

Summary
Concomitant profiling of transcriptome and chromatin accessibility in isolated nuclei can reveal gene regulatory control mechanisms in health and disease. We report a single nucleus multi-omics analysis protocol optimized for frozen archived postmortem human pituitaries that is also effective for frozen ovine and murine pituitaries and human skeletal muscle biopsies. Its main advantages are that (1) it is not limited to fresh tissue, (2) it avoids tissue dissociation-induced transcriptional changes, and (3) it includes a novel, automated quality control pipeline.
For complete details on the use and execution of this protocol, please refer to Ruf-Zamojski et al. (2021) and Zhang et al. (2022).
Subject areas: Cell Biology, Single Cell, Health Sciences, Genomics, Sequencing, RNAseq
Graphical abstract
Highlights
-
•
Isolation of nuclei from frozen archived tissues for single nucleus multi-omics
-
•
Using a fluorescent counter is beneficial for precise nuclei loading
-
•
An automated pipeline establishes clear passing/failing metrics of sequenced datasets
-
•
Quality control steps are critical to assess nuclei, libraries, and sequenced datasets
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Concomitant profiling of transcriptome and chromatin accessibility in isolated nuclei can reveal gene regulatory control mechanisms in health and disease. We report a single nucleus multi-omics analysis protocol optimized for frozen archived postmortem human pituitaries that is also effective for frozen ovine and murine pituitaries and human skeletal muscle biopsies. Its main advantages are that (1) it is not limited to fresh tissue, (2) it avoids tissue dissociation-induced transcriptional changes, and (3) it includes a novel, automated quality control pipeline.
Before you begin
Single cell (sc) assays have become a tool of choice for studying complex tissues. However, sc assays require the dissociation of fresh tissue, a process that can elicit artifactual gene expression (Nguyen et al., 2018), and cannot be applied to frozen archived samples. Unlike sc-based methods, single nuclei (sn) approaches are compatible with snap-frozen and biobank-archived tissue samples, and they minimize potential ex vivo expression changes.
Presently, frozen postmortem pituitary samples obtained from tissue banks represent the only source of normal human pituitary tissue. We previously reported the sn multi-omics analysis of snap-frozen murine pituitary tissue, and demonstrated that cell type identification was comparable using snRNAseq of snap-frozen whole pituitaries vs. scRNAseq of dissociated fresh pituitaries (Ruf-Zamojski et al., 2021). Recently, we successfully applied nuclei isolation and sn assays for the analysis of all major pituitary cell types in frozen archived postmortem human pituitaries (Zhang et al., 2022).
Consistent sn isolation coupled with mRNA and DNA preservation is a prerequisite for studying archived tissues at sc resolution. While several protocols have been developed to assay relatively large pieces of tissues (> 20 mg), no sn protocol has been described for as little as 1 mg of pituitary tissue (Corces et al., 2019; Mathys et al., 2019; Swiech et al., 2015).
Here, we describe a step-by-step protocol for 1) isolating nuclei from as little as 1 mg of tissue, 2) pooling libraries for deep sequencing, and 3) assessing data quality. Our method yields high quality sn datasets and achieves high data reproducibility. Although we focus on its application to frozen archived human pituitaries, this protocol has been successfully used on snap-frozen murine and ovine pituitaries, and on snap-frozen human skeletal muscle biopsies. To our knowledge, no other protocol, including the 10× Genomics protocol, has been designed to effectively conduct sn assays on frozen tissues from nuclei isolation all the way through data analysis.
Institutional permissions
Ethical compliance
We have complied with all ethical regulations and institutional protocols for studying human postmortem human samples. All human specimens were frozen samples from deceased donors obtained from the NIH NeuroBioBank. Donor anonymity was preserved, and guidelines were followed regarding consent, protection of human subjects, and donor confidentiality. Prior to being shared with the tissue biobank, collection of the pituitary samples upon death was approved by IRB #HP-00042077 for University of Maryland and consent was obtained from all donors or next of kin. Study # 13-00709 PS for tissue repository at the Bronx VA Medical Center was determined Human Research Exempt by the IRB at the Icahn School of Medicine at Mount Sinai (ISMMS), as defined by DHHS regulations (45 CFR 46.101(b) (2). As these human samples were obtained de-identified, the study itself was not considered human subject research by the IRB at the ISMMS, where the assays were performed. The following criteria were used to select samples: gender, absence of degenerative, neuroendocrine or endocrine disease, and coverage of a range of ages.
Sample procurement
Flash-frozen postmortem human pituitaries were obtained from the National Institutes of Health (NIH) NeuroBioBank, and kept at −80°C until processing. The tissues that were received varied from whole to pieces of pituitaries. For additional information refer to (Zhang et al., 2022).
Preparation of instruments and reagents
Timing: 30–40 min
Before starting the protocol, the researcher must ensure he/she has: one autoclaved 1-mL glass homogenizer and one set of autoclaved forceps per sample, a clean 6-well flat bottom plate, 4°C-cooled ultracentrifuge buckets, a dry-ice container and access to dry-ice, and −20°C-cooled mortar and pestle placed on dry-ice.
Preparation for the assay.
-
1.
Install the SW41 swinging-bucket rotor in the centrifuge and fast cool to 4°C. Cool down to 4°C the rotors and buckets of ultracentrifuge, as well as the cell culture centrifuge.
-
2.Prepare the following reagents and add RNase inhibitors on ice (see detailed description in the materials and equipment section):
-
a.Homogenizing buffer (HB).
-
b.50% OptiPrep Density Media Solution.
-
c.30% OptiPrep Density Media Solution.
-
d.35% OptiPrep Density Media Solution.
-
a.
-
3.
Obtain dry-ice.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Pediatric female pituitary (frozen from deceased) | NIH NeuroBioBank | #1275, NDAR_INVCK755EF3 |
| Adult female pituitary (frozen from deceased) | NIH NeuroBioBank | #5621, NDAR_INVXK717LJA |
| Aged female pituitary (frozen from deceased) | NIH NeuroBioBank | #187443 |
| Pediatric male pituitary (frozen from deceased) | NIH NeuroBioBank | #1674, NDAR_INVMV348UPL |
| Adult male pituitary (frozen from deceased) | NIH NeuroBioBank | #5818, NDAR_INVKF350UGX |
| Aged male pituitary (frozen from deceased) | NIH NeuroBioBank | #187438 |
| Chemicals, peptides, and recombinant proteins | ||
| RNAse inhibitor | New England Biolabs | # MO314L |
| OptiPrep | STEMCELL Technologies | 07820 |
| Sucrose | Sigma-Aldrich | S0389 |
| EDTA | Corning | 46-034-Cl |
| Tris-HCl, pH 7.4 | Sigma-Aldrich | T2663 |
| CaCl2 | Sigma-Aldrich | 21115 |
| Magnesium acetate | Boston Bioproducts | MT-190 |
| IGEPAL CA-630 | Sigma-Aldrich | I3021 |
| Tween-20 Surfact-Amps Detergent | Thermo Scientific | 85113 |
| Buffer EB | QIAGEN | 19086 |
| SPRI Select | Beckman Coulter | B23318 |
| 10% BSA | Sigma-Aldrich | SRE0036 |
| PBS | Gibco | 10010-023 |
| Critical commercial assays | ||
| Chromium Single Cell 3′ Reagents, including Chips G | 10× Genomics | V3, https://support.10xgenomics.com/single-cell-gene-expression/library-prep/doc/user-guide-chromium-single-cell-3-reagent-kits-user-guide-v31-chemistry |
| Chromium Single Cell ATAC Reagents, including Chips H | 10× Genomics | V1, https://support.10xgenomics.com/single-cell-atac/library-prep/doc/user-guide-chromium-single-cell-atac-reagent-kits-user-guide-v11-chemistry |
| Chromium Single Cell Multiome ATAC and Gene Expression Reagent, including Chips J | 10× Genomics | V1, https://support.10xgenomics.com/single-cell-multiome-atac-gex/index/doc/user-guide-chromium-next-gem-single-cell-multiome-atac-gene-expression-reagent-kits-user-guide |
| MiSeq Reagent Nano Kit (300 cycles) | Illumina | MS-103-1001, https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/miseq/miseq-system-guide-for-local-run-manager-15027617-05.pdf |
| Qubit dsDNA High Sensitivity kit | Invitrogen | Q32851, https://assets.fishersci.com/TFS-Assets/LSG/manuals/Qubit_dsDNA_HS_Assay_UG.pdf |
| High Sensitivity DNA Bioanalyzer Kit or TapeStation | Agilent | 5067-4626, https://www.agilent.com/cs/library/usermanuals/Public/G2938-90321_SensitivityDNA_KG_EN.pdf |
| PhiX control v3 | Illumina | FC-110-3001 |
| NovaSeq 6000 S4 Reagent Kit v1.5 (200 cycles) | Illumina | 20028313 |
| Deposited data | ||
| Raw and analyzed data | (Zhang et al., 2022) | GSE178454 |
| Software and algorithms | ||
| Cell Ranger | 10× Genomics | v5.0.0 |
| Seurat | (Butler et al., 2018; Stuart et al., 2019) | v.3.9.9.9024; v4.0.1; v3.1.5 |
| Cell Ranger-ATAC pipeline | 10× Genomics | v1.2.0 |
| Seurat/Signac | (Stuart et al., 2020) | v3.1.5/0.2.4 |
| Cell Ranger ARC | 10× Genomics | v1.0.0 |
| Automated QC pipeline | This paper and https://github.com/sealfonlab/snRNA_snATAC_snMultiome_QC | |
| Web portal | (Ruf-Zamojski et al., 2021; Zhang et al., 2022) | www.snpituitaryatlas.princeton.edu |
| Other | ||
| 1× TE Molecular Grade | Promega | V6231 |
| 1 mL Dounce tissue glass homogenizer | VWR | # 71000-514 |
| 40 μm cell strainer | Falcon | 352340 |
| Centrifuge SW41 rotor | Beckman Coulter | #331362 |
| Optima LE-80K ultracentrifuge | Beckman Coulter | Optima LE-80K |
| Cell culture centrifuge | Eppendorf | 5804R |
| Open top thin-wall ultra clear tubes for SW41 | Beckman Coulter | cat# 344059 |
| 14 mL polystyrene round-bottom tubes | Falcon | cat# 352057 |
| Cellometer PD100 Counting Chambers | Nexcelom | CHT4-PD100-002 |
| ViaStain Propidium Iodide (PI) staining Solution | Nexcelom | CS1-0109-5mL |
| Thin-wall clear 0.5-mL Qubit assay PCR tubes | Invitrogen | Q32856 |
| Fluorometer | Invitrogen | Qubit3 or Qubit4 |
| Bioanalyzer or TapeStation | Agilent | 2100 |
| Sequencer | Illumina | MiSeq |
| Sequencer | Illumina | NovaSeq 6000 |
| Cell counter | Nexcelom | Cellometer K2, https://www.nexcelom.com/knowledge-base/cellometer-k2-user-manaul/ |
| Falcon 6-well clear flat bottom cell culture plate | Corning | Cat# 353046 |
| HEPES buffer, 1 M | Corning | Cat# 255-060-CL |
| Magnesium chloride 1 M | Sigma-Aldrich | Cat# M1028 |
| KCl | Fisher Scientific | Cat# P217-3 |
| Glycerol | Sigma-Aldrich | Cat# G6279 |
| Nuclei preservation protocol | Collas Lab, University of Oslo | http://collaslab.org/wp-content/uploads/2011/07/Isolation_somatic_cell_nuclei.pdf |
Materials and equipment
All buffers can be prepared the day before the isolation. The RNase inhibitor will be added last, immediately prior to starting the isolation protocol.
Homogenizing Buffer (HB), prepare and store at 4°C for a maximum of 7 days
| Reagent | Final concentration | Amount for 50 mL |
|---|---|---|
| Sucrose | 0.32 M | 5.47 g |
| EDTA 0.5 M | 0.1 mM | 10 μL |
| Tris-HCL 1 M | 10 mM | 500 μL |
| CaCl2 1 M | 5 mM | 250 μL |
| Magnesium acetate 1 M | 3 mM | 150 μL |
| IGEPAL CA-630 10% | 0.1% | 500 μL |
| Water | Up to 50 mL | |
| Total | n/a | 50 mL |
Dilution Buffer, prepare and store at 4°C for a maximum of 7 days
| Reagent | Final concentration | Amount for 50 mL |
|---|---|---|
| Sucrose | 0.32 M | 5.47 g |
| EDTA 0.5 M | 6 mM | 600 μL |
| Tris-HCL pH7.5, 1 M | 60 mM | 3 mL |
| CaCl2 1 M | 30 mM | 1.5 mL |
| Magnesium acetate 1 M | 18 mM | 0.9 mL |
| Water | Up to 50 mL | |
| Total | n/a | 50 mL |
50% Optiprep Density Media Solution, prepare and store at 4°C for a maximum of 7 days
| Reagent | Final amount |
|---|---|
| OptiPrep 60% | 20 mL |
| Dilution Buffer | 4 mL |
| Total | 24 mL |
30% Optiprep Density Media Solution, prepare and store at 4°C for a maximum of 7 days
| Reagent | Final amount |
|---|---|
| OptiPrep 50% | 6 mL |
| Homogenizing Buffer | 4 mL |
| Total | 10 mL |
35% Optiprep Density Media Solution, prepare and store at 4°C for a maximum of 7 days
| Reagent | Final amount |
|---|---|
| OptiPrep 50% | 7 mL |
| Homogenizing Buffer | 3 mL |
| Total | 10 mL |
1× PBS, 0.04% BSA, prepare and store at 4°C for a maximum of 2 weeks
| Reagent | Amount for 50 mL |
|---|---|
| Phosphate-Buffered Saline (PBS) with 10% Bovine Albumin (BSA) | 200 μL |
| 1× PBS | 49.8 mL |
| Total | 50 mL |
Buffer N, prepare at 4°C and store at −20°C for a maximum of 1 year
| Reagent | Final concentration | Amount for 100 mL |
|---|---|---|
| HEPES pH 7.5, 1 M | 10 mM | 1 mL |
| MgCl2, 1 M | 2 mM | 200 μL |
| KCL, 3 M stock | 25 mM | 800 μL |
| Sucrose, 2 M stock in H2O | 250 mM | 12.5 mL |
| Water | Up to 100 mL | |
| Total | n/a | 100 mL |
Nuclei freezing medium, prepare at 4°C and store at −20°C for a maximum of 1 year
| Reagent | Amount for 50 mL |
|---|---|
| Buffer N | 15 mL |
| Glycerol | 35 mL |
| See note | |
| Total | 50 mL |
Note: It is possible to add 1 mM of DTT and protease inhibitor cocktail to the Buffer N. See http://collaslab.org/wp-content/uploads/2011/07/Isolation_somatic_cell_nuclei.pdf.
CRITICAL: The RNase inhibitor must be added at the very last moment. For details, see the next step, Frozen tissue pre-crushing or pulverization.
Step-by-step method details
Frozen tissue pre-crushing or pulverization
There are two alternate ways of conducting the first step of the method: 1) either by breaking the tissue into smaller pieces and processing one piece for nuclei extraction, 2) or by pulverizing the tissue and processing part of the powder for nuclei extraction. In either case, the remainder of the pituitary sample should be stored back at −80°C. Tissue pulverization is more advantageous than breaking a piece of tissue, as it yields a homogeneous nuclei preparation and thus precludes tissue regionalization.
-
1.
Pre-chill the homogenizer on ice.
-
2.
Pre-chill the ceramic mortar and pestle on dry-ice.
-
3.
Place aluminum foil in the pre-chilled mortar.
-
4.
Transfer the frozen tissue vial from −80°C to dry-ice.
-
5.
Add 0.2 U/μL of RNase inhibitor (NEB MO314L) to the Homogenizing Buffer (HB), 50% OptiPrep, 35% OptiPrep, and 30% OptiPrep solutions.
-
6.Frozen tissue crushing or pulverization:
-
a.Take out the frozen tissue sample from the vial and place it on aluminum foil in the pre-chilled mortar (Figure 1).
-
b.Use the pestle to either break the tissue into small chunks or smash it into powder.
-
c.Save an aliquot of the crushed or pulverized tissue (∼5–10 mg) for the assay, and transfer the remainder immediately on dry-ice for storage at −80°C.
-
a.
-
7.
Place the tissue chunk or powder to assay in the pre-chilled homogenizer containing 200 μL of HB.
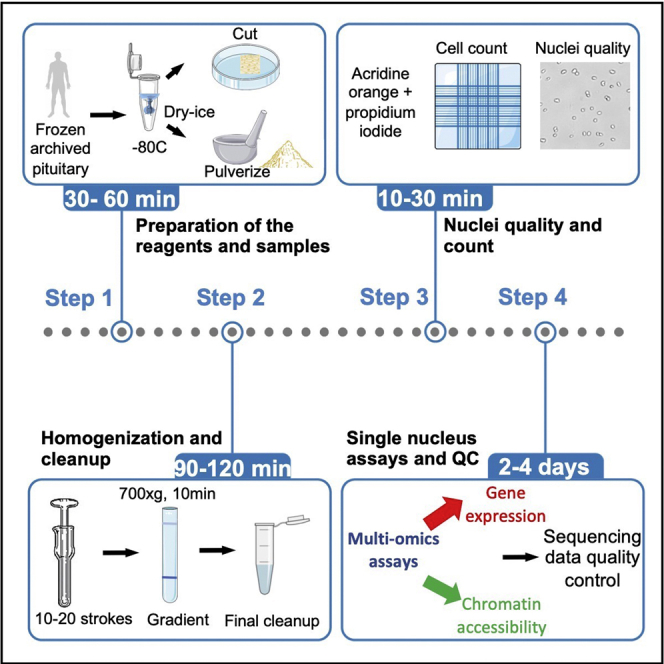
Figure 1.

Flowchart of the protocol
The key steps of the protocol from nuclei isolation to data analysis are depicted.
Tissue homogenization and nuclei extraction
During this 2nd step, nuclei are extracted from the tissue and purified for single nucleus (sn) multi-omics assays. All steps are done on ice using ice-cold reagents.
-
8.
Homogenize the tissue with 10–20 strokes of the Loose Pestle.
-
9.
Add another 300 μL of HB. Incubate on ice for 5 min, and apply another 10 strokes with the Loose Pestle.
-
10.
Place on ice a 50 mL Falcon tube with a 40 μm filter on the top, wet the filter with 100 μL of HB using standard tips. The 100 μL used to wet the filter should be retained in the 50 mL tube.
-
11.
Transfer the tissue homogenate onto the filter using standard tips.
-
12.
Add another 500 μL HB to the glass homogenizer. Collect the wash with a pipette and transfer it to the same 40 μm filter (Figure 1).
-
13.
Measure the volume of the flow-through and add an equal volume of 50% OptiPrep to bring the concentration of the homogenate to 25% and keep aside until the next step.
-
14.
Prepare a gradient in one of the clear centrifuge tube for SW41 (Beckman Coulter, cat# 344059). Dispense slowly 3 mL of 35% OptiPrep at the bottom of the tube; then add a layer of 4 mL of 30% OptiPrep, followed by about 2.5–4 mL of the mixed tissue homogenate with 50% OptiPrep (Figure 1). The volume in the tube will be between 9.5 to 11 mL at this stage.
-
15.
Balance the tubes and centrifuge in a Beckman ultracentrifuge SW41 rotor at 12,818 × g at 4°C for 25 min (Figure 1).
-
16.
After centrifugation, discard the remaining of the homogenate (25% OptiPrep part of the gradient, i.e., top layer) from the tube (Figure 2).
-
17.
Collect nuclei from the interphase between 30% OptiPrep and 35% OptiPrep; the volume from the SW41 tubes is about 2 mL–3 mL. Transfer nuclei to a 14 mL polystyrene round-bottom tubes (Falcon # 352057).
-
18.
Add 7 mL of ice-cold 1× PBS 0.04% BSA to the top, mix by inverting the tubes.
-
19.
Spin at 700 g for 10 min in a 4°C-pre-cooled cell culture centrifuge (Eppendorf 5804R) using a swinging-bucket rotor (A-4-44).
-
20.
Remove the top layer of supernatant, leaving 500 μL behind in the tube.
-
21.
Add 10 mL of ice-cold PBS 0.04% BSA to the top and repeat centrifugation at 500 g for 5 min at 4°C. Avoid pipetting of nuclei if possible; pipetting may result in clumping of nuclei. If pipetting, use 1 mL wide bore tips.
-
22.
Remove all but 200 μL of supernatant. Gently pipette 3 times with a 1-mL pipette tip.
Figure 2.
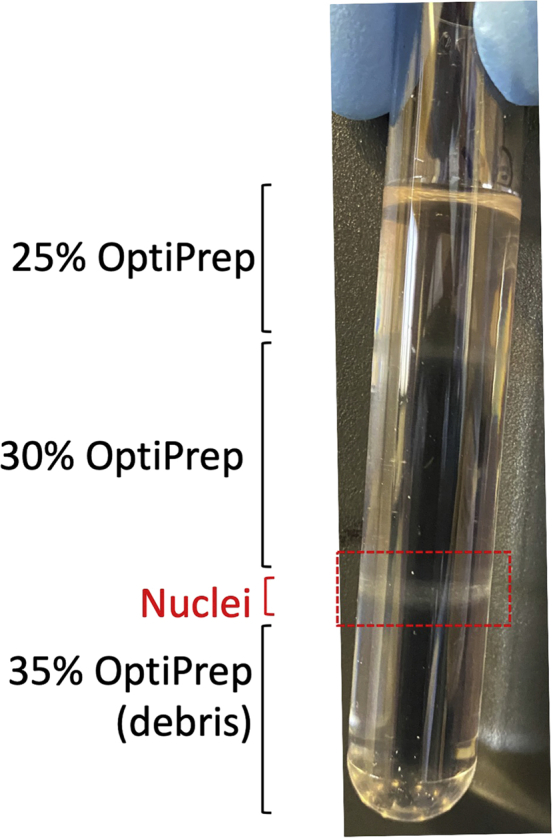
Purification of nuclei with gradient centrifugation
Following gradient centrifugation, nuclei are at the border between the 30% and 35% OptiPrep. The suspension around this band is taken for further cleanup of the nuclei in preparation for downstream sn assays.
Nuclei counting using fluorescent cell counter
Counting the nuclei will ensure correct loading onto the 10× Genomics chips.
-
23.Prepare counting slides and tubes (Figure 1):
-
a.Remove protective layers from Nexcelom counting slides.
-
b.Add 5 μL of nuclei suspension to 5 μL of PBS 0.04% BSA buffer in a 1.5 mL Eppendorf tube.
-
c.Add 10 μL of Nexcelom ViaStain propidium iodide (PI) staining solution to tube, pipette to mix.
-
a.
-
24.
Load the 20 μL onto the Nexcelom slide.
-
25.
Insert the slide into the Nexcelom K2 cell counter or an alternative fluorescence counter.
-
26.
Observe the brightfield and adjust the focus as per manufacturer’s recommendations (Figure 3).
-
27.
Make sure that PI emits fluorescence in the red channel.
-
28.
Count nuclei on the Nexcelom K2 instrument and record nuclei number and nuclei diameter.
-
29.Before proceeding to the next step, note your nuclei dilution in order to calculate your total number of nuclei. The nuclei can be preserved once isolated.
-
a.If freezing nuclei upon isolation, follow the protocol adapted from the Collas lab (University of Oslo), http://collaslab.org/wp-content/uploads/2011/07/Isolation_somatic_cell_nuclei.pdf.
-
i.Re-suspend the pelleted nuclei into 50 μL of PBS 0.04% BSA. Add up to 500 μL of Nuclei freezing medium. Mix by gently pipetting.
-
ii.Freeze aliquots directly in the −80°C freezer and store at –80°C.
-
i.
-
b.If not freezing nuclei at the previous stage, these nuclei can now be used for single nucleus assays (See following section for details).
-
c.If thawing frozen nuclei for further sn assays:
-
i.Take out the tube of frozen nuclei (500 μL), thaw it at 20°C–25°C temperature, and place on ice.
-
ii.Dilute with 1 mL of ice-cold Buffer N.
-
iii.Mix well but gently with a 1000 μL pipette tip.
-
iv.Centrifuge at 1,620 g for 15 min in a swing out rotor.
-
v.Discard supernatant and re-suspend nuclei in 500 μL of ice-cold buffer N.
-
vi.Centrifuge at 1,620 g for 10 min in a swing out rotor.
-
vii.Remove supernatant and wash nuclei in buffer suitable for the next sn application.
-
i.
-
a.
Note: The K2 cellometer is our cell counter of choice for nuclei. It is not a requirement, but in our experience, it is currently the most reliable and reproducible nuclei counter on the market for downstream sn assays. Trypan blue is not reliable to count nuclei.
Alternatives: Any other fluorescence-based cell counter could be used instead of the Nexcelom instrument.
Figure 3.

Quality assessment of nuclei preparation
Following nuclei isolation and cleanup steps, nuclei quality is assessed and nuclei are counted on a Nexcelom fluorescent cell counter.
(A) Image showing highly concentrated nuclei and clumps in the preparation.
(B) Image showing the same preparation after dilution and pipetting to remove large clumps.
Single nucleus assays
-
30.
Single nucleus assays are performed following the 10× Genomics protocols. Users should follow the links below for the specific protocol to be performed.
If users perform snRNAseq for gene expression, they should use the 10× Genomics Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 protocol: https://assets.ctfassets.net/an68im79xiti/1eX2FPdpeCgnCJtw4fj9Hx/7cb84edaa9eca04b607f9193162994de/CG000204_ChromiumNextGEMSingleCell3_v3.1_Rev_D.pdf.
If users perform snATACseq for chromatin accessibility, they should use the 10× Genomics Chromium Next GEM Single Cell ATAC Reagent Kits v1.1 protocol: https://assets.ctfassets.net/an68im79xiti/6Be7TWXkltEluNIaWgA0dj/37c815979660f6a718328d525914a5d5/CG000209_Chromium_NextGEM_SingleCell_ATAC_ReagentKits_v1.1_UserGuide_RevF.pdf.
If users perform same-cell sn multiome for both gene expression and chromatin accessibility from the same nuclei, they should use the 10× Genomics Chromium Next GEM Single Cell Multiome ATAC + Gene Expression protocol: https://assets.ctfassets.net/an68im79xiti/7x5E4P6xefQruTbFg0yr3a/1381fdcd2d2e7d667ef5b415119dab15/CG000338_ChromiumNextGEM_Multiome_ATAC_GEX_User_Guide_RevE.pdf.
Pause point: The 10× Genomics protocols include specific Pause points to use accordingly.
Pooling of libraries for sequencing
In this section, we will explain how we pool multiple libraries for sequencing. This step is only possible if each of the libraries is prepared with unique indexes, so as to distinguish them in sequencing.
-
31.
Accurately determine library concentration using the Qubit double stranded (ds) DNA high-sensitivity (HS) Assay in individual tubes with the Qubit3 or Qubit 4 fluorometers and following the manufacturer’s instructions (https://assets.fishersci.com/TFS-Assets/LSG/manuals/Qubit_dsDNA_HS_Assay_UG.pdf).
-
32.
Record the measured library concentration from the qubit assay for use in calculating the sample molarity.
-
33.
Determine the average fragment size in each sample by running a Bioanalyzer assay. Refer to Figures 4 and 5.
Alternatives: A TapeStation system may be used instead of the Bioanalyzer.
-
34.
Calculate the molarity of each sample using the following formula:
Figure 4.

Representative Bioanalyzer traces for snRNAseq QC
(A–C) Shown are representative traces of successful Bioanalyzer QC analyses for snRNAseq (A) and sn multiome (B), each at the cDNA amplification step (i) and following library preparation (ii). Failed QC traces are shown in (C).
Figure 5.

Representative Bioanalyzer traces for snATACseq QC
(A–C) Shown are representative traces of successful Bioanalyzer QC analyses for snATACseq (A) and sn multiome (B) libraries. Failed QC traces are shown in (C).
Molarity in nM = [(Concentration in ng/μL) / (Fragment size in bp × 660)] × 10ˆ6.
-
35.
Dilute all samples to a uniform concentration ≥ 2 nM (ex: 5 nM or 10 nM). MiSeq sequencing requires ≥ 2 nM of library concentration. Any sample whose concentration is already lower than the chosen concentration should not be diluted and kept as is.
Note: When including samples of a lower molarity ∼2–5 nM, we recommend avoiding dilution of the samples to the lowest molarity. Lower molarity samples are assumed to be of lower quality, and although they can be included for sequencing, they should be included as undiluted stock samples. Instead of diluting to the lowest molarity, the lower molarity samples should be added to the pool with a higher volume compared to the rest of the samples to account for the lower yield.
Note: We recommend keeping the sequencing pool concentration well above 2 nM. A higher starting pool concentration is better in case of any pool adjustments based on rounds of shallow sequencing. If the sample pool molarity decreases to less than 2 nM, it will be harder to continue with deep sequencing.
-
36.
Pool each of the diluted sample dilutions together, using the same volume for each sample.
-
37.
Measure the library pool concentration in duplicate using the Qubit assay, then calculate the pool molarity with the average measured concentration and the overall average fragment size.
-
38.
Use the calculated pool molarity to prepare the pool for MiSeq sequencing.
MiSeq sequencing of libraries for pool adjustment
-
39.
Thaw a MiSeq V2 nano kit 300 cycle reagent cartridge according to manufacturer’s instructions (See ‘Chapter 3 Sequencing’ of the Illumina MiSeq System Guide).
-
40.
Refer to the Illumina ‘MiSeq System Denature and Dilute Libraries Guide, Protocol A – Standard Normalization Method’ for preparing a library pool for MiSeq sequencing.
-
41.
Refer to Illumina ‘MiSeq System Denature and Dilute Libraries Guide, Denature and Dilute PhiX Control’ for preparing PhiX control to include in MiSeq sequencing. Follow instructions for a 1% PhiX spike-in.
-
42.
Refer to ‘Chapter 3 Sequencing’ of the Illumina MiSeq System Guide to load the MiSeq reagent kit for sequencing and prepare the MiSeq instrument for a sequencing run.
-
43.
Allow the necessary duration for the MiSeq sequencing run.
-
44.
After completion of the sequencing run, check the read distribution results. If needed, use the results to adjust the sample volumes in the pool to further balance the sequencing read distribution prior to deep sequencing.
Deep sequencing of libraries
-
45.
Run a final Bioanalyzer QC of the library pools prior to deep sequencing (Figure 6).
-
46.
We out-source NovaSeq sequencing to the New York Genome Center and follow their guidelines for sample submission.
The sequencing parameters that we use for deep sequencing of our sn libraries are the following:-
a.V1.5 FC.
-
b.100 × 10 × 24 × 100 with a total number of 234 cycles.
-
c.No need to accommodate the dark cycles.
-
a.
Figure 6.

Representative Bioanalyzer traces for library pool QC
(A and B) Shown are representative traces of successful Bioanalyzer QC analyses after the pooling of snRNAseq (A) or snATACseq (B) libraries. Twenty-four libraries were pooled in both (A and B).
Quality control of the sn sequencing data
The quality of the sequencing data is estimated for each dataset. We use the metrics from the CellRanger Summary output files as well as additional metrics (see Tables 1 and 2 below).
-
47.
After samples are sequenced, follow the 10× Genomics Cell Ranger pipeline to process the sequenced data (Cell Ranger for snRNAseq, Cell Ranger ATACseq for snATAC, and Cell Ranger ARC for multiome).
-
48.
Extract relevant metrics from the summary CSV file generated through the Cell Ranger pipeline (metrics_summary.csv for snRNAseq, summary.csv for snATAC, and summary.csv for multiome). The metrics used for QC and their appropriate threshold are listed in Table 1.
Table 1.
snRNAseq quality control (QC) rules
| snRNAseq Rules | Not Pass | Pass | Good |
|---|---|---|---|
| Estimated_Number_of_Cells | <500 | [500,1000) | ≥1000 |
| Mean_Reads_per_Cell | <500 | ≥2,000 | ≥2,000 |
| Median_Genes_per_Cell | <500 | ≥500 | ≥1,000 |
| Median_UMI_Counts_per_Cell | <1000 | [1000, 1500) | ≥1500 |
| Reads_Mapped_Confidently_to_Intronic_Regions | <15% | [15%, 20%) | ≥20% |
| Reads_Mapped_Confidently_to_Exonic_Regions | <20% | [20%, 25%) | ≥25% |
| Q30 Bases in RNA Read | <55% | [55%–65%) | ≥65% |
| Percent Reads in Cells | <40% | [40%,70%) | ≥70% |
| Fraction of cells with mitochondrial reads >= 20% | >50% | (20%,50%) | ≤20% |
| Dataset sex matches sample sex | No | Yes | Yes |
Presented in the table are the QC metrics rules used to assess sequencing quality of the snRNAseq libraries.
Table 2.
snATACseq quality control rules
| snATACseq Rules | Not Pass | Pass | Good |
|---|---|---|---|
| Fraction of transposition events in peaks in cells | <0.05 | [0.05,0.2) | >0.2 |
| Confidently mapped read pairs | <0.5 | [0.5, 0.75) | >0.75 |
| Q30 bases in read 1 | <55% | [55%–65%) | >65% |
| Q30 bases in read 2 | <55% | [55%–65%) | >65% |
| Median high-quality fragments per cell | <500 | ≥500 | ≥500 |
| TSS enrichment score | <2 | [2,4) | ≥4 |
| Additional QC reference metrics | Caution | Pass | Good |
| Nucleosome-free region (NFR) peak | No | Yes | Yes |
| Mononucleosome peak | No | Yes | Yes |
Presented in the table are the QC metrics rules used to assess sequencing quality of the snATACseq libraries.
For each assay modality, please follow the corresponding guidelines below:
snRNA.
-
a.
Use snrna_qc.R to calculate the percentage of cells with over 20 percent of mitochondrial read counts.
-
b.
Calculate the percentage of cells with expression of XIST gene. If the result is greater than 0.1, the sample is determined as female. Otherwise, the sample is determined as male.
snATAC.
Use snatac.R to calculate the nucleosome-free region (NFR) peak and mononucleosome peak.
Optional: Determine the presence of the NFR peak and mononucleosome peak. Use Chr1-1-200000000 to generate the fragment length histogram, retaining fragments that are shorter than 600 bp for analysis. If one of the highest two peaks in the resulting density plot is located between 0 and 150 bp, then the NFR peak is determined as TRUE. If the other of the highest two peaks is located between 150 and 300 bp, then the mononucleosome peak is determined as TRUE. NFR peak and mononucleosome peak are used as additional reference metrics for quality assurance.
snMultiome.
snMultiome QC pipeline consists of the snRNA and snATAC pipelines presented above. Use multiome_qc.R to calculate both snRNA and snATAC metrics listed individually in the snRNA and snATAC sections.
For nuclei, sequencing data quality is assessed following the criteria listed in Table 1 for snRNAseq and in Table 2 for snATACseq (Reprinted with permission from (Zhang et al., 2022) and from Elsevier under the license number 5302501441831).
The pipeline code has been deposited on Github and is accessible at https://github.com/sealfonlab/snRNA_snATAC_snMultiome_QC.
For the full QC outcomes of both male and female murine and human pituitaries, the reader should refer to the Supplementary Information and tables in (Zhang et al., 2022). We are reproducing below the QC metrics summary tables for the male human samples published in (Zhang et al., 2022) as examples. Reprinted with permission from (Zhang et al., 2022) and from Elsevier under the license number 5302501441831.
Preliminary cell type identification and quality assessment
-
49.
Once datasets have passed the QC pipeline, they are visualized as Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) for cluster identification. Preliminary analysis enables the identification of multiplets and debris in addition to the pituitary cell clusters (Figure 7A). Clusters with low RNA counts are usually debris or dead cells, whereas clusters with very high (>2) RNA counts are often multiplets (Figure 7B). Dying cells usually exhibit a higher than normal expression level of mitochondrial genes (Figure 7C). After removal of the dead cells and multiplets by filtering, one can obtain a clean UMAP (Figure 7D).
Figure 7.

Cluster identification and quality assessment
Results obtained for the frozen archived human pediatric pituitaries processed for sn multiome.
(A) Preliminary UMAP including multiplet and debris/dead cell clusters.
(B) UMI/RNA count.
(C) Mitochondrial gene content.
(D) Clean UMAP following the removal of multiplet and debris/dead clusters.
Expected outcomes
Following nuclei isolation from 1-3 mg of human pituitaries, we usually get 60,000–1,000,000 nuclei in 30 μL. This yield is dependent on the amount of material available for nuclei isolation. A minimum of 30,000 nuclei in 30 μL is needed for successful assays. The nuclei should be single and without visible clumps to prevent clogging of the 10× Genomics chips.
Expected outcomes for the sn assays are provided in each of the 10× Genomics protocols (Tables 1 and 2). All sequencing outcomes from male human samples are summarized in Tables 3 and 4. The human female data and all murine data QC are presented in the Supplementary Information from Zhang et al., Cell Reports 2022. In addition, all characterized pituitary cell types were detected using our multi-omics approach (Zhang et al., Cell Reports, 2022; Figure 7).
Table 3.
Human males, snRNAseq metrics
| snRNAseq Rules | Donor 1 | Donor 2 | Donor 3 |
|---|---|---|---|
| Estimated_Number_of_Cells | 12429 | 9022 | 1642 |
| Mean_Reads_per_Cell | 19448 | 27586 | 160560 |
| Median_Genes_per_Cell | 2570 | 2990 | 3408 |
| Median_UMI_Counts_per_Cell | 5075 | 6812 | 9007 |
| Reads_Mapped_Confidently_to_Intronic_Regions | 48.20% | 48.90% | 43.60% |
| Reads_Mapped_Confidently_to_Exonic_Regions | 38.20% | 37.50% | 34.10% |
| Q30 Bases in RNA Read | 93.20% | 93.20% | 93.20% |
| Percent Reads in Cells | 90.00% | 88.50% | 85.60% |
| Fraction of cells with mitochondrial reads >= 20% | 0.7804 | 1.1973 | 2.1354 |
| Dataset sex matches sample sex | TRUE | TRUE | TRUE |
| QC Result | GOOD | GOOD | GOOD |
Presented in the table is the snRNAseq QC metrics summary for the male human samples.
Table 4.
Human males, snATACseq metrics
| snATACseq Rules | Donor 1 | Donor 2 | Donor 3 |
|---|---|---|---|
| Fraction of transposition events in peaks in cells | 7745 | 6741 | 5476 |
| Confidently mapped read pairs | 0.573 | 0.22 | 0.413 |
| Q30 bases in read 1 | 0.816 | 0.81 | 0.799 |
| Q30 bases in read 2 | 0.93 | 0.931 | 0.933 |
| Median high-quality fragments per cell | 0.92 | 0.928 | 0.928 |
| TSS enrichment score | 8718 | 5780 | 11032 |
| Nucleosome-free region (NFR) peak | 6.02 | 3.26 | 4.78 |
| Mononucleosome peak | TRUE | TRUE | TRUE |
| QC Result | GOOD | PASS | GOOD |
Presented in the table is the snATACseq QC metrics summary for the male human samples.
Limitations
While snRNAseq analysis has significant advantages over scRNAseq, such as that i) it is less cell type-biased than scRNAseq (as some cell types are more vulnerable to the tissue dissociation process), ii) it is compatible with frozen samples, and iii) it circumvents dissociation-induced transcriptional artifacts, it also has a few limitations.
Whole-cell extracts contain cytoplasmic and nuclear transcripts, whereas in nuclei extracts only nuclear transcripts are detected (Bakken et al., 2018). Nuclear transcripts are notably depleted in ribosomal and mitochondrial genes (Bakken et al., 2018; Selewa et al., 2020). Moreover, due to the abundance of nascent RNAs in the nucleus, a higher proportion of long genes with wide intronic regions are detected in nuclei compared to whole cells (Lake et al., 2017). Nevertheless, the ability of snRNA-seq assays to capture RNA molecules in a cell is good and cell type discrimination is similar with either method (Bakken et al., 2018; Ding et al., 2020; Selewa et al., 2020).
Biospecimen quality represents a technical limitation, as it will determine the quality of nuclei preparations. Unfortunately, it cannot be evaluated on archived samples. Another issue is nuclei doublets/multiplets. These should be minimized as they may hinder the identification of cell types and/or cell states (Figure 7).
SnRNAseq data contain no information about spatial localization of the nuclei/cells within the tissue. FISH-based techniques or spatial transcriptomics could complement the snRNAseq data by mapping cell type subpopulations across the tissue regions ((Pincas et al., 2021) for review). Generally, snATACseq is a highly reliable protocol. It allows us to predict cis-regulatory DNA interactions through the measurement of co-accessibility between two or more open chromatin regions (Pliner et al., 2018). Our multi-omics approach allows us to reliably detect cell states in addition to cell types. Furthermore, with the development of a linear model (Zhang et al., Cell Reports, 2022), we have the opportunity to infer the transcription factors and chromatin accessible regions contributing to differential gene expression.
The QC pipeline, when classifying sn datasets according to the categories reported in Tables 1 and 2, robustly determines their quality. However, there exist good quality nuclei in overall “failed” datasets that can be rescued by careful sub-selection. For snATAC data, the absence of an NFR or mononucleosome peak does not necessarily indicate a failed dataset. Therefore, these metrics are included as additional metrics and not primary metrics. Furthermore, we find a high degree of variability in sample sex calling from snATAC data, therefore we currently don’t include it in the pipeline.
Troubleshooting
Problem 1
The number of isolated nuclei is too high. Refer to step 28.
Potential solution
Make sure to dilute your nuclei suspension and recount before loading the 10× Genomics chips. In case of doubt, always do a second count.
Problem 2
The number of isolated nuclei is too low. Refer to step 28.
Potential solution
If the number of nuclei is not sufficient to proceed, you may centrifuge and gently resuspend your nuclei in a smaller volume of buffer. Alternatively, you can run several GEM replicates. Although the various GEM replicates derived from the same starting sample cannot be combined experimentally, they may be combined at the analysis stage following sequencing.
Problem 3
Clumps are detected in the nuclei preparation. The presence of clumps in the nuclei preparation can result in clogging issues at the GEM step for any of the sn assays. Refer to step 28 and Figure 3.
Potential solution
Try to gently pipette up and down to obtain a better resuspension. Alternatively, you may filter your nuclei preparation using a 40 μm filter. Place your suspension on top of the filter and centrifuge gently at 100 g for a few seconds.
Problem 4
Low cDNA amount in the sn RNAseq or multiome assay based on the Bioanalyzer QC results. Refer to the single nucleus assays section step 30, 10× Genomics protocols, and Figure 4.
Potential solution
There is no solution. Whether the RNA has been lost or has low quality, it cannot be recovered. The only option is to re-run the sample, i.e., re-extract nuclei, if you have some left, or to assay a different sample to complete your series.
Problem 5
Peaks in the library QC Bioanalyzer profile instead of a smooth trace. Refer to the single nucleus assays section, 10× Genomics protocols, and Figure 4C ii.
Potential solution
There is no real solution, apart from repeating the library preparation from amplified cDNA, as long as not all was used for library preparation. The 10× Genomics protocol recommends to move forward only 25% of the original amplified cDNA. If this was done as suggested, then there should be enough cDNA and reagents left to repeat the failed library. Unfortunately, such QC traces are often the results of low starting RNA amount or low sample quality, and it may not be solvable. If one has additional samples available then the whole protocol can be repeated starting at nuclei isolation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Frederique Ruf-Zamojski (frederique.ruf-zamojski@mssm.edu).
Further information and requests for information about the protocol should be directed to and will be fulfilled by the technical contact, Frederique Ruf-Zamojski (frederique.ruf-zamojski@mssm.edu).
Materials availability
This study did not generate new unique reagents. Feel free to contact the lead contact with any questions.
Acknowledgments
This work was supported by funding from the National Institute of Health (NIH) Grant DK46943 (S.C.S.). We acknowledge the New York Genome Center for sequencing. Human tissue was obtained from the NIH NeuroBioBank. This work was supported in part through the computational and data resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Author contributions
N.M. performed research. H.P. drafted the manuscript. M.Z., W.S.C., and E.Z. performed the QC analysis. M.A.S.A. and V.D.N. sequenced the samples. S.C.S. designed the study, analyzed and interpreted data, and drafted the manuscript. F.R.Z. designed the study, performed the research, analyzed and interpreted data, and drafted the manuscript. All authors edited the manuscript and approved its final version.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Stuart C. Sealfon, Email: stuart.sealfon@mssm.edu.
Frederique Ruf-Zamojski, Email: frederique.ruf-zamojski@mssm.edu.
Data and code availability
-
•
The datasets (snRNAseq, snATACseq, sn multiome) generated in the present study are deposited in GEO (accession #GSE178454) and are publicly available as of date of publication (Zhang et al., 2022). The sn human pituitary multi-omics atlas can be browsed via a web-based portal accessible at http://snpituitaryatlas.princeton.edu/. All datasets will also be deposited with the Human Cell Atlas. Accession numbers and web-portal access are also listed in the key resources table.
-
•
All original code has been deposited in Github and is publicly available as of the date of publication. It is accessible at https://github.com/sealfonlab/snRNA_snATAC_snMultiome_QC. DOIs are listed in the text and in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bakken T.E., Hodge R.D., Miller J.A., Yao Z., Nguyen T.N., Aevermann B., Barkan E., Bertagnolli D., Casper T., Dee N., et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 2018;13:e0209648. doi: 10.1371/journal.pone.0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Greenleaf W.J., Chang H.Y. protocolsio; 2019. Isolation of Nuclei from Frozen Tissue for ATAC-Seq and Other Epigenomic Assays. [Google Scholar]
- Ding J., Adiconis X., Simmons S.K., Kowalczyk M.S., Hession C.C., Marjanovic N.D., Hughes T.K., Wadsworth M.H., Burks T., Nguyen L.T., et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020;38:737–746. doi: 10.1038/s41587-020-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake B.B., Codeluppi S., Yung Y.C., Gao D., Chun J., Kharchenko P.V., Linnarsson S., Zhang K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017;7:6031. doi: 10.1038/s41598-017-04426-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X., et al. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q.H., Pervolarakis N., Nee K., Kessenbrock K. Experimental considerations for single-cell RNA sequencing approaches. Front. Cell Dev. Biol. 2018;6:108. doi: 10.3389/fcell.2018.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincas H., Ruf-Zamojski F., Turgeon J.L., Sealfon S.C. In: Cellular Endocrinology in Health and Disease. Second Edition. Tao Y.-X., editor. Academic Press, Elsevier; 2021. Endocrinology of a single cell: tools and insights; pp. 1–25. [Google Scholar]
- Pliner H.A., Packer J.S., McFaline-Figueroa J.L., Cusanovich D.A., Daza R.M., Aghamirzaie D., Srivatsan S., Qiu X., Jackson D., Minkina A., et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol. Cell. 2018;71:858–871.e8. doi: 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf-Zamojski F., Zhang Z., Zamojski M., Smith G.R., Mendelev N., Liu H., Nudelman G., Moriwaki M., Pincas H., Castanon R.G., et al. Single nucleus multi-omics regulatory landscape of the murine pituitary. Nat. Commun. 2021;12:2677. doi: 10.1530/ey.18.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selewa A., Dohn R., Eckart H., Lozano S., Xie B., Gauchat E., Elorbany R., Rhodes K., Burnett J., Gilad Y., et al. Systematic comparison of high-throughput single-cell and single-nucleus transcriptomes during cardiomyocyte differentiation. Sci. Rep. 2020;10:1535. doi: 10.1038/s41598-020-58327-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Srivastava A., Lareau C., Satija R. Multimodal single-cell chromatin analysis with Signac. Preprint at bioRxiv. 2020 doi: 10.1101/2020.11.09.373613. [DOI] [Google Scholar]
- Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Zamojski M., Smith G.R., Willis T.L., Yianni V., Mendelev N., Pincas H., Seenarine N., Amper M.A.S., Vasoya M., et al. Single nucleus transcriptome and chromatin accessibility of postmortem human pituitaries reveal diverse stem cell regulatory mechanisms. Cell Rep. 2022;38:110467. doi: 10.1016/j.celrep.2022.110467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
The datasets (snRNAseq, snATACseq, sn multiome) generated in the present study are deposited in GEO (accession #GSE178454) and are publicly available as of date of publication (Zhang et al., 2022). The sn human pituitary multi-omics atlas can be browsed via a web-based portal accessible at http://snpituitaryatlas.princeton.edu/. All datasets will also be deposited with the Human Cell Atlas. Accession numbers and web-portal access are also listed in the key resources table.
-
•
All original code has been deposited in Github and is publicly available as of the date of publication. It is accessible at https://github.com/sealfonlab/snRNA_snATAC_snMultiome_QC. DOIs are listed in the text and in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.