

Abstract
Microglial cells serve as molecular sensors of the brain that play a role in physiological and pathological conditions. Under normal physiology, microglia are primarily responsible for regulating central nervous system homeostasis through the phagocytic clearance of redundant protein aggregates, apoptotic cells, damaged neurons, and synapses. Furthermore, microglial cells can promote and mitigate amyloid β phagocytosis and tau phosphorylation. Dysregulation of the microglial programming alters cellular morphology, molecular signaling, and secretory inflammatory molecules that contribute to various neurodegenerative disorders especially Alzheimer's disease (AD). Furthermore, microglia are considered primary sources of inflammatory molecules and can induce or regulate a broad spectrum of cellular responses. Interestingly, in AD, microglia play a double-edged role in disease progression; for instance, the detrimental microglial effects increase in AD while microglial beneficiary mechanisms are jeopardized. Depending on the disease stages, microglial cells are expressed differently, which may open new avenues for AD therapy. However, the disease-related role of microglial cells and their receptors in the AD brain remain unclear. Therefore, this review represents the role of microglial cells and their involvement in AD pathogenesis.
1. Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder that is well characterized by complex cellular and molecular alterations, such as loss of neurons and synapses, protuberant gliosis, dystrophic neuritis, formation of extracellular deposits of amyloid β (Aβ), and intracellular aggregated phosphorylated tau [1, 2]. Interestingly, reactive gliosis includes changes in function and morphology of astrocytes and microglia [3–5]. The neuroinflammatory process plays an important role in several neurological diseases, including autoimmune ailments [6]. In the case of AD development, the inflammatory response has been undoubtedly connected. Moreover, microglia have been found to have a pivotal role in the pathogenesis of sporadic AD [7–9]. M1 microglia produce inflammatory mediators, which cause inflammation and neurotoxicity, while M2 microglia produce anti-inflammatory mediators, resulting in anti-inflammatory and neuroprotective effects. Microglia-facilitated neuroinflammation is a dual-edged sword in neurodegenerative events, with both damaging and beneficial consequences [10].
Activated microglial cells surround the Aβ plaques during Aβ phagocytosis/compaction, which may play either a neuroprotective or neurodegenerative role that depends on microglial phenotype switching [11–14]. In fact, via a reduction in the levels of Aβ in amyloid precursor protein- (APP-) based models, chronic microglial activation might improve the AD pathology [15]. Nevertheless, it has been implicated that inflammatory response exerts harmful neurotoxic effects via the release of neurotoxins and proinflammatory chemokines/cytokines [16, 17]. Induction of inflammation is also likely associated with tau pathology [18]. Evidence suggests that microglia have been linked to tau pathology and spatial memory deficits [19].
Human genome-wide association studies (GWAS) further strengthened the relationship between microglia and AD pathology. GWAS data showed that the microglial immune response is associated with multiple polymorphisms [8, 20, 21]. On the other hand, within a diverse range of AD-related genes, the microglial triggering receptor expressed in the myeloid cell 2 (TREM2) gene appears to have a critical contribution in case of AD-related immune response [8]. TREM2 is a lipoprotein sensor and lipid that encourages reactive microgliosis via its DNAX activation protein of 12 kDa (DAP12, a transmembrane protein) [22, 23]. It has been exhibited that through its interaction with apolipoprotein E (APOE), TREM2 controls the transcriptional activation of microglial cells [24–26]. Nonetheless, the impact of TREM2-facilitated microglial activation in AD pathogenesis, or the activities of the microglial cell, is not yet well-explained [27].
In the case of AD individuals, the neuroinflammatory response is possibly not entirely beneficial or harmful. Indeed, an uncontrolled microglial reaction might be detrimental to the surrounding neuronal elements or neurons [28]. In AD mouse models, parabiosis experiments revealed that, with a negligible contribution of infiltrating macrophages, microglia are responsible for increasing the number of myeloid cells observed in brains with plaque pathology [29]. Furthermore, via the elimination of undesirable synapses and neurons (i.e., immature synaptic connectivity whereby less active synaptic connections are formed), microglia also contribute to the developmental sculpting of neural circuits [30, 31]. These microglial roles have been shown to be compromised in aging that contributes to AD progression [32–34]. Therefore, this review is aimed at discussing how microglia act as immune system cells and how this system is changed in AD pathogenesis.
2. Microglia in Brain Aging
Aging causes microglial morphology changes [35, 36]. It has been specified in mice that microglia surveying processes are not so dynamic and less critical because of age [32, 37]. This explains the impact of pathogenic response, response to accumulated protein, or delayed injury in aged brains rather than younger mouse brains. In a facial nerve axotomy study, microglial proliferation during aging remained significantly higher in response to neuronal injury, suggesting that regulation of microglial proliferation changes with aging [38]. Moreover, the migration rate of the microglial cell was affected by aging when microglia responded to injury [32, 39]. A study on the dynamic behavior and morphology of microglia with aging disclosed that microglial response significantly reduced with age [39], whereas the distal branches become thinner and contain major functions [40–42]. Most importantly, myelin fragmentation has a role in the formation of myelin inclusions [43]. In addition to this, aging can cause the reduction of the somatic volume of the microglial cell that reduces tissue distribution homogeneity [44, 45].
During aging, microglia show an increased inflammatory response and exhibit differential changes in expression level [35]. For example, the expression level of major histocompatibility complex (MHC) II and cluster of differentiation (CD) 68 was higher in the aged microglial cell [46, 47]. On the other hand, the CD200 shows a decreased expression [48]. In order to form the ramified microglia, the CX3CL/fractalkine cytokine also plays a similar role. CX3CL1 connects with C-X3-C motif chemokine receptor 1 (CX3CR1), which is expressed vastly in the microglial cell [49]. Generally, in the pathway of canonical signaling for transforming growth factor-beta (TGFβ), Smad3 takes part in signaling, and the aged brain cell shows reduced anti-inflammatory functions [50]. Moreover, in proinflammatory gene transcription, interferon-gamma (IFNγ) activates microglia [51, 52], and the activation increases in the aged brain. Microglial maturation is influenced by altering gene function, which is predicted as a principal regulator of aging-associated changes in the microglial cell [53]. Apart from this, Iba-1 is highly expressed in microglia, which exerts its lessened ramified structure of microglial cells during aging [54], and it also accomplishes the proliferation of microglia.
According to the previous literature, microglial age-related phenotypes vary based on central nervous system (CNS) compartments [55]. The current studies have also reported that the aging effects on the microglial transcriptome are predominantly reliable on the basis of CNS locations [56]. Normally, microglia become highly activated during aging, and it acts towards CNS and peripheral nervous system (PNS) insults combined. Caldeira et al. [57] have reported by in vitro experiment that the isolated microglial cell tends to show a reduced reaction in autophagic capability, chemotaxis, phagocytosis, and overall reactivity.
3. Microglia in Neurodegeneration
Microglial activation exacerbates the production of cytokines, chemokines, and other factors that trigger AD progression [58]. Not only do proliferative microglia correlate with disease severity in AD patients but also AD animal models [59]. Their gradual gathering and changing in signaling prompt the cognitive decline; thereby, targeting microglia and their signaling pathways would be a potential therapeutic strategy.
By using a gene expression profile, a study identified a newer type of microglia that extended the existent microglia classification, and investigators in this inquiry yclept this molecular signature of disease-associated microglia (DAM, distinctive microglia subgroups) [24]. Interestingly, Krasemann et al. [24] showed that microglial neurodegenerative phenotype (MGnD) upregulated 28 inflammatory molecules and diminished the expression of 68 homeostatic microglial genes; in contrast, a large segment of these activities disappeared in the microglia-specific knockout of APOE in mice. These findings indicate that the APOE strongly persuades phenotypic switching in disease-related microglia and is upregulated through the vicinity of plaques. Moreover, MGnD microglia remarkably increased in miR-155 expression resulting in a notable upregulation of microRNA (miRNA) in microglia after extreme provocation with an insult, which leads to the release of proinflammatory molecules, including interleukin-6 (IL-6), interleukin-1β (IL-1β), nitric oxide synthase-2 (NOS-2), and tumor necrosis factor-alpha (TNF-α) [60]. In 5XFAD transgenic mice, elevation of APOE, TREM-2, and leukocystatin (Cst7) gene expression was associated with the transition from homeostatic microglia to DAM activation [61]. Previously, it has been demonstrated that DAM activation is tightly linked with the loss of microglial homeostatic genes such as purinergic receptor P2Y (P2RY12) and CX3CR1 [61].
In addition, Runt-related transcription factor 1 (RUNX-1), Sal-like 1 (SALL-1), T-cell-acute-lymphocytic leukemia protein-1 (TAL-1), and interferon regulatory factor 8 (IRF8) genes acquainted with microglia maturation and ramification are also influenced by AD pathology [60]. Usually, MGnD is a consequence of chronic manifestation of disease pathology and can easily differentiate between M1 and M2 microglia through the appearance of ApoE, TREM2, chitinase-3-like protein (Ym1), arginase 1 (Arg1) as well as the nonappearance of a homeostatic transcription factor, namely, early growth response protein 1 (Egr1), respectively [24]. In the AD brain, both the MGnD and DAM phenotypes are upregulated in the microglia and influenced mainly by TREM2 expression [62, 63]. From these studies, the researchers propose that the APOE-TREM2 signaling pathway is mainly accountable for the remodelling of the gene expression profile that prompts the MGnD phenotype in microglia [24, 61, 63]. While the relationship between the MGnD phenotype and aging is questionable, how microglia with advancing age are responsible for making this transition needs to be solved. Advance research is warranted for better understanding to elucidate the close relationship between the time-dependent APOE-TREM2 signaling complex and the MGnD phenotype.
4. Activated Microglia and Alzheimer's Pathogenesis
Microglia have a dual role in AD pathogenesis; in AD, microglial detrimental effects are associated with proinflammatory mediators [64, 65]. Apparently, Aβ provoked the microglial activation, and deteriorated neuron-derived ingredients may exaggerate microglial neurotoxicity in AD [66]. Aβ subsists in several assembly forms, such as monomers, oligomers, and fibrils. However, from these three Aβ assemblies, only oligomeric Aβ (oAβ) and fibrillar Aβ (fAβ) have been implicated to microglial releases of proinflammatory mediators (Figure 1) such as cytokines (i.e., IL-1, IL-6, and TNF-α), chemokines (i.e., monocyte chemotactic-1 (MCP-1) and macrophage inflammatory protein-1 (MIP-1)), and reactive oxygen species (ROS) [67, 68].
Figure 1.
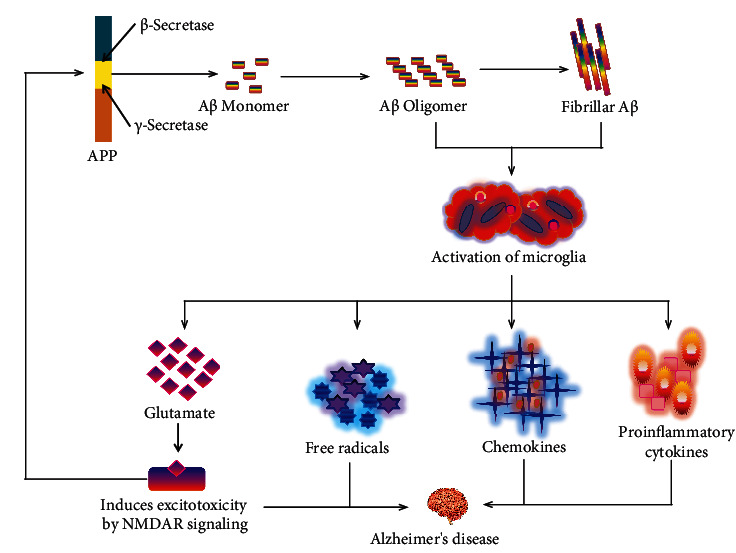
Role of Aβ in the activation of microglia to initiate Alzheimer's pathology. Aβ: amyloid beta; APP: amyloid precursor protein; IL-1: interleukin-1; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; MCP-1: monocyte chemotactic-1; MIP-1: macrophage inflammatory protein-1; HO: hydroxyl radical; H2O2: hydrogen peroxide; O2: oxygen radical.
The expression of nicotinamide-adenine-dinucleotide-phosphate-oxidase (NADPH oxidase) is stimulated to produce the ROS, which is correlated with the upregulation of AD [69]. fAβ is liable to microglial NADPH [70, 71], and activation of NADPH oxidase eventually leads to neurotoxicity. In fact, microglia produce extracellular ROS that has been directly harmful to neurons. Moreover, intracellular ROS act like a signaling molecule in microglia, which promotes the secretion of different proinflammatory cytokines and neurotoxic molecules [72].
Furthermore, glutaminase expression was disorganized by microglial activation; as a result, release of a large proportion of glutamate influenced excitoneurotoxicity through the N-methyl-D-aspartate (NMDA) receptor signaling pathway [73–75]. Previously, it has been demonstrated that persistent triggering of extrasynaptic NMDA receptors contributes to accelerated Aβ production [76]. Accumulating evidence supports that expression of Aβ itself disrupts the synaptic function, such as suppressing hippocampal long-term potentiating, the assistance of prolonged depression, and disturbance of synaptic plasticity [77, 78]. Hence, it is crucially important to examine the microglial neurotoxicity along with Aβ neurotoxicity. In addition, both the tau protein and Aβ pathology have been directly linked to the neuroinflammatory responses through the accumulation of reactive microglia and astrocytes, which are close to the amyloid deposits, an additional histological characteristic of AD [8, 79]. For example, in P301S tau, transgenic mice exhibit prominent microglial activation that ultimately disrupts hippocampal synaptic function [80]. Thus, microgliosis-induced hippocampal synaptic pathology may be the earliest expression of neurodegenerative tauopathies. Activated microglia can also reactivate astrocytes by releasing cytokines, including IL-1α, TNF-α, and C1q [81]. Reactivation of these astrocytes notably upregulates complement cascade genes, including C3, and fails to contribute to synaptogenesis and phagocytose synapses and myelin debris. In the prefrontal cortex of AD patients, nearly 60% of the astrocytes are C3-expressing astrocytes and may possibly cause neuronal injury [81]. During AD, reactive astrocytes interact with neuronal and nonneuronal (i.e., microglia and oligodendrocytes) cells by secreting feedforward signals and contributing to the vicious cycle that expedites neurodegeneration [82]. Although reactive astrocytes have both beneficial and harmful functions during AD, atrophic astrocytes (reduction of the surface area and volume of astroglial morphological profiles) might lose their homeostatic functions.
Microglia-neuron communication is bidirectional. Microglia-derived exosomes serve as a carrier for tau and Aβ in the brain. On the other hand, neuron-derived exosomes have similar effects on microglia. A study has shown that microglia act as scavengers by uptaking neuronal exosomes containing toxic proteins, including pTau and Aβ [83].
5. Microglia Receptors in the Amyloid Cascade of Alzheimer's Disease
5.1. Complement Receptors
Complement components (CRs) and their receptors are categorized as cell surface molecules on microglia that are located within or around Aβ cerebral plaques in AD [84]. Previously, it has been demonstrated that microglia not only express complement protein components such as complement component-1 (C1q) and complement component-3 (C3) but also precisely express complement receptors, including complement receptor type-1 (CR1), complement receptor type 3 (CR3), complement receptor type-4 (CR4), and complement component 5a receptor 1 (C5aR1), which support phagocytic uptake [85]. The imbalance of these complementary systems is correlated with the development of AD pathogenesis (Table 1). For instance, Aβ plaque formation was observed to be markedly increased with the suppression of these complement systems in the AD transgenic mouse model [86]. However, different proteins of the complement system and its analogous mRNAs are unregulated, resulting in Aβ-instigated inflammation, the emergence of senile plaque, and Aβ phagocytosis in AD patients [87]. C3 is denoted as a protein and an integral part of the complement system, which influences the phagocytosis of pathogens by interacting with the CR3 receptor. CR3 is also familiar as a macrophage-1 antigen and indisputably observed in microglia that have been upregulated by the AD brains [88]. In addition, both in vivo and in vitro studies have demonstrated that CR3 was responsible for the uptake and clearance of Aβ [89–91]. Likewise, this receptor is partially associated with Aβ-induced microglial activation and involved in Aβ-mediated microglia ROS generation [92], as stated in Figure 2. Furthermore, a study in AD mice showed that microglia were associated with synaptic pruning in a CR3-dependent pathway [93]. More clearly, oligomeric Aβ locally activated complement (i.e., C1q and C3) at vulnerable synapses, resulting in microglial engulfment of these synapses via C3/CR3 signaling. Nowadays, CR3 antagonists are widely accepted as potential therapeutics to treat AD owing to their potential to significantly decrease the Aβ-induced proinflammatory molecules and ROS in microglia [92].
Table 1.
Outline of microglia receptors and their function in Alzheimer's disease.
| Microglia receptors | Functions in Alzheimer's disease | References |
|---|---|---|
| Complement receptors (CRs) | (i) Phagocytic uptake (ii) Microglia activation (iii) Proinflammatory molecule generation (iv) Aβ clearance |
[89–92, 94–97] |
| Toll-like receptors (TLRs) | (i) Proinflammatory mediator generation (ii) Aβ clearance (iii) Microglia activation (iv) Synaptic plasticity (v) tau phosphorylation |
[15, 98–103] |
| Scavenger receptor type-A (SR-A) | (i) Aβ internalization and clearance (ii) Inflammatory response (iii) Maintain microglia immune response |
[104–107] |
| Cluster of differentiation 36 (CD36) | (i) Microglia recruitment (ii) Inflammatory response (iii) Activation of Aβ phagocytosis (iv) Modulates microglial Aβ42 phagocytosis |
[108–111] |
| Receptor for advanced glycation end products (RAGE) | (i) Microglia activation (ii) Stimulate IL-1β (iii) TNF-α production (iv) Intensify oxidative stress |
[112–116] |
| Triggering receptor expressed in the myeloid cell 2 (TREM2) | (i) Aβ clearance (ii) Regulates microglial mammalian target of rapamycin (mTOR) activation and metabolism (iii) Balanced microglial autophagy |
[117, 118] |
Figure 2.
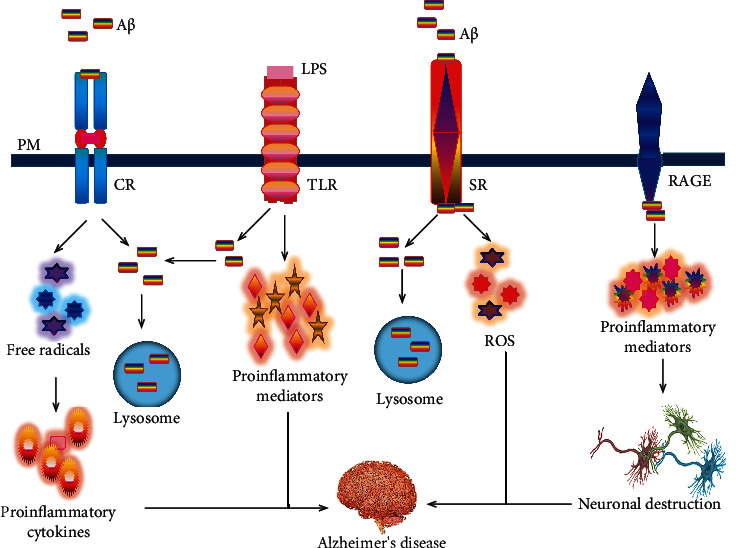
The linkage of microglia receptors in the pathogenesis of Alzheimer's disease. CR3 is responsible for the Aβ-induced microglial activation and involved in Aβ-mediated microglia free radical generation as well as uptake and clearance of Aβ. TLR2 is implicated in the generation of the inflammatory response. On the other hand, TLR4 (i.e., stimulated with LPS) is associated with the clearance of Aβ. Microglia cells showed an increase in Aβ uptake. The binding of Aβ to SRs internalizes Aβ and could activate inflammation responses and generate reactive species. Microglia RAGE-Aβ interaction triggers the genesis of proinflammatory molecules that causes neuronal destruction. PM: plasma membrane; Aβ: amyloid beta; CR: complement receptor; LPS: lipopolysaccharide; TLR: Toll-like receptor; SR: scavenger receptor; RAGE: receptor for advanced glycation end products; ROS: reactive oxygen species.
C5a is a protein fragment that can generate highly proinflammatory molecules via activating the complement system [41]. It is also called a CD88 and is located on the surface of the microglial cell. CD88 is involved in microglial recruitment and activation; an elevated level of CD88 has been observed in microglia and appeared close to the amyloid plaques in the AD mouse brains [119]. In addition, coinvigoration of human monocytes with Aβ and C5a encourages the promotion of IL-1β as well as IL-6 secretion [120]; mitigating the destructive role of CD88 would be a potential strategy for AD pathogenesis. Therefore, Fonseca et al. [97] conducted a study to assess the efficacy of this receptor antagonist, which markedly attenuated Aβ plaques, reduced glial triggering, and ameliorated context-dependent memory in double transgenic AD mouse models (Table 2).
Table 2.
Microglia in various Alzheimer's disease preclinical models.
| Species/studied material | Experimental model | Effects | References |
|---|---|---|---|
| Complement C3-deficient APP transgenic mouse (APP; C3−/−) | Mouse model of AD | (i) Increased Aβ levels (ii) Elevation of fibrillar amyloid plaque burden |
[89] |
| Homozygous C3-deficient and Mac-1-deficient mice (C3−/−; Mac-1−/−) | Mouse model of AD | (i) Implicated in the phagocytosis and removal of fAβ by microglia | [91] |
| hAPP transgenic mouse | Aβ-induced neurotoxicity | (i) Reduced Aβ accumulation | [121] |
| TLR2 knockdown mice | Aβ42-induced neuroinflammation | (i) Suppressed proinflammatory molecules and integrin markers in microglia | [100] |
| Mutation of TLR4 in C3H/HeJ mice | Aβ-mediated cognitive dysfunction and neurotoxicity | (i) Reduced microglial activation (ii) Increase Aβ deposits |
[122] |
| CD36-deficient C57BL/6J mice | Nitro blue tetrazolium induced ROS generation | (i) Decreased microglial recruitment to sites of fAβ (ii) Reduced the production of ROS, TNF-α, IL-1β, and several chemokines |
[110] |
| RAGE overexpressed mAPP transgenic mice | Mouse model of AD | (i) Increased the production of IL-1β and TNF-α (ii) Enhanced the infiltration of microglia as well as astrocytes (iii) Decreased acetylcholine esterase activity and causes Aβ accretion |
[115] |
| Mutation of TLR4 in Mo/Hu APPswe PS1dE9 mice | Amyloidogenesis | (i) Increases in diffuse and fAβ deposits (ii) Decreased Aβ uptake by microglia |
[102] |
| PS1-APP transgenic mice | Aging induced Aβ deposition | (i) Early microglial enrollment fosters Aβ clearance (ii) Aβ-mediated inflammation reduces microglial Aβ clearance |
[106] |
| Aged APP23 transgenic mice | Aβ induced neuroinflammation | (i) TREM2 is increased in microglia associated with amyloid plaques (ii) Lack of extracellular amyloid clearance due to TREM2 signaling |
[117] |
| AD-mutant hAPP transgenic mice | Complement receptor and microglia mediated synaptic loss early in AD | (i) Inhibition of CR3 decreases phagocytic microglia (ii) Adult brain microglia engulf synaptic material when exposed to soluble Aβ oligomers |
[93] |
| Triple transgenic mice that are deficient in TLR2 (TLR2−/−) | Aβ42-induced memory impairments | (i) Accelerated spatial and contextual memory impairments (ii) Increased levels of Aβ |
[123] |
| APP/PS1/SR-A−/− mice | Aβ induced cognitive decline and neuroinflammation | (i) Increased neuroinflammation (ii) Elevated Aβ accumulation (iii) Increased cognitive impairment |
[107] |
Although accumulating evidence indicates the complement system manifested detrimental effects, a few data claimed that it has beneficial effects too in AD. For instance, C3-deficient APP mice showed an elevated level of Aβ in the brain area linked with notable neuronal damage [89]. More interestingly, higher expression of C3 mRNA levels is linked with a depletion in Aβ deposition in hAPP/TGF-β1 transgenic mice [121]. Overall, activation of these receptors might encourage the Aβ clearance, therefore eventually decreasing the Aβ accumulation in the AD. Still, many issues remain unsolved, so future studies are warranted to expurgate the molecular mechanism of the complement system in the brain and evaluate its suitability to the design and development of novel AD treatments.
5.2. Toll-Like Receptors
In 1997, Toll-like receptors (TLRs) were first identified as membrane proteins found in different types of cells, such as microglia and astrocytes [124, 125]. Although in mammals, there are 12 TLRs that have been described, only TLR2 and TLR4 can recognize Aβ [126]. However, its activation stimulates several signaling pathways; as well as, the secretion of several cytokines, nitric oxide (NO), and ROS [98]. Surprisingly, animal and human brain microglia expressed among the TLRs 1-9 and maxima of these receptors were responsible for microglial activation and neurotoxicity [125, 127]. For example, aged APP23 transgenic mice showed an upregulation of TLR-2, TLR-4, TLR-5, TLR-7, and TLR-9 mRNA levels in plaque-related brain tissue [128]. Studies have demonstrated that TLRs stimulate the intracellular cascade that leads to either release of proinflammatory mediators or the uptake and clearance of Aβ [99, 102]. Likewise, TLR2 involvement in the activation of microglial proinflammatory signaling to Aβ has been shown in Figure 2. Both AD patients and AD murine models found an increase in mRNA levels for TLR2 in the brains [129, 130]. Additionally, it has been reported that deficiency of TLR2 promotes a reduction in both spatial and nonspatial memory [123]. Interestingly, knockdown of TLR2 mice has disclosed a depletion of Aβ-induced manifestation of proinflammatory molecules (i.e., TNF-α, iNOS, IL-1β, and IL-6) and integrin markers (i.e., CD11a, CD11b, and CD68) in microglia [99]. Likewise, Liu et al. [101] have demonstrated that TLR2 deficiency suppressed Aβ-induced inflammatory signaling and improved Aβ internalization by phagocytosis in cultured microglia and macrophages. So suppression of TLR2 would be a powerful scheme that could markedly dwindle the inflammatory response and notably enhance the Aβ clearance, consequently slowing the AD pathogenesis.
TLR4 can recognize LPS by microglia; previous studies have identified its influences on stimulating the microglia-Aβ activation [131]. For instance, an activated murine microglia cell demonstrates that TLR4 contributes to Aβ-induced microglial neurotoxicity combined with a CD14 and myeloid differentiation protein-2 (MD2) [131]. In an in vitro experiment, microglia cells invigorated with LPS (i.e., a TLR4 ligand) showed an upregulation of Aβ uptake [102], as shown in Figure 2. In addition, both in vivo and in vitro studies on an LPS-deficient response have revealed that microglia increased the Aβ load and decreased Aβ uptake [102]. Moreover, in early stages, the TLR4-mutated AD animal model expressed a deficiency of spatial learning and increased levels of Aβ42 in the brain [122]. Altogether, roundup evidence on TLR2 and TLR4 indicates that depending upon diverse microglial phenotypes, these receptors have a complex role in AD. However, consolidated evidence strongly suggests that activation of TLR2 and TLR4 contribute to AD progression, and their inhibition may suppress AD pathogenesis [132]. Maybe these receptors show their beneficial effects in the early stages of AD, and their opposite role is exhibited in the late stages of AD due to diverse microglial phenotypes. Therefore, microglial TLR2 and TLR4 represent an acceptable target for therapeutic intervention within the disease progression, and targeting them could increase Aβ phagocytosis or reduce inflammatory responses [133–135].
5.3. Scavenger Receptors
Two kinds of scavenger receptors (SRs) have been identified in the CNS. Scavenger receptor type-A (SR-A) is manifested on microglia and astrocytes, whereas scavenger receptor type-B (SR-B) receptors are manifested on microglial and endothelial cells [108]. Microglial adherence via SR-A binding to fibrillar Aβ causes microglial immobilization, the genesis of ROS, and secretion of cytokines [70]. Both of these SRs could bind and internalize Aβ (Figure 2), inducing an inflammatory response that leads to AD pathogenesis [104]. Furthermore, both the SR-AI expression levels and Aβ clearance have been attenuated by prolonged preservation of microglia activation [106]. In addition, SR-AI deficiency with a presenilin1 (PS1)/APP transgenic mouse brain showed that increased levels of Aβ deposition correlated with an increase in mortality [11].
CD36 is a pattern recognition receptor (PRR) found on many different kinds of cells. This receptor comprehends not only exogenous molecules, for example, microbial elements [136] but also endogenous molecules, such as low-density lipoproteins (LDL), oxidized phospholipids (oxPCCD36) [137], programmed cell death-related cells, and Aβ [138]. CD36 is responsible for the development of several diseases, including AD [139]. Furthermore, CD36 interacts with Aβ by microglia to generate ROS [110] and activation in response to fAβ [108, 110, 140]. For instance, reducing the expression of cytokine and chemokine such as monocyte chemoattractant protein-1 (MCP-1), IL-1β, macrophage inflammatory protein-1α (MIP-1α), macrophage inflammatory protein-1β (MIP1β), macrophage inflammatory protein-2 (MIP-2), and TNF-α has been seen in macrophages and microglia from CD36-deficient mice vivified with fAβ [110]. However, in human brains, CD36 was observed at an overexpressed level with Aβ deposits, but without Aβ deposition, CD36 has not been detected in healthy brains [141]. Moreover, CD36 configures complexes with other PRRs to bind to fibrillar proteins.
CD163 is an unclassified SR that is expressed on mature tissue macrophages and is involved in hemoglobin-haptoglobin clearance from the blood [142]. CD163 engagement caused macrophages to produce proinflammatory mediators, indicating that CD163 is involved in macrophage activation [143]. Fabriek et al. [144] also reported CD163 functions as an innate immunological sensor and modulator of local inflammation in the host's defense against both gram-positive and gram-negative bacteria. Interestingly, CD163 was found to be expressed on microglia in the brains of patients with HIV-associated dementia [145]. However, whether CD163 is involved in AD pathogenesis is still elusive.
5.4. Receptor for Advanced Glycation End Products
The receptor for advanced glycation end products (RAGE) is a multiligand receptor and a compelling factor in aging that identifies the Aβ peptides [146]. Previously, it has been observed that Aβ provokes nuclear factor kappa light chain enhancer of activated B cell (NF-κB) activation in several cells and stimulates the release of proinflammatory mediators by the dealings with RAGE [147, 148].
Different experimental data disclosed that microglial RAGE-dependent molecular signaling drives Aβ-induced inflammatory response and neuronal damage in the AD [112–114, 149]. In particular, the experimental result proposes that the p38 mitogen-activated protein kinase (MAPK) signaling pathways engage in the activation of microglia through the interaction between Aβ and RAGE receptor [113, 115]. Fang et al. [115] have documented microglial RAGE in the pathogenesis of AD and proposed that refraining of the RAGE signaling pathway may be a quintessential target for reducing the secretion of proinflammatory molecules like TNF-α and IL-1β after Aβ stimulation in the AD. Microglia RAGE-Aβ interaction stimulates to upregulate the proinflammatory response as a consequence; the neuronal destruction that directly influences a shortage in learning and memory is mentioned in Figure 2. Further exploration has been suggested to ascertain small molecules for the blocking of Aβ-RAGE interaction, which would be a possible therapeutic stratagem to deal with most devastating AD pathogenesis.
6. Microglia in the Spread of Tau Pathology in Alzheimer's Disease
The hyperphosphorylation and accumulation of microtubule-associated protein tau (MAPT) form the initial event before neurodegeneration [150]. In humans, neuroinflammation is positively linked with tau pathology and is involved in the production of tau hyperphosphorylation, accumulation, and neurodegeneration [151, 152]. In the P301S animal model of tauopathy, it has been shown that microglial activation is the earliest manifestation of tau pathology [80]. Notably, in this study, they administered FK506 (i.e., an immunosuppressant drug), which reduced the microglial activation and augmented the lifespan of tau (P301S) transgenic mice [80]. Later, Maphis et al. [19] demonstrated that activated microglia played a pivotal role in the proliferation of tau. Afterwards, Bolós et al. [153] reported that microglia phagocytose the tau. However, how microglia induced tau pathology is yet to be confirmed.
Interestingly, an in vivo humanized mouse model of tauopathy (hTau) showed that either chemical compound or genetically induced microglial triggering markedly manifested tau pathology and behavioral malformation [154]. Furthermore, in hTau mice, deficiency of microglia-specific CX3CR1 evolved in triggered microglial activation as a result of increased tau pathology and impaired working memory [154]. This effect is arbitrated through the IL-1/p38 MAPK signaling pathway. Another study showed that deleting CX3CR1 in hAPP mice promoted the expression of inflammatory mediators and enhanced plaque-independent neuronal abnormality as well as cognitive deficits [155]. The CX3CL1/CX3CR1 signaling pathway is an important neuron and microglial communication [156]. A study demonstrated that nonappearance of CX3CR1 weakens the microglial internalization of tau, which leads to AD progress [157]. Accumulating studies indicate that microglia-allocated neuroinflammation increases the tau pathology as a consequence of neurodegenerative disease. In hTau mice, Maphis et al. [19] evaluated that depending on the different disease stages, CX3CR1 deficiency is responsible for the onset and development of tau pathology. They suggest that these reactive microglia can influence the development of the tau pathology and be consistent with the propagation of pathological tau in the brain. In addition, a study reveals that lacking microglial TREM2 results in exacerbated tau pathology and a profound dysregulation of stress-related kinase pathways in a humanized mouse model of tauopathy [158]. On the other hand, TREM2 reduces neuronal tau hyperphosphorylation by reducing the microglial inflammatory response [159]. During the pathological investigation of human brains, it has been found that microglia morphologically degenerated and were associated with tau pathology [160]. These morphological changes are suggested to result from microglial senescence and chronologically precede the spread of tau pathology [161, 162].
7. Microglial Activation in Alzheimer's Stage
7.1. Activated Microglia in Early-Onset Alzheimer's
The amyloid cascade-neuroinflammation hypothesis characterized as an abnormal production of Aβ owing to the redundancy of Aβ synthesis or a dysfunctioning of Aβ clearance is the paramount causality of the AD, which consequently stimulates neuroinflammation-induced neuronal loss [163, 164]. Therefore, neuroinflammation is noticed as a critical factor in the development of AD pathogenesis [165]. In addition, activated microglia can be either proinflammatory or anti-inflammatory.
In AD at its early stages, it has been proposed that the initial microglial activation may have a beneficial function through the clearance of the amyloid and releasing potential nerve growth factors [166]. On the other hand, due to the failure of this process hence to promote the Aβ aggregation or other lethal products, therefore, activation of proinflammatory phenotypes leads to a rapid destruction of the neurons. However, the genetic data from GWAS propose microglial activation to be able to execute several critical functions in the early stages of AD and autonomous amyloid pathology [167, 168]. Although epidemiological analysis has demonstrated that people who take nonsteroidal anti-inflammatory drugs (NSAIDs) have an inferior frequency of AD, randomized control trials have not shown the effectiveness of these NSAIDs in subjects with later onset of AD [169]. Recently, one study hypothesized two particular stages of microglial activation in the AD trajectory, an early anti-inflammatory phase and an advanced proinflammatory phase [170]. In this case, targeting antimicroglial medications would be most favorable to protect against the battle of the proinflammatory phenotype in the advanced phase of this disease. In the early phase of AD, microglial activation is able to alleviate Aβ aggregation by augmenting its phagocytosis, clearance, and degradation properties [171, 172]. For instance, an investigation of amyloid plaques by electron microscopy demonstrated that microglia are efficiently engulfing Aβ, and Aβ appeared in the endosome-like cellular domain [173].
7.2. Activated Microglia in Late-Onset Alzheimer's
In late-onset AD (LOAD), microglia have been deprived of their beneficiary function due to a tenacious production of proinflammatory mediators [174]. A study has shown that the amyloid plaque burden upsurges with aging in human patients, indicating the relatively ineffective phagocytic potential of microglia [175]. In human AD, Aβ42 immunization improves the function of microglia by intensifying their phagocytic activity [176]. Based on the microglial dysfunctioning notion, there is a loss in microglial neuroprotective activity in AD, rather than an increase in an inflammatory role [177]. Previously, it has been reported that the microglial phagocytic capabilities are shifted with aging and similarly decrease this feature in neurodegenerative diseases. Likewise, these senescent (i.e., biological aging) microglia are linked with the onset of sporadic AD [178]. Furthermore, recent studies on TREM2 also ascertain both early and late stages of microglial activation during the AD trajectory [179, 180]. TREM2 expressed in microglia is supposed to link with microglial activation. Even though a few studies indicate opposed effects for TREM2 levels in AD, lately, a study reported the level of soluble TREM2 (sTREM) directly linked with the early and delayed stage of AD [181, 182], where the peak beneficial role of TREM2 has observed in the early stage and later stage; its salutary effect gradually decreased (Figure 3).
Figure 3.
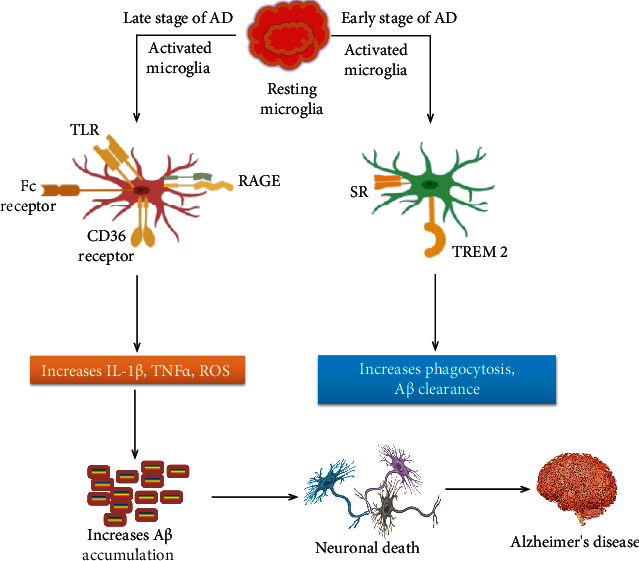
Possible mechanisms of action of activated microglia in different early and later stages of Alzheimer's disease. In the early stages of AD, activated microglia may increase Aβ clearance through TREM2 and scavenger receptors. On the other hand, in the late stage of the disease, continuous microglial activation induced by Aβ through various receptors triggers a vicious cycle of microglial activation, neuroinflammation, and Aβ buildup that leads to AD. AD: Alzheimer's disease; TLR: Toll-like receptor; RAGE: receptor for advanced glycation end products; IL-1β: interleukin-1β; TNF-α: tumor necrosis factor-α; ROS: reactive oxygen species; SR: scavenger receptor; TREM2: triggering receptor expressed on myeloid cell 2
8. Microglial Deterioration in Alzheimer's Patients
The activated microglial response has been extensively explored in AD brain regions by comparatively exalted Aβ subjects or in Aβ-rich transgenic models [183–185]. Fascinatingly, Aβ accumulation and neurofibrillary tangles (NFTs) do not appear in similar anatomical locus; in this sense, a direct pathogenic connection between amyloid plaques and neurodegenerative diseases is still elusive [186, 187]. In fact, cognitive disability is not compatible with an overabundance of amyloid plaque, even so with the presence of neurofibrillary pathology explicit as tau-positive morphology, including unmyelinated axons in the nervous system so-called neuropil threads, NFTs, and neuritic plaques [188, 189]. Additionally, an unambiguous determination of microglial activation in the human brain is extremely complicated since there is no effective biomarker for differentiating between activated and nonactivated cells. It is also surprising that microglial cells become progressively dysfunctional with aging in the human brain that displays morphologically senescence rather than activation, like fragmented cytoplasmic processes [190]. The identification of senescence microglia has imparted new aspects on the possible implication of microglia in aging-associated neurodegeneration; for example, aging causes loss of notable microglial cell function involved in the reduction of microglial neuroprotection [190, 191]. A study evaluating the microglial reaction in postmortem hippocampal human tissue demonstrated that microglia underwent a noticeable degenerative process in the dentate gyrus (DG) as well as CA3 of Braak V–VI samples, likely to be the case linked with the accumulation of soluble pTau [160].
Not only are microglial cells able to protect the synaptic integrity [192] but also they contribute to the learning ancillary synaptic formation [193]. Moreover, microglia induce Aβ-phagocytosis [194, 195] and senile plaque compaction and limit the Aβ toxicity [196, 197]. Furthermore, microglia contribute to removing depreciated neurons as well as neuronal stuff, for example, paranormal synaptic terminals or axonal demyelination. In this context, deficits in colony-stimulating factor 1 receptor (CSF1R) or TREM2 are correlated with a rare group of neurodegenerative disorder, for example, adult-onset leukoencephalopathy with axonal spheroids (i.e., characterized by excessive demyelinating lesions in the cerebral white matter) or Nasu-Hakola disease (i.e., characterized by multiple bone cysts linked to neurodegeneration), respectively [198–200]. These studies, together with Sanchez-Mejias [160] data, strongly indicated microglial pathology resulting in a deficient immunoprotection in DG and CA3 that leads to progressive AD pathology and cognitive damage. Furthermore, TREM2-knockout models show dystrophic microglial cells [201], shortages in microglial survival, and worsening in AD pathology [22].
Accumulating evidence demonstrates that AD is associated not only with microglial activation but also with microglial senescence, which might be considered the degeneration of these cells continuously [190]. These findings suggest that NSAIDs have become incapable of preventing or decreasing neurodegenerative disease like AD. Surprisingly, they reconstructed the conception regarding AD pathogenesis far away from inflammation-related impairment and proximate to an uninvestigated area of neuroscience, for instance, activities or events that can destroy microglial cells. Incredibly, it has become crystal clear that senescence microglia are responsible for age-related telomere length (TL) shortening [202, 203]. In addition, shortened TL in peripheral blood leukocytes is further recognized as early jeopardy of dementia [204]. Since microglia are indispensable for providing neuroprotection [191], aging-associated loss of a microglial protective role in neurodegenerative disease is likely to have detrimental repercussions for neurons.
9. Conclusion
Nowadays, studies started focusing on microglia to better understand the functional role of microglia in changing the progression of AD. Microglial cells are dynamic and reactive and change their surrounding environment rapidly, resulting in either proinflammatory or anti-inflammatory states. It has become apparent that microglia not only produce neurotoxic products but also need for phagocytic clearance of neurotoxic proteins associated with AD. The randomized control trials that employed nonspecific anti-inflammatory agents have not appeared to be significant in mitigating disease, possibly because of the inhibition of indispensable phagocytic functions that accumulate toxic proteins. Furthermore, depending on the clinical environment, microglia phenotypes may have a negative or positive effect. In fact, the ultimate beneficial role of TREM2 has been observed in the early stage and later stage, and its beneficial effect gradually decreased. AD pathogenesis is dependent on microglial cells and their receptors. Therefore, targeting microglial receptors to maintain microglial homeostasis would be a potential therapeutic strategy in AD.
Acknowledgments
The authors are grateful to the Pharmakon Neuroscience Research Network, Dhaka, Bangladesh, for supporting this project.
Conflicts of Interest
The authors proclaim no conflict of interest.
Authors' Contributions
MSU conceived the original idea and designed the outlines of the study. DMS, MSU, MTK, and SH wrote the draft of the manuscript. MSU and DMS prepared the figures for the manuscript. AP, ISA, GMA, MMA-D, and GhMA performed the literature review and aided in revising the manuscript. All authors have read and agreed to the published version of the manuscript.
References
- 1.Uddin M. S., Kabir M. T., Behl T., Ashraf G. M. Reconsidering and reforming the amyloid cascade hypothesis. Current Protein & Peptide Science . 2021;22(6):449–457. doi: 10.2174/1389203722666210322151627. [DOI] [PubMed] [Google Scholar]
- 2.Guo T., Zhang D., Zeng Y., Huang T. Y., Xu H., Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Molecular Neurodegeneration . 2020;15(1):p. 40. doi: 10.1186/s13024-020-00391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heneka M. T., Carson M. J., Khoury J., et al. Neuroinflammation in Alzheimer's disease. The Lancet Neurology . 2015;14(4):388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuello A. C., Ferretti M. T., Leon W. C., et al. Early-stage inflammation and experimental therapy in transgenic models of the Alzheimer-like amyloid pathology. Neurodegenerative Diseases . 2010;7(1-3):96–98. doi: 10.1159/000285514. [DOI] [PubMed] [Google Scholar]
- 5.Calsolaro V., Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimer’s & Dementia . 2016;12:719–732. doi: 10.1016/j.jalz.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Ibrahim A. M., Pottoo F. H., Dahiya E. S., Khan F. A., Kumar J. S. Neuron‐glia interactions: Molecular basis of alzheimer’s disease and applications of neuroproteomics. European Journal of Neuroscience . 2020;52(2):2931–2943. doi: 10.1111/ejn.14838. [DOI] [PubMed] [Google Scholar]
- 7.Streit W. J., Khoshbouei H., Bechmann I. The role of microglia in sporadic Alzheimer’s disease. Journal of Alzheimer’s Disease . 2021;79(3):961–968. doi: 10.3233/JAD-201248. [DOI] [PubMed] [Google Scholar]
- 8.Hansen D. V., Hanson J. E., Sheng M. Microglia in Alzheimer’s disease. Journal of Cell Biology . 2018;217(2):459–472. doi: 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hemonnot A. L., Hua J., Ulmann L., Hirbec H. Microglia in Alzheimer disease: well-known targets and new opportunities. Frontiers in Aging Neuroscience . 2019;11:p. 233. doi: 10.3389/fnagi.2019.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J., Liu L., Wang X., Jiang R., Bai Q., Wang G. Microglia: A double-edged sword in intracerebral hemorrhage from basic mechanisms to clinical research. Frontiers in Immunology . 2021;12 doi: 10.3389/FIMMU.2021.675660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frenkel D., Wilkinson K., Zhao L., et al. Scara1 deficiency impairs clearance of soluble amyloid- β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nature Communications . 2013;4(1):1–9. doi: 10.1038/ncomms3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarro V., Sanchez-Mejias E., Jimenez S., et al. Microglia in Alzheimer’s disease: activated, dysfunctional or degenerative. Frontiers in Aging Neuroscience . 2018;10 doi: 10.3389/fnagi.2018.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nizami S., Hall-Roberts H., Warrier S., Cowley S. A., Di Daniel E. Microglial inflammation and phagocytosis in Alzheimer’s disease: potential therapeutic targets. British Journal of Pharmacology . 2019;176(18):3515–3532. doi: 10.1111/bph.14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laversenne V., Nazeeruddin S., Källstig E. C., Colin P., Voize C., Schneider B. L. Anti-Aβ antibodies bound to neuritic plaques enhance microglia activity and mitigate tau pathology. Acta Neuropathologica Communications . 2020;8(1):1–19. doi: 10.1186/s40478-020-01069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michaud J. P., Hallé M., Lampron A., et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proceedings of the National Academy of Sciences . 2013;110(5):1941–1946. doi: 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helmut K., Hanisch U. K., Noda M., Verkhratsky A. Physiology of microglia. Physiological Reviews . 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 17.Sochocka M., Diniz B. S., Leszek J. Inflammatory response in the CNS: friend or foe? Molecular Neurobiology . 2017;54:8071–8089. doi: 10.1007/s12035-016-0297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogels T., Murgoci A.-N., Hromádka T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathologica Communications . 2019;7(1):p. ???. doi: 10.1186/S40478-019-0754-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maphis N., Xu G., Kokiko-Cochran O. N., et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain . 2015;138:1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerreiro R., Wojtas A., Bras J., et al. TREM2Variants in Alzheimer's Disease. New England Journal of Medicine . 2013;368(2):117–127. doi: 10.1056/nejmoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuyvers E., Sleegers K. Genetic variations underlying Alzheimer's disease: evidence from genome-wide association studies and beyond. The Lancet Neurology . 2016;15(8):857–868. doi: 10.1016/S1474-4422(16)00127-7. [DOI] [PubMed] [Google Scholar]
- 22.Yeh F. L., Wang Y., Tom I., Gonzalez L. C., Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron . 2016;91(2):328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Cella M., Mallinson K., et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer's Disease Model. Cell . 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krasemann S., Madore C., Cialic R., et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity . 2017;47(3):566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfe C. M., Fitz N. F., Nam K. N., Lefterov I., Koldamova R. The role of APOE and TREM2 in Alzheimer’s disease-current understanding and perspectives. International Journal of Molecular Sciences . 2019;20(1):p. 81. doi: 10.3390/ijms20010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao H., Coppola K., Schweig J. E., Crawford F., Mullan M., Paris D. Distinct signaling pathways regulate TREM2 phagocytic and NFκB antagonistic activities. Frontiers in Cellular Neuroscience . 2019;13:p. 457. doi: 10.3389/fncel.2019.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ulrich J. D., Ulland T. K., Colonna M., Holtzman D. M. Elucidating the Role of TREM2 in Alzheimer's Disease. Neuron . 2017;94(2):237–248. doi: 10.1016/j.neuron.2017.02.042. [DOI] [PubMed] [Google Scholar]
- 28.Matejuk A., Ransohoff R. M. Crosstalk between astrocytes and microglia: an overview. Frontiers in Immunology . 2020;11:p. 1416. doi: 10.3389/fimmu.2020.01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Ulland T. K., Ulrich J. D., et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. Journal of Experimental Medicine . 2016;213:667–675. doi: 10.1084/jem.20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldman L., Schafer A. I. Goldman’s Cecil Medicine: Twenty Fourth Edition . Vol. 1. Elsevier Inc.; 2012. Approach to Medicine, the Patient, and the Medical Profession: Medicine as a Learned and Humane Profession; pp. 1–2. [DOI] [Google Scholar]
- 31.Frost J. L., Schafer D. P. Microglia: architects of the developing nervous system. Trends in Cell Biology . 2016;26(8):587–597. doi: 10.1016/j.tcb.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Damani M. R., Zhao L., Fontainhas A. M., Amaral J., Fariss R. N., Wong W. T. Age-related alterations in the dynamic behavior of microglia. Aging Cell . 2011;10(2):263–276. doi: 10.1111/j.1474-9726.2010.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masliah E., Crews L., Hansen L. Synaptic remodeling during aging and in Alzheimer’s disease. Journal of Alzheimer's Disease . 2006;9(s3):91–99. doi: 10.3233/jad-2006-9s311. [DOI] [PubMed] [Google Scholar]
- 34.Yeh F. L., Hansen D. V., Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends in Molecular Medicine . 2017;23(6):512–533. doi: 10.1016/J.MOLMED.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Angelova D. M., Brown D. R. Microglia and the aging brain: are senescent microglia the key to neurodegeneration? Journal of Neurochemistry . 2019;151(6):676–688. doi: 10.1111/jnc.14860. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y., Jin M. Z., Yang Z. Y., Jin W. L. Microglia in neurodegenerative diseases. Neural Regeneration Research . 2021;16(2):270–280. doi: 10.4103/1673-5374.290881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sierra A., Gottfried-Blackmore A. C., Mcewen B. S., Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia . 2007;55(4):412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- 38.Conde J. R., Streit W. J. Effect of aging on the microglial response to peripheral nerve injury. Neurobiology of Aging . 2006;27(10):1451–1461. doi: 10.1016/j.neurobiolaging.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Hefendehl J. K., Neher J. J., Sühs R. B., Kohsaka S., Skodras A., Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell . 2014;13:60–69. doi: 10.1111/acel.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egensperger R., Kösel S., Von Eitzen U., Graeber M. B. Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathology . 1998;8(3):439–447. doi: 10.1111/j.1750-3639.1998.tb00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons D. A., Casale M., Alcon B., Pham N., Narayan N., Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia . 2007;55(10):1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- 42.Streit W. J., Braak H., Xue Q. S., Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathologica . 2009;118(4):475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safaiyan S., Kannaiyan N., Snaidero N., et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nature Neuroscience . 2016;19(8):995–998. doi: 10.1038/nn.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Euler Z., Schuitemaker H. Cross-reactive broadly neutralizing antibodies: timing is everything. Frontiers in Immunology . 2012;3 doi: 10.3389/fimmu.2012.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Bernhardi R., Eugenín-von Bernhardi L., Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Frontiers in Aging Neuroscience . 2015;7 doi: 10.3389/fnagi.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Godbout J. P., Chen J., Abraham J., et al. Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system. The FASEB Journal . 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- 47.Henry C. J., Huang Y., Wynne A. M., Godbout J. P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain, Behavior, and Immunity . 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker D. G., Dalsing-Hernandez J. E., Campbell N. A., Lue L. F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Experimental Neurology . 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuhrmann M., Bittner T., Jung C. K. E., et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nature Neuroscience . 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Bernhardi R., Cornejo F., Parada G. E., Eugenín J. Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Frontiers in Cellular Neuroscience . 2015;9 doi: 10.3389/fncel.2015.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klegeris A., Bissonnette C. J., McGeer P. L. Modulation of human microglia and THP-1 cell toxicity by cytokines endogenous to the nervous system. Neurobiology of Aging . 2005;26:673–682. doi: 10.1016/j.neurobiolaging.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Rock R. B., Gekker G., Hu S., et al. Role of microglia in central nervous system infections. Clinical microbiology reviews . 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wehrspaun C. C., Haerty W., Ponting C. P. Microglia recapitulate a hematopoietic master regulator network in the aging human frontal cortex. Neurobiology of Aging . 2015;36(2443):2443.e9–2443.e20. doi: 10.1016/j.neurobiolaging.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Streit W. J., Mrak R. E., Griffin W. S. T. Microglia and neuroinflammation: a pathological rerspective. Journal of Neuroinflammation . 2004;1:p. 14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hart A. D., Wyttenbach A., Hugh Perry V., Teeling J. L. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain, Behavior, and Immunity . 2012;26:754–765. doi: 10.1016/j.bbi.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grabert K., Michoel T., Karavolos M. H., et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nature Neuroscience . 2016;19:504–516. doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caldeira C., Oliveira A. F., Cunha C., et al. Microglia change from a reactive to an age-like phenotype with the time in culture. Frontiers in Cellular Neuroscience . 2014;8 doi: 10.3389/fncel.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uddin M. S., Kabir M., Jalouli M., et al. Neuroinflammatory signaling in the pathogenesis of Alzheimer’s disease. Current Neuropharmacology . 2021;20:126–146. doi: 10.2174/1570159X19666210826130210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olmos-Alonso A., Schetters S. T. T., Sri S., et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain . 2016;139:891–907. doi: 10.1093/brain/awv379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woodbury M. E., Freilich R. W., Cheng C. J., et al. MiR-155 is essential for inflammation-induced hippocampal neurogenic dysfunction. J. Neurosci. . 2015;35:9764–9781. doi: 10.1523/JNEUROSCI.4790-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keren-Shaul H., Spinrad A., Weiner A., et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell . 2017;169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 62.Ofengeim D., Mazzitelli S., Ito Y., et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proceedings of the National Academy of Sciences . 2017;114:E8788–E8797. doi: 10.1073/pnas.1714175114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yin Z., Raj D., Saiepour N., et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiology of aging . 2017;55:115–122. doi: 10.1016/j.neurobiolaging.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 64.Meda L., Cassatella M. A., Szendrei G. I., et al. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature . 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 65.Katsumoto A., Takeuchi H., Takahashi K., Tanaka F. Microglia in Alzheimer’s disease: risk factors and inflammation. Frontiers in Neurology . 2018;9:p. 978. doi: 10.3389/fneur.2018.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pais T. F., Figueiredo C., Peixoto R., Braz M. H., Chatterjee S. Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. J. Neuroinflammation . 2008;5:p. 43. doi: 10.1186/1742-2094-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGeer P. L., McGeer E. G. Inflammation, autotoxicity and Alzheimer disease. Neurobiology of aging . 2001;22:799–809. doi: 10.1016/S0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 68.Della Bianca V., Dusi S., Bianchini E., Dal Prà I., Rossi F. β-Amyloid activates the O2/- forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. Journal of Biological Chemistry . 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 69.Shimohama S., Tanino H., Kawakami N., et al. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochemical and biophysical research communications . 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 70.El Khoury J., Hickman S. E., Thomas C. A., Cao L., Silverstein S. C., Loike J. D. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature . 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 71.McDonald D. R., Brunden K. R., Landreth G. E. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. Journal of Neuroscience . 1997;17:2284–2294. doi: 10.1523/jneurosci.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Block M. L. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC neuroscience . 2008;9 doi: 10.1186/1471-2202-9-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takeuchi H., Mizuno T., Zhang G., et al. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. Journal of Biological Chemistry . 2005;280:10444–10454. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- 74.Piani D., Spranger M., Frei K., Schaffner A., Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor psitive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. European journal of immunology . 1992;22:2429–2436. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- 75.Barger S. W., Basile A. S. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. Journal of neurochemistry . 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 76.Bordji K., Becerril-Ortega J., Nicole O., Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. Journal of Neuroscience . 2010;30:15927–15942. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walsh D. M., Klyubin I., Fadeeva J. V., et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature . 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 78.Li S., Hong S., Shepardson N. E., Walsh D. M., Shankar G. M., Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron . 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor perspectives in medicine . 2011;1:a006189–a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoshiyama Y., Higuchi M., Zhang B., et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron . 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 81.Liddelow S. A., Guttenplan K. A., Clarke L. E., et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature . 2017;541:481–487. doi: 10.1038/NATURE21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Preman P., Alfonso-Triguero M., Alberdi E., Verkhratsky A., Arranz A. M. Astrocytes in Alzheimer’s disease: pathological significance and molecular pathways. Cells . 2021;10:1–19. doi: 10.3390/CELLS10030540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y., Balaji V., Kaniyappan S., et al. The release and trans-synaptic transmission of tau via exosomes. Molecular neurodegeneration . 2017;12 doi: 10.1186/S13024-016-0143-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walport M. J. Complement. Second of two parts. The New England journal of medicine . 2001;344(15):1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 85.Keene C. D., Cudaback E., Li X., Montine K. S., Montine T. J. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Current opinion in neurobiology . 2011;21:920–928. doi: 10.1016/j.conb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shen Y., Meri S. Yin and yang: complement activation and regulation in Alzheimer’s disease. Progress in neurobiology . 2003;70:463–472. doi: 10.1016/j.pneurobio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 87.Bonifati D. M., Kishore U. Role of complement in neurodegeneration and neuroinflammation. Molecular immunology . 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 88.Strohmeyer R., Ramirez M., Cole G. J., Mueller K., Rogers J. Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer’s disease brain. Journal of neuroimmunology . 2002;131:135–146. doi: 10.1016/s0165-5728(02)00272-2. [DOI] [PubMed] [Google Scholar]
- 89.Maier M., Peng Y., Jiang L., Seabrook T. J., Carroll M. C., Lemere C. A. Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. Journal of Neuroscience . 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Choucair-Jaafar N., Laporte V., Levy R., Poindron P., Lombard Y., Gies J. P. Complement receptor 3 (CD11b/CD18) is implicated in the elimination of β-amyloid peptides. Fundamental & clinical pharmacology . 2011;25:115–122. doi: 10.1111/j.1472-8206.2010.00811.x. [DOI] [PubMed] [Google Scholar]
- 91.Fu H., Liu B., Frost J. L., et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia . 2012;60:993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang D., Hu X., Qian L., et al. Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. Journal of Neuroinflammation . 2011;8:p. 3. doi: 10.1186/1742-2094-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hong S., Beja-Glasser V. F., Nfonoyim B. M., et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science . 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crehan H., Hardy J., Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiology of disease . 2013;54:139–149. doi: 10.1016/j.nbd.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 95.Rogers J., Li R., Mastroeni D., et al. Peripheral clearance of amyloid β peptide by complement C3-dependent adherence to erythrocytes. Neurobiology of disease . 2006;27:1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 96.Crehan H., Holton P., Wray S., Pocock J., Guerreiro R., Hardy J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology . 2012;217:244–250. doi: 10.1016/j.imbio.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 97.Fonseca M. I., Ager R. R., Chu S.-H., et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. The Journal of Immunology . 2009;183:1375–1383. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takeda K., Yamamoto M. Current views of Toll-like receptor signaling pathways. Gastroenterology research and practice . 2010;2010 doi: 10.1155/2010/240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jin J. J., Kim H. D., Maxwell J. A., Li L., Fukuchi K. I. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. Journal of neuroinflammation . 2008;5 doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jana M., Palencia C. A., Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. The Journal of Immunology . 1950;2008(181):7254–7262. doi: 10.4049/jimmunol.181.10.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu S., Liu Y., Hao W., et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. The Journal of Immunology . 2012;188:1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 102.Tahara K., Kim H. D., Jin J. J., Maxwell J. A., Li L., Fukuchi K. I. Role of Toll-like receptor signalling in Aβ uptake and clearance. Brain . 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Momtazmanesh S., Perry G., Rezaei N. Toll-like receptors in Alzheimer’s disease. Journal of Neuroimmunology . 2020;348 doi: 10.1016/J.JNEUROIM.2020.577362. [DOI] [PubMed] [Google Scholar]
- 104.Murgas P., Godoy B., Von Bernhardi R. Aβ potentiates inflammatory activation of glial cells induced by scavenger receptor ligands and inflammatory mediators in culture. Neurotoxicity research . 2012;22:69–78. doi: 10.1007/s12640-011-9306-3. [DOI] [PubMed] [Google Scholar]
- 105.Yang C. N., Shiao Y. J., Shie F. S., et al. Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiology of disease . 2011;42:221–230. doi: 10.1016/j.nbd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 106.Hickman S. E., Allison E. K., El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. Journal of Neuroscience . 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cornejo F., Vruwink M., Metz C., et al. Scavenger receptor-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer’s disease pathophysiology. Brain, behavior, and immunity . 2018;69:336–350. doi: 10.1016/j.bbi.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 108.Coraci I. S., Husemann J., Berman J. W., et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to β-amyloid fibrils. The American journal of pathology . 2002;160:101–112. doi: 10.1016/S0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stewart C. R., Stuart L. M., Wilkinson K., et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nature immunology . 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.El Khoury J. B., Moore K. J., Means T. K., et al. CD36 mediates the innate host response to β-amyloid. The Journal of experimental medicine . 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li X., Melief E., Postupna N., Montine K. S., Keene C. D., Montine T. J. Prostaglandin E2 receptor subtype 2 regulation of scavenger receptor CD36 modulates microglial Aβ42 phagocytosis. The American journal of pathology . 2015;185:p. 230. doi: 10.1016/J.AJPATH.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Deane R., Du Yan S., Submamaryan R. K., et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nature medicine . 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 113.Origlia N., Righi M., Capsoni S., et al. Receptor for advanced glycation end product-dependent activation of P38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. Journal of Neuroscience . 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Onyango I. G., Tuttle J. B., Bennett J. P. Altered intracellular signaling and reduced viability of Alzheimer’s disease neuronal cybrids is reproduced by β-amyloid peptide acting through receptor for advanced glycation end products (RAGE) Molecular and Cellular Neuroscience . 2005;29:333–343. doi: 10.1016/j.mcn.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 115.Fang F., Lue L. F., Yan S., et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Aβ accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. The FASEB Journal . 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shen C., Ma Y., Zeng Z., et al. RAGE-specific inhibitor FPS-ZM1 attenuates AGEs-induced neuroinflammation and oxidative stress in rat primary microglia. Neurochemical Research . 2017;42(10):2902–2911. doi: 10.1007/S11064-017-2321-X. [DOI] [PubMed] [Google Scholar]
- 117.Frank S., Burbach G. J., Bonin M., et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia . 2008;56:1438–1447. doi: 10.1002/glia.20710. [DOI] [PubMed] [Google Scholar]
- 118.Ulland T. K., Song W. M., Huang S. C., et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell . 2017;170:649–663.e13. doi: 10.1016/J.CELL.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ager R. R., Fonseca M. I., Chu S. H., et al. Microglial C5aR (CD88) expression correlates with amyloid-β deposition in murine models of Alzheimer’s disease. Journal of neurochemistry . 2010;113:389–401. doi: 10.1111/j.1471-4159.2010.06595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.O’Barr S., Cooper N. R. The C5a complement activation peptide increases IL-1β and IL-6 release from amyloid-β primed human monocytes: implications for Alzheimer’s disease. Journal of neuroimmunology . 2000;109:87–94. doi: 10.1016/S0165-5728(00)00291-5. [DOI] [PubMed] [Google Scholar]
- 121.Wyss-Coray T., Yan F., Lin A. H. T., et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proceedings of the National Academy of Sciences . 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Song M., Jin J. J., Lim J. E., et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Journal of neuroinflammation . 2011;8 doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Richard K. L., Filali M., Préfontaine P., Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. Journal of Neuroscience . 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia . 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- 125.Hanke M. L., Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clinical science . 2011;121:367–387. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Carty M., Bowie A. G. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochemical pharmacology . 2011;81:825–837. doi: 10.1016/j.bcp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 127.Olson J. K., Miller S. D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. The Journal of Immunology . 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 128.Frank S., Copanaki E., Burbach G. J., Müller U. C., Deller T. Differential regulation of Toll-like receptor MRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neuroscience letters . 2009;453:41–44. doi: 10.1016/j.neulet.2009.01.075. [DOI] [PubMed] [Google Scholar]
- 129.Bsibsi M., Ravid R., Gveric D., van Noort J. M. Broad expression of Toll-like receptors in the hman central nervous system. Journal of Neuropathology & Experimental Neurology . 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 130.Chen K., Iribarren P., Hu J., et al. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid β peptide. Journal of Biological Chemistry . 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 131.Walter S., Letiembre M., Liu Y., et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cellular Physiology and Biochemistry . 2007;20(6):947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 132.Su F., Bai F., Zhou H., Zhang Z. Reprint of: Microglial toll-like receptors and Alzheimer’s disease. Brain, Behavior, and Immunity . 2016;55:166–178. doi: 10.1016/j.bbi.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 133.Gambuzza M., Sofo V., Salmeri F., Soraci L., Marino S., Bramanti P. Toll-like receptors in Alzheimer’s disease: a therapeutic perspective. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders) . 2014;13:1542–1558. doi: 10.2174/1871527313666140806124850. [DOI] [PubMed] [Google Scholar]
- 134.Landreth G. E., Reed-Geaghan E. G. Toll-like receptors in Alzheimer’s disease. Journal of Neuroimmunology . 2009;336:137–153. doi: 10.1007/978-3-642-00549-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fiebich B. L., Batista C. R. A., Saliba S. W., Yousif N. M., de Oliveira A. C. P. Role of microglia TLRs in neurodegeneration. Frontiers in Cellular Neuroscience . 2018;12:p. 329. doi: 10.3389/fncel.2018.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hoebe K., Georgel P., Rutschmann S., et al. CD36 is a sensor of diacylglycerides. Nature . 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 137.Gao D., Ashraf M. Z., Kar N. S., Lin D., Sayre L. M., Podrez E. A. Structural basis for the recognition of oxidized phospholipids in oxidized low density lipoproteins by class B scavenger receptors CD36 and SR-BI. Journal of Biological Chemistry . 2010;285:4447–4454. doi: 10.1074/jbc.M109.082800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Savill J., Hogg N., Ren Y., Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. The Journal of clinical investigation . 1992;90:1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moore K. J., El Khoury J., Medeiros L. A., et al. A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. Journal of Biological Chemistry . 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 140.Bornemann K. D., Wiederhold K. H., Pauli C., et al. Aβ-induced inflammatory processes in microglia cells of APP23 transgenic mice. The American Journal of Pathology . 2001;158:63–73. doi: 10.1016/S0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ricciarelli R., D’Abramo C., Zingg J. M., et al. CD36 overexpression in human brain correlates with β-amyloid deposition but not with Alzheimer’s disease. Free Radical Biology and Medicine . 2004;36:1018–1024. doi: 10.1016/j.freeradbiomed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 142.Fabriek B. O., Dijkstra C. D., van den Berg T. K. The macrophage scavenger receptor CD163. Immunobiology . 2005;210:153–160. doi: 10.1016/J.IMBIO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 143.Polfliet M. M. J., Fabriek B. O., Daniëls W. P., Dijkstra C. D., van den Berg T. K. The rat macrophage scavenger receptor CD163: expression, regulation and role in inflammatory mediator production. Immunobiology . 2006;211:419–425. doi: 10.1016/J.IMBIO.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 144.Fabriek B. O., van Bruggen R., Deng D. M., et al. The macrophage scavenger receptor CD163 functions as an innate immune sensor for bacteria. Blood . 2009;113:887–892. doi: 10.1182/BLOOD-2008-07-167064. [DOI] [PubMed] [Google Scholar]
- 145.Roberts E. S., Masliah E., Fox H. S. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE) Journal of Neuropathology & Experimental Neurology . 2004;63:1255–1264. doi: 10.1093/JNEN/63.12.1255. [DOI] [PubMed] [Google Scholar]
- 146.Leclerc E., Fritz G., Vetter S. W., Heizmann C. W. Binding of S100 proteins to RAGE: an update. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research . 2009;1793:993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 147.Moreira P. I., Duarte A. I., Santos M. S., Rego A. C., Oliveira C. R. An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. Journal of Alzheimer's Disease . 2009;16:741–761. doi: 10.3233/JAD-2009-0972. [DOI] [PubMed] [Google Scholar]
- 148.Reddy V. P., Zhu X., Perry G., Smith M. A. Oxidative stress in diabetes and Alzheimer’s disease. Journal of Alzheimer's Disease . 2009;16:763–774. doi: 10.3233/JAD-2009-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Origlia N., Bonadonna C., Rosellini A., et al. Microglial receptor for advanced glycation end product-dependent signal pathway drives-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. Journal of Neuroscience . 2010;30:11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee V. M. Y., Goedert M., Trojanowski J. Q. Neurodegenerative tauopathies. Annual review of neuroscience . 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 151.Gebicke-Haerter P. J. Microglia in neurodegeneration: molecular aspects. Microscopy research and technique . 2001;54:47–58. doi: 10.1002/jemt.1120. [DOI] [PubMed] [Google Scholar]
- 152.Zilka N., Stozicka Z., Kovac A., Pilipcinec E., Bugos O., Novak M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. Journal of neuroimmunology . 2009;209:16–25. doi: 10.1016/j.jneuroim.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 153.Bolós M., Llorens-Martín M., Jurado-Arjona J., Hernández F., Rábano A., Avila J. Direct evidence of internalization of tau by microglia in vitro and in vivo. Journal of Alzheimer's Disease . 2016;50:77–87. doi: 10.3233/JAD-150704. [DOI] [PubMed] [Google Scholar]
- 154.Bhaskar K., Konerth M., Kokiko-Cochran O. N., Cardona A., Ransohoff R. M., Lamb B. T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron . 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cho S.-H., Sun B., Zhou Y., et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. Journal of Biological Chemistry . 2011;286:32713–32722. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ginhoux F., Greter M., Leboeuf M., et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science . 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bolós M., Llorens-Martín M., Perea J. R., et al. Absence of CX3CR1 impairs the internalization of tau by microglia. Molecular neurodegeneration . 2017;12:p. 59. doi: 10.1186/s13024-017-0200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bemiller S. M., McCray T. J., Allan K., et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Molecular neurodegeneration . 2017;12 doi: 10.1186/S13024-017-0216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jiang T., Zhang Y. D., Gao Q., et al. TREM2 ameliorates neuronal tau pathology through suppression of microglial inflammatory response. Inflammation . 2018;41(3):811–823. doi: 10.1007/S10753-018-0735-5. [DOI] [PubMed] [Google Scholar]
- 160.Sanchez-Mejias E., Navarro V., Jimenez S., et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta neuropathologica . 2016;132:897–916. doi: 10.1007/s00401-016-1630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Streit W. J., Braak H., Del Tredici K., et al. Microglial activation occurs late during preclinical Alzheimer’s disease. Glia . 2018;66:2550–2562. doi: 10.1002/GLIA.23510. [DOI] [PubMed] [Google Scholar]
- 162.Leng F., Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nature Reviews Neurology . 2021;17(3):157–172. doi: 10.1038/s41582-020-00435-y. [DOI] [PubMed] [Google Scholar]
- 163.McGeer P. L., McGeer E. G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta neuropathologica . 2013;126:479–497. doi: 10.1007/s00401-013-1177-7. [DOI] [PubMed] [Google Scholar]
- 164.Uddin M. S., Kabir M. T., Jeandet P., et al. Novel anti-Alzheimer’s therapeutic molecules targeting amyloid precursor protein processing. Oxidative Medicine and Cellular Longevity . 2020;2020:19. doi: 10.1155/2020/7039138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Amor S., Puentes F., Baker D., Van Der Valk P. Inflammation in neurodegenerative diseases. Immunology . 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hamelin L., Lagarde J., Dorothée G., et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain . 2016;139:1252–1264. doi: 10.1093/brain/aww017. [DOI] [PubMed] [Google Scholar]
- 167.Guerreiro R. J., Hardy J. Alzheimer’s disease genetics: lessons to improve disease modelling. Biochemical Society transactions . 2011;39:910–916. doi: 10.1042/BST0390910. [DOI] [PubMed] [Google Scholar]
- 168.Reitz C., Mayeux R. Alzheimer’s disease genetics consortium TREM2 and neurodegenerative disease. New England Journal of Medicine . 2013;369:1564–1565. doi: 10.1056/NEJMc1306509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Uddin M. S., Kabir M. T., Al Mamun A., et al. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. International Immunopharmacology . 2020;84:p. 106479. doi: 10.1016/j.intimp.2020.106479. [DOI] [PubMed] [Google Scholar]
- 170.Fan Z., Brooks D. J., Okello A., Edison P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain . 2017;140:792–803. doi: 10.1093/brain/aww349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Qiu W. Q., Walsh D. M., Ye Z., et al. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. Journal of Biological Chemistry . 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 172.Frautschy S. A., Yang F., Irrizarry M., et al. Microglial response to amyloid plaques in APPsw transgenic mice. The American journal of pathology . 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 173.Frackowiak J., Wisniewski H. M., Wegiel J., Merz G. S., Iqbal K., Wang K. C. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta neuropathologica . 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 174.Jimenez S., Baglietto-Vargas D., Caballero C., et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. Journal of Neuroscience . 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Okello A., Koivunen J., Edison P., et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study symbol. Neurology . 2009;73:754–760. doi: 10.1212/WNL.0b013e3181b23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zotova E., Holmes C., Johnston D., Neal J. W., Nicoll J. A., Boche D. Microglial alterations in human Alzheimer's disease following Aβ42 immunization. Neuropathology and applied neurobiology . 2011;37(5):513–524. doi: 10.1111/j.1365-2990.2010.01156.x. [DOI] [PubMed] [Google Scholar]
- 177.Polazzi E., Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Progress in neurobiology . 2010;92:293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 178.Streit W. J., Sammons N. W., Kuhns A. J., Sparks D. L. Dystrophic microglia in the aging human brain. Glia . 2004;45:208–212. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- 179.Heslegrave A., Heywood W., Paterson R., et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Molecular neurodegeneration . 2016;11:p. 3. doi: 10.1186/s13024-016-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Piccio L., Deming Y., Del-Águila J. L., et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta neuropathologica . 2016;131:925–933. doi: 10.1007/s00401-016-1533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Suárez‐Calvet M., Kleinberger G., Araque Caballero M. Á., et al. STREM 2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO molecular medicine . 2016;8:466–476. doi: 10.15252/emmm.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jiang T., Yu J. T., Zhu X. C., Tan L. TREM2 in Alzheimer’s disease. Molecular neurobiology . 2013;48:180–185. doi: 10.1007/s12035-013-8424-8. [DOI] [PubMed] [Google Scholar]
- 183.Serrano-Pozo A., Betensky R. A., Frosch M. P., Hyman B. T. Plaque-associated local toxicity increases over the clinical course of Alzheimer disease. The American journal of pathology . 2016;186:375–384. doi: 10.1016/j.ajpath.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Serrano-Pozo A., Mielke M. L., Gómez-Isla T., et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. The American journal of pathology . 2011;179:1373–1384. doi: 10.1016/j.ajpath.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Serrano-Pozo A., Gómez-Isla T., Growdon J. H., Frosch M. P., Hyman B. T. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. The American journal of pathology . 2013;182:2332–2344. doi: 10.1016/j.ajpath.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica . 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 187.Dickson D. W., Crystal H. A., Mattiace L. A., et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiology of aging . 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-U. [DOI] [PubMed] [Google Scholar]
- 188.Grober E., Dickson D., Sliwinski M. J., et al. Memory and mental status correlates of modified Braak staging. Neurobiology of aging . 1999;20:573–579. doi: 10.1016/S0197-4580(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 189.Riley K. P., Snowdon D. A., Markesbery W. R. Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Annals of neurology . 2002;51:567–577. doi: 10.1002/ana.10161. [DOI] [PubMed] [Google Scholar]
- 190.Streit W. J. Microglia and neuroprotection: implications for Alzheimer’s disease. Brain Research Reviews . 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 191.Streit W. J. Microglia and Alzheimer’s disease pathogenesis. Journal of neuroscience research . 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- 192.Wang X., Zhao L., Zhang J., et al. Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. Journal of Neuroscience . 2016;36:2827–2842. doi: 10.1523/JNEUROSCI.3575-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Parkhurst C. N., Yang G., Ninan I., et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell . 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Krauthausen M., Kummer M. P., Zimmermann J., et al. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer’s disease model. The Journal of clinical investigation . 2015;125:365–378. doi: 10.1172/JCI66771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yamanaka M., Ishikawa T., Griep A., Axt D., Kummer M. P., Heneka M. T. PPARγ/RXRA-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. Journal of Neuroscience . 2012;32:17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yuan P., Condello C., Keene C. D., et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron . 2016;90:724–739. doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Condello C., Yuan P., Schain A., Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nature communications . 2015;6 doi: 10.1038/ncomms7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chitu V., Gokhan Ş., Nandi S., Mehler M. F., Stanley E. R. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends in neurosciences . 2016;39:378–393. doi: 10.1016/j.tins.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Paloneva J., Kestilä M., Wu J., et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nature genetics . 2000;25:357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- 200.Paloneva J., Manninen T., Christman G., et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. The American Journal of Human Genetics . 2002;71(3):656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Poliani P. L., Wang Y., Fontana E., et al. TREM2 sustains microglial expansion during aging and response to demyelination. The Journal of clinical investigation . 2015;125(5):2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Flanary B. E., Sammons N. W., Nguyen C., Walker D., Streit W. J. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation research . 2007;10:61–74. doi: 10.1089/rej.2006.9096. [DOI] [PubMed] [Google Scholar]
- 203.Streit W. J. Microglial senescence: does the brain’s immune system have an expiration date? Trends in neurosciences . 2006;29:506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 204.Grodstein F., van Oijen M., Irizarry M. C., et al. Shorter telomeres may mark early risk of dementia: preliminary analysis of 62 participants from the nurses’ health study. PLoS One . 2008;3 doi: 10.1371/journal.pone.0001590. [DOI] [PMC free article] [PubMed] [Google Scholar]