

Abstract
BACKGROUND & AIMS:
Microbiota composition and mechanisms of host–microbiota interactions in the esophagus are unclear. We aimed to uncover fundamental information about the esophageal microbiome and its potential significance to eosinophilic esophagitis (EoE).
METHODS:
Microbiota composition, transplantation potential, and antibiotic responsiveness in the esophagus were established via 16S ribosomal RNA sequencing. Functional outcomes of microbiota colonization were assessed by RNA sequencing analysis of mouse esophageal epithelium and compared with the human EoE transcriptome. The impact of dysbiosis was assessed using a preclinical model of EoE.
RESULTS:
We found that the murine esophagus is colonized with diverse microbial communities within the first month of life. The esophageal microbiota is distinct, dominated by Lactobacillales, and demonstrates spatial heterogeneity as the proximal and distal esophagus are enriched in Bifidobacteriales and Lactobacillales, respectively. Fecal matter transplantation restores the esophageal microbiota, demonstrating that the local environment drives diversity. Microbiota colonization modifies esophageal tissue morphology and gene expression that is enriched in pathways associated with epithelial barrier function and overlapping with genes involved in EoE, including POSTN, KLK5, and HIF1A. Finally, neonatal antibiotic treatment reduces the abundance of Lactobacillales and exaggerates type 2 inflammation in the esophagus. Clinical data substantiated loss of esophageal Lactobacillales in EoE compared with controls.
CONCLUSIONS:
The esophagus has a unique microbiome with notable differences between its proximal and distal regions. Fecal matter transplantation restores the esophageal microbiome. Antibiotic-induced dysbiosis exacerbates disease in a murine model of EoE. Collectively, these data establish the composition, transplantation potential, antibiotic responsiveness, and host–microbiota interaction in the esophagus and have implications for gastrointestinal health and disease.
Keywords: Esophagus, Lactobacillales, Lactobacillus, Firmicutes, Bifidobacteria
Graphical Abstract
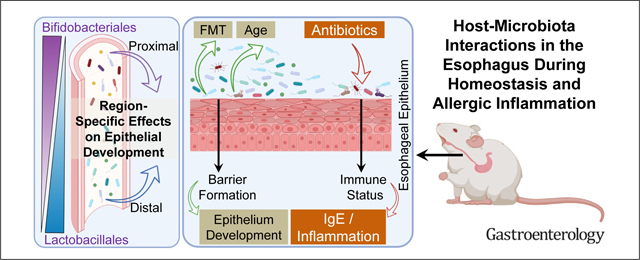
An increasing body of evidence supports a role for the microbiome in modulating immunity and susceptibility to a variety of diseases, including food allergy1–4 and asthma.5,6 Notably, the microbiota modulates inflammation in the gastrointestinal tract7,8 and airways,9 as bacteria-derived pathogen- and damage-associated molecular pattern derivatives stimulate epithelial cells to produce pro-inflammatory cytokines, such as thymic stromal lymphopoietin, thereby skewing T cell inflammatory responses.10–12 The ability of oral administration of specific bacterial strains to reduce inflammation demonstrates the therapeutic potential of microbiota manipulation.13–15
Focusing on the esophagus, there are multiple diseases, such as eosinophilic esophagitis (EoE), Barrett’s esophagus, and esophageal adenocarcinoma, that are associated with local esophageal dysbiosis and chronic inflammation.16–24 Antibiotic use in infancy and cesarean delivery, which modify microbial colonization, are risk factors for a number of these inflammatory diseases, including EoE.5,25–27 However, little knowledge exists regarding how the acquisition of microbiota influences esophageal tissue development and whether dysbiosis contributes to inflammation in this organ.16,17,28,29 We aimed to uncover fundamental properties of host–microbiota interactions in the esophagus during homeostasis and antibiotic-induced dysbiosis and to test their role in allergic inflammatory esophageal responses. Accordingly, we established the composition, transplantation potential, antibiotic responsiveness, and host–microbiota interactions of the esophagus. These findings have important implications for managing emerging inflammatory diseases of the esophagus, such as EoE.
Materials and Methods
Mice
C57BL/6 mice were maintained in a Pasteurella- and Helicobacter-free (PHF), specific pathogen–free (SPF) facility at the University of Chicago (UC) and at a standard SPF facility at Cincinnati Children’s Hospital Medical Center (CCHMC; Pasteurella- and Helicobacter-positive). In addition, we used conventionally housed (CNV) mice from outside of the CCHMC SPF facility for the histologic analysis only. All experiments were littermate controlled and performed in accordance with the Institutional Biosafety and Animal Care and Use Committees. The UC mouse colony, used in the first two main figures, was established from C57BL/6J. The CCHMC colony, used in the rest of the figures, was established from C57BL/6NCrl. Each experiment was controlled internally; data from the 2 facilities were not compared directly with each other. For housing details see the Supplementary Methods.
Fecal Matter Transplantation
For details about gnotobiotic mouse husbandry, please refer to the Supplementary Methods. All mice were weaned at 3 weeks of age and transitioned to plant-based mouse chow (Purina Lab Diet 5K67) and autoclaved sterile water. Germ-free (GF) mice received fecal transplants at weaning; cecal and fecal contents were collected from a PHF SPF donor, homogenized in 4.5 mL of sterile phosphate-buffered saline in a laminar flow hood, and delivered via an oral gavage of 500 μL per recipient GF mouse. Mice were housed in a gnotobiotic isolator (as described in the Supplementary Methods) for 4 weeks. The fecal matter transplantation (FMT) was littermate controlled, and the recipient mice were compared with PHF SPF mice from the same litter and euthanized at the same time (ie, 7–8 weeks of age).
Antibiotic Treatment
Neonatal antibiotic treatment was started at 14 days of age via intragastric gavage with 100 μL of sterile, filtered (0.22 μm) antibiotic mixture using a 24-gauge, 1-inch, flexible feeding needle.30 The antibiotics were vancomycin (10 mg/mL), neomycin (1 mg/mL), ampicillin (1 mg/mL), and metronidazole (1 mg/mL).31 After the mice were weaned, 10 mL of the 50X antibiotic mixture (with 0.5 mg/mL vancomycin instead of 10 mg/mL) was added to 500 mL of autoclaved water and supplemented with sucrose (adjusted to 2% sucrose solution) and the mice were allowed to drink freely for the time periods indicated. The water bottles were changed once every 2 weeks. The antibiotic mix was stored for a maximum of 2 weeks at 4°C.
Induction of Allergic Inflammation
Fourteen-day-old antibiotic-treated and drinking water– fed C57BL/6NCrl littermates were sensitized with 50 μg/ mouse Aspergillus fumigatus (Greer) and alum adjuvant (Fisher) via a single intraperitoneal injection. After 7 days, mice were challenged with intra-esophageal A fumigatus for 2 consecutive days at subsequent 2-day intervals (100 μg/mouse; 50% medium-chain triglyceride oil) over the course of another 3 weeks. Esophageal delivery was performed with an 18-gauge, 1.2-inch flexible, plastic feeding tube. The tube was perforated with an 18-gauge injection needle in 4 places at the proximal end and heat-sealed at the distal end to allow liquid to flow into the esophagus instead of the stomach. Mice were kept on antibiotics or regular autoclaved drinking water supplemented with 2% sucrose until euthanasia. To measure levels of A fumigatus–specific antibodies, enzyme-linked immunosorbent assay plates were precoated with 10 mg/mL of A fumigatus extracts (Greer) in phosphate-buffered saline (Fisher) and supplemented with complete EDTA-free protease inhibitors cocktail (Roche) for 16 hours at 4°C. Next, samples were processed according to the enzyme-linked immunosorbent assay manufacturer’s (Fisher) protocol, starting with the plate-washing step after the coating step. Serum cytokines were detected according to the enzyme-linked immunosorbent assay manufacturer’s (R&D) protocol.
16S Ribosomal RNA Sequencing
For tissue collection, DNA isolation, bacterial load assessment, and the 16S ribosomal RNA (rRNA) gene amplicon library preparation, please refer to the Supplementary Methods. Quantification of the operational taxonomic unit (OTU) abundance and taxonomy assignment were performed using Quantitative Insights into Microbial Ecology, version 1.9,32 following protocols similar to those described previously.3 After quality assessment of raw reads using FastQC (version 0.11.5),33 low-quality bases were trimmed and the 3’ ends of overlapping mates were merged using SeqPrep, version 1.2 (https://github.com/jstjohn/SeqPrep). De-multiplexed reads were clustered into OTUs using the open reference OTU picking protocol at 97% sequence identity against the Greengenes database (8/2013 release),34 followed by chimeric sequence removal using ChimeraSlayer, version 20110519, and taxonomy assignment using uclust. For the analyses of mouse samples only, both mouse and human samples, or human samples only, samples were rarefied to an even depth of 4081, 9505, or 9515 reads, respectively. The α-diversity (Shannon index) and β-diversity (weighted UniFrac distance35) were compared between groups using the Mann-Whitney U test (nonparametric) and permutational multivariate analysis of variance in the R package vegan, version 2.5.4,36 respectively. Taxa differentially abundant between groups were identified using discrete false-discovery rate37 with the parameters “transform_type=normdata, method=meandiff, alpha=0.10, numperm=1000, fdr_method=dsfdr” (accessed 2/26/2018). In each comparison, OTUs present in fewer than 8 samples were removed before the discrete false-discovery rate test. Taxa significantly enriched in 1 group compared with the other were detected using linear discriminant analysis effect size, version 1.0,38 and filtered by P < .05 (Kruskal-Wallis test) and by log10-transfomed linear discriminant analysis score ≥2.0 (or ≤ −2.0).
RNA Sequencing and Data Analysis
For tissue collection and RNA isolation, please refer to the Supplementary Methods. RNA-sequencing (RNA-seq) was performed at the Functional Genomics Facility at the University of Chicago. Libraries were prepared using the Illumina TruSeq Stranded mRNA kit for oligo dT library preparation, and samples were sequenced in 2 replicate lanes as 50-bp, single-end reads on a HiSeq4000 or a NovaSeq 6000 instrument. RNA-seq data from mouse esophageal tissues was analyzed using CLC Genomics Pro Suite (Qiagen), as described previously.39 Gene ontology and functional enrichment analyses were performed using ToppGene Suite (CCHMC), as described previously.40
Procurement and Processing of Human Esophageal Biopsies and Immunohistochemistry
This study was performed with the approval of the CCHMC Institutional Review Board. Informed consent was obtained from patients or their legal guardians to donate tissue samples for research and to have their clinical information entered into the Cincinnati Center for Eosinophilic Disorders database. Patients with active EoE were defined as those having 15 or more esophageal eosinophils per high-power microscopic field at the time of biopsy and not receiving swallowed glucocorticoid or dietary treatment at time of endoscopy. Controls were defined as those without history of EoE or esophageal eosinophilia and not receiving swallowed glucocorticoid or dietary treatment at time of endoscopy, but may have other gastrointestinal disorders. This study was performed retrospectively on the basis of existing samples and no patients were actively recruited. Consistent with the EoE Diagnostic Panel analysis, biopsies were derived from the distal esophagus (DE).41
Histology
Tissues were formalin-fixed and embedded in paraffin. Sample and slide preparation, H&E, and major basic protein (eosinophil marker) staining were performed as described elsewhere.42
Statistical Analysis
GraphPad Prism software was used for the indicated statistical analyses. P ≤.05 was considered statistically significant.
Author Statement
All authors had access to the study data and reviewed and approved the final manuscript.
Data Availability
The FastQ files of human and mouse 16S rRNA gene amplicon sequencing and mouse tissue RNA sequencing have been deposited in National Center for Biotechnology Information Sequence Read Archive database under BioProject accession numbers PRJNA743083, PRJNA694966, and PRJNA694967, respectively. Publicly available expression data were used for esophageal biopsies of patients with EoE or controls (GSE58640).43
Results
The Esophageal Microbiota Is Reconstituted by Transplantation of Feces to Germ-Free Mice
We first examined the role of the local tissue microenvironment in esophageal microbiota colonization. We evaluated whether FMT reconstituted the esophageal microbiota. GF mice were transplanted with feces from littermate control PHF SPF donor mice (see Materials and Methods). The FMT was carefully littermate controlled. FMT recipient mice were compared with PHF SPF mice from the same litter as the FMT donor mice and euthanized at the same time (ie, 7–8 weeks of age). The α-diversity (Shannon index) and bacterial load in all examined tissues showed no significant differences between the PHF SPF littermates and the GF recipients that received FMT in all tissues examined (Figure 1A and Supplementary Figure 1A). Principal coordinate analysis (PCoA), a measure of overall temporal variance in β-diversity among the same tissues, showed that the microbiota composition of GF mice after FMT was like the PHF SPF donor littermates (Figure 1B). Relative abundance analysis showed that the esophagus was enriched in Lactobacillales (Lactobacillaceae) and the colon was enriched in Bacteroidetes (Muribaculaceae). Taxa composition between PHF SPF and FMT mice was comparable in both the esophagus and colon; however, a larger variation was detected in taxa abundance in the oropharynx and outgrowth of Bacillales (Staphylococcaceae) was noted in the skin (Figure 1C). Taken together, these data indicate that the esophageal microbiota can be reconstituted successfully by FMT, demonstrating that the local microenvironment dictates the bacterial composition.
Figure 1.

Esophageal FMT feasibility and efficacy. 16S rRNA analysis of esophagus, colon, oropharynx, and skin from 8-week-old FMT mice compared with PHF SPF littermates of the FMT donor mice (inclusive of the donor): (A) α-diversity (Shannon index) of microbiota at indicated sites. (B) β-diversity PCoA of microbiota at indicated sites with weighted UniFrac distance. (C) Taxonomic composition of bacterial taxa at the family level. Each vertical bar or dot represents 1 individual mouse. Data are pooled from 2 independent experiments; n = 18 for each tissue (72 samples total): SPF, n = 9; FMT, n = 9 (esophagus, oropharynx, and skin) and n = 13 colon. Mann-Whitney U test was used in (A).
The Microbiota Shapes Epithelial Homeostatic Responses in the Esophagus
We next examined whether bacterial colonization might, in return, impact the local esophageal tissue microenvironment. We assessed how bacterial colonization affected esophageal gene expression. We first established that the GF esophageal transcriptome differed and clustered separately from the PHF SPF esophageal transcriptome (Figure 2A and B). FMT to GF mice greatly altered the transcriptome of both the proximal esophagus (PE) (Figure 2A) and DE (Figure 2B). RNA-seq gene expression analysis of differentially colonized PE (Figure 2C and Supplementary Table 1) and DE (Figure 2D and Supplementary Table 2) revealed 1229 and 1158 differentially expressed genes, respectively. Unsupervised clustering of differentially expressed genes showed that PHF SPF, GF, and FMT samples generated 3 functionally independent gene clusters in both segments of the esophagus (Figure 2C and D). The differentially expressed genes were enriched in pathways related to epithelial tissue homeostasis and cellular metabolic functions, including epithelium development, cell adhesion, nuclear body proteins, cellular respiration, and histone acetylation (Figure 2C and D and Supplementary Figure 2). Notably, esophageal microbiota colonization affected local transcriptional responses; distinct regional effects commonly affected 346 individual genes related to cell-to-cell interaction (cell adhesion) functions and differentially affected a total of 1695 common genes related to esophageal epithelial tissue structural functions, including epithelial differentiation (DE 812 genes) and extracellular matrix organization (PE 883 genes) (Supplementary Figure 3 and Supplementary Table 3). Taken together, these data revealed that the colonization affects tissue microenvironment, where esophageal transcriptome is affected by microbial colonization.
Figure 2.

The effect of the microbiota on epithelial homeostatic and immune-mediated responses in the esophagus. RNA-seq gene expression analysis of esophageal tissues from DEs and PEs of 8-week-old PHF SPF, GF, and FMT littermates: (A, B) PCoA of PE (A) and DE (B) samples. Direction of the shift from GF to FMT groups is shown with an arrow. (C, D) Heatmap of differentially expressed (>2 RPKM) and false discovery rate (FDR)–adjusted P < .05 genes with significantly enriched gene ontology terms annotation per cluster (Supplementary Figure 2) of proximal (C; Supplementary Table 2) and distal (D; Supplementary Table 3) esophagus samples. Data are representative from 2 independent experiments: SPF, n = 6; GF, n = 5; and FMT n = 6 mice.
Temporal Development of the Esophageal Microbiota
As mentioned, our initial experiments were performed using a PHF SPF barrier colony previously used in our studies of food allergy.30 We continued analysis of the temporal development of the esophageal microbiota in SPF colonies at CCHMC, which included Pasteurella and Helicobacter, thus more closely resembling the microbial diversity present in the human esophagus. Each experiment was internally controlled; data from the 2 facilities were not compared directly with each other.
First, we examined natural tissue colonization as a function of age. In contrast to the colon and skin, which showed a gradual and significant increase in α-diversity (Shannon index) over the first 4 weeks of life, the diversity of the esophageal and oropharynx microbiota did not change significantly over time (Figure 3A). However, growing variation in α-diversity (Shannon index) post weaning suggested increasing diversification among individuals as a function of age (Figure 3A). PCoA showed clear separation of samples as a function of age (Figure 3B). As further evident from the relative abundance analysis, the earliest transition toward stable colonization of microbial communities occurred before 3 days of age in the oropharynx and at 1 week after birth in the skin, 2 weeks after birth in the colon, and 4 weeks after birth in the esophagus (Figure 3C). Similar to what was observed in Figure 1, the adult esophageal microbiota was predominantly colonized by Firmicutes-Lactobacillales (Lactobacillaceae). However, in this SPF colony, we noted a transition from the early colonization with Proteobacteria (Pasteurellaceae), which was gradually replaced with other Firmicute taxa, such as Clostridiales and Bacteroidetes (Muribaculaceae). For comparison, we examined the age-dependent microbiota in several other tissues. Proteobacteria (Pasteurellaceae) predominantly colonized the oropharynx throughout life. The adult skin was colonized with Planococcaceae with less Bacteroidetes (Muribaculaceae) and Proteobacteria (Pasteurellaceae) than skin at earlier time points. Firmicutes (Clostridia) and Bacteroidetes (Muribaculaceae) colonized the adult colon, transitioning from Proteobacteria (Pasteurellaceae) after 1 week of age.
Figure 3.
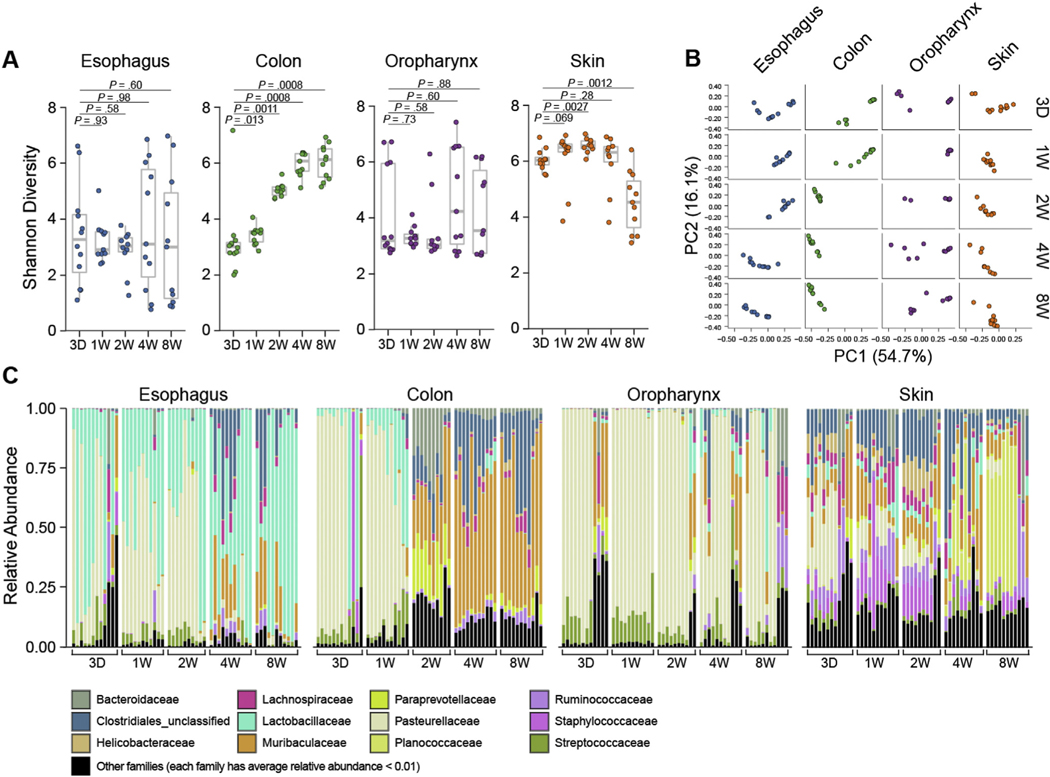
Temporal profile of the esophageal microbiota. 16S rRNA analysis of esophagus, colon, oropharynx, and skin from SPF littermates: (A) α-diversity (Shannon index) of microbiota over time. (B) β-diversity PCoA of microbiota over time with weighted UniFrac distance. (C) Taxonomic composition of bacterial taxa at the family level over time since birth (ie, mouse age). Each vertical bar or dot represents 1 individual mouse. Data are pooled from 3 independent experiments; n = 55 for each tissue (220 samples total); 3 days, n = 12; 1 week, n = 11; 2 weeks, n = 10; 4 weeks, n = 11 and 8 weeks, n = 11. Mann-Whitney U test was used in (A). D, day; W, week.
Taken together, these data indicate that the microbiota is fully established in the esophagus by 1 month (4 weeks) of age and that the murine esophageal microbiota is unique, diverse, and dominated by Lactobacillales, like the human esophagus.22,28
Distal and Proximal Esophageal Microbiota Profiles and Esophageal Tissue Morphology
Although most studies concerning inflammatory diseases in the esophagus focus on analysis of the esophageal biopsies from the DE, there are regional differences, such as the composition of muscle.44 We had already noted differences in the transcriptomes of the DE and PE post FMT in Figure 2. We therefore continued with an examination of microbial community structure in the DE and PE in adult SPF mice. Measurement of α-diversity (Shannon index) suggested diversification within the esophagus, with clear separation between distal and proximal sites (Figure 4A). Likewise, the overall variance in β-diversity among the samples showed clear separation of the DE and PE by PCoA (Figure 4B). Furthermore, differential abundance analysis showed distinct microbiota profiles with 34 and 112 OTUs distinguishing the DE and PE, respectively (Figure 4C and Supplementary Table 4). Differential abundance analysis indicated that the Firmicutes Lactobacillaceae, but not Streptococcaceae, were more prevalent in the DE, and Proteobacteria (Pasteurellaceae), Bacteroidetes (Muribaculaceae), and Actinobacteria (Bifidobacteriaceae) were more prevalent in the PE (Figure 4D). Similarly, phylogenetic analysis differentiated the taxonomic profiles of the PE and DE microbiota (Figure 4E). Taken together, these analyses demonstrate an impact of the regional microenvironment and anatomic location on microbiota colonization in the esophagus.
Figure 4.

Microbiota profiles of the DE and PE. 16S rRNA analysis from DE and PE of 8-week-old SPF littermates (see Supplementary Table 4): (A) α-diversity (Shannon index) of microbiota at indicated sites. (B) β-diversity PCoA of microbiota at indicated sites with weighted UniFrac distance. (C) Heatmap of differentially abundant OTUs between sites (discrete false discovery rate [DS-FDR] 0.10; Supplementary Table 1) labeled at the family level (right). (D) Linear discriminant analysis effect size (LEfSe) of bacterial taxa enriched at indicated sites. (E) Phylogenetic analysis of esophageal microbiota at indicated sites. (A–E) Data are pooled from 3 independent experiments; n = 24 for each site (48 total samples): 24 for DE and 24 for PE. Mann-Whitney U test was used in (A), DS-FDR in (C), and Kruskal-Wallis test in (D). LDA, linear discriminant analysis.
Finally, we assessed the effect of microbial colonization on esophageal tissue morphology in both proximal and distal anatomic locations (Supplementary Figure 4). Soon after birth (day 3), the PE epithelium was not keratinized and resembled oral buccal epithelium consisting of 2 layers—basal and superficial (Supplementary Figure 4A). The spinous layer was not clearly defined until week 2, and granular and stratum corneum layers were visible in young adult mice at the age of 4 weeks (Supplementary Figure 4A). We next compared GF and CNV mice, which contain potential pathobionts. Notably, adult GF mice had less defined and disorganized stratum corneum and mucosal layers compared with CNV counterparts (Supplementary Figure 4B), resembling earlier time points of PE tissue development (Supplementary Figure 4A). The DE already had less resemblance to the oral buccal epithelium at the age of 1 week; spinous and superficial layers were already visible (Supplementary Figure 4C) compared with the proximal anatomic location (Supplementary Figure 4A). However, like the proximal part, the DE granular and stratum corneum layers were clearly defined only in young adult mice at the age of 4 weeks (Supplementary Figure 4A and C). Like the PE, the DE epithelium in adult GF mice had less defined and disorganized stratum corneum and mucosal layers compared with CNV counterparts (Supplementary Figure 4D).
Taken together, these analyses are consistent with an effect of microbiota colonization on the esophageal transcriptome (Figure 2) and demonstrate that microbiota colonization promotes epithelium development and alters tissue morphology in both PE and DE.
Commonalities of Microbiota Colonization and Microbiota-Regulated Functions in the Mouse and Human Esophagus
We next aimed to determine whether there is overlap in bacterial communities of the murine and human esophagus. We compared the murine esophageal microbiota of the PE and DE (Figure 4) with non-EoE human esophageal biopsy–derived microbiota (Supplementary Figure 5; Supplementary Table 5– including disease status and additional patient information). Notably, human esophageal biopsies are derived almost exclusively from the DE.44 Relative abundance analysis demonstrated that both proximal and distal parts of the murine esophagus and the distal part of the human esophagus are colonized by Firmicutes (Lactobacillales). Lactobacillales (Streptococcaceae) and Proteobacteria (Pasteurellaceae) were dominant in the murine PE and human DE alike (Supplementary Figure 5A). Bacteroidetes were also present in human and both parts of murine esophagi, however, Bacteroidetes Prevotellaceae taxa in humans were instead represented as Bacteroidetes Muribaculaceae in mice (Supplementary Figure 5A). We next looked at the disease-associated changes of human esophageal microbiome in EoE compared with non-EoE human controls. Twentyseven OTUs were differentially abundant between human non-EoE controls and patients with EoE. The esophageal microbiome in patients with EoE was characterized by dysbiosis, where Firmicutes Lactobacillales (Streptococcaceae) were markedly less abundant in EoE (Supplementary Figure 5B).
We asked whether dysbiosis in EoE may be affecting disease-associated esophageal epithelial gene expression shifts seen in EoE.43,45 We integrated murine genes differentially expressed due to colonization (RNA-seq of murine esophageal samples of PHF SPF, GF, and FMT mice) (Figure 2) with differentially expressed genes in EoE (RNA-seq of esophageal biopsies from patients with EoE compared with healthy controls43) (Supplementary Table 6). We found a total of 336 common genes that were differentially expressed in the murine esophagus due to colonization status were also differentially expressed in tissues from patients with EoE compared with healthy controls (Supplementary Table 6). The overlapping genes were involved in functional processes related to extracellular matrix organization (124 genes, including kallikrein related peptidase 5 [KLK5) and collagens; PE), epithelial differentiation (146 genes, including hypoxia inducible factor 1 subunit alpha [HIF1A) and cytokeratins; DE), and cell migration (66 genes including periostin [POSTN]; commonly affected) (Supplementary Figure 6 and Supplementary Table 7).
Taken together, this analysis identified commonalities of microbiota colonization of human and murine esophagus, as well as disease-associated changes of esophageal microbiota in humans. Furthermore, we showed that esophageal epithelial barrier genes and pathways may be commonly affected due to colonization and depend on the disease status in mice and humans, respectively.
Antibiotic Treatment Leads to Tissue- and Time- Dependent Esophageal Microbiota Dysbiosis
We examined the temporal effect of exposure to a cocktail of broad-spectrum antibiotics that is documented to deplete the colonic microbiota30 on the community structure of the esophageal microbiota. Esophageal bacterial load decreased more than 10-fold in response to antibiotic treatment; the bacterial load of the colon served as a positive control (Supplementary Figure 1B). The α-diversity (Shannon index) decreased slightly (P = .056) over the first week and then bounced back at the end of the second week (Figure 5A). Similar trends were observed in oropharynx and skin, but not the colon, in which α-diversity decreased steadily over time (Figure 5A). β-diversity PCoA showed that the overall clustering of the esophageal microbiota in untreated control mice differed from that of antibiotic-treated mice at each time point examined, demonstrating antibiotic-induced dysbiosis in the esophagus (Figure 5B). Oropharynx and skin β-diversity undergo relatively modest changes and stabilized after 1 week of treatment, and colon β-diversity continued to change over time (Figure 5B). Relative abundance analysis demonstrated gradual depletion of endogenous taxa as a function of the duration of antibiotic exposure, with notable increases in Lachnospiraceae and Bacteroidaceae in all tissues examined (Figure 5C). The esophagi of antibiotic-treated mice demonstrated depletion of Firmicutes Lactobacillales (Lactobacillaceae) (Figure 5C). Indeed, differential abundance analysis showed relatively higher abundance of Mollicutes, Proteobacteria, and Actinobacteria (Supplementary Figure 7A). The microbiota in other tissues were also examined and found to be depleted, especially in the colon, where Muribaculaceae and Prevotellaceae, as well as some Clostridiales, were initially replaced with Porphyromonadaceae and Alcaligenaceae, which were eventually completely swapped with Mycoplasmataceae (Figure 5C). Differential abundance analysis further showed increased relative abundance of Verrucomicrobiae and Mollicutes in the colon (Supplementary Figure 7B). The oropharynx and skin were depleted of Muribaculaceae concurrent with relative outgrowth of Bacteroidaceae and Ruminococcaceae (Figure 5C). Differential abundance analysis demonstrated a relative increase of Pseudomonadales in the oropharynx (Supplementary Figure 7C) and of Methylobacteria and Porphyrobacteria in the skin (Supplementary Figure 7D).
Figure 5.
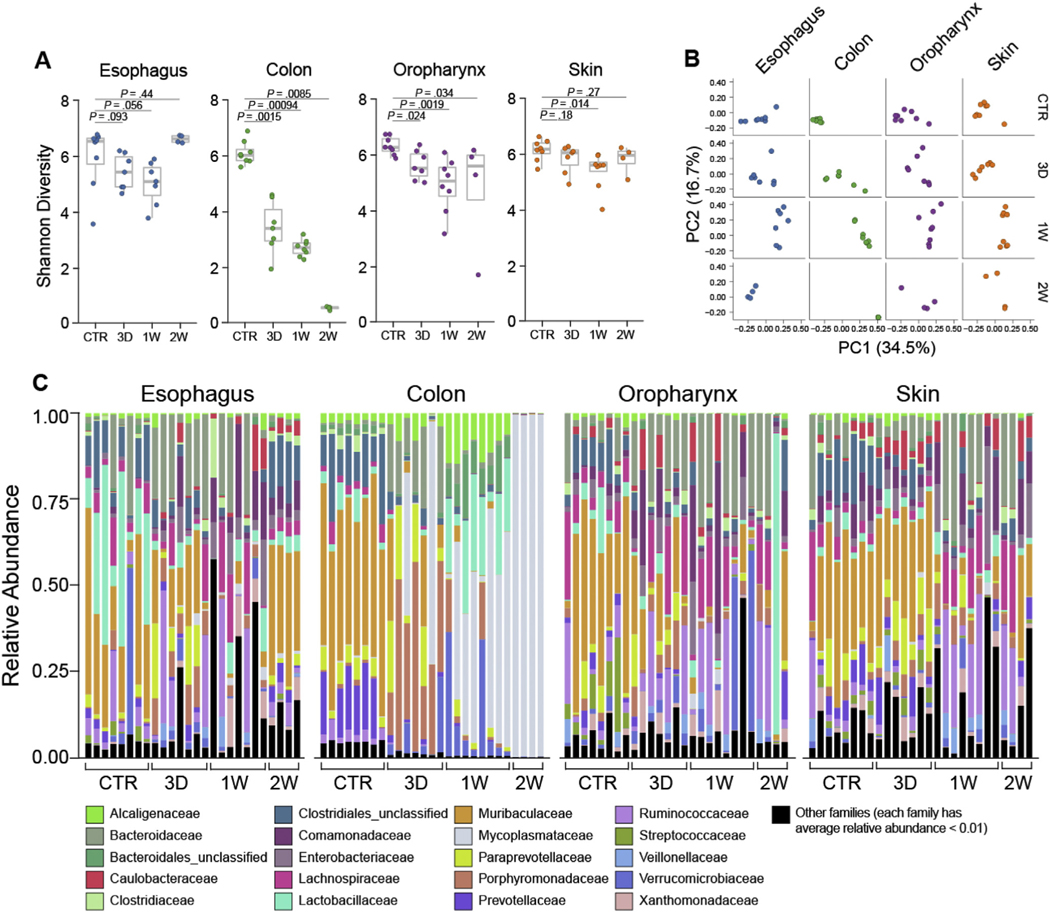
Effect of antibiotic treatment on tissue- and time-dependent esophageal microbiota.16S rRNA analysis of esophagus, colon, oropharynx, and skin from 8-week-old SPF littermates treated with antibiotics for the indicated period of time or untreated controls: (A) α-diversity (Shannon index) of microbiota at indicated sites. (B) β-diversity analysis PCoA of microbiota at indicated sites with weighted UniFrac distance. (C) Taxonomic composition of bacterial taxa at the family level over time. Each vertical bar or dot represents 1 individual mouse. Data are pooled from 2 independent experiments; n = 24 for each tissue (96 total samples): SPF control (CTR), n = 8; 3D, n = 7; 1W, n = 8; 2W, n = 4. Mann-Whitney U test was used in (A). CTR, control; D, day; W, week.
Overall, these data indicate that treatment with broad-spectrum systemic antibiotics induced dysbiosis, significantly affecting the Lactobacillales that were dominant under homeostatic conditions in the esophagus. Notably, EoE-associated dysbiosis was also characterized by the loss of Lactobacillales (Supplementary Figure 5B).
Neonatal Use of Antibiotics Promotes Type 2 Inflammation in a Murine Model of Eosinophilic Esophagitis
We contextualized atopy-associated microbial dysbiosis in infancy5,25,27 with the development of type 2 inflammation and investigated whether antibiotic exposure starting at the preweaning stage would exacerbate type 2–associated esophageal inflammation in a murine model of EoE later in life. This temporal scheme induces lasting dysbiosis30 and models early-life antibiotic exposure that has been shown to be a risk for the development of human EoE.26,27,46 Indeed, both antibiotic treatment and EoE-associated dysbiosis were characterized by the loss of Lactobacillales (Figure 5C and Supplementary Figures 5B and 7A). Accordingly, we administered broad-spectrum antibiotics by oral gavage during sensitization with an allergen (A fumigatus). Control mice were sensitized with A fumigatus but received water instead of antibiotics by oral gavage. Starting at weaning, antibiotics were added to the drinking water, and both experimental groups were repeatedly challenged with intra-esophageal allergen over a course of 3 weeks (Figure 6A). Allergen-challenged mice developed eosinophilia in esophageal tissue, irrespective of antibiotic treatment (Figure 6B–E). However, mice that received both antibiotic treatment and allergen challenge had higher esophageal eosinophil counts and allergen-specific IgE (Figure 6F and G), but lower allergen-specific IgG1 (Figure 6H) and IgG2a (Figure 6I) than control mice that were allergen challenged but not treated with antibiotics. In addition, antibiotic treatment increased the concentration of the type 2 cytokines interleukin (IL)-5 and IL-13 in the serum of allergen-challenged mice (Figure 6J–K). Allergen-challenged mice that did not receive antibiotics had increased levels of the type 1 cytokine interferon-gamma compared with non-antibiotic–treated mice (Figure 6L).
Figure 6.

Effect of neonatal use of antibiotics on type 2 inflammation in murine model of EoE. (A) Experimental procedure schematic. (B–E) Representative eosinophil levels as determined by anti–major basic protein (MBP) staining of esophageal tissue (×10 magnification; MBP-positive eosinophils are indicated with arrows) from control (regular drinking water) naïve mice (B), mice whose drinking water was supplemented with antibiotics (H2O Abx) and who were allergen challenged (Allergen; A fumigatus) (C), mice whose drinking water was supplemented with antibiotics (H2O Abx) but who were naïve (D), or mice who were provided regular drinking water but were allergen challenged (E). (F) Eosinophil counts from anti-MBP staining of esophageal tissue from mice treated as indicated. (G–I) Allergen–specific antibodies in serum of mice from above groups by enzyme-linked immunosorbent assay. (J) IL-5, (K) IL-13, and (L) Interferon-gamma in the mouse sera from the aforementioned groups. Each dot represents the mean from a technical duplicate of samples from each individual mouse: H2O control, n = 22; Allergen Abx, n = 17; H2O Abx alone, n = 13; Allergen alone, n = 14. Data are represented as mean ± SD and are pooled from 3 independent experiments. Statistics by 1-way analysis of variance. ****P ≤ .0001. Abx, antibiotics; E, epithelium; LP, lamina propria; ROD, relative optical density.
Taken together, these data indicate that antibiotic use in early life and esophageal dysbiosis, characterized by the loss of Lactobacillales, promote type 2 inflammation in the esophagus in response to environmental allergens.
Discussion
In this report, we aimed to identify the fundamental properties of the esophageal microbiota, including their potential function. Human clinical data demonstrate that the dominant early colonizers of the normal infant gut are maternal fecal bacteria and, therefore, FMT is a natural course of colonization in human infants47,48; however, the role of this route in the colonization of the esophagus has not yet been described. Notably, cesarean section deprives human infants of the natural course of colonization by the fecal–oral route47,48 and it is a risk factor for EoE.26 We demonstrated that the esophageal microbiota can be restored after FMT to GF mice. We report that the esophageal microbiota is unique compared with that of the oropharynx, skin, and colon and the murine esophagus is colonized with populations of Firmicutes, particularly Lactobacillales. We established that the local esophageal microenvironment dictates the diversification of bacterial colonization. Specifically, the DE was enriched in Lactobacillaceae and had less Bifidobacteriaceae than its proximal counterpart. Indeed, the esophagus is the uppermost gastrointestinal compartment and, as such, has no strict anaerobic conditions due to the proximity to the oropharynx and food/oral oxygenation, has higher pH, and has reduced salts (ie, no bile salts) and IgA levels49 compared with the lower gastrointestinal compartments. Subsequently, these anatomic and physiological properties of the esophageal tissue generate a unique selective and differentiating growth microenvironment. It is therefore plausible that regional microbiota colonization patterns parallel observed distinct changes in the host esophageal transcriptome as a function of location and colonization, modifying homeostatic pathways, including those associated with epithelial barrier function. The distinction between the DE and PE underscores the importance of evaluating these tissues separately, especially because regional differences in cellular constituents and tissue architecture have been reported.50 Notably, Streptococcaceae were not detectable in the esophagi of all of the recipients of FMT or their PHF SPF littermates, which is consistent with natural transient variation in colonization.51 However, it is plausible that Streptococcaceae are most abundant in the PE and not always detectable in whole tissue esophagus analysis, underscoring the significance of site-specific microbiota analysis. Indeed, there is evidence that the co-involvement of the PE and DE may be a more general feature of EoE than of gastroesophageal reflux disease50,52 and that the PE may have distinct responses to some medications, such as fluticasone.53 In line with these premises, we have integrated murine genes differentially expressed due to colonization in the PE and DE with differentially expressed esophageal genes in EoE. This intersection revealed that most common genes (212 of 336) were from the DE and functionally related to epithelial cellular homeostatic functions, such as differentiation and cell migration. We suggest that the murine DE is suited for study of human disease pathophysiology as a function of microbiota colonization, especially because most studies of esophageal inflammation in humans are performed on the DE. Interestingly, we have identified that periostin (POSTN) expression was universally affected in the DE and PE and in EoE as a function of microbiota colonization. Indeed, POSTN has been linked to the development of type 2 immunity, including EoE.46,54 We have also detected changes in genes shown to be cardinal in EoE pathogenesis, including KLK5 (encoding kallikrein related peptidase 5)55 and HIF1A (encoding hypoxia inducible factor 1 subunit alpha).56 Finally, we demonstrated that esophageal microbiota populations undergo dynamic changes from infancy into adulthood. Microbiota colonization impacts esophageal tissue morphology and promotes epithelial differentiation and transition from a simple bilayer in the neonatal stage to complex multilayer squamous epithelium in adulthood. Indeed, these features of keratinized squamous epithelium were partially lost in GF mice compared with CNV counterparts. We postulate that microbiota colonization is essential to the normal homeostatic function in the esophagus and the baseline accumulation of Lactobacillales adds to the intrinsic protective role of the homeostatic esophagus and its components. In both the UC and CCHMC SPF facilities, Firmicutes (Lactobacillales) was one of the major species in the esophageal microbiota. Our data also demonstrated that Lactobacillales are present in healthy human esophageal tissue. Furthermore, we showed that disease-associated dysbiosis in EoE is characterized by the profound loss of Lactobacillales, which may execute protective effects for allergies and antiinflammatory functions,13,57,58 consistent with an increasing body of evidence that substantiates the role of the environment and microbial dysbiosis on susceptibility to EoE.25,27 Notably, supplementation with Lactobacilli alleviated pathogenicity and decreased esophageal eosinophilia in a murine model of EoE.13
Neonatal use of antibiotics has been associated with increased risk for a number of inflammatory diseases, including EoE.25,27 We showed that microbiota dysbiosis characterized by the loss of Lactobacillales is a hallmark of the EoE esophageal microenvironment. We, therefore, aimed to test the effect of in vivo exposure to antibiotics on the esophageal microbiota in a model of neonatal induction of lasting dysbiosis.30 We showed that antibiotic treatment induced depletion of homeostatic microbiota associated with protective functions, including esophageal Lactobacillales,5,13,16,57–60 and a relative increase of colonization by inflammation-associated bacteria, including Actinobacteria and Mollicutes (Mycoplasmataceae).59–61 These data indicate that the esophageal microbiome is modified by systemic use of broad-spectrum antibiotics. Broad-spectrum antibiotics may cause depletion of protective bacteria and outgrowth of bacterial species associated with human atopic inflammation.58–61 Notably, the proportional increase in bacterial species, such as Mycoplasma and Actinobacteria, could be linked to amplification of eosinophil responses in EoE.5,62–65 We therefore assessed the effect of neonatal use of antibiotics on the development of allergen-induced inflammation in the esophagus. Indeed, use of antibiotics resulted in higher eosinophil infiltration in the esophagus and increased allergen-specific IgE and the pro-Th2 cytokines IL-5 and IL-13. Overall, these data suggest that neonatal use of antibiotics promotes type 2 responses, underscoring the role of microbial dysbiosis in esophageal eosinophilic inflammation.
In conclusion, we have described the basic properties of the esophageal microbiome in health and allergic states in mice and humans. We report that FMT has the capacity to restore the resident esophageal microbiota of GF mice, providing a basis for considering FMT for esophageal diseases. In addition, the finding that microbiota colonization promotes esophageal barrier function and that systemic antibiotic exposure induces esophageal dysbiosis and exacerbates type 2 allergic inflammation in the esophagus provides a framework to further understand why early-life antibiotic exposure is a risk factor for developing EoE. Collectively, these data establish host–microbiota interactions of the esophagus and have important implications for managing emerging inflammatory diseases of the esophagus, such as EoE.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Disruption of microbial colonization are risk factors for esophageal inflammatory diseases, including EoE; however, host–microbiota interaction in the esophagus remains unclear.
NEW FINDINGS
FMT can restore the esophageal microbiota, dominated by Lactobacillales. Microbial colonization shows spatial heterogeneity and modifies tissue morphology and gene expression, and dysbiosis exaggerates type 2 inflammation.
LIMITATIONS
Although these findings have implications for understanding EoE, deeper analysis of human systems is needed.
IMPACT
We provide a framework to understand the role of microbiota in the esophagus and lay the foundation for considering FMT for the treatment of esophageal diseases.
Acknowledgments
The authors are grateful to Stephanie M. Greenwald and Sarah Owens for 16S ribosomal RNA amplicon library preparation and sequencing at Argonne National Laboratory. The authors would like to thank Shawna Hottinger for editorial support.
These authors disclose the following: Cathryn R. Nagler is president and co-founder of ClostraBio, Inc. Marc E. Rothenberg is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celgene, Astra Zeneca, Arena Pharmaceuticals, Glaxo Smith Kline, Revolo Biotherapeutics, and Guidepoint and has an equity interest in the Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, and Allakos and royalties from Reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust), and UpToDate and is an inventor of patents owned by Cincinnati Children’s Hospital.
Funding
Marc E. Rothenberg was supported by NIH R01 A1045898, the CURED Foundation, and Dave and Denise Bunning Sunshine Foundation. Cathryn R. Nagler was supported by NIH R01 AI146099 and the Moss Family Foundation. The Center for Research Informatics is funded by the Biological Sciences Division at the University of Chicago, with additional support provided by The Institute for Translational Medicine, Clinical and Translational Award (NIH 5UL1 TR002389-02) and The University of Chicago Comprehensive Cancer Center Support Grant (NIH P30 CA014599).
Abbreviations used in this paper:
- CCHMC
Cincinnati Children’s Hospital Medical Center
- CNV
conventionally housed
- DE
distal esophagus
- EoE
eosinophilic esophagitis
- FMT
fecal matter transplantation
- GF
germ-free
- IL
interleukin
- OTU
operational taxonomic unit
- PCoA
principal coordinate analysis
- PE
proximal esophagus
- PHF
Pasteurella- and Helicobacter-free
- rRNA
ribosomal RNA
- RNA-seq
RNA sequencing
- SPF
specific pathogen-free
- UC
University of Chicago
Footnotes
Conflicts of interest
The remaining authors disclose no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http://doi.org/10.1053/j.gastro.2021.10.002.
Data Availability Statement
The FastQ files of human and mouse 16S ribosomal RNA gene amplicon sequencing and mouse tissue RNA sequencing have been deposited in National Center for Biotechnology Information Sequence Read Archive database under BioProject accession numbers PRJNA743083, PRJNA694966, and PRJNA694967, respectively. Publicly available expression data were used for esophageal biopsies of patients with EoE or controls (GSE58640).
References
- 1.Iweala OI, Nagler CR. The microbiome and food allergy. Annu Rev Immunol 2019;37:377–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vujkovic-Cvijin I, Sklar J, Jiang L, et al. Host variables confound gut microbiota studies of human disease. Nature 2020;587:448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feehley T, Plunkett CH, Bao R, et al. Healthy infants harbor intestinal bacteria that protect against food allergy. Nat Med 2019;25:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Gadir A, Stephen-Victor E, Gerber GK, et al. Microbiota therapy acts via a regulatory T cell MyD88/RORgammat pathway to suppress food allergy. Nat Med 2019;25:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujimura KE, Lynch SV. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe 2015;17:592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Depner M, Taft DH, Kirjavainen PV, et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat Med 2020;26:1766–1775. [DOI] [PubMed] [Google Scholar]
- 7.Zhou L, Chu C, Teng F, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 2019;568:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu SE, Hashimoto-Hill S, Woo V, et al. Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature 2020;586:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trompette A, Gollwitzer ES, Yadava K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 2014;20:159–166. [DOI] [PubMed] [Google Scholar]
- 10.Sherrill JD, Gao PS, Stucke EM, et al. Variants of thymic stromal lymphopoietin and its receptor associate with eosinophilic esophagitis. J Allergy Clin Immunol 2010;126:160–165.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Josefowicz SZ, Niec RE, Kim HY, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012;482:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Britton GJ, Contijoch EJ, Mogno I, et al. Microbiotas from humans with inflammatory bowel disease alter the balance of gut Th17 and RORgammat(+) regulatory T cells and exacerbate colitis in mice. Immunity 2019;50:212–224.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holvoet S, Doucet-Ladeveze R, Perrot M, et al. Beneficial effect of Lactococcus lactis NCC 2287 in a murine model of eosinophilic esophagitis. Allergy 2016;71:1753–1761. [DOI] [PubMed] [Google Scholar]
- 14.Planer JD, Peng Y, Kau AL, et al. Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 2016;534:263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shepherd ES, DeLoache WC, Pruss KM, et al. An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 2018;557:434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benitez AJ, Hoffmann C, Muir AB, et al. Inflammation-associated microbiota in pediatric eosinophilic esophagitis. Microbiome 2015;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amir I, Konikoff FM, Oppenheim M, et al. Gastric microbiota is altered in oesophagitis and Barrett’s oesophagus and further modified by proton pump inhibitors. Environ Microbiol 2014;16:2905–2914. [DOI] [PubMed] [Google Scholar]
- 18.Walker MM, Talley NJ. Review article: bacteria and pathogenesis of disease in the upper gastrointestinal tract—beyond the era of. Helicobacter pylori. Aliment Pharmacol Ther 2014;39:767–779. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Chaudhary N, Baghdadi J, et al. Microbiome in reflux disorders and esophageal adenocarcinoma. Cancer J 2014;20:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baghdadi J, Chaudhary N, Pei Z, et al. Microbiome, innate immunity, and esophageal adenocarcinoma. Clin Lab Med 2014;34:721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blackett KL, Siddhi SS, Cleary S, et al. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett’s and oesophageal carcinoma: association or causality? Aliment Pharmacol Ther 2013;37:1084–1092. [DOI] [PubMed] [Google Scholar]
- 22.Neto AG, Whitaker A, Pei Z. Microbiome and potential targets for chemoprevention of esophageal adenocarcinoma. Semin Oncol 2016;43:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L, Francois F, Pei Z. Molecular pathways: pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett esophagus. Clin Cancer Res 2012;18:2138–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Lu X, Nossa CW, et al. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009;137:588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen ET, Kappelman MD, Kim HP, et al. Early life exposures as risk factors for pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2013;57:67–71. [DOI] [PubMed] [Google Scholar]
- 26.Alexander ES, Martin LJ, Collins MH, et al. Twin and family studies reveal strong environmental and weaker genetic cues explaining heritability of eosinophilic esophagitis. J Allergy Clin Immunol 2014;134:1084–1092.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen ET, Kuhl JT, Martin LJ, et al. Early-life environmental exposures interact with genetic susceptibility variants in pediatric patients with eosinophilic esophagitis. J Allergy Clin Immunol 2018;141:632–637.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pei Z, Bini EJ, Yang L, et al. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A 2004;101:4250–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fillon SA, Harris JK, Wagner BD, et al. Novel device to sample the esophageal microbiome—the esophageal string test. PLoS One 2012;7:e42938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefka AT, Feehley T, Tripathi P, et al. Commensal bacteria protect against food allergen sensitization. Proc Natl Acad Sci U S A 2014;111:13145–13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgun A, Dzutsev A, Dong X, et al. Uncovering effects of antibiotics on the host and microbiota using trans-kingdom gene networks. Gut 2015;64:1732–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrews S. FastQC: A quality control application for high throughput sequence data. Babraham Institute. Accessed March 17, 2018. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- 34.McDonald D, Price MN, Goodrich J, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012;6:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lozupone C, Lladser ME, Knights D, et al. UniFrac: an effective distance metric for microbial community comparison. ISME J 2011;5:169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oksanen J, Blanchet FG, Friendly M, et al. vegan: Community Ecology Package. R package version 2.5–2, 2018. Accessed February 23, 2018. Available at: https://CRAN.R-project.org/package=vegan [Google Scholar]
- 37.Jiang L, Amir A, Morton JT, et al. Discrete false-discovery rate improves identification of differentially abundant microbes. mSystems 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621–628. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Bardes EE, Aronow BJ, et al. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Min S, Shoda T, Wen T, et al. Diagnostic merits of the Eosinophilic Esophagitis Diagnostic Panel from a single esophageal biopsy [published online ahead of print August 8, 2021]. J Allergy Clin Immunol doi: 10.1016/j.jaci.2021.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Straumann A, Conus S, Degen L, et al. Budesonide is effective in adolescent and adult patients with active eosinophilic esophagitis. Gastroenterology 2010; 139:1526–1537; 1537.e1. [DOI] [PubMed] [Google Scholar]
- 43.Sherrill JD, Kiran KC, Blanchard C, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014;15:361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adachi K, Mishiro T, Tanaka S, et al. Suitable biopsy site for detection of esophageal eosinophilia in eosinophilic esophagitis suspected cases. Dig Endosc 2016;28:139–144. [DOI] [PubMed] [Google Scholar]
- 45.Rochman M, Travers J, Miracle CE, et al. Profound loss of esophageal tissue differentiation in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2017; 140:738–749.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyles J, Rothenberg M. Role of genetics, environment, and their interactions in the pathogenesis of eosinophilic esophagitis. Curr Opin Immunol 2019;60:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helve O, Dikareva E, Stefanovic V, et al. Protocol for oral transplantation of maternal fecal microbiota to newborn infants born by cesarean section. STAR Protoc 2021;2:100271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korpela K, Helve O, Kolho KL, et al. Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: a proof-of-concept study. Cell 2020;183:324–334.e5. [DOI] [PubMed] [Google Scholar]
- 49.Rosenberg CE, Mingler MK, Caldwell JM, et al. Esophageal IgG4 levels correlate with histopathologic and transcriptomic features in eosinophilic esophagitis. Allergy 2018;73:1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodland P, Aktar R, Mthunzi E, et al. Distinct afferent innervation patterns within the human proximal and distal esophageal mucosa. Am J Physiol Gastrointest Liver Physiol 2015;308:G525–G531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wesemann DR, Nagler CR. The microbiome, timing, and barrier function in the context of allergic disease. Immunity 2016;44:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roman S, Savarino E, Savarino V, et al. Eosinophilic oesophagitis: from physiopathology to treatment. Dig Liver Dis 2013;45:871–878. [DOI] [PubMed] [Google Scholar]
- 53.Butz BK, Wen T, Gleich GJ, et al. Efficacy, dose reduction, and resistance to high-dose fluticasone in patients with eosinophilic esophagitis. Gastroenterology 2014;147:324–333.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanchard C, Mingler MK, McBride M, et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal Immunol 2008;1:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Azouz NP, Klingler AM, Pathre P, et al. Functional role of kallikrein 5 and proteinase-activated receptor 2 in eosinophilic esophagitis. Sci Transl Med 2020;12(545):eaaz7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masterson JC, Biette KA, Hammer JA, et al. Epithelial HIF1alpha/claudin-1 axis regulates barrier dysfunction in eosinophilic esophagitis. J Clin Invest 2019;129:3224–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ozdemir O. Various effects of different probiotic strains in allergic disorders: an update from laboratory and clinical data. Clin Exp Immunol 2010;160:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nance CL, Deniskin R, Diaz VC, et al. The role of the microbiome in food allergy: a review. Children (Basel) 2020;7(6):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bridgman SL, Kozyrskyj AL, Scott JA, et al. Gut microbiota and allergic disease in children. Ann Allergy Asthma Immunol 2016;116:99–105. [DOI] [PubMed] [Google Scholar]
- 60.Banskar S, Detzner AA, Juarez-Rodriguez MD, et al. The Pglyrp1-regulated microbiome enhances experimental allergic asthma. J Immunol 2019;203:3113–3125. [DOI] [PubMed] [Google Scholar]
- 61.Kumar S, Roy RD, Sethi GR, et al. Mycoplasma pneumoniae infection and asthma in children. Trop Doct 2019;49:117–119. [DOI] [PubMed] [Google Scholar]
- 62.Rosenberg HF, Masterson JC, Furuta GT. Eosinophils, probiotics, and the microbiome. J Leukoc Biol 2016; 100:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Defilippi AC, Silvestri M, Giacchino R, et al. Changes in blood eosinophil numbers during Mycoplasma pneumoniae infection in wheezing and non-wheezing, atopic and non-atopic children. Pediatr Int 2008;50:718–721. [DOI] [PubMed] [Google Scholar]
- 64.Medina JL, Coalson JJ, Brooks EG, et al. Mycoplasma pneumoniae CARDS toxin exacerbates ovalbumin-induced asthma-like inflammation in BALB/c mice. PLoS One 2014;9:e102613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Medina JL, Coalson JJ, Brooks EG, et al. Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am J Respir Cell Mol Biol 2012;46:815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The FastQ files of human and mouse 16S rRNA gene amplicon sequencing and mouse tissue RNA sequencing have been deposited in National Center for Biotechnology Information Sequence Read Archive database under BioProject accession numbers PRJNA743083, PRJNA694966, and PRJNA694967, respectively. Publicly available expression data were used for esophageal biopsies of patients with EoE or controls (GSE58640).43
The FastQ files of human and mouse 16S ribosomal RNA gene amplicon sequencing and mouse tissue RNA sequencing have been deposited in National Center for Biotechnology Information Sequence Read Archive database under BioProject accession numbers PRJNA743083, PRJNA694966, and PRJNA694967, respectively. Publicly available expression data were used for esophageal biopsies of patients with EoE or controls (GSE58640).