

Abstract
IL-38 is a recently discovered cytokine and member of the IL-1 Family. In the IL-1 Family, IL-38 is unique because the cytokine is primarily a B lymphocyte product and functions to suppress inflammation. Studies in humans with inflammatory bowel disease (IBD) suggest that IL-38 may be protective for ulcerative colitis or Crohn’s disease, and that IL-38 acts to maintain homeostasis in the intestinal tract. Here we investigated the role of endogenous IL-38 in experimental colitis in mice deficient in IL-38 by deletion of exons 1-4 in C57 BL/6 mice. Compared to WT mice, IL-38 deficient mice subjected to dextran sulfate sodium (DSS) showed greater severity of disease, more weight loss, increased intestinal permeability, and a worse histological phenotype including increased neutrophil influx in the colon. Mice lacking IL-38 exhibited elevated colonic Nlrp3 mRNA and protein levels, increased caspase-1 activation, and the concomitant increased processing of IL-1β precursor into active IL-1β. Expression of IL-1α, an exacerbator of IBD, was also upregulated. Colonic myleloperoxidase protein and Il17a, and Il17f mRNA levels were higher in the IL-38 deficient mice. Daily treatment of IL-38 deficient mice with an NLRP3 inhibitor attenuated diarrhea and weight loss during the recovery phase. These data implicate endogenous IL-38 as an anti-inflammatory cytokine that reduces DSS colitis severity. We propose that a relative deficiency of IL-38 contributes to IBD by disinhibition of the NLRP3 inflammasome.
Keywords: IL-38, NLRP3, IL-1α, IL-1β, colitis, IBD – inflammatory bowel diseases
Introduction
Ulcerative colitis (UC) and Crohn’s disease (CD) are characterized by chronic hyperinflammation of the gastrointestinal tract (1). An innate inflammatory response is elicited in healthy subjects upon recognition of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) via pattern recognition receptors (2). In inflammatory bowel disease (IBD) patients with susceptible genetics a disproportional, auto-inflammatory response is triggered by environmental factors. As a result, the mucosal barrier is impaired and leads to symptoms of abdominal pain, fatigue, weight loss, diarrhea, and intestinal bleeding (3, 4). Currently, IBD treatments range from glucocorticoids to thiopurines, antibodies targeting cytokines and integrins, small-molecule drugs, and fecal- and stem-cell transplants (5). However, these therapies fail to induce a response in a portion of patients and lose efficacy in those who initially benefit.
The nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3) forms an inflammasome with the adaptor molecule apoptosis-associated speck-like (ASC) protein containing a caspase recruitment domain and pro-caspase-1 (6). The NLRP3 inflammasome drives the innate immune response against pathogenic infections, by cleaving precursor IL-1β and IL-18 precursors into active cytokines (7). NLRP3 in the gut promotes intestinal health by contributing to barrier integrity and pathogen clearance (8). However, in several inflammatory diseases, NLRP3 activity is dysregulated and contributes to disease severity (9). In IBD patients, for example, disease severity is associated with upregulation of IL-1β, NLRP3, and caspase-1 (10). Indeed, murine colitis induced by dextran sodium sulfate (DSS) is exacerbated by Nrlp3 deficiency when compared to WT mice (11, 12).
IL-38 is a recently discovered anti-inflammatory IL-1 Family member that is abundantly expressed in keratinocytes of the skin and (circulating) B lymphocytes (13–15). In humans, the gene encoding IL-38 (IL1F10) is located within the IL-1 gene cluster on chromosome 2p13, adjacent to the genes encoding receptor antagonists IL-1Ra and IL-36Ra with which IL-38 shares 41% and 43% homology, respectively. Similar to IL-36Ra, IL-38 inhibits IL-36 signaling by inhibiting the IL-1R6, which reduces the Th17 response (16). Notably, IL-17 expression in the mucosa and serum of IBD patients is upregulated (17).
IL-38 expression in colon samples from patients with active UC and CD is increased compared to age-matched healthy controls (18). The IL-38 expression is confined to infiltrating immune cells in the lamina propria, mucosa, submucosa, muscular and serosa layers, epithelial and parenchymal cells (18), and CD123+ cells (19). Furthermore, IL-38 staining in the lamina propria was detected CD19+ B cells but absent in CD3+ T cells or CD68+ monocytes and CD14+ macrophages (18, 20). In a separate study, IL-38 expression is highest in colonic tissue from non-inflamed patients with UC, i.e., those in remission, compared to patients with active UC, CD and healthy subjects (19). This finding is in stark contrast to the expression of other members of the IL-36 subfamily including IL-36Ra, which was upregulated in inflamed colonic tissue from patients with active UC (19). Recently Xie et al. demonstrated that recombinant IL-38 is protective in a murine model of dextran sulfate sodium (DSS) colitis (18). Thus, endogenous IL-38 may have a role in the maintenance of gut homeostasis and tissue repair. We recently reported that recombinant IL-38 reduces Nlrp3 gene expression and promotor accessibility in mouse bone marrow (21) and can limit IL-1β production in the synovium of mice subjected to gouty arthritis (22). As of this writing, no relationship between IL-38 signaling and the NLRP3 inflammasome activity has been described in IBD.
Here, we studied the role of endogenous IL-38 in a murine model of DSS colitis by comparing responses in WT to those in IL-38 deficient mice, focusing particularly on the involvement of the NLRP3 signaling. We hypothesize that endogenous IL-38 contributes to IBD resolution and intestinal homeostasis by limiting NLRP3 expression and activity.
Materials and Methods
Ethics Statement
Animal protocols were reviewed and approved by the University of Colorado Animal Care and Use Committee.
Mice
Il1f10 (IL-38) deficient mice (GenBank accession number: NM_153077.2; Ensembl: ENSMUSG00000046845) were generated using CRISPR/Cas9 technology (Cyagen Biosciences, CA, USA) on a C57BL/6 background. Table 1 contains gRNA sequences used for the removal of Il1f10 exons 1-4 and nucleotides to confirm the deletion. Cas9 mRNA and gRNA were generated by in vitro transcription and injected into fertilized C57BL/6 eggs. Founders were genotyped by PCR using TaKaRa TaqTM Hot Start Version (Takara) and the PCR product was purified using the MiniBEST Universal Genomic DNA Extraction Kit Ver.5.0, 9765 (Takara). The IL-38 deficiency was confirmed by PCR and gel electrophoresis, and DNA sequencing analysis (Figure 1). DNA sequencing revealed that F0 Mouse-ID #19, and F1 Mouse-ID #2 and #8 were missing 4725 bases in one Il1f10 allele, indicating loss of all Il1f10 exons. After transportation to our animal facility and further breeding, presence of homozygote IL-38 deficient knockout or wild type (WT) alleles were confirmed by ear clippings through Transnetyx prior to 3 weeks of age. All mice used in this study were between 8 and 10 weeks of age. All national and institutional guidelines for the care and use of laboratory animals were adhered to.
Table 1.
gRNA and PCR primer sequences.
| Purpose | Sequence | Vector ID |
|---|---|---|
| gRNA before Il1f10 exon 1 (matches fwd. strand) | GCGAGAGAACAGTTACCGAATGG | VB180116-1110yca |
| gRNA after Il1f10 exon 4 (matches fwd. strand) | TTCAATATTGGTAGGCACCCCGG | VB180116-1112eat |
| Identification of IL-38 deficiency (Short product indicates deletion of exons 1-4) | Fwd: CCCATGCCGTAGAGCACATCTGT Rev: GGCTCATCTTGTGCTGTAGCTCTGC |
|
| PCR Identification of WT (Transnetyx) | Fwd: TCCAGGGTACCTGAGCTTCA Rev: GTCTTCTCATGAGGTAATTGAGGATGT |
|
| PCR Identification of knockout (Transnetyx) (presence indicates Il1f10 deficiency) | Fwd: CTGGCTCATTGCTTGTAACACTAC Rev: AGGAGAATCCCTAGATGTCTTTCCA |
Figure 1.

Generation of IL-38/Il1f10 deficient mice. (A) Representation of the IL-38 gene, exons are in black, gRNA and PCR primer locations are indicated with arrows. F0 (B) and F1 (C) Heterozygote Il1f10 deficient founder screening. PCR products were generated using primers in Table 1 and conditions listed in the Supplemental Material.
Dextran Sodium Sulfate Colitis
Male and female mice were subjected to DSS at 8-10 weeks of age. From day 0, mice received drinking water with 3% DSS (molecular weight 36,000–50,000; MP Biomedicals) to induce acute colitis, or H2O as a vehicle control. After 5 days of treatment, mice were allowed to recover for 4 days on regular drinking water before sacrifice. A disease activity index (DAI) score was assessed daily to evaluate the development of colitis based on the parameters of weight loss compared to initial weight, stool consistency, and rectal bleeding (23). Evaluation was performed by two researchers who were blinded to the experimental groups. Scores were defined as weight loss: 0 (0%), 1 (1-5%), 2 (5-10%), 3 (11-20%), and 4 (>20%); stool consistency: 0 (well-formed pellets), 2 (pasty, semi-formed pellets), and 4 (liquid stools); and rectal bleeding: 0 (no blood), 2 (hemoccult positive), and 4 (gross bleeding). The highest DAI score possible was 12 (24). Colon lengths were measured at time of sacrifice, and tissue collected for histology, immunofluorescence, RNA, and protein analyses. For select experiments, mice lacking IL-38 and exposed to DSS for 5 days were treated daily i.p. with 200 mg/kg OLT1177, a specific NLRP3 inhibitor, or saline as a control, for the duration of the experiment, based on a previous study (25).
Histological Scoring
Distal colon tissue was fixed in methacarn (methanol:chloroform:acetic acid, 60:30:10) and stained with hematoxylin and eosin. All histological scoring was performed blinded. Three independent parameters were assessed: severity of inflammation (0-3: none, slight, moderate, severe), depth of injury (0-3: none, mucosal, mucosal, and submucosal, transmural), and amount of crypt damage (0-4: none, basal 1/3 damaged, basal 2/3 damaged, only surface epithelium intact, entire crypt and epithelium lost). The independent parameter scores were multiplied by a factor reflecting the percentage of tissue involved (x1: 0-25%, x2: 26-50%, x3: 51-75%, x4: 76- 100%) and then added up. Maximum histological score possible was 40 (26).
Colon Permeability
Six hours before the sacrifice, mice received 100 μL of 100 mg/mL 70-kDa FITC-dextran (Sigma-Aldrich) by oral gavage, which only transits a permeable gut barrier. Blood was collected 4h later, centrifuged at 1000 x g for 10 min, and serum was analyzed for FITC fluorescence and compared to a FITC-dextran standard.
RT-qPCR
TRIzol reagent (ThermoFisher Scientific) was used to isolate total RNA from colon tissue. cDNA was prepared using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR analysis was performed using the Power SYBR Green master mix (Applied Biosystems) in a thermocycler. Fold change in expression of target mRNA relative to Gapdh mRNA was calculated using the delta-delta Ct method. Gapdh F: 5′-TTCAACAGCAACTCCCACTCTTCCA-3′, Gapdh R: 5′- ACCCTGTTGCTGTAGCCGTATTCA-3′ Il17a F: 5′- TTTAACTCCCTTGGCGCAAAA-3′, Il17a R, 5′-CTTTCCCTCCGCATTGACAC-3′ Il17f F: 5′- TGCTACTGTTGATGTTGGGAC-3′, Il17f R, 5′- AATGCCCTGGTTTTGGTTGAA-3′, Nlrp3 F: 5′- TGGTATGCCAGGAGGACAGCCT 3′, Nlrp3 R: 5′- AGACGCGCGTTCCTGTCCTT -3′.
ELISA
Colon samples were rinsed with PBS and lysed in 200 μL of radioimmunoprecipitation assay (RIPA, 50 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% Triton X-100, 10% SDS, 0.5% sodium deoxycholate, 150 mM NaCl, Sigma-Aldrich) buffer with protease inhibitors on ice. Samples were homogenized by sonication, and insoluble materials removed by centrifugation at 10,000 x g for 5 min at 4°C. Total protein was quantified using the Bradford Assay (Bio-Rad). ELISA on colon protein extracts was performed for IL-1α, IL-1β and myeloperoxidase (MPO), and on plasma for KC/CXCL1 according to the manufacturer’s instructions (Biotechne).
Western Blot
Colon protein extractss were electrophoresed on Mini-PROTEAN TGX 4−20% gels (Bio-Rad) and transferred to nitrocellulose 0.2 μM (GE Water & Process Technologies). Membranes were blocked in 5% dried milk in 0.5% PBS-T for 1 hour at room temperature. Primary antibodies for caspase-1 1:500 (sc-514 Santa Cruz Biotechnology, Dallas, TX, USA), and Nlrp3 1:1000 (Adipogen, San Diego CA) were used in combination with peroxidase-conjugated secondary antibodies and chemiluminescence to detect the protein. A primary antibody against β-actin (Santa Cruz Biotechnology) was used to assess protein loading. Bands were quantified using ImageJ (Maryland, USA).
Statistical Analysis
The data represents the mean ± SEM, #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-way ANOVA with Fisher’s multiple comparison, or Student’s T-test.
Results
Loss of IL-38 Exacerbates the Phenotype of DSS Colitis
To investigate the influence of endogenous IL-38 to outcomes of experimental colitis, WT and IL-38 deficient mice were subjected to 3% DSS or water control for 5 days, allowed to recover for 4 days and sacrificed on day 9. As shown in Figure 2A, IL-38 deficient mice displayed enhanced susceptibility to DSS colitis compared to WT mice, as demonstrated by significantly higher DAI scores at day 3 (P < 0.05) and days 5-9 (P < 0.0001). The DAI in WT and IL-38 deficient plateaued from days 6 to 8 and recovery began on day 8. Moreover, mice lacking IL-38 had a greater loss of bodyweight than WT mice from day 6 onwards (P < 0.01) (Figure 2B). Upon sacrifice, IL-38 deficient mice revealed a greater reduction in colon length/weight ratio compared to WT control (P < 0.01, Figure 2C), whereas this ratio was not dissimilar between vehicle-treated WT and IL-38 deficient mice (P = 0.27).
Figure 2.

IL-38 deficiency aggravates DSS colitis. (A) DAI scores of WT and IL-38 deficient mice [indicated as knock-out (KO) mice]. Mice were subjected to DSS or H2O only. Maximum DAI is 121. N = 11 for H2O, 14 for DSS. (B) Weight loss of WT and IL-38 KO mice subjected to DSS versus H2O only. Maximum loss is 20%. N = 11 for H2O, N = 14 for DSS. (C) Colon length to body mass ratios of WT and IL-38 KO mice subjected to DSS versus H2O only. N = 8-9. (D) Serum FITC-dextran in WT and IL-38 KO mice subjected to DSS versus H2O (n = 6-7). (E) Histological scoring of colon tissue of WT and IL-38 KO mice subjected to DSS versus H2O. Maximum histological score 40 (n = 6). (F-I) Representative H&E stained histological slide of colon tissue of WT mice subjected to H2O (F) or DSS (G), and IL-38 deficient mice subjected to H2O (H) and DSS (I). Data were pooled from 2-3 separate experiments. The data represents the mean ± SEM, #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-way ANOVA with Fisher’s multiple comparison. N.d., Not detectable; ns, not significant.
To assess intestinal barrier permeability, mice received 70 kDa FITC-dextran by oral gavage on the last day of the experiment, and FITC-dextran was measured in serum after 4h. DSS treated IL-38 deficient mice had an enhanced permeability defect compared to vehicle (H2O) treated IL-38 deficient mice (P < 0.05), and IL-38 deficient colitic mice trended towards having an enhanced barrier defect compared to WT colitic mice (P < 0.1) (Figure 2D).
Colons were assessed histologically after sacrifice. As presented in Figure 2E, colitic IL-38 deficient mice had increased histologic scores that reflected the severity of inflammation, depth of injury, and crypt damage compared to WT mice (P < 0.001). Representative images in Figures 2F–I demonstrate a complete loss of crypt morphology associated with inflammatory leukocyte infiltration and severe epithelial damage in mice lacking IL-38 mice. In contrast, DSS treated WT mice displayed a modest level of inflammation and associated epithelial and crypt damage. Overall, a deficiency in IL-38 amplified the disruption of the intestinal barrier function and intensified colitic disease. No histological differences between WT and IL-38 deficient mice were observed at baseline.
IL-38 Deficiency Enhances Inflammatory Cytokine Production in DSS Colitis
We investigated the influence of endogenous IL-38 on inflammatory cytokines during colitis. As presented in Figures 3A, B, pro-inflammatory IL-1α and IL-1β protein levels were increased in the IL-38 deficient mice compared to WT mice treated with DSS. Plasma concentrations of the neutrophil chemokine KC and colonic expression of the neutrophil activation marker MPO were upregulated in WT mice exposed to DSS in comparison to water controls, and further increased in DSS treated IL-38 deficient mice (Figures 3C, D). As shown in Figures 3E, F, gene expression of Th17 cytokines Ill17a and Il17f were upregulated only in IL-38 deficient mice.
Figure 3.
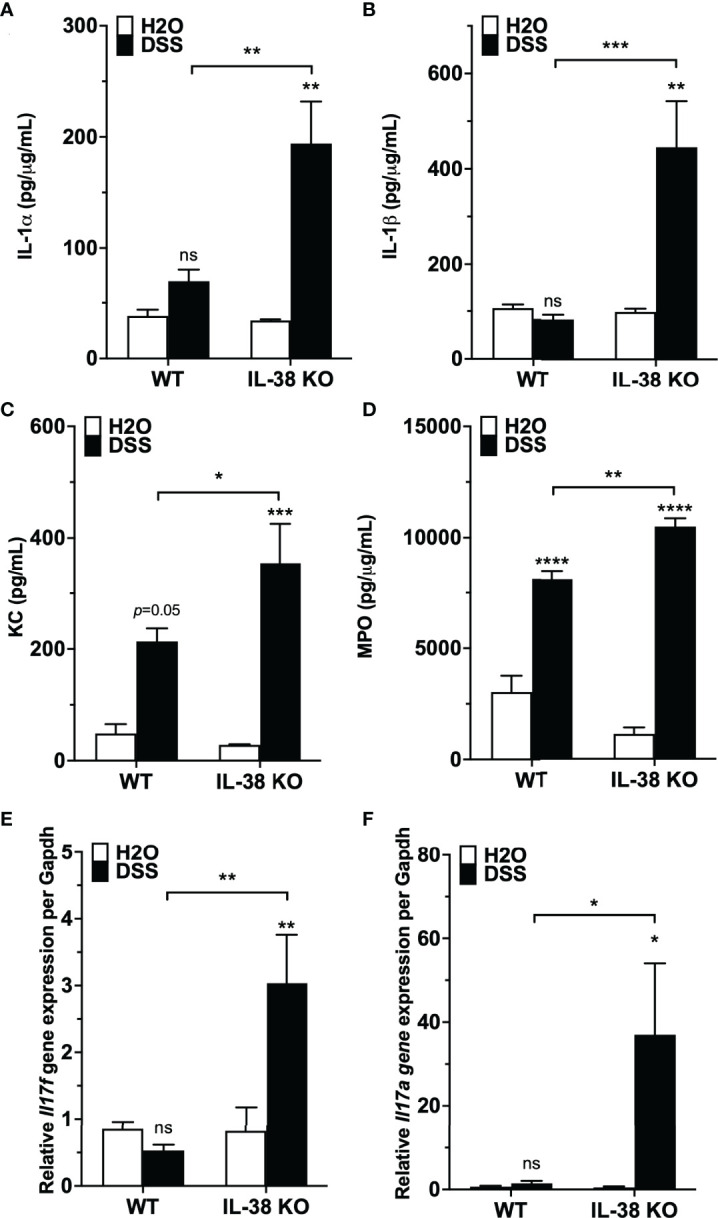
Mice lacking IL-38 have increased expression of inflammatory cytokines in colitis. WT and IL-38 KO mice were subjected to H2O or DSS. (A, B) IL-1α, IL-1β protein expression in colon tissue, (C) plasma KC concentration, (D) MPO protein expression in colon tissue, (E) mRNA levels of Il17a and (F) Il17f in colonic tissue measured by qPCR, normalized to GAPDH. Data was pooled from 2-3 separate experiments. The data represents the mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-way ANOVA with Fisher’s multiple comparison. IL-38 KO: IL-38 deficient. ns, not significant.
Endogenous IL-38 Reduces Inflammatory Signaling
Next, we investigated the effect of endogenous IL-38 on the expression of NLRP3 and caspase-1 in mice subjected to DSS. As shown in Figure 4A, Nlrp3 gene expression was significantly increased in IL-38 deficient mice. Western blot analysis revealed an increase in protein expression Nlrp3, pro-caspase 1 (p45), cleaved caspase-1 (p20) (Figures 4B, C) and the ratio of cleaved-to precursor caspase-1 (Figure 4D) in the colon of IL-38 deficient mice subjected to DSS in comparison to WT mice. These findings indicate that endogenous IL-38 is associated with inhibition of NLRP3 inflammasome. In H2O-treated WT and IL-38 deficient mice, Nlrp3 protein expression was nearly undetectable, whereas IL-38 deficient mice had increased baseline caspase-1 cleavage (Supplementary Figure 1). To investigate whether the exacerbated colitis phenotype in the IL-38 deficient mouse depended on NLRP3 activity, mice were treated with vehicle or an NLRP3 inhibitor for the duration of DSS treatment and recovery. As presented in Figures 5A–D, NLRP3 inhibition in IL-38 deficient mice leads to more DSS-induced bleeding and disease activity on day 4. However, less disease activity was observed during the last days of the recovery phase due to a reduction in diarrhea and weight loss. These data indicate that NLRP3 activation during the recovery phase delays the recovery from DSS-induced colitis in IL-38 deficient mice.
Figure 4.

Endogenous IL-38 inhibits NLRP3 activation in colitis. WT and IL-38 KO (IL-38 deficient) mice subjected to DSS. (A) mRNA expression of Nlrp3 in colonic tissue measured by qPCR, normalized to GAPDH. (B) NLRP3, Caspase-1 (p20), Caspase-1 (p45) and β-Actin protein abundance in colonic tissues quantified by Western Blot analysis. Lanes 1-3 contain colonic tissue of WT mice treated with DSS, lanes 4-7 from IL-38 deficient mice subjected to DSS. A representative blot is shown from two experiments with 3-4 mice per group. (C) Quantifications of blots presented in (B). (D) P45/p20 ratio in WT mice treated with DSS compared to IL-38 KO mice. The data represents the mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, by two-way ANOVA with Fisher’s multiple comparison in (A), and student’s T-test in (C, D).
Figure 5.

NLRP3 inhibition enhances recovery in IL-38 deficient mice. IL-38 deficient mice were subjected to DSS and treated daily i.p. with 200 mg/kg OLT1177, an NLRP3 inhibitor, or vehicle (N = 7 per group). (A) DAI scores, maximum DAI is 12. (B) Weight loss, maximum loss is 20%. (C) Bleeding score, maximum score is 4. (D) Diarrhea score, maximum score is 4. Data were pooled from 2 separate experiments with 3-4 mice each. The data represents the mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, by two-way ANOVA with Fisher’s multiple comparison. Ns: not significant.
Discussion
In the present paper, we examined whether IL-38 deficiency amplifies the colitis response using the DSS model. The data indicate that the IL-38 deficient mouse has an increased disease activity score, weight loss, histological damage, and intestinal permeability. The aggravated colitis in mice lacking IL-38 was accompanied by a greater increase in the pro-inflammatory cytokines IL-1α and IL-1β in the colon compared to WT mice. These observations are supported by a previous publication showing that treatment with recombinant IL-38 reduces DSS induced colitis (18).
We observed that the IL-38 deficient mouse exhibited consistently increased expression of the neutrophil chemokine KC in the plasma and the neutrophil activation marker MPO in the colon. We also observed an increase in colonic Il17a and Il17f mRNA expression (Figure 3). Relevant to these data, IL-1R1 deficient mice were reported to have a reduced colonic Th17 response and IL-17 secretion (10), which is in line with endogenous IL-38 reducing colonic IL-1β and IL-1α protein expression, thereby reducing IL-1R1 signaling. Neutrophil influx, here a factor contributing to the disease activity index, was also elevated in IL-38 deficient mice, as indicated by an increase in MPO expression and increased leukocyte influx as part of the histological scoring. This finding corroborates previous observations of recombinant IL-38 reducing leukocyte influx into the synovium of mice subjected to gouty arthritis (22) and neutrophils into lesional skin in a mouse model of psoriasis (14, 27).
The measurement of inflammatory cytokines was performed on samples obtained at the time of sacrifice, i.e., 4 days after the mice were withdrawn from DSS. At that time point, the WT mice subjected to DSS had an increase in KC and MPO in comparison to WT mice that received vehicle treatment with H2O, but were without an increase in IL-1α and IL-1β protein, and Il17a, and Il17b gene expression. To unequivocally determine the effect of endogenous IL-38 on either the induction or recovery phase of DSS-induced colitis, inducible knock-out/knock-in IL-38 mice may be generated in which IL-38 deficiency occurs only during each specific phase.
The IL-38 deficiency that aggravated DSS colitis was associated with elevated gene expression and protein levels of NLRP3 inflammasome and enhanced IL-1β signaling at the time of sacrifice. The IL-38 deficient mouse also had increased expression of pro- and active caspase-1, a prerequisite for IL-1β processing. At baseline, mice lacking IL-38 had increased colonic caspase-1 activation. In IL-38 deficient mice treated with DSS, inhibition of NLRP3 resulted in more bleeding on day 4, but overall less disease activity due to reduced weight loss and diarrhea in the last days of the recovery phase. Thus, the amplified disease phenotype observed during the recovery phase in the IL-38 deficient mice could be partially rescued by NLRP3 inhibition. Previous reports on blockade of NLRP3 with this specific NLRP3 inhibitor effectively limited the induction of DSS colitis in mice (28), and inhibits NF-κB, IL-1β, caspase-1 and MPO activity (25). In line with our observations, NLRP3 deficient mice treated with DSS were reported to have increased rectal bleeding on days 3 and 4 (29). Notably, a detrimental role of NLRP3 in DSS colitis has also been reported (11), indicating that the contribution of NLRP3 to DSS-induced colitis may be different between the induction- and recovery phase of this model.
Under homeostatic conditions, the NLRP3 inflammasome aids epithelial barrier integrity and has antimicrobial activity (8). In disease, excessive NLRP3 and the IL-1β precursor expression are induced by NF-κB through DAMPs and PAMPs (30). PAMPs associated with the gut microbiota make their way through the epithelial barrier and bind pattern recognition receptor expressing cells. DAMPs such as IL-1α are released from epithelial cells undergoing cell death (31). Relevantly, Bersudsky et al. reported that IL-1β from myeloid cells promotes healing and repair in DSS-induced colitis, unlike IL-1α which is primarily inflammatory (32). In IBD, colonic IL-1β expression correlates positively with disease activity, particularly in active lesions (33). Genetic polymorphisms in NLRP3 are associated with increased risk of CD (34) and UC (35, 36). The inhibition of NLRP3 by IL-38 has recently been demonstrated in an in vitro model of temporomandibular joint inflammation (37).
Several studies report on the role of IL-36 family members in mouse models of IBD. In mice subjected to DSS colitis, IL-36α/γ and IL-38 gene expression are increased in the colon during the peak of colitis, whereas IL-36β, IL-36Ra, and IL-1R6 remained stable (38, 39). Yang et al., recently reported that deficiency of the IL-1R6 agonist IL-36γ leads to hypo-responsiveness to DSS-induced colitis, whereas the IL-36Ra deficient mouse is hyper-responsive (40). These data are in line with our observations of amplified colitis in the IL-38 deficient mouse, and are consistent with IL-38 and IL-36Ra both inhibiting IL-1R6 signaling. Interestingly, IL-1R6 deficient mice display decreased DSS-induced colitis compared to WT mice through reduced infiltration of neutrophils and macrophages (38), yet also have a defective recovery (41). The detrimental effects of endogenous IL-1R6 signaling during the induction of colitis by DSS, and its beneficial role during the recovery phase, may be dependent on the balance of pro- and anti-inflammatory IL-36 cytokines during each phase, which requires further investigation.
In IBD, patients with no detectable serum IL-38 most often had measurable CRP concentrations, although this negative correlation was not significant (18). Circulating IL-38 levels of overweight individuals with chronic low-grade inflammation are inversely correlated to CRP, TNF, IL-6 and leptin (42). Relevantly, CRP itself can induce NLRP3 activation (43), and conversely IL-38 may reduce the induction of CRP by inhibiting NLRP3.
The gut microbiome also plays a key role in IBD (44). Blockade of IL-1α with a neutralizing antibody protects mice from acute DSS-induced colitis and modifies the gut microbiota into an anti-inflammatory flora (31). Here, we show that endogenous IL-38 reduces IL-1α expression in mice subjected to DSS. Hence, a thorough characterization of the microbiota of IL-38 deficient mice, or perhaps human subjects with a relative IL-38 deficiency, will help determine whether loss of endogenous IL-38 results in gut microbiota alterations that exacerbates the inflammatory phenotype reported in these animals. Our data indicate that mice lacking IL-38 that received normal drinking water did not show a difference from vehicle treated WT mice, indicating that there is no spontaneous inflammatory phenotype.
The expression of IL-38 in IBD patient samples is abundant throughout the mucosa, submucosa, muscular, and serosa layers (19). IL-38 gene expression is significantly higher in active UC compared to active CD (19). The analysis of colonic biopsy specimens showed that levels of IL-36α/γ and IL-38, but not IL-36β, are increased in active CD patients and related to IL-1β and IL-17A (39). IL-36 precursor levels are enhanced in active lesions in UC patients (19, 33). IL-36Ra expression in lamina propria mononuclear cells is significantly reduced in the colon of UC patients (38), and DNA microarray analysis has identified IL-36γ as the most preferentially expressed cytokine in inflammatory colonic macrophages (45).
IBD patients show increased risk for developing colorectal cancer (CRC), due to the pro-carcinogenic effects of chronic inflammation (46, 47). Wang et al. observed that IL-38 gene expression was reduced in CRC tissues in comparison to healthy tissue, and high IL-38 expression in CRC biopsies was associated with prolonged survival and smaller tumor size (48), indicating that therapeutic use of recombinant IL-38 in CRC deserves investigation. Furthermore, the IL-38 associated SNP rs6734328 was also related to a reduced risk for CRC (49). This implies that chronic reduced expression, or deficient endogenous IL-38 likely contributes to both IBD and CRC development and progression.
Concluding remarks
We demonstrate that endogenous IL-38 reduces the inflammatory phenotype associated with IBD in a mouse model of DSS colitis. Our data suggest that during the recovery from colitis, IL-38 deficient animals have detrimental NLRP3 activity, and NLRP3 inhibition attenuates the recovery process. We propose that a relative deficiency of IL-38 contributes to IBD by disinhibition of the NLRP3 inflammasome.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by University of Colorado Animal Care and Use Committee.
Author Contributions
DG, RW, SC, and CD designed research; DG, RW, JA-A, JL, AD, IT, and CM performed research; DG and RW analyzed data; DG drafted the manuscript paper. RW, JA-A, SC, LJ and CD corrected the manuscript. All authors contributed to the article and approved of the submitted version.
Funding
DG and CM are supported by the Interleukin Foundation. RW is supported by an NIH National Research Service Award (NRSA) fellowship F30DK120072 and NIH Medical Scientist Training Program (MSTP) training grant T32GM008497. JL is supported by NIH grant DK129410. CD is supported by NIH Grant AI-15614. SC is supported by NIH grants DK1047893, DK50189, and DK095491.
Conflict of Interest
LJ serves on Olatec’s scientific advisory board and receives compensation. CD serves as chairman of Olatec’s scientific advisory board, is co-chief scientific officer, receives compensation, and has equity in Olatec. CM serves as director for Olatec’s innovative science program and has equity in Olatec.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to Tania Azam for help with breeding of WT and IL-38 deficient mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.840719/full#supplementary-material
Endogenous IL-38 expression is associated with reduced colonic caspase-1 activation. WT and IL-38 KO (IL-38 deficient) mice subjected to H2O. NLRP3, Caspase-1 (p20), Caspase-1 (p45) and β-Actin protein abundance in colonic tissues quantified by Western Blot analysis. Lanes 1-3 contain colonic tissue of WT mice treated with H2O, lanes 4-6 from IL-38 deficient mice subjected to H2O. A representative blot is shown from two experiments with 3 mice per group.
Abbreviations
ASC, apoptosis-associated speck-like; CD, Crohn’s Disease; DAI, Disease Activity Index; DAMP, Danger Associated Molecular Pattern; DSS, Dextran Sulfate Sodium; IBD, Inflammatory Bowel Disease; NLRP3, nucleotide-binding oligomerization domain-like receptor family; pyrin domain-containing 3; PAMP, Pathogen Associated Molecular Pattern; UC, Ulcerative Colitis; WT, wild type.
References
- 1. Podolsky DK. Inflammatory Bowel Disease. N Engl J Med (2002) 347(6):417–29. doi: 10.1056/NEJMra020831 [DOI] [PubMed] [Google Scholar]
- 2. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 3. Goyette P, Labbé C, Trinh TT, Xavier RJ, Rioux JD. Molecular Pathogenesis of Inflammatory Bowel Disease: Genotypes, Phenotypes and Personalized Medicine. Ann Med (2007) 39(3):177–99. doi: 10.1080/07853890701197615 [DOI] [PubMed] [Google Scholar]
- 4. Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-Like Receptors in Host Defense and Disease. Immunity (2007) 27(4):549–59. doi: 10.1016/j.immuni.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 5. Hazel K, O'Connor A. Emerging Treatments for Inflammatory Bowel Disease. Ther Adv Chronic Dis (2020) 11:2040622319899297. doi: 10.1177/2040622319899297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR Gene Family: A Standard Nomenclature. Immunity (2008) 28(3):285–7. doi: 10.1016/j.immuni.2008.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rathinam VAK, Zhao Y, Shao F. Innate Immunity to Intracellular LPS. Nat Immunol (2019) 20(5):527–33. doi: 10.1038/s41590-019-0368-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharma D, Kanneganti TD. Inflammatory Cell Death in Intestinal Pathologies. Immunol Rev (2017) 280(1):57–73. doi: 10.1111/imr.12602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int J Mol Sci (2020) 21(12):4223. doi: 10.3390/ijms21124223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. IL-1β Mediates Chronic Intestinal Inflammation by Promoting the Accumulation of IL-17A Secreting Innate Lymphoid Cells and CD4(+) Th17 Cells. J Exp Med (2012) 209(9):1595–609. doi: 10.1084/jem.20111453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis Induced in Mice With Dextran Sulfate Sodium (DSS) is Mediated by the NLRP3 Inflammasome. Gut (2010) 59(9):1192–9. doi: 10.1136/gut.2009.197822 [DOI] [PubMed] [Google Scholar]
- 12. Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 Interacts With NLRP3 to Modulate the Pyroptosis in Inflammatory Bowel Disease via NF-κb Signaling. Cell Death Dis (2019) 10(12):906. doi: 10.1038/s41419-019-2157-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin H, Ho AS, Haley-Vicente D, Zhang J, Bernal-Fussell J, Pace AM, et al. Cloning and Characterization of IL-1HY2, a Novel Interleukin-1 Family Member. J Biol Chem (2001) 276(23):20597–602. doi: 10.1074/jbc.M010095200 [DOI] [PubMed] [Google Scholar]
- 14. Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, et al. IL-38 has an Anti-Inflammatory Action in Psoriasis and its Expression Correlates With Disease Severity and Therapeutic Response to Anti-IL-17A Treatment. Cell Death Dis (2018) 9(11):1104. doi: 10.1038/s41419-018-1143-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Graaf DM, Jaeger M, van den Munckhof ICL, Ter Horst R, Schraa K, Zwaag J, et al. Reduced Concentrations of the B Cell Cytokine Interleukin 38 are Associated With Cardiovascular Disease Risk in Overweight Subjects. Eur J Immunol (2021) 51(3):662–71. doi: 10.1002/eji.201948390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, et al. IL-38 Binds to the IL-36 Receptor and has Biological Effects on Immune Cells Similar to IL-36 Receptor Antagonist. Proc Natl Acad Sci USA (2012) 109(8):3001–5. doi: 10.1073/pnas.1121534109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased Expression of Interleukin 17 in Inflammatory Bowel Disease. Gut (2003) 52(1):65–70. doi: 10.1136/gut.52.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xie C, Yan W, Quan R, Chen C, Tu L, Hou X, et al. Interleukin-38 is Elevated in Inflammatory Bowel Diseases and Suppresses Intestinal Inflammation. Cytokine (2020) 127:154963. doi: 10.1016/j.cyto.2019.154963 [DOI] [PubMed] [Google Scholar]
- 19. Fonseca-Camarillo G, Furuzawa-Carballeda J, Iturriaga-Goyon E, Yamamoto-Furusho JK. Differential Expression of IL-36 Family Members and IL-38 by Immune and Nonimmune Cells in Patients With Active Inflammatory Bowel Disease. BioMed Res Int (2018) 2018:5140691. doi: 10.1155/2018/5140691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie L, Huang Z, Li H, Liu X, Zheng S, Su W. IL-38: A New Player in Inflammatory Autoimmune Disorders. Biomolecules (2019) 9(8):345. doi: 10.3390/biom9080345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Graaf DM, Teufel LU, van de Veerdonk FL, Joosten LAB, Netea MG, Dinarello CA, et al. IL-38 Prevents Induction of Trained Immunity by Inhibition of mTOR Signaling. J Leukoc Biol (2021) 110(5):907–15. doi: 10.1002/jlb.3a0220-143rrr [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Graaf DM, Maas RJA, Smeekens SP, Eisenmesser E, Redzic JS, Helsen MM, et al. Human Recombinant Interleukin-38 Suppresses Inflammation in Mouse Models of Local and Systemic Disease. Cytokine (2020) 137:155334. doi: 10.1016/j.cyto.2020.155334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating Intestinal Inflammation in DSS-Induced Model of IBD. J Vis Exp (2012) 60):e3678. doi: 10.3791/3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, et al. Paneth Cells as a Site of Origin for Intestinal Inflammation. Nature (2013) 503(7475):272–6. doi: 10.1038/nature12599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saber S, Youssef ME, Sharaf H, Amin NA, El-Shedody R, Aboutouk FH, et al. BBG Enhances OLT1177-Induced NLRP3 Inflammasome Inactivation by Targeting P2X7R/NLRP3 and MyD88/NF-κb Signaling in DSS-Induced Colitis in Rats. Life Sci (2021) 270:119123. doi: 10.1016/j.lfs.2021.119123 [DOI] [PubMed] [Google Scholar]
- 26. Dieleman LA, Palmen MJ, Akol H, Bloemena E, Peña AS, Meuwissen SG, et al. Chronic Experimental Colitis Induced by Dextran Sulphate Sodium (DSS) is Characterized by Th1 and Th2 Cytokines. Clin Exp Immunol (1998) 114(3):385–91. doi: 10.1046/j.1365-2249.1998.00728.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han Y, Mora J, Huard A, da Silva P, Wiechmann S, Putyrski M, et al. IL-38 Ameliorates Skin Inflammation and Limits IL-17 Production From Gammadelta T Cells. Cell Rep (2019) 27(3):835–46. doi: 10.1016/j.celrep.2019.03.082 [DOI] [PubMed] [Google Scholar]
- 28. Oizumi T, Mayanagi T, Toya Y, Sugai T, Matsumoto T, Sobue K. NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Onset of Inflammation in Mice With Dextran Sulfate Sodium-Induced Colitis. Dig Dis Sci (2021) 8(19):e2101501. doi: 10.1007/s10620-021-07184-y [DOI] [PubMed] [Google Scholar]
- 29. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 Inflammasome Protects Against Loss of Epithelial Integrity and Mortality During Experimental Colitis. Immunity (2010) 32(3):379–91. doi: 10.1016/j.immuni.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Próchnicki T, Mangan MS, Latz E. Recent Insights Into the Molecular Mechanisms of the NLRP3 Inflammasome Activation. F1000Research (2016) 5(F100 Faculty Rev):1469. doi: 10.12688/f1000research.8614.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Menghini P, Corridoni D, Buttó LF, Osme A, Shivaswamy S, Lam M, et al. Neutralization of IL-1α Ameliorates Crohn's Disease-Like Ileitis by Functional Alterations of the Gut Microbiome. Proc Natl Acad Sci USA (2019) 116(52):26717–26. doi: 10.1073/pnas.1915043116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, et al. Non-Redundant Properties of IL-1α and IL-1β During Acute Colon Inflammation in Mice. Gut (2014) 63(4):598–609. doi: 10.1136/gutjnl-2012-303329 [DOI] [PubMed] [Google Scholar]
- 33. Nishida A, Hidaka K, Kanda T, Imaeda H, Shioya M, Inatomi O, et al. Increased Expression of Interleukin-36, a Member of the Interleukin-1 Cytokine Family, in Inflammatory Bowel Disease. Inflammation Bowel Dis (2016) 22(2):303–14. doi: 10.1097/mib.0000000000000654 [DOI] [PubMed] [Google Scholar]
- 34. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common Variants in the NLRP3 Region Contribute to Crohn's Disease Susceptibility. Nat Genet (2009) 41(1):71–6. doi: 10.1038/ng.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang HX, Wang ZT, Lu XX, Wang YG, Zhong J, Liu J. NLRP3 Gene is Associated With Ulcerative Colitis (UC), But Not Crohn's Disease (CD), in Chinese Han Population. Inflamm Res (2014) 63(12):979–85. doi: 10.1007/s00011-014-0774-9 [DOI] [PubMed] [Google Scholar]
- 36. Zhou L, Liu T, Huang B, Luo M, Chen Z, Zhao Z, et al. Excessive Deubiquitination of NLRP3-R779C Variant Contributes to Very-Early-Onset Inflammatory Bowel Disease Development. J Allergy Clin Immunol (2021) 147(1):267–79. doi: 10.1016/j.jaci.2020.09.003 [DOI] [PubMed] [Google Scholar]
- 37. Luo P, Zhao T, He H. IL-38-Mediated NLRP3/caspase-1 Inhibition is a Disease-Modifying Treatment for TMJ Inflammation. Ann N Y Acad Sci (2021) 1508(1):92–104. doi: 10.1111/nyas.14704 [DOI] [PubMed] [Google Scholar]
- 38. Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, et al. IL-36α Expression is Elevated in Ulcerative Colitis and Promotes Colonic Inflammation. Mucosal Immunol (2016) 9(5):1193–204. doi: 10.1038/mi.2015.134 [DOI] [PubMed] [Google Scholar]
- 39. Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, et al. Distinct Expression of Interleukin (IL)-36alpha, Beta and Gamma, Their Antagonist IL-36Ra and IL-38 in Psoriasis, Rheumatoid Arthritis and Crohn's Disease. Clin Exp Immunol (2016) 184(2):159–73. doi: 10.1111/cei.12761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang W, Dong H-P, Wang P, Xu Z-G, Xian J, Chen J, et al. IL-36γ and IL-36ra Reciprocally Regulate Colon Inflammation and Tumorigenesis by Modulating the Cell–Matrix Adhesion Network and Wnt Signaling. Advanced Sci (2022)n/a(n/a):2103035. doi: 10.1002/advs.202103035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, et al. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J Immunol (2016) 196(1):34. doi: 10.4049/jimmunol.1501312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Graaf DM, Jaeger M, van den Munckhof ICL, Horst RT, Schraa K, Zwaag J, et al. Dinarello: Reduced concentrations of the B cell cytokine Interleukin 38 are associated with cardiovascular disease risk in overweight subjects. Eur J Immunol (2021) 51(3):662–71. doi: 10.1002/eji.201948390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bian F, Yang X-Y, Xu G, Zheng T, Jin S. CRP-Induced NLRP3 Inflammasome Activation Increases LDL Transcytosis Across Endothelial Cells. Front Pharmacol (2019) 10:40(40). doi: 10.3389/fphar.2019.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Glassner KL, Abraham BP, Quigley EMM. The Microbiome and Inflammatory Bowel Disease. J Allergy Clin Immunol (2020) 145(1):16–27. doi: 10.1016/j.jaci.2019.11.003 [DOI] [PubMed] [Google Scholar]
- 45. Harusato A, Medina-Contreras O, Nishio H, Chassaing B, Gewirtz A, Parkos C, et al. IL-36 Receptor Is Required for Resolution of Intestinal Damage. Inflamm Bowel Dis (2016) 22:S54–5. doi: 10.1097/01.MIB.0000480273.37247.26 [DOI] [Google Scholar]
- 46. Stidham RW, Higgins PDR. Colorectal Cancer in Inflammatory Bowel Disease. Clin Colon Rectal Surg (2018) 31(3):168–78. doi: 10.1055/s-0037-1602237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rutter MD, Saunders BP, Wilkinson KH, Rumbles S, Schofield G, Kamm MA, et al. Cancer Surveillance in Longstanding Ulcerative Colitis: Endoscopic Appearances Help Predict Cancer Risk. Gut (2004) 53(12):1813–6. doi: 10.1136/gut.2003.038505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen F, Zhang F, Tan Z, Hambly BD, Bao S, Tao K. Interleukin-38 in Colorectal Cancer: A Potential Role in Precision Medicine. Cancer Immunol Immunother (2020) 69(1):69–79. doi: 10.1007/s00262-019-02440-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Dai JY, Albanes D, Arndt V, Berndt SI, Bézieau S, et al. Mendelian Randomization Analysis of C-Reactive Protein on Colorectal Cancer Risk. Int J Epidemiol (2018) 48(3):767–80. doi: 10.1093/ije/dyy244 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Endogenous IL-38 expression is associated with reduced colonic caspase-1 activation. WT and IL-38 KO (IL-38 deficient) mice subjected to H2O. NLRP3, Caspase-1 (p20), Caspase-1 (p45) and β-Actin protein abundance in colonic tissues quantified by Western Blot analysis. Lanes 1-3 contain colonic tissue of WT mice treated with H2O, lanes 4-6 from IL-38 deficient mice subjected to H2O. A representative blot is shown from two experiments with 3 mice per group.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.