

Abstract
Lung cancer is the primary cause of cancer-related deaths worldwide. Recently, although the microbiome has emerged as the key modulator of the carcinogenesis, it has not been evaluated in lung cancer. Here, we evaluated the microbial composition of lung cancer tissues according to the histologic type and genetic mutation, compared it with that of the adjacent normal lung tissues, and investigated the association between the lung microbiome and clinical parameters. We collected lung tissue samples from 162 patients with non-small cell lung cancer (NSCLC, 162 cancer and 54 adjacent normal tissues), surgically resected between January 2018 and December 2019, and analyzed their microbiome using 16S rRNA gene amplicon sequencing, the QIIME2 pipeline, and statistical analyses. NSCLC tissues had significantly lower alpha diversity than the normal tissues, and their microbial composition differed according to the histologic type and cancer genetic mutation. The genera Romboutsia, Novosphingobium, Acinetobacter, and Prevotella were significantly overrepresented in NSCLC tissues. Alpha diversity steadily declined from a normal to a more advanced stage, and microbial compositional differences were noted along with recurrence. Stenotrophomonas was the most predominant genus in the NSCLC tissues of patients with recurrence. The pathways related to the tricarboxylic acid cycle and L-glutamate and L-glutamine biosynthesis were predominant in adenocarcinoma, whereas those related to purine and pyrimidine nucleotide degradation and formaldehyde assimilation were predominant in squamous cell carcinoma. Our findings suggest that the altered lung cancer microbial composition might be associated with cancer initiation and/or progression.
Keywords: Lung cancer, normal lung tissue, microbiome, Stenotrophomonas
Introduction
Lung cancer is the most frequently diagnosed cancer and the leading cause of cancer-related deaths worldwide, constituting approximately 11.6% of all cancer diagnoses and being responsible for 18.4% of all cancer-related deaths in 2020 [1]. Lung cancer is widely known as a complicated disease caused by the interactions between the host and the environment [2]. Among the diverse environmental risk factors, microbiota have emerged as key modulators of both the carcinogenic process and the immune response against cancer cells [3]. While healthy lungs were historically considered as a sterile environment, it has recently been suggested that certain microbiota exist in the lungs and microbiota changes are associated with the development of diseases, including lung cancer [4]. The microbiome can contribute to carcinogenesis via host inflammatory pathways, bacterial metabolites, and genotoxic pathways [4].
Previous studies on the lung cancer microbiome have identified the Firmicutes, Proteobacteria, and Bacteroidetes phyla, including the Streptococcus, Neisseria, and Prevotella genera, in lung cancer tissues [2,5-10]. A study by Yu et al. found that the Thermus and Legionella genera are highly abundant in tissues from advanced stage lung cancer patients and in patients who develop metastases, respectively, suggesting their role in tumor progression [8]. Another study showed that greater abundance of the families Bacteroidaceae, Lachnospiraceae, and Ruminococcaceae in the paired normal lung tissues is associated with reduced recurrence-free or disease-free survival and demonstrated the potential relationship between the normal lung microbiota and lung cancer prognosis [10].
Although there have been various studies to identify the lung cancer microbiome in respiratory samples such as saliva, sputum, bronchoalveolar lavage fluid, and bronchial brushing [2], little is known about the microbiota profile in lung cancer tissues, particularly the differences in the microbial composition according to the histologic type and/or genetic mutations. Moreover, few studies exist on the relationship between the microbiome and lung cancer prognosis.
Therefore, we aimed to identify the microbial composition of lung cancer tissues according to the histologic type and genetic mutations, compare it with that of the adjacent normal lung tissues, and investigate the association between the lung microbiome and the prognosis of lung cancer. Among lung cancers, non-small cell lung cancer (NSCLC) was selected for the analysis, which accounts for the majority (80-85%) of lung cancers and almost all surgically resected lung cancer cases, allowing for the collection of tissues from a large number of patients.
Material and methods
Study population and sample collection
We retrospectively and randomly selected 216 frozen lung tissue samples (162 cancer tissues and 54 adjacent normal tissues), which were surgically resected from 162 patients with NSCLC between January 2018 and December 2019 at the Asan Medical Center. Patients were included if they were 18 or more years of age and signed an informed consent form for the collection of tissues in the operating room. The exclusion criteria were as follows: patients with SCLC, NSCLC other than adenocarcinoma (AC) and squamous cell carcinoma (SCC), less than 18 years of age, and inability to provide informed consent.
We collected lung cancer samples by dividing them into AC and SCC, the most common subtypes of NSCLC. Then, the AC group was divided according to the presence of the epidermal growth factor receptor (EGFR) mutation, which is one of the most frequent mutations in NSCLC. Thus, the lung cancer samples were divided into the following three groups according to the histologic type and the presence of the EGFR mutation: AC with EGFR mutation (AC EGFR+), AC without EGFR mutation (AC EGFR-), and SCC. In total, 54 cancer tissues were included in each group, together with the paired normal lung tissues obtained from AC EGFR+ patients who were predicted to show relatively homogeneous features (Figure S1). All study patients consented to the collection of tissues in the operating room at the time of resection. Tissue samples were sterilely cut in the operating room, transferred to cryovials, and immediately snap-frozen in liquid nitrogen. Then, they were stored in the Bio-Resource Center at the Asan Medical Center until analysis. Clinical data, including baseline demographics, smoking status, postoperative stage, recurrence, and death, were retrospectively collected from medical records. The study protocol was approved by the Institutional Review Board of Asan Medical Center (IRB No. 2020-0194).
DNA extraction and bacterial 16S rRNA gene sequencing
We extracted the bacterial gDNA from 216 lung tissue samples. DNA extraction was performed using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. We cut each lung tissue using a flame-sterilized blade, and a piece of lung tissue (0.2 g) was transferred to a PowerBead tube. Horizontal vortexing was performed using Vortex Genie 2 (Scientific Industries, Bohemia, New York, USA) at the maximum speed. The gDNA concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Polymerase chain reaction (PCR) amplification was performed using the primers targeting the V4-V5 region of the 16S rRNA gene (forward: 5’-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCA GCA GCY GCG GTA AN-3’; reverse: 5’-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GCC GTC AAT TCN TTT RAG T-3’). The PCR conditions used were as follows: denaturation at 95°C for 3 min, followed by 30 cycles of amplification at 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min. The PCR products were purified using AMPure XP beads (Beckman Coulter, High Wycombe, UK). The quality of the final purified product was assessed using a Nanodrop 2000 spectrophotometer. The final products were sequenced using an Illumina MiseqTM platform (Illumina, San Diego, CA, USA).
Microbiome data analysis
The lung tissue microbiome sequences were analyzed using the QIIME™ (Quantitative Insights Into Microbial Ecology) 2 pipeline (2019.7) [11]. Primer sequences of the raw sequences were removed using the Cutadapt [12] plugin. In this step, the reads that did not include bacterial primer sequences or that included low-quality primer sequences were removed. We denoised the sequences using the DADA2 plugin implemented in QIIMETM 2 and identified the bacterial amplicon sequence variants (ASVs). The ASVs were classified using the SILVA database [13]. Chloroplast, mitochondrial, and unassigned ASVs were removed using a taxa filter. We aligned the ASVs using the phylogeny align-to-tree-mafft-fasttree plugin and performed microbial diversity analyses using rarefying samples to an even depth of 50 sequences per sample.
Alpha diversity was evaluated using Chao1, the Shannon index, and phylogenetic diversity [14,15]. Beta diversity was assessed using unweighted UniFrac distances [16]. The microbial compositional differences between groups were assessed using principal coordinates analysis (PCoA) with unweighted UniFrac distances. The unweighted intragroup UniFrac distances were represented by box-and-whiskers plots. Differential taxonomy was identifi-ed using linear discriminant analysis (LDA) effect size (LEfSe) [17]. We used the neutral community model-based dominance test to determine the cause of the lung microbiome differences between the normal and cancer groups [18]. Additionally, we conducted a stratified sub-analysis, based on the postoperative stage and the recurrence of lung cancer, to address the association between the lung microbiome and lung cancer prognosis [10]. The Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2) v.2.1.3-b software, which predicts the gene family abundance, was used to predict the functional profiles from the 16S rRNA data [19,20]. The ASV table resulting from bacterial community analysis and the representative sequences were used, and the predicted functional profiles were obtained using the picrust2_pipeline.py. We used the MetaCyc pathway, which was inferred using PICRUSt2.
Statistical analysis
All clinical data are presented as mean ± standard deviation or median (interquartile range [IQR]) for continuous variables and as numbers (%) for categorical variables. Data categorized according to the cancer type were compared using one-way analysis of variance or Kruskal-Wallis test (for continuous variables) and the χ2 or Fisher’s exact test (for categorical variables). If there was a significant difference among the three values, a post hoc analysis for multiple comparisons was performed using the Bonferroni correction. All analyses of the clinical data were performed using SPSS software (Version 24.0; SPSS, Chicago, IL, USA).
We performed an analysis of similarity to identify the differences in the microbial communities. To identify any significant differences in alpha diversity and the UniFrac dissimilarities, we performed the Wilcoxon rank-sum test or t-test in R. The Kruskal-Wallis rank-sum test was used to distinguish the significant differential abundance among different groups in LEfSe analysis. The Kaplan-Meier method was used to estimate the cumulative rate of recurrence. All significance tests were two-sided, and p-values < 0.05 indicated statistical significance.
Results
Clinical characteristics of the study population
The mean patient age was 65.3 years (range 21-83), with a preponderance of male (64.8%) patients. Upon comparing the three groups according to the cancer type, the proportions of males and smokers were significantly greater in the SCC group than in the AC EGFR+ and AC EGFR- groups (P < 0.001). Furthermore, the SCC group had a significantly higher median number of pack-years (39 pack-years, P < 0.001). Otherwise, there were no significant differences in age, postoperative stage, nodal stage, recurrence, and death among the three groups. Table 1 presents the clinical characteristics of the 162 study patients.
Table 1.
Clinical characteristics of the study population
| AC EGFR+ (n = 54) | AC EGFR- (n = 54) | SCC (n = 54) | p-value | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| AC EGFR+ vs. AC EGFR- | AC EGFR- vs. SCC | SCC vs. AC EGFR+ | |||||
| Age, yr | 64.9 ± 9.3 | 64.2 ± 9.2 | 66.7 ± 9.4 | 0.358 | |||
| Male gender, n (%) | 28 (51.9%) | 25 (46.3%) | 52 (96.3%) | < 0.001 | 0.564 | < 0.001 | < 0.001 |
| Body mass index (kg/m2) | 25.0 ± 2.5 | 25.0 ± 3.4 | 24.7 ± 3.0 | 0.882 | |||
| Smoking status | |||||||
| Non-smoker, n (%) | 30 (55.6%) | 31 (57.4%) | 3 (5.6%) | < 0.001 | 0.846 | < 0.001 | < 0.001 |
| Ever smoker, n (%) | 24 (44.4%) | 23 (42.6%) | 51 (94.4%) | ||||
| Pack-years, median (IQR) | 0 (0-30) | 0 (0-27) | 39 (20-46) | < 0.001 | 0.671 | < 0.001 | < 0.001 |
| Stage, n (%) | |||||||
| I-II | 41 (75.9%) | 44 (81.5%) | 45 (83.3%) | 0.603 | |||
| III-IV | 13 (24.1%) | 10 (18.5%) | 9 (16.7%) | ||||
| Nodal stage | |||||||
| N0 | 39 (72.2%) | 45 (83.3%) | 39 (72.2%) | 0.296 | |||
| N1-2 | 15 (27.8%) | 9 (16.7%) | 15 (27.8%) | ||||
| Follow-up duration months, median (IQR) | 27.5 (24.0-29.0) | 22.0 (18.8-25.3) | 25.0 (11.8-27.0) | < 0.001 | < 0.001 | 0.193 | 0.004 |
| Recurrence, n (%) | 16 (29.6%) | 13 (24.1%) | 9 (16.7%) | 0.280 | |||
| Death, n (%) | 2 (3.7%) | 1 (1.9%) | 6 (11.1%) | 0.085 | |||
Microbial diversity analysis
The lung tissue microbiome of the 216 tissues was analyzed using 16S rRNA gene amplicon sequencing. Twelve samples that had no or low sequencing data were excluded. In 204 samples, a total of 8,552,251 paired-end reads were merged into 326,385 reads from the tissue samples, resulting in an average of 1,599 merged reads per sample. As the rarefied depth was 50, we calculated the alpha diversity and beta diversity using 112 samples (Figure S1).
The alpha diversity indexes, including Chao1, Shannon, and phylogenetic diversity, were significantly higher in the normal group than in the cancer groups (P < 0.0001, P < 0.05, and P < 0.01, respectively, Figure 1A). When comparing the three histologic cancer types, Chao1 and phylogenetic diversity were higher in the normal group than in the AC and SCC groups (Figure 1B). The comparison among the four groups in accordance with the histologic type and EGFR mutation showed that the normal group had greater diversity than all the other groups, as assessed via the Chao1 index (Figure 1C). When we performed sub-analysis by rarefying samples to the depth of 25 and 100 sequences, according to the study flow of Figure S2, not only the Chao1 index but also the phylogenetic diversity was greater in the normal group than in all the other groups (Figure S3).
Figure 1.
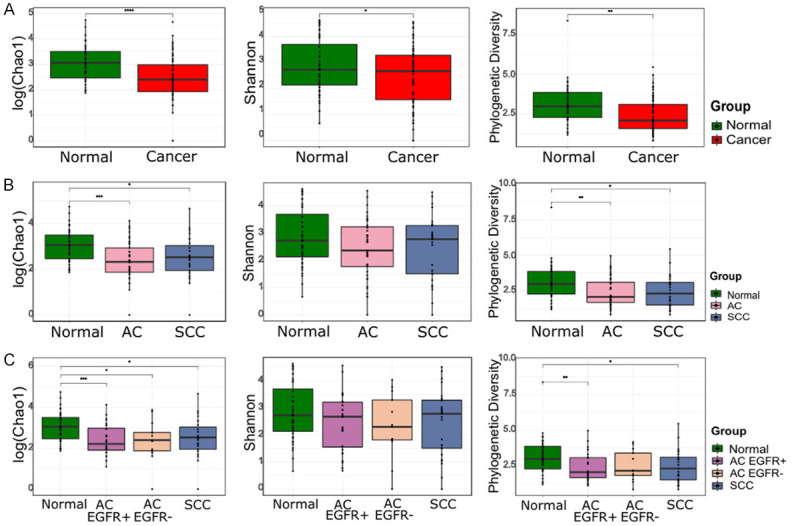
Comparison of the alpha diversity indexes (Chao1, Shannon, and phylogenetic diversity) between lung cancer and normal lung tissues. (A) Normal vs. Cancer, (B) Normal vs. AC vs. SCC, and (C) Normal vs. AC EGFR+ vs. AC EGFR- vs. SCC. Box plot represents the minimum, first quartile, median, third quartile, and maximum of the alpha indexes.
Unweighted UniFrac distances were calculated to evaluate the similarity of microbiota profiles. The PCoA based on the unweighted UniFrac distance revealed significant compositional differences among different groups, indicating a more heterogeneous microbial distribution in the cancer group (Figure S4B and S4C). The results of the sub-analysis, wherein samples were rarefied to a depth of 25 and 100 sequences, were similar (Figure S3). Among the cancer groups, there was also a compositional difference between AC and SCC (Figure S4D). The box-and-whisker plot of unweighted intragroup UniFrac distances showed that the cancer group had a significantly greater dissimilarity among samples than the normal group, in the following order: SCC, AC, and normal group (Figure S4E-S4H).
Differential abundance analysis
To detect the taxa with differential abundance among the different groups, we used LEfSe with an LDA value of 3.0. By comparing the cancer and normal groups, 25 bacterial taxa at the genus level with significant abundance differences were identified. The genera Romboutsia, Christensenellaceae R-7 group, Novosphingobium, Acinetobacter, Rhizobium, and Prevotella were significantly overrepresented in the cancer group, whereas other taxa, including Staphylococcus, Burkholderiaceae, and Cutibacterium, were enriched in the normal group. The taxa enriched in the different groups are displayed in Figure 2. Notably, compared with the normal group, Haemophilus influenzae (H. influenzae) was enriched in the SCC group (Figure 2). In total, four ASVs were classified as species H. influenzae and there were nine samples containing these ASVs. Seven of them (77.8%, 7/9) were in the SCC group, one in the normal group, and the other in the AC group. Table S1 presents the ASV count classified as species H. influenzae in each sample. We selected two SCC tissues with the most abundant H. influenzae ASVs. The tissues were disrupted with stainless steel beads, plated, and cultured on Chocolate agar in 5% CO2 at 37°C. After three days, we observed colonies in one of the two SCC tissues. We picked the colonies, spread them onto a fresh Chocolate agar plate using the dilution streaking method [21], and incubated the plate at 37°C and 5% CO2 for three days. We could identify H. influenzae from the pure culture using 16S rRNA sequencing.
Figure 2.

The linear discriminant analysis effect size plots of bacterial communities in cancer and normal lung tissues. Linear discriminant analysis scores were calculated, with higher scores indicating a greater effect size (significance determined by an LDA score > 3.0 and a p < 0.05 in the Kruskal-Wallis rank-sum test). Taxonomic categories include f = family, g = genus, and s = species. (A) Normal vs. Cancer, (B) Normal vs. AC EGFR+, (C) Normal vs. AC EGFR-, (D) Normal vs. SCC, (E) SCC vs. AC, and (F) AC EGFR+ vs. AC EGFR-.
Neutral community model-based dominance test
We used a neutral community model-based dominance test to determine the cause of the lung microbiome difference between the normal and cancer groups (Figure S5). The goodness-of-fit (R 2) values were 0.74 and 0.64 for the normal and cancer groups, respectively. Considering that an R 2 value close to 1 implies that the composition of the lung microbiome is consistent with the neutral processes of dispersal and ecological drift [18], the microbial community of the normal group was a better fit to the neutral model than that of the cancer group. Next, we determined whether any ASVs deviated from the predictions of the neutral model. While some ASVs, including the genera Micrococcus, Romboutsia, Novosphingobium, Bacteroides, Parasutterella, and Sphingobium, were overrepresented, H. influenzae species and Burkholderiaceae family were underrepresented in the cancer group.
Microbiome analysis according to the postoperative lung cancer stage
To address the association between the lung microbiome and the prognosis of lung cancer, microbial diversity and taxonomy were analyzed by dividing the samples according to the postoperative stage: stages l-ll and stages lll-lV of TNM classification. The alpha diversity, which was evaluated via Chao1, and the phylogenetic diversity steadily declined from normal to stages I-II and to stages lll-lV (Figure 3A). Compositional differences were noted based on the beta diversity analysis. The PCoA plot revealed that the microbial distribution differed according to the postoperative lung cancer stage (P = 0.014, Figure 3B). Although there was no statistical significance, lung cancer at an advanced stage tended to have greater dissimilarities (Figure 3C). In the LEfSe analysis of three subgroups according to the postoperative lung cancer stage, the Staphylococcus, Burkholderia-Caballeronia-Paraburkholderia, and Peptoniphilus genera were enriched in the advanced stage lung cancer group (Figure 3D).
Figure 3.
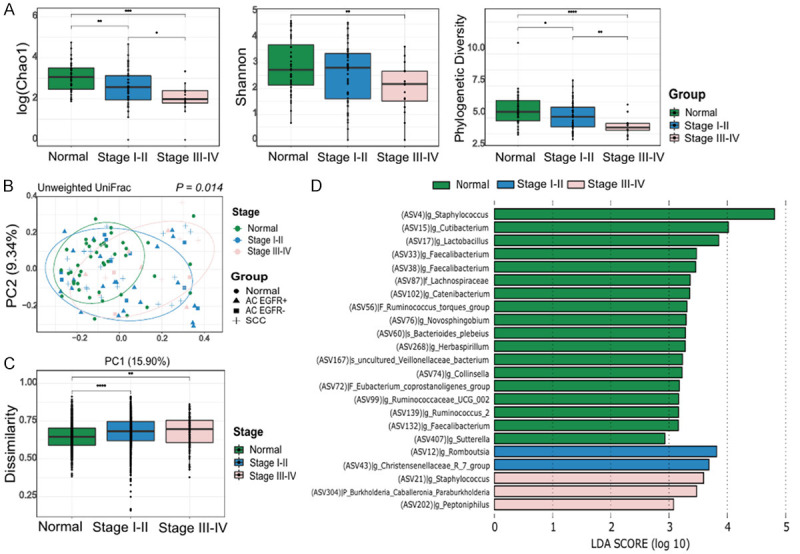
Microbiome analysis according to the postoperative lung cancer stage. (A) Comparison of the alpha diversity indexes (Chao1, Shannon, and phylogenetic diversity), (B) Principal coordinates analyses of the unweighted UniFrac distance, (C) Box-and-whisker plot of unweighted intragroup UniFrac distances, and (D) LEfSe analysis of the bacterial communities.
Microbiome analysis according to lung cancer recurrence
Next, we analyzed the microbial diversity and taxonomy by dividing the samples according to recurrence during the follow-up period. The alpha diversity indexes did not differ according to recurrence (Figure 4A). Compositional differences were noted based on beta diversity analysis. The PCoA plot revealed that the microbial distribution differed with recurrence, presenting a more heterogeneous microbial distribution in the cancer with recurrence group (P = 0.002, Figure 4B). Considering the unweighted intragroup UniFrac distance in Figure 4C, the dissimilarity among the samples steadily increased in the following order: normal, lung cancer without recurrence, and lung cancer with recurrence. In the LEfSe analysis of the three subgroups according to recurrence, several differential microbial taxa, including the Stenotrophomonas, Bacteroides, and Peptoniphilus genera, were noted in the cancer with recurrence group (Figure 4D). We used the Kaplan-Meier method to estimate the cumulative rate of recurrence according to the presence of the genus Stenotrophomonas, which was identified as the most predominant in the lung cancer with recurrence group. The cumulative rate of recurrence was significantly higher in the groups with Stenotrophomonas than in those without Stenotrophomonas (P = 0.013, Figure 4E).
Figure 4.

Microbiome analysis according to the recurrence of lung cancer. (A) Comparison of the alpha diversity indexes (Chao1, Shannon, and phylogenetic diversity), (B) Principal coordinates analyses of the unweighted UniFrac distance, (C) Box-and-whisker plot of unweighted intragroup UniFrac distance, (D) LEfSe analysis of the bacterial communities, and (E) The cumulative rate of recurrence according to the presence of the Stenotrophomonas genus.
Pathway analysis predicted using PICRUSt2
To better understand how the bacterial functional profiles differed according to the histologic type of cancer, we used PICRUSt2, a bioinformatic analysis tool, to predict the functional metagenomes from 16S rRNA gene profiling data. We found 18 differentially abundant MetaCyc pathways among the three groups (with an LDA score > 2.9) (Figure 5). The pathways related to the tricarboxylic acid (TCA) cycle, sulfur compound metabolism, fatty acid salvage, and L-glutamate and L-glutamine biosynthesis were predominantly observed in the AC group, whereas those related to adenine and adenosine salvage, acetylene degradation, purine and pyrimidine nucleotide degradation, L-arginine degradation, formaldehyde assimilation, biotin biosynthesis, and 2-carboxy-1,4-naphthoquinol biosynthesis were predominant in the SCC group.
Figure 5.

Heatmap of the differentially presented pathways according to the histologic type of cancer predicted using PICRUSt2. Red represents a high abundance, whereas blue represents a low abundance. Significance was determined based on an LDA score > 2.9 and P < 0.05.
Discussion
Although an increasing number of studies have profiled the microbiome of respiratory samples from lung cancer patients, the microbiota profiles of lung cancer tissues and their association with clinical and pathological parameters remain unexplored. One of the key findings in our study was that the alpha diversity was significantly higher in normal lung tissues than in the cancer tissues, and the microbial composition differed according to the histologic type of cancer and the EGFR mutation. When we conducted a stratified sub-analysis to address the association between the lung microbiome and lung cancer prognosis, microbial compositional differences were noted according to the postoperative lung cancer stage and the presence of recurrence. Finally, we found several inferred metagenomic functional pathways enriched in different histologic types of cancer from the 16S rRNA data.
Greathouse et al. showed a similar alpha diversity between non-tumor adjacent tissues and tumor tissues [6]. Another study showed that the alpha diversity was significantly lower in tumor tissues than in non-malignant lung tissues, but no significant differences were observed in the overall composition (beta diversity) [8]. We revealed the compositional differences between the normal and cancer groups, as well as a decreased bacterial diversity in lung cancer tissues compared to that in normal lung tissues. This suggested that the dysbiosis and altered microbial composition may be associated with cancer pathogenesis.
Only a few studies have reported the microbiome profile in lung cancer tissues. Liu et al. observed that lung cancer patients had a higher prevalence of Firmicutes (Streptococcus) and Bacteroidetes (Prevotella) than emphysema-only patients [22]. In our study, Provotella_9 was also predominant in cancer tissues, particularly in the AC EGFR+ group. Another study by Apopa et al. found that the predominant phyla in the formalin-fixed lung samples embedded in paraffin were Actinobacteria, Proteobacteria, and Bacteroidetes. The Cyanobacteria phylum was consistently observed in AC samples [22]. We found that the Romboutsia, Christensenellaceae R-7 group, Novo-sphingobium, Acinetobacter, Rhizobium, and Prevotella genera were significantly overrepresented in the cancer group, with some ASVs occurring more frequently than predicted using the neutral community model. A previous study reported that the Novosphingobium genus is present in more severe chronic obstructive pulmonary disease and increases inflammation in a smoke exposure mouse model [23]. It might also promote carcinogenesis by creating an inflammatory milieu. Previously, 16S rRNA gene sequencing and transcriptome analyses of bronchial brushing samples revealed that airway Veillonella, Streptococcus, and Prevotella are associated with the upregulation of extracellular signal-regulated kinase and phosphoinositide 3-kinase signaling pathways, which regulate cell proliferation, survival, and differentiation in the airway [24].
Here, H. influenzae was enriched in the SCC group compared to the normal group, and we demonstrated the presence of H. influenzae from a pure culture of SCC lung tissues. Ochoa et al. found that the exposure of the airway to smoke particulates and non-typeable H. influenzae promoted lung cancer cell proliferation via the release of interleukin-6 and other inflammatory cytokines, which further activated the signal transducer and activator of transcription 3 and nuclear factor kB pathways in the airway epithelium [25]. Additionally, several studies reported that airway inflammation induced by non-typeable H. influenzae provides a tumor microenvironment that favors lung tumor promotion and progression [26-28]. Thus, lung microbiota could lead to carcinogenesis via several mechanisms, such as the creation of an inflammatory milieu, metabolic effects of dysbiosis, and genotoxicity [4,29], and H. influenzae may be associated with these mechanisms.
We performed a stratified sub-analysis according to the postoperative stage and the recurrence of lung cancer to address the association between the lung microbiome and lung cancer prognosis. Although Kovaleva et al. did not find any significant differences in alpha diversity between tumors at different stages, they reported a tendency of alpha diversity increase in tumors at later stages [30]. In contrast, here, several alpha diversity indexes steadily declined from normal to more advanced stages, reflecting a trend toward better prognosis with higher alpha diversity. Moreover, a previous pilot study demonstrated that several microbiota in normal lung tissues were associated with recurrence-free survival; however, the association between tumor tissue microbiome and survival was not revealed [10]. We found that there was a microbial compositional difference along with the presence of recurrence during the follow-up period, with the cancer with recurrence group presenting a more heterogeneous microbial distribution. Representatively, the Stenotrophomonas genus, which is known as a significant pathogen, particularly in patients with obstructive lung cancer [31,32], was identified as the most predominant genus in the lung cancer with recurrence group.
We further investigated the inferred metabolic/metagenomic function of the lung microbiome from 16S rRNA data of lung tissues in different histologic types of lung cancer. Cheng et al. performed 16S rRNA sequencing to show that the pathways of ribosome, pyrimidine, and purine metabolism were overrepresented in the bronchoalveolar lavage samples of lung cancer patients [33]. Furthermore, Apopa et al. showed that the peroxisome proliferator-activated receptors signaling pathway and the Dglutamine and D-glutamate metabolism were more enriched in AC than in SCC [22]. In our study, pathways such as the TCA cycle, and L-glutamate and L-glutamine biosynthesis were predominant in the AC group. Emerging evidence demonstrates that certain cancer cells, particularly those with deregulated oncogene and tumor suppressor expression, rely heavily on the TCA cycle for energy production and macromolecule synthesis [34]. Lung cancer cells also require glutamine as a nitrogen source for various anabolic processes in cancer and as an anaplerotic carbon source that replenishes the TCA intermediates [35]. Moreover, the pathway related to purine and pyrimidine nucleotide degradation was predominant in the SCC group. Increased nucleotide metabolism, which is a critical pathway for DNA replication, RNA synthesis, and cellular bioenergetics, suggests uncontrolled growth of tumors and is a hallmark of cancer [36]. The microbiome of lung cancer might play a role in these associated pathways. Further studies of functional microbiomic approaches, focused on the metabolic activity and function of the lung microbiota, are needed.
This study has several limitations. First, this study has a cross-sectional design, from which it is difficult to infer causality. Second, only a single race (Asian) was included, and the microbiota composition can differ according to racial characteristics [37,38]. Third, although there were various subtypes of EGFR mutations, including exon 19 deletions and the L858R point mutation, we did not investigate which mutation occurred in each AC EGFR+ tissue sample. Finally, further studies with validation cohorts are needed for more confirmative results.
In conclusion, we showed that the lung cancer tissues had significantly lower alpha diversity than the normal tissues and that the microbial composition differed according to the cancer histologic type and a genetic mutation. Then, we revealed that alpha diversity steadily declined from normal to more advanced stages, and there were microbial compositional differences according to the presence of recurrence. Finally, we found several inferred metagenomic functional pathways enriched in different histologic types of lung cancer. These findings suggest that the altered microbial composition in lung cancer might be associated with cancer initiation and/or progression.
Acknowledgements
The biospecimen and data used in this study were provided by the Asan Bio-Resource Center, Korea Biobank Network (2020-05(202)). This study was supported by a grant (2020-IF0013) from the Asan Institute for Life, Asan Medical Center, Seoul, Korea, the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2020-R1A2C1008431, SWL) and a grant of the Korea Health Promotion R&D Project, funded by the Ministry of Health & Welfare, Republic of Korea (HS21C0096).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.WHO report on cancer: setting priorities, investing wisely and providing care for all. Geneva: World Health Organization; 2020. [Google Scholar]
- 2.Liu NN, Ma Q, Ge Y, Yi CX, Wei LQ, Tan JC, Chu Q, Li JQ, Zhang P, Wang H. Microbiome dysbiosis in lung cancer: from composition to therapy. NPJ Precis Oncol. 2020;4:33. doi: 10.1038/s41698-020-00138-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez-Labrada AG, Isla D, Artal A, Arias M, Rezusta A, Pardo J, Galvez EM. The influence of lung microbiota on lung carcinogenesis, immunity, and immunotherapy. Trends Cancer. 2020;6:86–97. doi: 10.1016/j.trecan.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Maddi A, Sabharwal A, Violante T, Manuballa S, Genco R, Patnaik S, Yendamuri S. The microbiome and lung cancer. J Thorac Dis. 2019;11:280–291. doi: 10.21037/jtd.2018.12.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, Whary MT, Meyerson M, Germain R, Blainey PC, Fox JG, Jacks T. Commensal microbiota promote lung cancer development via γδ T cells. Cell. 2019;176:998–1013. doi: 10.1016/j.cell.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greathouse KL, White JR, Vargas AJ, Bliskovsky VV, Beck JA, von Muhlinen N, Polley EC, Bowman ED, Khan MA, Robles AI, Cooks T, Ryan BM, Padgett N, Dzutsev AH, Trinchieri G, Pineda MA, Bilke S, Meltzer PS, Hokenstad AN, Stickrod TM, Walther-Antonio MR, Earl JP, Mell JC, Krol JE, Balashov SV, Bhat AS, Ehrlich GD, Valm A, Deming C, Conlan S, Oh J, Segre JA, Harris CC. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018;19:123. doi: 10.1186/s13059-018-1501-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, O’Brien JL, Ajami NJ, Scheurer ME, Amirian ES, Armstrong G, Tsavachidis S, Thrift AP, Jiao L, Wong MC, Smith DP, Spitz MR, Bondy ML, Petrosino JF, Kheradmand F. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am J Cancer Res. 2018;8:1775–1787. [PMC free article] [PubMed] [Google Scholar]
- 8.Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC, Caporaso NE, Goedert JJ, Ravel J, Landi MT. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016;17:163. doi: 10.1186/s13059-016-1021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu HX, Tao LL, Zhang J, Zhu YG, Zheng Y, Liu D, Zhou M, Ke H, Shi MM, Qu JM. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int J Cancer. 2018;142:769–778. doi: 10.1002/ijc.31098. [DOI] [PubMed] [Google Scholar]
- 10.Peters BA, Hayes RB, Goparaju C, Reid C, Pass HI, Ahn J. The microbiome in lung cancer tissue and recurrence-free survival. Cancer Epidemiol Biomarkers Prev. 2019;28:731–740. doi: 10.1158/1055-9965.EPI-18-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodriguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vazquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- 13.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gotelli NJ, Colwell RK. Estimating species richness. In: Magurran AE, McGill BJ, editors. Biological diversity: frontiers in measurement and assessment. Oxford: Oxford University Press; 2011. pp. 39–54. [Google Scholar]
- 15.Magurran AE. Measuring biological diversity. Hoboken, NJ: Wiley; 2004. [DOI] [PubMed] [Google Scholar]
- 16.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang D, Su X, Yuan M, Zhang S, He J, Deng Q, Qiu W, Dong H, Cai S. The characterization of lung microbiome in lung cancer patients with different clinicopathology. Am J Cancer Res. 2019;9:2047–2063. [PMC free article] [PubMed] [Google Scholar]
- 18.Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, Schmidt TM. Application of a neutral community model to assess structuring of the human lung microbiome. MBio. 2015;6:e02284-14. doi: 10.1128/mBio.02284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HJ, Kim JJ, Myeong NR, Kim T, Kim D, An S, Kim H, Park T, Jang SI, Yeon JH, Kwack I, Sul WJ. Segregation of age-related skin microbiome characteristics by functionality. Sci Rep. 2019;9:16748. doi: 10.1038/s41598-019-53266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia F, Zhou X, Liu Y, Li Y, Bai X, Zhou X. Composition and predictive functional analysis of bacterial communities inhabiting Chinese Cordyceps insight into conserved core microbiome. BMC Microbiol. 2019;19:105. doi: 10.1186/s12866-019-1472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poje G, Redfield RJ. General methods for culturing Haemophilus influenzae. In: Herbert MA, Hood DW, Moxon ER, editors. Haemophilus influenzae protocols. Totowa, NJ: Humana Press; 2003. pp. 51–56. [DOI] [PubMed] [Google Scholar]
- 22.Apopa PL, Alley L, Penney RB, Arnaoutakis K, Steliga MA, Jeffus S, Bircan E, Gopalan B, Jin J, Patumcharoenpol P, Jenjaroenpun P, Wongsurawat T, Shah N, Boysen G, Ussery D, Nookaew I, Fagan P, Bebek G, Orloff MS. PARP1 is up-regulated in non-small cell lung cancer tissues in the presence of the cyanobacterial toxin microcystin. Front Microbiol. 2018;9:1757. doi: 10.3389/fmicb.2018.01757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutebemberwa A, Stevens MJ, Perez MJ, Smith LP, Sanders L, Cosgrove G, Robertson CE, Tuder RM, Harris JK. Novosphingobium and its potential role in chronic obstructive pulmonary diseases: insights from microbiome studies. PLoS One. 2014;9:e111150. doi: 10.1371/journal.pone.0111150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goto T. Airway microbiota as a modulator of lung cancer. Int J Mol Sci. 2020;21:3044. doi: 10.3390/ijms21093044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ochoa CE, Mirabolfathinejad SG, Ruiz VA, Evans SE, Gagea M, Evans CM, Dickey BF, Moghaddam SJ. Interleukin 6, but not T helper 2 cytokines, promotes lung carcinogenesis. Cancer Prev Res (Phila) 2011;4:51–64. doi: 10.1158/1940-6207.CAPR-10-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King PT, Sharma R. The lung immune response to nontypeable Haemophilus influenzae (Lung Immunity to NTHi) J Immunol Res. 2015;2015:706376. doi: 10.1155/2015/706376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moghaddam SJ, Ochoa CE, Sethi S, Dickey BF. Nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease and lung cancer. Int J Chron Obstruct Pulmon Dis. 2011;6:113–123. doi: 10.2147/COPD.S15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sriram KB, Cox AJ, Sivakumaran P, Singh M, Watts AM, West NP, Cripps AW. Non-typeable Haemophilus influenzae detection in the lower airways of patients with lung cancer and chronic obstructive pulmonary disease. Multidiscip Respir Med. 2018;13:11. doi: 10.1186/s40248-018-0123-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu N, Wang L, Li C, Ding C, Li C, Fan W, Cheng C, Gu B. Microbiota dysbiosis in lung cancer: evidence of association and potential mechanisms. Transl Lung Cancer Res. 2020;9:1554–1568. doi: 10.21037/tlcr-20-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovaleva O, Podlesnaya P, Rashidova M, Samoilova D, Petrenko A, Zborovskaya I, Mochalnikova V, Kataev V, Khlopko Y, Plotnikov A, Gratchev A. Lung microbiome differentially impacts survival of patients with non-small cell lung cancer depending on tumor stroma phenotype. Biomedicines. 2020;8:349. doi: 10.3390/biomedicines8090349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Safdar A, Rolston KV. Stenotrophomonas maltophilia: changing spectrum of a serious bacterial pathogen in patients with cancer. Clin Infect Dis. 2007;45:1602–1609. doi: 10.1086/522998. [DOI] [PubMed] [Google Scholar]
- 32.Brooke JS. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev. 2012;25:2–41. doi: 10.1128/CMR.00019-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng C, Wang Z, Wang J, Ding C, Sun C, Liu P, Xu X, Liu Y, Chen B, Gu B. Characterization of the lung microbiome and exploration of potential bacterial biomarkers for lung cancer. Transl Lung Cancer Res. 2020;9:693–704. doi: 10.21037/tlcr-19-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson NM, Mucka P, Kern JG, Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9:216–237. doi: 10.1007/s13238-017-0451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanhove K, Derveaux E, Graulus GJ, Mesotten L, Thomeer M, Noben JP, Guedens W, Adriaensens P. Glutamine addiction and therapeutic strategies in lung cancer. Int J Mol Sci. 2019;20:252. doi: 10.3390/ijms20020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siddiqui A, Ceppi P. A non-proliferative role of pyrimidine metabolism in cancer. Mol Metab. 2020;35:100962. doi: 10.1016/j.molmet.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carbone C, Piro G, Di Noia V, D’Argento E, Vita E, Ferrara MG, Pilotto S, Milella M, Cammarota G, Gasbarrini A, Tortora G, Bria E. Lung and gut microbiota as potential hidden driver of immunotherapy efficacy in lung cancer. Mediators Inflamm. 2019;2019:7652014. doi: 10.1155/2019/7652014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta VK, Paul S, Dutta C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol. 2017;8:1162. doi: 10.3389/fmicb.2017.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.