

Abstract
Natural products have traditionally been a fruitful source of chemical matter that has been developed into novel therapeutics. Actinomycetes and several other bacterial taxa are especially gifted in biosynthesizing natural products. However, many decades of intense bioactivity-based screening led to a large rediscovery problem, rendering industrial natural product discovery pipelines uneconomical. Numerous methods for circumventing the rediscovery problem have been developed, among them various chemistry-focused strategies, including reactivity-based screening. Emerging from the field of chemical proteomics, reactivity-based screening relies on a reactive probe that chemoselectively modifies a functional group of interest in the context of a complex biological sample. Reactivity-based probes for several distinct functional groups have been deployed to discover new polyketide and peptidic natural products. This chapter describes the protocols to conduct a reactivity-based screening campaign, including bacteria cultivation and screening of cellular extracts with phenylglyoxal-, tetrazine-, thiol-, and aminooxy-functionalized probes, which respectively target primary uriedo, electron-rich olefins, Michael acceptors, and reactive carbonyls. In addition, a recent case study is presented that employs reactivity-based screening as a component of a forward genetics screen to identify a previously unknown peptidyl arginine deiminase. We anticipate that these methods will be useful for those interested in discovering natural products that evade detection by traditional, bioassay-guided methods and others who wish to rapidly connect metabolic chemotype with genotype.
Keywords: Natural products, genome mining, bioinformatics, reactivity-based screening, RiPPs, bioorthogonal chemistry, activity-based profiling
1. Introduction
Natural products (NPs) have long been a productive source of new bioactive molecules. Between 1981 and 2019, over half of all newly approved small molecule drugs were NPs or derivatives thereof (Newman & Cragg, 2020). Many NPs were discovered from bacteria, especially the genus Streptomyces (Katz & Baltz, 2016). During the so-called “golden era” of antibiotic discovery, new compounds were discovered via phenotypic screening of spent media or cellular extracts. While highly successful and enabling the discovery of many classes of drugs used today (Newman & Cragg, 2020), rediscovery of commonly occurring bioactive molecules became an ever increasing problem. Bioactive, small molecule metabolites are produced by Streptomyces at vastly differing frequencies (e.g. 1% of strains produce streptomycin while only 0.0001% produce daptomycin) (Baltz, 2005), meaning researchers are much more likely to discover an already known NP rather than a novel one. Early dereplication of known NPs is critical to the viability of continued microbial screening (Katz & Baltz, 2016). Unfortunately, the high costs and diminishing returns led numerous companies to discontinue their NP discovery efforts (Dougan et al., 2019). However, the genomics revolution has rekindled interest in NP discovery, given the small fraction of known compounds compared to the enormous, untapped biosynthetic potential of Nature (Kautsar et al., 2021). Innovative methods to discover new NPs that circumvent the longstanding issue of rediscovery are thus urgently needed (Ahmad & Khan, 2019).
In recent years, several strategies have been developed to address rediscovery, including: (i) deliberate efforts to screen new sources of biological diversity (Ling et al., 2015) (Chaudhary et al., 2019) (Cahn & Piel, 2021), (ii) genomics-guided structure prediction and prioritized screening (Blin et al., 2021) (Skinnider et al., 2020) (Tietz et al., 2017) (Mungan et al., 2020) (Goering et al., 2016) (Gross et al., 2007), (iii) construction of open-access mass spectrometry databases for rapid dereplication of known compounds (M. Wang et al., 2016) (Kersten et al., 2011), (iv) techniques to activate NP biosynthetic gene clusters (BGCs) that are dormant during laboratory cultivation (B. Wang et al., 2020) (Zhao et al., 2020), (v) improvements in heterologous expression of NPs in non-native hosts (Huo et al., 2019) (Zhang et al., 2019) (Xu & Wright, 2019), and (vi) various reactivity-based methods for probing NP structure. The use of reactivity-based methods to interrogate biology has a long history, perhaps beginning with the characterization of penicillin-binding proteins through covalent modification by radiolabeled β-lactam antibiotics (Blumberg & Strominger, 1972) (Suginaka et al., 1972). Subsequent work on electrophilic protease inhibitors inspired the development of what is broadly known as chemical proteomics, which use small molecule probes to interrogate protein function (Rudolf et al., 2013). Among the most widely used chemical proteomics strategies is activity-based protein profiling (ABPP), which interrogates small molecule-protein interactions using a probe that covalently modifies the protein of interest in complex biological settings (S. Wang et al., 2018). An advantage of ABPP is that the target must be catalytically active in order to be modified, which means biologically relevant phenotypes can be directly measured, rather than inferred based on protein abundance in the case of traditional proteomics. ABPP has been deployed to quantify activity for numerous proteins from several enzyme classes, such as serine and cysteine proteases, kinases, and glycosidases, and has seen wide use discovering and optimizing drug candidates (Deng et al., 2020). While initially detected through fluorescent reporters, methods such as ABPP-multidimensional protein identification technology (MudPIT) (Speers & Cravatt, 2009) and isotopic tandem orthogonal proteolysis (isoTOP)-ABPP (Weerapana et al., 2010) were developed to rapidly analyze ABPP experiments via mass spectrometry (MS). The use of isotopically labeled probes to quantify and identify which protein residue is modified has inspired isotope labeling in other reactivity-based strategies.
In addition to ABPP, advances in biorthogonal chemistry have been instrumental in the development of reactivity-based methods for NP discovery. Bioorthogonal reactions are those that can occur in living cells without disrupting biological processes, and ideally have no (or negligible) cross reactivity with endogenous functional groups (Sletten & Bertozzi, 2011). The first example of a bioorthogonal reaction applied in a biological setting was the Staudinger ligation, which was used to label engineered azido-sugars on the cell surface using a biotinylated triarylphosphine (Saxon & Bertozzi, 2000). Though revolutionary, the Staudinger ligation had several drawbacks, such as slow reaction kinetics and oxidation of the triarylphosphine in living cells, spurring the development of other bioorthogonal reactions (Sletten & Bertozzi, 2011). Inspired by the copper-catalyzed Huisgen cycloaddition (Rostovtsev et al., 2002), a strain-promoted azide-alkyne click reaction was developed to circumvent the in vivo toxicity of copper (Agard et al., 2004). To date, numerous bioorthogonal reactions have been developed in five broad reaction categories: [3+2] dipolar cycloadditions (azide-alkyne click reaction), phosphine ligation reactions (Staudinger ligation), 2-acylboronic acid condensation, [4+1] isonitrile cycloaddition, and the inverse electron demand Diels-Alder reaction (Smeenk et al., 2021). This last reaction utilizes an electron-poor dienophile, most commonly 1,2,4,5-tetrazine, and a strained cyclic alkene (Blackman et al., 2008) (Devaraj et al., 2008). Advantages of the tetrazine ligation reaction are the exceptionally high rate constant and the air- and solution-stability of the reactant and ligation products (Wu & Devaraj, 2018). Bioorthogonal chemistry has become a foundational tool in chemical biology, and fittingly, has seen wide application in cellular and whole organism imaging, metabolite engineering, drug conjugation/delivery, and synthesizing novel biomaterials (Porte et al., 2021) (Kim & Koo, 2019). Of relevance to this chapter, several bioorthogonal reactions have been coopted for reactivity-based NP discovery.
From critical advances in chemical proteomics, bio-orthogonal chemistry, and mass spectrometry emerged the idea to develop a reactivity-based strategy for NP discovery. Dubbed reactivity-based screening (RBS) (Cox et al., 2014), this method leverages robust, chemoselective warheads that selectively modify a functional group of interest in biological samples, including but not limited to bacterial extracts (Fig. 1). By deploying a probe with an appropriately reactive warhead and comparing mass spectra with and without the probe added, one gleans an additional layer of information about the metabolites present, specifically whether the functional group of interest is present. Using the mass of the unreacted parent peak and the functional group present, the compound can be rapidly dereplicated using one of many NP MS databases (Sorokina & Steinbeck, 2020). The RBS warheads are also amenable to modular tailoring, allowing for incorporation of bromine for rapid identification of labeled peaks via 1:1 79Br/81Br isotope distribution (Palaniappan et al., 2011) (Maxson et al., 2016), UV-absorbing arenes for increased sensitivity in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (Cabrera-Pardo et al., 2013) (Mandal et al., 2015), and selective enrichment via appended affinity tags. A pioneering example of an RBS-like strategy was reported by Frechét and coworkers, who selectively purified α,β-unsaturated lactone-containing NPs on a solid support (Cheminat et al., 1981). This report has inspired others to develop probes for numerous functional groups, such as thiols for the detection of epoxide, β-lactones, and other electrophilic NPs (Cox et al., 2014) (Georgiou et al., 2020) (Castro-Falcón et al., 2016) (Rudolf et al., 2015), nitrosopyridines for the detection of conjugated alkenes (Castro-Falcón et al., 2018), tetrazine probes for electron-rich alkenes (Guo et al., 2021) and isonitriles (Huang et al., 2020), azide probes for internal (Back et al., 2021) and terminal alkynes (Ross et al., 2014), aminooxy probes for reactive carbonyl-containing NPs (Maxson et al., 2016), silane (Y. Odendaal et al., 2011) and boronic acid probes (Gamoh et al., 1994) for alcohols and diols, respectively, disulfides for the capture of thiol- and disulfide-containing NPs (L. Capehart & E. Carlson, 2016), and phenylglyoxal probes for primary ureido containing NPs (Harris et al., 2020). To date, RBS has been used to discover numerous novel compounds from a variety of NP classes, among them ribosomally synthesized and post-translationally modified peptides (RiPPs), non-ribosomal peptides (NRPs), polyketides, and hybrids thereof (Tollefson & Carlson, 2019).
Figure 1. Overview of Reactivity-based Screening.
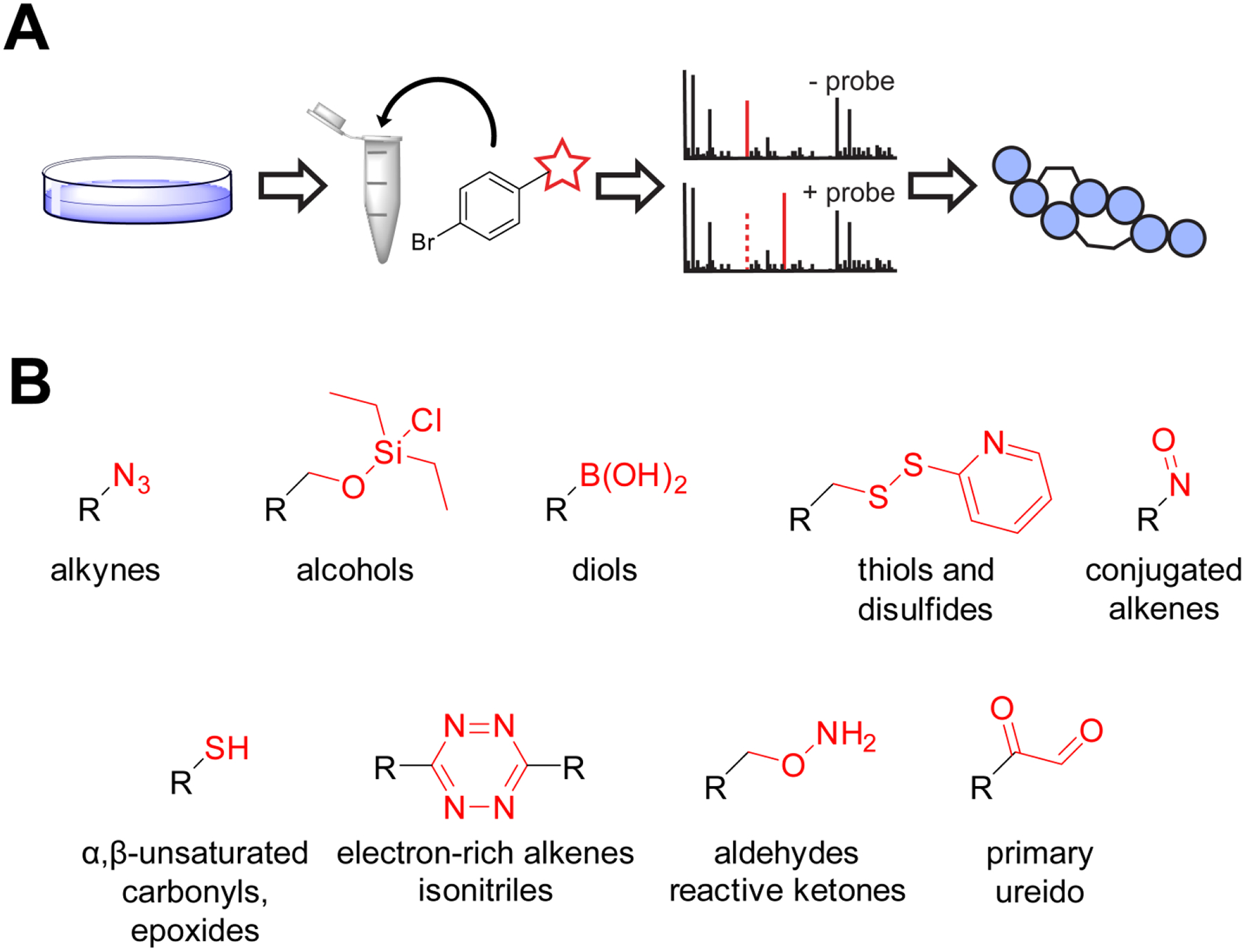
Overview of reactivity-based screening. (A) Workflow for reactivity-based NP discovery. Organisms are cultivated and their extracts reacted with a probe that will selectively modify a desired organic functional group. Mass spectra are collected of the extract before and after treatment with the probe. New masses that appear after probe reaction can be cross-referenced for novelty, potentially leading to the discovery of novel NPs. (B) Structures of the reactive warhead (red) that have been have been deployed in probes to discover/purify NPs, with the targeting functional group written below.
Herein, we describe in detail the use of phenylglyoxal- (Harris et al., 2020), tetrazine- (Guo et al., 2021), thiol- (Cox et al., 2014), and aminooxy-functionalized probes (Maxson et al., 2016). We include a brief overview of selecting and growing organisms to be screened, how to prepare the bacterial extract, perform the reaction with the RBS probe, and acquisition/analysis of mass spectral data. In addition, a case study in using RBS as a key component in a forward chemical genetic screen is presented to demonstrate that the utility of RBS extends beyond NP discovery. As the functional groups detected by these probes are important to biology outside of the realm of NP discovery, we believe that these methods may be of use to those interested in primary metabolic profiling.
2. RBS Screening Paradigms: Targeted vs Untargeted
RBS can be broadly divided into two types: targeted and untargeted. Several examples of RBS employed untargeted screens to discover new NPs with a functional group of interest (Maxson et al., 2016) (Reimer & Hughes, 2017). With open access to hundreds of thousands of bacterial genomes, RBS campaigns can also leverage genomic information in a targeted fashion, if prior work has elucidated the genes responsible for functional group installation. A curated list of strains with the capacity to biosynthesize NPs with the desired functional group can be bioinformatically predicted. An illustrative example was the co-deployment of bioinformatics and the tetrazine probe to discover new glycosylated polyene macrolides (GPMs) (Guo et al., 2021). Core features of the required polyketide synthase genes are conserved across GPM BGCs, and these insights were used to generate a list of putative GPM producers using Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1990) and genome neighborhood analysis (Zallot et al., 2019). Screening a subset of readily cultivable candidate GPM producers led to discovery of a known GPM from a new producer and the discovery of a new GPM kineosporicin (Guo et al., 2021). The targeted and untargeted RBS paradigms each carry advantages and disadvantages. A targeted screen may have a higher likelihood of generating useful hits, as the strains have been pre-selected for the ability to biosynthesize the functional group of interest. However, targeted screening is not feasible if the genes responsible for functional group installation are unknown. Moreover, a targeted screen is based on known biochemistry, meaning that discovery of novel structures and mechanisms of functional group installation could be less likely. Though untargeted screens often require the researcher to screen larger numbers of organisms for each hit, they require no prior biochemical knowledge of the functional group in question and are biased towards already known NPs.
3. Growth and Harvest of Screening Organisms
RBS can theoretically be applied to any culturable organism or environmental sample, but owing to ease of implementation, strain accessibility, and high biosynthetic potential, much of our published work centers on Actinobacteria, which contains the Streptomyces genus. Interested readers are referred to the book Practical Streptomyces Genetics as an excellent primer (Kieser et al., 2000), especially for working with these organisms in a laboratory setting. Briefly, each strain is grown under various nutritional conditions to elicit the fullest extent of NP production. For Streptomyces, NP production is often associated with sporulation and numerous media have been developed for this purpose (i.e., nutrient poor and/or metal and phosphate rich) (Kieser et al., 2000). Once grown, the cells are harvested, extracted, and subjected to RBS.
3.1. Materials, Reagents and Equipment
Equipment:
Bunsen burner
New Brunswick Scientific i2500 series incubator shaker
Fisher Scientific Isotemp incubator
CG Life Sciences versa-roll drumroller
Thermo Scientific Sorvall Legend micro 17
(optional) Fisher Scientific vortex mixer
(optional) Speed vacuum concentrator (Thermo Scientific)
Materials:
Frozen glycerol stock of organism(s) of interest, spore or cell
Sterile L-shaped spreaders
70% ethanol in a spray bottle
5 mL sterile pipettes
125 mL baffled flasks
(optional) 3 mm glass beads (Fisher), sterilized
Disposable razor blade
1.5 mL Eppendorf tube
15 cm petri dishes
10 mL culture tubes with plastic caps
Plastic tips, metal inoculation loop, or wooden toothpick, sterilized
Reagents:
ATCC172 growth medium (20 g/L soluble starch, 10 g/L glucose, 5 g/L yeast extract, 5 g/L N-Z amine, 1 g/L calcium carbonate, 15 g/L agar, pH 7.2)
ISP2 medium (4 g/L yeast extract, 10 g/L malt extract, 4 g/L glucose, 15 g/L agar, pH 7.2)
GUBC medium (6.25 g/L glycerol, 10 g/L sucrose, 10 g/L beef extract, 5 g/L casamino acids [Bacto], 20 mM Na2HPO4-KH2PO4, and 10 mL/L of a solution of Balch’s vitamins [described below], 15 g/L agar, pH 7.2)
CFood medium (25 g/L glucose, 15 g/L fish meal [Dr. Earth], 2 g/L yeast extract, 4 g/L calcium carbonate, 15 g/L agar, pH 7.2)
V8 medium (200 mL/L V8 brand vegetable juice, 3 g/L calcium carbonate, 15 g/L agar, pH 7.2)
MS medium (20 g/L mannitol, 20 g/L soya flour [Kinako], 10 mM magnesium chloride, 15 g/L agar, pH 7.2)
ISP4 medium (10 g/L soluble starch, 1 g/L potassium phosphate dibasic, 1 g/L magnesium sulfate heptahydrate, 1 g/L sodium chloride, 2 g/L ammonium sulfate, 2 g/L calcium carbonate, 1 mg iron (II) sulfate heptahydrate, 1 mg zinc sulfate heptahydrate, 1 mg manganese (II) chloride, 15 g/L agar, pH 7.2)
Balch’s vitamins (2 mg biotin, 2 mg folic acid, 10 mg pyridoxine hydrochloride, 5 mg thiamine hydrochloride, 5 mg riboflavin, 5 mg nicotinic acid, and 5 mg DL-calcium pantothenate in 100 mL deionized water)
Reagent grade methanol (VWR)
3.2. Growth and Extraction Procedure for Streptomyces strains
Timing: 10–16 days
Using sterile technique, inoculate a 15 cm plate of ATCC172 from a frozen stock of the organism of interest using a T-streak.
Incubate the plate at 30 °C for 2–4 days. Plates are ready for the next step once well-developed colonies form. For most Streptomyces strains in the DSMZ (https://www.dsmz.de/collection/catalogue/microorganisms/catalogue), morphology can be checked against photos of the strain grown on various media.
Using sterile technique, pick an isolated colony from the 15 cm plate using a sterile plastic pipette tip, etc., and use to inoculate a 10 mL culture tube containing 5 mL ATCC172 w/o agar.
Incubate tube at 30 °C (Fisher Scientific Isotemp incubator) with agitation (CG Life Sciences versa-roll drumroller, 40 rpm) for 1–3 days. Morphology in liquid culture can vary greatly for Actinomycetes, from diffuse, Escherichia coli like cultures to a few large, spherical cell masses; 50–100 masses of 2–4 mm in size is most common.
(Optional) Sterilized glass beads can be used to break up large masses of cells for better inoculation of subsequent plates. Using sterile technique, transfer approximately 1 mL of sterile glass beads to the 5 mL culture tube, and vortex until the colonies are broken up, typically 1–2 minutes is sufficient.
Using a sterile pipette, transfer the entire 5 mL from the 10 mL culture tube to 125 mL baffled flask containing 50 mL ATCC172 w/o agar. Incubate at 30 °C (New Brunswick Scientific i2500 series incubator shaker) with shaking at 220 rpm for 1–3 days. Resulting cultures should be visibly turbid.
Using sterile technique, transfer 1 mL of the culture to inoculate a 15 cm agar plate containing the desired medium and spread the inoculum using an L-shaped spreader. We typically use a representative set of six media that are detailed in the materials section above.
Incubate at 30 °C for 7–10 days. The plates should be stored face up for the first 1–2 days to allow the plates to dry. Afterwards, the plates should be placed face down. Bacterial growth should be confluent, and if sporulation occurs it should be apparent by day 7–10 (i.e. color change and the colony surface developing a fuzzy appearance).
Using a disposable razor blade, scrape the cell mass from the 15 cm plate and transfer to a 1.5 mL Eppendorf tube. For initial RBS screening, approximately 50 μL of cells is sufficient. For isolating larger quantities, collecting the cell mass from an entire plate or more may be needed (see section 5.2).
Add to the Eppendorf tube a volume of methanol equivalent to the volume of the cell mass. Extract for 1–2 hrs, then centrifuge at 13,000 × g using a Thermo Scientific Sorvall Legend micro 17. Decant the supernatant with an appropriately sized pipetteman and transfer to a new 1.5 mL Eppendorf tube, taking care not to transfer any residual cell mass.
(Optional) Extracts are typically used immediately for RBS; however, the methanol can be evaporated in a speed vacuum concentrator prior to storage at −20 °C or below.
4. Reactivity-Based Screening of Bacterial Extracts
This section details the methods used to react Streptomyces sp. extracts with one of four RBS probes followed by MALDI-TOF-MS analysis. The first reaction detailed is the phenylglyoxal probe reaction (Fig. 2), which is selective under the conditions reported for 1° ureido groups (unreactive towards 1° guanidino groups). An example reaction on the lasso peptide citrulassin F is shown (Fig. 3). The second example described uses a tetrazine probe, which reacts with electron-rich olefins via an inverse demand Diels-Alder reaction (Fig. 4). An example of this reaction on the GPM natamycin is provided (Fig. 5). The third reaction described is the dithiothreitol reaction, which under basic conditions undergoes a nucleophilic 1,4-addition to various electrophiles (Fig. 6), with the multiple labeling of pegvadin A shown (Fig. 7). Finally, the last detailed reaction is the aminooxy probe, which is selective for aldehydes and reactive ketones (Fig. 8), and its reaction with streptomycin is presented (Fig 9).
Figure 2. Phenylglyoxal Overview.
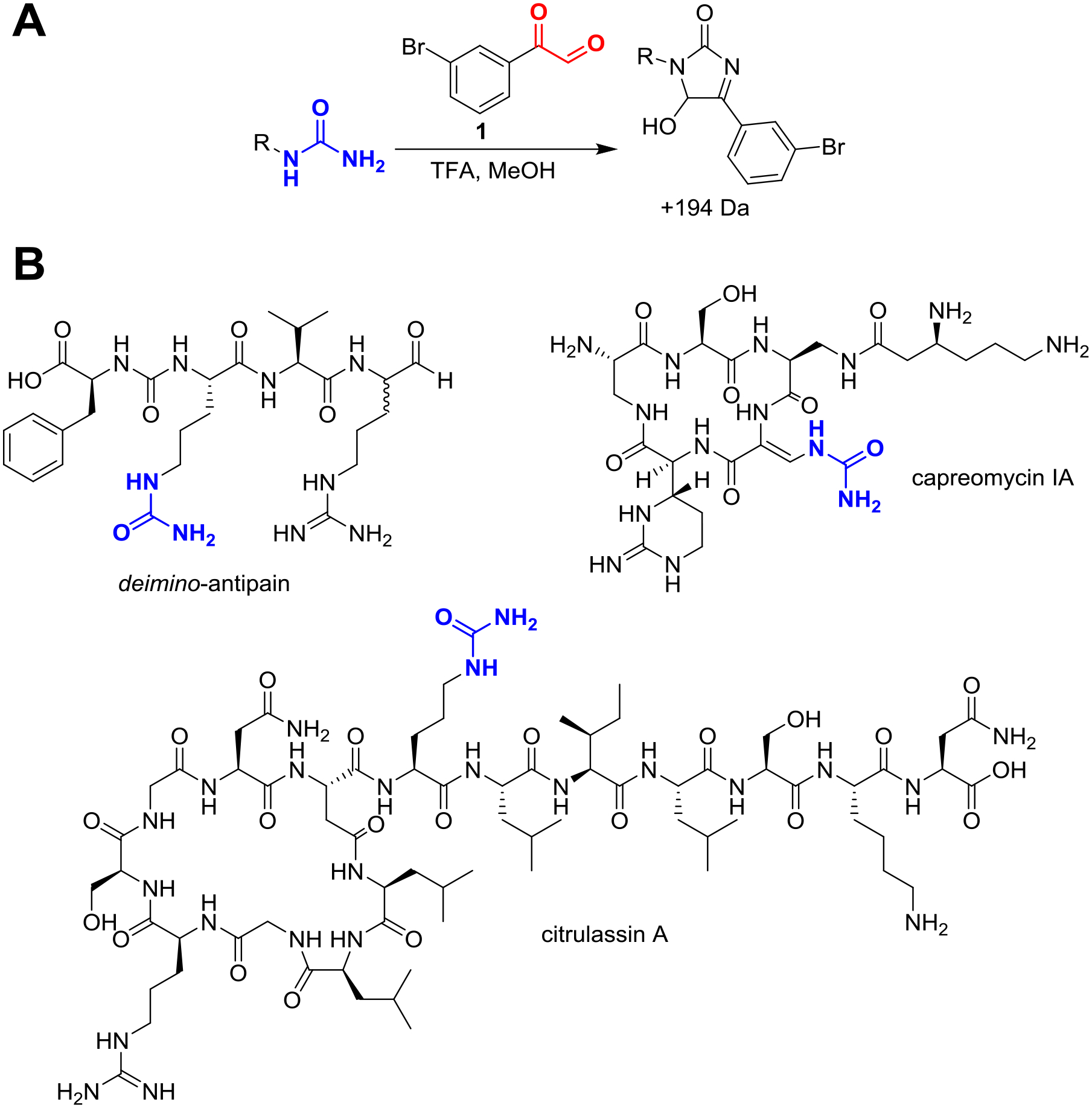
Overview of phenylglyoxal probe chemistry. (A) General conditions for labeling 1° ureido groups in a methanolic bacterial extract. Under acidic conditions, 3-bromo-phenylglyoxal (compound 1) is selective for 1° ureido over 1° guanidino groups. (B) Three representative NPs that are reactive towards 3-bromophenylglyoxal with the site of modification in blue. Natural citrulassin is found in a threaded “lasso” conformation but drawn in a branched-cyclic form for improved structural visualization.
Figure 3. Phenylglyoxal Reaction on Citrulassin F.
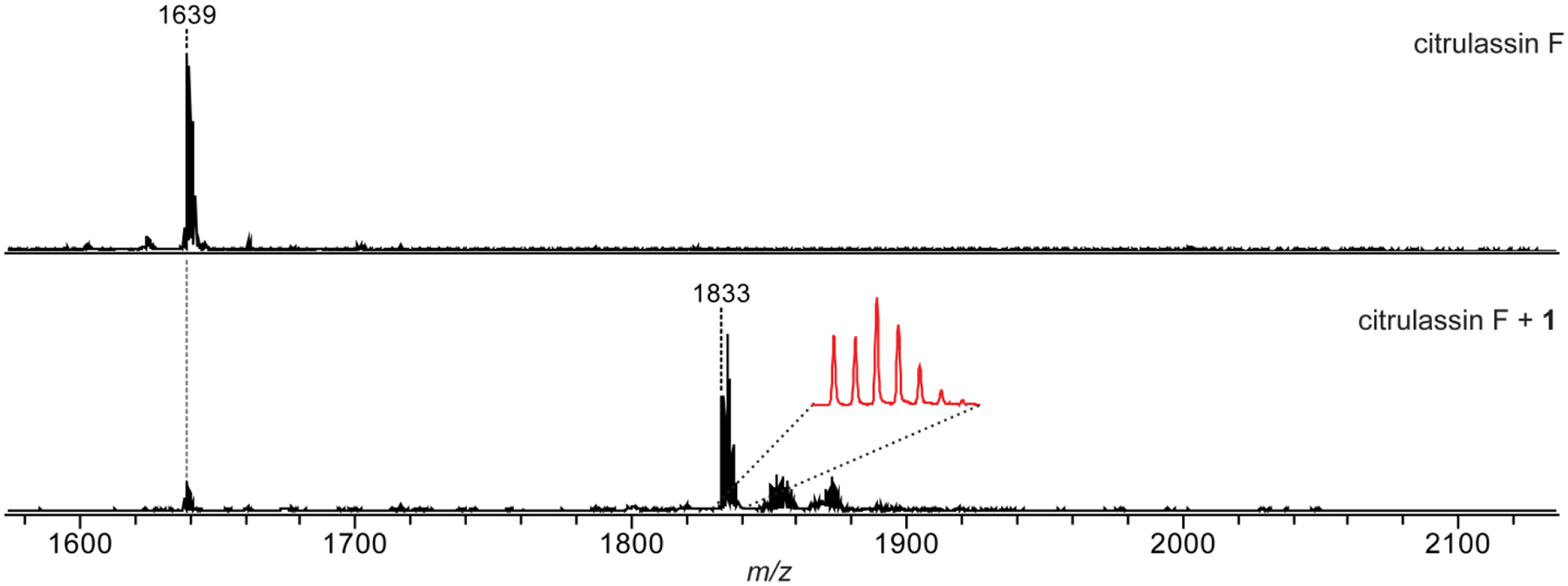
Phenylglyoxal reaction on citrulassin F (LLGRSGNDCitLILSKN). MALDI-TOF mass spectra of a methanolic extract of Streptomyces torulosus S-189 (citrulassin F producer) prior to the addition of compound 1 (top) and after reaction with 1 (bottom). Magnified inset shows the 79Br/81Br isotopic pattern. Note only one labeling event occurs, consistent with only the citrulline being modified.
Adapted from Harris, L. A., Saint-Vincent, P. M. B., Guo, X., Hudson, G. A., DiCaprio, A. J., Zhu, L., & Mitchell, D. A. (2020). Reactivity-Based Screening for Citrulline-Containing Natural Products Reveals a Family of Bacterial Peptidyl Arginine Deiminases. ACS Chemical Biology, 15(12), 3167–3175.
Figure 4. Tetrazine Overview.
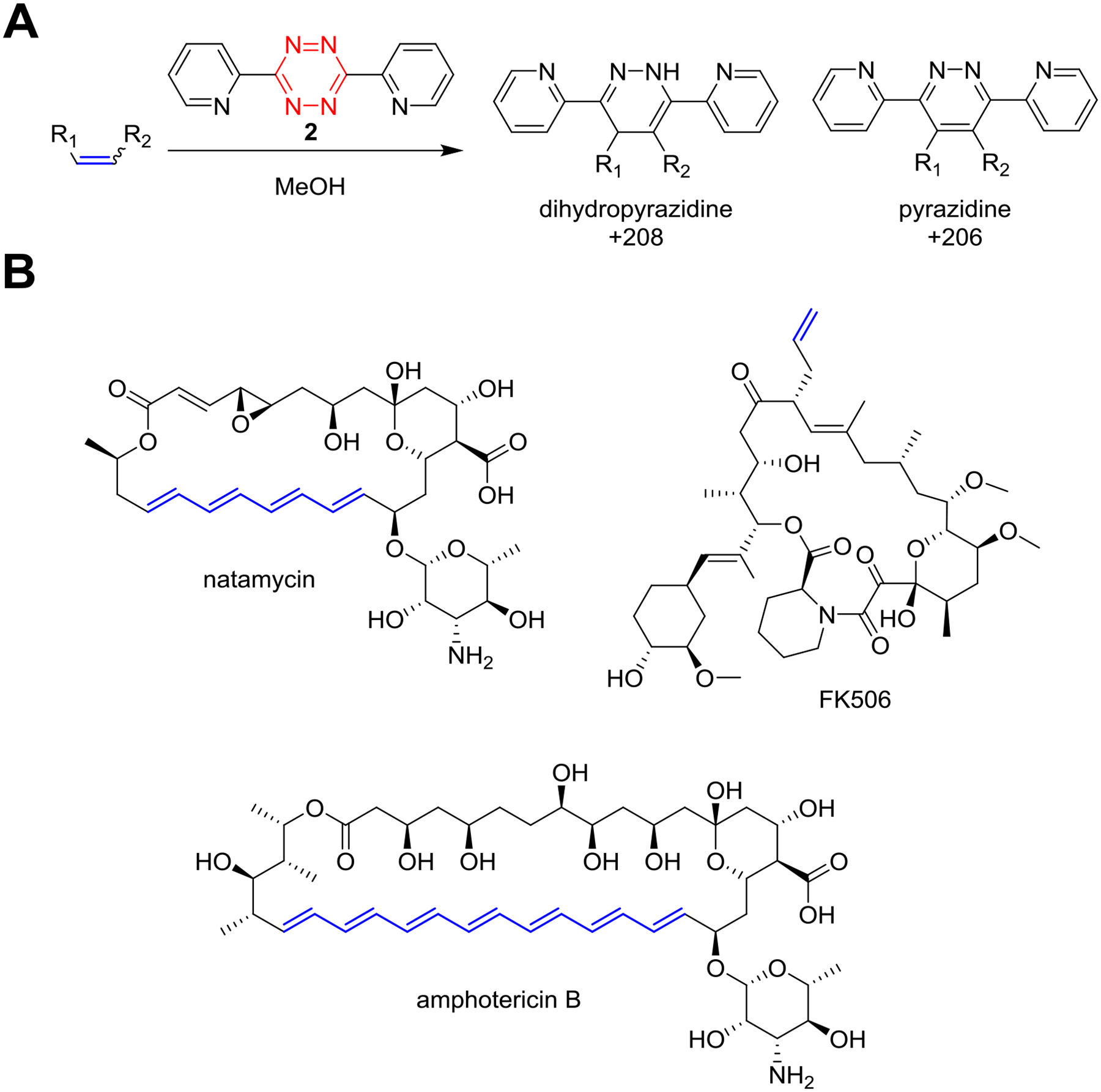
Overview of tetrazine probe chemistry. (A) General reaction conditions for labeling electron-rich olefins using 3,6-di-2-pyridyl-1,2,4,5-tetrazine (compound 2) in methanolic bacterial extract. (B) Representative NP substrates that are reactive towards 2 with the sites of modification indicated (red).
Figure 5. Tetrazine Reaction on Natamycin.
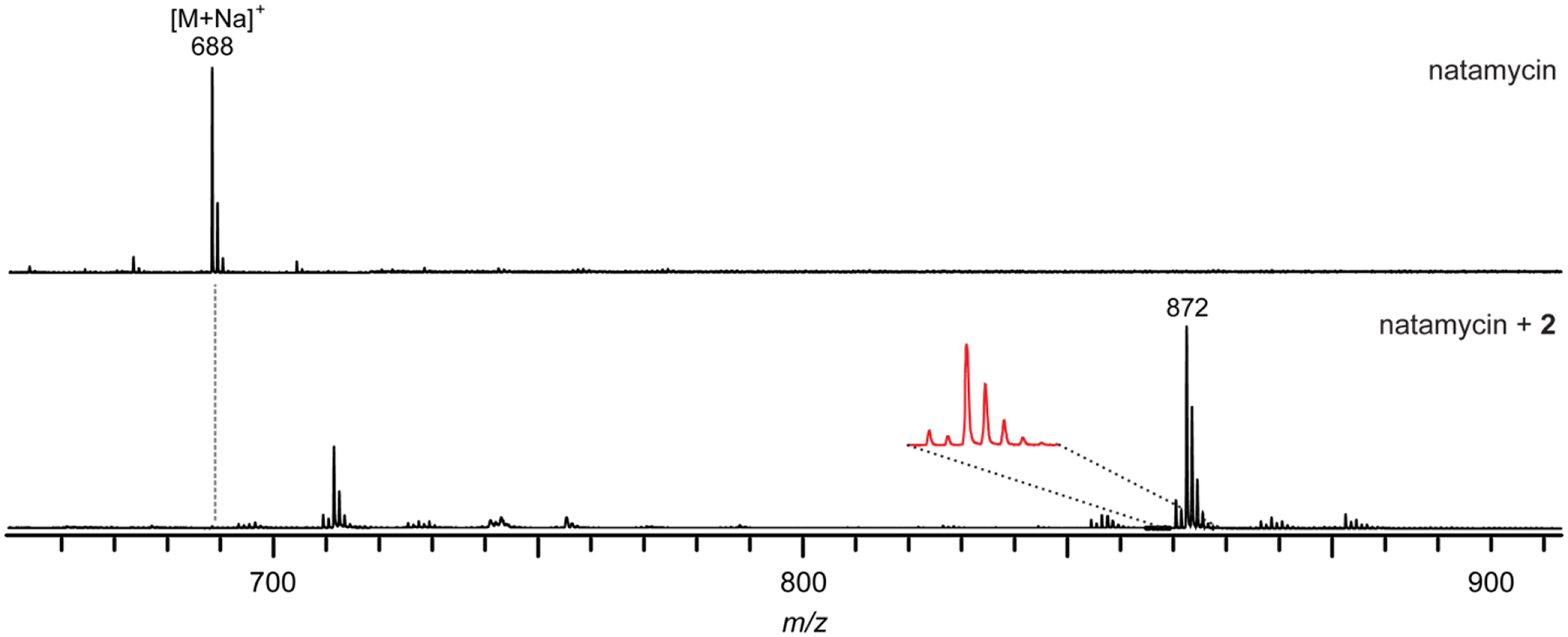
Tetrazine ligation reaction on natamycin. MALDI-TOF mass spectra of a methanolic extract of Streptomyces natalensis ISP-5314 (natamycin producer) prior to the addition of compound 2 (top) and after reaction with 2 (bottom). Note the distinct mass pattern due to the presence of dihydropyrazidine (+208) and pyrazidine (+206) adducts.
Adapted from Guo, X., Zhang, J., Li, X., Xiao, E., Lange, J. D., Rienstra, C. M., Burke, M. D., & Mitchell, D. A. (2021). Sterol Sponge Mechanism Is Conserved for Glycosylated Polyene Macrolides. ACS Central Science, 7(5), 781–791.
Figure 6. DTT Overview.
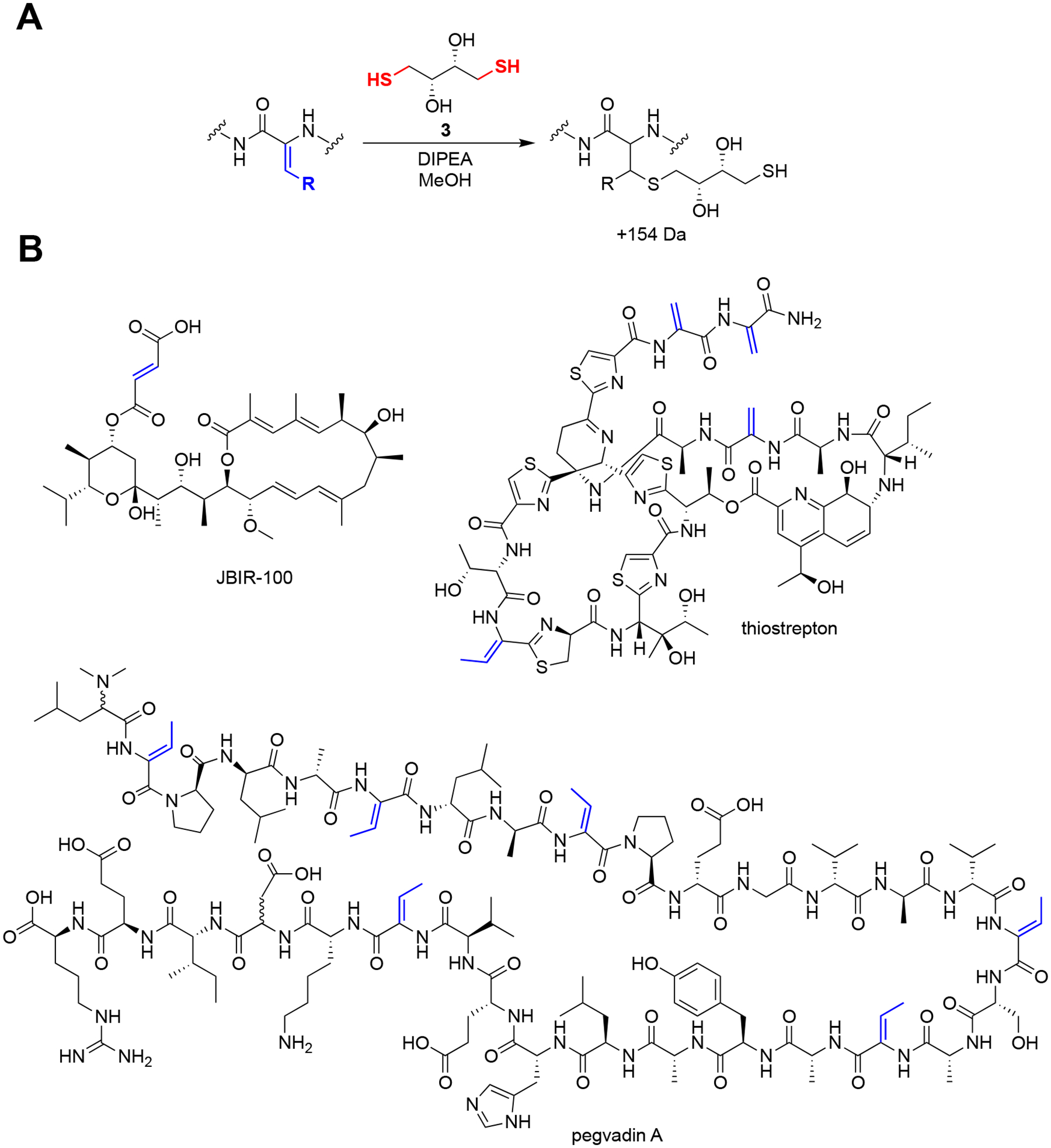
Overview of dithiothreitol probe chemistry. (A) General reaction conditions for labeling Michael acceptors with dithiothreitol (compound 3) in methanolic bacterial extract under basic conditions. (B) Representative NP substrates that are reactive to compound 3 with the sites of modification indicated (red). Note that dehydrobutyrines typically do not completely label under these conditions, likely due to steric hindrance.
Figure 7. Dithiothreitol Reaction on Pegvadin A.
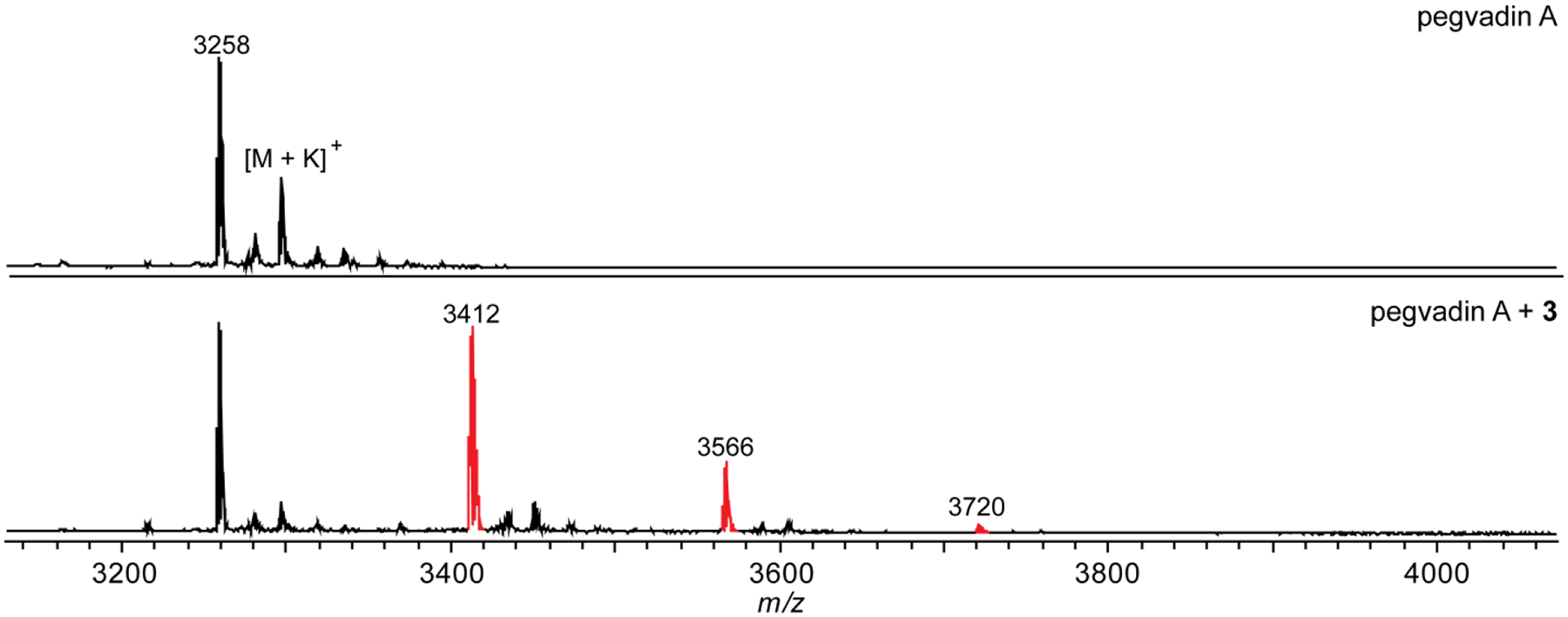
Dithiothreitol reaction on pegvadin A (see structure above). MALDI-TOF mass spectra of a methanolic extract of Streptomyces noursei NRRL B-1714 (pegvadin A producer) prior to the addition of compound 3 (top) and after reaction with 3 (bottom). Three labeling events out of six possible were evident (indicated in red) due to a reduced reactivity of dehydrobutyrine compared to dehydroalanine. In certain cases, extent of labeling can allow for the differentiation of the two electrophiles.
Adapted from Georgiou, M. A., Dommaraju, S. R., Guo, X., Mast, D. H., & Mitchell, D. A. (2020). Bioinformatic and Reactivity-Based Discovery of Linaridins. ACS Chemical Biology, 15(11), 2976–2985.
Figure 8. Aminooxy Overview.
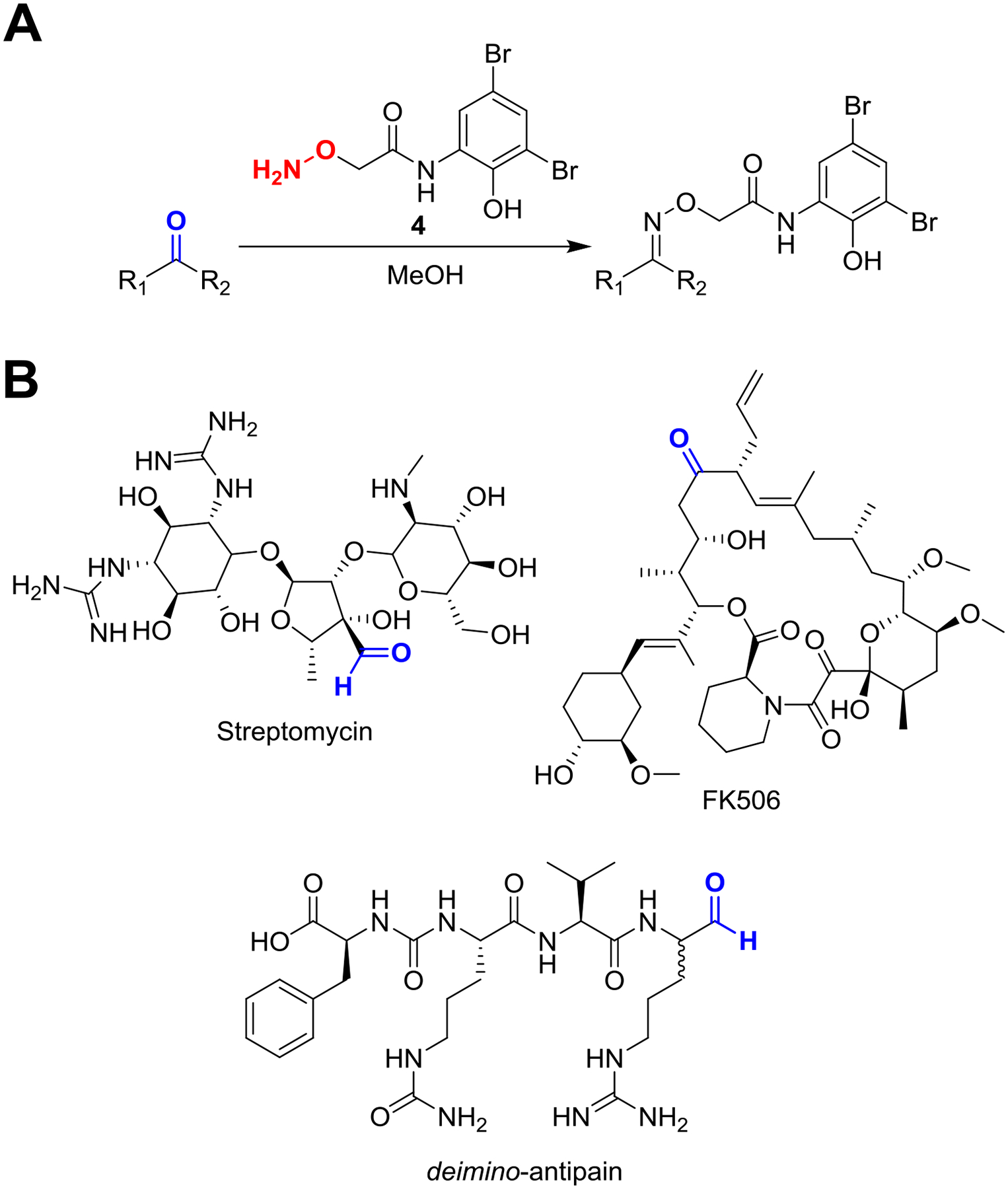
Overview of aminooxy probe chemistry. (A) General reaction conditions for labeling reactive carbonyls using the aminooxy probe (compound 4) in methanolic bacterial extract. (B) Representative NP substrates that are reactive to compound 4 and the site of modification indicated (red). Note that ketones typically react to a lesser extent than aldehydes, and the sterically occluded ketone of FK506 was not labeled.
Figure 9. Aminooxy Reaction on Streptomycin.
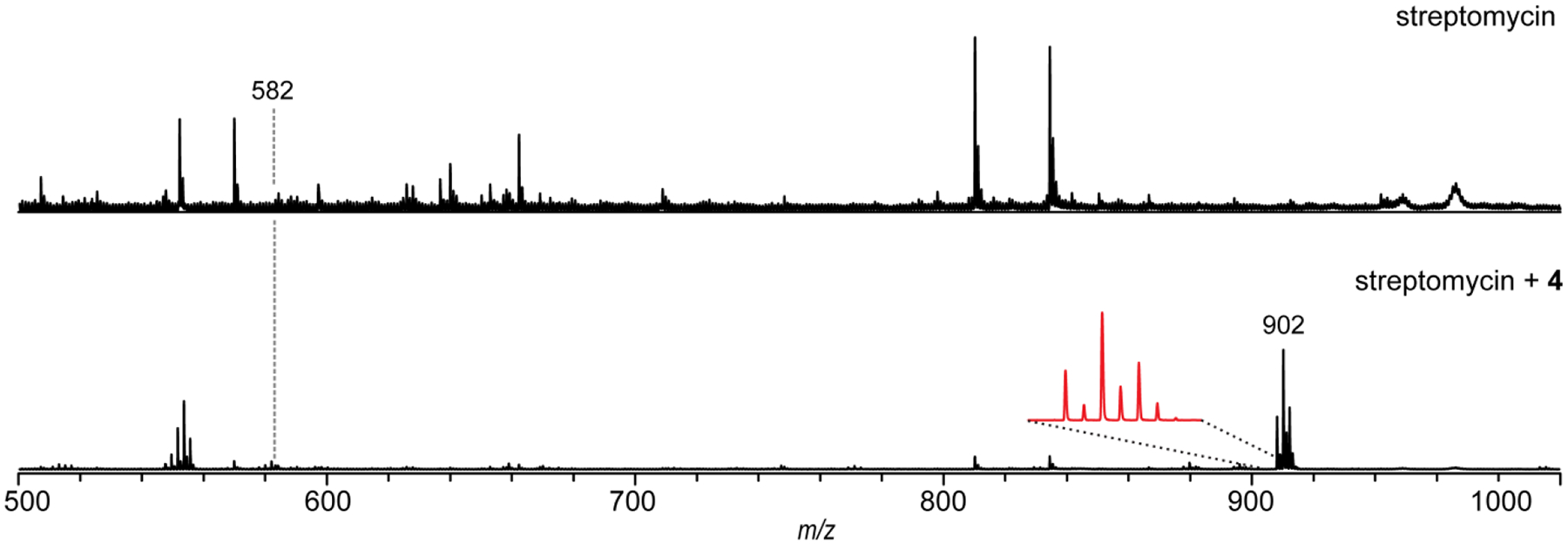
Aminooxy reaction on streptomycin. MALDI-TOF mass spectra of a methanolic extract of Streptomyces griseus WC-3480 (streptomycin producer) prior to the addition of compound 4 (top) and after reaction with 4 (bottom). Magnified inset shows the 79Br/81Br dibrominated isotopic pattern. Note the large signal enhancement of the labeled adduct compared to the parent compound due to label-assisted ionization.
Adapted from Maxson, T., Tietz, J. I., Hudson, G. A., Guo, X. R., Tai, H.-C., & Mitchell, D. A. (2016). Targeting Reactive Carbonyls for Identifying Natural Products and Their Biosynthetic Origins. Journal of the American Chemical Society, 138(46), 15157–15166.
4.1. Materials, Reagents and Equipment
Equipment:
New Brunswick Scientific i2500 series incubator shaker set to 37 °C
Speed vacuum concentrator (Thermo Scientific)
Mettler Toledo XA105 DualRange balance
Fisher Scientific vortex mixer
Bruker Daltonics UltrafleXtreme mass spectrometer
Computer with FlexAnalysis software installed (Bruker)
Materials and Reagents
1.5 mL Eppendorf tubes
Reagent grade methanol (VWR)
3-bromophenylglyoxal hydrate (1) (Ark-Pharm)
3,6-di-2-pyridyl-1,2,4,5-tetrazine (2) (Sigma)
Trifluoroacetic acid (TFA, Sigma)
Dithiothreitol (3) (GoldBio)
Diisopropyl ethyl amine (Fisher)
2-(aminooxy)-N-(3,5-dibromo-2-hydroxyphenyl)acetamide (4) (aminooxy-functionalized probe, chemically synthesized) (Maxson et al., 2016)
α-cyano-4-hydroxycinnamic acid (CHCA, Sigma)
HPLC grade acetonitrile (Fisher)
Milli-Q water
ProteoMass Peptide MALDI-MS Calibration kit (Millipore Sigma, Cat. No. MSCAL1-1KT)
4.2. Phenylglyoxal Probe Procedure
Timing: 3–4 h
Dissolve 3-bromophenylglyoxal hydrate (compound 1, 4.6 mg/mL, 20 mM) and TFA (2% v/v) in methanol. Vortex to dissolve.
Add 10 μL of previously prepared bacterial extract and 10 μL stock solution to a 1.5 mL Eppendorf tube. Pipette up and down to mix thoroughly.
Incubate in a 37 °C incubator for 1 h.
Evaporate reaction mixture to dryness using a speed vacuum concentrator, typically 1 h.
Reconstitute dried reaction mixture in 10 μL methanol.
Deposit 1 μL of the reaction mixture onto a stainless steel MALDI target plate, then 1 μL 50% MeCN/water saturated with CHCA and mix on the plate via repeated pipetting. A small fan can be used to aid in crystallization. Similar results can be obtained by placing the plate in a chemical fume hood and leaving the sash partially open.
Deposit a second sample as above with unreacted bacterial extract.
Analyze samples by MALDI-TOF-MS using a Bruker Daltonics UltrafleXtreme instrument in reflector positive mode. Our published MALDI-TOF analysis was conducted at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory, and the instrument was calibrated using the MALDI calibration kit prior to data acquisition.
Analyze the data using the Bruker FlexAnalysis software. Data similar to the example reaction on citrulassin F shown below (LLGRSGNDCitLILSKN) can be obtained (Fig. 3). Compare the reacted and unreacted spectra for new peaks that are offset from a parent peak by +194 Da. The distinctive 1:1 79Br/81Br isotope pattern will aid in finding true probe adducts.
4.3. Tetrazine Probe Procedure
Timing 13–14 h
Dissolve 3,6-di-2-pyridyl-1,2,4,5-tetrazine (compound 2, 2.4 mg/mL, 10 mM) in methanol. Vortex to dissolve.
Add 5 μL of previously prepared bacterial extract to a 1.5 mL Eppendorf tube then add 20 μL stock solution. Pipette up and down to mix thoroughly.
Incubate at room temperature for 12 h.
Deposit 1 μL of the reaction mixture onto a stainless steel MALDI target plate, then 1 μL 50% MeCN/water saturated with CHCA and mix on the plate via repeated pipetting. A small fan can be used to aid in crystallization. Similar results can be obtained by placing the plate in a chemical fume hood and leaving the sash partially open.
Deposit a second sample as above with unreacted bacterial extract.
Analyze samples by MALDI-TOF-MS using a Bruker Daltonics UltrafleXtreme instrument in reflector positive mode. Our published MALDI-TOF analysis was conducted at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory, and the instrument was calibrated using the MALDI calibration kit prior to data acquisition.
Analyze the data using the Bruker FlexAnalysis software. Data similar to the example reaction on natamycin shown below can be obtained (Fig. 5). Compare the reacted and unreacted spectra for new peaks that are offset from a parent peak by 206/208 Da. The mixture of pyridazine and dihydropyridazine adducts leads to a mass pattern that aids in identification.
4.4. Dithiothreitol Probe Procedure:
Timing: 17–18 h
Dissolve dithiothreitol (compound 3, 154 mg/mL, 1 M) and diisopropylethylamine (2.5 μL/mL, 20 mM) in methanol. Vortex to dissolve.
Add 10 μL of previously prepared bacterial extract to a 1.5 mL Eppendorf tube then add 10 μL stock solution. Pipette up and down to mix thoroughly.
Equilibrate at room temperature for 16 h.
Deposit 1 μL of the reaction mixture onto a stainless steel MALDI target plate, then 1 μL 50% MeCN/water saturated with CHCA and mix on the plate via repeated pipetting. A small fan can be used to aid in crystallization. Similar results can be obtained by placing the plate in a chemical fume hood and leaving the sash partially open.
Deposit a second sample as above with unreacted bacterial extract.
Analyze samples by MALDI-TOF-MS using a Bruker UltrafleXtreme instrument in reflector positive mode. Our published MALDI-TOF analysis was conducted at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory, and the instrument was calibrated using the MALDI calibration kit prior to data acquisition.
Analyze the data using the Bruker FlexAnalysis software. Data similar to the example reaction on pegvadin A can be obtained (Fig. 7). Compare the reacted and unreacted spectra for new peaks that are offset from a parent peak by 154 Da.
4.5. Aminooxy Probe Procedure:
Timing: 4–5 h
Dissolve 2-(aminooxy)-N-(3,5-dibromo-2-hydroxyphenyl)acetamide (compound 4, 3.4 mg/mL, 10 mM) in methanol. Vortex to dissolve.
Add 18 μL of previously prepared bacterial extract to a 1.5 mL Eppendorf tube then add 2 μL stock solution. Pipette up and down to mix thoroughly.
Incubate at room temperature for 3 h.
Deposit 1 μL of the reaction mixture onto a stainless steel MALDI target plate, then 1 μL 50% MeCN/water saturated with CHCA and mix on the plate via repeated pipetting. A small fan can be used to aid in crystallization. Similar results can be obtained by placing the plate in a chemical fume hood and leaving the sash partially open.
Deposit a second sample as above with unreacted bacterial extract.
Analyze samples by MALDI-TOF-MS using a Bruker UltrafleXtreme instrument in reflector positive mode. Our published MALDI-TOF analysis was conducted at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory, and the instrument was calibrated using the MALDI calibration kit prior to data acquisition.
Analyze the data using the Bruker FlexAnalysis software. Data similar to the example reaction on streptomycin can be obtained (Fig. 7). Compare the reacted and unreacted spectra for new peaks that are offset from a parent peak by 320 Da. The distinct isotope distribution of 79Br:81Br aid in identifying true labeling events.
5. RBS for Citrulline Identifies a Novel Peptidyl-Arginine Deiminase
While RBS has typically been leveraged to discover novel NPs, a recently published example uses probe chemistry to interrogate protein function. Previous work in our lab had discovered the novel lasso peptide citrulassin A, so named because of a citrulline residue in the isolated NP (Tietz et al., 2017). Lasso peptides are a class of ribosomally synthesized and post-translationally modified peptides (RiPP), meaning that all amino acids are proteinogenic in origin. Proteinaceous citrulline forms in eukaryotes through the deimination of the guanidino group of arginine by a peptidyl arginine deiminase (PAD), with the best-known substrates being histone tails. However, no bacterial PAD substrate had been characterized. Sequencing of the native producer of citrulassin A (Streptomyces albus NRRL B-3066) revealed no genes with predicted PAD activity within 20 kb of the citrulassin A BGC. Moreover, heterologous expression of the BGC and flanking regions in Streptomyces lividans led to the production of des-citrulassin A (contains arginine, not citrulline). Thus, it was hypothesized that a putative PAD was distally encoded, and identification would require an alternative approach (Fig. 10). To discriminate between Arg and Cit containing peptides, we deployed previously developed phenylglyoxal chemistry (Bicker et al., 2012) to probe the deimination state of the citrulassin group of lasso peptides. The following section details the bioinformatic and RBS methods we used to find additional members of the citrulassin group. This workflow may be useful to others in the field, as it demonstrates that RBS can be employed beyond NP discovery to assign new enzyme functions. In addition, the general workflow of confirming the initial hit through high resolution tandem mass spectrometry (HR-MS/MS) and purification of larger amounts of compound are generally useful in any NP discovery pipeline. Further experiments were conducted to confirm the role of PAD in citrulassin deimination but are beyond the scope of this chapter.
Figure 10. Strategy to Identify Citrulline-forming PAD.
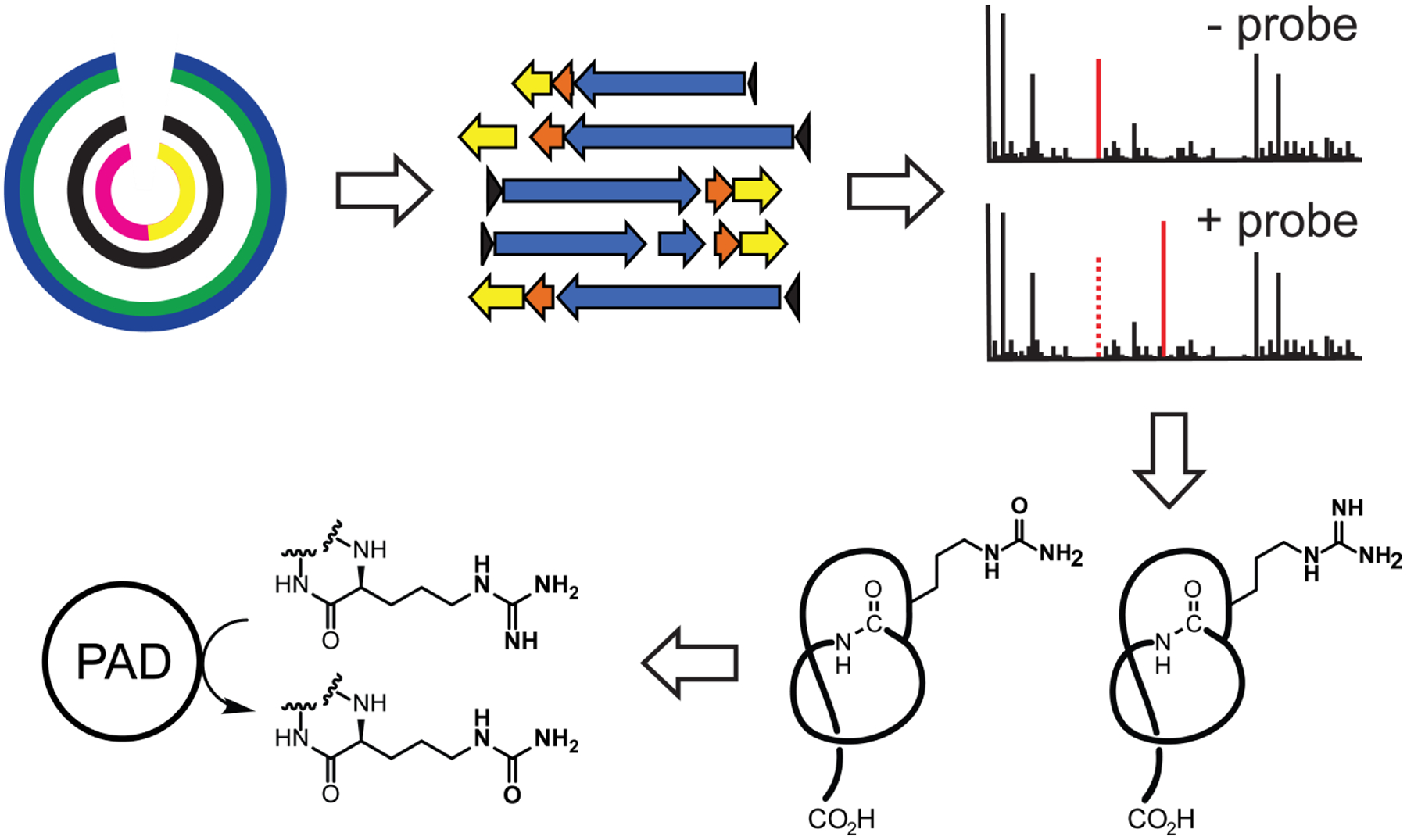
Strategy to identify citrulline-forming PAD. Genomes encoding citrulassin BGCs are first bioinformatically identified. Selected strains are then cultured and the presence of peptidic citrulline was monitored by reacting cellular extracts with compound 1. After a sufficient number of cases where the Cit vs. Arg phenotype was determined, partial phylogenetic profiling was then used to populate a list of candidate gene(s) correlating with citrulassin deimination.
Adapted from Harris, L. A., Saint-Vincent, P. M. B., Guo, X., Hudson, G. A., DiCaprio, A. J., Zhu, L., & Mitchell, D. A. (2020). Reactivity-Based Screening for Citrulline-Containing Natural Products Reveals a Family of Bacterial Peptidyl Arginine Deiminases. ACS Chemical Biology, 15(12), 3167–3175.
5.1. Bioinformatic Prioritization of Putative Citrulassin BGCs
To determine the PAD responsible for deimination of citrulassin congeners, the deimination phenotype needed to be determined for enough citrulassin producing organisms for comparative genomics to have sufficient resolving power. To do this, a list of putative citrulassin producers was generated via BLAST (Altschul et al., 1990) and precursors identified using the online webtool RODEO (www.ripprodeo.org/). In this section, the bioinformatics tools used to discover putative citrulassin producers will be detailed. Prioritized organisms were then grown, extracted and screened by RBS (see sections 3 and 4, respectively)
Timing: 2–3 h for bioinformatics only, additional analysis time may be required for larger datasets.
Perform a BLAST search using a protein of interest as a query, in our case WP_079136914.1, the Streptomyces albulus citrulassin A cyclase. Export the resulting protein accessions and save the protein list as a text file. Each protein accession ID must be separated by a line break.
Use the text file to initiate a RODEO run, selecting the lasso peptide module to perform lasso precursor prediction. For the approximately 250 proteins, RODEO will finish in around 1 h.
Open the results comma-separated values (csv) file, and manually examine the predicted precursors. Peptides were compared to the consensus motif: LLxxxxNDRLxxSKN (x=any proteinogenic amino acid).
Cross reference the protein accession identifier with the genome of the harboring organism. Include organisms with the same specific name even if there is no published genome, as they often harbor similar BGCs.
Note: In this case, a BLAST search that returned 250 proteins was sufficient to cover all putative citrulassins. For larger and more sequence diverse proteins, a position specific iterative (PSI)-BLAST (Altschul et al., 1997) may be more appropriate. For novel precursors, a sequence similarity network can be generated to determine groups with high similarity. See Tietz, et al. 2017 for more information on the initial bioinformatic discovery of the citrulassin lasso peptides.
5.2. Isolation and Initial Purification of Citrulassins
To further characterize members of the citrulassin family, larger scale isolation and purification from the native producers was undertaken. For some NPs, drying extract from a single plate and desalting using a C18 ziptip (Millipore Sigma) is sufficient for obtaining high quality HR-MS/MS data. In the case of the citrulassins, higher purities were required to obtain quality spectra. In this section, the methanolic extraction, solid phase extraction (SPE) and ion exchange chromatography of citrulassins is detailed.
5.21. Materials, Reagents and Equipment
Equipment:
Bruker Daltonics UltrafleXtreme mass spectrometer
−20 °C Freezer
Buchi (Switzerland) Rotavapor R-210
Buchi (Switzerland) heating bath B-491
Welch self-cleaning dry vacuum system model 2027
Thermo Scientific MaxQ 4450 set to 120 rpm
Computer with FlexAnalysis software
Materials:
Streptomyces strain of interest grown on 0.5 L solid media
2 L plastic buckets with lid
Disposable razor blade
Reagent grade methanol (VWR)
Spatula/paint scraper
Cotton
Powder funnel
CHCA (Sigma)
500 mL Ehrlenmeyer flask
5.22. Initial Isolation of Streptomyces Strains
Timing: 24–32 h (isolation only)
Grow the Streptomyces of interest on 0.5 L of solid production media as detailed in section 3. With 15 cm plates, 0.5 L is around 20 plates.
Use a disposable razor blade to cut the agar into approx. 0.5–1 cm squares and transfer the cubes into the 2 L bucket using a spatula.
Freeze cubed agar overnight at −20 °C. In some cases, over 24 h is necessary to completely freeze.
Thaw the agar in a heating bath (Buchi) set to 40 °C. Remove from water bath after thawing but before the resulting contents have warmed to 40 °C to prevent the resulting liquid from reabsorbing into the agar.
Decant the expressed liquid and manually squeeze the agar to remove the remainder. Gravity filter the resulting liquid through a funnel loosely packed with cotton.
Add 0.5 L of methanol to the remaining agar and nutate at 120 rpm for 2 h at room temperature.
Repeat steps 5 for the methanolic extract, but keep separate from the initial aqueous agar squeeze.
Remove the methanol from the extract using a rotary evaporator and combine with the aqueous extract.
Note: some researchers first scrape the cell mass from the agar and extract separately. This step was determined to be unnecessary for citrulassin and thus the steps are combined here. The agar can also be extracted twice, but one extraction was found to be sufficient. After step 8, the combined extract can be frozen for storage for a few days to weeks.
5.23. Materials, Reagents and Equipment
Equipment:
Sorvall RC6 Plus floor centrifuge, Rotor SLA-3000
C18 50 g Solid Phase Extraction column (Fisher, catalog # 3251292)
Rubber filter adapter
500 mL distillation bulb
Two utility clamps
Ground glass gas inlet-male adapter
Bruker Daltonics UltrafleXtreme mass spectrometer
Speed vacuum concentrator (Thermo Scientific)
Buchi (Switzerland) Rotavapor R-210
Buchi (Switzerland) heating bath B-491
Welch self-cleaning dry vacuum system model 2027
250 mL round bottom flask
Teledyne Isco Combiflash EZprep
Teledyne Isco 50 g strong cation exchange column
Materials and Reagents
HPLC grade acetonitrile (Fisher)
Milli-Q water
Whatman #1 filter paper
Scintillation vial w/ caps
TFA (Sigma)
Calcium chloride hexahydrate (Sigma)
CHCA (Sigma)
5.24. Solid Phase Extraction (SPE) and Ion-Exchange Purification of Citrulassins
Timing: 36–48 h
Dilute the combined agar squeeze/ methanolic extract previously prepared with acetonitrile sufficient to make a 5% (v/v) acetonitrile solution.
Use a high speed centrifuge to clarify the solution (30 min at 12,000 rpm is usually sufficient).
Decant solution and vacuum filter through Whatman #1 paper to remove any pellet disrupted by decanting.
Prime a new SPE C18 column with 50 mL 100% acetonitrile, 100 mL 50% acetonitrile/water, and 50 mL water.
Load the SPE column with the clarified extract, using a 500 mL distillation bulb, rubber filter adapter, and clamps to create a loading reservoir above the stationary phase. Pressurized air is usually used to load more quickly, but must be light to prevent separation of the SPE cartridge and the distillation bulb.
Desalt using 50 mL water, then elute using 50 mL each of 10, 20, 30, 40, 50, and 100% acetonitrile/water.
Spot and analyze each fraction by MALDI as detailed in section 4.
Combine fractions containing the citrulassin of interest in a 250 mL round bottom flask and dry to approx. 10 mL using a rotary evaporator.
Transfer solution to a scintillation vial, and dry overnight using a speed vacuum concentrator.
Reconstitute the crude material in 80% acetonitrile/water with 0.1% TFA. One half mL was typically sufficient.
Load onto a RediSep Rf strong cation exchange (SCX) column, washed with 2 column volumes of 100% A, and eluted using a 0–100% gradient from A to B, where A is 80% acetonitrile water w/ 0.1% TFA, and B is A supplemented with 0.5 M calcium chloride.
Spot and analyze each fraction by MALDI as detailed in section 4.
Pool fractions containing citrulassin and dry overnight using a speed vacuum concentrator.
Note: To improve separation, 0.1% (v/v) formic acid or TFA as an acidic buffer and 10 mM ammonium bicarbonate as a basic buffer are sometimes added to the loaded material, priming solutions and eluent for SPE. In this case however, it was found that neat water was optimal. For some analytes, the high concentration of calcium chloride can suppress the intensity of MALDI-MS signals. In these cases, desalting of samples is required.
5.3. Citrulassin Final Purification and HR-MS/MS
To confirm the bioinformatically predicted structures and use an orthogonal method to evaluate the Cit/Arg phenotype, high resolution tandem MS was performed on all newly discovered citrulassins. In several cases multiple arginines were present in the precursor peptide, only a subset of which was modified in the detected NP. Which residues were modified was of interest as a way to probe the selectivity of the putative PAD. In the case of an unbiased screen where structural information beyond the moiety probed by RBS is unknown, HR-MS/MS is critical for structural elucidation and, if necessary, more accurate dereplication through NP databases. In this section, the final HPLC purification and HR-MS/MS analysis of citrulassin is detailed.
5.31. Materials, Reagents and Equipment
Equipment
PerkinElmer Flexar HPLC
Betasil C18 (Thermo Scientific) reverse phase column (250 × 4.6 mm, 5 μm particle size, 100 Å pore size).
Speed vacuum concentrator (Thermo Scientific)
ThermoFisher Orbitrap Fusion Electrospray ionization mass spectrometer (ESI-MS)
Advion TriVersa NanoMate
Computer with Xcalibur software (Thermo Fisher) installed
Materials and Reagents
HPLC grade acetonitrile (Fisher)
Milli-Q water
LC-MS grade ammonium bicarbonate (Fisher)
LC-MS grade acetonitrile (Fisher)
HPLC grade glacial acetic acid (VWR)
Pierce LTQ Velos ESI Positive Ion Calibration Solution (ThermoFisher)
5.32. Citrulassin Purification and HR-MS/MS Procedure
Timing: 6–24 h
Reconstitute the semi-pure citrulassin in 0.4 mL 5% acetonitrile/water with 10 mM ammonium bicarbonate. Remove undissolved calcium chloride by centrifugation at 13,000 × g for 2 min. Transfer the clarified solution to a 1.5 mL Eppendorf tube, diluted with 0.1 mL more of the solvent detailed previously to avert LC system clogging by precipitating CaCl2.
Purify the sample using a Flexar HPLC equipped with a Betasil C18 reverse phase column with a mobile phase of 10 mM aq. NH4HCO3/acetonitrile at a flow rate of 1 mL/min with a method of: 5% acetonitrile (isocratic, 5 min), 5–50% acetonitrile (gradient, 30 min), 50% acetonitrile (isocratic, 5 min) 50–100% acetonitrile (gradient, 5 min), 100% acetonitrile (isocratic, 5 min).
Spot and analyze each fraction by MALDI as detailed in section 4.
Pool fractions that contain the citrulassin of interest and dry overnight using a speed vacuum concentrator.
Re-dissolve the dried reaction in 80% (v/v) aq. acetonitrile and 1% (v/v) acetic acid immediately prior to HR-MS analysis. If material does not completely dissolve, clarify by centrifugation at 13,000 × g for 5 min.
Infuse the samples using a TriVersa Nanomate 100 into a Thermo Fisher Scientific Orbitrap Fusion ESI-MS.
Prior to data acquisition, the mass spectrometer should be calibrated and tuned routinely. Our analysis was calibrated on the Pierce LTQ Velos ESI Positive Ion Calibration Solution.
HR-MS and MS/MS data can be obtained using the following parameters: resolution: 100,000; isolation width (MS/MS): 1 m/z; normalized collision energy (MS/MS): 35–70; activation q value (MS/MS): 0.4, activation time (MS/MS): 30 ms. Fragmentation can be performed using collision-induced dissociation (CID) at 35% or 70%.
Analyze data using the Qualbrowser application of Xcalibur software (Thermo Fisher Scientific). Data can be averaged across the time dimension prior to analysis. An example ESI-HR-MS/MS For the citrulassin F is shown below, with the site of deimination localized to Arg9 in the sequence (Figure 11). A CID fragmentation corresponding to a [Citrulline→Ornithine] was observed, and was diagnostic for the presence of citrulline in the peptides analyzed.
Figure 11. HR-MS/MS figure of Citrulassin F.
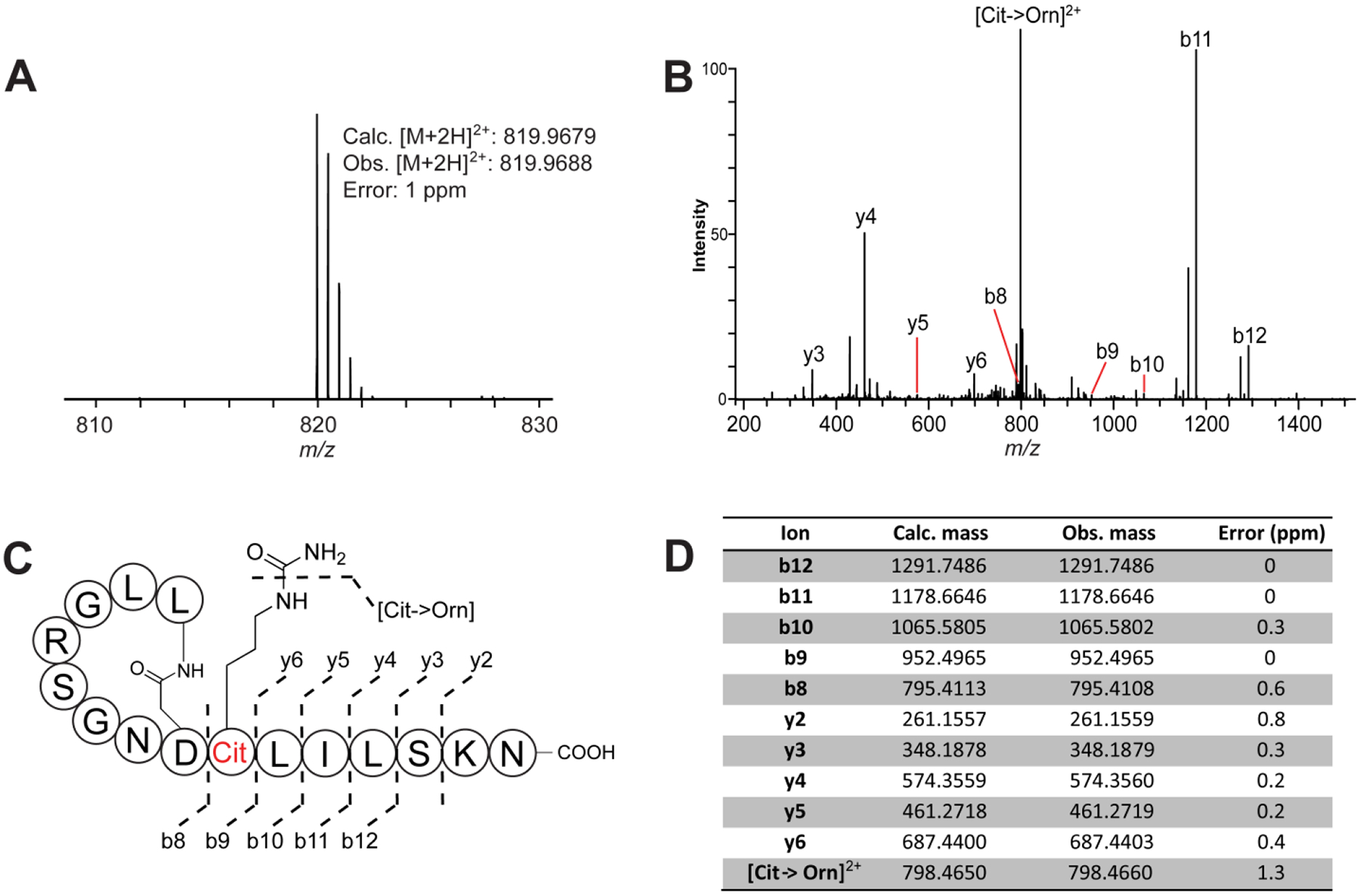
Peptide sequence confirmation and citrulline localization of citrulassin F by HR-ESI-MS/MS. (A) HR-ESI-MS of citrulassin F. The reported error is based on the doubly protonated species and the calculated molecular formula of C70H123N23O22. (B) HR-ESI-MS/MS spectrum of citrulassin F, with the observable b and y ions labeled. (C) Schematized lasso peptide diagram with identified fragment ions (shown unthreaded to aid interpretation). (D) Table of assigned daughter ions. Cit, citrulline; Orn, ornithine.
Adapted from Harris, L. A., Saint-Vincent, P. M. B., Guo, X., Hudson, G. A., DiCaprio, A. J., Zhu, L., & Mitchell, D. A. (2020). Reactivity-Based Screening for Citrulline-Containing Natural Products Reveals a Family of Bacterial Peptidyl Arginine Deiminases. ACS Chemical Biology, 15(12), 3167–3175.
5.4. Phylogenetic Profiling to Identify the Citrulassin-modifying PAD
With the arginine/citrulline phenotype determined for a sufficient number of sequenced organisms, we next sought to determine the gene(s) responsible for the post-translational deimination of arginine to citrulline. Though several phylogenetic profiling tools have been developed, we decided to use MicroScope phyloprofile tool, as it allows users to upload genomes of interest for annotation and use. The only major disadvantages of this tool is that in-house annotation takes 4–8 weeks and a single user can only submit 10 genomes at a time. The following section gives some details on how to use the phyloprofile tool (Vallenet et al., 2013), a more in depth tutorial can be found on the MicroScope webpage: (https://microscope.readthedocs.io/en/3.15.2/content/compgenomics/phyloprofil.html).
Timing: 4–8 weeks if desired genomes are not present in database; otherwise, 30 min. or less.
If desired genomes are not already annotated in the MicroScope database, download the requisite genomes from NCBI to a local hard drive (if the genome is not closed, download the WGS project).
Submit genomes to MicroScope pipeline. Annotation typically requires 4–8 weeks.
Use the phyloprofile tool to determine genes in S. albulus NRRL B-3066 with homologs in the genomes of citrulassin producers (Streptomyces glaucescens NRRL B-11408, Streptomyces torulosus S-189, Streptomyces sp. NRRL S-118, and Streptomyces pharetrae NRRL B-24333) but not in des-citrulassin producers (Streptomyces avermitilis NRRL B-16169, Streptomyces tricolor B-16925, Streptomyces katrae B-16271, and Streptomyces auratus NRRL 8097).
The algorithm identified 30 candidate genes (out of ~6,800 total genes) correlating with a citrulassin vs. des-citrulassin phenotype. Among the 30 candidates was a putative PAD. Additional experiments outside the scope of this chapter were conducted that confirmed the predicted function.
6. Summary
In this chapter, we have detailed the protocols for screening bacterial organisms via RBS, including considerations for organism selection, growth and extraction of these organisms, treating these extracts with phenylglyoxal, tetrazine, dithiothreitol, and aminooxy probes, and analyzing these reactions by MALDI-TOF-MS. In addition, a case study has been presented that utilizes RBS beyond its initially developed purpose of NP discovery to tie the deimination of citrulassin lasso peptides to a previously uncharacterized PAD. Protocols for the bioinformatic prioritization of strains based on the presence of a citrulassin BGC, larger scale isolation and purification of citrulassins by SPE, ion exchange chromatography and HPLC, confirmation of arginine/citrulline phenotype by HR-MS/MS, and identification of the responsible PAD by the MicroScope phylogenetic profiling tool were also detailed. These methods may be useful to researchers who wish to probe the functional groups targeted by the probes outlined here, and the general workflow can be easily adapted for those wishing to target other chemical functional groups present in natural products. Moreover, RBS can have a wider application beyond NP discovery, and could be used to interrogate protein function and other fundamental biological questions.
References
- Agard NJ, Prescher JA, & Bertozzi CR (2004). A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. Journal of the American Chemical Society, 126(46), 15046–15047. 10.1021/ja044996f [DOI] [PubMed] [Google Scholar]
- Ahmad M, & Khan AU (2019). Global economic impact of antibiotic resistance: A review. Journal of Global Antimicrobial Resistance, 19, 313–316. 10.1016/j.jgar.2019.05.024 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, & Lipman DJ (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, & Lipman DJ (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Research, 25(17), 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back D, Shaffer B, Loper J, & Philmus B (2021). Untargeted identification of alkyne containing natural products using ruthenium catalyzed azide alkyne cycloaddition reactions coupled to LC-MS/MS. 10.33774/chemrxiv-2021-hwwr6-v3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz RH (2005). Antibiotic discovery from actinomycetes: Will a renaissance follow the decline and fall? SIM News, 55(5), 186–196. Scopus. [Google Scholar]
- Bicker KL, Subramanian V, Chumanevich AA, Hofseth LJ, & Thompson PR (2012). Seeing Citrulline: Development of a Phenylglyoxal-Based Probe To Visualize Protein Citrullination. Journal of the American Chemical Society, 134(41), 17015–17018. 10.1021/ja308871v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman ML, Royzen M, & Fox JM (2008). Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity. Journal of the American Chemical Society, 130(41), 13518–13519. 10.1021/ja8053805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, & Weber T (2021). antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Research, 49(W1), W29–W35. 10.1093/nar/gkab335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg PM, & Strominger JL (1972). Five Penicillin-binding Components Occur in Bacillus subtilis Membranes. Journal of Biological Chemistry, 247(24), 8107–8113. 10.1016/S0021-9258(20)81815-8 [DOI] [PubMed] [Google Scholar]
- Cabrera-Pardo JR, Chai DI, Liu S, Mrksich M, & Kozmin SA (2013). Label-assisted mass spectrometry for the acceleration of reaction discovery and optimization. Nature Chemistry, 5(5), 423–427. 10.1038/nchem.1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahn JKB, & Piel J (2021). Opening up the Single-Cell Toolbox for Microbial Natural Products Research. Angewandte Chemie International Edition, 60(34), 18412–18428. 10.1002/anie.201900532 [DOI] [PubMed] [Google Scholar]
- Castro-Falcón G, Hahn D, Reimer D, & Hughes CC (2016). Thiol Probes To Detect Electrophilic Natural Products Based on Their Mechanism of Action. ACS Chemical Biology, 11(8), 2328–2336. 10.1021/acschembio.5b00924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Falcón G, Millán-Aguiñaga N, Roullier C, Jensen PR, & Hughes CC (2018). Nitrosopyridine Probe To Detect Polyketide Natural Products with Conjugated Alkenes: Discovery of Novodaryamide and Nocarditriene. ACS Chemical Biology, 13(11), 3097–3106. 10.1021/acschembio.8b00598 [DOI] [PubMed] [Google Scholar]
- Chaudhary DK, Khulan A, & Kim J (2019). Development of a novel cultivation technique for uncultured soil bacteria. Scientific Reports, 9(1), 6666. 10.1038/s41598-019-43182-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheminat A, Benezra C, Farrall MJ, & Fréchet JMJ (1981). Removal of allergens from natural oils by selective binding to polymer supports. II. Application of aminated resins to isoalantolactone and costus oil. Canadian Journal of Chemistry, 59(10), 1405–1414. 10.1139/v81-207 [DOI] [Google Scholar]
- Cox CL, Tietz JI, Sokolowski K, Melby JO, Doroghazi JR, & Mitchell DA (2014). Nucleophilic 1,4-Additions for Natural Product Discovery. ACS Chemical Biology, 9(9), 2014–2022. 10.1021/cb500324n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Lei Q, Wu Y, He Y, & Li W (2020). Activity-based protein profiling: Recent advances in medicinal chemistry. European Journal of Medicinal Chemistry, 191, 112151. 10.1016/j.ejmech.2020.112151 [DOI] [PubMed] [Google Scholar]
- Devaraj NK, Weissleder R, & Hilderbrand SA (2008). Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chemistry, 19(12), 2297–2299. 10.1021/bc8004446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan G, Dowson C, & Overington J (2019). Meeting the discovery challenge of drug-resistant infections: Progress and focusing resources. Drug Discovery Today, 24(2), 452–461. 10.1016/j.drudis.2018.11.015 [DOI] [PubMed] [Google Scholar]
- Gamoh K, Yamaguchi I, & Takatsuto S (1994). Rapid and Selective Sample Preparation for the Chromatographic Determination of Brassinosteroids from Plant Material Using Solid-Phase Extraction Method. Analytical Sciences, 10(6), 913–917. 10.2116/analsci.10.913 [DOI] [Google Scholar]
- Georgiou MA, Dommaraju SR, Guo X, Mast DH, & Mitchell DA (2020). Bioinformatic and Reactivity-Based Discovery of Linaridins. ACS Chemical Biology, 15(11), 2976–2985. 10.1021/acschembio.0c00620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang Y, Ju K-S, Thomson RJ, Metcalf WW, & Kelleher NL (2016). Metabologenomics: Correlation of Microbial Gene Clusters with Metabolites Drives Discovery of a Nonribosomal Peptide with an Unusual Amino Acid Monomer. ACS Central Science, 2(2), 99–108. 10.1021/acscentsci.5b00331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross H, Stockwell VO, Henkels MD, Nowak-Thompson B, Loper JE, & Gerwick WH (2007). The Genomisotopic Approach: A Systematic Method to Isolate Products of Orphan Biosynthetic Gene Clusters. Chemistry & Biology, 14(1), 53–63. 10.1016/j.chembiol.2006.11.007 [DOI] [PubMed] [Google Scholar]
- Guo X, Zhang J, Li X, Xiao E, Lange JD, Rienstra CM, Burke MD, & Mitchell DA (2021). Sterol Sponge Mechanism Is Conserved for Glycosylated Polyene Macrolides. ACS Central Science, 7(5), 781–791. 10.1021/acscentsci.1c00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LA, Saint-Vincent PMB, Guo X, Hudson GA, DiCaprio AJ, Zhu L, & Mitchell DA (2020). Reactivity-Based Screening for Citrulline-Containing Natural Products Reveals a Family of Bacterial Peptidyl Arginine Deiminases. ACS Chemical Biology, 15(12), 3167–3175. 10.1021/acschembio.0c00685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-B, Cai W, Del Rio Flores A, Twigg FF, & Zhang W (2020). Facile Discovery and Quantification of Isonitrile Natural Products via Tetrazine-Based Click Reactions. Analytical Chemistry, 92(1), 599–602. 10.1021/acs.analchem.9b05147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L, Hug JJ, Fu C, Bian X, Zhang Y, & Müller R (2019). Heterologous expression of bacterial natural product biosynthetic pathways. Natural Product Reports, 36(10), 1412–1436. 10.1039/C8NP00091C [DOI] [PubMed] [Google Scholar]
- Jia Zhang J, Tang X, & Moore BS, (2019). Genetic platforms for heterologous expression of microbial natural products. Natural Product Reports, 36(9), 1313–1332. 10.1039/C9NP00025A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L, & Baltz RH (2016). Natural product discovery: Past, present, and future. Journal of Industrial Microbiology and Biotechnology, 43(2–3), 155–176. 10.1007/s10295-015-1723-5 [DOI] [PubMed] [Google Scholar]
- Kautsar SA, van der Hooft JJJ, de Ridder D, & Medema MH (2021). BiG-SLiCE: A highly scalable tool maps the diversity of 1.2 million biosynthetic gene clusters. GigaScience, 10(1). 10.1093/gigascience/giaa154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten RD, Yang Y-L, Xu Y, Cimermancic P, Nam S-J, Fenical W, Fischbach MA, Moore BS, & Dorrestein PC (2011). A mass spectrometry–guided genome mining approach for natural product peptidogenomics. Nature Chemical Biology, 7(11), 794–802. 10.1038/nchembio.684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser T, Bibb MJ, Buttner MJ, Chater KF, & Hopwood DA (2000). Practical streptomyces genetics (Vol. 291). John Innes Foundation Norwich. [Google Scholar]
- Kim E, & Koo H (2019). Biomedical applications of copper-free click chemistry: In vitro, in vivo, and ex vivo. Chemical Science, 10(34), 7835–7851. 10.1039/C9SC03368H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capehart SL, & Carlson EE, (2016). Mass spectrometry-based assay for the rapid detection of thiol-containing natural products. Chemical Communications, 52(90), 13229–13232. 10.1039/C6CC07111B [DOI] [PubMed] [Google Scholar]
- Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, … Lewis K (2015). A new antibiotic kills pathogens without detectable resistance. Nature, 517(7535), 455–459. 10.1038/nature14098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Das AK, & Basak A (2015). Label-assisted laser desorption/ionization mass spectrometry (LA-LDI-MS): Use of pyrene aldehyde for detection of biogenic amines, amino acids and peptides. RSC Advances, 5(129), 106912–106917. 10.1039/C5RA20678B [DOI] [Google Scholar]
- Maxson T, Tietz JI, Hudson GA, Guo XR, Tai H-C, & Mitchell DA (2016). Targeting Reactive Carbonyls for Identifying Natural Products and Their Biosynthetic Origins. Journal of the American Chemical Society, 138(46), 15157–15166. 10.1021/jacs.6b06848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungan MD, Alanjary M, Blin K, Weber T, Medema MH, & Ziemert N (2020). ARTS 2.0: Feature updates and expansion of the Antibiotic Resistant Target Seeker for comparative genome mining. Nucleic Acids Research, 48(W1), W546–W552. 10.1093/nar/gkaa374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DJ, & Cragg GM (2020). Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. Journal of Natural Products, 83(3), 770–803. 10.1021/acs.jnatprod.9b01285 [DOI] [PubMed] [Google Scholar]
- Palaniappan KK, Pitcher AA, Smart BP, Spiciarich DR, Iavarone AT, & Bertozzi CR (2011). Isotopic Signature Transfer and Mass Pattern Prediction (IsoStamp): An Enabling Technique for Chemically-Directed Proteomics. ACS Chemical Biology, 6(8), 829–836. 10.1021/cb100338x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porte K, Riberaud M, Châtre R, Audisio D, Papot S, & Taran F (2021). Bioorthogonal Reactions in Animals. ChemBioChem, 22(1), 100–113. 10.1002/cbic.202000525 [DOI] [PubMed] [Google Scholar]
- Reimer D, & Hughes CC (2017). Thiol-Based Probe for Electrophilic Natural Products Reveals That Most of the Ammosamides Are Artifacts. Journal of Natural Products, 80(1), 126–133. 10.1021/acs.jnatprod.6b00773 [DOI] [PubMed] [Google Scholar]
- Ross C, Scherlach K, Kloss F, & Hertweck C (2014). The Molecular Basis of Conjugated Polyyne Biosynthesis in Phytopathogenic Bacteria. Angewandte Chemie International Edition, 53(30), 7794–7798. 10.1002/anie.201403344 [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, & Sharpless KB (2002). A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angewandte Chemie International Edition, 41(14), 2596–2599. [DOI] [PubMed] [Google Scholar]
- Rudolf GC, Heydenreuter W, & Sieber SA (2013). Chemical proteomics: Ligation and cleavage of protein modifications. Current Opinion in Chemical Biology, 17(1), 110–117. 10.1016/j.cbpa.2012.11.007 [DOI] [PubMed] [Google Scholar]
- Rudolf GC, Koch MF, Mandl FAM, & Sieber SA (2015). Subclass-Specific Labeling of Protein-Reactive Natural Products with Customized Nucleophilic Probes. Chemistry – A European Journal, 21(9), 3701–3707. 10.1002/chem.201405009 [DOI] [PubMed] [Google Scholar]
- Saxon E, & Bertozzi CR (2000). Cell Surface Engineering by a Modified Staudinger Reaction. Science, 287(5460), 2007–2010. 10.1126/science.287.5460.2007 [DOI] [PubMed] [Google Scholar]
- Skinnider MA, Johnston CW, Gunabalasingam M, Merwin NJ, Kieliszek AM, MacLellan RJ, Li H, Ranieri MRM, Webster ALH, Cao MPT, Pfeifle A, Spencer N, To QH, Wallace DP, Dejong CA, & Magarvey NA (2020). Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nature Communications, 11(1), 6058. 10.1038/s41467-020-19986-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten EM, & Bertozzi CR (2011). From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions. Accounts of Chemical Research, 44(9), 666–676. 10.1021/ar200148z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk MLWJ, Agramunt J, & Bonger KM (2021). Recent developments in bioorthogonal chemistry and the orthogonality within. Current Opinion in Chemical Biology, 60, 79–88. 10.1016/j.cbpa.2020.09.002 [DOI] [PubMed] [Google Scholar]
- Sorokina M, & Steinbeck C (2020). Review on natural products databases: Where to find data in 2020. Journal of Cheminformatics, 12(1), 20. 10.1186/s13321-020-00424-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers AE, & Cravatt BF (2009). Activity-Based Protein Profiling (ABPP) and Click Chemistry (CC)-ABPP by MudPIT Mass Spectrometry. Current Protocols in Chemical Biology, 1, 29–41. 10.1002/9780470559277.ch090138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suginaka H, Blumberg PM, & Strominger JL (1972). Multiple Penicillin-binding Components in Bacillus subtilis, Bacillus cereus, Staphylococcus aureus, and Escherichia coli. Journal of Biological Chemistry, 247(17), 5279–5288. 10.1016/S0021-9258(20)81102-8 [DOI] [PubMed] [Google Scholar]
- Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai H-C, Zakai UI, & Mitchell DA (2017). A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nature Chemical Biology, 13(5), 470–478. 10.1038/nchembio.2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefson EJ, & Carlson EE (2019). Chemoselective Tagging to Promote Natural Product Discovery. In Mass Spectrometry-Based Chemical Proteomics (pp. 187–206). John Wiley & Sons, Ltd. 10.1002/9781118970195.ch7 [DOI] [Google Scholar]
- Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin C, Mornico D, Roche D, Rouy Z, Salvignol G, Scarpelli C, Thil Smith AA, Weiman M, & Médigue C (2013). MicroScope—An integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Research, 41(D1), D636–D647. 10.1093/nar/gks1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Ren H, Tian Q, & Zhao H (2020). 6.07—Activation of Silent Natural Product Biosynthetic Gene Clusters Using Synthetic Biology Tools. In Liu H.-W. (Ben) & Begley TP (Eds.), Comprehensive Natural Products III (pp. 113–135). Elsevier. 10.1016/B978-0-12-409547-2.14725-0 [DOI] [Google Scholar]
- Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu W-T, Crüsemann M, Boudreau PD, Esquenazi E, Sandoval-Calderón M, … Bandeira N (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nature Biotechnology, 34(8), 828–837. 10.1038/nbt.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tian Y, Wang M, Wang M, Sun G, & Sun X (2018). Advanced Activity-Based Protein Profiling Application Strategies for Drug Development. Frontiers in Pharmacology, 9, 353. 10.3389/fphar.2018.00353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, & Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature, 468(7325), 790–795. 10.1038/nature09472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, & Devaraj NK (2018). Advances in Tetrazine Bioorthogonal Chemistry Driven by the Synthesis of Novel Tetrazines and Dienophiles. Accounts of Chemical Research, 51(5), 1249–1259. 10.1021/acs.accounts.8b00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, & Wright GD (2019). Heterologous expression-facilitated natural products’ discovery in actinomycetes. Journal of Industrial Microbiology and Biotechnology, 46(3–4), 415–431. 10.1007/s10295-018-2097-2 [DOI] [PubMed] [Google Scholar]
- Odendaal AY, Trader DJ, & Carlson EE, (2011). Chemoselective enrichment for natural products discovery. Chemical Science, 2(4), 760–764. 10.1039/C0SC00620C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R, Oberg N, & Gerlt JA (2019). The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry, 58(41), 4169–4182. 10.1021/acs.biochem.9b00735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Li G, Chen Y, & Lu Y (2020). Challenges and Advances in Genome Editing Technologies in Streptomyces. Biomolecules, 10(5), 734. 10.3390/biom10050734 [DOI] [PMC free article] [PubMed] [Google Scholar]