

Abstract
With recent advances in mass spectrometry-based proteomics technologies, deep profiling of hundreds of proteomes has become increasingly feasible. However, deriving biological insights from such valuable datasets is challenging. Here we introduce a systems biology-based software JUMPn, and its associated protocol to organize the proteome into protein co-expression clusters across samples and protein-protein interaction (PPI) networks connected by modules (e.g., protein complexes). Using the R/Shiny platform, the JUMPn software streamlines the analysis of co-expression clustering, pathway enrichment, and PPI module detection, with integrated data visualization and a user-friendly interface. The main steps of the protocol include installation of the JUMPn software, the definition of differentially expressed proteins or the (dys)regulated proteome, determination of meaningful co-expression clusters and PPI modules, and result visualization. While the protocol is demonstrated using an isobaric labeling-based proteome profile, JUMPn is generally applicable to a wide range of quantitative datasets (e.g., label-free proteomics). The JUMPn software and protocol thus provide a powerful tool to facilitate biological interpretation in quantitative proteomics.
SUMMARY:
We present a systems biology tool JUMPn to perform and visualize network analysis for quantitative proteomics data, with a detailed protocol including data pre-processing, co-expression clustering, pathway enrichment, and protein-protein interaction network analysis.
INTRODUCTION:
Mass spectrometry-based shotgun proteomics has become the key approach for analyzing proteome diversity of complex samples1. With recent advances in mass spectrometry instrumentation2,3, chromatography4,5, ion mobility detection6, acquisition methods (data-independent7 and data-dependent acquisition8), quantification approaches (multi-plex isobaric peptide labeling method, e.g., TMT9,10, and label-free quantification11,12) and data analysis strategies/software development13–18, quantification of the whole proteome (e.g., over 10,000 proteins) is now routine19–21. However, how to gain mechanistic insights from such deep quantitative datasets is still challenging22. Initial attempts for investigating these datasets relied predominantly upon the annotation of individual elements of the data, treating each component (protein) independently. However, biological systems and their behavior cannot be solely explained by examining individual components23. Therefore, a systems approach that places the quantified biomolecules in the context of interaction networks is essential for the understanding of complex systems and the associated processes such as embryogenesis, immune response, and pathogenesis of human diseases24.
Network-based systems biology has emerged as a powerful paradigm for analyzing large-scale quantitative proteomics data25–33. Conceptually, complex systems such as mammalian cells could be modeled as a hierarchical network34,35, in which the whole system is represented in tiers: first by a number of large components, each of which then iteratively modeled by smaller subsystems. Technically, the structure of proteome dynamics can be presented by inter-connected networks of co-expressed protein clusters (because co-expressed genes/proteins often share similar biological functions or mechanisms of regulation36) and physically interacting PPI modules37. As a recent example25, we generated temporal profiles of whole proteome and phosphoproteome during T cell activation and used integrative co-expression networks with PPIs to identify functional modules that mediate T-cell quiescence exit. Multiple bioenergetic-related modules were highlighted and experimentally validated (e.g., the mitoribosome and complex IV modules25, and the one-carbon module38). In another example26, we further extended our approach to study the pathogenesis of Alzheimer’s disease, and successfully prioritized disease progression associated protein modules and molecules. Importantly, many of our unbiased discoveries were validated by independent patient cohorts26,29 and/or disease mouse models26. These examples illustrated the power of the systems biology approach for dissecting molecular mechanisms with quantitative proteomics and other omics integrations.
Here we introduce JUMPn, a streamlined software that explores quantitative proteomics data using network-based systems biology approaches. JUMPn serves as the downstream component of the established JUMP proteomics software suite13,14,39, and aims to fill the gap from individual protein quantifications to biologically meaningful pathways and protein modules using the systems biology approach. By taking the quantification matrix of differentially expressed (or the most variable) proteins as input, JUMPn aims to organize the proteome into a tiered hierarchy of protein clusters co-expressed across samples and densely connected PPI modules (e.g., protein complexes), which are further annotated with public pathway databases by over-representation (or enrichment) analysis (Figure 1). JUMPn is developed with the R/Shiny platform40 for a user-friendly interface and integrates three major functional modules: co-expression clustering analysis, pathway enrichment analysis, and PPI network analysis (Figure 1). After each analysis, results are automatically visualized and are adjustable via the R/shiny widget functions and readily downloadable as publication tables in Microsoft Excel format. In the following protocol, we use quantitative whole proteome data as an example and describe the major steps of using JUMPn, including installation of the JUMPn software, the definition of differentially expressed proteins or the (dys)regulated proteome, co-expression network analysis, and PPI module analysis, result visualization and interpretation, and trouble shootings. JUMPn software is freely available on GitHub41.
Figure 1: Workflow of JUMPn.
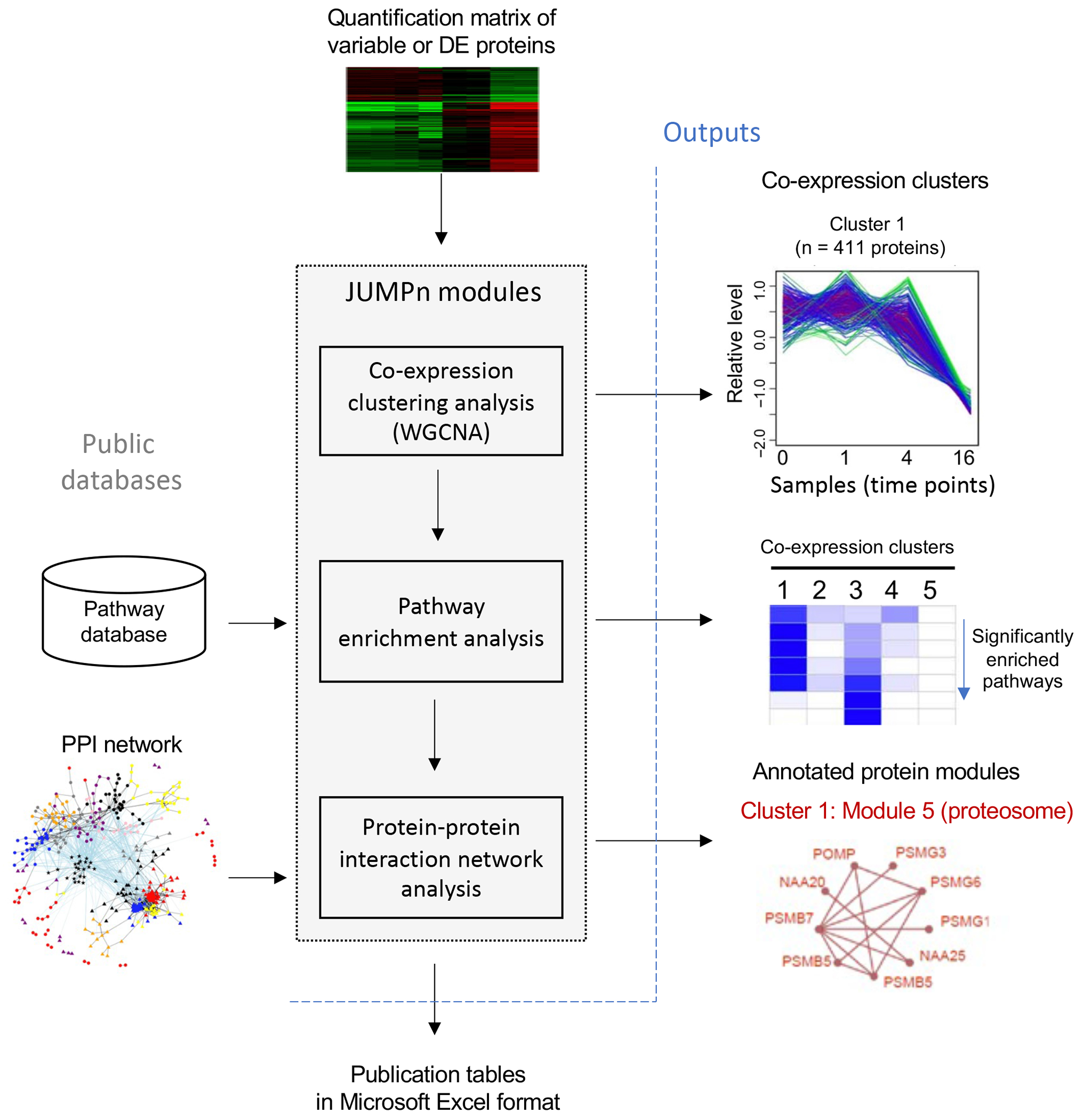
Quantification matrix of the top variable of differentially expressed (DE) proteins are taken as input, and proteins are grouped into co-expression clusters by the WGCNA algorithm. Each co-expression is then annotated by pathway enrichment analysis and further superimposed onto the protein-protein interaction (PPI) network for densely connected protein module identifications.
PROTOCOL
NOTE: In this protocol, the usage of JUMPn is illustrated by utilizing a published dataset of whole proteome profiling during B cell differentiation quantified by TMT isobaric label reagent27.
1. Setup of JUMPn software
NOTE: Two options are provided for setting up the JUMPn software: (i) installation on a local computer for personal use; and (ii) deployment of JUMPn on a remote Shiny Server for multiple users. For local installation, a personal computer with Internet access and ≥4 Gb of RAM is sufficient to run JUMPn analysis for a dataset with a small sample size (n < 30); larger RAM (e.g., 16 Gb) is needed for large-cohort analysis (e.g., n = 200 samples).
1.1. Install the software on a local computer. After installation, allow the web browser to launch JUMPn and let the analysis run on the local computer.
1.1.1. Install anaconda42 or miniconda43 following the online instructions.
1.1.2. Download the JUMPn source code41. Double click to unzip the downloaded file JUMPn_v_1.0.0.zip; a new folder named JUMPn_v_1.0.0 will be created.
1.1.3. Open command line terminal. On Windows, use the Anaconda Prompt. On MacOS, use the built-in Terminal application.
1.1.4. Create the JUMPn Conda environment: Get the absolute path of JUMPn_v_1.0.0 folder (e.g., /path/to/JUMPn_v_1.0.0). To create and activate an empty Conda environment type the following commands on the terminal
conda create -p /path/to/JUMPn_v_1.0.0/JUMPn -y
conda activate /path/to/JUMPn_v_1.0.0/JUMPn
1.1.5. Install JUMPn dependencies: Install R (on the terminal, type conda install -c conda-forge r=4.0.0 -y), change the current directory to the JUMPn_v_1.0.0 folder (on the terminal, type cd path/to/JUMPn_v_1.0.0), and install the dependency packages (on the terminal, type Rscript bootstrap.R)
1.1.6. Launch JUMPn on the web browser: Change the current directory to the execution folder (on the terminal, type cd execution) and launch JUMPn (on the terminal, type R -e “shiny::runApp()”)
1.1.7. Once the above is executed, the terminal screen will show up Listening on http://127.0.0.1:XXXX (here XXXX indicates 4 random numbers). Copy and paste http://127.0.0.1:XXXX onto the web browser, on which JUMPn welcome page will show up (Figure 2).
Figure 2:
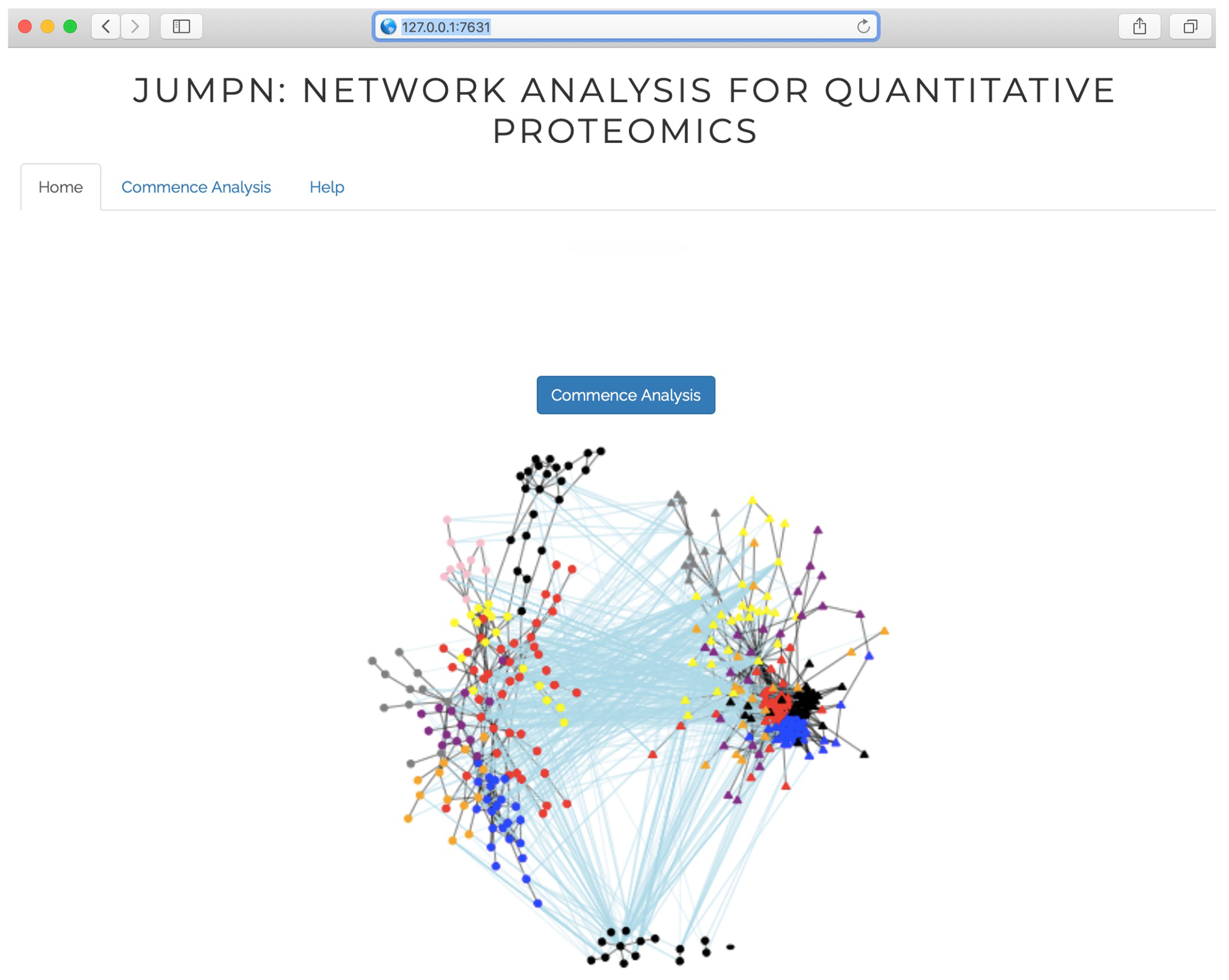
JUMPn welcome page.
1.2. Deployment on Shiny Server. Examples of Shiny Server include the commercial shinyapps.io server or any institutionally supported Shiny Servers.
1.2.1. Download and install RStudio following the instruction44.
1.2.2. Obtain the deployment permission for the Shiny Server. For the shinyapps.io server, set up the user account by following the instruction45. For institutional Shiny server, contact the server administrator for requesting permissions.
1.2.3. Download the JUMPn source code41 to the local machine; installation is not necessary. Open either the server.R or ui.R files in RStudio and click the Publish to Server drop-down menu in the top right of the RStudio IDE.
1.2.4. In the Publish to Account panel, type the server address. Press the Publish button. Successful deployment is validated upon automatic redirect from RStudio to the RShiny server where the application was deployed.
2. Demo run using an example dataset
NOTE: JUMPn offers a demo run using the published B cell proteomics dataset. The demo run illustrates a streamlined workflow that takes the quantification matrix of differentially expressed proteins as input and performs co-expression clustering, pathway enrichment, and PPI network analysis sequentially.
2.1. On the JUMPn home page (Figure 2), click on the Commence Analysis button to start JUMPn analysis.
2.2. In the bottom left corner of the Commence Analysis page (Figure 3), click on the Upload Demo B Cell Proteomic Data button; a dialog box will appear notifying the success of the data upload.
Figure 3: Input page of JUMPn.
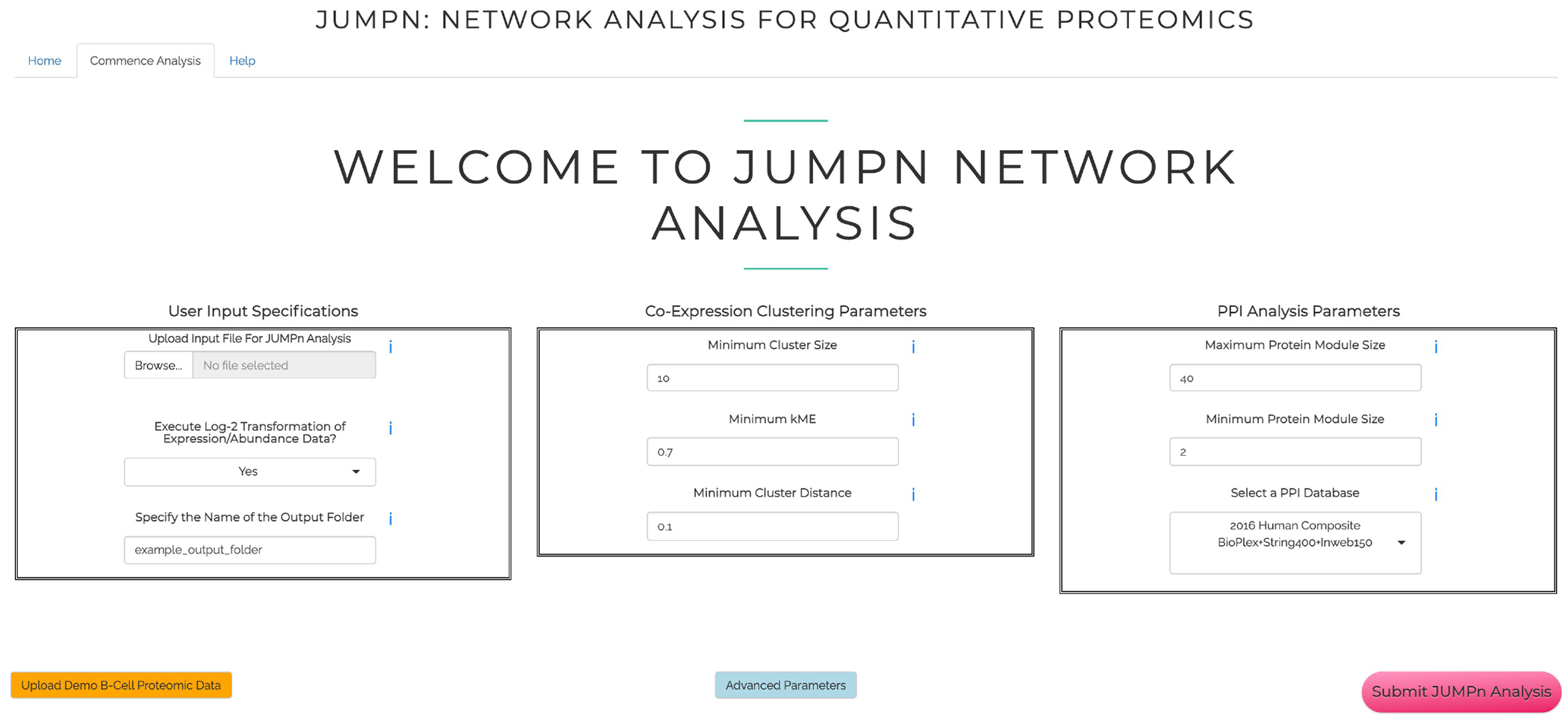
The page includes the input file upload panel and parameter configuration panels for co-expression clustering and PPI network analysis, respectively.
2.3. In the bottom right corner of the page, click on the Submit JUMPn Analysis button to initiate the demo run using default parameters; a progress bar will appear that denotes the course of the analysis. Wait until the progress bar is fulfilled (3 min expected).
2.4. Once the demo run is finished, a dialog box will appear with the success run message and the absolute path to the result folder. Click on Continue to Results to continue.
2.5. The webpage will first guide the user to the co-expression cluster results by WGCNA. Click on View Results on the dialog window to continue.
2.6. Find the protein co-expression patterns on the left of the Result Page 1: WGCNA Output page. Click on the Select the Expression Format drop-down box to navigate between two figure formats:
2.6.1. Select Trends to display the trends plot, with each line representing individual protein abundance across samples. The color of each line represents how close the expression pattern is to the co-expression cluster consensus (i.e., “eigengene” as defined by the WGCNA algorithm).
2.6.2. Select Boxplot to display co-expression patterns in boxplot format for each sample.
2.7. View the pathway/ontology enrichment heatmap on the right of the WGCNA output page. The most highly enriched pathways for each cluster are displayed together in a heatmap, with the color intensity reflecting the Benjamini-Hochberg adjusted p-value.
2.8. Scroll down the webpage to view the expression pattern for individual proteins.
2.8.1. Use the drop-down box Select the Co-Expression Cluster to view proteins from each cluster (default is Cluster 1). Select a specific protein in the table, upon which the bar plot below the table will be automatically updated to reflect its protein abundance.
2.8.2. Search specific protein names using the Search box on the right side of the table for a specific protein.
2.9. To view PPI results, click on the Results Page 2: PPI Output on the top.
2.10. Click on Select the Co-Expression Cluster to view the results for a specific co-expression cluster (default is cluster 1). The displays of all figure panels on this page will be updated for the newly selected cluster.
2.11. View the PPI networks for the selected co-expression cluster on the left figure panel:
2.11.1. Click the Select by Group drop-down box to highlight individual PPI modules within the network. Click on the Select a Network Layout Format drop-down box to change the network layout (default is by Fruchterman Reingold).
2.11.2. Use the mouse and the trackpad to perform steps 2.11.3–2.11.5.
2.11.3. Zoom in or zoom out the PPI network as needed. The gene names of each node in the network will be shown when zoomed in sufficiently.
2.11.4. When zoomed in, select and click a certain protein to highlight that protein and its network neighbors.
2.11.5. Drag a certain node (protein) in the network to change its position in the layout; thereby the network layout can be re-organized by the user.
2.12. On the right panel of the PPI result page, view the co-expression cluster-level information that assists interpretation of PPI results:
2.12.1. View the co-expression pattern of the selected cluster as boxplot by default.
2.12.2. Click on the Select the Expression Format drop-down box for more information or displays as mentioned in steps 2.12.3–2.12.5.
2.12.3. Select Trends to show trends plot for the co-expression pattern.
2.12.4. Select Pathway Barplot to show significantly enriched pathways for the co-expression cluster.
2.12.5. Select Pathway Circle Plot to show significantly enriched pathways for the co-expression cluster in the circle plot format.
2.13. Scroll down the Result Page 2: PPI Output webpage to view results on the individual PPI module level. Click on the Select the Module drop-down box to select a specific PPI module for display (Cluster1: Module 1 is shown by default).
2.14. View the PPI module on the left panel.
2.14.1. Use a mouse or trackpad to zoom in or zoom out the PPI network as needed. The gene names of each node in the network will be shown when zoomed in sufficiently.
2.14.2. When zoomed in, select and click a certain protein to highlight that protein and its network neighbors.
2.14.3. Drag a certain node (protein) in the network to change its position in the layout; thereby the network layout can be re-organized by the user.
2.15. View the pathway/ontology enrichment results on the right panel. Click on the Select the Pathway Annotation Style drop-down box for more information and displays:
2.15.1. Select Barplot to show significantly enriched pathways for the selected PPI module.
2.15.2. Select Circle Plot to show significantly enriched pathways for the selected PPI module in the format of a circle plot.
2.15.3. Select Heatmap to show significantly enriched pathways and the associated gene names from the selected PPI module.
2.15.4. Select Table to show the detailed pathway enrichment results, including the name of pathways/ontology terms, gene names, and the P-value by Fisher’s exact test.
2.16. View the publication table in a spreadsheet format: follow the absolute path (printed on the top of both results pages) and find the publication spreadsheet table named ComprehensiveSummaryTables.xlsx.
3. Preparation of the input file and upload to JUMPn
NOTE: JUMPn takes as input the quantification matrix of either the differentially expressed proteins (supervised method) or the most variable proteins (unsupervised method). If the goal of the project is to understand proteins changed across multiple conditions (e.g., different disease groups, or time-series analysis of biological process), the supervised method of performing DE analysis is preferred; otherwise, an unsupervised approach of selecting the most variable proteins may be used for the exploratory purpose.
3.1. Generate the protein quantification table, with each protein as rows and each sample as columns. Achieve this via modern mass spectrometry-based proteomics software suite (e.g., JUMP suite13,14,39, Proteome Discoverer, Maxquant15,46).
3.2. Define the variable proteome.
3.2.1. Use the statistical analysis results provided by the proteomics software suite to define differentially expressed (DE) proteins (for example, with adjusted p-value < 0.05).
3.2.2. Alternatively, users may follow the example R code47 to define either DE or most variable proteins.
3.3. Format the input file using the defined variable proteome.
NOTE: The required input file format (Figure 4) includes a header row; the columns include protein accession (or any unique IDs), GN (official gene symbols), protein description (or any user-provided information), followed by protein quantification of individual samples.
Figure 4: Example input file of quantification matrix.

Columns include protein accession (or any unique IDs), GN (official gene symbols), protein description (or any user-provided information), followed by protein quantification of individual samples.
3.3.1. Follow the order of the columns specified in step 3.1, but the column names of the header is flexible to the user.
3.3.2. For TMT (or similar) quantified proteome, use the summarized TMT reporter intensity as input quantification values. For label-free data, use either normalized spectral counts (e.g., NSAF48) or intensity-based method (e.g., LFQ intensity or iBAQ protein intensity reported by Maxquant46).
3.3.3. Missing values are allowed for JUMPn analysis. Ensure to label these as NA in the quantification matrix. However, it is recommended to only use proteins with quantification in more than 50% of the samples.
3.3.4. Save the resulting input file as .txt, .xlsx, or .csv format (all three are supported by JUMPn).
3.4. Upload input file:
3.4.1. Click the Browser button and select the input file (Figure 3, left panel); the file format (xlsx, csv, and txt are supported) will be automatically detected.
3.4.2. If the input file contains intensity-like quantification values (e.g., those generated by JUMP suite39) or ratio-like (e.g., from Proteome Discoverer), select Yes for the Execute Log2-Transformation of Data Option; otherwise, the data may have already been log-transformed, so select No for this option.
4. Co-expression clustering analysis
NOTE: Our group25–27 and others28,29,31 have proved WGCNA49 an effective method for co-expression clustering analysis of quantitative proteomics. JUMPn follows a 3-step procedure for WGCNA analysis25,50: (i) initial definition of co-expression gene/protein clusters by dynamic tree cutting51 based on the topological overlap matrix (TOM; determined by quantification similarities among genes/proteins); (ii) merging of similar clusters to reduce redundancy (based on dendrogram of eigengene similarities); and (iii) final assignment of genes/proteins to each cluster that exceed the minimal Pearson correlation cutoff.
4.1. Configure the WGCNA parameters (Figure 3, middle panel). The following three parameters control the three steps, respectively:
4.1.1. Set minimum cluster size as 30. This parameter defines the minimal number of proteins required for each co-expression cluster in the initial step (i) of TOM-based hybrid dynamic tree cutting. The larger the value, the smaller the number of clusters returned by the algorithm.
4.1.2. Set minimum cluster distance as 0.2. Increasing this value (e.g., from 0.2–0.3) may cause more cluster merging during step (ii), thus resulting in a fewer number of clusters.
4.1.3. Set minimum kME as 0.7. Proteins will be assigned to the most correlated cluster defined in step (ii), but only proteins with Pearson correlation passing this threshold will be retained. Proteins that fail in this step will not be assigned to any cluster (‘NA’ cluster for the failed proteins in the final report).
4.2. Initiate the analysis. There are two ways to submit the co-expression clustering analysis:
4.2.1. Click on the Submit JUMPn Analysis button in the bottom right corner to initiate the comprehensive analysis of WGCNA automatically followed by PPI network analysis.
4.2.2. Alternatively, select to execute the WGCNA step only (especially for the purpose of parameter tuning; see steps 4.2.3–4.2.4):
4.2.3. Click on the Advanced Parameters button at the bottom of the Commence Analysis page; a new parameter window will pop up. In the bottom widget, Select Mode of Analysis, select WGCNA Only, then click on Dismiss to continue.
4.2.4. On the Commence Analysis page, click on the Submit JUMPn Analysis button.
4.2.5. In either case above, a progress bar will appear upon analysis submission.
NOTE: Once the analysis is finished (typically < 1 min for WGCNA Only analysis and <3 min for comprehensive analysis), a dialog box will appear with a success run message and the absolute path to the result folder.
4.3. Examine the WGCNA results as illustrated in steps 2.4–2.8. Note that the absolute path to the file co_exp_clusters_3colums.txt is highlighted on the top of the Results Page: WGCNA Output to record the cluster membership of each protein and use it as input for the PPI Only analysis.
4.4. Troubleshooting. The following three common cases are discussed. Once the parameters are updated as discussed below, follow steps 4.2.2–4.2.4 to generate new WGCNA results.
4.4.1. If one important co-expression pattern is expected from the data but missed by the algorithm, follow steps 4.4.2–4.4.4
4.4.2. A missing cluster is especially likely for small co-expression clusters, i.e., only a limited number (e.g., <30) of proteins exhibiting this pattern. Before the re-analysis, re-examine the input file of protein quantification matrix and locate several positive control proteins that adhere to that important co-expression pattern.
4.4.3. To rescue the small clusters, decrease the Minimal Cluster Size (e.g., 10; cluster size less than 10 may not be robust thus not recommended), and decrease the Minimal Cluster Distance (e.g., 0.1; here setting as 0 is also allowed, which means automatic cluster merging will be skipped).
4.4.4. After executing the co-expression clustering step with the updated parameters, first, check if the cluster is rescued from the Co-Expression Pattern Plots, then check the positive controls by searching their protein accessions from Detailed Protein Quantification (make sure to select the appropriate co-expression cluster from the left side drop-down widget before the search).
NOTE: Multiple iterations of parameter tuning and rerun may be needed for the rescue.
4.4.5. If there are too many proteins that cannot be assigned to any cluster, follow steps 4.4.6–4.4.7.
NOTE: Usually, a small percentage (typically <10%) of proteins may not be assigned to any cluster as those may be outlier proteins that did not follow any of the common expression patterns of the dataset. However, if such percentage is significant (e.g., >30%), it suggests that there exist additional co-expression patterns that cannot be ignored.
4.4.6. Decrease both the Minimal Cluster Size and Minimal Cluster Distance parameters to alleviate this situation by detecting ‘new’ co-expression clusters.
4.4.7. In addition, decrease the Minimal Pearson Correlation (kME) parameter to shrink these ‘NA cluster’ proteins.
NOTE: Tuning this parameter will not generate new clusters but instead will increase the size of ‘existing’ clusters by accepting more previously failed proteins with the lower threshold; however, this will also increase the heterogeneity of each cluster, as more noisy proteins are now allowed.
4.4.8. Two clusters have a very minor difference of patterns; merge them into one cluster following steps 4.4.9–4.4.11.
4.4.9. Increase the Minimal Cluster Distance parameter to solve the issue.
4.4.10. However, in some situations, the algorithm may never return the desired pattern; in such an instant, manually adjust or edit cluster membership in the file co_exp_clusters_3colums.txt (file from step 4.3) to merge.
4.4.11. Take the post-edited file as input for the downstream PPI network analysis. In case of manual editing, justify the criteria of cluster assignment, and record the procedure of manual editing.
5. Protein-protein interaction network analysis
NOTE: By superimposing co-expression clusters onto the PPI network, each co-expression cluster is further stratified into smaller PPI modules. The analysis is performed for each co-expression cluster and includes two stages: in the first stage, JUMPn superimposes proteins from the co-expression cluster onto the PPI network and find all connected components (i.e., multiple clusters of connected nodes/proteins; as an example, see Figure 6A); then, communities or modules (of densely connected nodes) will be detected for each connected component iteratively using the topological overlap matrix (TOM) method52.
Figure 6: PPI network analysis results reported by JUMPn.
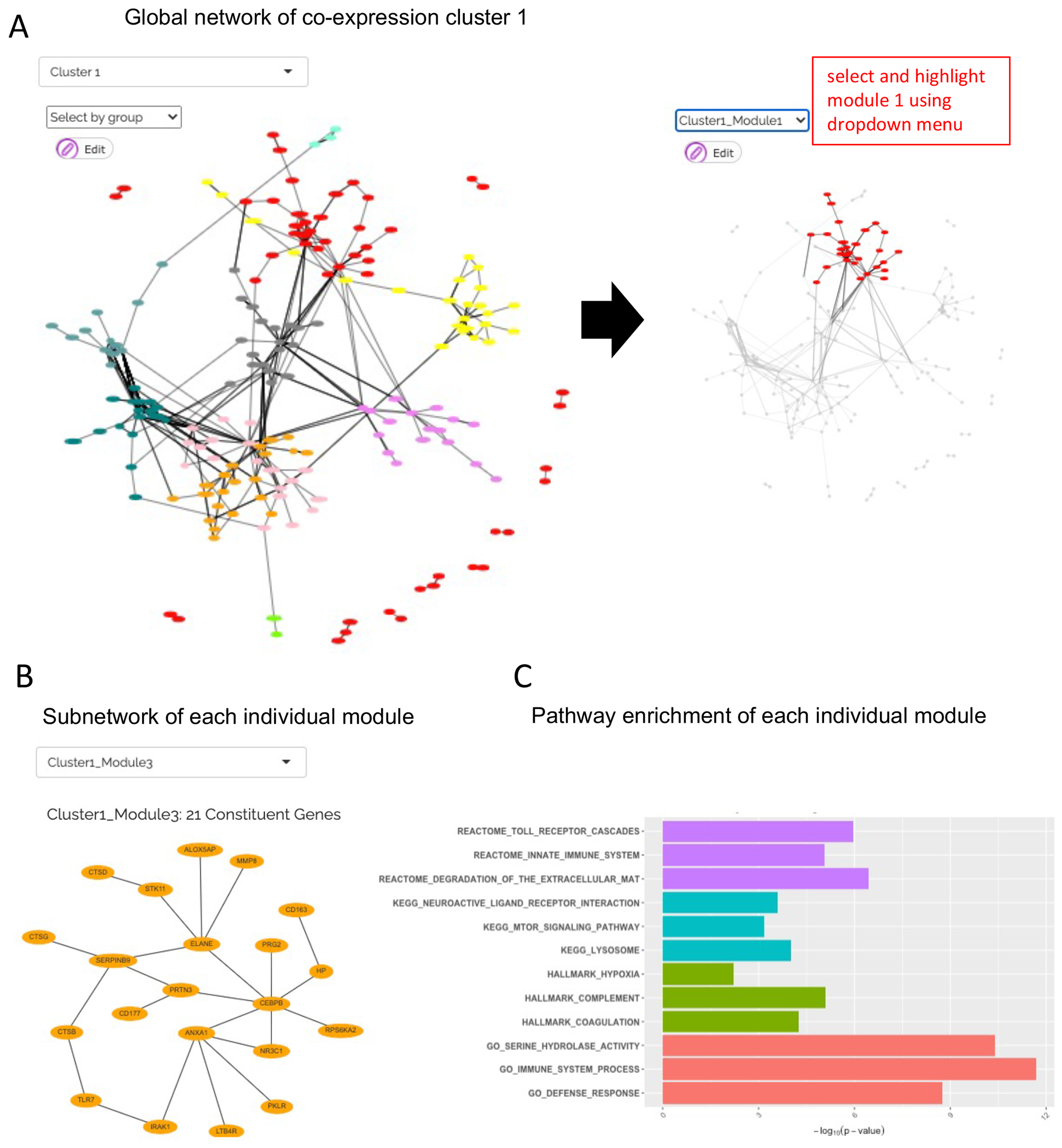
The global inter-module network is shown (A), followed by a subnetwork of individual modules (B) and its significantly enriched pathways (C). Users may select various display options and navigate between different clusters and modules via the selection box.
5.1. Configure parameters for PPI network analysis (Figure 3, right panel).
5.1.1. Set Minimal PPI Module Size as 2. This parameter defines the minimal size of the disconnected components from the first stage analysis. Any component smaller than the specified parameter will be removed from the final results.
5.1.2. Set Maximal PPI Module Size as 40. Large, disconnected components that pass this threshold will undergo second stage TOM-based analysis. The second stage analysis will further split each large component into smaller modules: each module presumably contains proteins more densely connected than the original component as a whole.
5.2. Initiate the analysis. There are two ways to submit the PPI network analysis:
5.2.1. Hit the Submit JUMPn Analysis button to automatically perform the PPI analysis following WGCNA analysis by default.
5.2.2. Alternatively, upload customized co-expression cluster results and perform PPI Only analysis following steps 5.2.3–5.2.5.
5.2.3. Prepare input file by following the format of the file co_exp_clusters_3colums.txt (see subsection 4.4).
5.2.4. Click on the Advanced Parameters button at the bottom of the Commence Analysis page; a new parameter window will pop up. In the upper session Upload Co-Expression Cluster Result for ‘PPI Only’ Analysis, click on Browser to upload the input file prepared by step 5.2.3.
5.2.5. In the bottom widget, Select Mode of Analysis, select PPI only, then click on Dismiss to continue. On the Commence Analysis page, click on the Submit JUMPn Analysis button.
5.3. Once the analysis is finished (typically <3 min), examine the PPI results as illustrated in steps 2.10–2.15.
5.4. (Optional advanced step) Adjust PPI modularization by tuning parameters:
5.4.3. Increase the Maximal Module Size parameter to allow more proteins included in the PPI results. Upload customized PPI network to cover undocumented interactions, following steps 5.4.2–5.4.3.
5.4.2. Click on the Advanced Parameters button at the bottom of the Commence Analysis page; a new parameter window will pop up. Prepare the customized PPI file, which contains three columns in the format of <Protein_A>, Connection, and <Protein_B>; here <Protein_X> are presented by the official gene names of each protein.
5.4.3. In Upload a PPI Database, click on the Browse button to upload the customized PPI file.
6. Pathway enrichment analysis
NOTE: The JUMPn-derived hierarchical structures of both co-expression clusters and PPI modules within are automatically annotated with over-represented pathways using Fisher’s exact test. The pathway/topology databases used include Gene Ontology (GO), KEGG, Hallmark, and Reactome. Users may use advanced options to upload customized databases for the analysis (e.g., in the case of analyzing data from non-human species).
6.1. By default, the pathway enrichment analysis is initiated automatically with co-expression clustering and PPI network analysis.
6.2. View the pathway enrichment results:
6.2.1. Follow steps 2.7, 2.12, and 2.15 to visualize different formats on the result pages. View detailed results in spreadsheet publication table in the ComprehensiveSummaryTables.xlsx file (step 2.16).
6.3. (Optional advanced step) Upload customized database for pathway enrichment analysis:
6.3.1. Prepare the gene background file, which typically contains the official gene names of all genes of a species.
6.3.2. Prepare the ontology library file following steps 6.3.3–6.3.4.
6.3.3. Download the ontology library files from public websites including EnrichR53, and MSigDB54. For example, download ontology from Drosophila from the EnrichR website55.
6.3.4. Edit the downloaded file for the required format with two columns: the pathway name as the first column, and then the official gene symbols (separated by “/”) as the second column. The detailed file format is described in the Help page of the JUMPn R shiny software.
NOTE: Find example files of gene background and ontology library (using Drosophila as an instance) in the JUMPn GitHub site56.
6.3.5. Click on the Advanced Parameters button at the bottom of the Commence Analysis page; a new parameter window will pop up.
6.3.6. Find Upload a Background File for Pathway Enrichment Analysis item and click on Browser to upload the background file prepared at step 6.3.1. Then in the session, Select The Background to be Used for Pathway Enrichment Analysis, click on User-Supplied Background.
6.3.7. Find Upload an Ontology Library File for Pathway Enrichment Analysis item and click on Browser to upload the background file prepared at steps 6.3.2–6.3.5. Then in the session, Select Databases for Pathway Enrichment Analysis, click on User-Supplied Database in .xlsx Format”.
6.4. Click on the Submit JUMPn Analysis button in the bottom right corner to initiate the analysis using the customized database.
7. Analysis of dataset with large sample size
NOTE: JUMPn supports analysis of dataset with large sample size (up to 200 samples tested). To facilitate the visualization of a large sample size, an additional file (named “meta file”) that specifies the sample group is needed to facilitate the display of co-expression clustering results.
7.1. Prepare and upload meta file.
7.1.1. Prepare the meta file that specifies group information (e.g., control and disease groups) for each sample following steps 7.1.2–7.1.3.
7.1.2. Ensure that the meta file contains at least two columns: column 1 must contain the sample names identical to the column names and order from the protein quantification matrix file (as prepared in step 3.3); Column 2 onwards will be used for group assignment for any number of features defined by the user. The number of columns is flexible.
7.1.3. Ensure that the first row of the meta file contains the column names for each column; from the second row onwards, individual sample information of groups or other features (e.g., sex, age, treatment, etc.) should be listed.
7.1.4. Upload the meta file by clicking on the Advanced Parameters button in the bottom of the Commence Analysis page; a new parameter window will pop up. Proceed to step 7.1.5
7.1.5. Find Upload a Meta File item and click on Browser to upload the background file. If the unexpected format or unmatched sample names are detected by JUMPn, an error message will pop up for further formatting of the meta file (steps 7.1.1–7.1.3).
7.2. Adjust the parameters for co-expression clustering analysis: set Minimal Pearson Correlation as 0.2. This parameter needs to be relaxed due to larger sample size.
7.3. Click on Submit JUMPn Analysis button in the bottom right corner to submit the analysis.
7.4. View analysis results: all the data output is the same except for displaying the co-expression cluster patterns.
7.4.1. In the Results Page 1: WGCNA Output page, visualize the co-expression clusters as boxplots with samples stratified by the user-defined sample groups or features. Each dot in the plot represents the eigengene (i.e., the consensus pattern of the cluster) calculated by the WGCNA algorithm.
7.4.2. If the user provided multiple features (e.g., age, sex, treatment, etc.) to group the samples, click on the Select the Expression Format drop-down box to select another feature for grouping the samples.
REPRESENTATIVE RESULTS:
We used our published deep proteomics datasets25–27,30 (Figures 5 and 6) as well as data simulations57 (Table 1) to optimize and evaluate JUMPn performance. For co-expression protein clustering analysis via WGCNA, we recommend utilizing proteins significantly changed across samples as the input (e.g., differentially expressed (DE) proteins detected by statistical analysis). While including non-DE proteins for the analysis may result in more co-expression clusters returned by the program (due to larger input size), we hypothesize that mixing the real signal (e.g., the DE proteins) with the background (the remaining non-DE) for systems-level analysis may dilute the signal and mask the underlying network structure. To test this, simulation analysis was performed under two different conditions: i) highly dynamic proteome (e.g., 50% altered in T cell activation25) and ii) relatively stable proteome (e.g., 2% proteome changed in AD26). For the highly dynamic proteome, six co-expression clusters were simulated from 50% proteome following the same cluster size and expression patterns (i.e., eigengenes) of our published results25. Similarly, for a relatively stable proteome, we simulated three clusters from 2% proteome following our recent AD proteomics study26. As expected, increasing the input number of proteins increases the number of detected clusters (Table 1). For the highly dynamic proteome, using all proteins as input can capture most of the true clusters (5 out of the 6 simulated bona fide clusters; 83% recall) with 63% precision (5 out of the 8 returned clusters are true positives; i.e., the remaining 3 clusters are false positives). However, for the relatively stable proteome, increasing the input size with non-DE proteins dramatically reduces precision (Table 1). For instance, using the whole proteome as input, 169 modules are detected, of which only 2 are correct (1.2% precision; the remaining 98.8% detected modules are false positives). These results thus indicate that choosing only the changed proteome as input will increase the precision of co-expression analysis, especially for relatively stable proteome.
Figure 5: Co-expression cluster results reported by JUMPn.
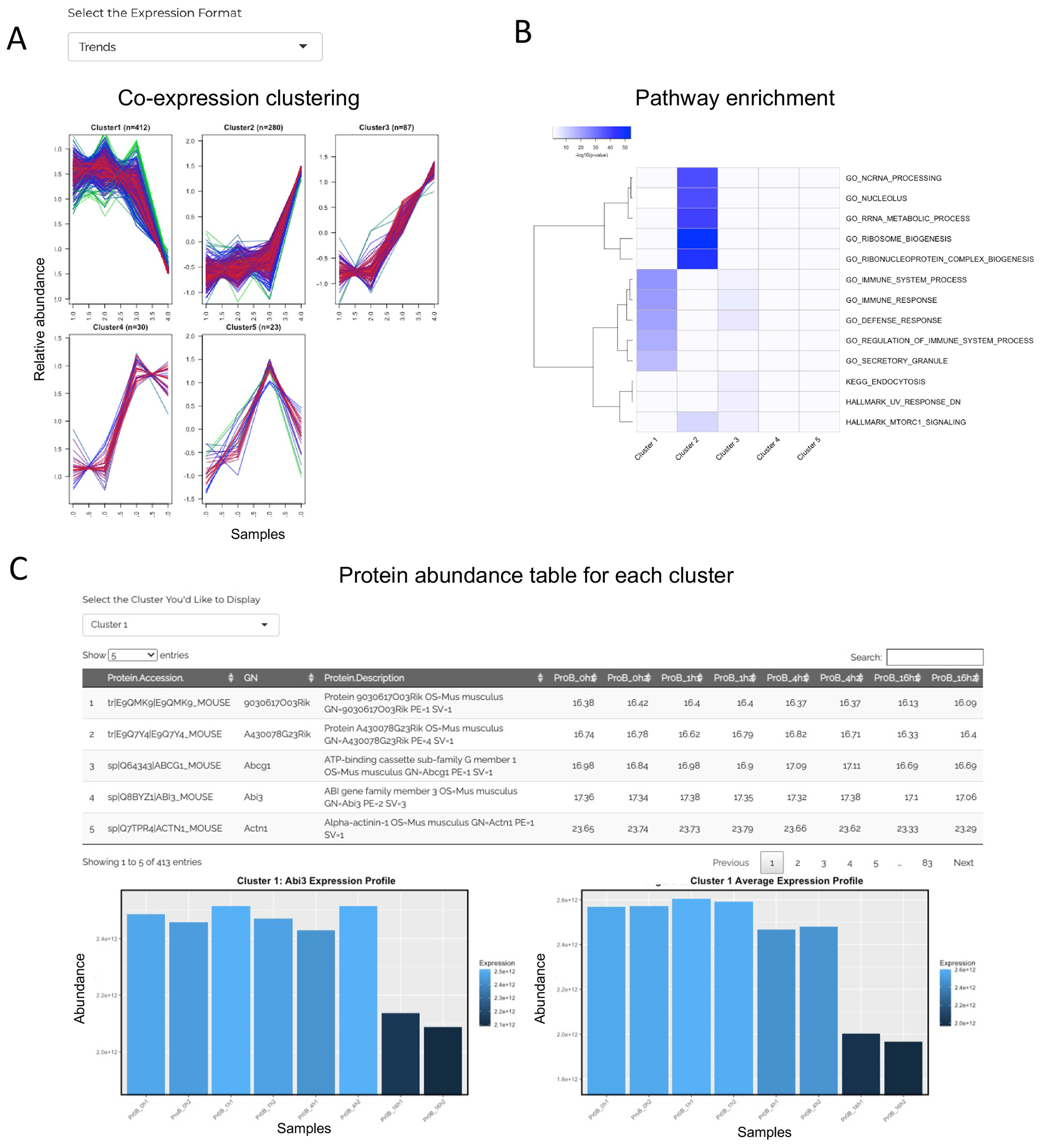
The co-expression clustering patterns (A), top enriched pathway heatmap across clusters (B), and detailed protein abundance for each cluster are shown (C). Users may select various display options and navigate between different clusters via the selection box.
Table 1. Simulation studies of co-expression cluster detection.
Conditions with optimal performance are highlighted in red.
| % top proteins for analysis | # simulated modules | # detected modules | # recaptured modules1 | precision2 | recall3 |
|---|---|---|---|---|---|
| Highly dynamic proteome (e.g., during T cell activation): 6 simulated modules from 50% proteome | |||||
|
| |||||
| 2 | 6 | 2 | 2 | 1 | 0.33 |
| 5 | 6 | 2 | 2 | 1 | 0.33 |
| 10 | 6 | 3 | 3 | 1 | 0.5 |
| 20 | 6 | 4 | 4 | 1 | 0.67 |
| 50 | 6 | 6 | 6 | 1 | 1 |
| 100 | 6 | 8 | 5 | 0.63 | 0.83 |
|
| |||||
| Relatively stable proteome (e.g., during pathogenesis of AD): 3 simulated modules from 2% proteome | |||||
|
| |||||
| 1 | 3 | 1 | 1 | 1 | 0.33 |
| 2 | 3 | 3 | 3 | 1 | 1 |
| 5 | 3 | 8 | 3 | 0.38 | 1 |
| 10 | 3 | 13 | 3 | 0.23 | 1 |
| 20 | 3 | 19 | 3 | 0.16 | 1 |
| 50 | 3 | 71 | 2 | 0.03 | 0.67 |
| 100 | 3 | 169 | 2 | 0.01 | 0.67 |
A recaptured module is a detected module whose eigengene highly correlates (Pearson R > 0.95) with one of the simulated eigengenes.
precision = # recaptured modules / # detected modules
recall = # recaptured modules / # simulated modules
Following the detection of co-expression protein clusters, each cluster will be annotated by JUMPn using the pathway enrichment analysis (Figure 1). The current version includes four commonly used pathway databases, including Gene Ontology (GO), KEGG, Hallmark, and Reactome. Users may also compile their own database in GMT format54, which can be uploaded into JUMPn. Integrating multiple databases for pathway enrichment analysis may provide more comprehensive views; however, the sizes of different pathway databases vary significantly, which may induce unwanted bias to certain (especially large) databases. Two solutions are provided within JUMPn. First, using a statistical approach, nominal p values are adjusted (or penalized) for multiple-hypothesis testing by the Benjamini-Hochberg method58, with a larger database requiring a more significant nominal p-value to reach the same adjusted p level than that from a small database. Second, JUMPn highlights the top significantly enriched pathway for each database separately, thus database-specific top enriched pathways are always displayed.
Similar to pathway enrichment analysis, a composite PPI network was compiled by combining STRING59,60, BioPlex61,62, and InWeb_IM63 databases. The BioPlex database was created using affinity purification followed by mass spectrometry in human cell lines, whereas the STRING and InWeb contain information from various sources. Therefore the STRING and InWeb databases were further filtered by the edge score to ensure high quality, with the cutoff determined by best fitting the scale-free criteria24. The final merged PPI network covers more than 20,000 human genes with ~1,100,000 edges (Table 2). This comprehensive interactome is included and published in a bundle with our JUMPn software for sensitive PPI analysis.
Table 2. Statistics of human protein-protein interaction (PPI) networks.
PPI networks are filtered by edge score to ensure high quality, with the score cutoff determined by best fitting the scale free criteria.
| PPI networks | No. of Nodes | No. of Edges |
|---|---|---|
| BioPlex 3.0 combined (293T+HCT116) | 14,551 | 167,399 |
| InBio_Map_core_2016_09_12 | 17,429 | 608,166 |
| STRING (v11.0) | 18,954 | 587,482 |
| Composite PPI network | 20,485 | 1,152,607 |
After the analysis is finished, JUMPn generates the publication table spreadsheet file ComprehensiveSummaryTables.xlsx, consisting of three individual sheets. The first sheet contains results of co-expression protein clusters with one protein per row: the first column indicates the cluster membership of each input protein, and the remaining columns are copied from the user-input file, which contains the protein accession, gene names, protein description, and quantification of individual samples. The second sheet contains results of pathway enrichment analysis, displaying significant pathways enriched in each co-expression cluster. This table is first organized by different pathway databases, then sorted by co-expression clusters, functional pathways, the total number of pathway genes, the total number of genes in the individual cluster, the overlapped gene numbers and names, enrichment fold, Fisher exact test derived P-values and Benjamini–Hochberg false discovery rate. The third sheet contains results of PPI module analysis with one PPI module per row; its columns include the module name (defined by its co-expression membership and module ID, for example, Cluster1_Module1), the mapped proteins and numbers, as well as functional pathways that are defined by searching the module proteins against the pathway databases.
DISCUSSION
Here we introduced our JUMPn software and its protocol, which have been applied in multiple projects for dissecting molecular mechanisms using deep quantitative proteomics data25–27,30,64. The JUMPn software and protocol have been fully optimized, including consideration of DE proteins for co-expression network analysis, a compilation of comprehensive and high-quality PPI network, stringent statistical analysis (e.g., by consideration of multiple hypothesis testing) with a streamlined and user-friendly interface. Multiple protein modules identified by JUMPn have been validated by functional experiment studies25,27 or independent patient cohorts26, exemplifying JUMPn as an effective tool for identifying key molecules and pathways underlying diverse biological processes.
Critical steps of this protocol include the generation of optimal results of co-expression clusters and PPI modules, which may require multiple iterations of parameter tuning, as well as upload of customized PPI network. In our protocol, we discussed common practical scenarios, including how to handle missing of important clusters, a high percentage of unassigned proteins, merging of two redundant clusters, and missing of important proteins within PPI modules. We recommend the user to prepare several positive control proteins and confirm their presence in the final co-expression clusters. Sometimes a positive control will never be included in the final PPI modules due to an incomplete PPI network database. To partially alleviate this, we have updated our PPI network with the latest versions of BioPlex V362 and STRING V1160. In addition, JUMPn allows users to upload customized PPI networks. For example, novel interactions derived from affinity purification-mass spectrometry (AP-MS) experiments using an important positive control protein as bait may be integrated with the current composite PPI network for more customized analysis.
By using the framework of pathway enrichment analysis for each co-expression protein cluster, JUMPn can be extended for inferring transcription factor (TF) activity. The assumption is that if there exists an over-representation of target genes of a specific TF in a co-expression cluster (i.e., these targets are differentially expressed and follow the same expression pattern), the activity of that TF is potentially altered across experimental conditions because its target protein abundance is changed consistently. Technically, this can be simply achieved via JUMPn by replacing the current pathway database with the TF-target database (e.g., from the ENCODE project65). Similarly, kinase activity may also be inferred by leveraging the kinase-substrate database, taking deep phosphoproteomics as input. As an example, we successfully identified dysregulated TFs and kinases underlying brain tumor pathogenesis64. Indeed, using the network approach for activity inference has emerged as a powerful approach for identifying dysregulated drivers for human diseases66,67.
The JUMPn software is readily applied to a wide range of data types. Even though isobaric labeling quantified proteome was used as an illustrative example, the same protocol is applicable also for label-free quantified proteomics data, as well as genome-wide expression profiles (e.g., quantified by RNA-seq or microarray; see our recent example of applying JUMPn for both gene and protein expression profiles27). Phosphoproteomics data could also be taken by JUMPn to identify co-expressed phosphosites, followed by kinase activity inference25. In addition, interactome data generated by the AP-MS approach will also be appropriate, by which prey proteins that follow similar bait interaction strength and stoichiometry will form co-expression clusters and further overlapped with known PPIs for data interpretation68.
Limitations exist for the current version of JUMPn. First, the installation procedure is command line-based and requires basic knowledge of computer science. This hinders wider usage of JUMPn, especially from biologists without computational background. A more ideal implementation is to publish JUMPn on an online server. Second, the current databases are human-centric because of our focus on human disease studies. Note that proteomics data generated by mice has also been analyzed by JUMPn using such human-centric databases25,27, assuming that most PPIs are conserved across both species69,70. Mouse-specific signaling will not be captured by this approach but is not of interest in those human studies. However, for non-mammalian model systems (e.g., zebrafish, fly, or yeast), species-specific databases should be prepared and uploaded to JUMPn using the advanced options. Resources of additional species may be provided via future JUMPn release. Third, the current step of ontology/pathway analysis takes significant time, which can be further optimized by parallel computing.
In conclusion, we present the JUMPn software and protocol for exploring quantitative proteomics data to identify and visualize co-expressed and potentially physically interacting protein modules by systems biology approach. The key features that distinguish JUMPn from others53,71,72 include: (i) JUMPn integrates and streamlines four major components of the pathway and network analysis (Figure 1); (ii) Different from most pathway analysis software that takes a simple gene list as input, JUMPn starts from quantification matrix, by which quantitative information can be seamlessly integrated with literature documented pathways and networks; (iii) Both co-expression protein clusters and interaction modules are automatically annotated by known pathways, and visualized via the R/shiny interacting platform using a user-friendly web browser; (iv) Final results are organized into three tables that are readily publishable in Excel format. Thus, we expect the JUMPn and this protocol will be widely applicable to many studies for dissecting mechanisms using quantitative proteomics data.
ACKNOWLEDGEMENTS
Funding support was provided by the National Institutes of Health (NIH) (R01AG047928, R01AG053987, RF1AG064909, RF1AG068581, and U54NS110435) and ALSAC (American Lebanese Syrian Associated Charities). The MS analysis was carried out in St. Jude Children’s Research Hospital’s Center of Proteomics and Metabolomics, which was partially supported by NIH Cancer Center Support Grant (P30CA021765). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/62796.
DISCLOSURES
The authors have nothing to disclose.
REFERENCES
- 1.Aebersold R, Mann M Mass-spectrometric exploration of proteome structure and function. Nature. 537, 347–355 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Senko MW et al. Novel parallelized quadrupole/linear ion trap/orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Analytical Chemistry. 85, 11710–11714 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Eliuk S, Makarov A Evolution of orbitrap mass spectrometry instrumentation. Annual Review of Analytical Chemistry. 8, 61–80 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Wang H et al. Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. Journal of Proteome Research. 14, 829–838 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blue LE et al. Recent advances in capillary ultrahigh pressure liquid chromatography. Journal of Chromatography A. 1523, 17–39 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Meier F et al. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Molecular & Cellular Proteomics. 17, 2534–2545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ludwig C et al. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Molecular Systems Biology. 14 (8), e8126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YY, Fonslow BR, Shan B, Baek MC, Yates JR Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113, 2343–2394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z et al. 27-Plex tandem mass tag mass spectrometry for profiling brain proteome in Alzheimer’s disease. Analytical Chemistry. 92, 7162–7170 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li JM et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nature Methods. 17 (4), 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins BC et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nature Communications. 8 (1), 291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarro P et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nature Biotechnology. 34, 1130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang XS et al. JUMP: A tag-based database search tool for peptide identification with high sensitivity and accuracy. Molecular & Cellular Proteomics. 13, 3663–3673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li YX et al. JUMPg: An integrative proteogenomics pipeline identifying unannotated proteins in human brain and cancer cells. Journal of Proteome Research. 15, 2309–2320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox J, Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nature Methods. 14, 513 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chi H et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nature Biotechnology. 36, 1059, (2018). [DOI] [PubMed] [Google Scholar]
- 18.Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nature Methods. 17, 41 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.High AA et al. Deep proteome profiling by isobaric labeling, extensive liquid chromatography, mass spectrometry, and software-assisted quantification. Journal of Visualized Experiments: JoVE. 129, 56474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z et al. High-throughput and deep-proteome profiling by 16-plex tandem mass tag labeling coupled with two-dimensional chromatography and mass spectrometry. Journal of Visualized Experiments: JoVE. 162, 61684 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meier F, Geyer PE, Winter SV, Cox J, Mann M BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nature Methods. 15, 440 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Sinitcyn P, Rudolph JD, Cox J Computational methods for understanding mass spectrometry-based shotgun proteomics data. Annual Review of Biomedical Data Science. 1, 207–234 (2018). [Google Scholar]
- 23.Ideker T, Galitski T, Hood L A new approach to decoding life: Systems biology. Annual Review of Genomics and Human Genetics. 2, 343–372 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Barabasi AL, Oltvai ZN Network biology: understanding the cell’s functional organization. Nature Reviews Genetics. 5, 101–113 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Tan H et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity. 46, 488–503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai B et al. Deep multilayer brain proteomics identifies molecular networks in alzheimer’s disease progression. Neuron. 105, 975–991 e977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H et al. Discrete roles and bifurcation of PTEN signaling and mTORC1-mediated anabolic metabolism underlie IL-7-driven B lymphopoiesis. Science Advances. 4, eaar5701 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seyfried NT et al. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Systems. 4, 60–72 e64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson ECB et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26, 769–780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart E et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell. 34, 411–426 e419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudolph JD, Cox J A network module for the perseus software for computational proteomics facilitates proteome interaction graph analysis. Journal of Proteome Research. 18, 2052–2064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 513, 382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petralia F et al. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell. 183, 1962 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutkowski J et al. A gene ontology inferred from molecular networks. Nature Biotechnology. 31, 38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu MK et al. Translation of genotype to phenotype by a hierarchy of cell subsystems. Cell Systems. 2, 77–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansen R, Greenbaum D, Gerstein M Relating whole-genome expression data with protein-protein interactions. Genome Research. 12, 37–46 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huttlin EL et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545, 505–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ron-Harel N et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metabolism. 24, 104–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niu MM et al. Extensive peptide fractionation and y(1) ion-based interference detection method for enabling accurate quantification by isobaric labeling and mass spectrometry. Analytical Chemistry. 89, 2956–2963 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang W et al. shiny: Web Application Framework for R. (2021).
- 41.JUMPn <https://github.com/VanderwallDavid/JUMPn_1.0.0> (2021)
- 42.Anaconda <https://docs.anaconda.com/anaconda/install/> (2021).
- 43.miniconda <https://docs.conda.io/en/latest/miniconda.html> (2021).
- 44.RStudio <https://www.rstudio.com/products/rstudio/download/> (2021)
- 45.Shiny Server <https://shiny.rstudio.com/articles/shinyapps.html> (2021)
- 46.Tyanova S, Temu T, Cox J The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 47.R code <https://github.com/VanderwallDavid/JUMPn_1.0.0/tree/main/JUMPn_preprocessing> (2021).
- 48.Florens L et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 40, 303–311 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang B, Horvath S A general framework for weighted gene co-expression network analysis. Statistical Applications in Genetics and Molecular Biology. 4, Article 17 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Voineagu I et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 474, 380 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langfelder P, Zhang B, Horvath S Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 24, 719–720 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL Hierarchical organization of modularity in metabolic networks. Science. 297, 1551–1555 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.FlyEn rich r <https://maayanlab.cloud/FlyEnrichr/#stats> (2021)
- 56.JUMPn GitHub <https://github.com/VanderwallDavid/JUMPn_1.0.0/tree/main/resources/example_fly_lib> (2021).
- 57.Langfelder P, Horvath S Eigengene networks for studying the relationships between co-expression modules. BMC Systems Biology. 1, 54 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benjamini Y, Hochberg Y Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B. 57, 289–300 (1995). [Google Scholar]
- 59.Szklarczyk D et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research. 43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szklarczyk D et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huttlin EL et al. The BioPlex network: A systematic exploration of the human interactome. Cell. 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huttlin EL et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell. 184, 3022–3040 e3028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li T et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nature Methods. 14, 61–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang H et al. Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nature Communications. 10, 3718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerstein MB et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 489, 91–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu J, Peng J, Chi H Systems immunology: Integrating multi-omics data to infer regulatory networks and hidden drivers of immunity. Current Opinion in Systems Biology. 15, 19–29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Califano A, Alvarez MJ The recurrent architecture of tumour initiation, progression and drug sensitivity. Nature Reviews Cancer. 17, 116–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hein MY et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163, 712–723 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Liang Z, Xu M, Teng MK, Niu LW Comparison of protein interaction networks reveals species conservation and divergence. BMC Bioinformatics. 7, 457 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shou C et al. Measuring the evolutionary rewiring of biological networks. PLOS Computational Biology. 7, e1001050 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature Communications. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cline MS et al. Integration of biological networks and gene expression data using Cytoscape. Nature Protocols. 2, 2366–2382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]