

Abstract
Current anti-VEGF-A therapies inhibit choroidal neovascularization (CNV) in a subset of patients with neovascular age-related macular degeneration (NV-AMD). However, long-term treatment with such anti-VEGF-A therapies may impair physiological functions of the choriocapillaris and retina for which VEGF-A is needed. Moreover, disease progression can occur despite continuous anti-VEGF-A treatment. Thus, novel therapies for NV-AMD are urgently needed that target specifically disease-associated mechanisms without impairing growth factors and cellular pathways that are required for homeostatic functions of the retina and choroid. Inhibiting the inflammatory pathways that promote CNV would be such a promising novel approach that would likely not interfere with the normal functions of healthy retinal and choroidal cells. In this context, the inflammasome, a proinflammatory protein complex that promotes pathologic angiogenesis largely through generation of IL-1β and which has been reported to be activated in AMD, has become an area of much interest in the AMD field. However, most studies have focused mainly on the NLRP3 inflammasome in retinal pigment epithelial cells (RPE), and conflicting findings have resulted in an unclear picture of the role of the inflammasome for AMD pathogenesis. Recent data suggest that inflammasome activation in activated macrophages and retinal microglia but not in RPE cells promotes CNV. Furthermore, inflammasome activation can occur in CNV macrophages and microglia despite lack of NLRP3. Thus, activation of both NLRP3 inflammasomes as well as non-NLRP3 inflammasomes in macrophages/microglia at sites of CNV formation likely promote NV-AMD.
Keywords: Inflammasome, age-related macular degeneration, caspase-1, NLRP3, VEGF-A, choroidal neovascularization
Introduction
Inflammation is a major factor in the pathogenesis of both nonexudative as well as neovascular age-related macular degeneration (NV-AMD). Inflammatory cells that infiltrate the site of choroidal neovascularization (CNV) in NV-AMD, such as macrophages and activated retinal microglia, express high levels of proangiogenic factors (including VEGF-A and IL-1β) that induce and promote CNV lesion formation. Current NV-AMD therapies target mainly the proangiogenic growth factor VEGF-A, but this approach is not effective in all patients with NV-AMD or its effectiveness may decline over time [1]. Moreover, the long-term safety profile of chronic anti-VEGF-A therapies for choroidal and retinal functions is not well established, and it is possible that chronic anti-VEGF-A therapies may impair the function of normal choroidal vessels or retinal cells, as VEGF-A has been implicated in homeostatic functions of the choroidal vasculature and retina [2–4]. In contrast to current anti-VEGF-A therapies that indiscriminately diminish VEGF-A levels both at sites of CNV as well as at sites devoid of CNV, preventing the production of proangiogenic factors only in activated inflammatory cells would likely inhibit CNV without affecting normal tissue functions in the eye where no inflammation occurs.
One particular key proinflammatory pathway that has been implicated in promoting AMD involves activation of inflammasomes, which are multiprotein complexes that lead to the proteolytic activation of proinflammatory IL-1β and IL-18 through the catalytic activity of caspase-1. Inflammasome activation is initiated by various different cytosolic pattern recognition receptors (PRRs) in response to specific triggers. PRRs comprise a group of different proteins, including the family of NOD-like receptors (including NLRP1, NLRP3, NLRP6, NLRC4), AIM2, IFI16, and pyrin. In response to specific stimuli, PRRs oligomerize and form together with the adaptor protein ASC scaffolds that activate caspase-1, which leads to autoproteolytic activation of pro-caspase-1 that results in release of their effector domains caspase-1 p10 and p20. These proteolytic caspase-1 activation products are necessary for the proteolytic processing of pro-IL-1β and pro-IL-18 into their active mature forms that stimulate inflammation [5–7] (Figure 1). Thus, caspase-1 is at the center of the inflammasome activation cascade, and caspase-1 p10/p20 levels correlate with inflammasome activity and are used to quantify the degree of inflammasome activation. For example, antibodies have been generated that allow to detect specifically p10 or p20 only when proteolytically released from caspase-1, thereby, distinguishing total caspase-1 levels from active caspase-1. As inflammasome activation has been linked to various chronic inflammatory conditions, several studies have aimed to determine whether inflammasome activation occurs also in human AMD.
Figure 1: Model of inflammasome activation that leads to IL-1b release in CNV macrophages and retinal microglia.
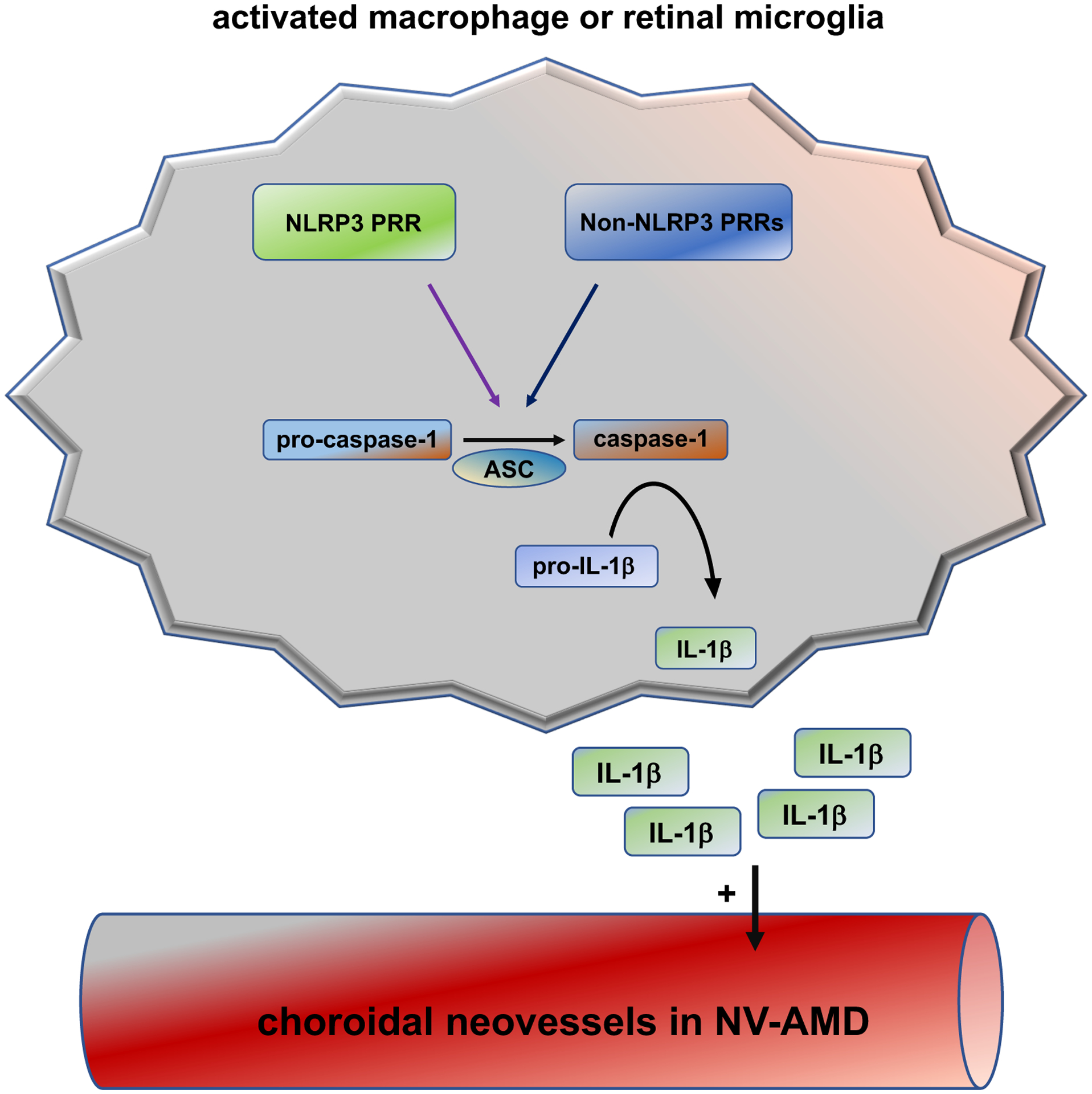
In response to specific stimuli, PRRs oligomerize and form together with the adaptor protein ASC scaffolds that activate caspase-1. Caspase-1 activation products are necessary for the proteolytic processing of pro-IL-1β to active IL-1β that once secreted can promote neovascularization. Whereas NLRP3 is an established PRR that can trigger the inflammasome in CNV macrophages/microglia, which non-NLRP3 PRRs can do this as well is currently unknown.
Inflammasome activation in human AMD
Various studies provided some evidence that inflammasome activation occurs in human AMD and promotes AMD-like pathologies in animal models as well [8–13]. However, the studies that assessed inflammasome activation in human AMD specimens have their limitations, including a small number of samples examined or a focus on gene expression rather than demonstration of actual inflammasome activation (Table 1). One study suggested increased gene expression of NLRP3, IL1B and IL18 in maculae of paraffin-embedded human AMD samples, but this study was limited by a small number of samples (e.g. only one NV-AMD sample showed NLRP3 expression), exclusion of a significant number of samples due to RNA degradation (raising concerns about the quality of the material), and a wide variability of values among samples [13]. Another study showed gene expression analysis of the RPE from eyes with geographic atrophy (GA) and found a modest increase (~2-fold) in expression of NLRP3 and IL18 (but the increase in IL1B was not significant). This study also showed increased caspase-1 p20 levels in Western blots of macular RPE lysates from patients with GA [8]. The study did not assess whether inflammasome activation occurs in other choroidal or retinal cells as well. A different study detected NLRP3 protein in RPE cells of two eyes with GA and in two eyes with NV-AMD by immunohistochemistry [12]. The images shown in the paper do not depict the retinal layers and no co-immunolabeling studies were performed to assess labeling for NLRP3 in other cell types, such as infiltrating macrophages. Moreover, immunolabeling for NLRP3 does not address the question whether inflammasome activation actually occurs. In contrast, a different study did not detect NLRP3 protein in RPE lysates from eye specimens with nonexudative AMD [14]. This study emphasized also that some commercially available NLRP3 antibodies lack specificity for NLRP3 and have not been sufficiently validated. Notably, measurements of IL-1β protein levels showed increased protein levels in the vitreous of patients with NV-AMD [15]. Collectively, these studies show that there has been mainly a focus on assessing NLRP3 expression in RPE cells of eyes with nonexudative AMD/GA, without clearly assessing overall inflammasome activation. Most studies did also not sufficiently address the question whether inflammasome activation occurs in non-RPE cells in human AMD, and data on inflammasome activation in NV-AMD are particularly sparse. Thus, while these studies indicate a potential role of inflammasome activation in AMD pathogenesis, to what extent inflammasome activation occurs in specific cell types during the different stages of human nonexudative AMD and NV-AMD remains to be conclusively demonstrated.
Table 1:
Studies examining inflammasome activation in human AMD.
| METHOD OF DETECTION | AMD TYPE | KEY FINDINGS | LIMITATIONS OF STUDY | REFERENCE |
|---|---|---|---|---|
| Semiquantitative RT-PCR | NV-AMD | upregulation of NLRP3, IL1B and IL18 | small sample size, high variability; high increase in NLRP3 and IL18, modest increase in IL1B | Wang et al, 2016 |
| GA | no assessment of caspase-1 p10 or p20; large proportion of samples excluded due to RNA degradation | |||
| photoreceptors and RPE cells microdissected; no other parts of retina examined | ||||
| Semiquantitative RT-PCR | GA | upregulation of NLRP3 and IL18 | only ~2-fold increase of NLRP3 and IL18; IL1B increase not statistically significant | Tarallo et al, 2012 |
| (not IL1B) | evaluation of only RPE and not other cells | |||
| WB of macular RPE lysates | GA | increase in caspase-1 p20 and NLRP3 | no evaluation of NV-AMD | Tarallo et al, 2012 |
| for caspase-1 p20 and NLRP3 | evaluation of only RPE and not other cells | |||
| IHC for NLRP3 | GA | increased NLRP3 in RPE | small sample size (only n=2 eyes with GA and n=2 eyes with NV-AMD) | Tseng et al., 2013 |
| NV-AMD | RPE cells; no evaluation of other cells (no co-immunolabeling); no evaluation of retina | |||
| WB for NLRP3 | GA/dry AMD | no NLRP3 protein detected | RPE cell lysates examined; no other parts of retina or choroid examined | Kosmidou et al., 2018 |
| ELISA for IL-1β | NV-AMD | increased IL-1β protein in vitreous | small sample number (n=10 patients with NV-AMD) | Zhao et al., 2015 |
BW, Western blot; IHC, immunohistochemistry
Similarly, in vitro studies have largely focused on assessing specifically NLRP3 inflammasome activation in human RPE cells. For example, one study showed NLRP3 expression and IL-1β secretion in primary human RPE cells [16]. Another study showed inhibition of IL-1β release with small molecule inhibitors of NLRP3 in human primary RPE cells as well as in the cell line ARPE-19, leading the authors to conclude that NLRP3 inflammasome activation occurs in human RPE cells [17]. However, various other studies were limited by the use of cell lines that poorly reflect human RPE cell behavior and function (such as ARPE-19 cells). Notably, other groups did not detect NLRP3 protein in ARPE-19 cells or even in primary human RPE cells cultures [14], challenging the relevance of in vitro data on the NLRP3 inflammasomes in RPE cells. Importantly, inflammasome activation in disease states in vivo occurs as a consequence of the interaction of various cell types in a complex microenvironment that often involves the accumulation of reactive oxygen species, extracellular lipids and other factors that promote inflammasome activation. Thus, in vitro studies have an inherent limitation in assessing the factors that promote inflammasome activation in the pathologic tissue microenvironment that occurs in vivo and that drives AMD pathologies. Therefore, this review focuses on in vivo studies that investigated pathomechanisms of inflammasome activation in NV-AMD mouse models (Table 2).
Table 2:
Targeting inflammasome components in mouse models of NV-AMD.
| MOUSE MODEL OF NV-AMD | KEY FINDINGS | LIMITATIONS OF STUDY | REFERENCE |
|---|---|---|---|
| laser-induced CNV | increased CNV area in Nlrp3−/− or IL18−/− mice but not Il1r1−/− mice | no detailed methods on how CNV lesions were quantitated and analyzed; | Doyle et al., 2012 |
| exact p-values not shown; no evaluation of gender differences | |||
| laser-induced CNV | IL-18 administration inhibits CNV area | no evaluation of gender differences | Doyle et al., 2014 |
| laser-induced CNV | IL-18 administration inhibits CNV area | barely significant decrease; no evaluation of gender differences | Shen et al., 2014 |
| laser-induced CNV | no effect of IL-18 administration on CNV area | no exact p-values shown; no evaluation of gender differences | Hirano et al., 2014 |
| IL18−/− mice and IL18r1−/− mice have reduced CNV area | |||
| laser-induced CNV | pharmacologic inhibitor of caspase-1 inhibits CNV area | no evaluation of gender differences | Marneros, 2016 |
| laser-induced CNV | IL1R antagonist treatment inhibits CNV area | no evaluation of gender differences | Lavalette et al., 2011 |
| small number of mice in study | |||
| CNV in Vegfahyper mice | fewer CNV lesions in Vegfahyper mice lacking Nlrp3 or Il1r1 | only CNV lesion numbers but no CNV area determined | Marneros, 2013 |
| no significant difference with Il18 deficiency | no evaluation of gender differences | ||
| CNV in Vegfahyper mice | Vegfahyper mice show fewer CNV lesions when deficient for Nlrp3, | no evaluation of gender differences | Marneros, 2016 |
| Il1r1 and caspase-1/caspase-11, but not Il18; | |||
| strongest decrease in CNV numbers and CNV area with | |||
| caspase-1/caspase-11 deficiency | |||
| CNV in Vegfahyper mice | CNV lesion numbers reduced in Vegfahyper mice lacking Nlrp3 or | Malsy et al., 2020 | |
| caspase-1/caspase-11; no effect of only caspase-11 deficiency on CNV; | |||
| no effect of Nlrp3 activation specifically in the RPE on CNV; | |||
| evaluation of gender effects on CNV lesion numbers and sizes |
Assessing the role of inflammasome activation in mouse models of NV-AMD
Notably, most studies that implicated inflammasomes in AMD pathogenesis have focused mainly on NLRP3-mediated inflammasome activation and did not consider whether PRRs other than NLRP3 may promote inflammasome activation in AMD. The almost exclusive focus on the NLRP3 inflammasome can be explained by the observation that various stimuli that are risk factors for AMD, including increased oxidative stress or lipid accumulations, have been shown to promote NLRP3 inflammasome activation [5–7]. Thus, these studies hypothesized that AMD risk factors promote NLRP3 inflammasome activation, which further exacerbates AMD pathologies through activation of proinflammatory cytokines, such as IL-1β that is known to be increased in human eyes with NV-AMD and that has been shown to stimulate CNV in both laser-induced CNV and VEGF-A-induced CNV mouse models [10, 11, 15, 18]. This hypothesis has been supported by the abovementioned studies that provided some evidence of NLRP3 inflammasome activation in human AMD (Table 1), as well as animal studies that showed a role of the NLRP3 inflammasome for the manifestation of AMD-like pathologies [8–12].
To test the effects of targeting inflammasomes by either genetic or pharmacologic approaches in vivo and determine the effects on CNV, animal models for NV-AMD need to be utilized. Any mouse model for NV-AMD has inherent limitations, especially as the mouse has no macula. However, it is also important to emphasize that many of the key findings in human AMD could be recapitulated in mouse models, and specific therapeutic approaches that are currently being pursued in AMD patients were previously validated in AMD mouse models. The two mouse models used so far to assess the role of the inflammasome for CNV formation are a laser-induced CNV model of NV-AMD and a VEGF-A-induced genetic mouse model of NV-AMD (Vegfahyper mice) [10, 11, 19–21]. In the laser-induced CNV model, injury with a laser to the RPE induces an acute wound healing response that leads to neovascular lesion formation at the site of injury within a few days, and the area of these CNV lesions can be quantitated in choroidal flat mounts [19]. Thus, this model allows only to assess CNV lesion size after experimentally-induced injury, which means we can determine the effects of genetic or pharmacologic interventions only on CNV growth, but we cannot assess factors that influence CNV lesion induction. Thus, the laser-induced CNV model is more of an acute injury model and not reflective of the progressive age-dependent development of CNV seen in human NV-AMD that occurs without an acute injury [19]. Another limitation of the model is that there can be significant variability in the results that strongly depends on the laser settings used for induction of CNV lesions and the genetic background of mouse strains utilized [22]. There can be also considerable variability in the outcomes depending on the experimental user [22]. This explains that often reproducibility of results is poor and interobserver variability is high if the reported differences between experimental groups are not very significant. This may also in part be the reason for some conflicting findings on the role of inflammasome components in laser-induced CNV experiments performed by different groups, which often used not the same laser settings and mouse strains.
In contrast, Vegfahyper mice form spontaneous CNV lesions without experimental injury, and CNV lesions can be assessed already in young adult mice [10]. CNV lesions in Vegfahyper mice enlarge with progressive age [10, 11, 23], similarly as observed in human NV-AMD [24]. These mice form multifocal spontaneous CNV due to an increase in VEGF-A as a consequence of an insertion of a lacZ cassette in their 3’-UTR, which leads to a modest increase in VEGF-A protein levels in the RPE of these mice [10, 25]. Thus, this NV-AMD mouse model allows us to test effects on both CNV lesion induction (CNV lesion numbers), as well as on CNV lesion growth (CNV lesion area). Importantly, in Vegfahyper mice increased VEGF-A levels are also associated with an increase in factors that are known to promote inflammasome activation, such as sub-RPE lipid deposits and increased oxidative stress [10]. Their increase as well as the elevated VEGF-A levels in Vegfahyper mice are linked to increased inflammasome activation products in RPE/choroids of these mice, and the progressive extent of inflammasome activation is associated with the age-dependent progression of CNV lesion formation in these mice [10, 11, 20, 23]. Thus, Vegfahyper mice may be regarded as a more disease-relevant animal model of NV-AMD than the laser-injury CNV model and may be particularly well suited to assess effects of targeting inflammasome activation on CNV lesion formation.
One group reported that NLRP3 inflammasome activation occurs in laser-induced CNV in macrophages and results in IL-18 activation that inhibits laser-induced CNV (albeit the data did not show individual values to assess outliers that commonly occur in this assay, and detailed methods on how CNV lesions were quantitated and analyzed were not reported) [9, 26, 27]. The authors found that laser-induced CNV lesions were smaller in mice lacking either Nlrp3 or Il18 genes but not when the Il1r1 was inactivated [9]. Similarly, this group reported that laser-induced CNV lesions were attenuated with administration of IL-18 [27]. However, another group showed a barely significant decrease in laser-induced CNV lesions with intraocular injection of IL-18 (p-value 0.0497) [28]. Moreover, other groups could not detect an effect of IL-18 on laser-induced CNV [29]. Instead, they reported a reduction of laser-induced CNV in mice lacking Il18 or Il18r1 [29]. These contrasting findings may be in part explained by different vehicles used in which IL-18 was formulated but may also in part be explained by the inherent variability of this CNV model, especially when the effects on CNV are not large. Even if administration of recombinant IL-18 would have a moderate inhibitory effect on laser-induced CNV, this does not answer the question whether endogenously produced IL-18 in conditions of CNV formation without experimental injury, such as in NV-AMD, does affect CNV severity. In fact, it is possible that exogenous administration of high doses of recombinant IL-18 has indeed antiangiogenic activity in various ocular neovascular conditions, such as has been reported in an ischemia-induced retinal neovascularization model [28]. Based on the published studies it may be concluded that even if IL-18 inhibits laser-induced CNV, then the effect is rather minor, in contrast to the strongly proangiogenic effects of IL-1β that promote CNV. This suggests that inflammasome blockade would likely have an overall CNV inhibitory antiangiogenic effect through inhibition of IL-1β production that would dominate the effects of loss of IL-18. Indeed, pharmacologic blockade of the inflammasome with a caspase-1 inhibitor (thereby blocking both caspase-1-mediated IL-1β as well as IL-18 activation) resulted in a highly significant reduction of laser-induced CNV (p-value: 0.0025). Similarly, genetic inactivation of caspase-1 resulted in a highly significant reduction in VEGF-A-induced CNV lesion formation (Vegfahyper mice) as well (p-value <0.0001) [11, 20].
In the initial study that described Vegfahyper mice as a model for spontaneous CNV lesion formation it was shown that inactivation of Il18 does not significantly affect CNV lesion formation in Vegfahyper mice, whereas inactivation of Nlrp3 or Il1r potently inhibits CNV lesion formation in these mice [10]. As this initial study did not have large enough numbers of mice to conclusively rule out a role of IL-18 in CNV lesion formation in this model, two subsequent studies expanded the analysis of CNV lesion formation in the various experimental groups with a large number of mice: these data showed unequivocally that inactivation of Nlrp3, Il1r1 or caspase-1 potently reduced CNV lesion numbers in Vegfahyper mice, whereas inactivation of Il18 had no effect on CNV lesion numbers in this mouse model of NV-AMD [11, 20]. Thus, inactivation of NLRP3 inflammasomes, of IL-1β signaling or of all canonical inflammasomes (caspase-1 deletion) potently inhibits VEGF-A-induced CNV in Vegfahyper mice [10, 11, 20]. Collectively, these findings provide strong evidence that activation of IL-1β through the inflammasome promotes VEGF-A-induced CNV in mice, whereas IL-18 has no major effect on CNV in this VEGF-A-induced CNV mouse model of NV-AMD. The debate whether IL-18 has a role for CNV should not cloud the clear observation that targeting IL-1β signaling, NLRP3 inflammasomes or all canonical inflammasomes effectively inhibits spontaneous VEGF-A-induced CNV and, thus, may have significant clinical relevance for NV-AMD therapies. These data provide strong preclinical evidence suggesting that pharmacologic blockade of inflammasomes would be expected to have an overall inhibitory effect on CNV lesion formation (as shown with a caspase-1 inhibitor for laser-induced CNV [11]).
The questions over the effects of IL-18 on CNV has resulted in a careful reevaluation whether the NLRP3 inflammasome plays indeed an import role in AMD pathogenesis. However, most studies had a narrow focus and assessed mainly whether NLRP3 inflammasome activation occurs in RPE cells. For example, a recent study tested a small number of anti-NLRP3 antibodies and could not detect NLRP3 protein in the RPE of human eyes with AMD [14]. This study did not investigate whether inflammasome activation occurs in the RPE, as read-outs of inflammasome activation (cleaved caspase-1 p10 or p20 levels or IL-1β secretion) were not assessed [14]. In contrast, other groups claimed that NLRP3 inflammasome activation occurs in human RPE cells [12, 16, 17]. It is important to emphasize that lack of NLRP3 expression does not allow the conclusion that inflammasome activation does not occur, as inflammasomes could be activated through PRRs other than NLRP3. Moreover, these studies did not answer the question whether NLRP3 or non-NLRP3 inflammasome activation occurs in specific cell types in situ in the context of the pathologic microenvironment that leads to CNV.
Importantly, recent data in Vegfahyper mice provide now evidence that high-level inflammasome activation occurs predominantly in activated macrophages and retinal microglia cells that infiltrate the site of CNV lesion formation but not to a significant degree in RPE cells (Figure 2) [20]. Inactivation of Nlrp3 in all cells strongly reduced CNV lesions in Vegfahyper mice. In contrast, constitutive activation of Nlrp3 specifically in the RPE, through expression of an Nlrp3 activating mutation (Nlrp3A350V) that is found in patients with cryopyrin-associated periodic syndrome (CAPS) mutation only in RPE cells, had no effect on CNV lesion formation in Vegfahyper mice that lacked Nlrp3 in all other cells [20]. Together with the observation that inflammasome activation markers were predominantly detected in activated macrophages and microglia and that deficiency of Il1r1 or caspase-1 strongly attenuated CNV lesion formation in Vegfahyper mice, this strongly suggests that NLRP3 inflammasomes promote VEGF-A-induced CNV via its activation in macrophages/microglia and not RPE cells through secretion of IL-1β. Furthermore, targeting caspase-1, and thereby blocking all canonical inflammasomes, led to a much stronger reduction of CNV lesion numbers compared to targeting only NLRP3 inflammasomes by inactivating Nlrp3. In contrast, targeting caspase-11 that may lead to activation of non-canonical inflammasomes had no effect. Strikingly, inactivation of Nlrp3 did not prevent the detection of inflammasome activation products caspase-1 p10 and p20 in RPE/choroid lysates of Vegfahyper mice. Similarly, caspase-1 p10 immunolabeling was still detected in macrophages/microglia in CNV lesions of Vegfahyper mice that lacked Nlrp3 [20]. Collectively, these experiments clearly demonstrate that in CNV lesions of Vegfahyper mice both NLRP3 inflammasome activation as well as non-NLPR3 inflammasome activation occur, and that both promote CNV. Which PRRs other than NLRP3 promote CNV in Vegfahyper mice has yet to be determined.
Figure 2: Both activated retinal microglia and macrophages show inflammasome activation in CNV lesions of Vegfahyper mice.
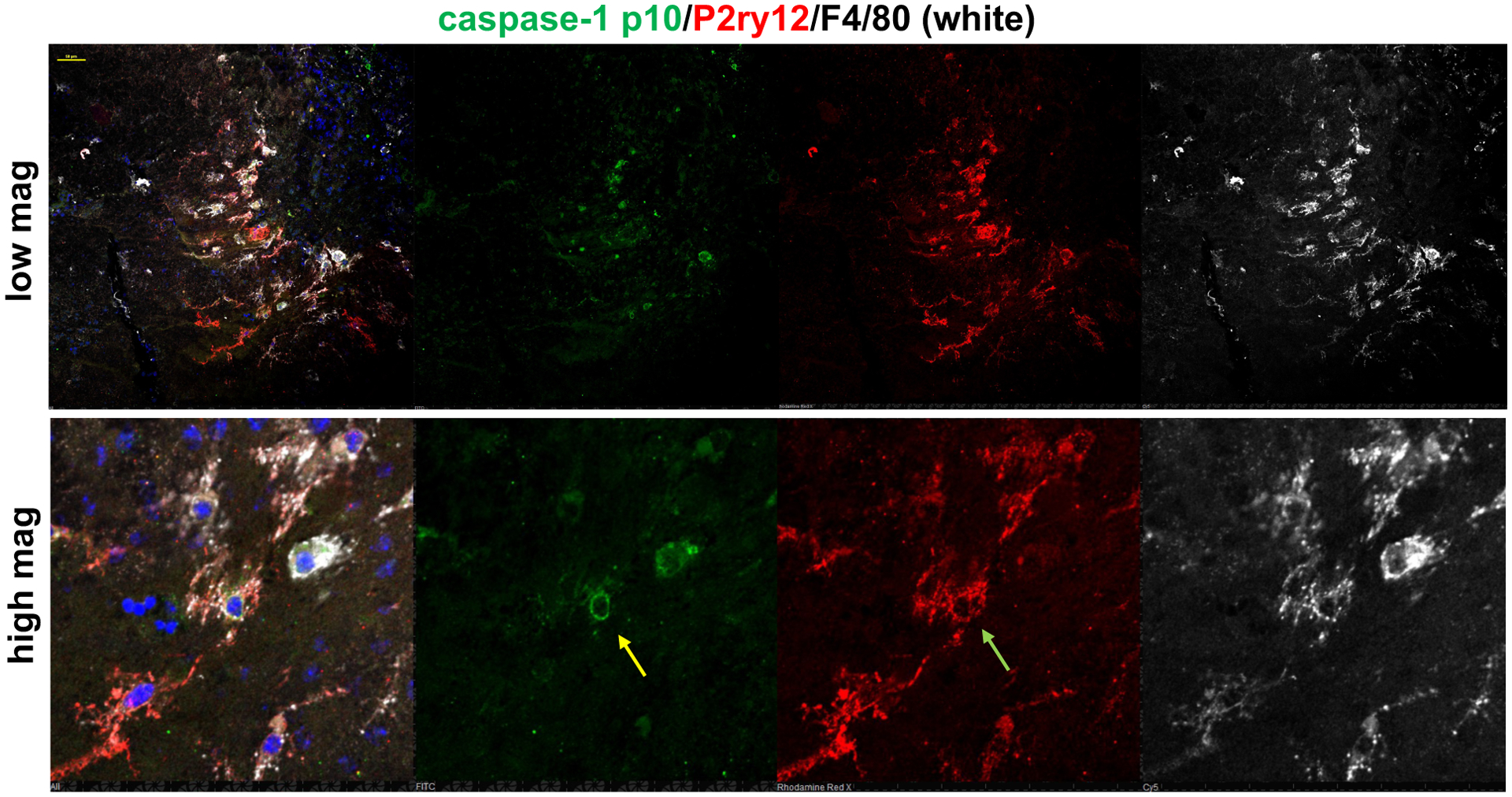
Choroidal flat mounts show a CNV lesion in a Vegfahyper mouse eye.
F4/80 is a marker that is expressed by macrophages and retinal microglia cells. F4/80+ cells (white) that infiltrate the subretinal space and are found in CNV lesions are often also staining for the microglia marker P2ry12 (red). Co-immunolabeling for cleaved caspase-1 p10 (inflammasome activation marker; green) shows that F4/80+P2ry12negp10+ cells, representing CNV macrophages (yellow arrow) show inflammasome activation, as well as activated retinal microglia cells that infiltrate CNV lesions (F4/80+P2ry12+p10+; green arrow). In contrast, these high levels of caspase-1 p10 immunolabeling are not detected in RPE cells of CNV lesions. Top shows lower magnification image (scale bar, 50mm), and lower panel shows a higher magnification of part of that image.
Moreover, this study showed that blockade of complement-mediated inflammation through inactivation of complement C3 reduced CNV lesion numbers without preventing inflammasome activation [20]. This suggests that complement-mediated and inflammasome-mediated inflammation occur largely independently of each other and promote CNV in Vegfahyper mice, raising the question of potential synergistic therapeutic effects when targeting both pathways [20]. In summary, these recent findings demonstrated an unexpected role of non-NLRP3 inflammasomes for CNV and a role of inflammasome activation in activated retinal microglia as well as macrophages for CNV. That inflammasome activation occurs mainly in macrophages/activated microglia at sites of RPE/retinal pathologies has also been shown with other experimental in vivo models. For example, Wortmannin-induced RPE damage in vivo resulted in inflammasome activation (detection of cleaved caspase-1) mainly in subretinal Iba1+ cells (a marker of macrophages/microglia) rather than in RPE cells [30].
Concluding remarks and future directions
Whereas much attention has been paid on the role of NLRP3 inflammasome activation in RPE cells for AMD pathogenesis, recent findings shift the focus towards determining mechanisms of NLRP3 and non-NLRP3 inflammasome activation in macrophages and activated retinal microglia cells for NV-AMD pathogenesis. Open key questions that remain to be answered are which other non-NLRP3 PRRs may promote inflammasome activation in NV-AMD and in which specific cell types. This remains not only to be determined in NV-AMD mouse models, but a more detailed evaluation of which inflammasomes are activated in which specific cell types in human NV-AMD remains to be undertaken as well. Furthermore, it will be important to assess to what extent similarities exist between inflammasome activation in nonexudative and neovascular AMD and whether inflammasomes have similar pathogenic relevance in both forms of AMD in humans. Finally, it will be important to assess how pharmacologic targeting of inflammasomes may be utilized for novel AMD therapies. In a proof-of-principle approach, recent data show that a pharmacologic caspase-1 inhibitor could attenuate laser-induced CNV growth in mice [11]. Alternatively, IL-1β/IL1R1 blockade is currently being pursued in various conditions promoted by inflammasome activation, such as cryopyrin-associated periodic syndromes, and may have clinical relevance in the treatment of NV-AMD as well.
METHODS
Detailed Methods for the immunolabeling of choroidal flat mounts shown in Figure 2 have previously been reported [20]. Whole eyes were enucleated and fixed in 4% paraformaldehyde overnight at 4°C and then washed in PBS. For choroidal flat mounts anterior segment, lens and retina were removed. RPE/choroid tissues were permeabilized in 0.5% Triton X-100 and blocked with 5% serum in which the secondary antibodies were raised for 30 minutes. Incubation with primary antibodies in blocking solution was performed overnight at 4°C. Tissues were then washed in PBS. The following primary antibodies were used at a 1:50 dilution: Alexa-647-conjugated rat anti-mouse F4/80 (BioLegend Cat# 123101, RRID:AB_893504), goat anti-cleaved-caspase-1 p10 (Santa Cruz Biotechnology Cat# sc-22166, RRID:AB_2068884), and a rabbit anti-mouse P2ry12 antibody (obtained from Dr. Oleg Butovsky). Secondary anti-goat Alexa-488 and anti-rabbit Alexa-555 antibodies (at a 1:100 dilution) (Life Technologies) were incubated for 3 hours at room temperature in the dark. DAPI (Life Technologies) was used for staining of nuclei. Subsequently to immunolabeling, RPE/choroid tissues were radially cut eight times and mounted on a slide for microscopic analysis.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (R21EY027104) and the BrightFocus foundation to AGM.
Abbreviations:
- NV-AMD
neovascular age-related macular degeneration
- CNV
choroidal neovascularization
- VEGF-A
vascular endothelial growth factor A
- IL-18
interleukin 18
- IL-1β
interleukin 1β
- RPE
retinal pigment epithelium
- GA
geographic atrophy
Footnotes
CONFLICT OF INTEREST
The author declares no conflict of interest.
REFERENCES
- 1.Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR & Zhang K (2013) Seven-Year Outcomes in Ranibizumab-Treated Patients in ANCHOR, MARINA, and HORIZON: A Multicenter Cohort Study (SEVEN-UP), Ophthalmology. 120, 2292–9. [DOI] [PubMed] [Google Scholar]
- 2.Kurihara T, Westenskow PD, Bravo S, Aguilar E & Friedlander M (2012) Targeted deletion of Vegfa in adult mice induces vision loss, The Journal of clinical investigation. 122, 4213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE & D’Amore PA (2009) An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris, Proceedings of the National Academy of Sciences of the United States of America. 106, 18751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakri SJ, Thorne JE, Ho AC, Ehlers JP, Schoenberger SD, Yeh S & Kim SJ (2019) Safety and Efficacy of Anti-Vascular Endothelial Growth Factor Therapies for Neovascular Age-Related Macular Degeneration: A Report by the American Academy of Ophthalmology, Ophthalmology. 126, 55–63. [DOI] [PubMed] [Google Scholar]
- 5.Schroder K & Tschopp J (2010) The inflammasomes, Cell. 140, 821–32. [DOI] [PubMed] [Google Scholar]
- 6.Latz E, Xiao TS & Stutz A (2013) Activation and regulation of the inflammasomes, Nature reviews Immunology. 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo H, Callaway JB & Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics, Nat Med. 21, 677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Nunez G, Baffi JZ, Kleinman ME & Ambati J (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88, Cell. 149, 847–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, O’Neill LA, Hollyfield JG & Humphries P (2012) NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components, Nat Med. 18, 791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marneros AG (2013) NLRP3 Inflammasome Blockade Inhibits VEGF-A-Induced Age-Related Macular Degeneration, Cell reports. 4, 945–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marneros AG (2016) Increased VEGF-A promotes multiple distinct aging diseases of the eye through shared pathomechanisms, EMBO Mol Med. 8, 208–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D’Amore PA & Ksander BR (2013) NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration, Investigative ophthalmology & visual science. 54, 110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Hanus JW, Abu-Asab MS, Shen D, Ogilvy A, Ou J, Chu XK, Shi G, Li W, Wang S & Chan CC (2016) NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration, Int J Mol Sci. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosmidou C, Efstathiou NE, Hoang MV, Notomi S, Konstantinou EK, Hirano M, Takahashi K, Maidana DE, Tsoka P, Young L, Gragoudas ES, Olsen TW, Morizane Y, Miller JW & Vavvas DG (2018) Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration, Sci Rep. 8, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao M, Bai Y, Xie W, Shi X, Li F, Yang F, Sun Y, Huang L & Li X (2015) Interleukin-1beta Level Is Increased in Vitreous of Patients with Neovascular Age-Related Macular Degeneration (nAMD) and Polypoidal Choroidal Vasculopathy (PCV), PLoS One. 10, e0125150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollborn M, Ackmann C, Kuhrt H, Doktor F, Kohen L, Wiedemann P & Bringmann A (2018) Osmotic and hypoxic induction of the complement factor C9 in cultured human retinal pigment epithelial cells: Regulation of VEGF and NLRP3 expression, Mol Vis. 24, 518–535. [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Schmidt S, Larsen PP, Meyer JH, Roush WR, Latz E, Holz FG & Krohne TU (2019) Efficacy of novel selective NLRP3 inhibitors in human and murine retinal pigment epithelial cells, J Mol Med (Berl). 97, 523–532. [DOI] [PubMed] [Google Scholar]
- 18.Lavalette S, Raoul W, Houssier M, Camelo S, Levy O, Calippe B, Jonet L, Behar-Cohen F, Chemtob S, Guillonneau X, Combadiere C & Sennlaub F (2011) Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration, Am J Pathol. 178, 2416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L & Marneros AG (2013) Macrophages are essential for the early wound healing response and the formation of a fibrovascular scar, Am J Pathol. 182, 2407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malsy J, Alvarado AC, Lamontagne JO, Strittmatter K & Marneros AG (2020) Distinct effects of complement and of NLRP3- and non-NLRP3 inflammasomes for choroidal neovascularization, Elife, e60194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang A-S, Humphries MM, Lavelle EC, O’Neill LAJ, Hollyfield JG & Humphries P (2012) NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components, Nature Medicine. 18, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poor SH, Qiu Y, Fassbender ES, Shen S, Woolfenden A, Delpero A, Kim Y, Buchanan N, Gebuhr TC, Hanks SM, Meredith EL, Jaffee BD & Dryja TP (2014) Reliability of the mouse model of choroidal neovascularization induced by laser photocoagulation, Invest Ophthalmol Vis Sci. 55, 6525–34. [DOI] [PubMed] [Google Scholar]
- 23.Ablonczy Z, Dahrouj M & Marneros AG (2014) Progressive dysfunction of the retinal pigment epithelium and retina due to increased VEGF-A levels, FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 28, 2369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grossniklaus HE & Green WR (2004) Choroidal neovascularization, Am J Ophthalmol. 137, 496–503. [DOI] [PubMed] [Google Scholar]
- 25.Miquerol L, Gertsenstein M, Harpal K, Rossant J & Nagy A (1999) Multiple developmental roles of VEGF suggested by a LacZ-tagged allele, Dev Biol. 212, 307–22. [DOI] [PubMed] [Google Scholar]
- 26.Doyle SL, Lopez FJ, Celkova L, Brennan K, Mulfaul K, Ozaki E, Kenna PF, Kurali E, Hudson N, Doggett T, Ferguson TA, Humphries P, Adamson P & Campbell M (2015) IL-18 Immunotherapy for Neovascular AMD: Tolerability and Efficacy in Nonhuman Primates, Invest Ophthalmol Vis Sci. 56, 5424–30. [DOI] [PubMed] [Google Scholar]
- 27.Doyle SL, Ozaki E, Brennan K, Humphries MM, Mulfaul K, Keaney J, Kenna PF, Maminishkis A, Kiang AS, Saunders SP, Hams E, Lavelle EC, Gardiner C, Fallon PG, Adamson P, Humphries P & Campbell M (2014) IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration, Sci Transl Med. 6, 230ra44. [DOI] [PubMed] [Google Scholar]
- 28.Shen J, Choy DF, Yoshida T, Iwase T, Hafiz G, Xie B, Hackett SF, Arron JR & Campochiaro PA (2014) Interleukin-18 has antipermeablity and antiangiogenic activities in the eye: reciprocal suppression with VEGF, J Cell Physiol. 229, 974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirano Y, Yasuma T, Mizutani T, Fowler BJ, Tarallo V, Yasuma R, Kim Y, Bastos-Carvalho A, Kerur N, Gelfand BD, Bogdanovich S, He S, Zhang X, Nozaki M, Ijima R, Kaneko H, Ogura Y, Terasaki H, Nagai H, Haro I, Nunez G, Ambati BK, Hinton DR & Ambati J (2014) IL-18 is not therapeutic for neovascular age-related macular degeneration, Nat Med. 20, 1372–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Copland DA, Theodoropoulou S, Chiu HA, Barba MD, Mak KW, Mack M, Nicholson LB & Dick AD (2016) Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis, Sci Rep. 6, 20639. [DOI] [PMC free article] [PubMed] [Google Scholar]