

Abstract
The fascination and fear of snakes dates back to time immemorial, with the first scientific treatise on snakebite envenoming, the Brooklyn Medical Papyrus, dating from ancient Egypt. Owing to their lethality, snakes have often been associated with images of perfidy, treachery and death. However, snakes did not always have such negative connotations. The curative capacity of venom has been known since antiquity, also making the snake a symbol of pharmacy and medicine. Today, there is renewed interest in pursuing snake-venom-based therapies. This Review focuses on the chemistry of snake venom and the potential for venom to be exploited for medicinal purposes in the development of drugs. The mixture of toxins that constitute snake venom is examined, focusing on the molecular structure, chemical reactivity and target recognition of the most bioactive toxins, from which bioactive drugs might be developed. The design and working mechanisms of snake-venom-derived drugs are illustrated, and the strategies by which toxins are transformed into therapeutics are analysed. Finally, the challenges in realizing the immense curative potential of snake venom are discussed, and chemical strategies by which a plethora of new drugs could be derived from snake venom are proposed.
Subject terms: Biochemistry, Drug discovery
Snake venom contains a diversity of bioactive compounds. This Review highlights the complex chemistry of snake venom and shows how venom can be used to derive new therapeutic drugs.
Introduction
More than 220,000 species, or approximately 15% of all animal diversity on earth, are venomous1. Venom endows predators with a chemical weapon far more potent than physical force. Animal venoms are complex and sophisticated bioactive cocktails, the main components of which are proteins and peptides2. The best characterized animal venoms are probably those derived from cone snails, spiders, scorpions and snakes. The composition of the venoms of the first three is dominated by short (3–9-kDa) disulfide-rich peptides that contain the inhibitor cysteine knot (ICK) motif, although heavier proteins, including enzymes, are also present. ICK peptides are structurally very stable and mainly target the nervous system, acting primarily on membrane channels or neuronal receptors3–5. The venom of a spider or cone snail might contain thousands of different peptides, whereas a scorpion’s venom might contain several hundred3,6. The large number of spider species (possibly >100,000) further increases venom diversity.
Snake venoms typically consist of a mixture of 20 to >100 components, of which the majority (>90%) are peptides and proteins7, with the dominant bioactivities including neurotoxicity, haemotoxicity and cytotoxicity, depending on the snake species. Venom composition varies widely between species and even within the same species8–15. Other factors, such as environmental conditions, age, sex or type of prey available, can also affect venom composition10.
This diversity is a double-edged sword. The only efficient treatment for a snakebite is the administration of the specific antivenom, but the variability in venom composition limits the availability and the upscaling of the production of antivenoms16–18. There are an estimated 2.7 million envenomings each year, which result in >100,000 deaths and leave >400,000 victims with severe and permanent sequelae16–19. However, the compositional diversity is a rich playground for medicinal chemists, providing a collection of highly specific and bioactive compounds that offer many paths towards developing new therapeutic drugs1,4,20–22.
In this Review, we first examine the chemical composition of snake venom, analysing the venoms of >200 snake species. We then discuss the mechanistic details of the chemistry of the principal enzymatic venom toxins. Next, we review the development, structure and mode of action of approved snake-venom-based drugs as well as those of compounds in clinical and preclinical testing. Finally, we conclude with an analysis of the vast therapeutic potential of snake venom, pointing out chemical strategies for the transformation of venom into a repertoire of new drugs.
The chemical composition of snake venom
From the middle of the twentieth century, researchers observed the richness in the constituents of venom and began to isolate and analyse the structures and activities of its toxins, as many of them have the potential to be turned into medicines. Most snake venom toxins belong to one of ~30 families23,24, although the venom of a given snake species can contain hundreds of bioactive compounds15,25. In the snake venoms of known composition, some protein families have many hundreds of isoforms26. For example, the UniProt database27 contains almost 3,000 isoforms of snake toxins within the reviewed entries. Although there is usually extensive functional redundancy among isoforms of the same protein, there are cases in which different isoforms have different biological activities, which makes their characterization relevant and essential28.
Advances in transcriptomics and proteomics, enabled by advances in mass spectrometry, reverse-phase high-performance liquid chromatography and next-generation sequencing15,26,29–31, have enabled scientists to determine the composition of the venom of hundreds of snake species, giving rise to a field named ‘venomics’25 and unveiling the chemical richness of venomous snakes (see the Reptile Database).
We have reviewed the venomics studies conducted in the past 15 years (2007–2021) for the two most relevant families of venomous snake: Elapidae (elapids) and Viperidae (viperids, commonly referred to as vipers, and further divided into the true viper and pit viper subfamilies). Elapids and vipers include almost all medically important snakes, although there are also some examples in the Colubridae (colubrids) family. Figure 1 depicts the averaged venom composition of 76 species and subspecies of the ~400 known elapids and 117 of the ~400 known vipers. Only protein families with an average abundance of >1% of the total venom proteome were considered here. However, because evolutionary and ecological factors can lead to considerable interpopulation and intrapopulation variation in the chemical composition of venom, with numerous exceptions and dichotomies adding to the molecular richness at all taxonomic levels, we also analysed the venom composition of individual snake genera (only the most well-studied were considered), which illustrates the venom diversity. Supplementary Table 1 further illustrates the chemical richness of snake venom by providing the detailed venom composition for each species and the respective bibliographic sources.
Fig. 1. Composition of the venom of snakes from the Elapidae and Viperidae families.

The large charts show the averaged composition of the venom of snake species from the Elapidae (elapids) or Viperidae (viperids) families. Each entry in the charts corresponds to a protein family, in which we group tens to hundreds of isoforms. Only protein families with an average abundance of >1% of the total venom proteome are represented, except for the SVSPs in elapids, which are included for comparison with the viperids, and defensins, which although seldom present, can be abundant in the venom of certain species. The distribution of the proportion of the most abundant protein families is shown in Supplementary Fig. 1. Data are from the proteomic studies of the past 15 years; 143 entries for 2007–2017 are from Isbister and Tasoulis’s database of snake venom proteomes7; we assembled the additional entries for 2017–2021 from the literature. The Atractaspididae and Colubridae snake families are not included in the study because most are non-venomous or their venoms are weak, not medically important and poorly studied (for venomics studies on colubrids see ref.15). Each species contributes with the same weight to the average; subspecies or species from different locations were averaged within the entry for the species. The entry ‘Other’ corresponds to unidentified components or components with an average abundance of <1%. The smaller charts decompose the snake venom composition at the genus level, which reveals the compositional diversity. Only the most well-studied genera are included, which comprise almost all the medically relevant snakes. Supplementary Table 1 details the composition of the venom of each species included in the study together with the relevant reference. 3FTx, three-finger toxin; CRiSP, cysteine-rich secretory protein; CTL/SNACLEC, C-type lectin and C-type lectin-like protein; DEF, defensin; DIS, disintegrin; KSPI, Kunitz-type serine protease inhibitor; LAAO, l-amino acid oxidase; NP, natriuretic peptide; PLA2, phospholipase A2; SVMP, snake venom metalloproteinase; SVSP, snake venom serine protease.
Some toxins act synergistically, and the combination and proportion of each toxin determine the pathophysiology of snakebite envenomation17,28. The difference in the chemical composition of venom from elapids and vipers (Fig. 1) leads to different clinical manifestations. Envenoming by elapids mostly induces neurotoxic, cytotoxic and cardiotoxic manifestations, whereas envenoming by vipers typically induces myotoxicity and haemotoxicity17.
Elapid venoms mainly comprise peptides and proteins from seven families; secreted phospholipases A2 (PLA2s)32–36 and three-finger toxins (3FTxs)36–39 are often major constituents and have a dominant role in the action of the venom, although there are many exceptions and considerable diversity at the species level (Supplementary Table 1). For example, the venom of snakes from the Dendroaspis genus (mambas) and many Australian snakes show notable exceptions in the composition and action of these toxins. The former lack PLA2 and the latter have a very low content (<6%) of 3FTx. Interestingly, all these snakes have highly potent venoms.
Other toxins — namely snake venom metalloproteinases (SVMPs)34,36,40–42, snake venom serine proteases (SVSPs)34,36,43 and l-amino acid oxidases (LAAOs)34,44–47 — represent an average of 6% of elapid venom. Kunitz-type peptides48 are a family of serine protease inhibitors with the Kunitz domain fold; these peptides constitute an average of ~5% of elapid venom and are potent and selective K+-channel blockers. Kunitz-type peptides are particularly prevalent in mambas. The remaining protein families that have been identified appear in smaller amounts.
Viperid venoms mostly include toxins from nine protein families. Again, there are many exceptions and substantial diversity at the individual species and subspecies level (Supplementary Table 1). In most species, the PLA2 (refs32–36), SVMP34,36,40–42,49 and SVSP34,36,43,50,51 toxins are dominant, representing an average of ~70% of the whole venom proteome. Most viperid PLA2s are myotoxic, despite sharing extensive sequence identity with the PLA2s of elapids, many of which are neurotoxic. Other toxins that are present in smaller proportions (4–7%) are LAAOs34,44–47, C-type lectins and C-type lectin-like proteins52–54, and natriuretic peptides55–57.
This analysis shows only part of the complexity of snake venom, as hundreds of additional proteins, enzymes and peptides can be present in the venom of each species.
Interspecies variation
Snake venom shows both considerable intraspecies (Box 1) and interspecies variation. The fraction of PLA2s and 3FTx in the venom of each elapid species varies widely (Supplementary Fig. 1), with the percentage of each ranging from almost 0% to nearly 100%. Interestingly, in most species, a lower fraction of PLA2 is compensated by a higher fraction of 3FTx, and vice versa. Thus, together, they represent, on average, >80% of the total venom proteome in most elapid species. Kunitz-type peptides generally represent <10%, with a more even distribution across species.
An exception is the black mamba (Dendroaspis polylepis) — the most feared snake of the African continent. It is very aggressive when threatened, extremely fast, intelligent and has highly toxic and fast-acting venom. Despite this, few fatalities are attributed to this snake, mainly because its habitat is generally far from densely inhabited areas. Black mamba venom primarily consists of Kunitz-type peptides and 3FTxs (61% and 31%, respectively) and lacks PLA2s58. Thus, its venom composition is highly atypical, similar to almost no other snake’s venom, except for that of the closely related eastern green mamba (Dendroaspis angusticeps), which is also devoid of PLA2s and rich in Kunitz-type peptides and 3FTxs, but with the opposite proportions (16% and 69%, respectively). These two mambas may represent the most outstanding examples of the chemical diversity of elapid venoms.
The composition of viperid venom varies widely across genera (Fig. 1) and species (Supplementary Fig. 1 and Supplementary Table 1). The PLA2, SVSP and SVMP enzymatic families represent an average of ~70% of the viperid proteome. The proportion of PLA2 ranges from almost zero to >90%, with the distribution peaking at ~10% (Supplementary Fig. 1). The SVMP ratio also has a broad distribution, with a peak at ~40%. SVSPs generally constitute <20%, although there are species, such as the Okinawa pit viper (Ovophis okinavensis), whose venom consists almost entirely of SVSPs59. Although present in low quantities on average, C-type lectins and C-type lectin-like proteins, defensins or natriuretic peptides can represent >37% of the viperid proteome in specific species.
Defensins are small proteins ubiquitous across life that function as host defence peptides and have antimicrobial and/or immune signalling activities. The defensins found in viperid venom act on the Na+ and K+ channels of plasma membranes, including that of muscle cells (the sarcolemma), and accumulate in the lysosomes, causing analgesic, neurotoxic, myotoxic and cytotoxic effects60,61. The accumulation in the lysosomes is an unusual mechanism of cytotoxicity among snake toxins. Furthermore, the structures of defensins are unlike any other channel-binding toxins. Although they can be more abundant in specific venoms, defensins are typically rare, and constitute on average 1% of venom.
In conclusion, our analysis confirms and further reinforces the understanding of the incredible diversity of snake venom. This diversity of highly bioactive proteins and peptides, which recognize essential biological targets with exquisite specificity and affinity, constitutes a unique pharmacological database for drug discovery.
Box 1 Intraspecies variation of snake venom.
Venom composition changes within a species owing to age, gender, prey availability, diet and geographic location, among other factors. The venom of the Russell’s viper (Daboia russelii) is a well known example of this. Its venom has been widely studied owing to the medical importance of this snake. The combination of the high toxicity of its venom, its discreet but aggressive habits and, most importantly, its prevalence in densely inhabited areas mean that the Russell’s viper is one of the two snake species that cause more fatalities than any other in the Indian subcontinent, with the other being the saw-scaled viper (Echis carinatus), for the same reasons. Together, they make a considerable contribution to the global snakebite burden.
By comparing the abundance of the three major enzymatic toxins in the venom of Russell’s vipers from different locations (Supplementary Table 1), it can be seen that the abundance of secreted phospholipases A2 (PLA2s) varies by threefold, that of snake venom serine proteases by threefold and that of snake venom metalloproteinases (SVMPs) by 19-fold.
Another species that shows notable variation in its venom is the common lancehead viper (Bothrops atrox), which is the leading cause of snakebite accidents in the Amazon. The venom of common lancehead vipers from Venezuela is composed chiefly of SVMPs (85%), whereas in individuals from Amazonian Peru, Colombia and Pará (Brazil), it constitutes approximately half of the venom only. In individuals in the latter regions, the lack of SVMPs is compensated by an increased abundance of PLA2s233–236. Even among siblings, there is sex-based variation in the venom237.
Chemistry of the major enzymatic toxins
Since the first identification and characterization of the structure of a snake venom toxin, crotoxin from Crotalus durissus terrificus in 1938 (ref.62), there has been intense research aimed at elucidating the structure, reactivity and target of venom toxins, in order to understand their function and determine their molecular determinants of recognition and therapeutic potential. Table 1 summarizes the information currently available for several prominent toxin families and provides suggestions for further reading. In this section, we discuss the reaction chemistry of the three principal enzymatic toxin families: PLA2s, SVMPs and SVSPs. Some of the other abundant toxins without catalytic activity are discussed in the next section.
Table 1.
Characteristics of the main families of snake venom toxins
| Toxin family | Snake family | Enzymatic activity | Principal biological targets | Major pathophysiological activities | Most promising therapeutic applications | Representative toxin structures | Further reading |
|---|---|---|---|---|---|---|---|
| PLA2 | Elapids and viperids | Yes | Plasma membrane of myocytes and various receptors in the axolemma (undetermined molecular target) | Acute skeletal muscle necrosis, flaccid paralysis, local inflammatory reactions (oedema, leukocyte influx into tissues and pain) | Antibacterial activity against Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa and Enterobacter aerogenes192; anti-parasite effects155; antiviral activity against HIV193,194 and dengue195 | 5TFV (basic); 1JIA (basic); 1Y4L (PLA2 homologue) | 32–36 |
| SVMP | Elapids (P-III SVMPs) and viperids (P-I, P-II and P-III SVMPs) | Yes | Wide variety of targets; most notable are collagen IV and blood coagulation factors | Predominantly, haemorrhagic activity but can cause the proteolytic degradation of fibrinogen and fibrin, induce apoptosis and inhibit platelet aggregation | Haemostasis; blood coagulation, fibrinolysis and platelet aggregation78,168,196 | 2W15 (P-I bound to a peptidomimetic); 2M75 (P-II, DIS domain); 3DSL (P-III) | 34,36,40–42,49 |
| SVSP | Elapids and viperids | Yes | Mostly blood coagulation factors | Imbalances the haemostatic system through action in the coagulation cascade on the fibrinolytic and kallikrein–kinin systems | Prevention of thrombus formation through fibrinogen depletion; anticoagulant197,198; applied in neurosurgical199 and vertebro-spinal200 procedures as a fibrin sealant | 1OP0 (glycosylated); 3S9B (RVV-V in open form); 3S9C (RVV-V bound to factor V) | 34,36,43,50,51 |
| 3FTx | Elapids | No | Nicotinic and muscarinic acetylcholine receptors, acetylcholinesterase and cardiomyocytes (undetermined molecular target) | Neurotoxic effects, which cause paralysis; muscle fasciculations; and cardiac arrest through lysis of cardiomyocytes | Regulation of blood pressure201; treatment of coagulation disorders202–204; analgesic action146,151,154 | 1QKD (short-chain α-neurotoxin); 1YI5 (long-chain α-neurotoxin); 1KBA (κ-neurotoxin dimer); 3PLC (β-cardiotoxin); 1F8U (receptor-bound fasciculin); 4DO8 (muscarinic toxin) | 36–39 |
| LAAO | Elapids and viperids | Yes | l-amino acids; substrate varies among species | Haemorrhagic or anticoagulant effects, induction of apoptosis, oedema and platelet aggregation or inhibition | Antimicrobial against P. aeruginosa, Candida albicans, S. aureus; antiparasitic against Leishmania chagasi and Leishmania amazonensis205,206; potential antiviral against HIV-1 (ref.207) | 2IID (with l-phenylalanine and its cofactor FAD, and glycosylated); 4E0V (with cofactor FAD); 5TS5 (glycosylated, and with cofactor FAD) | 34,44–47 |
| CRiSP | Elapids and viperids | No | Ca2+ channels, K+ channels, and signalling cascades involved in cell adhesion | Inhibits angiogenesis, increases vascular permeability and promotes inflammatory responses (leukocyte and neutrophil infiltration) | Antiparasitic against Leishmania and trypanosome strains208; antimicrobial against Gram-negative bacteria and filamentous fungus209 | 6IMF (bound to inhibitor peptide); 1WVR (Ca2+-channel blocker triflin); 3MZ8 (zinc-bound natrin) | 210 |
| CTL/SNACLEC | Viperids | No | Platelet and cellular receptors, as well as coagulation factors, such as factor IX and factor X | Diverse effects, including haemagglutination, mitogenic activity, platelet aggregation, oedema, elevated vascular permeability, renal effects, hypotension, cytotoxicity and modulation of Ca2+ release from skeletal muscle sarcoplasmic reticulum | Use in anticoagulant therapies211–213 | 1JZN (CTL in complex with galactose); 1UOS (CTL convulxin); 3UBU (SNALEC bound to platelet glycoprotein Ib) | 52–54 |
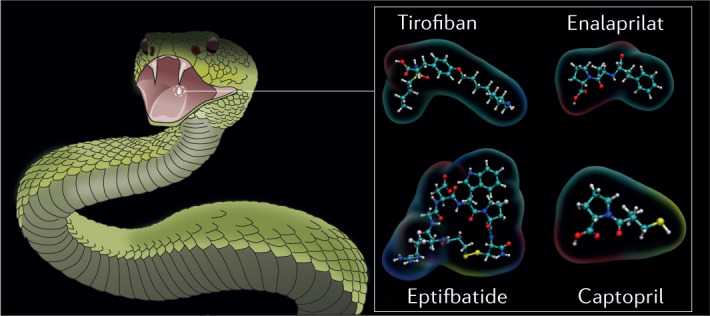
| DIS | Viperids | No | Integrins | Disrupts cell–cell adhesion and cell–matrix adhesion, and inhibits angiogenesis | Anti-inflammatory and antiangiogenic for chronic inflammatory processes214; template in the development of the anti-platelet marketed drugs eptifibatide and tirofiban to treat thrombosis215; treatment for neoplastic processes216 | 1J2L (trimestatin); 3C05 (acostatin); 1RMR (schistatin) | 107,217–219 |
| NPs | Elapids and viperids | No | NP receptors A, B and C | Potent hypotensive effects (vascular relaxation and a decrease in myocardial contractility), leading to rapid loss of consciousness | Cardiorenal diseases; heart failure144 | 4AA2 (BPPb bound to an ACE-I homologue); 4APJ (BPPb bound to ACE-I) | 55–57 |
| KSPI | Elapids and viperids | No | Proteases and K+ channels | Inhibition effects on a range of serine proteases, including plasmin and trypsin, leading to anticoagulation, fibrinolysis, inflammation and ion-channel blocking | Reduction of cyst development in polycystic kidney diseases through inhibition of vasopressin type 2 receptor pathways220 | 3BYB (textilinin-1); 3D65 (textilinin-1 in complex with trypsin); 5M4V (mambaquaretin-1) | 48 |
| DEF | Elapids and viperids | No | Skeletal muscle Na+ and K+ channels, lipid membranes and sarcolemma | Myotoxic damage through depolarization of skeletal muscles, and analgesic activity | Anti-proliferative, anti-nociceptive, anti-inflammatory, antifungal against C. albicans, anti-Plasmodium, anti-Leishmanial and anthelmintic129,221–224 | 4GV5 (crotamine, X-ray); 1H5O (crotamine, NMR) | 60,61 |
Only toxins from the elapid and viperid families are included. Within each family, there are tens or hundreds of different isoforms. Although there are numerous species-specific and isoform-specific exceptions, the most common biological target(s) and pathophysiologic manifestations are reported. Representative structures, when available, are taken from the Protein Data Bank. Additional details on each toxin can be found in the referenced articles. We also refer readers to ref.225, which is an excellent and detailed source of information on the structure, bioactivity, pathophysiology and therapeutic applications of snake venom toxins. 3FTx, three-finger toxin; ACE-I, angiotensin-1 converting enzyme; BPPb, bradykinin potentiating peptide b; CRiSP, cysteine-rich secretory protein; CTL/SNACLEC, C-type lectin and C-type lectin-like; DEF, defensin; DIS, disintegrin; KSPI, Kunitz-type serine protease inhibitor; LAAO, l-amino acid oxidase; NMR, nuclear magnetic resonance; NP, natriuretic peptide; PLA2, phospholipase A2; RVV-V, factor-V activating enzyme from Russell’s viper venom; SVSP, snake venom serine protease; SVMP, snake venom metalloproteinase.
Secreted phospholipases A2
PLA2s exist as active enzymes and as inactive PLA2-like proteins in snake venom. PLA2 enzymes catalyse the hydrolysis of the sn-2 ester bond of cell-membrane phospholipids and are classified into 14 groups, from which the groups IA and IIA are present in elapid and viperid venoms33,35. The enzymes in these groups have a molecular mass of 13–19 kDa, contain 5–8 disulfide bridges and form dimers in aqueous solution63. In cell membranes, PLA2s dissociate and bind as monomers63,64, and show an affinity for membrane regions in which at least 15% of the phospholipids are negatively charged65 (Fig. 2a). PLA2s can also be divided into acidic and basic isoforms, according to the isoelectric point (pI), with the basic isoforms having a higher membrane affinity and thus higher toxicity32.
Fig. 2. The three main types of PLA2 bound to their targets.

a | Myotoxin I (MT-I), a strongly myotoxic phospholipase A2 (PLA2) from the venom of a terciopelo viper (Bothrops asper), attached to the sarcolemma. MT-I (PDB ID: 5TFV)226 is shown with a phospholipid substrate bound to the active centre. In the phospholipid, oxygen is red, phosphorus is orange, nitrogen is blue, carbon is grey and hydrogen is white; the enzyme is shown in light red, and the Ca2+ ion is shown in light green. The residues that form the protein–membrane interface and the PLA2–membrane binding geometry were identified through mutagenesis, fluorescence and X-ray crystallography studies64,65. b | The PLA2 homologue myotoxin II (MT-II), also from terciopelo venom (PDB ID: 1Y4L)227, bound to the sarcolemma. The C-terminal region destabilizes and permeabilizes the membrane70. The protein is shown in light green, and the C-terminal KKYRYYLKPLCKK sequence is shown in pink. c | β-Bungarotoxin (PDB ID: 1 BUN)75 from the Taiwan banded krait (Bungarus multicinctus) bound to a neuronal membrane. The toxin travels silently through the victim’s body until its Kunitz (KUN) domain (green) recognizes and binds a presynaptic voltage-gated K+ channel (violet, PDB ID:6PBX) with high specificity, trapping the PLA2 domain (light blue) at the neuronal membrane, where its active site, otherwise occluded, opens and starts degrading the adjacent phospholipids (bound phospholipid coloured by element)75–77.
The catalytic mechanism of PLA2s is still unclear at the atomic level. Nevertheless, it is a two-stage process, with the first corresponding to the binding of the PLA2 to the membrane and the second to the chemical reaction. The first stage determines the enzyme–target specificity, whereas both stages determine the enzyme efficiency63. The catalytic activity of PLA2s depends on Ca2+, which stabilizes the tetrahedral transition state of the reaction65,66. An essential aspartate residue (Asp49) coordinates the Ca2+ cofactor, the mutation of which renders the enzyme inactive. As metadata analyses have shown, it is unusual for Ca2+ cofactors to participate in the catalytic cycle67,68. Other divalent metal ions that are larger or smaller, or harder or softer, than Ca2+ lead to a notable drop in activity69 for a reason not yet understood. In the most-accepted mechanism65, the active site histidine residue (His48) abstracts a proton from a water molecule bound to the Ca2+ ion, and the resulting hydroxy group attacks the sn-2 ester bond. A chain of water molecules probably mediates the proton transfer to His48.
In a twist of evolution, viper PLA2s split between enzymes and catalytically inactive proteins, known as PLA2 homologues. The latter lack the Ca2+ cofactor owing to substitution of the Asp49 residue by lysine or, less commonly, by serine, arginine, glutamine or asparagine. The (Lys49) PLA2 homologues are highly myotoxic, despite having no enzymatic activity. A sequence of ~12 positively charged and hydrophobic residues at the C-terminal region, with positive ends and a mixed positive and hydrophobic core (such as the KKYRYYLKPLCKK sequence in MT-II from the venom of terciopelo (Bothrops asper)), is believed to penetrate and destabilize the sarcolemma (Fig. 2b), and thus promote an influx of Ca2+ ions, which starts a chain of harmful events that leads to myotoxicity32,70,71. Further investigation is needed to achieve an atomic-level understanding of this effect. This stretch of residues alone often has similar bioactivity to that of the whole protein and is intensively investigated as a model to construct antimicrobial peptides69,72–74.
Some neurotoxic PLA2s have even more refined molecular mechanisms of action that serve as lessons for drug delivery. An example is β-bungarotoxin from the venom of the Taiwan banded krait (Bungarus multicinctus), which comprises a PLA2–Kunitz-type peptide heterodimer. The toxin travels silently through the victim’s body, avoiding off-target membranes owing to the partially occluded PLA2 active site and its low membrane affinity. When β-bungarotoxin reaches the pre-synaptic region of the neuromuscular junction, the Kunitz-type peptide recognizes and binds a specific K+ channel, trapping the toxin at this location. This event exerts a first neurotoxic action. The PLA2 monomer, once firmly anchored at the membrane, opens the active site and initiates hydrolysis of the membrane phospholipids, near the K+ channel, further enhancing the neurotoxic effect75–77 (Fig. 2c).
In summary, PLA2 enzymes and their inactive homologues share extensive sequence and structural similarity, and both induce myotoxicity but through surprisingly diverse molecular mechanisms. The great diversity of PLA2 isoforms translates into a surprising variety of biological activities, underlying molecular machinery and recognition targets, and, consequently, a variety of drug discovery opportunities73.
Snake venom metalloproteinases
SVMPs are mostly haemorrhagic and are classified into three groups (P-I to P-III) according to the number of domains (1–3), with further division into subgroups40,42. P-III SVMPs are the largest, more ancient and most complex enzymes, from which the P-II and P-I enzymes evolved through domain loss. Elapid venoms contain only P-III SVMPs. By contrast, viperid venoms contain SVMPs from each of the three groups, with SVMPs being a prominent toxin, and often the most abundant one (Fig. 1).
P-I SVMPs comprise the catalytic domain only, which catalyses the hydrolysis of a vast array of physiologically relevant enzymes and structural proteins. This domain is common to the three SVMP groups. Its hydrolytic targets include collagen IV, fibrinogen and coagulation factors, with extensive haemorrhagic consequences41,49. Hydrolysis of collagen IV weakens capillary walls, which causes them to collapse under otherwise normal haemodynamic pressure41. Continuous hydrolysis of fibrinogen in vivo leads to weak, inefficient fibrin clots and hypofibrinogenaemia, and the hydrolysis of blood coagulation factors deregulates blood clotting78,79.
P-II SVMPs have an additional disintegrin domain that inhibits platelet aggregation through specific binding to the blood platelet αIIBβ3 integrin — a vital protein that triggers fibrinogen binding and platelet aggregation80. This action reinforces the haemorrhagic effect of collagen IV hydrolysis.
P-III SVMPs (Fig. 3) have a catalytic domain, a disintegrin-like domain with a collagen-binding three-amino acid Glu–Cys–Asp (ECD) motif (instead of the typical P-II Arg–Gly–Asp (RGD) motif) and a cysteine-rich domain. The prominent role of the latter is substrate recognition and binding. Nevertheless, the catalytic domain is also involved in substrate recognition through an interesting conformational selection mechanism (Box 2). In some isoforms, a C-type lectin-like domain is also present40.
Fig. 3. Structures of SVMPs and their substrates.

a | The structure of the factor X activating enzyme RVV-X (PDB ID: 2E3X)81 from the eastern Russell’s viper (Daboia siamensis). RVV-X is a P-III snake venom metalloproteinase (SVMP) isoform that is ubiquitous in species from the Indian subcontinent228. RVV-X activates blood coagulation factor X by hydrolysing the Arg194–Ile195 position with such high specificity that it is used as a diagnostic tool for haematologic disorders26,93,94,229. The catalytic domain (MET) is coloured yellow, the disintegrin (DIS) domain is coloured green and the cysteine-rich domain (CR) is coloured pink. RVV-X has an additional C-type lectin and C-type lectin-like protein (CLT/SNACLEC) domain, which is shown in blue. The inset shows the Zn2+ cofactor with its coordination shell and the peptidomimetic inhibitor GM6001, whose two coordinated oxygen atoms mimic the positions of the water molecule and the carbonyl of the substrate (superimposed on top of GM6001 with a translucent ball and stick representation). b | Illustrative scheme of daborhagin230, a highly haemorrhagic SVMP from Russell’s viper venom, bound to collagen IV at the basement membrane of capillaries. The colour scheme of the enzyme domains is the same as that of RVV-X in part a. A collagen IV fibre is shown in light green, with a tropocollagen unit emphasized in dark green and drawn in a cartoon and tube representation. The hydrolysis of collagen IV weakens the mechanical stability of the capillary wall, which breaks down under regular haemodynamic forces, leading to massive haemorrhage. Daborhagin was modelled with the active site facing collagen IV.
The reaction mechanism of SVMPs is not fully clarified despite a wealth of X-ray structures81–83. The mechanism is proposed82 to begin with the coordination of the scissile carbonyl of the substrate to the Zn2+ ion in the active site, such that the carbonyl group is held at an attacking distance from a water molecule also bound to the Zn2+ centre. Following deprotonation of the water molecule by a conserved glutamate residue (Glu146), the resulting hydroxide ion attacks the carbonyl carbon to form a Zn2+-bound gem-diolate. Finally, the neutral Glu146 protonates the peptide amine, leading to cleavage of the peptide bond.
Box 2 Substrate recognition by snake venom metalloproteinases.
Despite extensive sequence identity, only some snake venom metalloproteinases (SVMPs) bind collagen IV and have haemorrhagic activity. Molecular dynamics simulations explained this paradox by revealing that in haemorrhagic SVMPs, the first half of the Ω-loop (residues 156–165) is highly flexible, whereas in non-haemorrhagic SVMPs, the second part of the Ω-loop (residues 166–175) is flexible instead. On the basis of this observation, the authors proposed that the flexibility of this loop is crucial for collagen IV recognition238 and, more recently, this hypothesis has been experimentally confirmed239. Thus, the lesson here is that target recognition might rely on protein dynamics and not only on the static X-ray structure; this constitutes a formidable challenge for drug discovery.
Snake venom serine proteases
SVSPs are primarily haemotoxic and interfere with blood coagulation, blood fibrinogen levels, blood pressure and platelet aggregation34,36,43,50,51 (Fig. 4a,b), although there is one known example of an SVSP with K+-channel blocking activity84 (Fig. 4c). The resistance of SVSPs to endogenous serine protease inhibitors endows them with their toxic effects. Many of the activities of SVSPs mimic those of the enzyme thrombin, which is a vital component of the blood coagulation cascade. Each SVSP exhibits one or more of the activities of thrombin and sometimes has bioactivities that thrombin does not. But no SVSP possesses all the bioactivities of thrombin85, which makes them toxic and, in contrast to thrombin, SVSPs deregulate homeostasis43,85. SVSPs that share some of the fibrinogenolytic activities of thrombin have been named thrombin-like enzymes.
Fig. 4. Structure of SVSPs and their substrates.

a | The factor V activating enzyme from Russell’s viper venom (RVV-V; PDB ID: 3S9C), which is a snake venom serine protease (SVSP) with specificity for blood coagulation factor V. RVV-V is depicted in a complex with the 14-residue terminal fragment of factor Va (residues 1533–1546), called FV14. RVV-V releases the last 61 residues of factor V by hydrolysing its Arg1545–Ser1546 bond, generating procoagulant factor Va and mimicking one of the physiological roles of thrombin231. The inset shows the active site and factor V hydrolysis product. RVV-V recognizes factor V through a selective induced-fit mechanism that opens an otherwise closed subpocket. The strict specificity of RVV-V for factor V makes it a useful diagnostic tool for measuring factor V levels, lupus anticoagulant levels and resistance to activated protein C93,94. b | Illustrative representation of the thrombin-like Brazilian lancehead pit viper (Bothrops moojeni) SVSP batroxobin (Defibrase)4,21,118,119,232 bound to fibrinogen. Thrombin cleaves the Aα and the Bβ chains of fibrinogen and converts factor XIII into factor XIIIa, which generates crosslinked fibrin, whereas most SVSPs cleave either the Aα or the Bβ chain only85. Batroxobin cleaves only the Aα chain118,232. SVSPs therefore form abnormal, easily degradable fibrin clots that lead to fibrinogen depletion and hypofibrinogenaemia. The clotting time in the presence of batroxobin (reptilase time) is used in the clinic to diagnose several diseases93,94. Batroxobin was modelled from the homologue saxthrombin (PDB ID: 3S69). c | Collinein-1 from the neotropical rattlesnake (Crotalus durissus collilineatus) is the first example of an SVSP with specific K+-channel blocking activity84. Through a mechanism that is independent of its enzyme activity, collinein-1 selectively inhibits the oncogenic hEAG1 channel (PDB ID:6PBX) among 12 tested voltage-gated K+-channels, with obvious antitumour implications. As K+ channels are known targets for many animal neurotoxins, the discovery of collinein-1 makes it tempting to speculate that some yet unknown SVSP isoforms might have found a neurotoxic role. Collinein-1 was modelled from the homologue thrombin-like enzyme AhV_TL-I (PDB ID: 4E7N) and is illustratively bound to the oncogenic hEAG1 channel.
SVSPs are monomeric glycoproteins with ~228–239 residues and a molecular mass of 26–67 kDa (refs34,43). This wide range of molecular masses is due to different patterns of N-glycosylation and O-glycosylation. The enzymes share the typical trypsin fold and the highly conserved Ser195–His57–Asp102 catalytic triad (chymotrypsin numbering) (Fig. 4). Six disulfide bonds stabilize the structures.
Most SVSPs share the classical reaction mechanism of serine proteases. However, >20 SVSPs with variations in the canonical catalytic triad have been found in snake venom transcripts86. Among the few of these that have been characterized, the horned viper (Vipera ammodytes ammodytes) serine protease VaSP1, which bears the rare Ser195–Lys57–Asp102 triad, was surprisingly found to be catalytically active87, illustrating an unexpected richness in SVSP chemistry.
In contrast to thrombin, which activates many different coagulation factors (factor V, factor VIII, factor XI and factor XIII, as well as fibrinogen), each SVSP is highly substrate-specific50. However, as different SVSPs are specific for different sets of targets, a group of isoforms can induce diverse physiological manifestations. This recognition diversity is striking given their extensive mutual sequence identity (50–85%), which is a phenomenon known as the identity–selectivity paradox50: their specificity cannot be understood from the primary sequence. Instead, the specificity appears to depend on a combination of subtle structural epitopes, primary and secondary binding sites, enzyme flexibility, glycosylation and water organization50. The precise specificity and intense haemoactivity of SVSPs make them potential diagnostic and therapeutic tools in the cardiovascular area.
The chemistry of snake venoms is partially understood for the major enzymes, but further understanding at the atomic level is required. Computer simulations are one of the best ways to answer remaining questions, particularly given the recent advances in quantum mechanical and classical mechanics methods, which enable the reliable prediction and determination of complex chemical reaction mechanisms88–91.
Drugs from snake venom toxins
Snake venom finds three major therapeutic applications: pharmaceutical drugs4,92, toxin-based diagnostic methods92–94 and biological markers for understanding human physiology26. We focus here on pharmaceutical drugs based on snake venom. This section discusses the snake venom toxins and toxin-inspired molecules that are being used to develop new drugs, focusing on the drugs approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as well as drugs under development in preclinical and clinical trials.
Approved drugs
Snake venoms are typically cytotoxic, neurotoxic and haemotoxic. The anticancer potential of cytotoxins has long been recognized92. Neurotoxins are of interest for the treatment of neurological diseases. However, no drug derived from a snake venom neurotoxin has yet reached the market. The complexity of the human neurological system, our insufficient understanding of this system and the difficulty in delivering medications to the nervous system contribute to the slow progress of this line of drug discovery20. Nevertheless, the FDA and EMA approved ziconotide, a ω-conotoxin peptide from the magic cone snail (Conus magus), as an analgesic for severe chronic pain4,21,95,96. The main limitation of this drug is its intrathecal administration route. In contrast to neurotoxins, haemotoxins have given rise to numerous drugs approved by the FDA and EMA, in part because they affect a system whose physiology is well known and easier to manipulate. As cardiovascular diseases are the leading cause of death globally, the development of snake-venom-derived drugs that target the cardiovascular system is appealing.
Captopril
The antihypertensive drug captopril was the first drug based on a bioactive component from snake venom that was approved in the US by the FDA in 1981 and in European countries from 1984 onwards. The realization that envenoming by the South American pit viper jararaca (Bothrops jararaca) caused notable hypotension led to the discovery of the vasodilator peptide bradykinin in its venom97. Subsequent studies led Sérgio Ferreira and colleagues to discover a set of nine peptides in the venom of jararaca that potentiated the effect of bradykinin, named bradykinin potentiating factors (BPFs)98–100. BPFs inhibit the angiotensin-converting enzyme (ACE)101, which otherwise degrades bradykinin. The therapeutic potential of BPFs led the pharmaceutical company Squibb to develop a drug against hypertension using BPF peptides (BPP5a and BPP9a, in particular) as templates102 (Fig. 5a). The result was captopril, a small, synthetic, orally bioavailable and potent bioactive molecule with a structure and electrostatics that mimic the BPP5a Pro–Ala–Trp recognition motif for ACE.
Fig. 5. Approved drugs derived from snake venoms.

Several drugs derived from snake venoms have been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). The chemical structures of these drugs are shown, with the region that mimics the snake toxin highlighted in grey. a | Nine hypotensive bradykinin potentiating peptides (BPPs) were isolated from the venom of the jararaca viper; they inspired the design of the antihypertensive drugs captopril and enalapril. These drugs mimic the Trp–Ala–Pro (WAP) motif by which BPP5a (top right) recognizes its target: the angiotensin-converting enzyme (ACE). ACE is shown on the left in a complex with BPP5b, another BPP (PDB ID: 6QS1). The ACE Zn2+ cofactor is shown in orange. b | The drug tirofiban was inspired by a disintegrin called echistatin found in the venom of the saw-scaled viper. Echistatin, shown on the left (PDB ID: 6LSQ), binds specifically to the αIIBβ3 integrin through its Arg–Gly–Asp (RGD) motif (coloured spheres and top right), which prevents platelet aggregation. In tirofiban, the piperidine moiety replicates arginine, the aliphatic linker replicates glycine, and the tyrosine carboxyl group replicates the aspartic acid carboxylate. The (S)-NHSO2nC4H9 group increases the affinity of tirofiban for its αIIBβ3 target. c | Eptifibatide is an antiplatelet drug inspired by the disintegrin babourin purified from the venom of Barbour’s pygmy rattlesnake. A homology model of babourin (template PDB ID: 1J2L) is shown on the left. Most disintegrins recognize the αIIBβ3 integrin through the RGD motif, but babourin uses a Lys–Gly–Asp (KGD) motif (coloured spheres and top right). Eptifibatide achieves maximum selectivity owing to the fusion of the two motifs into the unnatural homoRGD motif. Additional peripheral residues and cyclization endow further molecular recognition capabilities and resistance to proteolysis.
Captopril was a milestone in many ways: it was the first drug developed from animal venom; it was created by converting toxic action into therapeutic action; it was one of the first examples and a paradigm of ligand-based drug discovery; and it was the first drug targeting ACE, rapidly becoming a blockbuster and saving countless lives100.
To overcome the side effects of captopril caused by its thiol group, Merck developed enalapril102,103 (Fig. 5a). The thiol group in captopril was replaced by a carboxylate, leading to a loss of potency, which was compensated with additional modifications. The resulting compound (enalaprilat) lacked oral bioavailability, most probably owing to the ionic carboxylate. Enalaprilat was converted into its ethyl ester to overcome the problem, giving rise to enalapril, a prodrug with very good oral bioavailability21,104 and approved by the FDA and EMA. Enalapril became Merck’s first billion-dollar-selling drug in 1988.
Many ACE-inhibitor drugs based on the BPP5a binding motif were subsequently developed and approved. Examples include lisinopril, quinapril, ramipril, trandolapril and moexipril105,106, which, despite being frequently dismissed in the snake-based drug discovery world, deserve to be considered snake-venom-based drugs. These drugs are among the most prescribed globally and showcase the immense therapeutic potential of venoms, which is yet to be fully realized.
Tirofiban
Tirofiban is an antiplatelet drug approved by the FDA in 1998 and the EMA in 1999 for treating acute coronary syndrome107–110. Its structure is derived from the toxin echistatin111, a 49-residue disintegrin from saw-scaled viper (Echis carinatus) venom. Echistatin competes with fibrinogen for binding to the αIIBβ3 integrin, which inhibits the final step in platelet aggregation92. Echistatin thus reinforces the haemorrhagic activity of saw-scaled viper SVMPs.
Echistatin shares the RGD motif of the disintegrin domains of many P-II-type SVMPs, which is the minimal sequence for αIIBβ3 recognition. It binds several integrins with sub-nanomolar affinity, with selectivity for αIIBβ3 over others107. In high concentrations, the isolated RGD tripeptide also inhibits platelet aggregation. Tirofiban was modelled to replicate the RGD motif of echistatin within a small synthetic molecule92. The affinity of tirofiban for αIIBβ3 was enhanced by the (S)-NHSO2nC4H9 extension, which interacts with an αIIBβ3 exosite with which echistatin does not interact112 (Fig. 5b). The affinity and specificity of tirofiban thus surpass those of echistatin. Tirofiban is another example of the transformation of a venom toxin into a life-saving drug. It is also one of the first documented successful pharmacophore-based drug discovery applications113.
Eptifibatide
Eptifibatide is another antiplatelet drug approved by the FDA in 1998 and EMA in 1999 that was developed from a disintegrin (barbourin) found in the venom of Barbour’s pygmy rattlesnake (Sistrurus miliarius barbourin)107,108,114,115. Barbourin binds the αIIBβ3 integrin through a Lys–Gly–Asp (KGD) motif, rather than the more common but less specific RGD motif. The KGD motif provides excellent specificity for the αIIBβ3 integrin over other integrins115.
Residues adjacent to the KGD motif greatly affect the affinity of barbourin. Therefore, these neighbouring regions were also elucidated during the development of eptifibatide115,116. The final form of the drug consists of a heptapeptide cyclized through a disulfide bridge. Cyclization provides superior resistance to proteolysis21,116,117. Interestingly, the motif presenting the highest affinity and specificity for the αIIBβ3 integrin was neither RGD nor KGD, but a ‘hybrid’ of these, homoRGD (Fig. 5c). This surprising result indicates that there are limits to the structural versatility of protein toxins based on a small number of genetically encoded amino acids. The optimal structural solutions for molecular recognition might not be achievable through genetically encoded amino acids only, and might instead require complex and metabolic post-translational modifications that are too expensive for a secretion that a snake frequently depletes and reproduces. The versatility of synthetic chemistry presents an advantage that can be exploited to achieve affinity and specificity beyond what is observed in nature.
In addition to the drugs approved by the FDA and the EMA, other snake venom toxins have been approved for clinical use in other countries and are described below.
Batroxobin
Batroxobin (Defibrase) is a thrombin-like serine protease purified from the venom of the Brazilian lancehead pit viper (Bothrops moojeni) that induces defibrinogenation4,21,118,119. This toxin is marketed in China and Japan for the treatment of acute cerebral infarction, ischaemia caused by vascular occlusive diseases, and peripheral and microcirculation dysfunctions.
Haemocoagulase
Haemocoagulase (Reptilase)21,120 is an enzyme system purified from the venom of the common lancehead pit viper (Bothrops atrox). The enzyme system includes batroxobin and an SVMP that activates factor X, which results in anti-haemorrhagic activity. Haemocoagulase is approved for use in Japan, India and South Korea to treat internal and external haemorrhages.
α-Cobrotoxin
α-Cobrotoxin, which is purified from the venom of the Chinese cobra (Naja atra)21,121,122, is a 3FTx α-neurotoxin that binds nicotinic acetylcholine receptors at the neuromuscular junction. α-Cobrotoxin is approved for use in China as an analgesic for moderate to severe pain. However, its high bioactivity might lead to side effects, such as respiratory arrest.
Drugs in preclinical and clinical trials
Several compounds based on components from snake venom are in preclinical and clinical trials21,120,123. We focus on selected examples that are among the most promising and advanced in preclinical or clinical trials.
Anfibatide
Anfibatide is an anticoagulant C-type lectin-like protein purified from the venom of the sharp-nosed viper (Deinagkistrodon acutus). The protein is heterodimeric, comprising α-subunits and β-subunits linked by seven disulfide bonds. The anticoagulant activity of anfibatide is due to its strong binding to human platelet glycoprotein Ib α-chain (GPIbα), which inhibits the binding of GPIbα with von Willebrand factor (VWF) and thrombin124,125 (Fig. 6a). The binding of GPIbα and VWF is key to triggering platelet adhesion and thrombosis, particularly under the high shear stress conditions at sites of arterial stenosis126, which lead to myocardial infarction and stroke. In addition, anfibatide decreases thrombus volume and stability124.
Fig. 6. Drugs derived from snake venoms in clinical or preclinical trials.

a | Anfibatide (blue cartoon) is a snake C-type lectin-like protein that is predicted to bind to platelet glycoprotein Ib α-chain GPIbα (orange surface)124 at a site that partially overlaps with the GPIbα–von Willebrand factor binding surface (PDB ID: 1SQ0), thus inhibiting the association of von Willebrand factor and consequently platelet aggregation. Anfibatide is a promising anticoagulation candidate that has passed phase I clinical trials. b | Crotamine is an amphipathic and highly basic defensin that penetrates cells and is resistant to proteolysis. Crotamine exhibits antiproliferative, antinociceptive and analgesic activity in vivo upon oral administration. Cationic residues are shown as sticks and the disulfide bonds are shown in yellow. c | Dendroaspis natriuretic peptide (DNP) from the eastern green mamba (ochre tube with the disulfide bond in yellow) bound to the dimeric particulate guanylyl cyclase A receptor (shown as a lime surface and a green transparent cartoon) (PDB ID: 7BRI). Cenderitide is a natriuretic peptide chimaera resulting from the fusion of human C-type natriuretic peptide (CNP) to DNP and co-activates both DNP and CNP transmembrane receptors. d | The three-finger toxins mambalgin-1 and mambalgin-2 bind to the acid-sensing ion channels 1a and 1b, locking the channels in the closed state and impairing their function, with an analgesic effect as potent as that of morphine but with much lower toxicity in rodents. The complex of mambalgin-1 (green) with the transmembrane (light yellow) acid-sensing ion channel 1a (violet) is shown (PDB ID: 7CFT). The mambalgins are promising scaffolds for the development of a new generation of analgesics.
Recombinant anfibatide was produced at a pilot scale in yeast127, avoiding issues relating to quality control and the limited supply of snake venom. Anfibatide might become the first drug to target GPIbα, which would be a game-changer for anticoagulant therapy, as anfibatide does not interfere with haemostasis and thus does not seem to cause the haemorrhages that currently marketed drugs do. So far, anfibatide has passed phase I clinical trials124.
Crotamine
Another toxin with tremendous therapeutic potential is crotamine. This toxin is a small defensin purified from the venom of some populations of the South American neotropical rattlesnake (Crotalus durissus)128. Although the venom is very toxic, crotamine has low myotoxicity and neurotoxicity129. Crotamine is a very basic (with a pI of 10.3 and a charge of +8) amphipathic 42-residue peptide with three disulfide bridges and structural folds similar to those of human α-defensins and β-defensins60,130 (Fig. 6b).
Crotamine is a cell-penetrating peptide that is rapidly internalized into almost all cell types131. The primary cytotoxicity mechanism is accumulation in and disruption of lysosomes132. Crotamine has very high selectivity for actively proliferating cells133, such as cancer cells, making it a promising antitumour agent132. Its anti-melanoma activity was demonstrated in mice134 without toxicity to healthy cells and it can even be administered orally129,135 owing to its excellent resistance to proteolysis and its cell-penetrating ability.
Among other bioactivities (Table 1), crotamine also exhibits antinociceptive activity (and is 500 times more potent than morphine (mol mol–1))136 and anti-inflammatory activity in in vivo mouse models and upon oral administration129,135 without toxic side effects. Furthermore, its chemical and recombinant syntheses were recently reported137–139, which are fundamental steps required for upscaling crotamine production. Thus, the therapeutic future of crotamine looks promising.
Cenderitide
Cenderitide is a natriuretic peptide based on one purified from the venom of the eastern green mamba (Dendroaspis angusticeps) and is under clinical trials for the treatment of heart failure55,140.
Natriuretic peptides are regulators of body fluid volume and induce natriuresis, diuresis, vasodilation and hypotension, as well as inhibiting fibrosis, among other bioactivities. Natriuresis and diuresis are essential for the treatment of heart failure141. Three natriuretic peptides (atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C-type natriuretic peptide (CNP)) are endogenous to humans. They are small peptides of 28, 32 and 22 (or 53) residues, respectively, with a highly conserved 17-residue cyclic structure (Fig. 6c). Natriuretic peptides exert their effects by activating the particulate guanylyl cyclase (pGC)-A and pGC-B transmembrane receptors. ANP and BNP activate pGC-A, whereas CNP activates pGC-B55. Recombinant ANP and BNP were approved for use in Japan (1995) and the USA (2001), respectively, as a therapy for acute decompensated heart failure. However, later studies suggested that their efficacy is questionable142,143.
Snake venoms are a rich natriuretic peptide source, as hypotension leads to a rapid loss of consciousness in their prey. One of the first natriuretic peptides discovered in snake venom was the 38-residue Dendroaspis natriuretic peptide (DNP) from the venom of the eastern green mamba (Fig. 6c). DNP activates pGC-A and is as potent as ANP but is more resistant to metabolic ring opening. Cenderitide is a DNP–CNP chimaera that results from the addition of the 15 C-terminal residues of DNP to the C-terminal residue of CNP. It was designed to have the ability to co-activate both pGC receptors. Clinical trials have shown that receptor co-activation gives cenderitide natriuretic, diuretic and, possibly, anti-fibrotic activities without the undesirable hypotension effect of ANP and BNP. Cenderitide is safe and well tolerated in people with stable chronic heart failure144,145.
Mambalgin-1 and mambalgin-2
Mambalgins are 57-residue members of the 3FTx family purified from the venom of the black mamba that inhibit isoforms 1a and 1b of the acid-sensing ion channels (ASICs)146. ASICs are Na+ transporters activated by a decrease in the extracellular pH147. These channels, expressed in nociceptive neurons, have a central role in pain pathways and other critical pathophysiological processes, such as ischaemic strokes and tumour growth148. Mambalgins inhibit ASIC1a and ASIC1b in the central and peripheral nervous systems with nanomolar affinity, both in vitro (rat and human) and in vivo (rat)146,149,150. In rodents, administration of mambalgins into the peripheral or central nervous systems strongly abolishes acute and inflammatory pain146,151,152, with an analgesic effect as potent as that of morphine but with much less tolerance and without the respiratory arrest typical of morphine and toxic side effects146. Thus, mambalgins represent molecular scaffolds for a new generation of strong, non-toxic analgesics.
Mambalgin-1 and mambagalin-2 have been chemically synthesized and their structures determined149,150. These mambalgins represent a new family of 3FTx; they share the core of a typical 3FTx but with short first and third fingers and an elongated middle finger. The structure of the human ASIC1a channel, both free and bound to mambalgin-1, was determined in 2020 (ref.153). Experimental and computational studies154 used this structure to refine earlier proposals for the working model of ASIC inhibition, that is, the locking of the channel in the closed state (Fig. 6d). The recent wealth of activity and structural data have laid a solid foundation for the structure-based rational design of mambalgin analogues with favourable delivery routes.
Toxins targeting SARS-CoV-2 virus
We conclude this section with a review of early-stage in vitro tests of toxins that target the SARS-CoV-2 virus, which have directed considerable attention to the medicinal potential of snake venom.
Given the well documented antiviral and antimicrobial activity of snake venom PLA2s4,21,73,123,155,156, the activity of eight snake venom PLA2s against SARS-CoV-2 was tested in Vero cells156. The PLA2s were purified from the venoms of the banded krait (Bungarus fasciatus), the steppe viper (Vipera renardi) and the Nikolsky’s viper (Vipera nikolskii). The most active was the heterodimeric PLA2 HDP-2 from the Nikolsky’s viper, which exhibited nanomolar virucidal activity. The phospholipolytic activity of HDP-2 probably destroyed the viral envelope. HDP-2 also inhibited virus–host cell fusion. Direct interaction between the catalytically active subunit of HDP-2, HDP-2P, and the essential cellular ACE2 receptor was confirmed by surface plasmon resonance. All the tested PLA2s exhibit low cytotoxicity156.
Small peptides derived from the C-terminus of the myotoxin bothropstoxin-I (KKYRYHLKPFCKK), a PLA2-like protein purified from the venom of the Brazilian pit viper jararacussu (Bothrops jararacussu), have shown antimicrobial activity against Gram-positive, Gram-negative and multidrug-resistant bacterial strains157. Moreover, these peptides plus several analogues were tested against SARS-CoV-2 in Vero cells158. Three peptide dimers showed notable activity and selectivity against SARS-CoV-2. In addition, their cytotoxicity was low. The peptides targeted the viral papain-like cysteine protease with low, micromolar potency (with a binding affinity of 0.9–7 μM). Viral papain-like cysteine protease is an attractive SARS-CoV-2 target owing to its fundamental role in the cleavage and processing of viral polyproteins159.
Thus, although these studies are still in their infancy, the use of snake venom to treat SARS-CoV-2 infection is of increasing interest.
A drug repertoire from snake venom
It is evident that snake venom possesses immense therapeutic potential but that it is far from being fully exploited. This observation raises questions regarding the challenges that need to be overcome to transform snake venom into a drug repertoire.
Identifying drug candidates
Characterizing the toxins in the proteome of each species is fundamental, and this task has been facilitated by various technological advances. Snakes are the animals for which this characterization is most advanced, in part owing to a large amount of venom each individual produces compared with that of smaller animals, such as scorpions, spiders, centipedes or cone snails, whose peptide-based venoms are also promising from a therapeutic perspective3,4,20. However, the chemical structural diversity found in an animal venom is frequently less vast than that of large chemical compound libraries160–164, such as ZINC162, which is a widely used and ever-growing database that contains more than nine hundred million drug-like compounds. Pharmaceutical companies invest vast sums of capital in maintaining and expanding massive, private compound libraries. These infrastructures aim to fulfil a basic need of modern drug discovery: chemical diversity. In other words, if the goal is to conduct high-throughput ligand screening for a target of interest, it will not be easy to justify that the use of animal venoms is advantageous over the more extensive and more diverse traditional chemical libraries.
The advantage of animal venom toxins is their high, specific and inherent bioactivity, which enables a drug-candidate toxin to be chosen on the basis of a previously observed bioactivity and not through high-throughput screening. The selection process has to be rational, not automated. It is easy to glimpse the wealth of diseases we can eventually treat by looking at the summarized list of bioactivities reported in Table 1. Nevertheless, this approach implies the need for flexibility in the pharmaceutical industry, which is currently focusing on automation164.
There are two paths for developing drugs from snake venoms: the use of toxins without modifications or the design of small synthetic compounds that mimic the recognition motifs of toxins, which are called toxinomimetics.
The use of unmodified snake toxins has not been very successful. Neither the FDA nor the EMA has approved batroxobin, haemocoagulase or cobrotoxin. However, the FDA and the EMA have approved unmodified peptide toxins from the venom of other animals4, such as bivalirudin (a toxin from the medicinal leech used to prevent coagulation during surgery)165, ziconotide (a toxin from the magic cone snail used to treat chronic pain)96 and exenatide (a toxin from the Gila monster used to treat type 2 diabetes)166.
The use of unmodified toxins as prescription drugs comes at a cost: it hinders their administration, stability and large-scale production. To circumvent these issues, toxinomimecry, a technique that involves complex, rational transformations of the toxin core, has been successful for deriving drugs from snake venoms, as shown by the development of captopril and its analogues, as well as the development of tirofiban and eptifibatide. This path is not always preferable to the use of unmodified toxins. The preferable path is simply the one that provides more efficient, safer and cheaper drugs.
Routes of administration
As most toxins are peptides or proteins, their administration is usually problematic. Despite considerable resistance to proteolysis, owing to the numerous disulfide bridges within most toxins34,167, oral administration is generally inefficient, in part owing to difficulties in crossing cell membranes. This issue is evident in the lack of oral bioavailability of most snake-venom-based drugs21. Snake venom toxins are usually efficient in vivo upon parenteral injection, which is not surprising considering that they have evolved to be bioactive when injected through the snake’s fangs into their predators and prey.
Intravenous administration is generally the least invasive administration route that works, constraining the appeal of toxins for drug discovery. The cost and complexity of their large-scale synthesis, extraction and purification or heterologous expression is a further constraint. Toxinomimicry is a possible way to overcome these problems by using the toxin as a reporter rather than a drug. In this strategy, the role of the toxin is to reveal the molecular determinants of activity and specificity for the target, which are then mimicked as extensively as possible with a small synthetic molecule that is affordable to synthesize and orally bioavailable.
Target identification
The realization of the full power of toxinomimicry requires the resolution of the structures of toxin–target complexes of interest. Identification of the targets of haemotoxins in the well known coagulation cascade has been quite successful168; conversely, identification of the targets of neurotoxins in the less understood and more complex human neurological system has proved more challenging20. Venom toxins are probably the best lead sources for drugs directed at ion channels4, which are central targets in modern drug discovery, accounting for almost a fifth of today’s total drug targets169. However, the functions of many of these channels are poorly understood, if not uncharacterized. Their molecular structures are often unknown and challenging to determine. The many isoforms of ion channels further complicate their selective targeting169.
If a toxin binds an unknown target, identification of the target is challenging owing to the technical difficulty of screening target pools. For example, myotoxic PLA2s possess a region named the ‘pharmacologic site’ that binds a membrane protein prevalent in the sarcolemma. The binding of PLA2s to such a membrane protein narrows its biodistribution, focusing its hydrolytic action on muscle tissues170,171. However, even after many years of study, the identity of the myotoxic PLA2 pharmacologic target is still unknown.
When target identification is successful, determining the target structure and the target–toxin complex is still challenging. Knowing the 3D structure of the complex is a requisite to the understanding of molecular recognition, without which the design of toxinomimetics within a structure-based paradigm is not possible. In this case, toxinomimicry has to resort to a ligand-based paradigm, supported by measurements of ligand affinity for toxin mutants, which is less efficient than structure-based drug design because it is rooted in less molecular information.
Computational chemistry in toxinomimicry
The path forward should entail, at least in part, a much deeper involvement of computational chemistry. The increase in computational power allows for the more exact implementation of physical principles, which, together with the greater involvement of deep learning and artificial intelligence, is powering advances in computational fields important for snake-venom-based chemistry and drug discovery, such as protein homology modelling172–177; protein–protein docking178–185; computational mutagenesis, in particular alanine scanning186–191; and the determination of enzymatic mechanisms88–91. Computational chemistry can thus have a decisive role in speeding up the process of drug discovery based on snake venom toxins.
Computational chemistry can intervene whenever a toxin with the bioactivity of interest acts on an unknown target. Today, it is possible to assemble a database of biological targets for which the molecular structures have been determined by X-ray or cryogenic electron microscopy and homology modelling and then to screen the database according to toxin–target affinity. In several cases, the uncertainties associated with homology modelling172 and docking178 do not allow for the identification of a single and robust target. Nevertheless, computational chemistry reduces the target pool to a set small enough to be feasible for experimental testing.
It is also challenging to determine target–toxin complex geometries with atomic-level accuracy through computation alone178, particularly when modelled structures are involved. Despite this, computational chemistry can narrow down the target and toxin regions that contact each other to the point at which experimental mutagenesis (and other techniques) can be applied to provide the final atomic-level information. As an example, computational and experimental methodologies were used together to clarify the mechanism by which mambalgins inhibit ASICs146,154.
In summary, high-level computational chemistry has the power to advance target identification and target–toxin structural determination if conducted in synergy with experiments; together they could facilitate either the use of unmodified toxins or the modelling of toxin-based small ligands. For the latter, traditional medicinal chemistry can be employed to reduce the toxins into small, synthetic, bioavailable molecules while keeping most of the determinants for recognition and affinity. This approach follows the ‘captopril way’, which was a lesson of success at every level in drug discovery.
Conclusion
In modern Western civilization, the snake represents deceit and triggers both fascination and fear. However, ancient civilizations respected the snake owing to the healing power of its venom. It is becoming evident that the ancients were right, as the venom of this splendid animal is an extraordinary library of bioactive compounds that has great medicinal potential.
Our survey of hundreds of studies conducted over the past 15 years reveals the diversity in the chemical composition of snake venom, which is increasingly being explored for the development of drugs. Efforts to elucidate the chemical reactivity of the principal toxins within venom is helping to increase understanding of how toxins act on their prey targets, and how one can engineer toxin action to achieve a therapeutic goal. Furthermore, understanding of venom chemistry allows for the rational design of transition-state small-molecule analogue inhibitors for primary enzymatic toxins that are today the most promising candidates for replacing the difficult-to-manage and expensive antibody-based treatments for snakebite envenoming. The molecular recognition features of snake venom toxins are also being explored at a molecular level. As we have emphasized, these recognition motifs can be mimicked by small-molecule drugs directed to the toxin’s targets, which is a promising approach for developing cheap, stable, easily scalable and orally available medicines in drug discovery.
The drugs already approved and under development derived from snake venom demonstrate that toxic bioactivity can be transformed into a therapy for the right disease. Large toxin molecules can be redesigned and reduced to their recognition motifs for oral delivery while maintaining affinity and specificity. Of the many drugs in preclinical development, mambalgins in particular reflect the contrast between their therapeutic promise (in this case, to relieve pain) and their origin from one of the most feared snakes on the planet.
In terms of the future of venom-based drug development, we assert that toxinomimicry is an exciting alternative and a complement to the use of unmodified toxins. Furthermore, computational chemistry, which is still underused in the field, can accelerate the understanding of snake venom chemistry and hence the development of new drugs. We hope that this Review will inspire a new generation of scientists to explore and realize the immense potential of snake venoms.
Supplementary information
Acknowledgements
The authors acknowledge financial support from FCT/MCTES — the Portuguese Foundation for Science and Technology — through project PTDC/QUI-OUT/1401/2020 and from the Associate Laboratory for Green Chemistry (LAQV), which is financed by FCT/MCTES within the scope of project UIDB/50006/2020. M.F.V. acknowledges FCT/MCTES for PhD grant SFRH/BD/119206/2016. The authors thank the technological platforms network of the Oswaldo Cruz Foundation (FIOCRUZ) for the support and financing of the services provided by the Flow Cytometry and the Bioprospection and Molecular Interaction facilities/FIOCRUZ Rondônia and o Programa de Excelência em Pesquisa (PROEP) da FIOCRUZ Rondônia. The authors also thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Estado de Rondônia (FAPERO) and Instituto Nacional de Ciência e Tecnologia em Epidemiologia da Amazônia Ocidental (INCT-EpiAmO) from Brazil for financial support.
Glossary
- Neurotoxicity
The ability of a substance to negatively affect the structure or function of the central or peripheral nervous system.
- Haemotoxicity
The ability of a substance to negatively affect the cardiovascular system or disrupt haemostasis.
- Cytotoxicity
The ability of a substance to negatively affect the structure or function of cells.
- Toxins
Toxic compounds produced by a living organism or a virus.
- Elapidae
Family of >300 venomous snakes with fixed front fangs. Their venom is often neurotoxic. This family includes the mambas, cobras, coral snakes and most Australian snakes, among others.
- Viperidae
Family of >300 venomous heavy-body snakes with long, retractable front fangs. Their venom is frequently haemotoxic and cytotoxic. This family includes Old World vipers, rattlesnakes and lanceheads, among others.
- Medically important snakes
Snake species that cause notable morbidity and mortality. This classification depends on the venom toxicity, the frequency of snake–human interactions, the aggressiveness of the snake and the health-care facilities.
- Myotoxicity
Cytotoxicity specifically directed to myocytes (muscle cells).
- C-type lectins
Superfamily of >1,000 proteins, most of which bind carbohydrates in a Ca2+-dependent manner. The proteins share a C-type lectin-like domain in their carbohydrate-binding region. In snake venoms, they are haemotoxic.
- C-type lectin-like proteins
A protein family whose members feature a domain with the C-type lectin fold, which lacks critical structural elements to recognize and bind sugars. In snake venoms, these proteins are haemotoxic.
- Sarcolemma
Specialized type of cell plasma membrane that surrounds muscle cells. It is frequently the target of snake venom myotoxins.
- Lysosomes
Membrane-bound organelle containing digestive, hydrolytic enzymes whose function is primarily the degradation of macromolecules, old cell parts and microorganisms. Lysosomes represent the waste disposal of a cell.
- Neuromuscular junction
A specialized synapse established between a motor neuron and a muscle fibre through which signals for muscle contraction are transmitted.
- Fibrinogen
A protein complex in the plasma of vertebrates that is enzymatically and sequentially converted into fibrin and a fibrin-based blood clot. Fibrinogen is responsible for stopping bleeding from blood vessels.
- Intrathecal administration
Invasive drug administration by injection through the skull or the spine, allowing the drug to reach the cerebrospinal fluid, and thus the brain, without crossing the blood–brain barrier.
- Natriuresis
The process of excretion of sodium in the urine.
Author contributions
All authors contributed to the discussion of content and edited the article prior to submission. P.A.F., A.L.O., M.F.V. and M.J.R. also researched the data and contributed to the writing of the article.
Peer review
Peer review information
Nature Reviews Chemistry thanks Juan J. Calvete and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
The Reptile Database: http://www.reptile-database.org/
Supplementary information
The online version contains supplementary material available at 10.1038/s41570-022-00393-7.
References
- 1.Holford M, Daly M, King GF, Norton RS. Venoms to the rescue. Science. 2018;361:842–844. doi: 10.1126/science.aau7761. [DOI] [PubMed] [Google Scholar]
- 2.Casewell NR, Wüster W, Vonk FJ, Harrison RA, Fry BG. Complex cocktails: the evolutionary novelty of venoms. Trends Ecol. Evol. 2013;28:219–229. doi: 10.1016/j.tree.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 3.King GF. Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011;11:1469–1484. doi: 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- 4.Herzig V, et al. Animal toxins — nature’s evolutionary-refined toolkit for basic research and drug discovery. Biochem. Pharmacol. 2020;181:114096. doi: 10.1016/j.bcp.2020.114096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pineda SS, et al. Structural venomics reveals evolution of a complex venom by duplication and diversification of an ancient peptide-encoding gene. Proc. Natl Acad. Sci. USA. 2020;117:11399–11408. doi: 10.1073/pnas.1914536117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cid-Uribe JI, Veytia-Bucheli JI, Romero-Gutierrez T, Ortiz E, Possani LD. Scorpion venomics: a 2019 overview. Expert Rev. Proteom. 2020;17:67–83. doi: 10.1080/14789450.2020.1705158. [DOI] [PubMed] [Google Scholar]
- 7.Tasoulis T, Isbister GK. A review and database of snake venom proteomes. Toxins. 2017;9:290. doi: 10.3390/toxins9090290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casewell NR, et al. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc. Natl Acad. Sci. USA. 2014;111:9205–9210. doi: 10.1073/pnas.1405484111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massey DJ, et al. Venom variability and envenoming severity outcomes of the Crotalus scutulatus scutulatus (Mojave rattlesnake) from southern Arizona. J. Proteom. 2012;75:2576–87. doi: 10.1016/j.jprot.2012.02.035. [DOI] [PubMed] [Google Scholar]
- 10.Casewell NR, Jackson TNW, Laustsen AH, Sunagar K. Causes and consequences of snake venom variation. Trends Pharmacol. Sci. 2020;41:570–581. doi: 10.1016/j.tips.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durban J, et al. Integrated venomics and venom gland transcriptome analysis of juvenile and adult Mexican rattlesnakes Crotalus simus, C. tzabcan, and C. culminatus revealed miRNA-modulated ontogenetic shifts. J. Proteome Res. 2017;16:3370–3390. doi: 10.1021/acs.jproteome.7b00414. [DOI] [PubMed] [Google Scholar]
- 12.Pla D, et al. Phylovenomics of Daboia russelii across the Indian subcontinent. Bioactivities and comparative in vivo neutralization and in vitro third-generation antivenomics of antivenoms against venoms from India, Bangladesh and Sri Lanka. J. Proteom. 2019;207:103443. doi: 10.1016/j.jprot.2019.103443. [DOI] [PubMed] [Google Scholar]
- 13.Senji Laxme RR, et al. Beyond the ‘big four’: venom profiling of the medically important yet neglected Indian snakes reveals disturbing antivenom deficiencies. PLoS Negl. Trop. Dis. 2019;13:e0007899. doi: 10.1371/journal.pntd.0007899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chanda A, Kalita B, Patra A, Senevirathne WDST, Mukherjee AK. Proteomic analysis and antivenomics study of Western India Naja naja venom: correlation between venom composition and clinical manifestations of cobra bite in this region. Expert Rev. Proteom. 2018;16:171–184. doi: 10.1080/14789450.2019.1559735. [DOI] [PubMed] [Google Scholar]
- 15.Tasoulis, T., Pukala, T. L. & Isbister, G. K. Investigating toxin diversity and abundance in snake venom proteomes. Front. Pharmacol. (2022). A review of the proteomic methods used to separate and quantify snake venom toxins, comparing their merits and limitations. [DOI] [PMC free article] [PubMed]
- 16.Editorial. Snake-bite envenoming: a priority neglected tropical disease. Lancet. 2017;390:2. doi: 10.1016/S0140-6736(17)31751-8. [DOI] [PubMed] [Google Scholar]
- 17.Gutierrez JM, et al. Snakebite envenoming. Nat. Rev. Dis. Primers. 2017;3:17063. doi: 10.1038/nrdp.2017.63. [DOI] [PubMed] [Google Scholar]
- 18.Williams D. The Global Snake Bite Initiative: an antidote for snake bite. Lancet. 2010;375:89–91. doi: 10.1016/S0140-6736(09)61159-4. [DOI] [PubMed] [Google Scholar]
- 19.Kasturiratne A, et al. The global burden of snakebite: a literature analysis and modelling based on regional estimates of envenoming and deaths. PLoS Med. 2008;5:1591–1604. doi: 10.1371/journal.pmed.0050218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDermott A. Venom back in vogue as a wellspring for drug candidates. Proc. Natl Acad. Sci. USA. 2020;117:10100–10104. doi: 10.1073/pnas.2004486117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bordon K, et al. From animal poisons and venoms to medicines: achievements, challenges and perspectives in drug discovery. Front. Pharmacol. 2020;11:1132. doi: 10.3389/fphar.2020.01132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almeida JRR, et al. Snake venom peptides and low mass proteins: molecular tools and therapeutic agents. Curr. Med. Chem. 2017;24:3254–3282. doi: 10.2174/0929867323666161028155611. [DOI] [PubMed] [Google Scholar]
- 23.Fry BG. From genome to “venome”: molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 2005;15:403–420. doi: 10.1101/gr.3228405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ojeda PG, et al. Computational studies of snake venom toxins. Toxins. 2018;10:8. doi: 10.3390/toxins10010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calvete JJ, Sanz L, Angulo Y, Lomonte B, Gutiérrez JM. Venoms, venomics, antivenomics. FEBS Lett. 2009;583:1736–1743. doi: 10.1016/j.febslet.2009.03.029. [DOI] [PubMed] [Google Scholar]
- 26.Modahl CM, Brahma RK, Koh CY, Shioi N, Kini RM. Omics technologies for profiling toxin diversity and evolution in snake venom: impacts on the discovery of therapeutic and diagnostic agents. Annu. Rev. Anim. Biosci. 2020;8:91–116. doi: 10.1146/annurev-animal-021419-083626. [DOI] [PubMed] [Google Scholar]
- 27.The Uniprot Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:480–489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simoes-Silva R, et al. Snake venom, a natural library of new potential therapeutic molecules: challenges and current perspectives. Curr. Pharm. Biotechnol. 2018;19:308–335. doi: 10.2174/1389201019666180620111025. [DOI] [PubMed] [Google Scholar]
- 29.Calvete JJ. Next-generation snake venomics: protein-locus resolution through venom proteome decomplexation. Expert Rev. Proteom. 2014;11:315–329. doi: 10.1586/14789450.2014.900447. [DOI] [PubMed] [Google Scholar]
- 30.Brahma RK, McCleary RJR, Kini RM, Doley R. Venom gland transcriptomics for identifying, cataloging, and characterizing venom proteins in snakes. Toxicon. 2015;93:1–10. doi: 10.1016/j.toxicon.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Liu L. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutiérrez JM, Lomonte B. Phospholipases A2: unveiling the secrets of a functionally versatile group of snake venom toxins. Toxicon. 2013;62:27–39. doi: 10.1016/j.toxicon.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang TS, et al. Enzymatic toxins from snake venom: structural characterization and mechanism of catalysis. FEBS J. 2011;278:4544–4576. doi: 10.1111/j.1742-4658.2011.08115.x. [DOI] [PubMed] [Google Scholar]
- 35.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 2006;1761:1246–59. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Ferraz CR, et al. Multifunctional toxins in snake venoms and therapeutic implications: from pain to hemorrhage and necrosis. Front. Ecol. Evol. 2019;7:218. doi: 10.3389/fevo.2019.00218. [DOI] [Google Scholar]
- 37.Kini RM, Koh CY. Snake venom three-finger toxins and their potential in drug development targeting cardiovascular diseases. Biochem. Pharmacol. 2020;181:114105. doi: 10.1016/j.bcp.2020.114105. [DOI] [PubMed] [Google Scholar]
- 38.Fry BG, et al. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 2003;57:110–129. doi: 10.1007/s00239-003-2461-2. [DOI] [PubMed] [Google Scholar]
- 39.Kini RM, Doley R. Structure, function and evolution of three-finger toxins: mini proteins with multiple targets. Toxicon. 2010;56:855–867. doi: 10.1016/j.toxicon.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Olaoba OT, Karina dos Santos P, Selistre-de-Araujo HS, Ferreira de Souza DH. Snake venom metalloproteinases (SVMPs): a structure–function update. Toxicon X. 2020;7:100052. doi: 10.1016/j.toxcx.2020.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gutiérrez JM, Escalante T, Rucavado A, Herrera C. Hemorrhage caused by snake venom metalloproteinases: a journey of discovery and understanding. Toxins. 2016;8:93. doi: 10.3390/toxins8040093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda S. ADAM and ADAMTS family proteins and snake venom metalloproteinases: a structural overview. Toxins. 2016;8:155. doi: 10.3390/toxins8050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ullah A, et al. Thrombin-like enzymes from snake venom: structural characterization and mechanism of action. Int. J. Biol. Macromol. 2018;114:788–811. doi: 10.1016/j.ijbiomac.2018.03.164. [DOI] [PubMed] [Google Scholar]
- 44.Hiu JJ, Yap MKK. Cytotoxicity of snake venom enzymatic toxins: phospholipase A(2) and l-amino acid oxidase. Biochem. Soc. Trans. 2020;48:719–731. doi: 10.1042/BST20200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan KK, Bay BH, Gopalakrishnakone P. l-amino acid oxidase from snake venom and its anticancer potential. Toxicon. 2018;144:7–13. doi: 10.1016/j.toxicon.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Paloschi MV. An update on potential molecular mechanisms underlying the actions of snake venom l-amino acid oxidases (LAAOs) Curr. Med. Chem. 2018;25:2520–2530. doi: 10.2174/0929867324666171109114125. [DOI] [PubMed] [Google Scholar]
- 47.Ullah A. Structure–function studies and mechanism of action of snake venom l-amino acid oxidases. Front. Pharmacol. 2020;11:110. doi: 10.3389/fphar.2020.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inagaki, H. in Snake Venoms (eds Gopalakrishnakone, P., Inagaki, H., Vogel, C.-V., Mukherjee, A. K. & Rashed Rahmy, T.) (Springer, 2017).
- 49.Markland FS, Swenson S. Snake venom metalloproteinases. Toxicon. 2013;62:3–18. doi: 10.1016/j.toxicon.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Serrano SM, Maroun RC. Snake venom serine proteinases: sequence homology vs. substrate specificity, a paradox to be solved. Toxicon. 2005;45:1115–1132. doi: 10.1016/j.toxicon.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 51.Serrano SM. The long road of research on snake venom serine proteinases. Toxicon. 2013;62:19–26. doi: 10.1016/j.toxicon.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Arlinghaus FT, Eble JA. C-type lectin-like proteins from snake venoms. Toxicon. 2012;60:512–519. doi: 10.1016/j.toxicon.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Morita T. Structures and functions of snake venom CLPs (C-type lectin-like proteins) with anticoagulant-, procoagulant-, and platelet-modulating activities. Toxicon. 2005;45:1099–1114. doi: 10.1016/j.toxicon.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 54.Lu Q, Navdaev A, Clemetson JM, Clemetson KJ. Snake venom C-type lectins interacting with platelet receptors. Structure–function relationships and effects on haemostasis. Toxicon. 2005;45:1089–1098. doi: 10.1016/j.toxicon.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 55.Vink S. Natriuretic peptide drug leads from snake venom. Toxicon. 2012;59:434–445. doi: 10.1016/j.toxicon.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Sridharan S, Kini RM, Richards AM. Venom natriuretic peptides guide the design of heart failure therapeutics. Pharmacol. Res. 2020;155:104687. doi: 10.1016/j.phrs.2020.104687. [DOI] [PubMed] [Google Scholar]
- 57.Munawar A. Snake venom peptides: tools of biodiscovery. Toxins. 2018;10:474. doi: 10.3390/toxins10110474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laustsen AH, Lomonte B, Lohse B, Fernández J, Gutiérrez JM. Unveiling the nature of black mamba (Dendroaspis polylepis) venom through venomics and antivenom immunoprofiling: identification of key toxin targets for antivenom development. J. Proteom. 2015;119:126–142. doi: 10.1016/j.jprot.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 59.Damm M, Hempel BF, Nalbantsoy A, Süssmuth RD. Comprehensive snake venomics of the Okinawa Habu pit viper, Protobothrops flavoviridis, by complementary mass spectrometry-guided approaches. Molecules. 2018;23:1893. doi: 10.3390/molecules23081893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coronado MA, et al. Structure of the polypeptide crotamine from the Brazilian rattlesnake Crotalus durissus terrificus. Acta Crystallogr. D. 2013;69:1958–1964. doi: 10.1107/S0907444913018003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Falcao CB, Radis-Baptista G. Crotamine and crotalicidin, membrane active peptides from Crotalus durissus terrificus rattlesnake venom, and their structurally-minimized fragments for applications in medicine and biotechnology. Peptides. 2020;126:170234. doi: 10.1016/j.peptides.2019.170234. [DOI] [PubMed] [Google Scholar]
- 62.Slotta KH, Fraenkel-Conrat H. Two active proteins from rattlesnake venom. Nature. 1938;13:213–213. doi: 10.1038/142213a0. [DOI] [Google Scholar]
- 63.Berg OG, Gelb MH, Tsai MD, Jain MK. Interfacial enzymology: the secreted phospholipase A(2)-paradigm. Chem. Rev. 2001;101:2613–54. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]
- 64.Tsai YC, Yu BZ, Wang YZ, Chen J, Jain MK. Desolvation map of the i-face of phospholipase A2. Biochim. Biophys. Acta. 2006;1758:653–665. doi: 10.1016/j.bbamem.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Bahnson BJ. Structure, function and interfacial allosterism in phospholipase A2: insight from the anion-assisted dimer. Arch. Biochem. Biophys. 2005;433:96–106. doi: 10.1016/j.abb.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 66.Scott DL, et al. Interfacial catalysis: the mechanism of phospholipase A2. Science. 1990;250:1541–1546. doi: 10.1126/science.2274785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sérgio S, Ramos MJ, Lim C, Fernandes PA. Relationship between enzyme/substrate properties and enzyme efficiency in hydrolases. ACS Catal. 2015;5:5877–5887. doi: 10.1021/acscatal.5b00923. [DOI] [Google Scholar]
- 68.Sousa SF, et al. Activation free energy, substrate binding free energy, and enzyme efficiency fall in a very narrow range of values for most enzymes. ACS Catal. 2020;10:8444–8453. doi: 10.1021/acscatal.0c01947. [DOI] [Google Scholar]
- 69.Resende LM, et al. Structural, enzymatic and pharmacological profiles of AplTX-II — a basic sPLA2 (D49) isolated from the Agkistrodon piscivorus leucostoma snake venom. Int. J. Biol. Macromol. 2021;175:572–585. doi: 10.1016/j.ijbiomac.2021.01.187. [DOI] [PubMed] [Google Scholar]
- 70.Lomonte B, Rangel J. Snake venom Lys49 myotoxins: from phospholipases A(2) to non-enzymatic membrane disruptors. Toxicon. 2012;60:520–30. doi: 10.1016/j.toxicon.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 71.Fernández J, et al. Muscle phospholipid hydrolysis by Bothrops asper Asp49 and Lys49 phospholipase A2 myotoxins — distinct mechanisms of action. FEBS J. 2013;280:3878–3886. doi: 10.1111/febs.12386. [DOI] [PubMed] [Google Scholar]
- 72.Almeida JR, et al. CoaTx-II, a new dimeric Lys49 phospholipase A2 from Crotalus oreganus abyssus snake venom with bactericidal potential: insights into its structure and biological roles. Toxicon. 2016;120:147–58. doi: 10.1016/j.toxicon.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 73.Almeida JR, et al. Harnessing snake venom phospholipases A(2) to novel approaches for overcoming antibiotic resistance. Drug Dev. Res. 2019;80:68–85. doi: 10.1002/ddr.21456. [DOI] [PubMed] [Google Scholar]
- 74.Almeida JR, et al. A novel synthetic peptide inspired on Lys49 phospholipase A2 from Crotalus oreganus abyssus snake venom active against multidrug-resistant clinical isolates. Eur. J. Med. Chem. 2018;149:248–256. doi: 10.1016/j.ejmech.2018.02.055. [DOI] [PubMed] [Google Scholar]
- 75.Kwong PD, McDonald NQ, Sigler PB, Hendrickson WA. Structure of β2-bungarotoxin — potassium channel binding by kunitz modules and targeted phospholipase action. Structure. 1995;3:1109–1119. doi: 10.1016/S0969-2126(01)00246-5. [DOI] [PubMed] [Google Scholar]
- 76.Rowan EG. What does β-bungarotoxin do at the neuromuscular junction? Toxicon. 2001;39:107–118. doi: 10.1016/S0041-0101(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 77.Doley R, Kini RM. Protein complexes in snake venom. Cell. Mol. Life Sci. 2009;66:2851–2871. doi: 10.1007/s00018-009-0050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kini RM, Koh CY. Metalloproteases affecting blood coagulation, fibrinolysis and platelet aggregation from snake venoms: definition and nomenclature of interaction sites. Toxins. 2016;8:284. doi: 10.3390/toxins8100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanchez EF, Flores-Ortiz RJ, Alvarenga VG, Eble JA. Direct fibrinolytic snake venom metalloproteinases affecting hemostasis: structural, biochemical features and therapeutic potential. Toxins. 2017;9:392. doi: 10.3390/toxins9120392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bledzka K, Smyth SS, Plow EF. Integrin αIIbβ3 from discovery to efficacious therapeutic target. Circ. Res. 2013;112:1189–1200. doi: 10.1161/CIRCRESAHA.112.300570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takeda S, Igarashi T, Mori H. Crystal structure of RVV-X: an example of evolutionary gain of specificity by ADAM proteinases. FEBS Lett. 2007;581:5859–5864. doi: 10.1016/j.febslet.2007.11.062. [DOI] [PubMed] [Google Scholar]
- 82.Lingott T, Schleberger C, Gutiérrez JM, Merfort I. High-resolution crystal structure of the snake venom metalloproteinase BaP1 complexed with a peptidomimetic: Insight into inhibitor binding. Biochemistry. 2009;48:6166–6174. doi: 10.1021/bi9002315. [DOI] [PubMed] [Google Scholar]
- 83.Akao PK, et al. Structural studies of BmooMPα-I, a non-hemorrhagic metalloproteinase from Bothrops moojeni venom. Toxicon. 2010;55:361–368. doi: 10.1016/j.toxicon.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 84.Boldrini-França J, et al. Beyond hemostasis: a snake venom serine protease with potassium channel blocking and potential antitumor activities. Sci. Rep. 2020;10:4476. doi: 10.1038/s41598-020-61258-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mackessy, S. P. in Toxins and Hemostasis (eds Kini, R., Clemetson, K., Markland, F., McLane, M. & Morita, T.) 519–557 (Springer, 2010).
- 86.Vaiyapuri, S., Thiyagarajan, N., Hutchinson, E. G. & Gibbins, J. M. Sequence and phylogenetic analysis of viper venom serine proteases. Bioinformation8, 763–772 2012). [DOI] [PMC free article] [PubMed]
- 87.Kurtović T, et al. VaSP1, catalytically active serine proteinase from Vipera ammodytes ammodytes venom with unconventional active site triad. Toxicon. 2014;77:93–104. doi: 10.1016/j.toxicon.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 88.Sousa SF, et al. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wiley Interdisc. Rev. Mol. Sci. 2017;7:1281. [Google Scholar]
- 89.Chung LW, et al. The ONIOM method and its applications. Chem. Rev. 2015;115:5678–5796. doi: 10.1021/cr5004419. [DOI] [PubMed] [Google Scholar]
- 90.Amaro RE, Mulholland AJ. Multiscale methods in drug design bridge chemical and biological complexity in the search for cures. Nat. Rev. Chem. 2018;2:148. doi: 10.1038/s41570-018-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Himo F. Recent trends in quantum chemical modeling of enzymatic reactions. J. Am. Chem. Soc. 2017;139:6780–6786. doi: 10.1021/jacs.7b02671. [DOI] [PubMed] [Google Scholar]
- 92.Estevão-Costa MI, Sanz-Soler R, Johanningmeier B, Eble JA. Snake venom components in medicine: from the symbolic rod of Asclepius to tangible medical research and application. Int. J. Biochem. Cell Biol. 2018;104:94–113. doi: 10.1016/j.biocel.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 93.Marsh NA. Diagnostic uses of snake venom. Haemostasis. 2001;31:211–217. doi: 10.1159/000048065. [DOI] [PubMed] [Google Scholar]
- 94.Francischetti, I. M. B. & Gil, M. R. in Transfusion Medicine and Hemostasis (eds Shaz, B. H., Hillyer, C. D. & Reyes Gil, M.) 969–975 (Elsevier, 2019).
- 95.Schmidtko A, Lötsch J, Freynhagen R, Geisslinger G. Ziconotide for treatment of severe chronic pain. Lancet. 2010;375:1569–1577. doi: 10.1016/S0140-6736(10)60354-6. [DOI] [PubMed] [Google Scholar]
- 96.Miljanich GP. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- 97.Rocha ESM, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am. J. Physiol. 1949;156:261–273. doi: 10.1152/ajplegacy.1949.156.2.261. [DOI] [PubMed] [Google Scholar]
- 98.Ferreira SH. A bradykinin-potentiating factor (BPF) present in the venom of Bothrops jararaca. Br. J. Pharmacol. Chemother. 1965;24:163–169. doi: 10.1111/j.1476-5381.1965.tb02091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferreira SH, Bartelt DC, Greene LJ. Isolation of bradykinin-potentiating peptides from Bothrops jararaca venom. Biochemistry. 1970;9:2583–2593. doi: 10.1021/bi00815a005. [DOI] [PubMed] [Google Scholar]
- 100.McCleary RJR, Kini RM. Non-enzymatic proteins from snake venoms: a gold mine of pharmacological tools and drug leads. Toxicon. 2013;62:56–74. doi: 10.1016/j.toxicon.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 101.Ferreira SH, Greene LJ, Alabaster VA, Bakhle YS, Vane JR. Activity of various fractions of bradykinin potentiating factor against angiotensin-I converting enzyme. Nature. 1970;225:379–380. doi: 10.1038/225379a0. [DOI] [PubMed] [Google Scholar]
- 102.Cushman DW, Ondetti MA. History of the design of captopril and related inhibitors of angiotensin converting enzyme. Hypertension. 1991;17:589–592. doi: 10.1161/01.HYP.17.4.589. [DOI] [PubMed] [Google Scholar]
- 103.Bryan J. From snake venom to ACE inhibitor — the discovery and rise of captopril. Pharm. J. 2009;282:455–456. [Google Scholar]
- 104.Patchett AA. The chemistry of enalapril. Br. J. Clin. Pharmacol. 1984;18:201–207. doi: 10.1111/j.1365-2125.1984.tb02599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Acharya KR, Sturrock ED, Riordan JF, Ehlers MRW. ACE revisited: a new target for structure-based drug design. Nat. Rev. Drug Discov. 2003;2:891–902. doi: 10.1038/nrd1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Acharya, G., Wang, W., Vavilala, D. T., Mukherji, M. & Lee, C. H. in Advanced Drug Delivery (eds Mitra, A. K., Lee, C. H. & Cheng, K.) 341–364 (Wiley, 2014).
- 107.Lazarovici P, Marcinkiewicz C, Lelkes PI. From snake venom’s disintegrins and C-type lectins to anti-platelet drugs. Toxins. 2019;11:303. doi: 10.3390/toxins11050303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Topol EJ, Byzova TV, Plow EF. Platelet GPIIb-IIIa blockers. Lancet. 1999;353:227–231. doi: 10.1016/S0140-6736(98)11086-3. [DOI] [PubMed] [Google Scholar]
- 109.Lang SH, et al. Treatment with tirofiban for acute coronary syndrome (ACS): a systematic review and network analysis. Curr. Med. Res. Opin. 2012;28:351–370. doi: 10.1185/03007995.2012.657299. [DOI] [PubMed] [Google Scholar]
- 110.Barrett JS, et al. Pharmacokinetics and pharmacodynamics of MK-383, a selective nonpeptide platelet glycoprotein-IIb/IIIa receptor antagonist, in healthy men. Clin. Pharmacol. Ther. 1994;56:377–388. doi: 10.1038/clpt.1994.152. [DOI] [PubMed] [Google Scholar]
- 111.Gan ZR, Gould RJ, Jacobs JW, Friedman PA, Polokoff MA. Echistatin — a potent platelet-aggregation inhibitor from the venom of the viper Echis carinatus. J. Biol. Chem. 1988;263:19827–19832. doi: 10.1016/S0021-9258(19)77710-2. [DOI] [PubMed] [Google Scholar]
- 112.Hartman GD. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992;35:4640–4642. doi: 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
- 113.Van Drie JH. Computer-aided drug design: the next 20 years. J. Comput. Aided Mol. Des. 2007;21:591–601. doi: 10.1007/s10822-007-9142-y. [DOI] [PubMed] [Google Scholar]
- 114.Scarborough RM, et al. Design of potent and specific integrin antagonists — peptide antagonists with high specificity for glycoprotein-IIb–IIIa. J. Biol. Chem. 1993;268:1066–1073. doi: 10.1016/S0021-9258(18)54042-4. [DOI] [PubMed] [Google Scholar]
- 115.Scarborough RM, et al. Barbourin — a GPIIb–IIIa-specific integrin antagonist from the venom of Sistrurus m. barbouri. J. Biol. Chem. 1991;266:9359–9362. doi: 10.1016/S0021-9258(18)92826-7. [DOI] [PubMed] [Google Scholar]
- 116.Scarborough RM. Development of eptifibatide. Am. Heart J. 1999;138:1093–1104. doi: 10.1016/S0002-8703(99)70075-X. [DOI] [PubMed] [Google Scholar]
- 117.O’Shea JC, Tcheng JE. Eptifibatide: a potent inhibitor of the platelet receptor integrin glycoprotein IIb/IIIa. Expert Opin. Pharmacother. 2002;3:1199–1210. doi: 10.1517/14656566.3.8.1199. [DOI] [PubMed] [Google Scholar]
- 118.Masuda H. Batroxobin accelerated tissue repair via neutrophil extracellular trap regulation and defibrinogenation in a murine ischemic hindlimb model. PLoS ONE. 2019;14:e0220898. doi: 10.1371/journal.pone.0220898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vu TT. Batroxobin binds fibrin with higher affinity and promotes clot expansion to a greater extent than thrombin. J. Biol. Chem. 2013;288:16862–16871. doi: 10.1074/jbc.M113.464750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Waheed H, Moin SF, Choudhary MI. Snake venom: from deadly toxins to life-saving therapeutics. Curr. Med. Chem. 2017;24:1874–1891. doi: 10.2174/0929867324666170605091546. [DOI] [PubMed] [Google Scholar]
- 121.Gazerani P, Cairns BE. Venom-based biotoxins as potential analgesics. Expert Rev. Neurother. 2014;14:1261–1274. doi: 10.1586/14737175.2014.962518. [DOI] [PubMed] [Google Scholar]
- 122.Lin F, Reid PF, Qin Z-H. Cobrotoxin could be an effective therapeutic for COVID-19. Acta Pharmacol. Sin. 2020;41:1258–1260. doi: 10.1038/s41401-020-00501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pérez-Peinado C, et al. Hitchhiking with nature: snake venom peptides to fight cancer and superbugs. Toxins. 2020;12:255. doi: 10.3390/toxins12040255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li BX, et al. In vitro assessment and phase I randomized clinical trial of anfibatide, a snake-venom-derived anti-thrombotic agent targeting human platelet GPIbα. Sci. Rep. 2021;11:11663. doi: 10.1038/s41598-021-91165-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gao Y, et al. Crystal structure of agkisacucetin, a GPIb-binding snake C-type lectin that inhibits platelet adhesion and aggregation. Proteins. 2012;80:1707–1711. doi: 10.1002/prot.24060. [DOI] [PubMed] [Google Scholar]
- 126.Jackson SP. The growing complexity of platelet aggregation. Blood. 2007;109:5087–5095. doi: 10.1182/blood-2006-12-027698. [DOI] [PubMed] [Google Scholar]
- 127.Guo Y, et al. Balancing the expression and production of a heterodimeric protein: recombinant agkisacutacin as a novel antithrombotic drug candidate. Sci. Rep. 2015;5:11730. doi: 10.1038/srep11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tasima LJ, et al. Crotamine in Crotalus durissus: distribution according to subspecies and geographic origin, in captivity or nature. J. Venom. Anim. Toxins Incl. Trop. Dis. 2020;26:20190053. doi: 10.1590/1678-9199-jvatitd-2019-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moreira LA, et al. Acute toxicity, antinociceptive, and anti-inflammatory activities of the orally administered crotamine in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2021;394:1703–1711. doi: 10.1007/s00210-021-02103-4. [DOI] [PubMed] [Google Scholar]
- 130.Nicastro G, et al. Solution structure of crotamine, a Na+ channel affecting toxin from Crotalus durissus terrificus venom. Eur. J. Biochem. 2003;270:1969–1979. doi: 10.1046/j.1432-1033.2003.03563.x. [DOI] [PubMed] [Google Scholar]
- 131.Kerkis A, et al. Crotamine is a novel cell-penetrating protein from the venom of rattlesnake Crotalus durissus terrificus. FASEB J. 2004;18:1407–1409. doi: 10.1096/fj.03-1459fje. [DOI] [PubMed] [Google Scholar]
- 132.Hayashi MAF, et al. Cytotoxic effects of crotamine are mediated through lysosomal membrane permeabilization. Toxicon. 2008;52:508–517. doi: 10.1016/j.toxicon.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 133.Kerkis A, Hayashi MAF, Yamane T, Kerkis I. Properties of cell penetrating peptides (CPPs) IUBMB Life. 2006;58:7–13. doi: 10.1080/15216540500494508. [DOI] [PubMed] [Google Scholar]
- 134.Pereira A, et al. Crotamine toxicity and efficacy in mouse models of melanoma. Expert Opin. Investig. Drugs. 2011;20:1189–1200. doi: 10.1517/13543784.2011.602064. [DOI] [PubMed] [Google Scholar]
- 135.Campeiro JD, et al. Oral treatment with a rattlesnake native polypeptide crotamine efficiently inhibits the tumor growth with no potential toxicity for the host animal and with suggestive positive effects on animal metabolic profile. Amino Acids. 2018;50:267–278. doi: 10.1007/s00726-017-2513-3. [DOI] [PubMed] [Google Scholar]
- 136.Mancin AC, et al. The analgesic activity of crotamine, a neurotoxin from Crotalus durissus terrificus (South American rattlesnake) venom: a biochemical and pharmacological study. Toxicon. 1998;36:1927–37. doi: 10.1016/S0041-0101(98)00117-2. [DOI] [PubMed] [Google Scholar]
- 137.de Carvalho Porta L, et al. Biophysical and pharmacological characterization of a full-length synthetic analog of the antitumor polypeptide crotamine. J. Mol. Med. 2020;98:1561–1571. doi: 10.1007/s00109-020-01975-y. [DOI] [PubMed] [Google Scholar]
- 138.Mambelli-Lisboa NC, Sciani JM, Silva ARBPD, Kerkis I. Co-localization of crotamine with internal membranes and accentuated accumulation in tumor cells. Molecules. 2018;23:968. doi: 10.3390/molecules23040968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Park JY, et al. Antinociceptive and anti-inflammatory effects of recombinant crotamine in mouse models of pain. Toxins. 2021;13:707. doi: 10.3390/toxins13100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schweitz H, Vigne P, Moinier D, Frelin C, Lazdunski M. A new member of the natriuretic peptide family is present in the venom of the green mamba (Dendroaspis angusticeps) J. Biol. Chem. 1992;267:13928–13932. doi: 10.1016/S0021-9258(19)49658-0. [DOI] [PubMed] [Google Scholar]
- 141.Volpe M, Rubattu S, Burnett J. Natriuretic peptides in cardiovascular diseases: current use and perspectives. Eur. Heart J. 2014;35:419–425. doi: 10.1093/eurheartj/eht466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.O’Connor CMM, et al. Effect of nesiritide in patients with acute decompensated heart failure. N. Engl. J. Med. 2011;365:32–43. doi: 10.1056/NEJMoa1100171. [DOI] [PubMed] [Google Scholar]
- 143.Matsue Y, et al. Carperitide is associated with increased in-hospital mortality in acute heart failure: a propensity score-matched analysis. J. Card. Fail. 2015;21:859–864. doi: 10.1016/j.cardfail.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 144.Ichiki T, Dzhoyashvili N, Burnett JC., Jr Natriuretic peptide based therapeutics for heart failure: cenderitide. A novel first-in-class designer natriuretic peptide. Int. J. Cardiol. 2019;281:166–171. doi: 10.1016/j.ijcard.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kawakami R, et al. A human study to evaluate safety, tolerability, and cyclic GMP activating properties of cenderitide in subjects with stable chronic heart failure. Clin. Pharmacol. Ther. 2018;104:546–552. doi: 10.1002/cpt.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Diochot S, et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature. 2012;490:552–557. doi: 10.1038/nature11494. [DOI] [PubMed] [Google Scholar]
- 147.Yoder N, Yoshioka C, Gouaux E. Gating mechanisms of acid-sensing ion channels. Nature. 2018;555:397–401. doi: 10.1038/nature25782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013;14:461–471. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schroeder CI, et al. Chemical synthesis, 3D structure, and ASIC binding site of the toxin mambalgin-2. Angew. Chem. Int. Ed. 2014;53:1017–1020. doi: 10.1002/anie.201308898. [DOI] [PubMed] [Google Scholar]
- 150.Mourier G, et al. Mambalgin-1 pain-relieving peptide, stepwise solid-phase synthesis, crystal structure, and functional domain for acid-sensing ion channel 1a Inhibition. J. Biol. Chem. 2015;291:2616–2629. doi: 10.1074/jbc.M115.702373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Diochot S, et al. Analgesic effects of mambalgin peptide inhibitors of acid-sensing ion channels in inflammatory and neuropathic pain. Pain. 2016;157:552–559. doi: 10.1097/j.pain.0000000000000397. [DOI] [PubMed] [Google Scholar]
- 152.Verkest C, et al. Effects of systemic inhibitors of acid-sensing ion channels 1 (ASIC1) against acute and chronic mechanical allodynia in a rodent model of migraine. Br. J. Pharmacol. 2018;175:4154–4166. doi: 10.1111/bph.14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sun DM, et al. Structural insights into human acid-sensing ion channel 1a inhibition by snake toxin mambalgin1. eLife. 2020;9:57096. doi: 10.7554/eLife.57096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Salinas M, et al. Mambalgin-1 pain-relieving peptide locks the hinge between α4 and α5 helices to inhibit rat acid-sensing ion channel 1a. Neuropharmacology. 2021;185:108453. doi: 10.1016/j.neuropharm.2021.108453. [DOI] [PubMed] [Google Scholar]
- 155.Yacoub T. Antimicrobials from venomous animals: an overview. Molecules. 2020;25:2402. doi: 10.3390/molecules25102402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Siniavin AE, et al. Snake venom phospholipase A2s exhibit strong virucidal activity against SARS-CoV-2 and inhibit the viral spike glycoprotein interaction with ACE2. Cell Mol. Life Sci. 2021;78:7777–7794. doi: 10.1007/s00018-021-03985-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Santos-Filho NA, et al. Antibacterial activity of the non-cytotoxic peptide (p-BthTX-I)2 and its serum degradation product against multidrug-resistant bacteria. Molecules. 2017;22:1898. doi: 10.3390/molecules22111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Freire MCLC, et al. Non-toxic dimeric peptides derived from the bothropstoxin-I are potent SARS-CoV-2 and papain-like protease inhibitors. Molecules. 2021;26:4896. doi: 10.3390/molecules26164896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Domling A, Gao L. Chemistry and biology of SARS-CoV-2. Chem. 2020;6:1283–1295. doi: 10.1016/j.chempr.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yang J, et al. Freely accessible chemical database resources of compounds for in silico drug discovery. Curr. Med. Chem. 2019;26:7581–7597. doi: 10.2174/0929867325666180508100436. [DOI] [PubMed] [Google Scholar]
- 161.Mendez D, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47:930–940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sterling T, Irwin JJ. ZINC 15-ligand discovery for everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.van Hilten N, et al. Virtual compound libraries in computer-assisted drug discovery. J. Chem. Inf. Model. 2019;59:644–651. doi: 10.1021/acs.jcim.8b00737. [DOI] [PubMed] [Google Scholar]
- 164.Schneider G. Automating drug discovery. Nat. Rev. Drug Discov. 2018;17:97–113. doi: 10.1038/nrd.2017.232. [DOI] [PubMed] [Google Scholar]
- 165.Warkentin TE, Greinacher A, Koster A. Bivalirudin. Thromb. Haemost. 2008;99:830–9. doi: 10.1160/TH07-10-0644. [DOI] [PubMed] [Google Scholar]
- 166.Barnett A. Exenatide. Expert Opin. Pharmacother. 2007;8:2593–2608. doi: 10.1517/14656566.8.15.2593. [DOI] [PubMed] [Google Scholar]
- 167.King, G. F. in Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics (ed. King, G. F.) 37–79 (Royal Society of Chemistry, 2015).
- 168.Kini RM. Toxins in thrombosis and haemostasis: potential beyond imagination. J. Thromb. Haemost. 2011;9:195–208. doi: 10.1111/j.1538-7836.2011.04279.x. [DOI] [PubMed] [Google Scholar]
- 169.Santos R, et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kini RM, Evans HJ. A model to explain the pharmacological effects of snake venom phospholipases A2. Toxicon. 1989;27:613–35. doi: 10.1016/0041-0101(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 171.Kini RM. Excitement ahead: structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon. 2003;42:827–840. doi: 10.1016/j.toxicon.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 172.Croll TI, Sammito MD, Kryshtafovych A, Read RJ. Evaluation of template-based modeling in CASP13. Proteins Struct. Funct. Bioinform. 2019;87:1113–1127. doi: 10.1002/prot.25800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Alford RF, et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017;13:3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Das R, Baker D. Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 175.Wang Y, Virtanen J, Xue Z, Zhang Y. I-TASSER-MR: automated molecular replacement for distant-homology proteins using iterative fragment assembly and progressive sequence truncation. Nucleic Acids Res. 2017;45:429–434. doi: 10.1093/nar/gkx349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics. 2020;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lensink MF, Nadzirin N, Velankar S, Wodak SJ. Modeling protein–protein, protein–peptide, and protein–oligosaccharide complexes: CAPRI. Proteins. 2020;88:916–938. doi: 10.1002/prot.25870. [DOI] [PubMed] [Google Scholar]
- 179.Moreira IS, Fernandes PA, Ramos MJ. Protein–protein docking dealing with the unknown. J. Comput. Chem. 2010;31:317–342. doi: 10.1002/jcc.21276. [DOI] [PubMed] [Google Scholar]
- 180.Simões ICM, et al. Properties that rank protein–protein docking poses with high accuracy. Phys. Chem. Chem. Phys. 2018;20:20927–20942. doi: 10.1039/C8CP03888K. [DOI] [PubMed] [Google Scholar]
- 181.Moreira IS, Martins JM, Coimbra JTS, Ramos MJ, Fernandes PA. A new scoring function for protein–protein docking that identifies native structures with unprecedented accuracy. Phys. Chem. Chem. Phys. 2015;17:2378–2387. doi: 10.1039/C4CP04688A. [DOI] [PubMed] [Google Scholar]
- 182.Quignot C, et al. InterEvDock2: an expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs. Nucleic Acids Res. 2018;46:408–416. doi: 10.1093/nar/gky377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kozakov D, et al. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017;12:255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Park T, Baek M, Lee H, Seok C. GalaxyTongDock: symmetric and asymmetric ab initio protein–protein docking web server with improved energy parameters. J. Comput. Chem. 2019;40:2413–2417. doi: 10.1002/jcc.25874. [DOI] [PubMed] [Google Scholar]
- 185.Zundert GCP, et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 186.Moreira IS, Fernandes PA, Ramos MJ. Hot spots — a review of the protein–protein interface determinant amino-acid residues. Proteins Struct. Funct. Bioinform. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 187.Martins SA, et al. Computational alanine scanning mutagenesis: MM-PBSA vs TI. J. Chem. Theory Comput. 2013;9:1311–1319. doi: 10.1021/ct4000372. [DOI] [PubMed] [Google Scholar]
- 188.Simões ICM, Costa IPD, Coimbra JTS, Ramos MJ, Fernandes PA. New parameters for higher accuracy in the computation of binding free energy differences upon alanine scanning mutagenesis on protein–protein interfaces. J. Chem. Inf. Model. 2017;57:60–72. doi: 10.1021/acs.jcim.6b00378. [DOI] [PubMed] [Google Scholar]
- 189.Geng C, Xue LC, Roel-Touris J, Bonvin AMJJ. Finding the ΔΔG spot: are predictors of binding affinity changes upon mutations in protein–protein interactions ready for it? Wiley Interdiscip. Rev. Mol. Sci. 2019;9:1410. [Google Scholar]
- 190.Barlow KA, et al. Flex ddG: Rosetta ensemble-based estimation of changes in protein–protein binding affinity upon mutation. J. Phys. Chem. B. 2018;122:5389–5399. doi: 10.1021/acs.jpcb.7b11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein–protein complexes. Proc. Natl Acad. Sci. USA. 2002;99:14116–21. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sribar J, et al. The neurotoxic secreted phospholipase A2 from the Vipera a. ammodytes venom targets cytochrome c oxidase in neuronal mitochondria. Sci. Rep. 2019;9:283. doi: 10.1038/s41598-018-36461-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Meenakshisundaram R, Sweni S, Thirumalaikolundusubramanian P. Hypothesis of snake and insect venoms against human immunodeficiency virus: a review. AIDS Res. Ther. 2009;6:25. doi: 10.1186/1742-6405-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fenard D, et al. Secreted phospholipases A2, a new class of HIV inhibitors that block virus entry into host cells. J. Clin. Invest. 1999;104:611–618. doi: 10.1172/JCI6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Muller VDM, et al. Crotoxin and phospholipases A2 from Crotalus durissus terrificus showed antiviral activity against dengue and yellow fever viruses. Toxicon. 2012;59:507–15. doi: 10.1016/j.toxicon.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 196.Ahmed NK, Tennant KD, Markland FS, LACZ JP. Biochemical characteristics of fibrolase, a fibrinolytic protease from snake venom. Haemostasis. 1990;20:147–154. doi: 10.1159/000216121. [DOI] [PubMed] [Google Scholar]
- 197.Boldrini-Franca J, Pinheiro-Junior EL, Arantes EC. Functional and biological insights of rCollinein-1, a recombinant serine protease from Crotalus durissus collilineatus. J. Venom. Anim. Toxins Incl. Trop. Dis. 2019;25:147118. doi: 10.1590/1678-9199-jvatitd-1471-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Funk C, Gmür J, Herold R, Straub PW. Reptilase-R — a new reagent in blood coagulation. Br. J. Haematol. 1971;21:43–52. doi: 10.1111/j.1365-2141.1971.tb03415.x. [DOI] [PubMed] [Google Scholar]
- 199.Graziano F, et al. Autologous fibrin sealant (Vivostat(R)) in the neurosurgical practice. Part I: intracranial surgical procedure. Surg. Neurol. Int. 2015;6:77. doi: 10.4103/2152-7806.156871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Graziano F, Maugeri R, Basile L, Meccio F, Iacopino DG. Aulogous fibrin sealant (Vivostat(R)) in the neurosurgical practice. Part II: vertebro-spinal procedures. Surg. Neurol. Int. 2016;7:77–82. doi: 10.4103/2152-7806.174894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Koivula K, Rondinelli S, Nasman J. The three-finger toxin MTα is a selective α2B-adrenoceptor antagonist. Toxicon. 2010;56:440–447. doi: 10.1016/j.toxicon.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 202.Barnwal B, et al. Ringhalexin from Hemachatus haemachatus: a novel inhibitor of extrinsic tenase complex. Sci. Rep. 2016;6:25935. doi: 10.1038/srep25935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Banerjee Y, et al. Biophysical characterization of anticoagulant hemextin AB complex from the venom of snake Hemachatus haemachatus. Biophys. J. 2007;93:3963–76. doi: 10.1529/biophysj.106.100164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chanda C, Sarkar A, Sistla S, Chakrabarty D. Anti-platelet activity of a three-finger toxin (3FTx) from Indian monocled cobra (Naja kaouthia) venom. Biochem. Biophys. Res. Commun. 2013;441:550–554. doi: 10.1016/j.bbrc.2013.10.125. [DOI] [PubMed] [Google Scholar]
- 205.Fernandez-Gomez R, et al. Growth inhibition of Trypanosoma cruzi and Leishmania donovani infantum by different snake venoms — preliminary identification of proteins from Cerastes cerastes venom which interact with the parasites. Toxicon. 1994;32:875–882. doi: 10.1016/0041-0101(94)90366-2. [DOI] [PubMed] [Google Scholar]
- 206.Costa Torres AF, et al. Antibacterial and antiparasitic effects of Bothrops marajoensis venom and its fractions: phospholipase A2 and l-amino acid oxidase. Toxicon. 2010;55:795–804. doi: 10.1016/j.toxicon.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 207.Sakurai Y, et al. Anticoagulant activity of M-LAO, l-amino acid oxidase purified from Agkistrodon halys blomhoffii, through selective inhibition of factor IX. Biochim. Biophys. Acta. 2003;1649:51–57. doi: 10.1016/S1570-9639(03)00157-2. [DOI] [PubMed] [Google Scholar]
- 208.Adade CM, et al. Crovirin, a snake venom cysteine-rich secretory protein (CRISP) with promising activity against trypanosomes and Leishmania. PLoS Negl. Trop. Dis. 2014;8:3252. doi: 10.1371/journal.pntd.0003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Badari JC, Díaz-Roa A, Rocha MMT, Mendonça RZ, Silva Junior PID. Patagonin-CRISP: antimicrobial activity and source of antimicrobial molecules in Duvernoy’s gland secretion (Philodryas patagoniensis snake) Front. Pharmacol. 2021;11:586705. doi: 10.3389/fphar.2020.586705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tadokoro T, Modahl CM, Maenaka K, Aoki-Shioi N. Cysteine-rich secretory proteins (CRISPs) from venomous snakes: an overview of the functional diversity in a large and underappreciated superfamily. Toxins. 2020;12:175. doi: 10.3390/toxins12030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Assafim M, et al. Exploiting the antithrombotic effect of the (pro)thrombin inhibitor bothrojaracin. Toxicon. 2016;119:46–51. doi: 10.1016/j.toxicon.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 212.Shen D-K, et al. Ca2+-induced binding of anticoagulation factor II from the venom of Agkistrodon acutus with factor IX. Biopolymers. 2012;97:818–824. doi: 10.1002/bip.22078. [DOI] [PubMed] [Google Scholar]
- 213.Zhang Y, et al. Anticoagulation factor I, a snaclec (snake C-type lectin) from Agkistrodon acutus venom binds to FIX as well as FX: Ca2+ induced binding data. Toxicon. 2012;59:718–723. doi: 10.1016/j.toxicon.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 214.Rabelo LFG, et al. Alternagin-C, a disintegrin-like protein from Bothrops alternatus venom, attenuates inflammation and angiogenesis and stimulates collagen deposition of sponge-induced fibrovascular tissue in mice. Int. J. Biol. Macromol. 2019;140:653–660. doi: 10.1016/j.ijbiomac.2019.08.171. [DOI] [PubMed] [Google Scholar]
- 215.Gilchrist IC. Platelet glycoprotein IIb/IIIa inhibitors in percutaneous coronary intervention: focus on the pharmacokinetic–pharmacodynamic relationships of eptifibatide. Clin. Pharmacokinet. 2003;42:703–720. doi: 10.2165/00003088-200342080-00001. [DOI] [PubMed] [Google Scholar]
- 216.Lucena SE, et al. Anti-invasive and anti-adhesive activities of a recombinant disintegrin, r-viridistatin 2, derived from the Prairie rattlesnake (Crotalus viridis viridis) Toxicon. 2012;60:31–9. doi: 10.1016/j.toxicon.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kuo YJ, Chung CH, Huang TF. From discovery of snake venom disintegrins to a safer therapeutic antithrombotic agent. Toxins. 2019;11:372. doi: 10.3390/toxins11070372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Cesar PHS, Braga MA, Trento MVC, Menaldo DL, Marcussi S. Snake venom disintegrins: an overview of their interaction with integrins. Curr. Drug Targets. 2019;20:465–477. doi: 10.2174/1389450119666181022154737. [DOI] [PubMed] [Google Scholar]
- 219.Calvete JJ. The continuing saga of snake venom disintegrins. Toxicon. 2013;62:40–49. doi: 10.1016/j.toxicon.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 220.Ciolek J, et al. Green mamba peptide targets type-2 vasopressin receptor against polycystic kidney disease. Proc. Natl Acad. Sci. USA. 2017;114:7154–7159. doi: 10.1073/pnas.1620454114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Colombo AL, et al. Effects of the natural peptide crotamine from a South American rattlesnake on Candida auris, an emergent multidrug antifungal resistant human pathogen. Biomolecules. 2019;9:205. doi: 10.3390/biom9060205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.El Chamy Maluf S, et al. Inhibition of malaria parasite Plasmodium falciparum development by crotamine, a cell penetrating peptide from the snake venom. Peptides. 2016;78:11–16. doi: 10.1016/j.peptides.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 223.Dal Mas C, et al. Anthelmintic effects of a cationic toxin from a South American rattlesnake venom. Toxicon. 2016;116:49–55. doi: 10.1016/j.toxicon.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 224.Macedo SRA, et al. Biodegradable microparticles containing crotamine isolated from Crotalus durissus terrificus display antileishmanial activity in vitro. Pharmacology. 2015;95:78–86. doi: 10.1159/000371391. [DOI] [PubMed] [Google Scholar]
- 225.Mackessy S. P. (ed.) Handbook of Venoms and Toxins of Reptiles (CRC, 2021). This book describes the composition, bioactivity, pathophysiology and medicinal applications of snake venom.
- 226.Salvador GHM, dos Santos JI, Lomonte B, Fontes MRM. Crystal structure of a phospholipase A2 from Bothrops asper venom: insights into a new putative “myotoxic cluster”. Biochimie. 2017;133:95–102. doi: 10.1016/j.biochi.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 227.Murakami MT, et al. Inhibition of myotoxic activity of Bothrops asper myotoxin II by the anti-trypanosomal drug suramin. J. Mol. Biol. 2005;350:416–426. doi: 10.1016/j.jmb.2005.04.072. [DOI] [PubMed] [Google Scholar]
- 228.Kalita B, Mackessy SP, Mukherjee AK. Proteomic analysis reveals geographic variation in venom composition of Russell’s viper in the Indian subcontinent: implications for clinical manifestations post-envenomation and antivenom treatment. Expert Rev. Proteomics. 2018;15:837–849. doi: 10.1080/14789450.2018.1528150. [DOI] [PubMed] [Google Scholar]
- 229.Favaloro EJ. The Russell viper venom time (RVVT) test for investigation of lupus anticoagulant (LA) Am. J. Hematol. 2019;94:1290–1296. doi: 10.1002/ajh.25606. [DOI] [PubMed] [Google Scholar]
- 230.Chen H-S, Tsai H-Y, Wang Y-M, Tsai I-H. P-III hemorrhagic metalloproteinases from Russell’s viper venom: cloning, characterization, phylogenetic and functional site analyses. Biochimie. 2008;90:1486–1498. doi: 10.1016/j.biochi.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 231.Nakayama D, Ben Ammar Y, Miyata T, Takeda S. Structural basis of coagulation factor V recognition for cleavage by RVV-V. FEBS Lett. 2011;585:3020–3025. doi: 10.1016/j.febslet.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 232.You W-K, et al. Functional characterization of recombinant batroxobin, a snake venom thrombin-like enzyme, expressed from Pichia pastoris. FEBS Lett. 2004;571:67–73. doi: 10.1016/j.febslet.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 233.Sousa LF, et al. Functional proteomic analyses of Bothrops atrox venom reveals phenotypes associated with habitat variation in the Amazon. J. Proteom. 2017;159:32–46. doi: 10.1016/j.jprot.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 234.Núñez V, et al. Snake venomics and antivenomics of Bothrops atrox venoms from Colombia and the Amazon regions of Brazil, Perú and Ecuador suggest the occurrence of geographic variation of venom phenotype by a trend towards paedomorphism. J. Proteom. 2009;73:57–78. doi: 10.1016/j.jprot.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 235.Calvete JJ, et al. Snake population venomics and antivenomics of Bothrops atrox: paedomorphism along its transamazonian dispersal and implications of geographic venom variability on snakebite management. J. Proteom. 2011;74:510–527. doi: 10.1016/j.jprot.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 236.Kohlhoff M, et al. Exploring the proteomes of the venoms of the Peruvian pit vipers Bothrops atrox, B. barnetti and B. pictus. J. Proteom. 2012;75:2181–2195. doi: 10.1016/j.jprot.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 237.Hatakeyama DM, et al. Venom complexity of Bothrops atrox (common lancehead) siblings. J. Venom. Anim. Toxins Incl. Trop. Dis. 2020;26:20200018. doi: 10.1590/1678-9199-jvatitd-2020-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wallnoefer HG, Lingott T, Gutiérrez JM, Merfort I, Liedl KR. Backbone flexibility controls the activity and specificity of a protein–protein interface: specificity in snake venom metalloproteases. J. Am. Chem. Soc. 2010;132:10330–10337. doi: 10.1021/ja909908y. [DOI] [PubMed] [Google Scholar]
- 239.Camacho E, Escalante T, Remans K, Gutiérrez JM, Rucavado A. Site mutation of residues in a loop surrounding the active site of a PI snake venom metalloproteinase abrogates its hemorrhagic activity. Biochem. Biophys. Res. Commun. 2019;512:859–863. doi: 10.1016/j.bbrc.2019.03.152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.