

Abstract
Background
Schaaf‐Yang syndrome (SYS) is a rare hereditary disease caused by truncating point mutations of the paternal allele of melanoma antigen L2 (MAGEL2), one of five protein‐coding genes within the Prader‐Willi syndrome (PWS) critical domain. SYS shares many clinical and molecular characteristics with PWS but has some distinct features, such as joint contractures and autism. Patients with PWS show abnormal electroencephalography (EEG) patterns. However, there are very few reports on EEG findings in patients with SYS.
Methods
A SYS patient was included in this study. Detailed neurological examinations and EEG were performed from neonate to infant ages. Sanger sequencing was performed.
Results
Our patient presented abnormal EEG findings and had diffuse brain dysfunction symptoms including a reduced level of consciousness, diminished spontaneous movements, hypotonia, feeding difficulties, and hypoventilation from early after birth. As she grew older and her background activity of EEG normalized, her neurodevelopmental symptoms remained but improved. Sanger sequencing of this patient revealed a novel, heterozygous c.2005C > T, truncating mutation in the MEGAL2 gene.
Conclusions
We described an SYS‐associated, time‐dependent, EEG pattern in a patient with SYS. Our findings of longitudinal EEG changes in a patient with SYS revealed a specific pattern of how affected individuals develop brain function.
Keywords: electroencephalography, MAGEL2, neurodevelopmental disorders, Schaaf‐Yang syndrome
We describe a Schaaf‐Yang syndrome‐associated, time‐dependent, EEG pattern. Sanger sequencing of this patient revealed a novel, heterozygous c.2005C>T, truncating mutation in the MEGAL 2 gene. Our findings of longitudinal changes in EEG in a patient with Schaaf‐Yang syndrome showed a specific pattern of how affected individuals develop brain function.

1. INTRODUCTION
Patients with neonatal hypotonia, difficulty in feeding, intellectual disability, hypogonadism, joint contractures, and autism were reported by Schaaf et al. (2013) to have a “Prader‐Willi‐like syndrome”; those individuals share partial phenotypic characteristics with patients with Prader‐Willi syndrome (PWS [OMIM # 176270]) (Schaaf et al., 2013). As the distinctive phenotypic presentation of these patients became clearer, this disease was named Schaaf‐Yang syndrome (SHFYNG [OMIM # 615547]). Complete genome or whole‐exome sequencing revealed that SYS is caused by truncating point mutations in the maternally imprinted, paternally expressed melanoma antigen L2 (MAGEL2) gene, in the Prader‐Willi critical region on chromosome 15q11–15q13 (Sherry et al., 2001).
SYS typically manifests at birth with muscular hypotonia in almost all affected individuals and distal joint contractures in the majority of affected individuals (McCarthy et al., 2018; Patak et al., 2019). Many patients with SYS experience respiratory distress at birth, feeding difficulties in infancy and childhood, scoliosis, autism spectrum disorder, and developmental delay (McCarthy et al., 2018; Patak et al., 2019). Other findings, such as short stature, seizures, eye anomalies, endocrine dysfunction, hypogonadism, gastroesophageal reflux disease (GERD), sleep apnea, and temperature instability, are also commonly reported (McCarthy et al., 2018; Patak et al., 2019). However, there are no reports that describe seizure characteristics or EEG patterns in patients with SYS.
Neonatal EEG is an objective assessment of the functional integrity of the neonatal brain in a variety of neurodevelopmental disorders. The MAGEL2 protein is expressed in the supraoptic, paraventricular, and suprachiasmatic nuclei of the hypothalamus in some patients with PWS (Bervini & Herzog, 2013; Hiroi et al., 2000; Swaab, 1997). However, to our knowledge, there are no EEG reports on time‐dependent changes in patients with SYS. The present study shows, for the first time, that a heterozygous c.2005C > T, p.Gln669 variant, a new truncating mutation in MAGEL2, causes a characteristic longitudinal EEG pattern in SYS from the neonatal to infant age period.
2. MATERIALS AND METHODS
2.1. Case presentation
The patient was the first girl from a dichorionic diamniotic twin pregnancy born to a nonconsanguineous couple. The family histories of the patient were noncontributory. The pregnancy was uncomplicated, and the baby was delivered by Cesarean section at a gestational age of 36 weeks and 0 days. Apgar scores were 8 and 9, at 1 and 5 min, respectively. The birth weight was 2286 g (25–50th), cranial circumferences were 31.6 cm (50th), and birth length was 45.0 cm (25–50th).
The patient was admitted to the neonatal intensive care unit because of frequent apneic episodes and hematemesis. She presented with hypotonia in her upper and lower limbs with distal joint contractures, typical facial dysmorphism, and weak suction with difficulty in swallowing shortly after birth (Figure 1). On day 2, the baby was intubated and placed on an artificial ventilator because of frequent apneic episodes. As a neonate on day 23, her brain magnetic resonance imaging (MRI) results were normal (Figure 2). Following two unsuccessful attempts at extubation, we performed a tracheostomy on day 52. On day 65, the patient suddenly developed tachycardia above 200 beats/min; the tachycardia lasted over 24 h and was accompanied by fever and sweating. On day 66, her blood test showed high levels (1771.6 pg/ml) of brain natriuretic peptide. The patient’s echocardiogram showed no findings for congenital heart disease, but we diagnosed heart failure caused by tachycardia. Heart failure was alleviated by the administration of the beta‐blocker propranolol hydrochloride and the sodium‐potassium ATPase pump inhibitor digoxin. On day 139, she was discharged from the hospital with a tracheostomy tube and a feeding tube.
FIGURE 1.
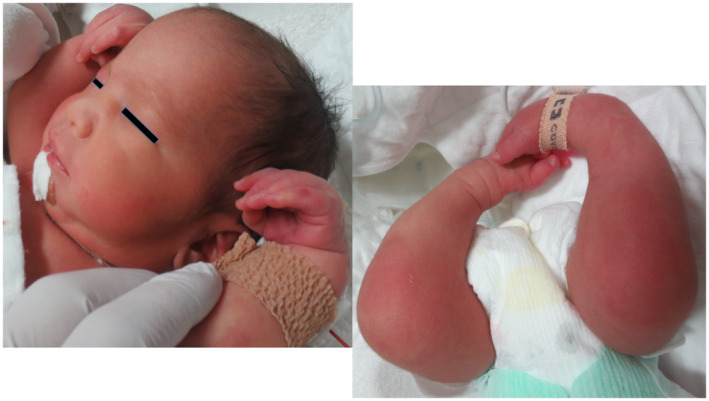
Extremities with distal joint contractures and characteristic facial dysmorphism
FIGURE 2.
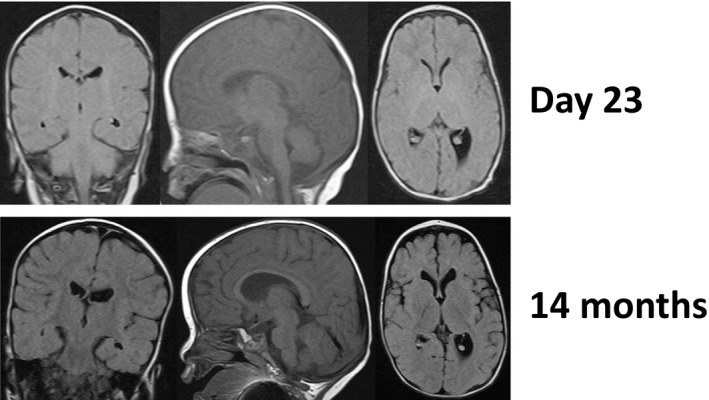
MRI at day 23 and 14 months
The first EEG, on day 17 (38 weeks gestational age), was performed without the patient being given a sedative or an antiepileptic drug. That test revealed that background activity was depressed with low voltage and discontinuous activity, as well as a lack of tracé alternant and a high voltage slow wave quiet sleep pattern, suggesting moderate depression of brain function (Figure 3). The EEG did not show an epileptic discharge. However, EEG recordings on day 65 (45 weeks gestational age) showed improved background wave activity (Figure 3). The patient’s neurological examination revealed improvements in her level of consciousness, movement, apnea, and swallowing. Brain MRIs at the ages of 2 months and 1 year revealed age‐appropriate myelination and slight enlargement of the lateral ventricle (Figure 2). At 4 months old, her background wave pattern returned to normal and was appropriately active; however, the patient still exhibited developmental delay but was slowly improving (Figure 3). At 9 and 16 months old, the EEG showed generalized rhythmic delta activity of 2–3 Hz and sometimes continuous beta activity, mainly from the posterior regions. At the ages of 9 months and 1 year, we tested for insulin‐thyrotropin‐releasing hormone (TRH)‐luteinizing hormone‐releasing hormone (LHRH) (ITL). The patient showed a delayed response to TRH stimulation, indicating hypothalamic disease. Moreover, a deficiency in growth hormone was confirmed, and she then began growth hormone therapy. Her younger twin has been growing, exhibiting none of the symptoms described.
FIGURE 3.

EEG time‐course at day 17, day 65, 4 months, 9 months, and 16 months. The EEG on day 17 shows that background activity is depressed with low voltage and discontinuous activity, and a lack of tracé alternant and high voltage slow wave quiet sleep pattern. The EEG on day 65 shows improved background wave activity. The EEG at 4 months shows normal background activity. At 9 and 16 months old, the EEG shows generalized rhythmic delta activity of 2–3 Hz and sometimes continuous beta activity, mainly from the posterior regions
2.2. Methods
The patient was clinically assessed by experienced clinical geneticists. After obtaining informed consent from the patient’s parents, genomic DNA was extracted from the peripheral blood samples of the patient. On the basis of the clinical scenario, Sanger sequencing was used to sequence only the MAGEL2 gene. The study protocol was approved by the Institutional Review Board of the Japanese Red Cross Wakayama Medical Center (approved number: 923). The parents of the patient provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki.
3. RESULTS
Sanger sequencing identified a heterozygous c.2005C > T, p.Gln669 variant in MAGEL2. This variant has not been reported previously in the dbSNP8 and gnomAD9 variant databases (Karczewski et al., 2020; Sherry et al., 2001). The cytosine at nucleotide position 2005 and the amino acid Gln669 are highly conserved. Therefore, the variant could be classified as potentially pathogenic.
4. DISCUSSION
The present study shows, for the first time, that a novel truncating mutation in MAGEL2 may be associated with characteristic EEG patterns in SYS. EEG recordings in a patient with SYS showed improved background wave activity from the neonatal to infant age periods; a neurological assessment confirmed these findings of improved development. Although other studies have reported neurodevelopmental delay in patients with SYS, changes in the background EEG wave have never been previously reported. Therefore, our data may provide insight into the mechanism underlying SYS‐related brain dysfunction.
PWS is caused by dysfunction in several paternal‐expressed genes, including makorin ring finger protein 3 (MKRN3), MAGEL2, necdin (NDN), small nuclear ribonucleoprotein polypeptide N (SNPRPN)‐upstream open reading frame (SNURF) (SNPRPN‐SNURF), nuclear pore‐associated protein 1 (NPAP1), and a cluster of small nucleolar RNA genes (Holm et al., 1993). Abnormal EEG findings are not common in patients with PWS but do occur; the most frequent observations are generalized and focal discharges (Elia et al., 2021; Sherry et al., 2001). PWS is associated with hypothalamic dysfunction, such as central hypoventilation, autonomic neuropathy, and temperature instability (Cohen et al., 2014; Felix et al., 2016; Miller & Wagner, 2013). Intellectual disability, dyspnea, and hypopituitarism also occur in patients with SYS (Chitayat et al., 1990; Yanjie et al., 2021). The MAGEL2 protein is highly expressed in the supraoptic, paraventricular, and suprachiasmatic nuclei of the hypothalamus in the embryonic and newborn brain and contributes to hypothalamic dysfunction in some patients with PWS (Bervini & Herzog, 2013; Hiroi et al., 2000; Swaab, 1997). Hypothalamic dysfunction, associated with EEG abnormalities, may occur in patients with SYS. To confirm this hypothesis, further evidence from a large number of patients with SYS is required.
The most important finding in the current study was the observation of characteristic changes in the background EEG wave pattern. These changes may be useful in assessing cerebral function in patients with SYS. Clinical presentations, including disturbed consciousness, loss of respiratory and circulatory autoregulation, hypotonia, feeding difficulty, acute gastric mucosal lesion, and temperature instability, may be attributed to hypothalamic immaturity like in patients with PWS. In the present study, the result of the ITL test supported this hypothesis. Our patient presented abnormal EEG findings and had diffuse brain dysfunction symptoms including a reduced level of consciousness, diminished spontaneous movements, hypotonia, feeding difficulties, and hypoventilation from early after birth. As she grew older and her background activity of EEG normalized, her neurodevelopmental symptoms remained but improved. Beta activity in the posterior regions has never been previously reported and may represent a unique observation in patients with SYS. Additional EEG findings from SYS cases are needed to confirm this point.
Second, a heterozygous c.2005C > T, p.Gln669 variant in the MAGEL2 gene is a novel mutation and may be related to the clinical presentation of SYS in our study. Some point mutations have been reported, and a heterozygous variant is the most frequent mutation of SYS (McCarthy et al., 2018; Patak et al., 2019). To our knowledge, a heterozygous c.2005C > T, p.Gln669 variant in the MAGEL2 gene has never been previously reported.
In conclusion, our findings of longitudinal EEG changes, from neonate to infant ages, indicate the specific pattern of how patients with SYS develop brain function and may be beneficial in managing their neurodevelopmental difficulties.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
Dr. Shinsuke conceptualized and designed this study, drafted the initial version of this paper, and reviewed and revised this paper. Dr. Yuka and Dr. Takayuki designed the data collection instruments, collected the data, and reviewed and revised this paper. Dr. Koji, Dr. Atsushi, and Dr. Shigeto interpreted the clinical data and critically reviewed this paper or important intellectual content. All the authors approved the final version of this paper as submitted and agree to be accountable for all aspects of this work.
ACKNOWLEDGMENT
We are grateful to Osaka Women’s and Children’s Hospital for technical help.
Mizuno, S. , Yokoyama, K. , Yokoyama, A. , Nukata, T. , Ikeda, Y. & Hara, S. (2022). Longitudinal analysis of electroencephalography pattern changes in an infant with Schaaf‐Yang syndrome and a novel mutation in melanoma antigen L2 (MAGEL2). Molecular Genetics & Genomic Medicine, 10, e1932. 10.1002/mgg3.1932
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Bervini, S. , & Herzog, H. (2013, April). Mouse models of Prader‐Willi syndrome: A systematic review. Frontiers in Neuroendocrinology, 34(2), 107–119. [DOI] [PubMed] [Google Scholar]
- Chitayat, D. , Hall, J. G. , Couch, R. M. , Phang, M. S. , & Baldwin, V. J. (1990). Syndrome of mental retardation, facial anomalies, hypopituitarism, and distal arthrogryposis in sibs. American Journal of Medical Genetics, 37(1), 65–70. [DOI] [PubMed] [Google Scholar]
- Cohen, M. , Hamilton, J. , & Narang, I. (2014). Clinically important age‐related differences in sleep related disordered breathing in infants and children with Prader‐Willi syndrome. PLoS One, 9(6), e101012 Epub 2014 Jun 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia, M. , Rutigliano, I. , Sacco, M. , Madeo, S. F. , Wasniewska, M. , Pomi, A. L. , Trifirò, G. , Di Bella, P. , De Lucia, S. , Vetri, L. , & Iughetti, L. (2021, August). EEG patterns in patients with Prader‐Willi syndrome. Brain Sciences, 11(8), 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix, O. , Amaddeo, A. , Olmo Arroyo, J. , Zerah, M. , Puget, S. , Cormier‐Daire, V. , Baujat, G. , Pinto, G. , Fernandez‐Bolanos, M. , & Fauroux, B. (2016, September). Central sleep apnea in children: Experience at a single center. Sleep Medicine, 25, 24–28. [DOI] [PubMed] [Google Scholar]
- Hiroi, H. , Kozuma, S. , Hayashi, N. , Unno, N. , Fujii, T. , Tsutsumi, O. , Okai, T. , & Taketani, Y. (2000). A fetus with Prader‐Willi syndrome showing normal diurnal rhythm and abnormal ultradian rhythm on heart rate monitoring. Fetal Diagnosis and Therapy, 15, 304–307. [DOI] [PubMed] [Google Scholar]
- Holm, V. A. , Cassidy, S. B. , Butler, M. , Hanchett, J. M. , Greenswag, L. R. , Whitman, B. Y. , & Greenberg, F. (1993). Prader‐Willi syndrome: Consensus diagnostic criteria. Pediatrics, 91, 398–402. [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , & Gauthier, L. D. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv, 581, 531210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, J. , Lupo, P. J. , Kovar, E. , Rech, M. , Bostwick, B. , Scott, D. , Kraft, K. , Roscioli, T. , Charrow, J. , Schrier Vergano, S. A. , Lose, E. , Smiegel, R. , Lacassie, Y. , & Schaaf, C. P. (2018, December). Schaaf‐Yang syndrome overview: Report of 78 individuals. American Journal of Medical Genetics. Part A, 176(12), 2564–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J. , & Wagner, M. (2013). Prader‐Willi syndrome and sleep‐disordered breathing. Pediatric Annals, 42(10), 200. [DOI] [PubMed] [Google Scholar]
- Patak, J. , Gilfert, J. , Byler, M. , Neerukonda, V. , Thiffault, I. , Cross, L. A. , Amudhavalli, S. M. , Pacio‐Miguez, M. , Palomares‐Bralo, M. , Garcia‐Minaur, S. , Santos‐Simarro, F. , Powis, Z. , Alcaraz, W. , Tang, S. , Jurgens, J. , Barry, B. , England, E. , Engle, E. C. , Hess, J. , & Lebel, R. R. (2019, December). MAGEL2‐related disorders: A study and case series. Clinical Genetics, 96(6), 493–505. 10.1111/cge.1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf, C. P. , Gonzalez‐Garay, M. L. , Xia, F. , Potocki, L. , Gripp, K. W. , Zhang, B. , Peters, B. A. , McElwain, M. A. , Drmanac, R. , Beaudet, A. L. , Caskey, C. T. , & Yang, Y. (2013, November). Truncating mutations of MAGEL2 cause Prader‐Willi phenotypes and autism. Nature Genetics, 45(11), 1405–1408 Epub 2013 Sep 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab, D. F. (1997). Prader‐Willi syndrome and the hypothalamus. Acta Peadiatrica Supplement, 423, 50–54. [DOI] [PubMed] [Google Scholar]
- Yanjie Duan, L. , Liu, X. Z. , Jiang, X. , Jin, X. , & Guan, Q. (2021, June 18). Phenotypic spectrum and mechanism analysis of Schaff Yang syndrome: A case report on new mutation of MAGEL2 gene. Medicine, 100(24), e26309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.