

Abstract

Multispecific biologics are an emerging class of drugs, in which antibodies and/or proteins designed to bind pharmacological targets are covalently linked or expressed as fusion proteins to increase both therapeutic efficacy and safety. Epitope mapping on the target proteins provides key information to improve the affinity and also to monitor the manufacturing process and drug stability. Solid-state NMR has been here used to identify the pattern of the residues of the programmed cell death ligand 1 (PD-L1) ectodomain that are involved in the interaction with a new multispecific biological drug. This is possible because the large size and the intrinsic flexibility of the complexes are not limiting factors for solid-state NMR.
Introduction
Drug discovery is a long and costly process that has a very low success rate. Structural biology is the game-changer for the identification and optimization of new lead compounds, but the relevance of the structural information that can be gathered is causing structural biology to emerge also for the development of biotherapeutics.1,2
As defined by international guidelines, pharmaceutical development should adhere to the Quality by Design paradigm (QbD), described by ICH Q8 (R2)3 from the European Medicine Agency (EMA) as a “systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management”. This important concept has revolutionized drug development by highlighting the importance of new analytical strategies based on advanced product and process knowledge. Developing a drug under the QbD paradigm not only aims at improving the quality and safety of pharmaceutical products but also at increasing the success rate by improving Critical Quality Attributes risk assessments, leading to more focused control strategies and release testing panels.
Monoclonal antibodies (mAbs) are, to date, the major class of biological drugs approved for the treatment of a large variety of pathologies, and new engineering solutions have solved most of the serious problems encountered in the therapeutic use of these proteins, improving the interactions with the effector cells, leading to less immunogenic molecules and allowing the selection of high-affinity species.4,5 Among these drugs, multispecific biologics obtained by fusing full-length antibodies, fragment antigen-binding (FAB), or other proteins together represent the next generation of biotherapeutics.6−12 This entire class of drugs can benefit from structural information obtained by investigating their complexes with the targets, for example, to reshape and optimize the interaction site.13,14
Structural information at the atomic level about the macromolecular complexes is routinely obtained using X-ray crystallography,15,16 much less so by NMR17,18 and, more recently, cryo-electron microscopy.19,20 However, the large molecular weight and the flexibility of fusion-derived biotherapeutics often prevent the structural characterization of their complexes with the targets. For instance, a large inherent flexibility makes it difficult to obtain crystals of diffraction quality or cryo-EM reconstruction. At the same time, the large molecular weight of these systems hampers a deep structural characterization by NMR in solution, although NMR is successfully used in the higher-order structure (HOS) assessment.21−29 Relevant and complementary information can be obtained from hydrogen–deuterium exchange coupled to mass spectrometry (HDX-MS): characterization of interaction surfaces in protein complexes is one of the strengths of this technique, but complex and extensive method optimization is needed, and data interpretation is not straightforward.30,31
Thanks to advances in the instrumentation and in sample preparation, solid-state NMR has reached sufficient maturity to start tackling systems of outstanding complexity, such as biological drugs, vaccine formulations, etc. A few years ago, a pioneering work by the group of Lewandowski reported the solid-state NMR characterization of a precipitated macromolecular complex between the first immunoglobulin binding domain of streptococcal protein G (GB1) and a full-length antibody.32 GB1 is a 6 kDa protein33 that is extensively used as a standard in solid-state NMR,34 and is reported to bind strongly to the crystallizable region fragment and weakly to the antigen-binding fragment of human immunoglobulin G. These results and previous studies on noncrystalline systems suggest that also very large macromolecular systems involving fusion-derived biologics can be characterized by solid-state NMR spectroscopy.35−62 One of the advantages of the noncrystalline samples, obtained by sedimentation or equivalently by rehydrating freeze-dried proteins,63 is the absence of crystalline (ordered) packing.45 Indeed, the shift perturbations due to the contacts among the different protein molecules are averaged over several poses with no energetic preferences and the hydration state of the biomolecules is closer to that present in solution.63,64 Therefore, a rehydrated freeze-dried material corresponds to an extremely concentrated solution of the protein, which is intrinsically comparable, for the scope of chemical shift mapping, to the diluted sample used for acquiring solution spectra.65
The observation of well-resolved spectra on a noncrystalline system of a small protein is not trivial: in our experience, noncrystalline samples of small proteins—including domains or fragments of therapeutic targets—can provide poor-quality solid-state NMR spectra63 that have discouraged so far the use of this strategy in the investigation of pharmaceutical relevant systems and in the development of biologics. Local structural inhomogeneity under magic angle spinning (MAS) conditions is among the possible reasons of the unsatisfactory quality of solid-state spectra recorded on noncrystalline samples of some small proteins. In the case of antibodies, however, since they usually bind a target with very high affinity by establishing an extensive network of interactions, a structural stabilization of the interacting protein is expected.
Programmed cell death 1 (PD-1)/programmed cell death ligand 1 (PD-L1) axis is one of the immune checkpoints that under healthy conditions promote self-tolerance and protect the host from autoimmunity.66 However, the PD-1/PD-L1 cascade is also used by several cancer cell lines to avoid the immune response by overexpressing the PD-L1 transmembrane protein on the surface.67,68 The ectodomain of PD-L1 is therefore the target for several in-use and in-development antibodies employed in the therapy of cancers overexpressing this protein.69−72 In this respect, the assignment of the target protein in complex with biotherapeutics provides the way for a structure-based approach to drug development and manufacturing.
This study explores the interaction between the PD-L1 receptor and an anti-PD-L1 biotherapeutic: an IgG1 fusion protein of about 190 kDa, composed of an extracellular domain (ECD) protein covalently linked via a flexible linker to the C-terminus of each heavy chain of an anti-PD-L1 antibody (Figure 1). Here, we show that the epitope mapping of this Fc-fusion protein on the PD-L1 ectodomain can be achieved by integrating solution and solid-state NMR studies and that the structural information obtained with our approach can be used to provide usable knowledge to develop a biotherapeutic under the Quality by Design paradigm (QbD).
Figure 1.

Schematic representation of the anti-PD-L1 fusion protein.
Methods
Expression and Purification of [U-13C, 15N] and [U-2H, 13C, 15N] PD-L1
Escherichia coli BL21 (DE3) cells were transformed with pET-21a (+) plasmid encoding PD-L1 gene (residues Ala18-Tyr134). To obtain uniformly isotopically enriched PD-L1 [U-13C, 15N], the cells were cultured in M9 Minimal Medium supplied with 3 g of 13C-glucose, 1.1 g of 15N-NH4Cl, 1 cm3 of 0.1 mg·cm–3 solution of ampicillin, 1 cm3 of 1 mg·cm–3 thiamine, 1 cm3 of 1 mg·cm–3 biotin, 1 mmol·dm–3 MgSO4, 0.3 mmol·dm–3 CaCl2, grown at 310 K, until OD600 reached 0.8, then induced with 1 mmol·dm–3 isopropyl β-d-1-thiogalactopyranoside. They were further grown at 310 K overnight and then harvested by centrifugation at 7500g (JA-10 Beckman Coulter) for 15 min at 277 K.
For uniformly isotopically enriched PD-L1 [U-2H, 13C, 15N], the cells were cultured in 2H-13C-15N-enriched medium (E. coli-OD2 rich growth media) containing 1 cm3 of 0.1 mg·cm–3 solution of ampicillin, grown at 310 K, until OD600 reached 0.6, then induced with 1 mmol·dm–3 isopropyl β-d-1-thiogalactopyranoside; all reagents were previously dissolved in 2H2O. The cells were further grown at 310 K overnight and then harvested by centrifugation at 7500g (JA-10 Beckman Coulter) for 15 min at 277 K. In all instances, the pellet was suspended, first, in 50 mmol·dm–3 Tris-HCl pH 8.0, 200 mmol·dm–3 NaCl, 10 mmol·dm–3 β-mercaptoethanol, and 10 mmol·dm–3 ethylenediaminetetraacetic acid (EDTA) (50 cm3 per dm3 of culture) and sonicated for 30 s 10 times on ice at 277 K. The suspension was centrifuged at 115,000g (Beckman Optima LE-80K Ultracentrifuge) for 40 min and the supernatant discarded. The recovered pellet was resuspended in 50 mmol·dm–3 Tris-HCl pH 8.0, 200 mmol·dm–3 NaCl, 10 mmol·dm–3 β-mercaptoethanol, 6 mol·dm–3 guanidinium chloride (25 cm3 per dm3 of culture) and newly incubated at 277 K overnight under magnetic stirring. Again, the suspension was centrifuged at 115,000g (Beckman Optima LE-80K Ultracentrifuge) for 40 min. The pellet was discarded, whereas the supernatant containing the denatured protein solution was diluted in a refolding buffer containing 0.1 mol·dm–3 Tris-HCl, pH 8.5, 1 mol·dm–3 arginine, 0.25 mmol·dm–3 reduced glutathione, and 0.25 mmol·dm–3 oxidized glutathione.73 The solution was incubated at 277 K under stirring, for 12–18 h, cleared by passing a 0.45 μm filter, and then dialyzed extensively against 10 mmol·dm–3 Tris, pH 8.0, 20 mmol·dm–3 NaCl. The protein solution was concentrated with an Amicon Stirred Cell and then purified by size exclusion chromatography on HiLoad Superdex 26/60 75pg (GE Healthcare) previously equilibrated in 0.1 mol·dm–3 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 6.8 and 20 mmol·dm–3 NaCl.
NMR Measurements
Solution NMR experiments for backbone resonance assignment [three-dimensional (3D) HNCA,74−76 HNCACB,77,78 CBCA(CO)NH,78,79 HNCO74−76] were performed on [U-13C, 15N] samples of PD-L1 (at the concentration of 150 μmol·dm–3) in the water buffer solution where the protein was more stable [10 mmol·dm–3 Tris, pH 8, 20 mmol·dm–3 NaCl, 0.1% NaN3, protease inhibitors (Roche)]. For 3D HNCACB nonuniform random sampling at 64% and compressed-sensing reconstruction was used.80 A 3D HNCA was also recorded at a lower pH [buffer: 20 mmol·dm–3 HEPES, pH 6.8, 20 mmol·dm–3 NaCl, 0.1% NaN3, protease inhibitors (Roche)] to identify a higher number of spin systems and to transfer the protein assignment to buffer conditions closer to those of the anti-PD-L1 fusion protein. All solution spectra were recorded at 298 K on Bruker AVANCE III and AVANCE NEO NMR spectrometers, operating at 1200, 950, and 900 MHz, 1H Larmor frequency (28.2, 22.3, and 21.1 T), respectively, equipped with triple-resonance cryo-probes.
Complexes of [U-15N], [U-13C, 15N] or [U-2H, 13C, 15N] PD-L1 with the anti-PD-L1 fusion protein were prepared by adding increasing aliquots of product [10 mg·cm–3 (∼50 μmol·dm–3)] to the solution of PD-L1 [50 μmol·dm–3 in 100 mmol·dm–3 HEPES, 20 mmol·dm–3 NaCl, pH 6.8] to reach the concentrations of 2.5, 5, and 7.5 μmol·dm–3 of anti-PD-L1 fusion protein. Each addition of the anti-PD-L1 fusion protein was monitored by two-dimensional (2D) 1H-15N SOFAST HMQC spectra.81 The excess of unbound PD-L1 was then purified from the complex by HiLoad Superdex 16/60 200pg gel filtration (GF) chromatography and buffer-exchanged to 1 mmol·dm–3 HEPES and 4 mmol·dm–3 NaCl. The solutions of the complexes (containing ∼10 mg of material) were freeze-dried and the materials used to pack 3.2 mm zirconia thin-wall rotors (open-ended, with bottom and top Vespel caps, Bruker Biospin). The dry samples were then rehydrated by multiple additions of Milli-Q H2O until the resolution of the one-dimensional (1D) {1H}13C CP82 (ωH = 70 kHz; ωC = 42 kHz) spectra stopped changing. Silicon plugs (courtesy of Bruker Biospin) placed below the turbine cap were used to close the rotor and preserve hydration. The complex between [U-2H, 13C, 15N] PD-L1 and anti-PD-L1 fusion protein was subsequently transferred in a 1.3 mm zirconia rotor (Bruker Biospin).
A sample of PD-L1 in the presence of a nonbinding antibody (nb-mAb) was also prepared as reference sample. Increasing aliquots of this product [25 mg cm–3 (∼170 μmol·dm–3)] to reach the concentrations of 12.5 and 25 μmol·dm–3 nb-mAb were added to the solution of [U-13C, 15N] PD-L1 [50 μmol·dm–3 in 100 mmol·dm–3 HEPES, 20 mmol·dm–3 NaCl, pH 6.8]. PD-L1 and nb-mAb were co-lyophilized, and the material (∼13.4 mg) used to fill thick walls 3.2 mm zirconia rotor. Also in this case, the dry material was rehydrated with Milli-Q H2O and the spectra acquired.
Another control sample of [U-13C, 15N] free PD-L1 was prepared by lyophilization in the presence of PEG, and spectra were acquired before and after rehydration, for reference to the SSNMR.
The SSNMR spectra of PD-L1 in the presence of mAbs were collected on a Bruker Avance III spectrometer operating at 800 MHz, 1H Larmor frequency (18.8 T, 201.2 MHz 13C Larmor frequency), equipped with a Bruker 3.2 mm Efree, and Bruker 1.3 mm NCH probe-heads. The spectra of the free protein were, instead, acquired on a Bruker Avance III 850 MHz, 1H Larmor frequency, wide-bore spectrometer (20 T, 213.6 MHz 13C Larmor frequency), equipped with a 3.2 mm DVT MAS probe head in triple-resonance mode. The spectra were recorded at 14 and 60 kHz MAS frequencies, for the 3.2 and 1.3 mm rotors, respectively, and the sample temperature was kept at ∼290 K.
Standard 13C-detected SSNMR spectra [2D 15N-13C NCA, 15N-13C NCO, and 13C-13C DARR, mixing time 50 ms]83−87 were acquired on the samples in 3.2 mm rotors, while 1H-detected SSNMR spectra [2D 15N-1H (H)NH CP-heteronuclear single quantum coherence (HSQC), 3D (H)CANH, 3D (H)CONH, and the 1H-13C 2D plane of 3D (H)(CA)CB(CA)NH]88 were acquired on the sample in 1.3 mm rotor, using the pulse sequences reported in the literature.89−92 Experimental details are reported in Tables S1 and S2. For comparison, two-dimensional carbon-detected solution NMR spectra [13C-15N CON (best-version), CACO and CBCACO]93,94 were acquired using a Bruker AVANCE NEO 700 spectrometer equipped with a triple-resonance Cryo-Probe optimized for 13C-direct detection, on a sample of free PD-L1 (50 μmol·dm–3 in 100 mmol·dm–3 MES, pH 6.8, 20 mmol·dm–3 NaCl).
All of the spectra were processed with the Bruker TopSpin 3.2 software and analyzed with the program CARA.95
Results
First, we proceeded to an extensive NMR characterization of the isolated PD-L1 ectodomain in solution and in the solid state to evaluate the quality of the spectra and to perform the backbone assignment. Isotopically enriched samples of PD-L1 ectodomain can be expressed in E. coli, while the labeling of full-length antibodies is still extremely challenging, although not impossible in principle.
NMR Characterization of the Isolated PD-L1 Ectodomain
The 2D 1H-15N HSQC of free PD-L1 in solution shows sharp and well-resolved signals, as expected for a structured low-molecular-weight protein (∼13.5 kDa). The backbone assignment of free PD-L1 was obtained from the analysis of triple-resonance spectra acquired on samples of [U-13C, 15N] PD-L1 in solution. All residues but the first three and Asp-61 could be assigned on the spectra (percentage of assignment 97%, Figure 2). In total, 114 signals could be identified and assigned for the free protein in solution. This is, to the best of our knowledge, the only available assignment of PD-L1. The assignment has been deposited on the bmrb under the accession code 51169.
Figure 2.

2D 1H 15N HSQC spectrum of the free PD-L1 in solution [at the concentration of 50 μmol·dm–3 in 10 mmol·dm–3 Tris, pH 8, 20 mmol·dm–3 NaCl, 0.1% NaN3, and with protease inhibitors (Roche)] with the assignment of the resonances reported in black. Dashed black circles indicate the missing peaks at pH 8 that conversely were assigned at pH 6.8. The spectrum was acquired on a spectrometer operating at 950 MHz and 298 K.
Then, the isolated PD-L1 ectodomain was freeze-dried and the sample was analyzed by SSNMR. As expected for a small protein, the 1D {1H}13C CP spectrum of the dry material displays broad signals (Figure S1). Also the controlled hydration of the material41,42 did not improve the quality and resolution of the spectra in the solid state (Figures S1 and S2).
NMR Analysis of PD-L1 in the Presence of the Anti-PD-L1 Fusion Protein
Samples of the PD-L1/anti-PD-L1 fusion protein complex were prepared by adding a solution of the product to solutions of the isotopically enriched PD-L1, and the titration was monitored by NMR. The addition of the anti-PD-L1 fusion protein to the solution of [U-13C, 15N] PD-L1 caused a global decrease in the intensity of the target protein’s signals in the 1D 1H and 2D 1H-15N SOFAST HMQC NMR spectra (Figures S3 and S4). This effect is due to the severe broadening of resonances resulting from the increase of the reorientation correlation time experienced by PD-L1, upon binding to the fusion protein.
Substoichiometric concentrations of the anti-PD-L1 drug were added to the PD-L1 solutions. The large PD-L1/anti-PD-L1 fusion protein complex was then purified from the residual free PD-L1 protein by gel filtration (GF) chromatography and characterized by solution NMR. Only a few signals (Gln/Asn side chains and the C-terminal HN), corresponding to atoms that preserve internal mobility after binding to the anti-PD-L1 fusion protein, were observed in the 2D 1H-15N SOFAST HMQC NMR spectrum acquired after GF (Figure S5), while signals of the free PD-L1 protein were completely disappeared.
Then, the PD-L1/anti-PD-L1 fusion protein complex was freeze-dried and analyzed by SSNMR. The 1D {1H}13C CP spectrum collected on the freeze-dried sample was of poor quality. However, the stepwise hydration of the material leads to a significant improvement in quality and resolution of the spectrum (Figure 3A).
Figure 3.

1D {1H}13C CP SSNMR spectra of freeze-dried [U-13C, 15N] PD-L1 in complex with the anti-PD-L1 fusion protein (black, A) and in the presence of the nonbinding mAb (red, B). The spectra were recorded on the dried materials and after the addition of increasing amounts of Milli-Q H2O. Spectra were acquired on a spectrometer operating at 800 MHz (1H Larmor frequency) with a MAS of 14 kHz and a temperature of ∼290 K.
Hetero- and homonuclear correlation spectra were recorded on the rehydrated sample (Figure S6) and used for resonance assignment. The assignment of the 2D 15N 13C NCA spectrum (Figure 4) was obtained starting from the data collected in solution on the isolated PD-L1 and complemented by the analysis of the 2D 15N 13C NCO and 13C-13C DARR (Figure 5A) spectra of the complex which allowed us, at the same time, to obtain side-chain assignments. First, the assignment of free PD-L1 in solution was superimposed on the 2D 15N 13C NCA spectrum (Figure S7A,B). The assignment was then matched to the closest signals in the spectrum by identifying the Cα frequencies of the neighboring signals also in the 2D 13C-13C DARR spectrum (Figure S7C). The pattern of carbon resonances correlated to the Cα frequencies in the 2D 13C-13C DARR spectrum allowed us to identify the spin systems characteristic of each residue type and distinguish among possible ambiguities. The resolution of 2D 15N 13C NCO was lower with respect to the other spectra; however, some signals in the 2D 15N 13C NCO were helpful in confirming the 15N chemical shift values of some residues obtained from the 2D 15N 13C NCA spectrum.
Figure 4.

2D 15N 13C NCA spectrum of [U-13C, 15N] PD-L1 in complex with the anti-PD-L1 fusion protein. The assignment of the resonances is reported in black. The spectrum was acquired on a spectrometer operating at 800 MHz (1H Larmor frequency) with a MAS of 14 kHz and a temperature of ∼290 K.
Figure 5.

2D 13C-13C DARR spectra acquired on rehydrated samples of freeze-dried complex of [U-13C, 15N] PD-L1 with the anti-PD-L1 fusion protein (black, A) and of the mixture of [U-13C, 15N] PD-L1 with the nonbinding mAb (red, B). The assignment of I65 side chain is indicated by a blue box. Spectra were acquired on a spectrometer operating at 800 MHz (1H Larmor frequency) with a MAS of 14 kHz and a temperature of ∼290 K.
Finally, a total of 99 spin systems could be identified and assigned in 13C-detected spectra. Interestingly, in addition to the three signals missing in solution NMR spectra, the signals of other residues located in flexible loops of PD-L1 (K25, L48, Q66, G70, L74-V76, K89, M115, G120, A132-Y134) are missing in the SSNMR spectra of the complex.
To improve the assignment of the resonances and the quality of the chemical shift mapping, a set of 1H-detected spectra was also acquired on a sample of [U-2H,13C, 15N] PD-L1 in complex with the anti-PD-L1 fusion protein, prepared under the same experimental conditions of the previously described complex ([U-13C, 15N] PD-L1/anti-PD-L1 drug). The sample was then transferred in a 1.3 mm rotor. The 2D 15N-1H (H)NH CP-HSQC spectrum of the PD-L1/anti-PD-L1 fusion protein complex is of high quality (Figure 6). Also in this case, the assignment of the SSNMR spectrum was obtained starting from the available assignment of the free protein in solution and confirmed by the analysis of 3D spectra [(H)CANH, (H)CONH, 2D 13C-1H plane of (H)(CA)CBNH]. Also in the 1H-detected spectra, some signals of residues belonging to flexible loops of PD-L1 (K41, K46, M59-D61, Q66, G70, L74-V76, Q83, L106, Y134) are missing. Summarizing, a total of 99 spin systems could be identified and assigned also in the 1H-detected spectra. Interestingly, in the solid state, some signals could be identified in the 13C-detected spectra, while others in the 1H-detected spectra.
Figure 6.
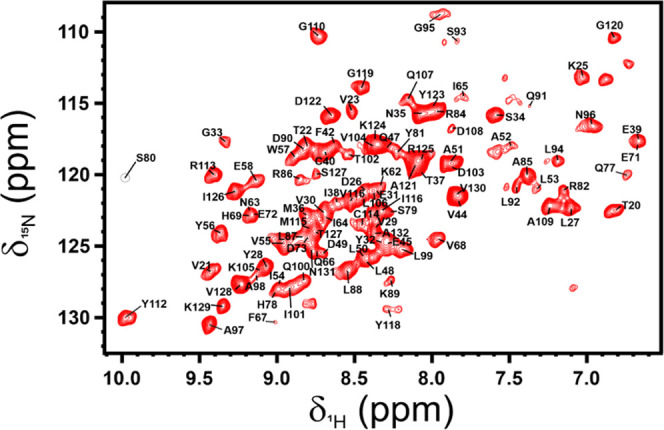
2D 15N 1H (H)NH CP-HSQC spectrum of [U-2H, 13C, 15N] PD-L1 in complex with the anti-PD-L1 fusion protein. The assigned resonances are reported in black. The spectrum was acquired on the rehydrated freeze-dried material, using a spectrometer operating at 800 MHz (1H Larmor frequency) with a MAS of 60 kHz and a temperature of ∼290 K.
Chemical Shift Perturbation (CSP) Can Map the Binding Regions of PD-L1
The availability of protein assignment for the isolated PD-L1 ectodomain in solution and for the same protein in complex with the anti-PD-L1 fusion protein in the solid state allows for the analysis of the chemical shift perturbation (CSP). The CSP of 13Cα/15N and 1H/15N resonances was calculated from the assignment of 13C- and 1H-detected SSNMR spectra, respectively, using the assignment of the isolated [U-2H, 13C, 15N] PD-L1 obtained in solution as reference. Although all residues experience a chemical shift variation moving from solution to solid-state experiments,34 those experiencing the largest chemical shift variations (Q47, E58, E60, I65, E72, Q77, H78, Q83, A93, C114, I116, Y118, and Y123 according to 13Cα/15N chemical shift values; M36, C40, V44, I64, I65, F67, V68, Q77, H78, S80, D108, G110, C114, I116, Y118, D122, R125, and I126 according to 1H/15N chemical shift values) are located on PD-L1 β-sheets and form a large interaction surface (Figure 7).
Figure 7.

Chemical shift perturbation (CSP) of free PD-L1
in solution with
respect to rehydrated freeze-dried PD-L1/anti-PD-L1 fusion protein
complex in the solid state, evaluated according to the formula (A) 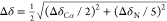 and (B)
and (B) 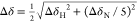 . The residues experiencing the largest
variations (> mean + σ) have been highlighted in magenta
and
blue, respectively. The secondary structure representation is reported
on the top of the plot. The β-strands facing Avelumab in the
structure of the complex are highlighted in yellow. (C, D) CSP mapping
(on the structure with PDB code: 5GRJ) with all of the residues experiencing
the largest perturbations colored in magenta and blue, respectively.
(E, F) Interacting surface of PD-L1 in 5GRJ with only the solvent-exposed residues
experiencing the largest CSP highlighted in magenta and blue, respectively.
The solvent-exposed residues are labeled in yellow. The residues missing
in the 2D 15N 13C NCA and in the 2D 15N 1H (H)NH CP-HSQC spectra are colored in light gray.
. The residues experiencing the largest
variations (> mean + σ) have been highlighted in magenta
and
blue, respectively. The secondary structure representation is reported
on the top of the plot. The β-strands facing Avelumab in the
structure of the complex are highlighted in yellow. (C, D) CSP mapping
(on the structure with PDB code: 5GRJ) with all of the residues experiencing
the largest perturbations colored in magenta and blue, respectively.
(E, F) Interacting surface of PD-L1 in 5GRJ with only the solvent-exposed residues
experiencing the largest CSP highlighted in magenta and blue, respectively.
The solvent-exposed residues are labeled in yellow. The residues missing
in the 2D 15N 13C NCA and in the 2D 15N 1H (H)NH CP-HSQC spectra are colored in light gray.
The CSP values were also analyzed using different thresholds obtained from the iterative procedure proposed by Schumann and co-workers.96 Interestingly, this analysis showed that residues below the new calculated threshold are located in regions noninteracting with the anti-PDL-1 fusion protein (see the Supporting Information for more details, Figure S8).
Comment about Spectral Quality
To confirm that the observed improvement in quality of the solid-state spectra of PD-L1 was due to its binding to the anti-PD-L1 fusion protein, the target was titrated with a noninteracting monoclonal antibody (nb-mAb). As expected, also at high concentrations (PD-L1: nb-mAb, 1:0.5 molar ratio, Figure S9), this antibody does not affect the signals of PD-L1 in a 2D 1H-15N SOFAST HMQC NMR spectrum. Then, the PD-L1/nb-mAb mixture was freeze-dried and analyzed by SSNMR in a 3.2 mm rotor. The experiments recorded on the sample show that in the presence of the nonbinding mAb, the stepwise rehydration does not improve sizably the quality and resolution of the solid-state spectra (Figures 3B and 5B). However, in some regions of this DARR spectrum, the signals are sufficiently resolved to be assigned and compared with those present in the 2D DARR spectrum recorded on the PD-L1/anti-PD-L1 fusion protein complex (Figure 8). The analysis of the two spectra allowed us to evaluate the occurrence of a meaningful chemical shift perturbation for some signals. Most of the signals experiencing the largest shift are indeed located on PD-L1 β-sheets that form the binding surface for anti-PD-L1 fusion protein. Conversely, the signals experiencing negligible effects are located on the opposite face of the PD-L1 protein. In this respect, it is interesting to point out that the signals of Ile54, Ile64, and Ile65, placed on the binding interface, are missing in the DARR spectrum of PD-L1 in the presence of nonbinding mAb, while they are present in the DARR spectrum of the PD-L1/anti-PD-L1 fusion protein complex. The appearance of these signals is consistent with a unique and more rigid conformation of the related residues due to the interaction with the anti-PD-L1 fusion protein.
Figure 8.

(A) Selected regions (C/Cα of Gly, Cβ/Cγ2 and Cβ/Cα of Thr, Cα/Cβ of Ala, Cδ1/Cali of Ile) of 2D 13C-13C DARR spectra acquired on samples of rehydrated freeze-dried complex of [U-13C, 15N] PD-L1 with the anti-PD-L1 fusion protein PD-L1 (black) and rehydrated freeze-dried mixture of [U-13C, 15N] PD-L1 with nonbinding mAb (red). The assignment of the two spectra is reported. In the Ile region of PD-L1/nb-mAb spectrum, some signals are missing. The signals of the assigned Ile in this region are labeled in red. (B) Cartoon representation of the structure of PD-L1 in complex with Avelumab-scFv (PDB code: 5GRJ), with highlighted in blue the residues experiencing the largest shift and in red the nonshifting/nonpresent residues in the spectra of PD-L1/nb-mAb with respect to the spectra of PD-L1 in the presence of anti-PD-L1 fusion protein.
Discussion
The last advances in antibody engineering have led to the development of complex fused biologics with multispecific activity and increased structural complexity. Understanding such a structural complexity and how it impacts the function of a biotherapeutic is, on the one hand, not a trivial task, but, on the other hand, it is of paramount importance during drug development because it is strictly linked to the QbD concept. Indeed, detailed product knowledge is instrumental to the production of safer and more effective drugs and to improve process control strategies.
The epitope mapping on a target can provide the structural information needed to understand the mechanism of action of biologics by supporting structure–activity relationship (SAR) studies, that are critical during pharmaceutical development. SAR can indeed be used to explain the different ways in which a ligand interacts with a receptor: this, in turn, can be used to optimize the physicochemical and functional properties of a biotherapeutic (e.g., solubility, potency, pharmacokinetics, etc.) and can support the design of mutants with larger interacting surfaces and affinities or capable of binding mutated targets.
The results here reported prove that a detailed characterization of the binding to the target of very large and flexible biologics can be achieved by integrating solution and solid-state NMR experiments. The epitope mapping on PD-L1 obtained by this NMR approach nicely matches with the interacting surface previously observed in the X-ray structure of the PD-L1 in complex with Avelumab-scFv (PDB code: 5GRJ),97 another anti-PD-L1 mAb that shares with the tested fusion protein the same Fab sequence (only three amino acids are mutated). Most of the residues experiencing the largest effects are hydrophobic amino acids: aromatic and aliphatic residues forming a wide hydrophobic patch on PD-L1 that is targeted by the anti-PD-L1 fusion protein. At the same time, residue R125 of PD-L1 that in the crystallographic complex97 is close to residue S95 of Avelumab, as well as E58 that is involved in hydrogen bonding with residue Y52 of mAb experience a large chemical shift variation in the presence of our tested anti-PD-L1 fusion protein.
An additional aspect that should be considered is the importance of the characterization of a protein structure per se and not necessarily when the molecule is bound to its target. Indeed, the higher-order structure (HOS) of a protein—intended as secondary, tertiary, and quaternary structures—is a fingerprint covering structural quality attributes potentially linked to the function of a biologic that is constantly monitored during its development. Unwanted perturbations of the folding introduced during the manufacturing process or formulation optimization may in fact lead, for example, to loss of function and/or immunogenicity. The dependence of the binding mechanism on the structural features of the interacting proteins suggests the use of our epitope mapping approach in HOS comparative studies, as the solid-state NMR spectra of the complex allow us to map the fingerprint of a biologic “left” on the target. The chemical shift perturbation (CSP) experienced by the target in the complex is sensitive to the HOS of the antibody—or at least of its binding domain—and it can be used as an “indirect” measure of the ligand structure.
Overall, this approach opens new ways to monitor HOS during pharmaceutical development, allowing us to focus on the structural alterations that may affect target recognition and binding affinity, thus linking HOS assessment to the drug mechanism of action.
The experimental protocol used here to prepare the sample is simple and every step is easily controlled. The methodology does not require the isotopic enrichment of the biological drug, which is usually expressed in eukaryotic cells and where the labeling is highly expensive, although feasible. Conversely, targets can often be obtained in E. coli expression system where the labeling is easy, inexpensive, and with high yields.
Acknowledgments
This work was supported by Regione Toscana (CERM-TT and BioEnable), the Italian Ministero dell’Istruzione, dell’Università e della Ricerca through PRIN 2017A2KEPL, the “Progetto Dipartimenti di Eccellenza 2018-2022” to the Department of Chemistry “Ugo Schiff” of the University of Florence, the Recombinant Proteins JOYNLAB laboratory, and the project FISR2021_SYLCOV. The authors acknowledge the support and the use of resources of Instruct-ERIC, a landmark ESFRI project, and specifically the CERM/CIRMMP Italy center. They also acknowledge H2020-INFRAIA iNEXT-Discovery—Structural Biology Research Infrastructures for Translational Research and Discovery (contract no. 871037), EOSC-Life “Providing an open collaborative space for digital biology in Europe” (H2020, contract no. 824087), “Glytunes” Marie Sklodowska-Curie Action (MSCA) Innovative Training Networks (ITN) H2020-MSCA-ITN-2020 (contract no. 956758), and PANACEA “A Pan-European Solid-State NMR Infrastructure for Chemistry-Enabling Access”, (H2020, contract no. 101008500101008500). The authors acknowledge also Mestrelab Research for providing Mnova software and Bruker BioSpin for AssureNMR software.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c03232.
1D {1H}13C, 2D 13C-13C, and 2D 15N-13C solid-state NMR spectra; 2D 1H-15N solution NMR spectra; details of assignment and CSP; acquisition parameters for SSNMR spectra; and assignment tables (PDF)
Author Contributions
⊥ D.R. and L.C. contributed equally to this paper.
The authors declare the following competing financial interest(s): L.I., C.P., A.P., and F.B. were employees of Merck Serono S.p.a, Guidonia, RM, Italy, an affiliate of Merck KGaA, at the date of the analyses. This research was performed using as case study sample a product in development by Merck KGaA. While Merck KGaA has filed for patent protection regarding the product in development, the technology described in this manuscript is independent from this product of Merck KGaA. No patents or patent applications have been filed for the technology described in this manuscript.
Notes
NMR assignment in solution of PD-L1 ectodomain (residues Ala18-Tyr134) generated during the current study is available in the BMRB database under the accession code: 51169. The raw data are available at https://zenodo.org under the DOI: 10.5281/zenodo.6363169.
Supplementary Material
References
- Zost S. J.; Gilchuk P.; Case J. B.; Binshtein E.; Chen R. E.; Nkolola J. P.; Schäfer A.; Reidy J. X.; Trivette A.; Nargi R. S.; Sutton R. E.; Suryadevara N.; Martinez D. R.; Williamson L. E.; Chen E. C.; Jones T.; Day S.; Myers L.; Hassan A. O.; Kafai N. M.; Winkler E. S.; Fox J. M.; Shrihari S.; Mueller B. K.; Meiler J.; Chandrashekar A.; Mercado N. B.; Steinhardt J. J.; Ren K.; Loo Y.-M.; Kallewaard N. L.; McCune B. T.; Keeler S. P.; Holtzman M. J.; Barouch D. H.; Gralinski L. E.; Baric R. S.; Thackray L. B.; Diamond M. S.; Carnahan R. H.; Crowe J. E. Potently Neutralizing and Protective Human Antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Sun Y.; Feng R.; Wang Y.; Guo Y.; Zhang L.; Deng Y.-Q.; Wang L.; Cui Z.; Cao L.; Zhang Y.-J.; Li W.; Zhu F.-C.; Qin C.-F.; Wang X. Structure-Based Development of Human Antibody Cocktails against SARS-CoV-2. Cell Res. 2021, 31, 101–103. 10.1038/s41422-020-00446-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. ICH Q8 (R2) Pharmaceutical Development, 2021. https://www.ema.europa.eu/en/ich-q8-r2-pharmaceutical-development.
- Lu R.-M.; Hwang Y.-C.; Liu I.-J.; Lee C.-C.; Tsai H.-Z.; Li H.-J.; Wu H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J.; Mathias S.; Kube S.; Otte K.; Garidel P.; Gamer M.; Blech M.; Fischer S.; Karow-Zwick A. R. Rational Optimization of a Monoclonal Antibody Improves the Aggregation Propensity and Enhances the CMC Properties along the Entire Pharmaceutical Process Chain. mAbs 2020, 12, 1787121 10.1080/19420862.2020.1787121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson J. S.; Demarest S. J.; Miller B.; Amatucci A.; Snyder W. B.; Wu X.; Huang F.; Phan S.; Gao S.; Doern A.; Farrington G. K.; Lugovskoy A. A.; Joseph I.; Bailly V.; Wang X.; Garber E.; Browning J.; Glaser S. M. Anti-Tumor Activity of Stability-Engineered IgG-like Bispecific Antibodies Targeting TRAIL-R2 and LTβR. mAbs 2009, 1, 128–141. 10.4161/mabs.1.2.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet D. R.; Atkins W. M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. 10.1016/j.xphs.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löfblom J.; Frejd F. Y.; Ståhl S. Non-Immunoglobulin Based Protein Scaffolds. Curr. Opin. Biotechnol. 2011, 22, 843–848. 10.1016/j.copbio.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Clarke S. C.; Ma B.; Trinklein N. D.; Schellenberger U.; Osborn M. J.; Ouisse L.-H.; Boudreau A.; Davison L. M.; Harris K. E.; Ugamraj H. S.; Balasubramani A.; Dang K. H.; Jorgensen B.; Ogana H. A. N.; Pham D. T.; Pratap P. P.; Sankaran P.; Anegon I.; van Schooten W. C.; Brüggemann M.; Buelow R.; Force Aldred S. Multispecific Antibody Development Platform Based on Human Heavy Chain Antibodies. Front. Immunol. 2019, 9, 3037 10.3389/fimmu.2018.03037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrup O. A.; Ong S. C.; Lykkemark S.; Dinesen A.; Rudnik-Jansen I.; Dagnæs-Hansen N. F.; Andersen J. T.; Alvarez-Vallina L.; Howard K. A. Programmable Half-Life and Anti-Tumour Effects of Bispecific T-Cell Engager-Albumin Fusions with Tuned FcRn Affinity. Commun. Biol. 2021, 4, 310 10.1038/s42003-021-01790-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X.; D’Antona A. M. Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics. Antibodies 2021, 10, 13 10.3390/antib10020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidle U. H.; Kontermann R. E.; Brinkmann U. Tumor-Antigen–Binding Bispecific Antibodies for Cancer Treatment. Semin. Oncol. 2014, 41, 653–660. 10.1053/j.seminoncol.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Renaud J.-P.; Chari A.; Ciferri C.; Liu W.; Rémigy H.-W.; Stark H.; Wiesmann C. Cryo-EM in Drug Discovery: Achievements, Limitations and Prospects. Nat. Rev. Drug Discovery 2018, 17, 471–492. 10.1038/nrd.2018.77. [DOI] [PubMed] [Google Scholar]
- Goswami D.; Zhang J.; Bondarenko P. V.; Zhang Z. MS-Based Conformation Analysis of Recombinant Proteins in Design, Optimization and Development of Biopharmaceuticals. Methods 2018, 144, 134–151. 10.1016/j.ymeth.2018.04.011. [DOI] [PubMed] [Google Scholar]
- Zhou D.; Duyvesteyn H. M. E.; Chen C.-P.; Huang C.-G.; Chen T.-H.; Shih S.-R.; Lin Y.-C.; Cheng C.-Y.; Cheng S.-H.; Huang Y.-C.; Lin T.-Y.; Ma C.; Huo J.; Carrique L.; Malinauskas T.; Ruza R. R.; Shah P. N. M.; Tan T. K.; Rijal P.; Donat R. F.; Godwin K.; Buttigieg K. R.; Tree J. A.; Radecke J.; Paterson N. G.; Supasa P.; Mongkolsapaya J.; Screaton G. R.; Carroll M. W.; Gilbert-Jaramillo J.; Knight M. L.; James W.; Owens R. J.; Naismith J. H.; Townsend A. R.; Fry E. E.; Zhao Y.; Ren J.; Stuart D. I.; Huang K.-Y. A. Structural Basis for the Neutralization of SARS-CoV-2 by an Antibody from a Convalescent Patient. Nat. Struct. Mol. Biol. 2020, 27, 950–958. 10.1038/s41594-020-0480-y. [DOI] [PubMed] [Google Scholar]
- Kallewaard N. L.; Corti D.; Collins P. J.; Neu U.; McAuliffe J. M.; Benjamin E.; Wachter-Rosati L.; Palmer-Hill F. J.; Yuan A. Q.; Walker P. A.; Vorlaender M. K.; Bianchi S.; Guarino B.; De Marco A.; Vanzetta F.; Agatic G.; Foglierini M.; Pinna D.; Fernandez-Rodriguez B.; Fruehwirth A.; Silacci C.; Ogrodowicz R. W.; Martin S. R.; Sallusto F.; Suzich J. A.; Lanzavecchia A.; Zhu Q.; Gamblin S. J.; Skehel J. J. Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell 2016, 166, 596–608. 10.1016/j.cell.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Rossi P.; Kalodimos C. G. Structural Basis for Client Recognition and Activity of Hsp40 Chaperones. Science 2019, 365, 1313–1319. 10.1126/science.aax1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H.; van Ingen H.; Zhou B.-R.; Feng H.; Bustin M.; Kay L. E.; Bai Y. Architecture of the High Mobility Group Nucleosomal Protein 2-Nucleosome Complex as Revealed by Methyl-Based NMR. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12283–12288. 10.1073/pnas.1105848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshun-Wilson L.; Zhang R.; Portran D.; Nachury M. V.; Toso D. B.; Löhr T.; Vendruscolo M.; Bonomi M.; Fraser J. S.; Nogales E. Effects of α-Tubulin Acetylation on Microtubule Structure and Stability. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 10366–10371. 10.1073/pnas.1900441116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J.; Le Bas A.; Ruza R. R.; Duyvesteyn H. M. E.; Mikolajek H.; Malinauskas T.; Tan T. K.; Rijal P.; Dumoux M.; Ward P. N.; Ren J.; Zhou D.; Harrison P. J.; Weckener M.; Clare D. K.; Vogirala V. K.; Radecke J.; Moynié L.; Zhao Y.; Gilbert-Jaramillo J.; Knight M. L.; Tree J. A.; Buttigieg K. R.; Coombes N.; Elmore M. J.; Carroll M. W.; Carrique L.; Shah P. N. M.; James W.; Townsend A. R.; Stuart D. I.; Owens R. J.; Naismith J. H. Neutralizing Nanobodies Bind SARS-CoV-2 Spike RBD and Block Interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. 10.1038/s41594-020-0469-6. [DOI] [PubMed] [Google Scholar]
- Arbogast L. W.; Brinson R. G.; Marino J. P. Mapping Monoclonal Antibody Structure by 2D 13C NMR at Natural Abundance. Anal. Chem. 2015, 87, 3556–3561. 10.1021/ac504804m. [DOI] [PubMed] [Google Scholar]
- Brinson R. G.; Marino J. P.; Delaglio F.; Arbogast L. W.; Evans R. M.; Kearsley A.; Gingras G.; Ghasriani H.; Aubin Y.; Pierens G. K.; Jia X.; Mobli M.; Grant H. G.; Keizer D. W.; Schweimer K.; Ståhle J.; Widmalm G.; Zartler E. R.; Lawrence C. W.; Reardon P. N.; Cort J. R.; Xu P.; Ni F.; Yanaka S.; Kato K.; Parnham S. R.; Tsao D.; Blomgren A.; Rundlöf T.; Trieloff N.; Schmieder P.; Ross A.; Skidmore K.; Chen K.; Keire D.; Freedberg D. I.; Suter-Stahel T.; Wider G.; Ilc G.; Plavec J.; Bradley S. A.; Baldisseri D. M.; Sforça M. L.; Zeri A. C.; de M.; Wei J. Y.; Szabo C. M.; Amezcua C. A.; Jordan J. B.; Wikström M. Enabling Adoption of 2D-NMR for the Higher Order Structure Assessment of Monoclonal Antibody Therapeutics. mAbs 2019, 11, 94–105. 10.1080/19420862.2018.1544454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinson R. G.; Ghasriani H.; Hodgson D. J.; Adams K. M.; McEwen I.; Freedberg D. I.; Chen K.; Keire D. A.; Aubin Y.; Marino J. P. Application of 2D-NMR with Room Temperature NMR Probes for the Assessment of the Higher Order Structure of Filgrastim. J. Pharm. Biomed. Anal. 2017, 141, 229–233. 10.1016/j.jpba.2017.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasriani H.; Hodgson D. J.; Brinson R. G.; McEwen I.; Buhse L. F.; Kozlowski S.; Marino J. P.; Aubin Y.; Keire D. A. Precision and Robustness of 2D-NMR for Structure Assessment of Filgrastim Biosimilars. Nat. Biotechnol. 2016, 34, 139–141. 10.1038/nbt.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbogast L. W.; Brinson R. G.; Formolo T.; Hoopes J. T.; Marino J. P. 2D 1HN, 15N Correlated NMR Methods at Natural Abundance for Obtaining Structural Maps and Statistical Comparability of Monoclonal Antibodies. Pharm. Res. 2016, 33, 462–475. 10.1007/s11095-015-1802-3. [DOI] [PubMed] [Google Scholar]
- Arbogast L. W.; Delaglio F.; Tolman J. R.; Marino J. P. Selective Suppression of Excipient Signals in 2D 1H-13C Methyl Spectra of Biopharmaceutical Products. J. Biomol. NMR 2018, 72, 149–161. 10.1007/s10858-018-0214-1. [DOI] [PubMed] [Google Scholar]
- Arbogast L. W.; Delaglio F.; Brinson R. G.; Marino J. P. Assessment of the Higher-Order Structure of Formulated Monoclonal Antibody Therapeutics by 2D Methyl Correlated NMR and Principal Component Analysis. Curr. Protoc. Protein Sci. 2020, 100, e105 10.1002/cpps.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxholm G. W.; Petersen B. O.; Malmstrøm J. Higher-Order Structure Characterization of Pharmaceutical Proteins by 2D Nuclear Magnetic Resonance Methyl Fingerprinting. J. Pharm. Sci. 2019, 108, 3029–3035. 10.1016/j.xphs.2019.04.032. [DOI] [PubMed] [Google Scholar]
- Rößler P.; Mathieu D.; Gossert A. D. Enabling NMR Studies of High Molecular Weight Systems Without the Need for Deuteration: The XL-ALSOFAST Experiment with Delayed Decoupling. Angew. Chem., Int. Ed. 2020, 59, 19329–19337. 10.1002/anie.202007715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ständer S.; Grauslund L.; Scarselli M.; Norais N.; Rand K. Epitope Mapping of Polyclonal Antibodies by Hydrogen–Deuterium Exchange Mass Spectrometry (HDX-MS). Anal. Chem. 2021, 93, 11669–11678. 10.1021/acs.analchem.1c00696. [DOI] [PubMed] [Google Scholar]
- Ozohanics O.; Ambrus A. Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life 2020, 10, 286 10.3390/life10110286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamley J. M.; Iuga D.; Öster C.; Sass H.-J.; Rogowski M.; Oss A.; Past J.; Reinhold A.; Grzesiek S.; Samoson A.; Lewandowski J. R. Solid-State NMR of a Protein in a Precipitated Complex with a Full-Length Antibody. J. Am. Chem. Soc. 2014, 136, 16800–16806. 10.1021/ja5069992. [DOI] [PubMed] [Google Scholar]
- Gronenborn A. M.; Filpula D. R.; Essig N. Z.; Achari A.; Whitlow M.; Wingfield P. T.; Clore G. M. A Novel, Highly Stable Fold of the Immunoglobulin Binding Domain of Streptococcal Protein G. Science 1991, 253, 657–661. 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- Franks W. T.; Zhou D. H.; Wylie B. J.; Money B. G.; Graesser D. T.; Frericks H. L.; Sahota G.; Rienstra C. M. Magic-Angle Spinning Solid-State NMR Spectroscopy of the B1 Immunoglobulin Binding Domain of Protein G (GB1): 15N and 13C Chemical Shift Assignments and Conformational Analysis. J. Am. Chem. Soc. 2005, 127, 12291–12305. 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Ravera E.; Reif B.; Turano P. Solid-State NMR of Proteins Sedimented by Ultracentrifugation. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10396–10399. 10.1073/pnas.1103854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainz A.; Jehle S.; van Rossum B. J.; Oschkinat H.; Reif B. Large Protein Complexes with Extreme Rotational Correlation Times Investigated in Solution by Magic-Angle-Spinning NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 15968–15969. 10.1021/ja904733v. [DOI] [PubMed] [Google Scholar]
- Mainz A.; Religa T. L.; Sprangers R.; Linser R.; Kay L. E.; Reif B. NMR Spectroscopy of Soluble Protein Complexes at One Mega-Dalton and Beyond. Angew. Chem., Int. Ed. 2013, 52, 8746–8751. 10.1002/anie.201301215. [DOI] [PubMed] [Google Scholar]
- Giuntini S.; Balducci E.; Cerofolini L.; Ravera E.; Fragai M.; Berti F.; Luchinat C. Characterization of the Conjugation Pattern in Large Polysaccharide–Protein Conjugates by NMR Spectroscopy. Angew. Chem., Int. Ed. 2017, 56, 14997–15001. 10.1002/anie.201709274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuntini S.; Cerofolini L.; Ravera E.; Fragai M.; Luchinat C. Atomic Structural Details of a Protein Grafted onto Gold Nanoparticles. Sci. Rep. 2017, 7, 17934 10.1038/s41598-017-18109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerofolini L.; Giuntini S.; Ravera E.; Luchinat C.; Berti F.; Fragai M. Structural Characterization of a Protein Adsorbed on Aluminum Hydroxide Adjuvant in Vaccine Formulation. npj Vaccines 2019, 4, 20 10.1038/s41541-019-0115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerofolini L.; Giuntini S.; Carlon A.; Ravera E.; Calderone V.; Fragai M.; Parigi G.; Luchinat C. Characterization of PEGylated Asparaginase: New Opportunities from NMR Analysis of Large PEGylated Therapeutics. Chem. - Eur. J. 2019, 25, 1984–1991. 10.1002/chem.201804488. [DOI] [PubMed] [Google Scholar]
- Ravera E.; Ciambellotti S.; Cerofolini L.; Martelli T.; Kozyreva T.; Bernacchioni C.; Giuntini S.; Fragai M.; Turano P.; Luchinat C. Solid-State NMR of PEGylated Proteins. Angew. Chem., Int. Ed. 2016, 55, 2446–2449. 10.1002/anie.201510148. [DOI] [PubMed] [Google Scholar]
- Cerofolini L.; Fragai M.; Ravera E.; Diebolder C. A.; Renault L.; Calderone V. Integrative Approaches in Structural Biology: A More Complete Picture from the Combination of Individual Techniques. Biomolecules 2019, 9, E370 10.3390/biom9080370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq L.; Fogeron M.-L.; Meier B. H.; Nassal M.; Böckmann A. Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies. Viruses 2020, 12, E1069 10.3390/v12101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand T.; Lacabanne D.; Torosyan A.; Boudet J.; Cadalbert R.; Allain F. H.-T.; Meier B. H.; Böckmann A. Sedimentation Yields Long-Term Stable Protein Samples as Shown by Solid-State NMR. Front. Mol. Biosci. 2020, 7, 17 10.3389/fmolb.2020.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.; Quinn C. M.; Struppe J.; Sergeyev I. V.; Zhang C.; Guo C.; Runge B.; Theint T.; Dao H. H.; Jaroniec C. P.; Berbon M.; Lends A.; Habenstein B.; Loquet A.; Kuemmerle R.; Perrone B.; Gronenborn A. M.; Polenova T. Sensitivity Boosts by the CPMAS CryoProbe for Challenging Biological Assemblies. J. Magn. Reson. 2020, 311, 106680 10.1016/j.jmr.2019.106680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Russell R. W.; Bryer A. J.; Quinn C. M.; Hou G.; Zhang H.; Schwieters C. D.; Perilla J. R.; Gronenborn A. M.; Polenova T. Atomic-Resolution Structure of HIV-1 Capsid Tubes by Magic-Angle Spinning NMR. Nat. Struct. Mol. Biol. 2020, 27, 863–869. 10.1038/s41594-020-0489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy M. T.; Yu T.-Y.; Wagner G.; Griffin R. G. Structural Characterization of the Human Membrane Protein VDAC2 in Lipid Bilayers by MAS NMR. J. Biomol. NMR 2019, 73, 451–460. 10.1007/s10858-019-00242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Zhang H.; Lu M.; Hou G.; Caporini M.; Rosay M.; Maas W.; Struppe J.; Ahn J.; Byeon I.-J. L.; Oschkinat H.; Jaudzems K.; Barbet-Massin E.; Emsley L.; Pintacuda G.; Lesage A.; Gronenborn A. M.; Polenova T. Dynamic Nuclear Polarization Magic-Angle Spinning Nuclear Magnetic Resonance Combined with Molecular Dynamics Simulations Permits Detection of Order and Disorder in Viral Assemblies. J. Phys. Chem. B 2019, 123, 5048–5058. 10.1021/acs.jpcb.9b02293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Paige U. B.; Xiang S.; Hendrix M. M. R. M.; Zhang Y.; Folkers G. E.; Weingarth M.; Bonvin A. M. J. J.; Kutateladze T. G.; Voets I. K.; Baldus M.; van Ingen H. Characterization of Nucleosome Sediments for Protein Interaction Studies by Solid-State NMR Spectroscopy. Magn. Reson. 2021, 2, 187–202. 10.5194/mr-2-187-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroue K. H.; MacKinnon N.; Xu J.; Zhu P.; McNerny E.; Kohn D. H.; Morris M. D.; Ramamoorthy A. High-Resolution Structural Insights into Bone: A Solid-State NMR Relaxation Study Utilizing Paramagnetic Doping. J. Phys. Chem. B 2012, 116, 11656–11661. 10.1021/jp307935g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaïs T.; Von Euw S.; Ajili W.; Auzoux-Bordenave S.; Bertani P.; Gajan D.; Emsley L.; Nassif N.; Lesage A. Structural Description of Surfaces and Interfaces in Biominerals by DNP SENS. Solid State Nucl. Magn. Reson. 2019, 102, 2–11. 10.1016/j.ssnmr.2019.06.001. [DOI] [PubMed] [Google Scholar]
- Cerofolini L.; Giuntini S.; Louka A.; Ravera E.; Fragai M.; Luchinat C. High-Resolution Solid-State NMR Characterization of Ligand Binding to a Protein Immobilized in a Silica Matrix. J. Phys. Chem. B 2017, 121, 8094–8101. 10.1021/acs.jpcb.7b05679. [DOI] [PubMed] [Google Scholar]
- Louka A.; Matlahov I.; Giuntini S.; Cerofolini L.; Cavallo A.; Pillozzi S.; Ravera E.; Fragai M.; Arcangeli A.; Ramamoorthy A.; Goobes G.; Luchinat C. Engineering L-Asparaginase for Spontaneous Formation of Calcium Phosphate Bioinspired Microreactors. Phys. Chem. Chem. Phys. 2018, 20, 12719–12726. 10.1039/c8cp00419f. [DOI] [PubMed] [Google Scholar]
- Ravera E.; Cerofolini L.; Martelli T.; Louka A.; Fragai M.; Luchinat C. (1)H-Detected Solid-State NMR of Proteins Entrapped in Bioinspired Silica: A New Tool for Biomaterials Characterization. Sci. Rep. 2016, 6, 27851 10.1038/srep27851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli T.; Ravera E.; Louka A.; Cerofolini L.; Hafner M.; Fragai M.; Becker C. F. W.; Luchinat C. Atomic-Level Quality Assessment of Enzymes Encapsulated in Bioinspired Silica. Chem. - Eur. J. 2016, 22, 425–432. 10.1002/chem.201503613. [DOI] [PubMed] [Google Scholar]
- Fragai M.; Luchinat C.; Martelli T.; Ravera E.; Sagi I.; Solomonov I.; Udi Y. SSNMR of Biosilica-Entrapped Enzymes Permits an Easy Assessment of Preservation of Native Conformation in Atomic Detail. Chem. Commun. 2014, 50, 421–423. 10.1039/c3cc46896h. [DOI] [PubMed] [Google Scholar]
- Ravera E.; Schubeis T.; Martelli T.; Fragai M.; Parigi G.; Luchinat C. NMR of Sedimented, Fibrillized, Silica-Entrapped and Microcrystalline (Metallo)Proteins. J. Magn. Reson. 2015, 253, 60–70. 10.1016/j.jmr.2014.12.019. [DOI] [PubMed] [Google Scholar]
- Viger-Gravel J.; Paruzzo F. M.; Cazaux C.; Jabbour R.; Leleu A.; Canini F.; Florian P.; Ronzon F.; Gajan D.; Lesage A. Atomic-Scale Description of Interfaces between Antigen and Aluminum-Based Adjuvants Used in Vaccines by Dynamic Nuclear Polarization (DNP) Enhanced NMR Spectroscopy. Chem. - Eur. J. 2020, 26, 8976–8982. 10.1002/chem.202001141. [DOI] [PubMed] [Google Scholar]
- Jaudzems K.; Kirsteina A.; Schubeis T.; Casano G.; Ouari O.; Bogans J.; Kazaks A.; Tars K.; Lesage A.; Pintacuda G. Structural Analysis of an Antigen Chemically Coupled on Virus-Like Particles in Vaccine Formulation. Angew. Chem., Int. Ed. 2021, 60, 12847–12851. 10.1002/anie.202013189. [DOI] [PubMed] [Google Scholar]
- Rizzo D.; Cerofolini L.; Pérez-Ràfols A.; Giuntini S.; Baroni F.; Ravera E.; Luchinat C.; Fragai M. Evaluation of the Higher Order Structure of Biotherapeutics Embedded in Hydrogels for Bioprinting and Drug Release. Anal. Chem. 2021, 93, 11208–11214. 10.1021/acs.analchem.1c01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Tycko R. Segmental Isotopic Labeling of HIV-1 Capsid Protein Assemblies for Solid State NMR. J. Biomol. NMR 2018, 70, 103–114. 10.1007/s10858-017-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragai M.; Luchinat C.; Parigi G.; Ravera E. Practical Considerations over Spectral Quality in Solid State NMR Spectroscopy of Soluble Proteins. J. Biomol. NMR 2013, 57, 155–166. 10.1007/s10858-013-9776-0. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Ravera E. SedNMR: On the Edge between Solution and Solid-State NMR. Acc. Chem. Res. 2013, 46, 2059–2069. 10.1021/ar300342f. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E.; Huang C. T.; Daebel V.; Hsu S. T.; Reif B. Site-Specific Solid-State NMR Studies of “Trigger Factor” in Complex with the Large Ribosomal Subunit 50S. Angew. Chem., Int. Ed. 2015, 54, 4367–4369. 10.1002/anie.201409393. [DOI] [PubMed] [Google Scholar]
- Sharpe A. H.; Pauken K. E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- Gao Q.; Wang X.-Y.; Qiu S.-J.; Yamato I.; Sho M.; Nakajima Y.; Zhou J.; Li B.-Z.; Shi Y.-H.; Xiao Y.-S.; Xu Y.; Fan J. Overexpression of PD-L1 Significantly Associates with Tumor Aggressiveness and Postoperative Recurrence in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2009, 15, 971–979. 10.1158/1078-0432.CCR-08-1608. [DOI] [PubMed] [Google Scholar]
- Darvin P.; Toor S. M.; Sasidharan Nair V.; Elkord E. Immune Checkpoint Inhibitors: Recent Progress and Potential Biomarkers. Exp. Mol. Med. 2018, 50, 1–11. 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D. M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian S. L.; Hodi F. S.; Brahmer J. R.; Gettinger S. N.; Smith D. C.; McDermott D. F.; Powderly J. D.; Carvajal R. D.; Sosman J. A.; Atkins M. B.; Leming P. D.; Spigel D. R.; Antonia S. J.; Horn L.; Drake C. G.; Pardoll D. M.; Chen L.; Sharfman W. H.; Anders R. A.; Taube J. M.; McMiller T. L.; Xu H.; Korman A. J.; Jure-Kunkel M.; Agrawal S.; McDonald D.; Kollia G. D.; Gupta A.; Wigginton J. M.; Sznol M. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow M. A.; Callahan M. K.; Wolchok J. D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A.; Wolchok J. D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak K. M.; Grudnik P.; Guzik K.; Zieba B. J.; Musielak B.; Dömling A.; Dubin G.; Holak T. A. Structural Basis for Small Molecule Targeting of the Programmed Death Ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. 10.18632/oncotarget.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S.; Bax A. Improved 3D Triple-Resonance NMR Techniques Applied to a 31 KDa Protein. J. Magn. Reson. 1992, 96, 432–440. 10.1016/0022-2364(92)90099-S. [DOI] [Google Scholar]
- Schleucher J.; Sattler M.; Griesinger C. Coherence Selection by Gradients without Signal Attenuation: Application to the Three-Dimensional HNCO Experiment. Angew. Chem., Int. Ed. 1993, 32, 1489–1491. 10.1002/anie.199314891. [DOI] [Google Scholar]
- Kay L. E.; Xu G. Y.; Yamazaki T. Enhanced-Sensitivity Triple-Resonance Spectroscopy with Minimal H2O Saturation. J. Magn. Reson., Ser. A 1994, 109, 129–133. 10.1006/jmra.1994.1145. [DOI] [Google Scholar]
- Wittekind M.; Mueller L. HNCACB, a High-Sensitivity 3D NMR Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha- and Beta-Carbon Resonances in Proteins. J. Magn. Reson., Ser. B 1993, 101, 201–205. 10.1006/jmrb.1993.1033. [DOI] [Google Scholar]
- Muhandiram D. R.; Kay L. E. Gradient-Enhanced Triple-Resonance Three-Dimensional NMR Experiments with Improved Sensitivity. J. Magn. Reson., Ser. B 1994, 103, 203–216. 10.1006/jmrb.1994.1032. [DOI] [Google Scholar]
- Grzesiek S.; Bax A. Amino Acid Type Determination in the Sequential Assignment Procedure of Uniformly 13C/15N-Enriched Proteins. J. Biomol. NMR 1993, 3, 185–204. 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- Bostock M.; Nietlispach D. Compressed Sensing: Reconstruction of Non-Uniformly Sampled Multidimensional NMR Data. Concepts Magn. Reson., Part A 2017, 46, e21438 10.1002/cmr.a.21438. [DOI] [Google Scholar]
- Schanda P.; Brutscher B. Very Fast Two-Dimensional NMR Spectroscopy for Real-Time Investigation of Dynamic Events in Proteins on the Time Scale of Seconds. J. Am. Chem. Soc. 2005, 127, 8014–8015. 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- Pines A.; Gibby M. G.; Waugh J. S. Proton-Enhanced Nuclear Induction Spectroscopy. A Method for High Resolution NMR of Dilute Spins in Solids. J. Chem. Phys. 1972, 56, 1776–1777. 10.1063/1.1677439. [DOI] [Google Scholar]
- Baldus M.; Petkova A. T.; Herzfeld J.; Griffin R. G. Cross Polarization in the Tilted Frame: Assignment and Spectral Simplification in Heteronuclear Spin Systems. Mol. Phys. 1998, 95, 1197–1207. 10.1080/00268979809483251. [DOI] [Google Scholar]
- Hong M.; Griffin R. G. Resonance Assignments for Solid Peptides by Dipolar-Mediated 13C/15N Correlation Solid-State NMR. J. Am. Chem. Soc. 1998, 120, 7113–7114. 10.1021/ja980775w. [DOI] [Google Scholar]
- Szeverenyi N. M.; Sullivan M. J.; Maciel G. E. Observation of Spin Exchange by Two-Dimensional Fourier Transform 13C Cross Polarization-Magic-Angle Spinning. J. Magn. Reson. 1982, 47, 462–475. 10.1016/0022-2364(82)90213-X. [DOI] [Google Scholar]
- Takegoshi K.; Nakamura S.; Terao T. 13C–1H Dipolar-Assisted Rotational Resonance in Magic-Angle Spinning NMR. Chem. Phys. Lett. 2001, 344, 631–637. 10.1016/S0009-2614(01)00791-6. [DOI] [Google Scholar]
- Morcombe C. R.; Gaponenko V.; Byrd R. A.; Zilm K. W. Diluting Abundant Spins by Isotope Edited Radio Frequency Field Assisted Diffusion. J. Am. Chem. Soc. 2004, 126, 7196–7197. 10.1021/ja047919t. [DOI] [PubMed] [Google Scholar]
- Knight M. J.; Webber A. L.; Pell A. J.; Guerry P.; Barbet-Massin E.; Bertini I.; Felli I. C.; Gonnelli L.; Pierattelli R.; Emsley L.; Lesage A.; Herrmann T.; Pintacuda G. Fast Resonance Assignment and Fold Determination of Human Superoxide Dismutase by High-Resolution Proton-Detected Solid-State MAS NMR Spectroscopy. Angew. Chem., Int. Ed. 2011, 123, 11901–11905. 10.1002/ange.201106340. [DOI] [PubMed] [Google Scholar]
- Andreas L. B.; Le Marchand T.; Jaudzems K.; Pintacuda G. High-Resolution Proton-Detected NMR of Proteins at Very Fast MAS. J. Magn. Reson. 2015, 253, 36–49. 10.1016/j.jmr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E.; Pell A. J.; Jaudzems K.; Franks W. T.; Retel J. S.; Kotelovica S.; Akopjana I.; Tars K.; Emsley L.; Oschkinat H.; Lesage A.; Pintacuda G. Out-and-Back 13C-13C Scalar Transfers in Protein Resonance Assignment by Proton-Detected Solid-State NMR under Ultra-Fast MAS. J. Biomol. NMR 2013, 56, 379–386. 10.1007/s10858-013-9757-3. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E.; Pell A. J.; Retel J. S.; Andreas L. B.; Jaudzems K.; Franks W. T.; Nieuwkoop A. J.; Hiller M.; Higman V.; Guerry P.; Bertarello A.; Knight M. J.; Felletti M.; Le Marchand T.; Kotelovica S.; Akopjana I.; Tars K.; Stoppini M.; Bellotti V.; Bolognesi M.; Ricagno S.; Chou J. J.; Griffin R. G.; Oschkinat H.; Lesage A.; Emsley L.; Herrmann T.; Pintacuda G. Rapid Proton-Detected NMR Assignment for Proteins with Fast Magic Angle Spinning. J. Am. Chem. Soc. 2014, 136, 12489–12497. 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A.; Wasmer C.; Habenstein B.; Verel R.; Greenwald J.; Riek R.; Böckmann A.; Meier B. H. Protocols for the Sequential Solid-State NMR Spectroscopic Assignment of a Uniformly Labeled 25 KDa Protein: HET-s(1-227). ChemBioChem 2010, 11, 1543–1551. 10.1002/cbic.201000124. [DOI] [PubMed] [Google Scholar]
- Bermel W.; Bertini I.; Duma L.; Felli I. C.; Emsley L.; Pierattelli R.; Vasos P. R. Complete Assignment of Heteronuclear Protein Resonances by Protonless NMR Spectroscopy. Angew. Chem., Int. Ed. 2005, 44, 3089–3092. 10.1002/anie.200461794. [DOI] [PubMed] [Google Scholar]
- Gil S.; Hošek T.; Solyom Z.; Kümmerle R.; Brutscher B.; Pierattelli R.; Felli I. C. NMR Spectroscopic Studies of Intrinsically Disordered Proteins at Near-Physiological Conditions. Angew. Chem., Int. Ed. 2013, 52, 11808–11812. 10.1002/anie.201304272. [DOI] [PubMed] [Google Scholar]
- Keller R.The Computer Aided Resonance Assignment Tutorial (CARA); The CARA/Lua Programmers Manual; DATONAL AG; CANTINA Verlag: Goldau. Switzerland, 2004. [Google Scholar]
- Schumann F. H.; Riepl H.; Maurer T.; Gronwald W.; Neidig K.-P.; Kalbitzer H. R. Combined Chemical Shift Changes and Amino Acid Specific Chemical Shift Mapping of Protein–Protein Interactions. J. Biomol. NMR 2007, 39, 275–289. 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- Liu K.; Tan S.; Chai Y.; Chen D.; Song H.; Zhang C. W.-H.; Shi Y.; Liu J.; Tan W.; Lyu J.; Gao S.; Yan J.; Qi J.; Gao G. F. Structural Basis of Anti-PD-L1 Monoclonal Antibody Avelumab for Tumor Therapy. Cell Res. 2017, 27, 151–153. 10.1038/cr.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.