

Abstract
Significant proof suggests an essential role played by the bile microbiota in biliary diseases. This study retrospectively analyzed the differences in biliary microbes among patients with perihilar cholangiocarcinoma (pCCA), distal cholangiocarcinoma (dCCA), pancreatic cancer (PC), and cholelithiasis (CH). Bile samples were obtained from 53 patients who underwent endoscopic retrograde cholangiopancreatography (ERCP), and the bile microbiota was analyzed through 16S rRNA gene analysis and next-generation sequencing. Based on the results of linear discriminant analysis effect size (LEfSe), the top three biomarkers for pCCA at the genus level were Pseudomonas, Sphingomonas, and Halomonas; for dCCA were Streptococcus, Prevotella, and Halomonas; and for PC were Pseudomonas, Chloroplast, and Acinetobacter. The top five genera in the pCCA, dCCA, and PC groups showed predictive values with areas under the receiver operating characteristic curves of 91.56%, 95.56%, and 96.59%, respectively. The PICRUSt2 analysis outcomes displayed the diversities of fifteen pathways between the CH and pCCA groups, 22 pathways between the CH and dCCA groups, and eighteen pathways between the CH and PC groups. As this pilot study identified specific microbial bile markers for patients with CH, pCCA, dCCA, and PC, the clinical implications are vast. Further study focusing on distinct bacterial populations in bile will help differentiate biliary diseases.
Keywords: Bile microbiota, perihilar cholangiocarcinoma, distal cholangiocarcinoma, pancreatic cancer, cholelithiasis
Introduction
Cholangiocarcinoma and pancreatic cancer (PC) are common clinical malignant tumors of the biliary and pancreatic systems [1,2]. Additionally, cholelithiasis (CH) is a common clinically benign disease of the biliary and pancreatic systems [3]. As the etiologies of these three diseases are complex and often difficult to differentiate, specific diagnostic capabilities are required.
Traditionally, the biliary system was considered a sterile environment in normal circumstances [4], however Jiménez et al. demonstrated healthy pigs having bacteria in the biliary tract [5]. The study of biliary tract microecology has been bereft of progress due to the difficulty of obtaining bile specimens from patients. Fortunately, the development of endoscopic retrograde cholangiopancreatography (ERCP) technology has made acquiring bile specimens easier.
As metagenomics via high-throughput next-generation sequencing (NGS) was gradually applied to human microbiome research through the years, a more comprehensive understanding of human microecology was revealed [6]. Furthermore, research on the relationship between diseases of the biliary and pancreatic systems, along with the composition of intestinal microbiota has been steadily progressing [7,8]. Han et al. determined that the top three key microbiota of choledocholithiasis at the genus level were Escherichia-Shigella, Fusobacterium, and Enterococcus [9]. Moreover, Lee et al. found that predictive models for biliary tract cancer [10] may be developed by adopting diversities in composing the Corynebacteriaceae Corynebacterium, Oxalobacteraceae Ralstonia, and Comamonadaceae Comamonas species. Nevertheless, the relationship between PC and bile microecology is still uncertain.
Similarly, we believe that there may be microbiome differences among biliary tract diseases. Our study aimed to compare the differences in biliary microbes in patients with CH, perihilar cholangiocarcinoma (pCCA), distal cholangiocarcinoma (dCCA), and PC. The research sought to discover specific bacteria in the CH, pCCA, dCCA, and PC, potentially as biomarkers for the differential diagnosis of these four diseases. Furthermore, our study revealed the relationship between micro-ecological changes and the occurrence of diseases from the perspective of microorganisms.
Materials and methods
Patients and biospecimens
Totally, 53 patients aged 43-80 years who were diagnosed with either CH, pCCA, dCCA, or PC at the Cancer Hospital of the Chinese Academy of Medical Sciences (Beijing, China) and Dongfang Hospital Beijing University of Chinese Medicine (Beijing, China) from March 2017 to March 2019 were retrospectively enrolled in this study. The pathological diagnoses were performed by the pathologists. Clear notice and signed written informed consent were obtained from all participants of this research. The research was approved and overseen by the Clinical Trials Center of the National Cancer Center (Ethical approval number: NCC2018JJJ-001).
The patients were included according to following inclusion criteria: (1) 43-80 years old; (2) diagnosed with either CH, pCCA, dCCA, or PC within one month; (3) able to communicate normally and join the study voluntarily.
The patients were excluded according to following exclusion criteria: (1) with other systemic diseases; (2) had received proton pump inhibitors, antibiotics, prebiotics, or other procedures in the past months; (3) complicated with or had a history of other types of neoplastic diseases; (4) a history of oral ulcers in the past months.
Sample collection
During the ERCP examination, a guidewire (Cook Group, Bloomington, IN, USA) was inserted into the duodenum of patients. Then the three-chamber sphincterotomy device (Cook Group) was inserted along the guidewire, and a 10-mL syringe was used to draw out 5 mL bile for cytology. Then, the bile was placed in a 15-mL round-bottom tube (Falcon-A; CORNING, Corning, NY, USA), and immediately refrigerated at -80°C for storage before using.
DNA extraction, amplification, and sequencing
The CTAB method [11] was employed for the extraction of total genomic DNA from the bile. DNA concentration and purity were checked on 1% agarose gels, and sterile water was used to dilute the DNA samples. The V4 area of the 16S ribosomal RNA (rRNA) gene was amplified by polymerase chain reaction (PCR) using bacterial and reverse primers. The 30 µL PCR reactions composed of 15 µL Phusion® High-Fidelity PCR master mix (New England Biolabs, Ipswich, MA, USA), 0.2 µM each of the forward and reverse primers, and 10 ng template DNA.
Thermal cycling was composed of initial denaturation at 98°C for 1 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s, and elongation at 72°C for 30 s, with a final extension at 72°C for 5 min. Next, the PCR outcomes were mixed with an equal volume of 1× loading buffer (containing SYBR green) and separated in 2% agarose gels for amplicon detection. The PCR mixtures in equidensity ratios were purified with a GeneJETTM gel extraction kit (Thermo Scientific, Waltham, MA, USA). Sequencing libraries were generated using the Ion Plus Fragment Library Kit 48 rxns (Thermo Scientific), following the manufacturer’s instruction. The samples were then evaluated with a Qubit 2.0 Fluorometer (Thermo Scientific), and the sequence library quality was made on an Ion S5TM XL platform to generate 600 bp single-end reads [12].
Sequence processing and taxonomic classification
A total of 53 collected specimens were successfully amplified and processed using the Quantitative Insights into Microbial Ecology (QIIME2, https://qiime2.org/) platform. Cutadapt [13] (v1.9.1, http://cutadapt.readthedocs.io/en/stable/) was employed for subject raw sequences to strictly control the quality and construct feature tables, and reads were analyzed using Usearch v11 [14,15]. After filtering the quality and removing the chimeras, Unoise3 was used for resolving the clean sequences and exacting the amplicon sequence variants (ASVs). The ‘otutab’ function was used to count the ASV abundance table. The feature-classifier ‘classify-sklearn’ plug-in of QIIME2 (QIIME2-2021.4) was used to annotate species based on the silva138.1 database, and the ‘taxa collapse’ plug-in of QIIME2 was used to count the species abundance. The Chao1, Shannon and Simpson index were calculated with the ‘scikit-bio’ module of Python. Lastly, differential biomarkers between various groups [16] were observed using linear discriminant analysis effect size (LEfSe).
Statistical analysis
Demographic and other features among patients with CH, pCCA, dCCA, or PC were compared using t-tests and chi-squared tests and analyzed in the R Studio (v4.1.0) program. The α diversity indicator of the ASV (P=0.05) was analyzed with the ‘LSD.test’ function of the ‘agricolae’ package of the R software. The Bray-Curtis distance matrix at the genus level was calculated with the ‘vegdist’ function of the ‘vegan’ package. Thereafter, principal coordinate analysis was performed using the ‘cmdscale’ function, and the ‘adonis’ function with PERMANOVA. Microbes associated with tumor status were identified by LEfSe. Microbiota with linear discriminant analysis (LDA) scores of more than 3.0 was regarded as various genera. The ‘random forest’ and ‘pROC’ packages were used to find biomarkers based on the relative abundance of species at the genus level. The Stamp software was used to analyze the difference of KEGG metabolites (level 2) predicted by Picrust2 [17]. The R packages adopted in the above data processing process were ‘data.table’, ‘reshape2’, ‘stringr’, and ‘aplot’. Unless stated otherwise, the ‘ggplot2’ package of the R software was adopted to create all figures [18].
Results
To determine the differences in microorganism composition and corresponding biomarkers in the bile of patients with either CH, pCCA, dCCA, or PC, the functional abundance of the various microbes was predicted using PICRUSt2.
Baseline characteristics of patients
The 53 patients enrolled were divided into four groups based on their pathology: pCCA (14 patients), dCCA (9 patients), PC (8 patients), and CH (22 patients). As shown in Table 1, there was no major difference in age or gender among the groups.
Table 1.
The baseline characteristics of patients
| Variables | CH (n=22) | pCCA (n=14) | dCCA (n=9) | PC (n=8) | P-value |
|---|---|---|---|---|---|
| Age, years (x̅±s) | 60.3±14.7 | 68.1±6.0 | 61.6±4.3 | 58.6±10.6 | 0.154 |
| Sex, n (%) | 0.146 | ||||
| Male | 6 (27.3) | 9 (64.3) | 5 (55.6) | 4 (50.0) | |
| Female | 16 (72.7) | 5 (35.7) | 4 (44.4) | 4 (50.0) |
Differences in microbial composition between groups
The relative abundances of microbiota in all groups
Figure 1A, 1B shows the microbiota compositions of the four groups. An upward trend of the Chao1 indicator, Shannon index, and Simpson indicator was observed in the CH as compared to PC groups, as shown in Figure 1C. Obvious diversities were observed between the CH and PC groups in the Chao1 index, as well as the CH and dCCA groups in the Shannon index and Simpson index (P<0.05). In terms of β diversity, significant differences were found in the Bray-Curtis distance matrix at the genus level among the CH, pCCA, dCCA, and PC groups (P=0.001; Figure 1D).
Figure 1.

The relative abundance of microbiota in all groups. A. The abundances of phyla in the CH, pCCA, dCCA, and PC groups. B. The abundances of genera in the CH, pCCA, dCCA, and PC groups. C. The α diversity indices of the CH, pCCA, dCCA, and PC groups. D. The β diversity indices of the CH, pCCA, dCCA, and PC groups.
The microbial biomarkers of all groups
The LEfSe outcomes (Figure 2A-C) at the genus level revealed that the biomarkers for pCCA were Pseudomonas, Sphingomonas, Halomonas, Acinetobacter, Prevotella, Shewanella, Bacillus, Corynebacterium, Chloroplast, Lactococcus, Ellin516, Catenisphaera, f_67_14, and Catenibacterium; for dCCA they were Streptococcus, Prevotella, Halomonas, Helicobacter, Rikenellaceae_RC9_gut_group, Actinobacillus, Lactococcus, Pseudomonas, Catenisphaera, Devosia, Mobiluncus, Sphingobacterium, Comamonas and SC_I_84; and for PC they were Pseudomonas, Chloroplast, Acinetobacter, Allorhizobium_Neorhizobium_Pararhizobium_Rhizobium, Exiguobacterium, Halomonas, Staphylococcus, Cupriavidus, Bacillus, Cutibacterium, Cloacibacterium, Vicinamibacteraceae, Shewanella, SC_I_84, Geobacter, Corynebacterium, MND1, Brevundimonas, f_67_14, Rubellimicrobium, Nitrospira, Massilia, Rokubacteriales, Gaiella, and RB41.
Figure 2.

Linear discriminant analysis effect size (LEfSe) analysis of the abundance patterns of bacterial taxa of bile samples. The LEfSe analysis of the (A) CH and pCCA groups, (B) CH and dCCA groups, and (C) CH and PC groups.
The area under the receiver operating characteristic curves (ROCs) of the top 3-30 genera in pCCA, dCCA, and PC groups
The genera for the random forest plot were chosen for species based on the ‘MeanDecreaseAccuracy’ function values as an analysis outcome of the ‘varImpPlot’ function in declining order. As shown in Figures 3, 4 and 5, the top 5 genera in the pCCA, dCCA, and PC groups showed predictive values with ROCs of 91.56%, 95.56%, and 96.59%, respectively.
Figure 3.

Random forest model and ROC results of CH and pCCA groups. A. The random forest model outcomes of 30 genera of CH and pCCA groups. B. ROC analysis of CH and pCCA groups tested top 3-30 genera.
Figure 4.

Random forest model and ROC results of CH and dCCA groups. A. Thirty genera of CH and dCCA groups tested for the random forest model outcomes. B. ROC analysis of CH and dCCA groups tested for the top 3-30 genera.
Figure 5.

Random forest model and ROC results of CH and PC groups. A. Thirty genera of CH and PC groups tested for the random forest model outcomes. B. ROC analysis of CH and PC groups tested for the top 3-30 genera.
The functional prediction of microbiomes among the pCCA, dCCA, and PC groups by PICRUSt2
The PICRUSt2 outcomes showed diversities in 15 pathways between the CH and pCCA groups, including amino acid metabolism, protein families, metabolism, drug resistance, and antimicrobial characteristics. There were differences in 22 pathways between the CH and dCCA groups, which includes protein families, genetic information processing, membrane transport, as well as signaling and cellular processes. There were differences in 18 pathways between the CH and PC groups, which included amino acid metabolism, xenobiotics biodegradation and metabolism, as well as lipid metabolism (Figures 6, 7 and 8).
Figure 6.
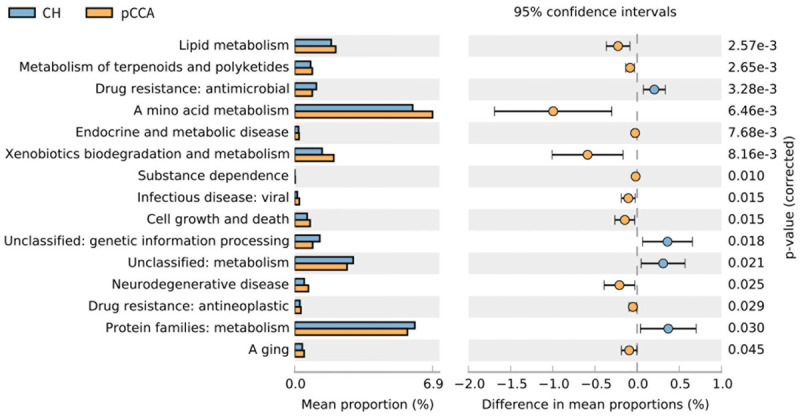
Forecast of functional abilities of CH and pCCA groups. KEGG pathway analysis and the related abundance of genes.
Figure 7.
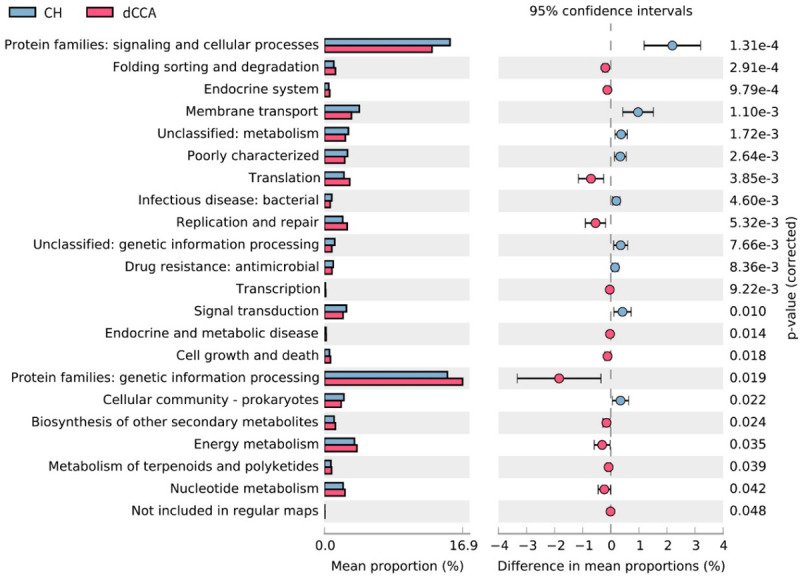
Forecast of functional abilities of CH and dCCA groups. KEGG pathway analysis and the related abundance of genes.
Figure 8.
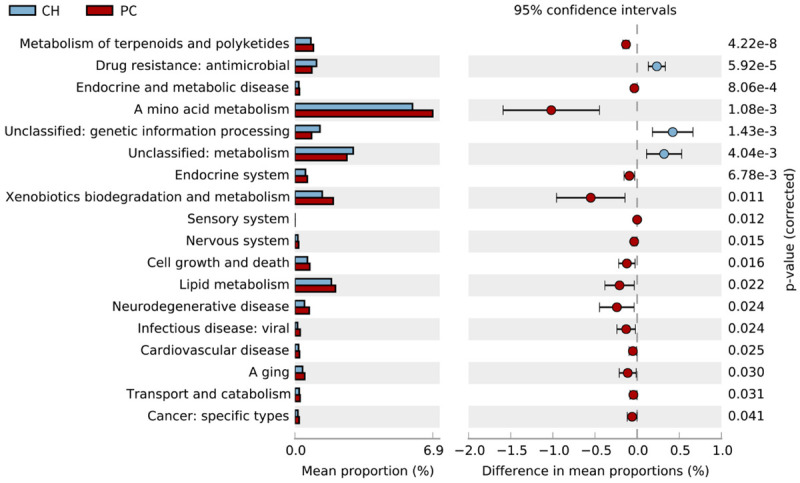
Forecast of functional abilities of CH and PC groups. KEGG pathway analysis and the related abundance of genes.
Discussion
The associations between microbes and tumors were suggested with the development of microbiome research; however the limitation of techniques to obtain bile specimens stalled major research in the field [6]. Fortunately, the development of NGS technology enables the use of 16s rRNA sequencing to find the species and functions of microorganisms present in bile [19]. Some studies have even used second-generation sequencing technology to characterize the microorganisms present in bile, primarily in relation to different disease conditions [20,21].
This was the first study to compare the bile microecology of patient groups with CH, pCCA, dCCA, and PC. We found an increase in α diversity of the dCCA and PC groups compared to the CH group, respectively. This may be due to the disease state where dysfunction of the sphincter of Oddi (SO) enables bacteria from the intestine to enter the biliary system.
A study by Chen et al. pointed out that microbial ecological disturbance may be the crucial element in forming dCCA. Compared with patients with choledocholithiasis, the phyla Gemmatimonadetes, Nitrospirae, Chloroflexi, Latescibacteria, and Planctomycetes were significantly increased in patients with dCCA [22]. Their results differ from the conclusions drawn by our study, which could be due to the relatively small number of cases studied in both studies. Aviles-Jimenez et al. observed that in patients with dCCA, the levels of Methylophilaceae, Fusobacterium, Prevotella, Actinomyces, Novosphingobium and H. pylori were elevated. Furthermore, their study predicted a growing abundance of functions related to virulence genes by H. pylori in dCCA [23]. In consensus, our LEfSe results showed that the genera Prevotella, Helicobacter, and Actinobacillus are biomarkers of dCCA. So far, no studies have specifically reported pCCA and PC microorganisms as bile markers. Therefore, our research can serve as a key resource for future research.
The top five genera in the pCCA, dCCA, and PC groups showed predictive values with ROCs of 91.56%, 95.56%, and 96.59%, respectively. This discovery suggests that the bile specimens collected from patients for microbiological testing during the ERCP examination can aid the differential diagnosis of bile diseases. The clinical application of this finding can prove to be highly valuable for future basic research and translational studies.
Lee et al. found that predictive models for biliary tract cancer may be developed by diversities composing the Bifidobacteriaceae and Pseudomonaceae families, as well as the species, Corynebacteriaceae Corynebacterium, Oxalobacteraceae Ralstonia, and Comamonadaceae Comamonas [10]. In our research, the microorganisms involved in the construction of the diagnostic model did not contain these same types of bacteria. Nevertheless, the proof-of-concept has been established to build a model for diagnostic purposes using the principles of our study.
The PICRUSt2 results showed that there were differences in the metabolic pathways between the CH and pCCA groups, the CH and dCCA groups, and the CH and PC groups, respectively. Although some studies have shown that PICRUSt2 is more precise than PICRUSt1 and other competitors [17], further research should combine metagenomics and metabolomics.
The current research has limitations. First, for ethical reasons, obtaining bile specimens from healthy people is not possible. Therefore, there were no bile specimens from healthy controls in this study and only patients with gallstones could be used as controls. Second, this is a single-center research study with only a small number of cases, which should be expanded in the future.
Conclusion
This study compared the bile microecology of CH, pCCA, dCCA, and PC for the first time, revealing the characteristic bacteria present in the bile specimens of patients with CH, pCCA, dCCA, and PC, which was used to construct diagnostic models for the respective bile diseases. The enrichment of species from the respective groups may progressively take part in the pathogenesis of cancer. Therefore, our study provides new insight into a better understanding the link between the bile microbiome and the mechanism of disease progression in pCCA, dCCA, and PC.
Acknowledgements
This project was supported by the National Key Research and Development Program of China (grant No. 2016YFC1302800, 2016YFC0901402, 2018YFC1313103); the Beijing Science and Technology Project (D17110002617002); the CAMS Innovation Fund for Medical Sciences (CIFMS) (grant No. 2016-I2 M-001, 2017-I2 M-1-001, 2019-I2 M-2-004); the Sanming Project of Medicine in Shenzhen (No. SZSM2019110080); the PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (grant No. 2017320012); and the PUMC Graduate Innovation Fund (grant No. 2019-1002-81).
Disclosure of conflict of interest
None.
Abbreviations
- ASVs
Amplicon sequence variants
- AUCs
Area under the curves
- CH
Cholelithiasis
- dCCA
Distal cholangiocarcinoma
- ERCP
Endoscopic retrograde cholangiopancreatography
- LDA
Linear discriminant analysis
- LEfSe
Linear discriminant analysis effect size
- NGS
Next generation sequencing
- PC
Pancreatic cancer
- pCCA
Perihilar cholangiocarcinoma
- PCR
Polymerase chain reaction
- rRNA
Ribosomal RNA
References
- 1.Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22:9694–9705. doi: 10.3748/wjg.v22.i44.9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15:95–111. doi: 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tazuma S, Unno M, Igarashi Y, Inui K, Uchiyama K, Kai M, Tsuyuguchi T, Maguchi H, Mori T, Yamaguchi K, Ryozawa S, Nimura Y, Fujita N, Kubota K, Shoda J, Tabata M, Mine T, Sugano K, Watanabe M, Shimosegawa T. Evidence-based clinical practice guidelines for cholelithiasis 2016. J Gastroenterol. 2017;52:276–300. doi: 10.1007/s00535-016-1289-7. [DOI] [PubMed] [Google Scholar]
- 4.Csendes A, Burdiles P, Maluenda F, Diaz JC, Csendes P, Mitru N. Simultaneous bacteriologic assessment of bile from gallbladder and common bile duct in control subjects and patients with gallstones and common duct stones. Arch Surg. 1996;131:389–394. doi: 10.1001/archsurg.1996.01430160047008. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez E, Sanchez B, Farina A, Margolles A, Rodriguez JM. Characterization of the bile and gall bladder microbiota of healthy pigs. Microbiologyopen. 2014;3:937–949. doi: 10.1002/mbo3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helmink BA, Khan M, Hermann A, Gopalakrishnan V, Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med. 2019;25:377–388. doi: 10.1038/s41591-019-0377-7. [DOI] [PubMed] [Google Scholar]
- 7.Nicoletti A, Ponziani FR, Nardella E, Ianiro G, Gasbarrini A, Zileri DVL. Biliary tract microbiota: a new kid on the block of liver diseases? Eur Rev Med Pharmacol Sci. 2020;24:2750–2775. doi: 10.26355/eurrev_202003_20548. [DOI] [PubMed] [Google Scholar]
- 8.Giordano DM, Pinto C, Maroni L, Benedetti A, Marzioni M. Inflammation and the Gut-Liver axis in the pathophysiology of cholangiopathies. Int J Mol Sci. 2018;19:3003. doi: 10.3390/ijms19103003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han J, Wu S, Fan Y, Tian Y, Kong J. Biliary microbiota in choledocholithiasis and correlation with duodenal microbiota. Front Cell Infect Mi. 2021;11:625589. doi: 10.3389/fcimb.2021.625589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H, Lee HK, Min SK, Lee WH. 16S rDNA microbiome composition pattern analysis as a diagnostic biomarker for biliary tract cancer. World J Surg Oncol. 2020;18:19. doi: 10.1186/s12957-020-1793-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Wang S. Bacterial community diversity on in-shell walnut surfaces from six representative provinces in China. Sci Rep. 2017;7:10054. doi: 10.1038/s41598-017-10138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z, Liu Y, Dou L, Zhang Y, He S, Zhao D, Zhang W, Wang G. The effects of smoking and drinking on the oral and esophageal microbiota of healthy people. Ann Transl Med. 2021;9:1244. doi: 10.21037/atm-21-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin M. Cut adapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- 14.Edgar RC. Accuracy of taxonomy prediction for 16S rRNA and fungal its sequences. PeerJ. 2018;6:e4652. doi: 10.7717/peerj.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–688. doi: 10.1038/s41587-020-0548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Dou L, Zhang Y, He S, Zhao D, Hao C, Song G, Zhang W, Liu Y, Wang G. Characterization of the oral and esophageal microbiota in esophageal precancerous lesions and squamous cell carcinoma. Front Cell Infect Mi. 2021;11:714162. doi: 10.3389/fcimb.2021.714162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swanson KS, de Vos WM, Martens EC, Gilbert JA, Menon RS, Soto-Vaca A, Hautvast J, Meyer PD, Borewicz K, Vaughan EE, Slavin JL. Effect of fructans, prebiotics and fibres on the human gut microbiome assessed by 16S rRNA-based approaches: a review. Benef Microbes. 2020;11:101–129. doi: 10.3920/BM2019.0082. [DOI] [PubMed] [Google Scholar]
- 20.Shen H, Ye F, Xie L, Yang J, Li Z, Xu P, Meng F, Li L, Chen Y, Bo X, Ni M, Zhang X. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci Rep. 2015;5:17450. doi: 10.1038/srep17450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye F, Shen H, Li Z, Meng F, Li L, Yang J, Chen Y, Bo X, Zhang X, Ni M. Influence of the biliary system on biliary bacteria revealed by bacterial communities of the human biliary and upper digestive tracts. PLoS One. 2016;11:e150519. doi: 10.1371/journal.pone.0150519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen B, Fu SW, Lu L, Zhao H. A preliminary study of biliary microbiota in patients with bile duct stones or distal cholangiocarcinoma. Biomed Res Int. 2019;2019:1–12. doi: 10.1155/2019/1092563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aviles-Jimenez F, Guitron A, Segura-Lopez F, Mendez-Tenorio A, Iwai S, Hernandez-Guerrero A, Torres J. Microbiota studies in the bile duct strongly suggest a role for Helicobacter pylori in extrahepatic cholangiocarcinoma. Clin Microbiol Infect. 2016;22:111–178. doi: 10.1016/j.cmi.2015.10.008. [DOI] [PubMed] [Google Scholar]