nosecone to maintain delivery of anesthetic throughout the course of the procedure) every 3 days and considered to be in compensatory hypertrophy when the EF was increased significantly above the mean of the SHAM group, and in heart failure when the EF was statistically depressed below the mean of the SHAM group. After diagnosis, the mice were sacrificed by a lethal dose of sodium pentobarbital (80–100 mg/kg) administered intraperitoneally, a method approved by the UCLA Animal Research Committee.
2.2. Inducible cardiac-specific CTCF knockout model
Adult (8 weeks) cardiac-specific Ctcfflox/flox-αMHCMerCreMer+/− mice [3] were fed a diet containing 0.4 mg tamoxifen citrate per gram of diet (Tekland) for five weeks to deplete CTCF followed by one week on normal chow prior to experimentation.
2.3. Cardiomyocyte isolation
The isolation of left ventricle cardiomyocytes from adult mice was performed as described in our previous work [15]. Briefly, adult mice were treated with heparin (100 USP units) for 20 min to prevent blood coagulation and then anesthetized with sodium pentobarbital (100 μl of 50 mg/ml dilution, intra-peritoneal). Upon loss of rearfoot reflex, the heart was excised, instantaneously arrested in ice-cold phosphate buffered saline (PBS), and mounted on a modified Langendorff apparatus. After 5 min of perfusion with Tyrode’s solution (130 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 0.6 mM Na2HPO4, 10 mM glucose, and 10 mM HEPES, pH 7.37, oxygenated with 95% (v/v) O2-5% (v/v) CO2) at 37° C, the heart was perfused for 15–30 min with 30 ml Tyrode’s containing 20 mg collagenase type-II and 3 mg protease type-XIV and then washed for an additional 10 min with Krebs buffer (KB) (25 mM KCl, 10 mM KH2PO4, 2 mM MgSO4, 20 mM glucose, 20 mM taurine, 5 mM creatine, 100 mM potassium glutamate, 10 mM aspartic acid, 0.5 mM EGTA, 5 mM HEPES, pH 7.18) oxygenated with 95% O2-5% (v/v) CO2. Cardiomyocytes were dissociated in KB solution, filtered (100 μm strainer), and centrifuged 2 min at 1000 ×g for further usage.
2.4. Cell culture
Neonatal rat ventricular myocytes (NRVM) were isolated using enzymatic digestion as in [18], collected and plated with DMEM supplemented with 17% M199, 1× Penicillin-Streptomycin-Glutamine (Gibco), 10% Horse Serum (Gibco) and 5% Newborn Calf Serum (Gibco). After 24 h plating, media was switched to serum-free (1% Penicillin/Streptomycin [Gibco] and 1:1000 ITS [BD] in DMEM). The next day cells were treated with 10 μM phenylephrine for 48 h to induce hypertrophy.
2.5. siRNA analysis
To silence Cebpd expression, NRVMs were transfected using Lipofectamine RNAiMAX (Thermo Fisher) following the manufacturers’ protocols. Briefly, 500 μl of Opti MEM Reduced Serum Media (Thermo Fisher) containing 50 nM of a mix of three Thermo Fisher’s siRNA targeting Cebpd transcripts (catalog numbers: 197886, 46610, 46706) or Dharmacom’s siRNA negative control (D-001206-14) were mixed with 500 μl of the same media containing 20 μl of Lipofectamine RNAiMAX. After 10 min at 25°C, the solution was added to the cells and slightly agitated to mix. After 24 h at 37°C, the solution was replaced with 37°C medium containing 10% FBS. 48 h after transfection, cells RNA was isolated using phenol/chloroform extraction, cDNA generated using the iScript cDNA synthesis kit (BioRad) and CEBPD levels measured by qPCR using BioRad commercial CEBPD, Rat primers (qRnoCED0004314).
2.6. Cell size measurements
NRVMs were fixed with formalin and then washed with PBS and permeabilized with 0.1% Triton X-100/PBS. Cells were then stained with Phalloidin/DAPI/PBS, washed with PBS and mounted using Prolong Gold. Area and circularity measurements were performed using FIJI/ImageJ [19] and statistical analyses were performed using a Wilcoxon rank-sum test in R.
2.7. Reduced representation bisulfite sequence (RRBS) procedure and data analysis
RRBS was performed as described in our previous work [15] with some modifications. DNA from left ventricular isolated adult cardiomyocytes was isolated using the QIAshredder kit (Qiagen). Initial digestion into CpG rich regions was done by the Msp1 enzyme (NEB). The 3′ → 5′ Klenow exonuclease was used to repair sticky ends and promote poly-A tail synthesis. To facilitate multiplexing, individual TruSeq Illumina adapters were ligated to samples using the QIAseq Methyl Library Kit (Qiagen). Samples were then filtered to enrich for DNA fragments within the desired size range using magnetic AMPure XP beads (Beckman). Bisulfite conversion was performed using the Zymo EZ DNA Methylation-Lightning kit (Zymo). This reaction distinguishes methylated from unmethylated CpGs by converting unmethylated cytosines to uracil. After the final amplification of DNA fragments, sequencing was performed by the Broad Stem Cell Research Center High Throughput Sequencing Core (UCLA) using an Illumina HiSeq 3000 or NovaSeq 6000 sequencer and resulting in 40 million reads per sample.
Data analysis with a minimum of three samples per condition were performed in our lab using custom pipelines. Single-end 100 bp reads were demultiplexed into sample-specific *.fastq files using custom scripts and then aligned to the mouse genome using the BSseeker2 v2.0.10 [20]. Briefly, we built an mm10-based reference index—simply 40—400 bp MspI restriction fragments digested in silico and consistent with our enriched fragment size—using the bs_seeker2-build.py script. We then aligned single-end reads to the reference index with bs_seeker2-align.py, using the 40–400 bp fragment size range and -r flag to indicate RRBS libraries, and allowing up to 5 mismatches. We selected Bowtie2 [21] as the aligner within BSseeker2 for this step. Samtools [22] was used to sort the resulting .bam files, and methylation was called using bs_seeker2-call_methylation.py, using a 10 read coverage minimum for any given cytosine, and removing all reads that did not undergo full bisulfite conversion. The resulting .CGmap files were reformatted for use with the methylKit [23] package in R, and once the data for each sample were loaded into memory, we retained cytosines that exist within a CpG (cytosine followed by guanine) context and then used the methylKit function filterByCoverage() to remove cytosines with read coverage above the 99.9th percentile (likely PCR artifacts with abnormally high coverage). To determine differential methylation between control and diseased sample groups, we ran the calculateDiffMeth() function from methylKit to generate q-values (corrected p-values) for each cytosine measured in both sample sets. Significantly differentially methylated CpGs had q < 0.05 and average methylation difference > 10% between groups.
2.8. RNA-seq procedure and data analysis
RNA-seq was performed as described in our previous work [15] with some modifications. RNA from left ventricular isolated adult cardiomyocytes was isolated using the RNeasy Mini Kit (Qiagen). RNA quality was assessed using the Agilent 2100 Bioanalyzer. Ribosomal RNA was then removed using an Illumina Ribo-Zero rRNA Removal Kit and the RNA-seq libraries prepared by the Technology Center for Genomics & Bioinformatics Core (UCLA) following standard protocols. Libraries were sequenced in an Illumina NovaSeq 6000 sequencer using as many lanes as are needed to ensure 40 million reads per sample.
Data analysis with three samples per condition were performed in our lab using custom pipelines. Paired-end 150 bp reads were demultiplexed into sample-specific *_R1.fastq and *_R2.fastq files using custom scripts and then pseudoaligned to the mm10 genome using the ultra-fast pseudoalignment tool, Salmon [24]. Briefly, we ran salmon index to generate a mm10 (mouse) transcriptome index using the appropriate “known cDNA” FASTA file from Ensembl release 81 [25]. Corresponding read pairs were pseudoaligned to the appropriate transcriptome using salmon quant with mm10-specific index, *_R1.fastq and *_R2.fastq files as inputs. This ultra-fast process results in a transcript count for each known gene in the genome and facilitates downstream calculation of count-based statistics. Salmon transcript quantifications (*.sf files) for all samples were loaded into a single R object using the tximport package [26], and then differential expression analysis was performed using DESeq2 [27]: First, any genes with no transcript counts in any sample were excluded from the analysis because they registered 0 measurements in the entire RNA-seq experiment. Then, we converted loaded Salmon quantifications into a DESeqDataSet and filtered to keep genes that have at least 10 observed reads amongst the samples. Differential expression analysis was performed on the filtered DESeqDataSet using the DESeq() function with default parameters, providing p-values and adjusted p-values (adjusted using the Benjamini-Hochberg method [28]) for each gene. Significantly differentially expressed genes were those with adjusted p-value <0.05. Heatmaps were generated using the heatmap.2() function within the gplots package in R (https://CRAN.R-project.org/package=gplots), with unbiased hierarchical clustering on the rows. Principal component analysis was performed using the DESeq2 package. Gene ontology analysis was performed using g:Profiler [29] with the default g:SCS multiple testing correction method.
For visualization purposes, we also ran alignments of raw RNA-seq data to mm10 using STAR [30] v. 2.5.4b on combined replicates for each group, using a reference genome based on Ensembl release 81 and generating sorted .bam files as output. We then generated bigWig browser tracks using the bamCoverage function of deeptools [31] v3.0.2, with the parameters –normalizeUsing RPGC and –effectiveGenomeSize 2652783500.
2.9. ATAC-seq procedure and data analysis
Tagmentation of nuclei isolated from adult cardiac myocytes was attempted several times in collaboration with Active Motif. The technical challenge of generating a complex library, probably due to the cell isolation buffer composition, led us to use frozen tissue. Flash frozen left ventricles from control, 3 days TAC, 3 weeks TAC, and CTCFKO animals (n = 3/group) were sent to Active Motif for ATAC-seq library preparation and sequencing [16]. The tissue was manually disassociated, isolated nuclei were quantified using a hemocytometer, and 100,000 nuclei were tagmented as previously described [16], with some modifications based on [32] using the enzyme and buffer provided in the Nextera Library Prep Kit (Illumina). Tagmented DNA was then purified using the MinElute PCR purification kit (Qiagen), amplified with 10 cycles of PCR, and purified using Agencourt AMPure SPRI beads (Beckman Coulter). Resulting material was quantified using the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems), and sequenced with paired-end 42-bp sequencing on the NextSeq 500 sequencer (Illumina).
Paired-end 42 bp reads measured on an Illumina NextSeq 500 were demultiplexed and mapped to the mm10 genome using BWA [33] in mem mode with default parameters, allowing for 2 mismatches. Uniquely mapped reads from matched pairs were retained for further analysis, and PCR duplicates were removed before peak calling. Peaks were called using MACS [34] v2.1.0 at a cutoff of p-value 1e-7 on sorted. bam files from each sample, using the –nomodel parameter. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. To calculate differential accessibility between biological groups, we used the R package DiffBind [35] from Bioconductor [36]. Briefly, quantified fragment counts within a consensus peak set that exists within 6/12 samples from the experiment, using the dba.count() function with parameters minOverlap = 6, fragmentSize = 0, and score = DBA_SCORE_TMM_READS_EFFECTIVE. We then performed differential accessibility testing using the dba.analyze() function in DiffBind, with method = DBA_EDGER and bFullLibrarySize = F as parameters. Differentially accessible peaks were those that had FDR < 0.05 after this process. Principal component analysis was performed on the consensus peaks using the DiffBind library.
In addition to performing differential accessibility analysis on peaks from this experiment, we repeated this process on autosomal cardiac enhancers [37] (lifted over to mm10) and gene promoters (from the Bioconductor package EnsDb.Mmusculus.v79). Promoters were defined as −2000 to +200 bp from the TSS. We also performed this analysis on 5 kb bins that either change compartment (AtoB or BtoA) or remain in the same compartment (AtoA, BtoB) with 3 weeks TAC or CTCFKO, based on Hi-C data. Enhancers, promoters, or bins with FDR < 0.05 were defined as statistically differentially accessible. Heatmaps were generated using the heatmap.2() function within the gplots package in R (https://CRAN.R-project.org/package=gplots), with unbiased hierarchical clustering on the rows. We generated bigWig visualizations by first combining all 3 replicate .bam files into a larger merged file for each condition, and then using that as input for the bamCoverage function of deepTools [31] v3.0.2, with the parameters –normalizeUsing RPGC and –effectiveGenomeSize 2652783500.
2.10. ChIP-qPCR
Chromatin immunoprecipitation was performed using the ChIP-IT High Sensitivity Kit (Active Motif) on 10 million cardiomyocytes isolated from the left ventricle of control, 3 weeks TAC, and CTCFKO mice. Briefly, cells were fixed, lysed and sonicated using a Covaris E220 sonicator to generate fragments between 500 and 1000 bp. DNA-bound protein was immunoprecipitated using anti-H3K27ac (ab4729), anti-GATA (sc1237), anti-Nkx2.5 (ab35842) or IgG (sc2027). ChIP-qPCR values were expressed as fold-enrichment over IgG. Average of three independent experiments is shown. See Supplemental Table 1 for primer sequences.
2.11. ChIP-seq data analysis
To visualize H3K27ac signal as a bigWig track within Integrated Genome Browser [38], we performed alignments on previously published raw H3K27ac ChIP-seq data from isolated cardiac myocytes from SHAM hearts [4], using Bowtie2 [21] v. 2.3.4.1 with an mm10 Bowtie2 index based on Ensembl version 81 (downloaded from Illumina iGenomes). The resulting .sam file was sorted and indexed using samtools [22] v. 1.7 and then used as input for the bamCoverage function of deepTools, using the parameters described above, to generate a browser track.
Published mm10 ChIP-seq peak sets were downloaded from the ENCODE Project [39], using file accession numbers ENCFF616HYA, ENCFF383PSV, ENCFF733HUI, ENCFF887SZF, ENCFF950UIP, ENCFF481GRM, ENCFF599BFW, ENCFF206SZJ, and ENCFF454UOO. We also downloaded adult cardiac transcription factor bioChIP-seq peak coordinates [40], corresponding to GATA4, MEF2A, NKX2.5, SRF, TBX5, and TEAD1 biochip experiments, and lifted the coordinates over to mm10 in R. To determine significance of transcription factor or histone mark overlap with specific subsets of enhancers, we performed hypergeometric significance tests in R. For each factor, we calculated a cumulative probability of k or more successes (that is, the probability of observing k or more enhancers overlapping with a given factor) given our enhancer subset and our global list of autosomal enhancers. This resulted in a metric that described whether a given transcription factor or histone mark was overrepresented within our subset of enhancers.
2.12. Hi-C analysis
Hi-C data from our published work [3] was used for this study (GSE96692). For each group, paired-end reads (*_R1 and *_R2 fastqs) from all biological replicates were combined and run through the HiC-Pro [41] v2.10.0 pipeline with default parameters and an MboI in silico digested genome file to perform alignments to the mm10 reference, generate 5 kb resolution contact matrices, and carry out iterative correction and eigenvector decomposition-based normalization of matrices. To call significant interactions from our Hi-C data, we first ran our original 5 kb contact matrices and ICE bias files through the hicpro2fithic.py script that comes with HiC-Pro and retained only autosomal data. Using the resulting properly formatted tables, we ran Fit-Hi-C [42] version 2.0.6 without lower or upper distance limits and under the following parameters: -b 200 -r 5000 -p 2. From these results, we filtered to keep bin pairs with q value <0.01 and interaction count of at least 10, and then retained only interactions more than 5 kb apart to avoid examining adjacent 5 kb bins. A/B compartmentalization was calculated as described [3]. The significant Fit-Hi-C interactions and bin compartment statuses from each biological group were then loaded into R for custom analyses and visualizations.
3. Results
3.1. Examination of chromatin accessibility, DNA methylation and gene expression in early and late phase disease
To investigate the dynamics of chromatin accessibility in cardiac myocytes during the different phases of pressure overload, we performed ATAC-seq at 3 days and 3 weeks after TAC surgery when the animals have begun compensatory hypertrophic growth with sustained EF and when the pathologic growth has further advanced to ventricular dilation and impaired EF, respectively (these physiological parameters were characterized by echocardiography and post-mortem histological and gene expression analyses, Supplemental Fig. 1). We [3] and others [4] have previously shown that depletion of the chromatin structural protein CTCF induces a heart failure phenotype that shares some but not all gene expression, chromatin structural, and histopathological features with the TAC model: ergo, we sought to compare chromatin accessibility changes between these two models. The initial phase (day 3) of the syndrome was associated with widespread change in chromatin accessibility, with a tendency toward decreased accessibility (5372 less accessible versus 4128 more accessible regions), some of which persisted into the later phase at week 3 (973 less accessible versus 2391 more accessible regions; Fig. 1A; see Supplemental Fig. 2A for PCA). The 3 week time point was also associated with novel accessibility changes not observed at 3 days (2891 less accessible versus 3895 more accessible regions; Fig. 1A, fourth panel) and CTCF depleted chromatin—examined here after CTCF depletion for 5 weeks and 1 additional week on regular chow—also showed a slight trend toward decreased accessibility (2129 less accessible versus 1349 more accessible regions) reflecting the role of CTCF to stabilize heterochromatin [43,44]. Interestingly, 3 weeks of TAC induced increased accessibility compared to the basal condition (Fig. 1A, second panel) suggesting that chromatin opens during progression of pressure overload-induced disease.
Fig. 1.
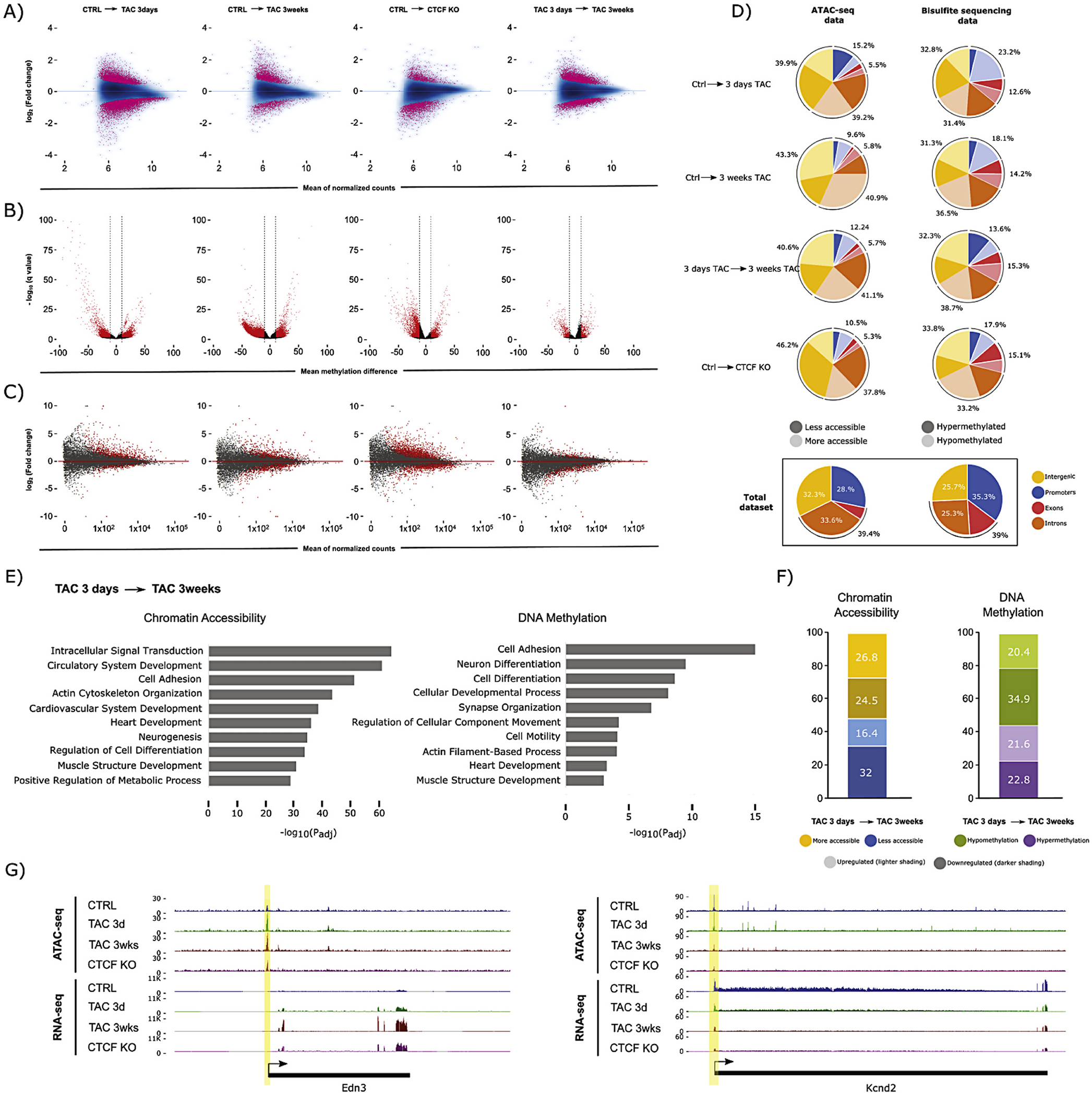
Chromatin accessibility, DNA methylation, and gene expression dynamics during heart failure. (A) Chromatin accessibility (ATAC-seq) MA plots comparing CTRL with 3 days TAC, 3 weeks TAC, and CTCFKO, (n = 3 animals per biological condition). The difference between 3 days and 3 weeks TAC is also shown. Pink dots indicate peaks with significant change in accessibility (FDR < 0.05). (B) DNA methylation (RRBS) volcano plots showing differentially methylated CpG sites (red, average DNA methylation difference > 10% and q-value <0.05) between conditions, (n = 3 animals per biological condition). (C) Gene expression (RNA-seq) MA plots for the same comparisons as in (A), (n = 3 animals per biological condition). Each dot represents an annotated transcript. Grey dots reflect no significant change. Red dots represent transcripts with adjusted p-value <0.05 (D) Differentially accessible peaks and differentially methylated CpGs occur preferentially in intergenic and intronic regions (top 4 pie charts) as judged by comparison to the total set of measured peaks and CpGs in our datasets (bottom pie charts). Promoter/exon/intron/intergenic distribution of differentially accessible ATAC-seq peaks (left) after 3 days TAC, 3 weeks TAC, and CTCFKO, in addition to the comparison between 3 days and 3 weeks TAC. Dark and light coloring indicates less and more accessibility, respectively. Right, Pie charts showing distribution of hypo- and hypermethylated CpGs in darker and lighter shading, respectively. Promoter regions were defined as −2000 to +200 bp from the transcription start site. (E) Gene ontology analysis of genes plus promoters (genes+2000 bp) that overlapping differentially accessible peaks (left) and differentially methylated CpGs (right) between 3 days and 3 weeks TAC. Terms associated with cardiovascular processes such as heart development, cytoskeletal and neuronal organization were significantly enriched when we study changes in chromatin accessibility (left) and DNA methylation (right). The x-axis indicates −logl0(adjusted p-value) for the analysis. (F) Charts indicating significant expression change (padj <0.05) for genes undergoing differential chromatin accessibility (left, FDR < 0.05) or DNA methylation (right, methylation difference > 10% and q-value <0.05). Left, Blue and yellow coloring indicate decrease and increase in chromatin accessibility between 3 days and 3 weeks TAC, respectively. Right, green and purple colour indicate hypo- and hypermethylation, respectively. For both stacked bar charts, shading indicates up- (lighter) or downregulation (darker) at the transcript level, respectively. (G) Browser tracks of genes that have change in chromatin accessibility between 3 days and 3 weeks TAC. Each track contains a combined signal from 3 replicates for each condition. Example loci showing increase in chromatin accessibility and transcription (Edn3, encoding Endothelin-3, a member of the endothelin family implicated in cardiovascular disease [57], left), and decrease in accessibility and transcription (Kcnd2, encoding Potassium voltagegated channel subfamily D member 2 which is associated with sudden cardiac death [58], right). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
When DNA methylation was examined in these same animals, the early and late stage disease (and CTCF knockout) showed a trend toward demethylation, whereas the transition from early to late stage did not (Fig. 1B). In contrast to accessibility, DNA methylation changes were smaller in number early after injury (5646 differentially methylated CpGs after 3 days TAC versus 27,275 and 13,224 after 3 weeks TAC and CTCF depletion, respectively). When we compared early and late stage pressure overload, we detected that 65% of the differentially methylated CpGs were shared between conditions (Supplemental Fig. 3A), suggesting that changes in DNA methylation are established early in disease and preserved during chronic pathology. Comparison between 3 weeks TAC and CTCF KO (both late phase disease conditions) demonstrated that 56% of differentially methylated CpGs change with both pathological stimuli (Supplemental Fig. 3B), showing similarities in methylome remodeling mediated by TAC and CTCF depletion. Strikingly, nearly all shared DNA methylation changes (~99% in both cases; Supplemental Fig. 3A and B) that changed at 3 weeks and 3 days when compared to control, or with 3 weeks TAC and CTCFKO compared to control, did so in the same direction.
Changes in accessibility and CpG methylation were associated with gene expression changes (Fig. 1C; see also Supplemental Fig. 2B for PCA and 2C and D for overlap of differentially expressed genes): the relationship between these phenomena are explored in greater detail for the remainder of the manuscript.
An interesting pattern was unearthed when examining the location of regions with altered accessibility or DNA methylation: the changes preferentially localized to intergenic and intronic regions (Fig. 1D). This observation was true for all comparisons. The fact that reduced representation bisulfite sequencing (RRBS) selects for CpG islands (which are enriched in promoters and TSSs) further suggests that the preponderance of change in non-coding regions was worthy of further investigation. Gene ontology analyses of the ATAC peaks or differentially methylated CpGs that were in or near genes revealed enrichment in similar subsets of terms (Fig. 1E; as did CTCF knockout cells, Supplemental Fig. 4A) including general cellular processes, heart function, neurogenesis and neuron differentiation, suggesting a role of the nervous system in cardiac compensation to pressure overload. The left panel of Fig. 1F shows that increased accessibility at a locus is associated with increased RNA transcription in cardiac cells after pressure overload (~60% of the loci show correlation between gene expression and chromatin accessibility), but only slightly, with many loci exhibiting the antithetical behavior. The same was true for DNA methylation and expression after pressure overload (Fig. 1F, right panel) and in CTCF depleted hearts (Supplemental Fig. 4B). Remodeling of accessibility at individual loci known to be involved in cardiac pathology is shown in granular detail in Fig. 1G.
3.2. DNA methylation at enhancers plays a role in gene transcription
The observation that intergenic and intronic regions were differentially methylated led us to hypothesize that enhancer elements may be subject to altered DNA methylation as a mechanism to regulate gene expression. To test this hypothesis, we selected a set of enhancers discovered in the mouse heart [37] via H3K27ac ChIP-seq and used chromatin architectural data from Hi-C experiments [3] to determine the genes these enhancers interact with in the myocyte nucleus. Changes in DNA methylation at these enhancers trended toward demethylation (Supplemental Fig. 5A) but did not predict gene expression behavior at either time point after pressure overload or following CTCF depletion (Fig. 2A; see also Supplemental Fig. 6), leading us to hypothesize several scenarios by which DNA methylation may interact with other factors at the TSS or enhancer to regulate transcription (Supplemental Fig. 7A). An exemplar of one of these models is shown in Supplemental Fig. 7B, wherein the gene Rtn4rl1 is up regulated in response to enhancer methylation. To determine the mechanism by which differential methylation is read to influence transcription, we performed a motif discovery analysis (Hypergeometric Optimization of Motif Enrichment, or HOMER [45]) on those enhancers with significant change in DNA methylation at 3 weeks after TAC or CTCF depletion. This study discovered motifs for several cardiac regulators (Supplemental Fig. 7C). We then used published ChIP-seq data to confirm these findings (Fig. 2B), focusing on occupancy of cardiac transcription factors in adult hearts [40], including GATA4, TEAD1, TBX5, SRF, NKX2.5, and MEF2A, which have multiple binding sites around genes undergoing altered expression in TAC (Supplemental Fig. 7B is an example), as well as histone marks and other factors from ENCODE, including: H3K27ac (active), H3K27me3 (repressed), H3K36me3 (gene body), H3K4me1 (enhancer), H3K4me3 (active), H3K79me2 (active), H3K9ac (active), p300 and CTCF. When the aforementioned subset of enhancers is examined (i.e. those enhancers making long range contacts with differentially expressed genes in either TAC or CTCF depletion) we observe a striking enrichment of colocalization of GATA4, TBX5, TEAD1 and p300 (MEF2A, NKX2.5, SRF and CTCF are also enriched but to a lesser degree) in the subset of these enhancers that are differentially methylated in TAC, and to a lesser degree in the subset of these enhancers differentially methylated after CTCF depletion (Fig. 2B; see also Supplemental Fig. 7D). These enhancers also seem to be preferentially targeted by acetylation marks (H3K27ac and H3K9ac; Fig. 2B), implying coordination between DNA methylation, transcription factors and histone marks to determine the transcriptional activity of enhancer interacting genes. To validate H3K27ac, GATA4 and NKX2.5 occupancy at candidate promoters and enhancers, we performed ChIP-qPCR for these factors around the Itga9, Nppa, and Mtss1 loci (Fig. 2C). The Itga9 enhancer and promoter exhibited an increase in H3K27ac and GATA4; the Nppa enhancer and promoter exhibited an increase in H3K27ac and NKX2.5; and the Mtss1 enhancer and promoter exhibited a decrease in H3K27ac occupancy with TAC 3 weeks and CTCFO. The differential occupancy detected by ChIP-qPCR is consistent with increased transcription of Itga9 and Nppa observed with 3 weeks TAC or CTCFKO as well as with decreased transcription in the case of Mtss1, however all three promoter and enhancer pairs examined by ChIP-qPCR undergo increased DNA methylation with pressure overload and CTCFKO. This suggests the effect of DNA methylation is complemented by nuclear effectors such as transcription factor and histone mark occupancy in regulation of important cardiac genes during disease.
Fig. 2.
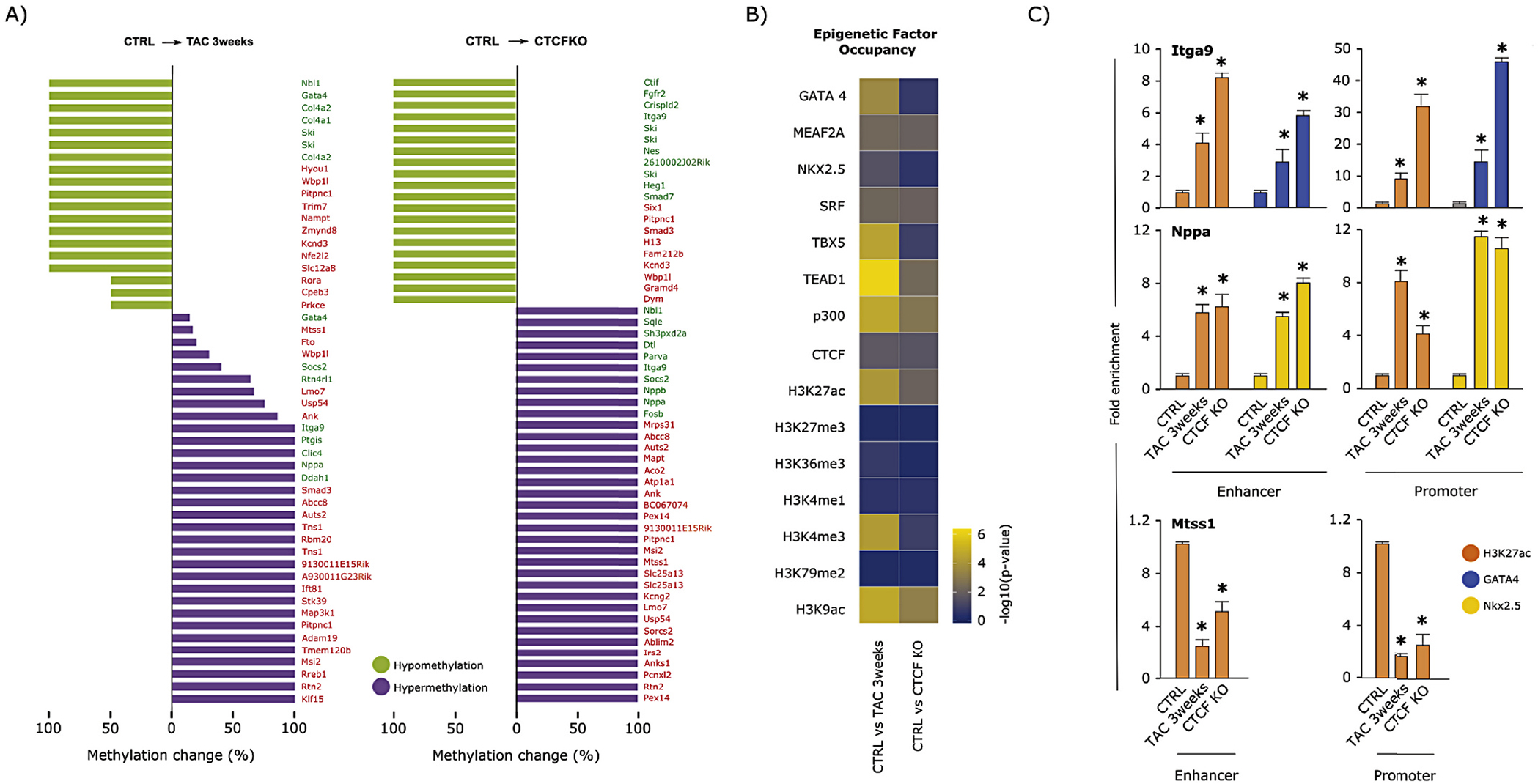
Enhancer methylation plays a role in the transcriptional regulation of subsets of cardiac genes. (A) Analysis of enhancers containing differentially methylated CpGs. Bar charts indicating percent of significantly hyper- (purple) or hypomethylated (green) CpGs in enhancers after 3 weeks TAC (left) or with CTCFKO (right). Of these, all enhancers that interact with differentially expressed (padj <0.05) genes based on our significant Fit-Hi-C interaction data are shown. Green and red text indicate up- and downregulation of transcript levels, respectively. (B) Heatmap showing -log10(p-value) (calculated as cumulative hypergeometric probabilities) as a metric to summarize overrepresentation of transcription factor binding or histone mark deposition (rows of heatmap) at the subset of enhancers that undergoes significant (average methylation difference > 10% and q-value <0.05) change in DNA methylation in 3 weeks TAC or with CTCFKO (left and right columns, respectively) when compared to control, and whose 3D interacting genes undergo significant (padj <0.05) transcriptional change. These data suggest that active histone marks from ENCODE and cardiac transcription factor peaks are statistically overrepresented at this subset of enhancers, while inactive marks such as H3K27me3 and H3K79me2 are not. (C) ChIP-qPCR experiments showing enrichment of the active mark H3K27ac and the cardiac transcription factors GATA4 or Nkx2.5 at the methylated promoters and respective enhancers of the Itga9 and Nppa loci during heart failure (3 weeks TAC and CTCFKO). Loss of H3K27ac was detected at both the promoter and interacting enhancer of the Mtss1 gene after TAC 3wks and CTCFKO. These experiments confirm that DNA methylation at the enhancer level influences pathological gene expression by participating with other epigenetic factors that can reinforce (e.g., Mtss1 locus) or prevent (e.g., Itga9 and Nppa loci) gene repression. Data show the average ± SD of three independent experiments; ChIP-qPCR values were expressed as fold-enrichment over IgG. Two-tailed Student’s t-test assessed statistical significance: *p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In addition to known cardiac transcription factor motifs, investigation of motifs within differentially accessible enhancers with 3 weeks TAC or CTCFKO revealed enrichment of CCAAT/Enhancer-binding protein (C/EBP) family (Supplemental Fig. 7C). Notably, Cebpd, the only differentially expressed C/EBP family gene at early and late phase disease, was upregulated with TAC (3 days and 3 weeks) and CTCFKO (Supplemental Fig. 8A). To test whether Cebpd function is important for cardiomyocyte response to hypertrophic stimulus, we transfected isolated neonatal rat ventricular myocytes with either siRNA negative control or siRNA against Cebpd transcripts and found a 78% decrease in Cebpd mRNA levels (Supplemental Fig. 8B) and marked abrogation of phenylephrine-induced hypertrophic phenotype upon Cebpd knockdown (70% of the average cell area in the phenylephrine-treated condition) (Supplemental Fig. 8C), suggesting that this gene is necessary for the hypertrophic response to phenylephrine.
3.3. Changes in chromatin accessibility and transcription are established in early disease
Most of the measured changes in chromatin accessibility at peaks were similar between TAC and CTCF depletion (late phase disease samples), with the changes beginning early (3 days) after pressure overload and progressing along with pathology (Fig. 3A). The majority of these changes in chromatin accessibility in the early phase are not yet significant, but the direction of change is determined by this time and continues into late pathology. This result is supported by comparing only the peaks with significant change in accessibility at early and at late phase TAC: 28% of those regions were shared in both conditions (Supplemental Fig. 3C), yet 99% of the shared differentially accessible peaks changed in the same direction, suggesting a time dependent shift in chromatin accessibility with chronic pressure overload. Comparison between the two late disease conditions (3 weeks TAC and CTCF KO) showed 50% shared peaks (Supplemental Fig. 3D), providing evidence for more common regions of differential chromatin accessibility between the two late stage pathologies (Supplemental Fig. 3D) than between early and late stage disease (Fig. 3A, right column; Supplemental Fig. 3C). Similar to what was detected with methylation, ~99% of all shared accessibility changes switched in the same direction (Supplemental Fig. 3C and D).
Fig. 3.
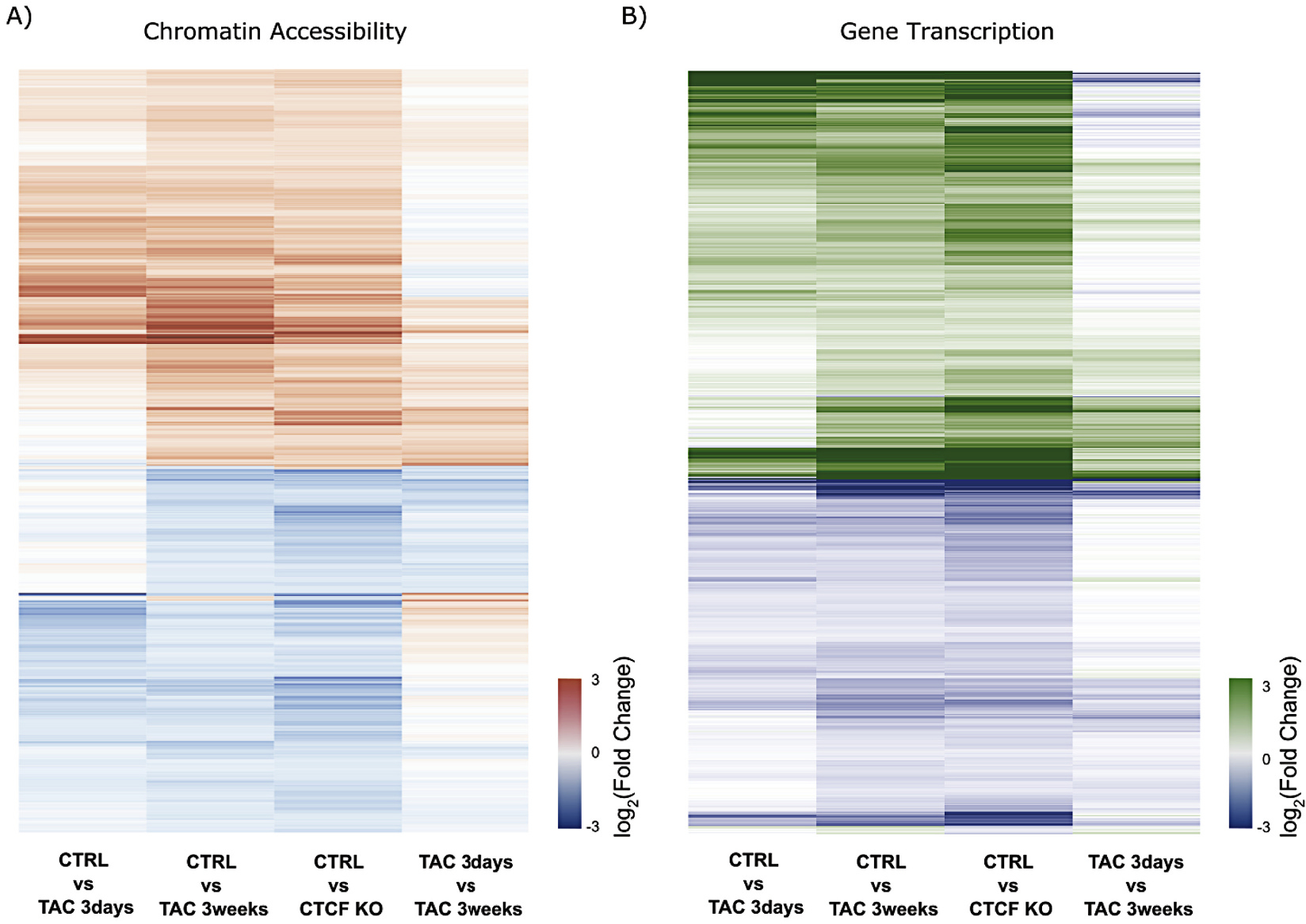
Transcription and accessibility change with chronic pathology are preceded by subtle architectural and transcriptional changes during acute perturbation. (A) Heatmap showing log2(Fold Change) of ATAC-seq peaks with significant change in chromatin accessibility (FDR < 0.05) at 3 weeks TAC and with CTCFKO. Accessibility changes occurring in late stage disease are preceded by minor changes during early pathology (1st column) that progress toward late stage disease (4th column shows continuation from 3 days to 3 weeks TAC). Red and blue coloring denote increase and decrease in accessibility, respectively. (B) Heatmap of log2(Fold Change) in transcription of differentially expressed genes (padj <0.05) at 3 weeks TAC and with CTCFKO when compared to Control. Green colour denotes upregulation of transcripts in treatment while blue denotes downregulation of mRNA levels. As in (A) changes that occur in later stages of pathology are not fully established by 3 days TAC (1st column), yet they progress from early to late pathology (4th column shows progression from 3 days to 3 weeks TAC), suggesting a time dependent shift in transcription with chronic pressure overload or CTCFKO. Panels (A) and (B) highlight progressive changes that result in significantly differential accessibility and transcription at late-stage pathology. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The same behavior was observed at the transcriptional level when we compared early and late phase disease (Fig. 3B, right column). The majority of changes in expression detected at the decompensated stage (3 weeks TAC and CTCF KO) were already appearing by the compensatory phase (3 days) and progressed toward the decompensatory phase (3 weeks), indicating that most of the transcriptome remodeling occurs prior to decompensatory changes in the phenotype.
A similar observation was found when we focused our analyses on promoters: late changes to promoter accessibility were established early after insult and continued down a path toward the late pathological state (with the late stage disease resembling the CTCF depleted scenario, where the animals are in heart failure; Fig. 4A). Accessibility of promoters (64.4%) was positively correlated with gene expression in a greater percentage of cases (Fig. 4B) than when the entire peak set was considered (Fig. 1F), and the genes whose promoters were rendered more accessible during heart failure were associated cardiac development and contraction, glucose metabolism, cytoskeletal organization and general processes like cell motility (Fig. 4C). Example changes in promoter accessibility for differentially expressed genes are shown in Fig. 4D.
Fig. 4.
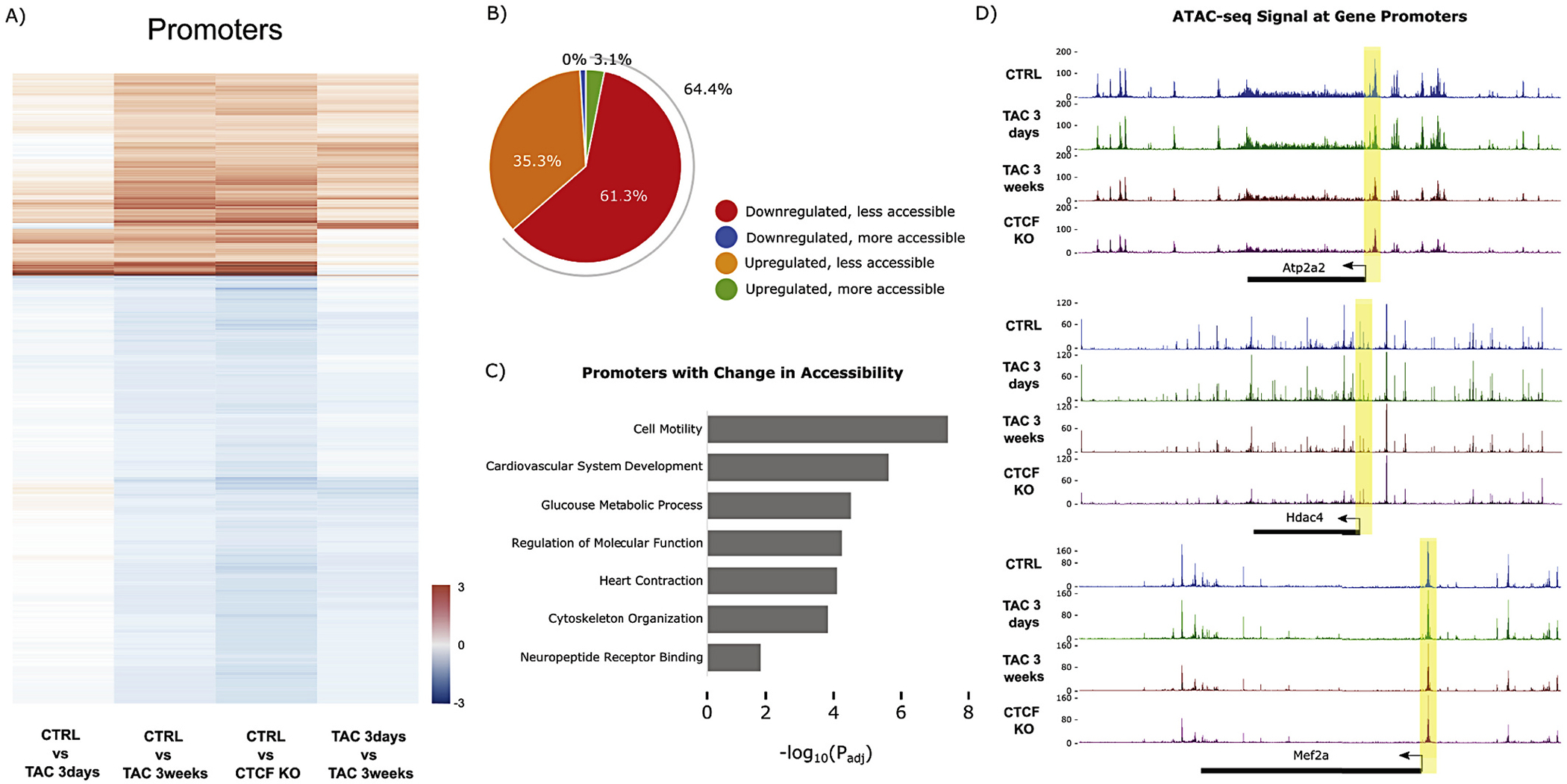
Promoter accessibility dynamics with pressure overload and CTCFKO. (A) Heatmap showing promoters with significant change (FDR < 0.05) in chromatin accessibility after 3 weeks TAC and with CTCFKO when compared to Control. Promoter accessibility dynamics with chronic pressure overload and long-term CTCF depletion are preceded by subtler changes during early/acute pathology (1st column) that progress into late pathology (further progression shown in 4th column, resulting in the sustained differences observed in 2nd and 3rd columns). Red and blue coloring denote gain and loss of accessibility, respectively. This panel highlights the gradual promoter accessibility changes at 3 days TAC that become significant at 3 weeks. (B) Pie chart summarizing the subset of promoters from (A) with significant (FDR < 0.05) change in chromatin accessibility and in transcription (padj <0.05) in both 3 weeks TAC and with CTCFKO. (C) Gene ontology analysis showing significant terms related to promoters with differential accessibility (FDR < 0.05) in 3 weeks TAC and with CTCFKO. The x-axis indicates -log10(adjusted p-value) for the analysis. (D) Chromatin accessibility tracks showing decreases in accessibility between control (blue), 3 days TAC (green), 3 weeks TAC (red) and CTCF depletion (purple) at the Atp2a2, Hdac4 and Mef2a loci (expression of all three genes are downregulated at early and late stage disease). Each track contains a combined signal from 3 replicates for each condition. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Enhancer accessibility obeyed the same logic: directions of common late changes in accessibility were observed in early stage disease, progressing between early and late pathology, and late stage TAC resembled CTCF depletion (Fig. 5A). During individual comparisons, a slight trend was seen toward decreased accessibility between 3 days and 3 weeks TAC and between control and CTCF knockout, but this trend was smaller when comparing control and 3 days TAC and was slightly reversed between control and 3 weeks TAC (Supplemental Fig. 5B). As with the promoters, a subset of those enhancers (65.5%) with differential accessibility was physically associated in 3D with genes whose expression increased, and vice versa (Fig. 5B), with cardiac terms again enriched in the gene ontology analyses (Fig. 5C). Interestingly, transcription factors and histone marks showed differential association with this subset of enhancer loci exhibiting differential accessibility (Fig. 5D; see also Supplemental Fig. 7D), although the enrichment pattern was quite distinct from the comparison between these marks and DNA methylation (Fig. 2D). Fig. 5E shows an example locus surrounding Nppa, the gene that encodes atrial natriuretic factor, which is upregulated in heart disease.
Fig. 5.
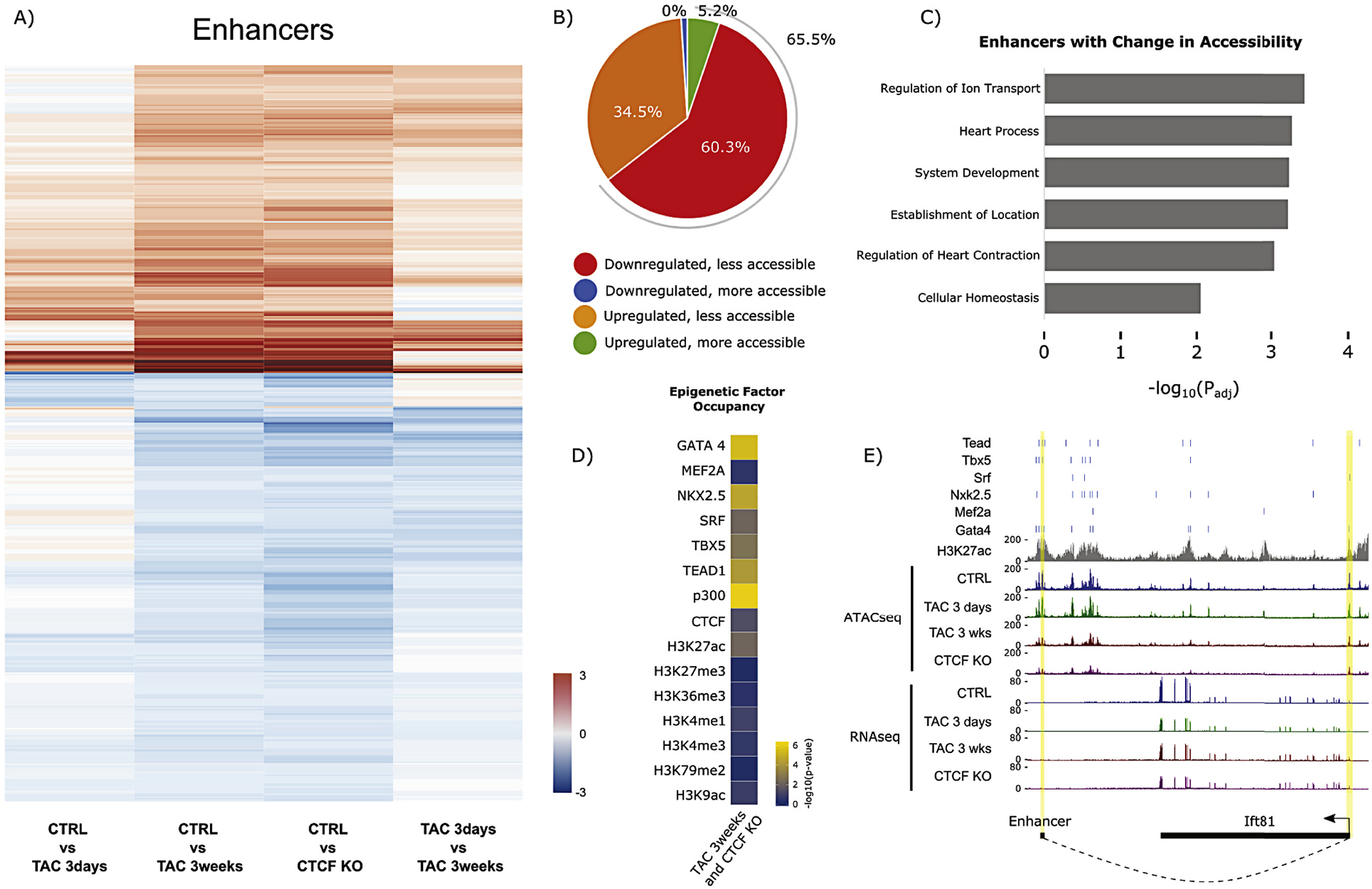
Enhancer accessibility dynamics with pressure overload and CTCFKO. (A) Heatmap showing enhancers with significant change (FDR < 0.05) in chromatin accessibility after 3 weeks TAC and with CTCFKO when compared to Control. Enhancer accessibility dynamics with chronic pressure overload and long-term CTCF depletion are preceded by subtler changes during early/acute pathology (1st column) that progress into late pathology (4th column shows progression to late-stage heart failure in 2nd and 3rd columns). Red and blue coloring denote gain and loss of accessibility, respectively. This panel highlights the gradual enhancer accessibility changes at 3 days TAC that become significant at 3 weeks. (B) Pie chart summarizing the subset of enhancers from (A) with significant (FDR < 0.05) change in chromatin accessibility, and that have significant Fit-Hi-C interactions (from Hi-C data) with differentially expressed (padj <0.05) genes. (C) Gene ontology analysis of the genes summarized in (B). The x-axis indicates -log10(adjusted p-value) for the indicated terms. (D) Heatmap showing -log10(p-value) of hypergeometric tests as a metric to summarize overrepresentation of transcription factor binding at the subset of enhancers that undergoes significant (FDR < 0.05) change in chromatin accessibility in 3 weeks TAC and with CTCFKO when compared to Control, and whose 3D interacting genes undergo significant (padj <0.05) transcriptional change. These data suggest that active histone marks from ENCODE and cardiac transcription factor peaks from [40] are overrepresented at this subset of enhancers, while inactive marks such as H3K27me3 and H3K79me2 are not. (E) Example locus where an enhancer with decreased accessibility interacts with a nearby gene, Ift81, also exhibiting decreased accessibility and decreased expression. This gene, also known as carnitine deficiency associated gene expressed in ventricle 1 (Cdv1), has been implicated in cardiac hypertrophy associated with carnitine deficiency [59]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. Chromatin accessibility and DNA methylation are poor predictors of genomic compartmentalization
To integrate these observations, we examined the totality of regions undergoing altered accessibility and DNA methylation, quantifying each of the four possible behaviors (increased/decreased accessibility, increased/decreased methylation). As shown in Fig. 6A, promoters had overall greater levels of change in each quadrant (owing to the larger number of promoters queried in the study) compared to enhancers (5188 regions in promoters compared to the 1333 detected in enhancers), yet both regulatory regions had all four accessibility-methylation behaviors represented. Some very intriguing differences were revealed: while the late stage of disease (3 weeks TAC and CTCF knockout) is associated with a greater concordance between hypomethylation and decreased accessibility at promoters, this stage of disease was accompanied by hypermethylation and decreased accessibility at enhancers (Fig. 6A, right panel). The physiological importance of this distinct temporal behavior between early and late stage disease was determined by analyzing gene expression: at the early time point, accessibility and DNA methylation at promoters are imperfect predictors of gene expression, however as the disease progresses, the relationship gets much stronger (compare TAC 3 days to TAC 3 weeks; Fig. 6B; the comparable analysis for enhancers resulted in only two genes with altered expression) These findings suggest that DNA methylation and accessibility preserve chromatin landscaping, thereby entrenching diseased gene expression. At promoters with hypermethylated CpGs and decrease in chromatin accessibility, the discordance between behaviors in the either 3 day or 3 week TAC condition with the CTCF knockout heart exemplifies this as a distinct model of heart failure resulting from direct disruption of chromatin (rather than through pressure overload; right panels in Fig. 6A and B).
Fig. 6.
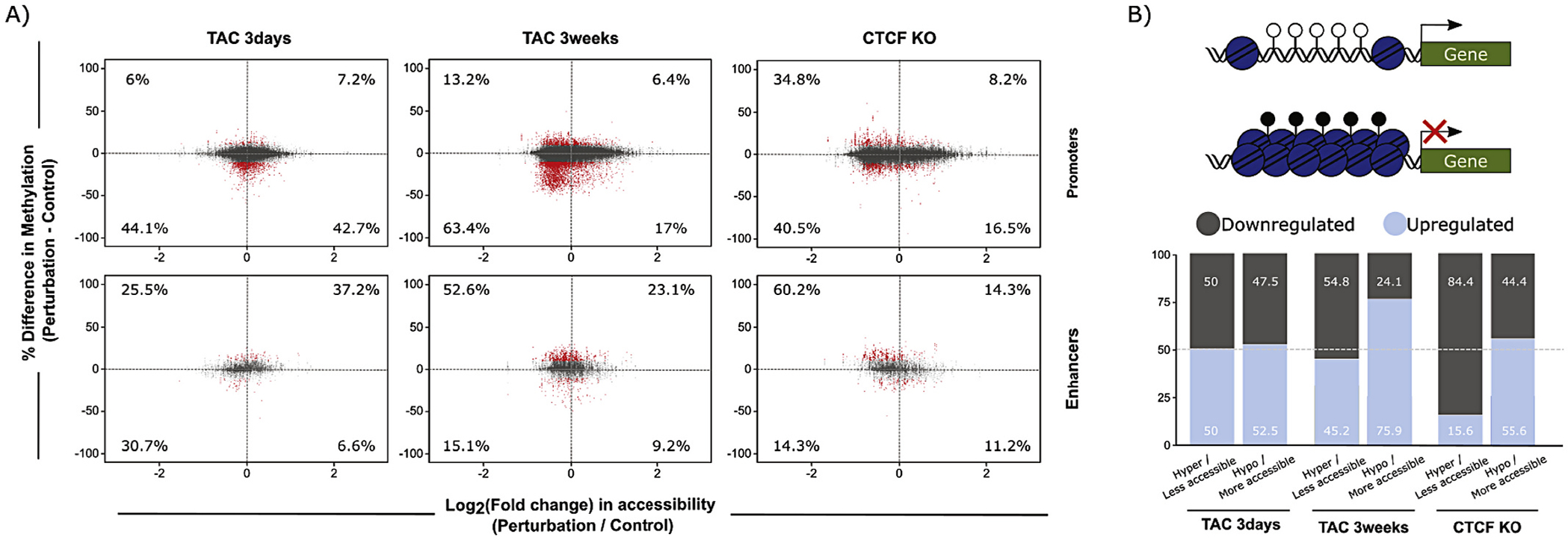
Integration of DNA methylation and chromatin accessibility data at promoters and enhancers. (A) DNA methylation and ATAC-seq integration charts, showing percent change in DNA methylation (y-axis) and log2(Fold Change) of chromatin accessibility (x-axis) of CpGs in promoters (top), and enhancers (bottom). Changes with 3 days TAC (left), 3 weeks TAC (center), and CTCFKO (right) are shown. Numbers within the charts indicate percentage of significantly differentially methylated CpGs (red points, average methylation difference > 10% and q-value <0.05) within each quadrant that overlap with regions of significant change in chromatin accessibility (FDR < 0.05). These findings demonstrate that the relationship between accessibility and DNA methylation varies between different regulatory elements (enhancers and promoters), suggesting the involvement of distinct epigenetic mechanisms at these different regulatory elements to determine their involvement in transcription. (B) Percent of up- and downregulated genes detected in the promoter analysis in (A). Top, hypothesis diagram showing a more accessible, hypomethylated promoter, and higher downstream transcription, or less accessible, hypermethylated promoter with no transcription. Bottom, summary charts indicating percent up- (lighter) and downregulated (darker shading) genes whose promoters underwent accessibility and methylation changes shown in top diagram. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Lastly, we sought to further explore the role of DNA methylation and accessibility changes in specifying, or responding to, chromatin compartmentalization changes. Chromatin compartmentalization is determined from the eigenvector of the contact matrix and is a method to segment the genome into active (A) and inactive (B) regions [46]. We classified dynamic regions of chromatin compartmentalization (which were ~4–7% of the total genome [3]) according to their direction of change (A to B or B to A), examining regions that did not change compartment as a control, and plotted the change in DNA methylation (Fig. 7A). Fig. 7B demonstrates that regions moving to, or remaining in, a given compartment showed varying accessibility changes that do not follow a specific logic. Fig. 7C shows a global analysis integrating all three datasets (ATAC-seq, RRBS, and Hi-C) for accessibility, methylation, and structure/compartmentalization, similar to Fig. 6A. Taken together, these integrative analyses of DNA methylation and/or chromatin accessibility changes with compartment dynamics reveal that while accessibility and DNA methylation play a powerful role in gene expression, they are poor predictors of changes in chromatin compartmentalization, consistent with previous observations [2]. These data suggest that while A/B compartmentalization correlates with gene expression at the genome-wide level, local DNA methylation and chromatin accessibility mediate exceptions to this relationship.
Fig. 7.
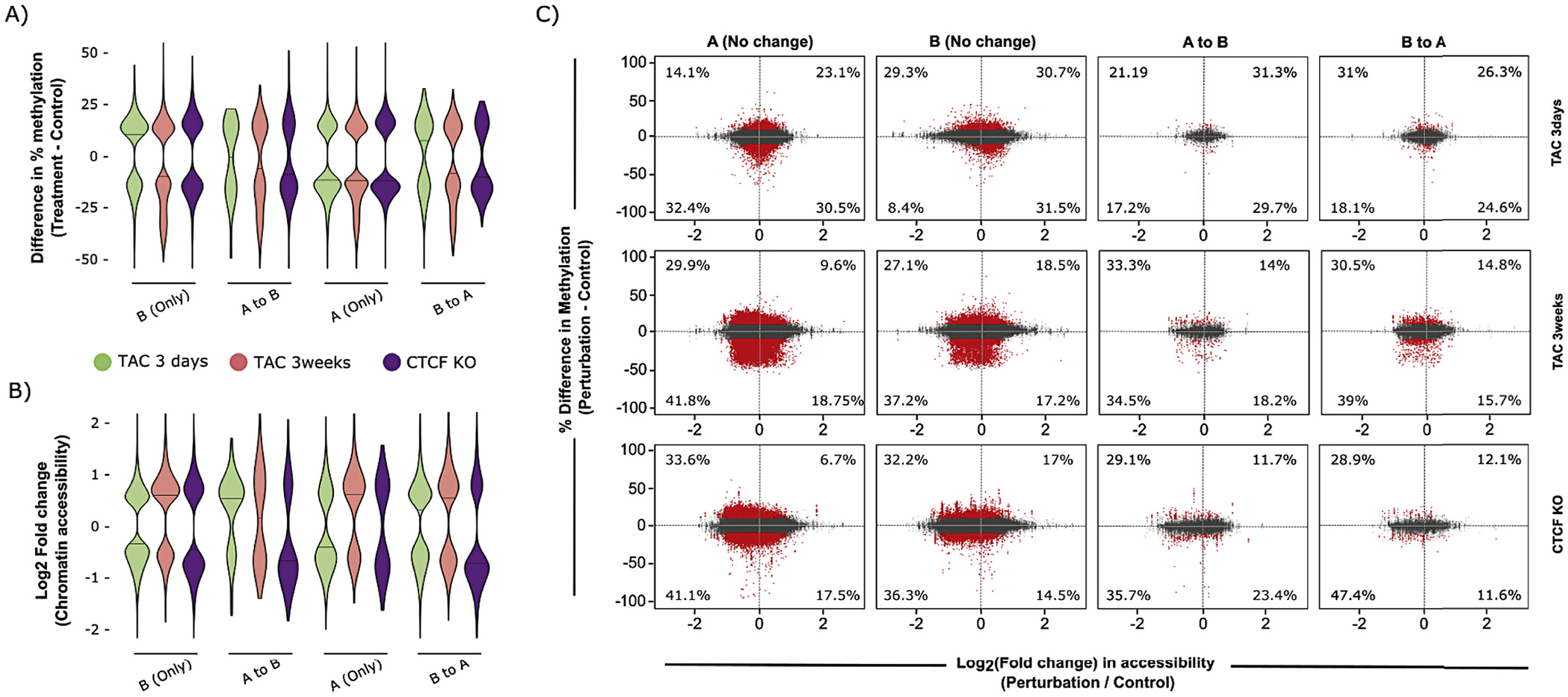
Integration of DNA methylation and chromatin accessibility data in regions that change compartmentalization with disease. (A) Percent methylation differences of differentially methylated CpGs after 3 days TAC (green), 3 weeks TAC (red) or CTCFKO (purple), when compared to control, that remain in the same compartment (A only, B only) and that change compartments (A to B, B to A) reveal no relationship between DNA methylation changes and compartment dynamics after cardiac stimulus. (B) Log2(fold change) in chromatin accessibility for differentially accessible (FDR < 0.05) ATAC-seq peaks after 3 days TAC, 3 weeks TAC or CTCFKO (when compared to control), for peaks changing or remaining in the same compartment, reveals no relationship between chromatin accessibility changes and compartment dynamics after cardiac stimulus. (C) Data integration chart with each point representing percent methylation change of CpGs that lie within 5 kb bins remaining in the same compartment (A or B), or changing compartmentalization (A to B or B to A) with disease along the y-axis (significant changes indicating average methylation difference of >10% and q < 0.05 shown in red). The x-axis indicates log2(Fold Change) of chromatin accessibility of the 5 kb bins corresponding to these CpGs. Percentages within the charts indicate proportion of significantly differentially methylated CpGs (red points, average methylation difference > 10% and q-value <0.05) within each quadrant that overlap with regions undergoing significant changes in chromatin accessibility (FDR < 0.05). These data demonstrate that local accessibility and DNA methylation cues are not deterministic of global chromatin structure (and vice versa) and that these features do not predict changes in compartmentalization with heart failure. As an example, the expected decrease in chromatin accessibility and DNA methylation was not prominent in regions that changed from active (A) to inactive (B) compartmentalization with cardiac stress (instead, all possible behaviors were detected without any leading trend). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
The role of chromatin modifying enzymes in controlling cardiac phenotype has received substantial attention in the last few years [47]. At the same time, the long-standing recognition that transcription factors can exert powerful effects on cell fate and disease pathogenesis has gained precise genomic context with the deployment of sequencing technologies to map transcription factor binding and histone occupancy [40], as well as chromatin accessibility [48]. We undertook the present study to address several issues that remained unknown: How are the actions of histone modifiers and transcription factors integrated at distinct loci, and what are the effects on chromatin accessibility? And, how do epigenomic signals change over time in vivo during the progression of disease? These questions must be answered if epigenomic medicine is to be operationalized: we need to know which proteins to target, which loci they modulate (and how to select for specific loci in the targeting regime) and at what point in disease. Heart failure, as with most chronic diseases, has distinct phases of compensation and decompensation and the involvement of epigenomic reprogramming at different phases is presumably distinct. The pressure overload mouse model of cardiac hypertrophy and failure recapitulates many features of heart failure in humans. The CTCF knockout mouse enables direct interrogation of the role chromatin structure plays in maintaining gene expression and cardiac phenotype. The tools of bisulfite sequencing and ATAC-seq allow us to examine how the totality of modifications confers alterations in chromatin accessibility—a cumulative readout of the actions of all epigenomic cues leveled by the cell at a given locus.
We find that while accessibility (defined by increased ATAC-seq signal) generally correlates positively with transcription, there are many exceptions to this rule. Likewise with DNA methylation: the overall trend is that increased methylation at regulatory regions corresponds to less gene expression, but when enhancers or promoters are examined, all possible distinct behaviors are observed (increase/decreased methylation and increased/decreased expression). Some loci have altered accessibility or methylation and no changes in expression. Graded DNA methylation change was observed in differentially methylated enhancers that physically interact with differentially expressed genes after 3 weeks TAC, but not after CTCFKO (Fig. 2A), suggesting that CTCF tightly regulates methylation at regulatory elements. A second key observation in this study is that accessibility changes—whether observed across the genome (Fig. 3), or focused at promoters (Fig. 4) or enhancers (Fig. 5)—are established early during the heart’s compensatory response to pressure overload: the trend at 3 days is, for nearly all loci, the same as that seen at 3 weeks. This observation also suggests that much of the epigenomic reorganization associated with fulminant disease occurs before the heart is in failure, with implications for biomarker development or epigenomic therapy.
Previous examination of H3K27ac by ChIP-seq revealed the landscape of putative enhancers in the cardiac myocyte [37]. We interrogated these reference enhancers in the present study, bringing to bear Hi-C data (to link them with genes), accessibility data and DNA methylation to examine their regulation. It is important to note that other histone marks (including H3K4me1) are known to contribute to enhancer function and may regulate gene expression in cardiac myocytes through mechanisms not measured in our study. The availability of high-resolution chromatin structure data from myocytes allows these regulatory elements to examined in the context of their structural orientation in the nucleus—rather than just examining them in the context of the nearest gene on a chromosome. Such analyses reveal precise regions where distal enhancers are brought into close proximity of gene promoters, and altered methylation, accessibility, and binding of transcription factors and histone modifications facilitate silencing or activation. Importantly, the combinatorial logic for how binding of cardiac transcription factors like GATA4, TEAD1, TBX5, SRF, NKX2.5, and MEF2A, along with histone marks including H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3, H3K79me2, and H3K9ac is distinct for enhancers, genes and promoter regions, implying that evolution has used the same raw materials to fashion different regulatory mechanisms for different coding, noncoding and regulatory regions of the genome (we identify and validate by ChIP-qPCR three example enhancer and promoter loci showing the regulatory interplay between DNA methylation, transcription factor binding and histone mark deposition in these regions; Fig. 2C). We interrogated Cebpd, an upregulated gene after 3 weeks TAC or CTCFKO, and found that this gene’s transcription is necessary for the phenylephrine-induced hypertrophic phenotype in isolated myocytes (Supplemental Fig. 8). This observation is consistent with a previous study indicating that inhibition of C/EBP proteins provides cardioprotection against ischemia-reperfusion injury in mice [49].
Our integration of DNA methylation and accessibility with chromatin compartmentalization data advances previous work in this area [2], and indicates that chromatin structure at the level of compartmentalization (i.e. A versus B compartmentalization, a metric driven primarily by gene expression and DNA sequence which is not itself a structural feature [actual structural features are influenced by other DNA binding proteins and can be studied with greater 3D precision using modeling techniques that incorporate Hi-C with orthogonal epigenomic and non-epigenomic methods such as microscopy [50,51]]) is exclusively determined by neither DNA methylation nor accessibility, but is rather an emergent property of both these features along with histone marks and other chromatin binding proteins. Because of technical limitations with the procedure in isolated myocytes, ATAC-seq was performed on ventricular tissue. Future studies with single cell approaches will be required to deconvolve cell type-specific changes in accessibility across the heterogeneous populations of cells in the mammalian heart following pressure overload. Future experiments will also need to examine the extent to which local and long range chromatin structural features participate in gene regulation of other cell types such as fibroblasts, which are known to deposit scar tissue after injury [52].
In the present study, we used a combination of male and female mice but did not design the study to examine sex as a biological variable. Biological sex is known to contribute to heart disease susceptibility [53] and chromatin features may be differentially regulated in males versus females, contributing to altered gene expression in disease [54]. Thus, future studies will be needed to determine whether the differences in chromatin accessibility and DNA methylation observed in this study are sex-dependent and if so, how these differences contribute to heart failure progression.
Heart failure is a progressive syndrome, affecting millions of people worldwide and in need of novel therapies, especially given the spectrum of phenotypes exhibited clinically across diverse patient populations [55,56]. For example, many patients have high blood pressure, are obese/overweight, and experience ischemic episodes and arrhythmias (heart failure patients present with a spectrum of systolic and diastolic dysfunction) yet no single combination of these factors defines the syndrome. While animal models are by definition imperfect, the insights from the pressure overload model in this study have important implications: changes in chromatin accessibility and DNA methylation mobilized early in the compensatory phase after TAC usually persist and often amplify. These findings support a model in which epigenetic mechanisms that promote and sustain cardiac pathology are established at an early stage of the disease. This observation has two implications: 1) Detection of specific chromatin changes in early disease may guide discovery of potential markers for prediction of cardiac pathology, and 2) Early epigenetic reversion of chromatin remodeling may constitute a valid strategy to prevent subsequent structural pathology and abrogate disease progression. The extent of regions with alteration in global accessibility (Fig. 3), promoter accessibility (Fig. 4) or enhancer accessibility (Fig. 5), observed in both CTCFKO and 3 week TAC model suggest a common landscape of disease-associated chromatin remodeling arising from distinct perturbations. In other words, CTCFKO and pressure overload cause heart failure by different mechanisms, but those mechanisms converge on many shared chromatin remodeling events. Aggressive modification of risk factors for heart failure, which can improve the symptoms of the disease in many patients, may reduce the parameter space of epigenomic targets that must be modified by new drugs to treat pathology. In other words, rigorous control of blood pressure may pave the way for adjuvant therapies targeting those chromatin regions identified in this study as newly altered (as small subset of the total altered loci) as the heart progresses from compensation (3 days in this study) to decompensation (3 weeks). An open question is the extent to which the early compensatory changes in accessibility and DNA methylation are reversed if the pressure overload is released and how the systemic effects of high blood pressure act in a non-cell autonomous manner to regulate the cardiac epigenome.
Supplementary Material
Acknowledgements
The authors would like to thank the past and current members of the Vondriska laboratory for helpful discussions.
Funding
This work was supported by National Institutes of Health (HL105699, HL143058 and HL150225 to TMV), an American Heart Association Career Development Award (19CDA34660084 to MRG), the Department of Anesthesiology & Perioperative Medicine and the David Geffen School of Medicine at UCLA.
Footnotes
Disclosures
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2021.07.002.
Data availability
Raw and processed ATAC-seq, RNA-seq, and RRBS data have been uploaded to the National Center for Biotechnology Information’s Gene Expression Omnibus under accession number GSE154521.
References
- [1].Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, Wurch A, Bonisch U, Gunther S, Backofen R, Fleischmann BK, Schubeler D, Hein L, Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease, Nat. Commun 5 (2014) 5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nothjunge S, Nuhrenberg TG, Gruning BA, Doppler SA, Preissl S, Schwaderer M, Rommel C, Krane M, Hein L, Gilsbach R, DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes, Nat. Commun 8 (2017) 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rosa-Garrido M, Chapski DJ, Schmitt AD, Kimball TH, Karbassi E, Monte E, Balderas E, Pellegrini M, Shih TT, Soehalim E, Liem D, Ping P, Galjart NJ, Ren S, Wang Y, Ren B, Vondriska TM, High-resolution mapping of chromatin conformation in cardiac myocytes reveals structural remodeling of the epigenome in heart failure, Circulation 136 (2017) 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee DP, Tan WLW, Anene-Nzelu CG, Lee CJM, Li PY, Luu TDA, Chan CX, Tiang Z, Ng SL, Huang X, Efthymios M, Autio MI, Jiang J, Fullwood MJ, Prabhakar S, Lieberman Aiden E, Foo RS, Robust CTCF-based chromatin architecture underpins epigenetic changes in the heart failure stress-gene response, Circulation 139 (2019) 1937–1956. [DOI] [PubMed] [Google Scholar]
- [5].Chang CP, Bruneau BG, Epigenetics and cardiovascular development, Annu. Rev. Physiol 74 (2012) 41–68. [DOI] [PubMed] [Google Scholar]
- [6].Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, Moon RT, Stamatoyannopoulos J, Murry CE, A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development, Cell 151 (2012) 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG, Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage, Cell 151 (2012) 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Haldar SM, McKinsey TA, BET-ting on chromatin-based therapeutics for heart failure, J. Mol. Cell. Cardiol 74 (2014) 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McKinsey TA, Olson EN, Dual roles of histone deacetylases in the control of cardiac growth, Novartis Found. Symp 259 (2004) 132–141, discussion 141–5, 163–9. [PubMed] [Google Scholar]
- [10].Lehmann LH, Worst BC, Stanmore DA, Backs J, Histone deacetylase signaling in cardioprotection, Cell. Mol. Life Sci 71 (2014) 1673–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Keating ST, El-Osta A, Epigenetics and metabolism, Circ. Res 116 (2015) 715–736. [DOI] [PubMed] [Google Scholar]
- [12].Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H, Return to the fetal gene program protects the stressed heart: a strong hypothesis, Heart Fail. Rev 12 (2007) 331–343. [DOI] [PubMed] [Google Scholar]
- [13].Matkovich SJ, Dorn GW 2nd., Deep sequencing of cardiac microRNA-mRNA interactomes in clinical and experimental cardiomyopathy, Methods Mol. Biol 1299 (2015) 27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gilsbach R, Schwaderer M, Preissl S, Gruning BA, Kranzhofer D, Schneider P, Nuhrenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dressen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L, Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo, Nat. Commun 9 (2018) 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen H, Orozco L, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM, DNA methylation indicates susceptibility to isoproterenol-inducd cardiac pathology and is associated with chromatin states, Circ. Res 118 (2016) 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position, Nat. Methods 10 (12) (2013) 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Franklin S, Chen H, Mitchell-Jordan S, Ren S, Wang Y, Vondriska TM, Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth, Mol. Cell. Proteomics 11 (2012). M111 014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Karbassi E, Rosa-Garrido M, Chapski DJ, Wu Y, Ren S, Wang Y, Stefani E, Vondriska TM, Direct visualization of cardiac transcription factories reveals regulatory principles of nuclear architecture during pathological remodeling, J. Mol. Cell. Cardiol 128 (2019) 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW, ImageJ2: ImageJ for the next generation of scientific image data, BMC Bioinforma. 18 (2017) 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen PY, Pellegrini M, BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data, BMC Genomics 14 (2013) 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2, Nat. Methods 9 (2012) 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S, The sequence alignment/map format and SAMtools, Bioinformatics 25 (2009) 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE, methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles, Genome Biol. 13 (2012) R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression, Nat. Methods 14 (2017) 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, Billis K, Boddu S, Marugan JC, Cummins C, Davidson C, Dodiya K, Fatima R, Gall A, Giron CG, Gil L, Grego T, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, Kay M, Lavidas I, Le T, Lemos D, Martinez JG, Maurel T, McDowall M, McMahon A, Mohanan S, Moore B, Nuhn M, Oheh DN, Parker A, Parton A, Patricio M, Sakthivel MP, Abdul Salam AI, Schmitt BM, Schuilenburg H, Sheppard D, Sycheva M, Szuba M, Taylor K, Thormann A, Threadgold G, Vullo A, Walts B, Winterbottom A, Zadissa A, Chakiachvili M, Flint B, Frankish A, Hunt SE, IIsley G, Kostadima M, Langridge N, Loveland JE, Martin FJ, Morales J, Mudge JM, Muffato M, Perry E, Ruffier M, Trevanion SJ, Cunningham F, Howe KL, Zerbino DR, Flicek P, Ensembl 2020, Nucleic Acids Res. 48 (2020) D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Soneson C, Love MI, Robinson MD, Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences, F1000Res 4 (2015) 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15 (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benjamini Y, Hochberg Y, Controlling the false discovery rate: a practical and powerful approach to multiple testing, J. R. Stat. Soc. B 57 (1995) 289–300. [Google Scholar]
- [29].Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J, g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update), Nucleic Acids Res. 47 (2019) W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics 29 (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T, deepTools2: a next generation web server for deepsequencing data analysis, Nucleic Acids Res. 44 (2016) W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, Kathiria A, Cho SW, Mumbach MR, Carter AC, Kasowski M, Orloff LA, Risca VI, Kundaje A, Khavari PA, Montine TJ, Greenleaf WJ, Chang HY, An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues, Nat. Methods 14 (2017) 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform, Bioinformatics 25 (2009) 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS), Genome Biol. 9 (2008) R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS, Differential oestrogen receptor binding is associated with clinical outcome in breast cancer, Nature 481 (2012) 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J, Bioconductor: open software development for computational biology and bioinformatics, Genome Biol. 5 (2004) R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, Vigano V, Stirparo GG, Latronico MV, Hasenfuss G, Chen J, Condorelli G, Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 20164–20169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Freese NH, Norris DC, Loraine AE, Integrated genome browser: visual analytics platform for genomics, Bioinformatics 32 (2016) 2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, Onate KC, Graham K, Miyasato SR, Dreszer TR, Strattan JS, Jolanki O, Tanaka FY, Cherry JM, The Encyclopedia of DNA elements (ENCODE): data portal update, Nucleic Acids Res. 46 (2018) D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, Wasson L, Wen T, Liu J, Dong K, Conlon FL, Zhou J, Yuan GC, Zhou P, Pu WT, A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers, Nat. Commun 10 (2019) 4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E, HiC-Pro: an optimized and flexible pipeline for Hi-C data processing, Genome Biol. 16 (2015) 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ay F, Bailey TL, Noble WS, Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts, Genome Res. 24 (2014) 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM, CCCTC-binding factor acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection, mBio 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ohlsson R, Bartkuhn M, Renkawitz R, CTCF shapes chromatin by multiple mechanisms: the impact of 20 years of CTCF research on understanding the workings of chromatin, Chromosoma 119 (2010) 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities, Mol. Cell 38 (2010) 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science 326 (2009) 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gillette TG, Hill JA, Readers, writers, and erasers chromatin as the whiteboard of heart disease, Circ. Res 116 (2015) 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER, Multicellular transcriptional analysis of mammalian heart regeneration, Circulation 136 (2017) 1123–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huang GN, Thatcher JE, McAnally J, Kong Y, Qi X, Tan W, DiMaio JM, Amatruda JF, Gerard RD, Hill JA, Bassel-Duby R, Olson EN, C/EBP transcription factors mediate epicardial activation during heart development and injury, Science 338 (2012) 1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hua N, Tjong H, Shin H, Gong K, Zhou XJ, Alber F, Producing genome structure populations with the dynamic and automated PGS software, Nat. Protoc 13 (2018) 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chapski DJ, Rosa-Garrido M, Hua N, Alber F, Vondriska TM, Spatial principles of chromatin architecture associated with organ-specific gene regulation, Front. Cardiovasc. Med 5 (2018) 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tallquist MD, Molkentin JD, Redefining the identity of cardiac fibroblasts, Nat. Rev. Cardiol 14 (2017) 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, VanWagner LB, Wang NY, Tsao CW, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S, Heart disease and stroke statistics-2021 update: a report from the American Heart Association, Circulation 143 (2021) e254–e743. [DOI] [PubMed] [Google Scholar]
- [54].Khramtsova EA, Davis LK, Stranger BE, The role of sex in the genomics of human complex traits, Nat. Rev. Genet 20 (2019) 173–190. [DOI] [PubMed] [Google Scholar]
- [55].Katz AM, Rolett EL, Heart failure: when form fails to follow function, Eur. Heart J 37 (2016) 449–U38. [DOI] [PubMed] [Google Scholar]
- [56].Mentz RJ, Kelly JP, von Lueder TG, Voors AA, Lam CS, Cowie MR, Kjeldsen K, Jankowska EA, Atar D, Butler J, Fiuzat M, Zannad F, Pitt B, O’Connor CM, Noncardiac comorbidities in heart failure with reduced versus preserved ejection fraction, J. Am. Coll. Cardiol 64 (2014) 2281–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Agapitov AV, Haynes WG, Role of endothelin in cardiovascular disease, J. Renin-Angiotensin-Aldosterone Syst. 3 (2002) 1–15. [DOI] [PubMed] [Google Scholar]
- [58].Perrin MJ, Adler A, Green S, Al-Zoughool F, Doroshenko P, Orr N, Uppal S, Healey JS, Birnie D, Sanatani S, Gardner M, Champagne J, Simpson C, Ahmad K, van den Berg MP, Chauhan V, Backx PH, van Tintelen JP, Krahn AD, Gollob MH, Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death, Circ. Cardiovasc. Genet 7 (2014) 782–789. [DOI] [PubMed] [Google Scholar]
- [59].Jalil A, Horiuchi M, Nomoto M, Kobayashi K, Saheki T, Catecholamine metabolism inhibitors and receptor blockades only partially suppress cardiac hypertrophy of juvenile visceral steatosis mice with systemic carnitine deficiency, Life Sci. 64 (1999) 1137–1144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed ATAC-seq, RNA-seq, and RRBS data have been uploaded to the National Center for Biotechnology Information’s Gene Expression Omnibus under accession number GSE154521.