

Abstract
Background
Glaucoma is a group of optic neuropathies characterized by progressive degeneration of the retinal ganglion cells, axonal loss and irreversible visual field defects. Glaucoma is classified as primary or secondary, and worldwide, primary glaucoma is a leading cause of irreversible blindness. Several subtypes of glaucoma exist, and primary open‐angle glaucoma (POAG) is the most common. The etiology of POAG is unknown, but current treatments aim to reduce intraocular pressure (IOP), thus preventing the onset and progression of the disease. Compared with traditional antiglaucomatous treatments, rho kinase inhibitors (ROKi) have a different pharmacodynamic. ROKi is the only current treatment that effectively lowers IOP by modulating the drainage of aqueous humor through the trabecular meshwork and Schlemm's canal. As ROKi are introduced into the market more widely, it is important to assess the efficacy and potential AEs of the treatment.
Objectives
To compare the efficacy and safety of ROKi with placebo or other glaucoma medication in people diagnosed with open‐angle glaucoma (OAG), primary open‐angle glaucoma (POAG) or ocular hypertension (OHT).
Search methods
We used standard Cochrane methods and searched databases on 11 December 2020.
Selection criteria
We included randomized clinical trials examining commercially available ROKi‐based monotherapy or combination therapy compared with placebo or other IOP‐lowering medical treatments in people diagnosed with (P)OAG or OHT. We included trials where ROKi were administered according to official glaucoma guidelines. There were no restrictions regarding type, year or status of the publication.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. Two review authors independently screened studies, extracted data, and evaluated risk of bias by using Cochrane's RoB 2 tool.
Main results
We included 17 trials with 4953 participants diagnosed with (P)OAG or OHT. Fifteen were multicenter trials and 15 were masked trials. All participants were aged above 18 years. Trial duration varied from 24 hours to 12 months. Trials were conducted in the USA, Canada and Japan. Sixteen trials were funded by pharmaceutical companies, and one trial provided no information about funding sources. The trials compared ROKi monotherapy (netarsudil or ripasudil) or combination therapy with latanoprost (prostaglandin analog) or timolol (beta‐blocker) with placebo, timolol, latanoprost or netarsudil. Reported outcomes were IOP and safety. Meta‐analyses were applied to 13 trials (IOP reduction from baseline) and 15 trials (ocular AEs).
Of the trials evaluating IOP, seven were at low risk, three had some concerns, and three were at high risk of bias. Three trials found that netarsudil monotherapy may be superior to placebo (mean difference [MD] 3.11 mmHg, 95% confidence interval [CI] 2.59 to 3.62; I2 = 0%; 155 participants; low‐certainty evidence). Evidence from three trials found that timolol may be superior to netarsudil with an MD of 0.66 mmHg (95% CI 0.41 to 0.91; I2 = 0%; 1415 participants; low‐certainty evidence). Evidence from four trials found that latanoprost may be superior to netarsudil with an MD of 0.97 mmHg (95% CI 0.67 to 1.27; I2 = 4%; 1283 participants; moderate‐certainty evidence).
Evidence from three trials showed that, compared with monotherapy with latanoprost, combination therapy with netarsudil and latanoprost probably led to an additional pooled mean IOP reduction from baseline of 1.64 mmHg (95% CI −2.16 to −1.11; 1114 participants). Evidence from three trials showed that, compared with monotherapy with netarsudil, combination therapy with netarsudil and latanoprost probably led to an additional pooled mean IOP reduction from baseline of 2.66 mmHg (95% CI −2.98 to −2.35; 1132 participants). The certainty of evidence was moderate. One trial showed that, compared with timolol monotherapy, combination therapy with ripasudil and timolol may lead to an IOP reduction from baseline of 0.75 mmHg (95% −1.29 to −CI 0.21; 208 participants). The certainty of evidence was moderate.
Of the trials assessing total ocular AEs, three were at low risk, four had some concerns, and eight were at high risk of bias.
We found very low‐certainty evidence that netarsudil may lead to more ocular AEs compared with placebo, with 66 more ocular AEs per 100 person‐months (95% CI 28 to 103; I2 = 86%; 4 trials, 188 participants). We found low‐certainty evidence that netarsudil may lead to more ocular AEs compared with latanoprost, with 29 more ocular AEs per 100 person‐months (95% CI 17 to 42; I2 = 95%; 4 trials, 1286 participants).
We found moderate‐certainty evidence that, compared with timolol, netarsudil probably led to 21 additional ocular AEs (95% CI 14 to 27; I2 = 93%; 4 trials, 1678 participants). Data from three trials (1132 participants) showed no evidence of differences in the incidence rate of AEs between combination therapy with netarsudil and latanoprost and netarsudil monotherapy (1 more event per 100 person‐months, 95% CI 0 to 3); however, the certainty of evidence was low. Similarly, we found low‐certainty evidence that, compared with latanoprost, combination therapy with netarsudil and latanoprost may cause 29 more ocular events per 100 person‐months (95% CI 11 to 47; 3 trials, 1116 participants). We found moderate‐certainty evidence that, compared with timolol monotherapy, combination therapy with ripasudil and timolol probably causes 35 more ocular events per 100 person‐months (95% CI 25 to 45; 1 trial, 208 participants). In all included trials, ROKi was reportedly not associated with any particular serious AEs.
Authors' conclusions
The current evidence suggests that in people diagnosed with OHT or (P)OAG, the hypotensive effect of netarsudil may be inferior to latanoprost and slightly inferior to timolol. Combining netarsudil and latanoprost probably further reduces IOP compared with monotherapy. Netarsudil as mono‐ or combination therapy may result in more ocular AEs. However, the certainty of evidence was very low or low for all comparisons except timolol. In general, AEs were described as mild, transient, and reversible upon treatment discontinuation. ROKi was not associated with any particular serious AEs. Future trials of sufficient size and follow‐up should be conducted to provide reliable information about glaucoma progression, relevant IOP measurements and a detailed description of AEs using similar terminology. This would ensure the robustness and confidence of the results and assess the intermediate‐ and long‐term efficacy and safety of ROKi.
Keywords: Humans; Glaucoma, Open-Angle; Glaucoma, Open-Angle/drug therapy; Ocular Hypertension; Ocular Hypertension/drug therapy; Randomized Controlled Trials as Topic; rho-Associated Kinases; rho-Associated Kinases/antagonists & inhibitors; rho-Associated Kinases/therapeutic use; Treatment Outcome
Plain language summary
Rho kinase inhibitors for primary open‐angle glaucoma and ocular hypertension
Question
What are the benefits and risks of rho kinase inhibitor eye drops to treat people with either glaucoma or increased eye pressure?
Key messages
Antiglaucomatous eye drops such as latanoprost and timolol may reduce the eye pressure more compared with treatment with a rho kinase inhibitor, but the difference with timolol is small. When combining rho kinase inhibitors with different types of medicine, the eye pressure may be reduced more. People treated with a rho kinase inhibitor experience more adverse events (side effects) compared with other treatments. Future research in this area should focus on reporting disease progression (how the glaucoma gets worse over time).
What is glaucoma?
Glaucoma is a sight‐threatening eye disease that can lead to blindness if left untreated. There are different types of glaucoma and the most common is called primary open‐angle glaucoma. High eye pressure is a known risk factor for developing glaucoma.
Medical glaucoma treatment
There are different types of eye drops that can be used to treat glaucoma. All medical treatments of glaucoma work by reducing eye pressure. Latanoprost and timolol are two glaucoma medications, and one of the new types of glaucoma medicine is called a rho kinase inhibitor.
What did we want to find out?
We wanted to examine whether the effectiveness and safety of rho kinase inhibitor eye drops were better or worse than other medicines.
What did we do?
We searched for studies that compared:
‐ rho kinase inhibitor with placebo (a treatment with no therapeutic effect);
‐ rho kinase inhibitor with other types of glaucoma treatments (latanoprost and timolol).
Search date
We searched medical databases on 11 December 2020.
What did we find?
We found 17 studies examining 4953 people aged at least 18 years diagnosed with primary open‐angle glaucoma or high eye pressure and treated with a rho kinase inhibitor. The studies varied in treatment duration from 24 hours to 12 months. They were conducted in the USA, Canada and Japan. Of the studies, 16 were funded by pharmaceutical companies and one did not provide information about potential funding sources. The effect of treatment was evaluated by measuring the eye pressure and assessing the adverse events of treatment.
The studies did not report data disease progression, but they reported data on the lowering of the pressure within the eye and adverse events. Treatment with latanoprost may be better than rho kinase inhibitor. Treatment with timolol may be slightly better than treatment with rho kinase inhibitor. Furthermore, treatment with both rho kinase inhibitor and latanoprost or timolol probably reduces the eye pressure even more. Overall, the studies reported adverse events very differently. More people treated with rho kinase inhibitors may have experienced eye‐related adverse events; however, we are not very certain about these findings. There were no serious adverse events reported for treatment with rho kinase inhibitor.
Main limitations of the evidence
The studies did not report all the outcomes that we were interested in. The studies focused on specific outcomes such as eye pressure and adverse events, whereas we wanted to answer other questions as well. The current evidence was based on few studies. Some studies were conducted in a way that may have introduced errors into the results. Studies varied in the way they measured the outcomes and thus may not be comparable to each other.
Summary of findings
Summary of findings 1. Rho kinase inhibitor compared to placeboa.
|
Population: people with primary open‐angle glaucoma or ocular hypertension Settings: ophthalmology clinics Intervention: netarsudil 0.02% once per daya Comparison: placebo | ||||||
| Outcomes | Illustrative absolute effect or risk* (95% CI) | Absolute difference (95% CI) | No. of participants (RCTs) | Certainty of the evidence (GRADE) | Comments | |
|
Assumed effect with placebo |
Corresponding effect with netarsudil |
|||||
| Glaucoma progression at 12 months, measured by additional visual field defects | — | — | — | — | — | Not measured |
| Difference in mean IOP from baseline at < 6 months | 1.20 mmHg (0.62 to 1.77) lower | 4.31 mmHg (3.79 to 4.82) lower | 3.11 mmHg (2.59 to 3.62) lower | 155 (3 RCTs) | ⊕⊕⊝⊝ Lowb,c | — |
| Glaucoma progression at 12 months, defined by anatomic (structural) criteriad | — | — | — | — | — | Not measured |
| Patient‐reported outcome at the longest follow‐up | — | — | — | — | — | Not measured |
| Mean change in the number of glaucoma medications at the longest follow‐up | — | — | — | — | — | Not measured |
| Need for additional treatment at the longest follow‐up | — | — | — | — | — | Not measured |
| Average number of ocular adverse events at the longest follow‐up | 60 events per 100 person‐months | 126 events per 100 person‐months (88 to 163) | 66 more events per 100 person‐months (28 to 103) | 188 (4 RCTs) | ⊕⊝⊝⊝ Very lowc,e | — |
|
CI: confidence interval; IOP: intraocular pressure; RD: rate difference; ROKi: rho kinase inhibitor *The basis for the assumed effect (or risk) is the effect (or risk) in the placebo group across studies. The corresponding effect (or risk and its 95% confidence interval) is based on the assumed risk in the comparison group and the difference in the effect (or risk) of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDue to heterogeneity between subgroups, only netarsudil versus placebo is represented in this summary of findings table. bDowngraded one level for risk of bias. cDowngraded one level for imprecision: small sample sizes. dAnatomic criteria may include thinning of neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer. eDowngraded two level for high risk of bias in outcome measurement and selective outcome reporting.
Summary of findings 2. Rho kinase inhibitor compared to beta‐blocker.
|
Population: people with primary open‐angle glaucoma or ocular hypertension Settings: ophthalmology clinics Intervention: netarsudil 0.02% once per day Comparison: timolol 0.5% twice per day | ||||||
| Outcomes | Illustrative absolute effect (or risk) * (95% CI) | Risk difference (95% CI) | No. of participants (RCTs) | Certainty of the evidence (GRADE) | Comments | |
|
Assumed risk with timolol |
Corresponding risk with netarsudil | |||||
| Glaucoma progression at 12 months, measured by additional visual field defects | — | — | — | — | — | Not measured |
| Difference in mean IOP from baseline at < 6 months | 4.60 mmHg lower (3.91 to 5.29) | 3.94 mmHg lower (3.69 to 4.19) | 0.66 mmHg higher (0.41 to 0.91) | 1415 (3 RCTs) | ⊕⊕⊝⊝ Lowa | — |
| Glaucoma progression at 12 months, defined by anatomic (structural) criteriab | — | — | — | — | — | Not measured |
| Patient‐reported outcome at the longest follow‐up | — | — | — | — | — | Not measured |
| Mean change in the number of glaucoma medications at the longest follow‐up | — | — | — | — | — | Not measured |
| Need for additional treatment at the longest follow‐up | — | — | — | — | — | Not measured |
| Number of ocular adverse events at the longest follow‐up | 9 events per 100 person‐months | 30 events per 100 person‐months (23 to 36) | 21 more events per 100 person‐months (14 to 27) | 1678 (4 RCTs) | ⊕⊕⊕⊝ Moderatec |
— |
| BB: beta‐blocker; CI: confidence interval; IOP: intraocular pressure; RD: rate difference; ROKi: rho kinase inhibitor. *The basis for the assumed effect (or risk) is the effect (or risk) in the Timolol group across studies. The corresponding effect (or risk and its 95% confidence interval) is based on the assumed risk in the comparison group and the difference in the effect (or risk) of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded two levels for risk of bias: high risk of bias due to deviation from the intended intervention and missing outcome. bAnatomic criteria may include thinning of neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer. cDowngraded one level for risk of bias in incomplete outcome reporting or selective outcome reporting.
Summary of findings 3. Rho kinase inhibitor compared to prostaglandin analog.
|
Population: people with primary open‐angle glaucoma or ocular hypertension Settings: ophthalmology clinics Intervention: netarsudil 0.02% once per day (4 studies) Comparison: latanoprost 0.005% twice per day | ||||||
| Outcomes | Illustrative absolute effect (or risk)* (95% CI) | Difference (95% CI) | No. of participants (RCTs) | Certainty of the evidence (GRADE) | Comments | |
| With latanoprost | With netarsudil | |||||
| Glaucoma progression at 12 months, measured by additional visual field defects | — | — | — | — | — | Not measured |
| Difference in mean IOP from baseline at < 6 months | 6.44 mmHg lower (6.24 to 6.64) | 5.47 mmHg lower (5.18 to 5.76) | 0.97 mmHg higher (0.67 to 1.27) | 1283 (4 RCTs) | ⊕⊕⊕⊝ Moderatea | — |
| Glaucoma progression at 12 months, defined by anatomic (structural) criteriab | — | — | — | — | — | Not measured |
| Patient‐reported outcome at the longest follow‐up | — | — | — | — | — | Not measured |
| Mean change in the number of glaucoma medications at the longest follow‐up | — | — | — | — | — | Not measured |
| Need for additional treatment at the longest follow‐up | — | — | — | — | — | Not measured |
| Number of ocular adverse events at the longest follow‐up | 14 events per 100 person‐months | 43 events per 100 person‐months (31 to 56) | 29 more events per 100 person‐months (17 to 42) | 1286 (4 RCTs) | ⊕⊕⊝⊝ Lowc | — |
|
CI: confidence interval; IOP: intraocular pressure; RD: rate difference; ROKi: rho kinase inhibitor. *The basis for the assumed effect (or risk) is the effect (or risk) in the placebo group across studies. The corresponding effect (or risk and its 95% confidence interval) is based on the assumed risk in the comparison group and the difference in the effect (or risk) of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded one level for risk of bias. bAnatomic criteria may include thinning of neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer. cDowngraded two levels for high risk of bias in selective outcome reporting and unclear bias in outcome measurement.
Summary of findings 4. Rho kinase inhibitor and prostaglandin analog compared to prostaglandin analoga.
|
Population: people with primary open‐angle glaucoma or ocular hypertension Settings: ophthalmology clinics Intervention: netarsudil 0.02% + latanoprost 0.005% (FDC) once per daya Comparison: latanoprost 0.005% once per day | ||||||
| Outcomes | Illustrative absolute effect (or risk) * (95% CI) | Difference (95% CI) | No. of participants (RCTs) | Certainty of the evidence (GRADE) | Comments | |
| With latanoprost | With netarsudil + latanoprost (FDC) | |||||
| Glaucoma progression at 12 months, measured by additional visual field defects | — | — | — | — | — | Not measured |
| Difference in mean IOP from baseline at < 6 months | 6.62 mmHg (5.67 to 7.57) lower | 8.26 mmHg (7.73 to 8.78) lower | 1.64 mmHg (1.11 to 2.16) lower | 1114 (3 RCTs) | ⊕⊕⊕⊝ Moderatea | — |
| Glaucoma progression at 12 months, defined by anatomic (structural) criteriab | — | — | — | — | — | Not measured |
| Patient‐reported outcome at the longest follow‐up | — | — | — | — | — | Not measured |
| Mean change in the number of glaucoma medications at the longest follow‐up | — | — | — | — | — | Not measured |
| Need for additional treatment at the longest follow‐up | — | — | — | — | — | Not measured |
| Number of ocular adverse events at the longest follow‐up | 11 events per 100 person‐months | 37 events per 100 person‐months (24 to 51) | 26 more events per 100 person‐months (13 to 40) | 1321 (4 RCTs) | ⊕⊕⊝⊝ Lowc | — |
|
CI: confidence interval; FDC: fixed‐dose compound; IOP: intraocular pressure; RD: rate difference; ROKi: rho kinase inhibitor. *The basis for the assumed effect (or risk) is the effect (or risk) in the placebo group across studies. The corresponding effect (or risk and its 95% confidence interval) is based on the assumed risk in the comparison group and the difference in the effect (or risk) of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDue to heterogeneity between subgroups, only netarsudil + latanoprost versus latanoprost is represented in this summary of findings table. bDowngraded one level for risk of bias in selective outcome reporting: not all studies reported uncorrected mean changes in diurnal IOP from baseline. cAnatomic criteria may include thinning of neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer. dDowngraded two levels for high risk of bias in selective reporting of adverse outcomes.
Summary of findings 5. Rho kinase inhibitor and prostaglandin analog compared to rho kinase inhibitor.
|
Population: people with primary open‐angle glaucoma or ocular hypertension Settings: ophthalmology clinics Intervention: netarsudil 0.02% + latanoprost 0.005% (FDC) once per day (3 studies) Comparison: netarsudil 0.02% once per day | ||||||
| Outcomes | Illustrative absolute effect (or risk) * (95% CI) | Difference (95% CI) | No. of participants (RCTs) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with netarsudil | Corresponding risk with netarsudil + latanoprost (FDC) | |||||
| Glaucoma progression at 12 months, measured by additional visual field defects | — | — | — | — | — | Not measured |
| Difference in mean IOP from baseline at < 6 months | 5.47 mmHg (5.23 to 5.70) lower | 8.13 mmHg (7.82 to 8.45) lower | 2.66 mmHg (2.35 to 2.98) lower | 1132 (3 RCTs) | ⊕⊕⊕⊝ Moderatea | — |
| Glaucoma progression at 12 months, defined by anatomic (structural) criteriab | — | — | — | — | — | Not measured |
| Patient‐reported outcome at the longest follow‐up | — | — | — | — | — | Not measured |
| Mean change in the number of glaucoma medications at the longest follow‐up | — | — | — | — | — | Not measured |
| Need for additional treatment at the longest follow‐up | — | — | — | — | — | Not measured |
| Number of ocular adverse events at the longest follow‐up | 38 events per 100 person‐months | 39 events per 100 person‐months (38 to 41) | 1 more event per 100 person‐months (0 to 3 more) | 1131 (3 RCTs) | ⊕⊕⊝⊝ Lowc | — |
|
CI: confidence interval; FDC: fixed‐dose compound; IOP: intraocular pressure; RD: rate difference; ROKi: rho kinase inhibitor. *The basis for the assumed effect (or risk) is the effect (or risk) in the placebo group across studies. The corresponding effect (or risk and its 95% confidence interval) is based on the assumed risk in the comparison group and the difference in the effect (or risk) of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded one level for risk of bias in selective outcome reporting in some of the included studies. bAnatomic criteria may include thinning of neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer. cDowngraded two levels for high risk of bias in selective outcome reporting in all the included studies.
Summary of findings 6. Rho kinase inhibitor and beta‐blocker compared to beta‐blocker.
|
Population: people with primary open‐angle glaucoma or ocular hypertension Settings: ophthalmology clinics Intervention: ripasudil 0.4% + timolol 0.5% twice per day (1 study) Comparison: timolol 0.5% twice per day | ||||||
| Outcomes | Illustrative absolute effect (or risk) * (95% CI) | Risk difference (95% CI) | No. of participants (RCTs) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with timolol | Corresponding risk with ripasudil + timolol | |||||
| Glaucoma progression at 12 months, measured by additional visual field defects | — | — | — | — | — | Not measured |
| Difference in mean IOP from baseline at < 6 months | 1.67 mmHg lower (SD 1.99) | 2.42 mmHg (1.88 to 2.96) lower | 0.75 mmHg (0.21 to 1.29) lower | 208 (1 RCT) | ⊕⊕⊕⊝ Moderatea |
— |
| Glaucoma progression at 12 months, defined by anatomic (structural) criteriab | — | — | — | — | — | Not measured |
| Patient‐reported outcome at the longest follow‐up | — | — | — | — | — | Not measured |
| Mean change in the number of glaucoma medications at the longest follow‐up | — | — | — | — | — | Not measured |
| Need for additional treatment at the longest follow‐up | — | — | — | — | — | Not measured |
| Number of ocular adverse events at the longest follow‐up | 6 events per 100 person‐months | 41 events per 100 person‐months (31 to 51) | 35 more events per 100 person‐months (25 to 45) | 208 (1 RCT) | ⊕⊕⊕⊝ Moderatea |
— |
|
BB: beta‐blocker; CI: confidence interval; IOP: intraocular pressure; RD: rate difference; ROKi: rho kinase inhibitor; SD: standard deviation. *The basis for the assumed effect (or risk) is the effect (or risk) in the Timolol group across studies. The corresponding effect (or risk and its 95% confidence interval) is based on the assumed risk in the comparison group and the difference in the effect (or risk) of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded one level for imprecision due to small sample size. bAnatomic criteria may include thinning of neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer.
Background
Description of the condition
Glaucoma is a group of optic neuropathies characterized by progressive degeneration of the retinal ganglion cells, axonal loss and characteristic irreversible visual field defects (Chang 2012; Kolko 2015; Weinreb 2014). Glaucoma is classified as primary or secondary, the latter term describing glaucoma as a complication to another identifiable eye disease, systemic condition or medical treatment. Based on the anatomy of the anterior chamber, primary glaucoma is subclassified as primary open‐angle glaucoma (POAG) or angle‐closure glaucoma. Based on the intraocular pressure (IOP), POAG is further subclassified as normal‐tension glaucoma or high‐tension glaucoma (Quigley 2011)
The exact pathogenesis of POAG is unknown. However, several risk factors have been identified including age, gender, familial disposition, ethnicity and IOP (Jonas 2017; Kwon 2009). POAG may be asymptomatic until the late stages of the disease (Weinreb 2014).
Epidemiology
Glaucoma is among the leading causes of irreversible blindness worldwide (Cedrone 2008; Quigley 2006), with an estimated global prevalence of 3.54% (95% confidence interval [CI] 2.09 to 5.82) for people aged 40 to 80 years, which is equivalent to 76.02 million people (Tham 2014). As a result of an increasing elderly population, the number is expected to rise to 111.8 million by 2040 (Tham 2014). Literature concerning the prevalence of ocular hypertension (OHT) is limited, but published studies report a prevalence of OHT between 2.7% (OHT greater than 25 mmHg, for those aged 52 to 82 years) and 3.56% (95% CI 3.12% to 4.06%) (OHT greater than 21 mmHg, for those aged 49 years and older) (Kreuger 1980; Varma 2004).
Glaucoma treatment
Current treatments work by reducing IOP (Mehran 2020), although axon loss and retinal ganglion cell death are only partly addressed by these treatments (Chang 2012). Evidence suggests that lowering IOP reduces the conversion from OHT to glaucoma and additionally slows disease progression in people with glaucoma (Heijl 2002). Thus, treatment initiation and adherence to treatment are vital to avoid significant damage to the ocular structures and impairment of the visual field.
The most common treatment used in preventing glaucoma progression and OHT is daily administration of IOP‐lowering eye drops, followed by laser treatment or surgery (Weinreb 2014), although laser treatment is now being advocated as a primary treatment (Gazzard 2019). Marketed conventional antiglaucomatous eye drops generally work by either lowering the production or increasing the outflow of the aqueous humor (AH) (Mehran 2020). Pharmacologically active agents in conventional eye drops include prostaglandin analogs (PA), beta‐adrenoceptor antagonists (beta‐blockers; BB), carbonic anhydrase inhibitors, alfa‐adrenoceptor agonists (alfa‐agonists) and cholinergic agonists. They are used as mono‐ or combination therapy (Conlon 2017). Of these topical treatments, PA is the most effective initial treatment for reducing IOP (Li 2016).
Description of the intervention
Rho kinase inhibitors (ROKi) are generally administered as one eye drop, once or twice per day in the affected eye(s). Once applied, the drugs are effectively absorbed (Isobe 2014; Lin 2018a). Netarsudil (AR‐13324) is metabolized to netarsudil‐M1 (AR‐13503) by esterases, which is a more potent ROKi (Lin 2018a).
ROKi antiglaucomatous eye drops were approved in Japan in 2014 (Garnock‐Jones 2014), in the USA by the Food and Drug Administration (FDA) in 2017 (FDA 2017) and in Europe by the European Medicines Agency (EMA) in 2019 (Aerie 2019).
How the intervention might work
Rho kinase (ROCK) is a serine/threonine‐protein kinase found downstream of the rho GTPase/ROCK signaling pathway. Currently, two isoenzymes ROCK1 and ROCK2 are identified. Upon activation, ROCK has been shown to regulate actin cytoskeletal dynamics, actomyosin contraction, cell adhesion, cell stiffness, cell morphology and extracellular matrix (ECM) reorganization (Rao 2017). It is evident that ROKis reduce IOP. Moreover, ROKis may possess neuroprotective (Rao 2017), antifibrotic (Rao 2017), as well as cornea protective properties (Okumura 2017). In general, ROKis reduce IOP by increasing the AH outflow through the trabecular (conventional) outflow pathway as a result of decreased contractility of the trabecular meshwork (TM) endothelial cells and the cells of Schlemm's canal (SC) (Braunger 2015; Rao 2017). The ROKi and norepinephrine transporter inhibitor netarsudil may also reduce the episcleral venous pressure (Kazemi 2018; Sit 2021) and the production of AH (Wang 2015).
The increase in AH outflow in response to ROKi application is associated with several factors such as (Rao 2017):
relaxation of the TM and expansion of the juxtacanalicular network;
possible increased formation of giant vacuoles in the inner wall of SC;
widening of SC;
washout of extracellular material in the TM.
On a cellular level, ROCK inhibition has been shown to inhibit the transdifferentiation of TM cells into a fibrogenic myofibroblast‐like phenotype and induce relaxation of TM and SC cells associated with decreased formation of actin stress fibers, focal adhesions and cell–cell interactions. Moreover, ROKis decreases pore formation in SC cells and decreases ECM production of TM and SC cells (Braunger 2015; Rao 2017). Increased drainage resistance in the trabecular pathway is a major cause of elevated IOP in POAG (Gabelt 2005; Lütjen‐Drecoll 1999; Stamer 2012). In vivo experiments have established that the rho GTPase/signaling pathway plays an important role in the regulation of IOP through modulation of AH outflow. Several clinical trials, involving healthy as well as people diagnosed with OHT or POAG, have demonstrated that ROKis effectively reduce IOP. Therefore, ROKi‐based treatments may prove to be important in the regulation of IOP, and thus prevention of onset and progression of glaucoma.
Why it is important to do this review
ROKis demonstrate different pharmacodynamic properties compared with traditional glaucoma drugs, as they are the only treatment that effectively reduce IOP through modulation of AH outflow by targeting the TM and SC cells (Tanna 2018). Therefore, ROKi may be an effective alternative or additional treatment, when conventional glaucoma medication is insufficient to prevent glaucoma progression and to control IOP. Adverse effects (AE) of treatment are one of many possible reasons that discourage people from following the prescribed treatment regimen (Wolfram 2019). Adherence to medical treatment is essential to lower IOP effectively and avoid the progression of glaucoma. As ROKi is introduced in the market more widely, it is important to assess the efficacy and potential AEs of this drug.
Objectives
To compare the efficacy and safety of ROKi with placebo or other glaucoma medication in people diagnosed with open‐angle glaucoma (OAG), primary open‐angle glaucoma (POAG) or ocular hypertension (OHT).
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) comparing monotherapy or combination therapy with ROKi to placebo or other IOP‐lowering medical treatments. RCTs were included regardless of type, year or status of publication.
Types of participants
Inclusion criteria
People diagnosed with (P)OAG or OHT, whether previously treated or recently diagnosed, and no prior exposure to ROKis. We applied no restrictions regarding age, gender or geography.
Exclusion criteria
People diagnosed with secondary OAG.
Types of interventions
We included RCTs comparing topical ROKi monotherapy or combination therapy with either placebo or other topical medical glaucoma treatments. The interventions and comparators were required to be administered as prescribed according to official clinical guidelines for the indication of glaucoma.
Types of outcome measures
Primary outcomes
The purpose of IOP‐lowering drugs is ultimately to prevent the development and progression of glaucoma. Thus, the primary outcome of this review was glaucoma progression.
Glaucoma progression, defined as additional visual field defects after at least 12 months of follow‐up compared with baseline. Glaucoma progression data were planned to be collected as a dichotomous outcome.
Secondary outcomes
Difference in mean IOP (mmHg) measured at baseline, compared with IOP at follow‐up, as it is the standard surrogate outcome in glaucoma research and known to be associated with glaucoma progression. As ROKis have only recently been approved, we expected most studies to report untreated IOP (after washout) at baseline, as they aimed to investigate the net IOP‐lowering effect of ROKi. The expected IOP‐lowering effect of a medication is used for decision‐making in clinical practice, but IOP may be influenced by other medications or treatments. We collected the change of medicated IOP when available. IOP change was collected as a continuous measure at an early (less than six months) or medium‐ to long‐term (six months or more) follow‐up, or both.
Glaucoma progression, defined by the investigators using valid anatomic (structural) criteria, such as thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer, or thinning of the macular ganglion cell layer, after at least 12 months of follow‐up compared with baseline.
Participant‐reported outcomes, including quality of life and preferences, medication adherence measured with validated questionnaires comparing the intervention groups at baseline to the longest follow‐up (continuous or categorical measure).
Mean change in the number of glaucoma medications between baseline and the longest follow‐up (continuous measure).
Need for IOP‐lowering medications, or additional laser, or surgical treatment measured as the need for additional medications, number of needed medications, laser or surgery at the longest follow‐up (dichotomous measure).
Adverse effects (AE), measured as the severity and number of AE related to the drug (continuous measures) at the longest follow‐up. Examples of AEs related to the use of ROKis included conjunctival hyperemia, conjunctival hemorrhage, corneal verticillata and instillation site pain.
Search methods for identification of studies
Electronic searches
The Cochrane Eyes and Vision (CEV) information specialist searched the following electronic databases for RCTs and controlled clinical trials. There were no restrictions to language or year of publication.
Cochrane Central Register of Controlled Trials (CENTRAL) (which contains the CEV Trials Register) in the Cochrane Library (2020, Issue 11) (Appendix 1).
MEDLINE Ovid (1946 to 11 December 2020) (Appendix 2).
Embase.com (1947 to 11 December 2020) (Appendix 3).
PubMed (1948 to 11 December 2020) (Appendix 4).
Latin American and Caribbean Health Sciences Literature Database (LILACS) (1982 to 11 December 2020) (Appendix 5).
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov; searched 11 December 2020) (Appendix 6).
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp; searched 11 December 2020) (Appendix 7).
Searching other resources
We searched the reference lists of included studies for additional trials. We did not search conference abstracts for this review, as many eyes and vision conference abstracts are included in Embase, which was accessed as part of the electronic searches.
Data collection and analysis
We published the protocol in December 2020 (Freiberg 2020). We collected the data in accordance with the standards and methods provided by Cochrane using Covidence to facilitate independent screening of records and data extraction from study reports by two or more review authors and adjudication after comparisons (Covidence).
Selection of studies
The information specialist from CEV removed duplicates from the search results. Two review authors (JCF and AVS) independently screened each title and abstract of the remaining records and classified it as 'relevant', 'possibly relevant' or 'not relevant'. In case of disagreements, a third review author adjudicated. The two review authors (JCF and AVS) read the full‐text reports of the records classified as 'possibly relevant' and 'relevant' and evaluated their final eligibility. We generated a PRISMA flow diagram to illustrate and document the process of identifying eligible studies (see Figure 1; Moher 2009).
1.
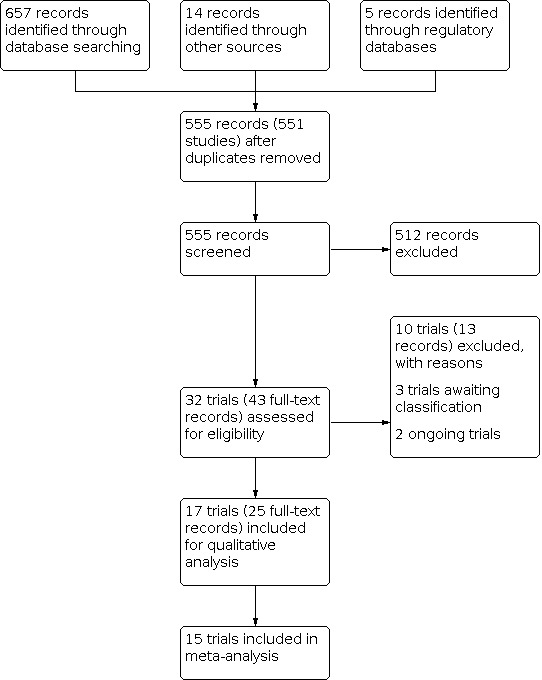
Study flow diagram.
Data extraction and management
We collected and organized data following guidance from Chapter 5 of the Cochrane Handbook for Systematic Reviews of Interventions (Li 2021a). Two review authors (JCF and AVS) independently extracted data from each full‐text article, including title; names of authors; study methods; descriptions of participants, interventions and outcomes; study results; and other relevant information (e.g. key conclusions of the study authors, reference to other relevant studies, identification and notes on funding and support, financial disclosures). In cases of disagreements in the data extracted between the two review authors, a third review author adjudicated. Whenever possible, we extracted observed data in preference to statistically corrected data.
In case of missing, incomplete or unclear information, the CEV methodologist contacted study investigators directly to request details. Whenever investigators did not respond within two weeks, the review authors proceeded with the existing information. We exported the collected data to RevMan Web (RevMan Web 2022).
Assessment of risk of bias in included studies
Two review authors (JCF and AVS) independently assessed the risk of bias of included studies using the RoB 2 tool (Sterne 2019), and according to Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a). In case of any disagreement, a third review author adjudicated.
The tool is structured into five domains through which bias may be introduced into outcomes.
Bias arising from the randomization process.
Bias due to deviations from intended interventions.
Bias due to missing outcome data.
Bias in measurement of the outcome.
Bias in selection of the reported result.
We evaluated the risk of bias for every domain as either low risk of bias, some concerns or high risk of bias. The assessment of each domain was guided by signaling questions. The overall assessment of the risk of bias for a given outcome was based on the sum of potential biases in each domain. We considered that a study or trial was at:
low risk of bias when the study was at low risk of bias for all domains with respect to an outcome;
some concerns when there was some concern for at least one domain for the specified outcome, but none of the domains was at high risk of bias;
high risk of bias when at least one domain was judged at high risk of bias, or there were some concerns for multiple domains in a way that substantially lowered confidence in the result.
Measures of treatment effect
We conducted the data analysis using guidance from Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (McKenzie 2021). We estimated the mean difference (MD) with 95% CIs for continuous measures and incidence rate difference (RD) with 95% CIs for ocular AEs. We provided a narrative description for sparse or heterogeneous outcome data. When numerical IOP data were not reported in either a registry record or publication, we derived IOP values and standard deviations from graphs (Rohatgi 2021). When mean diurnal IOP was not available, we calculated the mean IOP difference between baseline and follow‐up, and imputed a change‐from‐baseline standard deviation using the correlation coefficient (Higgins 2011).
To quantify the overall risks associated with treatment AEs on the eyes, we categorized the reported number of incidents of similar or clinically related adverse symptoms and signs to prespecified types of ocular AEs (Appendix 8), and then summed the numbers of incidents by type before estimating the incidence rates of total ocular AEs for each treatment group. We estimated standard errors for incidence RDs according to guidance from Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b).
Unit of analysis issues
The participants were the primary unit of analysis. We included one study where participants received different treatments in each eye (Sit 2021). We investigated the impact of this study through a sensitivity analysis.
Dealing with missing data
In case of missing, incomplete or unclear data, the CEV methodologist contacted study investigators directly to request or clarify data. Whenever the investigators did not respond within two weeks, the review authors proceeded with the available data. Furthermore, in cases of substantial quantitative discrepancies between reported data in a full‐text publication and trial registry, we contacted the authors to clarify the discrepancies. We conducted analyses using complete cases; we did not impute missing data. Instead, when relevant, we performed sensitivity analyses excluding studies at high risk of bias for missing data.
Assessment of heterogeneity
We assessed clinical or methodological heterogeneity across studies by comparing the study and participant characteristics and risk of bias assessment. We evaluated statistical heterogeneity by observing and analyzing the forest plot and using the I² statistic. We interpreted the values of I² by applying the following overlapping categories and individual judgment:
0% to 40%: may not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
To assess the risk of reporting bias, we compared the outcomes defined in the protocol of the trials with those in the full‐text publications from the trials. We included fewer than 10 studies in each analysis, too few to use a funnel plot to assess small‐study effects.
Data synthesis
We conducted quantitative synthesis when we had enough similar trials that had reported an outcome to combine their data. This decision was mainly based on the type of comparison. Intervention as a source of clinical heterogeneity and thus statistical heterogeneity were investigated in subgroup analyses. In cases where we did not have enough comparable studies to conduct a meta‐analysis, we provide a narrative summary of data.
We analyzed data using fixed‐effect or random‐effects statistical models. In cases where we included three or fewer studies, we used a fixed‐effect model. If there were more than three studies, the type of statistical model used depended on clinical judgment and the statistical heterogeneity among the included studies. We did not combine data in a meta‐analysis when the I² statistic was greater than 75% unless effects in the same direction and of similar magnitude were consistent across studies.
Subgroup analysis and investigation of heterogeneity
When there was considerable heterogeneity, we conducted a subgroup analysis based on the type of ROKi intervention. The decision was based on a judgment of the clinical heterogeneity across the included studies as well as the degree of statistical heterogeneity. We used statistical methods provided within Review Manager Web as a tool for formal testing of subgroup differences (RevMan Web 2022).
Sensitivity analysis
When relevant, we re‐ran the meta‐analyses by excluding studies at high risk of bias and studies that did not correctly manage the unit of analysis issue.
Summary of findings and assessment of the certainty of the evidence
We created summary of findings tables for each comparison using guidelines in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2019). The tables present the key information concerning the certainty of the evidence, the magnitude of the effect of the interventions examined and the sum of available data for the main outcomes. Two review authors independently analyzed the certainty of the evidence using the GRADE approach as high, moderate, low or very low (GRADE Handbook).
We included the following outcomes in the summary of findings tables.
Glaucoma progression, defined as additional visual field changes after at least 12 months of follow‐up compared with baseline.
Difference in mean IOP from baseline to the longest available follow‐up according to the predefined study outcomes.
Glaucoma progression, defined by the investigators using valid anatomic (structural) criteria after at least 12 months.
Participant‐reported outcomes at the longest available follow‐up (short or medium‐ to long‐term, as defined above).
Mean change in the number of glaucoma medications between baseline and the longest available follow‐up (short or medium‐to‐long term, as defined above).
Need for IOP‐lowering medications, or additional laser, or surgical treatment at the longest available follow‐up (short or medium‐to‐long term, as defined above).
Number of adverse events at the longest available follow‐up.
Results
Description of studies
A detailed description of each included trial is available in the Characteristics of included studies table.
Results of the search
We conducted a search of the electronic databases in December 2020 and identified 676 records (Figure 1). We removed duplicates and screened 555 records corresponding to 551 studies. We excluded 512 records, leaving 43 articles for full‐text screening. We excluded 13 full‐text articles with reasons, three trials awaited classification (CTRI/2018/04/013091; CTRI/2020/01/022619; NCT03284853), and two trials were ongoing (JapicCTI‐194920; UMIN000019017). We included 17 trials in the qualitative synthesis, of which we included 15 trials in one or more meta‐analysis.
Included studies
We included 17 RCTs in this review (Aerie 2017; Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Inoue 2018; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; NCT02246764 (ROCKET‐3); Peace 2021; Serle 2018 (ROCKET‐1); Sit 2021; Tanihara 2013; Tanihara 2015a; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2)).
Thirteen trials were included in the meta‐analysis concerning efficacy in IOP reduction (Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; Peace 2021; Serle 2018 (ROCKET‐1); Sit 2021; Tanihara 2013; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2)). Two additional trials were included in the meta‐analysis concerning safety defined as 'total ocular AEs', 'conjunctival hyperemia' and 'ocular pain and irritation' (Aerie 2017; NCT02246764 (ROCKET‐3)).
Study design
Fifteen trials were multicenter and two were single center (Peace 2021; Sit 2021). One was a cross‐over trial (Tanihara 2015a), and the rest were parallel‐group studies. Nine trials were in phase 2 and eight trials were in phase 3. Thirteen trials were double‐masked according to the publications. According to the trial registry, one trial was triple masked (Aerie 2017), and one trial was quadruple‐masked (NCT02246764 (ROCKET‐3)). Two trials were open‐label (Inoue 2018; Tanihara 2015a).
Participants
The 17 trials randomized 4953 participants. Participants were required to be a minimum of 18 years of age in 10 trials (Aerie 2017; Asrani 2019 (MERCURY‐1); Bacharach 2015; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; Peace 2021; Serle 2018 (ROCKET‐1); Sit 2021; Walters 2019 (MERCURY‐2)), a minimum of 19 years of age in one trial (NCT02246764 (ROCKET‐3)), and a minimum of 20 years of age in five trials (Araie 2021; Tanihara 2013; Tanihara 2015a; Tanihara 2015b; Tanihara 2015c). Two trials allowed participants to be aged from birth to two years but did not include any participants in this age group (Kahook 2019 (ROCKET‐2); Serle 2018 (ROCKET‐1)). One trial did not report any restrictions regarding age (Inoue 2018). All participants were diagnosed with POAG, OAG or OHT.
Twelve trials required participants to have a corrected visual acuity equal to or better than +1.0 logMAR on the ETDRS (Early Treatment Diabetic Retinopathy Study) chart equivalent to 20/200 on the Snellen chart or best‐corrected visual acuity (BCVA) 0.1 on a Landolt‐C Chart (Aerie 2017; Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; NCT02246764 (ROCKET‐3); Peace 2021; Serle 2018 (ROCKET‐1); Sit 2021; Walters 2019 (MERCURY‐2)). In 15 studies, the trial investigators applied restrictions to unmedicated IOP as an inclusion criterion, whereas three trials did not report sufficient information regarding unmedicated or medicated IOP restrictions (Araie 2021; Inoue 2018; Lewis 2016). One trial included only participants with poorly controlled IOP after three months of treatment with PAs (Inoue 2018). Twelve trials excluded people with pseudoexfoliation or pigment dispersion glaucoma (Aerie 2017; Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; NCT02246764 (ROCKET‐3); Peace 2021; Serle 2018 (ROCKET‐1); Sit 2021; Walters 2019 (MERCURY‐2)). People who had previously undergone intraocular surgery were excluded from all trials except from one trial, which did not specify any exclusion criteria (Inoue 2018).
Interventions
The 17 trials differed in both interventions and comparisons.
Netarsudil‐based interventions
Twelve trials evaluated the ROKi netarsudil monotherapy; four trials compared netarsudil with placebo (Aerie 2017; Araie 2021; Peace 2021; Sit 2021), four trials compared netarsudil with timolol (Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); NCT02246764 (ROCKET‐3); Serle 2018 (ROCKET‐1)), and four trials compared netarsudil with latanoprost (Asrani 2019 (MERCURY‐1); Bacharach 2015; Lewis 2016; Walters 2019 (MERCURY‐2)).
Three trials compared combination therapy of netarsudil and latanoprost with either netarsudil or latanoprost monotherapy (Asrani 2019 (MERCURY‐1); Lewis 2016; Walters 2019 (MERCURY‐2)).
Ripasudil‐based interventions
Five trials evaluated the ROKi ripasudil; two trials compared ripasudil with placebo (Tanihara 2013; Tanihara 2015a), two trials compared combination therapy of ripasudil and timolol or ripasudil and latanoprost with timolol or latanoprost (Tanihara 2015b; Tanihara 2015c), and one trial compared combination therapy of ripasudil and latanoprost/travoprost/tafluprost with timolol and latanoprost/travoprost/tafluprost (Inoue 2018).
Outcomes
Per protocol (Freiberg 2020), the primary review outcome was glaucoma progression, defined as additional visual field defects quantified after at least 12 months of follow‐up from baseline. We also sought to evaluate secondary outcomes, such as changes in mean IOP from baseline; glaucoma progression based on other anatomic or structural criteria defined by the included trials; participant‐reported outcomes; documented needs for IOP‐lowering medications or surgical treatment; treatment‐related adverse events. However, none of the included trials reported outcomes other than 'changes in IOP' and 'ocular adverse events'.
Intraocular pressure
Nine trials reported time point‐matched mean IOP at multiple time points during the day at baseline and follow‐up or the time point‐matched mean IOP change at follow‐up compared with baseline, or both (Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; Peace 2021; Serle 2018 (ROCKET‐1); Walters 2019 (MERCURY‐2). Of these, six trials reported the corresponding mean diurnal or nocturnal IOP change at follow‐up compared with baseline (Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Lewis 2016; Peace 2021; Walters 2019 (MERCURY‐2)). One trial reported the mean diurnal IOP change but not the single time point measurements (Sit 2021). One trial reported the mean diurnal IOP at follow‐up but not the baseline or the change from baseline values (Aerie 2017). Four trials reported the time point‐matched adjusted mean IOP change at follow‐up compared with baseline (Tanihara 2013; Tanihara 2015a; Tanihara 2015b; Tanihara 2015c). One trial reported single time point mean IOP at baseline and follow‐up and the mean IOP change at follow‐up compared with baseline. However, the mean single‐time point IOP values were based on unspecified, distinct single‐time point measures during the day (Inoue 2018).
Safety
All trials evaluated safety in the form of ocular adverse events. The terminology, the degree of detail in the reporting and the reporting threshold varied among the trials.
Overall, the follow‐up period varied from 24 hours to 12 months. Further characteristics of the trials are included in the Characteristics of included studies table.
Excluded studies
After full‐text screening, we excluded 10 trials, mainly because of ineligible study design. See Characteristics of excluded studies table.
Studies awaiting classification
Three studies are awaiting classification (CTRI/2018/04/013091; CTRI/2020/01/022619; NCT03284853; Characteristics of studies awaiting classification table).
Ongoing studies
Two studies are ongoing (JapicCTI‐194920; UMIN000019017; Characteristics of ongoing studies table).
Risk of bias in included studies
We assessed the risk of bias using RoB 2 (Higgins 2021a).
RoB 2 was applied to two critical outcomes: IOP and total ocular AE. Considering IOP, 13 trials were included in the risk of bias assessment (Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Lewis 2016; Peace 2021; Serle 2018 (ROCKET‐1); Sit 2021; Tanihara 2013; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2)); two additional trials were included in the risk of bias assessment considering total ocular AEs (Aerie 2017; NCT02246764 (ROCKET‐3)).
Domain 1 – randomization process
IOP and ocular AE: of the trials included in the quantitative synthesis, all trials except one (Bacharach 2015) provided sufficient information on the randomization process, the concealment of allocation, and the baseline characteristics of the participants, and thus were judged at low risk of bias.
Domain 2 – deviations from intended interventions
IOP: eight trials reported sufficient information about masking of participants and trial site personnel and methods to avoid deviation from assigned intervention (Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Lewis 2016; Peace 2021; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2)), thus were judged at low risk of bias. Another two trials were judged as having some concerns due to exclusion of participants after randomization, only including responders of treatment (Sit 2021), and excluding participants who experienced adverse events to treatments, from the analysis (Tanihara 2013). The rest of the trials examined a per‐protocol population defined as "subjects without major protocol violation (that was) likely to seriously affect the primary outcome" (Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Serle 2018 (ROCKET‐1)) and displayed differential completion rates between the interventions. The trials were thus evaluated as having high risk of bias.
Ocular AEs: all trials provided adequate information on masking and analysis used for effect estimation. We judged all as having low risk of bias in this domain.
Domain 3 – missing outcome data
IOP: 10 trials had no issue of missing outcome data (Araie 2021; Asrani 2019 (MERCURY‐1); Bacharach 2015; Lewis 2016; Peace 2021; Sit 2021; Tanihara 2013; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2)). Three trials were judged as having high risk of bias due to the per‐protocol analysis based on participants of differential completion rates between the comparison groups. The three studies lacked a detailed description of reasons for excluding participants from analysis, and thus the missing data might have affected the reported outcomes (Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Serle 2018 (ROCKET‐1)).
Ocular AEs: all trials had no issue of missing outcome data, and were considered as low risk of bias.
Domain 4 – measurement of the outcome
IOP: all trials provided sufficient information on the method used for outcome measurement, and were judged as having low risk of bias in this domain.
Ocular AEs: 11 trials provided sufficient information on the methods used for measurement of ocular AEs (Aerie 2017; Araie 2021; Asrani 2019 (MERCURY‐1); Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); NCT02246764 (ROCKET‐3); Serle 2018 (ROCKET‐1); Tanihara 2013; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2). The other two trials were judged as prompting some concerns due to limited information available for assessment, lacking a detailed description of how AEs were measured, that is, relying mostly on participant‐reported outcomes and thus at risk of being subjective (Bacharach 2015; Lewis 2016). Another two trials were at high risk of bias due to very limited information, with no description or information about how AEs were measured and reported (Peace 2021; Sit 2021).
Domain 5 – selection of the reported result
IOP: all trials reported sufficient information on the prespecified analysis plan/a priori analysis and were considered at low risk of bias.
Ocular AEs: five trials reported all detectable ocular AEs and, thus, were at low risk of bias (Araie 2021; Peace 2021; Sit 2021; Tanihara 2015b; Tanihara 2015c). The remaining trials varied in the degree of under‐reporting. Four trials were judged as causing some concerns due to intermediate reporting thresholds (3%) (Kahook 2019 (ROCKET‐2); Serle 2018 (ROCKET‐1)), or the combination of high reporting thresholds (5%) with low numbers of participants in the treatment arms (40 or fewer) (Aerie 2017; NCT02246764 (ROCKET‐3)). Six trials had high (5% or greater) or unknown reporting thresholds resulting in the judgment of having high risk of bias (Asrani 2019 (MERCURY‐1); Bacharach 2015; Khouri 2019 (ROCKET‐4); Lewis 2016; Tanihara 2013; Walters 2019 (MERCURY‐2)).
Overall bias judgment
IOP: seven trials were overall at low risk of bias (Araie 2021; Asrani 2019 (MERCURY‐1); Lewis 2016; Peace 2021; Tanihara 2015b; Tanihara 2015c; Walters 2019 (MERCURY‐2)), three trials were judged as some concerns (Bacharach 2015; Sit 2021; Tanihara 2013), and three trials were at high risk of bias (Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Serle 2018 (ROCKET‐1)).
Ocular AE: three trials were overall at low risk of bias (Araie 2021; Tanihara 2015b; Tanihara 2015c), four trials were overall judged as some concerns (Aerie 2017; Kahook 2019 (ROCKET‐2); NCT02246764 (ROCKET‐3); Serle 2018 (ROCKET‐1)), and eight trials were overall at high risk of bias (Asrani 2019 (MERCURY‐1); Bacharach 2015; Khouri 2019 (ROCKET‐4); Lewis 2016; Peace 2021; Sit 2021; Tanihara 2013; Walters 2019 (MERCURY‐2)).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
It was not possible to include all trials in meta‐analyses due to insufficient data (Tanihara 2015a) or non‐comparable interventions (Inoue 2018).
Rho kinase inhibitor versus placebo
Primary outcome
Glaucoma progression
None of the trials comparing ROKi versus placebo reported quantifiable data on glaucoma progression such as visual field defects, thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer or thinning of the macular ganglion cell layer.
Secondary outcomes
Difference in mean intraocular pressure
Four trials (258 participants) examined the efficacy of ripasudil 0.4% (twice per day, a.m. and p.m.) (Tanihara 2013) and netarsudil 0.02% (once per day, p.m.) (Araie 2021; Peace 2021; Sit 2021) compared with placebo. Due to substantial heterogeneity (I2 = 92.1%), we did not combine data of netarsudil and ripasudil, neither did we draw any conclusions on ROKi compared with placebo (Analysis 1.1). In a sensitivity analysis that excluded Sit 2021 in which the unit of randomization and analysis was eye and not person, we found no evidence that the heterogeneity was reduced substantially (I2 = 89.9%; Analysis 1.2).
1.1. Analysis.

Comparison 1: Rho kinase inhibitor versus placebo, Outcome 1: Mean intraocular pressure (IOP) changes from baseline
1.2. Analysis.

Comparison 1: Rho kinase inhibitor versus placebo, Outcome 2: Mean IOP changes from baseline: sensitivity analysis
In an indirect comparison, netarsudil 0.02% reduced IOP from baseline more than ripasudil (netarsudil: MD 3.11 mmHg, 95% CI 2.59 to 3.62; I2 = 0%; ripasudil: MD 1.30 mmHg, 95% CI 0.45 to 2.15; Figure 2). When excluding Sit 2021, the MD for netarsudil versus placebo was 2.96 mmHg (95% CI 2.37 to 3.56; I2 = 0%).
2.

Overall, we judged the evidence for the estimated MD in IOP reduction from baseline to be low certainty after downgrading one level for risk of bias and one level for imprecision due to small sample sizes (Table 1).
Glaucoma progression using valid anatomic (structural) criteria
No trials comparing ROKi versus placebo reported glaucoma progression using valid anatomic (structural) criteria.
Participant‐reported outcomes
No trials comparing ROKi versus placebo reported participant‐reported outcomes.
Mean change in the number of glaucoma medications
No trials comparing ROKi versus placebo reported mean change in the number of glaucoma medications.
Need for intraocular pressure‐lowering medications
No trials comparing ROKi versus placebo reported need for IOP‐lowering medications.
Adverse effects
Five trials (291 participants) examined AEs to treatment comparing either ripasudil 0.4% (twice per day, a.m./p.m.) (Tanihara 2013) or netarsudil 0.02% (once per day, p.m.) to placebo (Aerie 2017; Araie 2021; Peace 2021; Sit 2021).
Ocular adverse events: treatment with netarsudil may lead to an increased rate of ocular AEs compared with placebo, with 66 more ocular AEs per 100 person‐months (95% CI 28 to 103; I2 = 81.5%; Analysis 1.3). We applied a random‐effects model though there was substantial heterogeneity within and between the subgroups. Peace 2021 detected no ocular AEs in either of the group, whereas Sit 2021 detected no ocular AEs in the placebo group, which was considered as the major source of the observed heterogeneity (Figure 3). We found no evidence that excluding trials at high risk of bias overall (Peace 2021; Sit 2021) changed the mean incidence RD in ocular AEs (RD 67, 95% CI 55 to 79; I2 = 0%; Analysis 1.4). Treatment with ripasudil may lead to 27 more events (95% CI 13 to 41) compared with placebo. Overall, the evidence for the estimated difference in incidence rates of ocular AEs was very low certainty after downgrading one level for imprecision due to small sample sizes and two levels for high risk of bias in outcome measurement and selective outcome reporting.
1.3. Analysis.

Comparison 1: Rho kinase inhibitor versus placebo, Outcome 3: Total ocular adverse events (per person‐month) – incidence risk difference
3.

1.4. Analysis.

Comparison 1: Rho kinase inhibitor versus placebo, Outcome 4: Total ocular adverse events (per person‐month): sensitivity analysis
In general, treatment with ROKi reported more events of conjunctival hyperemia compared with placebo (Analysis 1.5). There were no events of cornea verticillata, and no evidence of a difference between treatments in terms of ocular pain and irritation (Analysis 1.6). Less than 5% of participants reported serious adverse events (SAEs). For a detailed description, see Appendix 9.
1.5. Analysis.

Comparison 1: Rho kinase inhibitor versus placebo, Outcome 5: Conjunctival hyperemia as adverse event (per person‐month)*
1.6. Analysis.

Comparison 1: Rho kinase inhibitor versus placebo, Outcome 6: Ocular pain or irritation as adverse event (per person‐month)*
Rho kinase inhibitor versus beta‐blocker
Primary outcome
Glaucoma progression
None of the trials comparing ROKi versus BB reported quantifiable data on glaucoma progression such as visual field defects, thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer or thinning of the macular ganglion cell layer.
Secondary outcomes
Difference in mean intraocular pressure
Three trials (1415 participants) compared netarsudil 0.02% (once per day, p.m.) with timolol 0.5% (twice per day, a.m. and p.m.) (Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); Serle 2018 (ROCKET‐1)).
After three months of treatment, the mean IOP reduction from baseline in the netarsudil group may be slightly smaller compared with timolol (MD 0.66 mmHg, 95% CI 0.41 to 0.91; I2 = 0%; Analysis 2.1). As all trials were at overall high risk of bias, we did not conduct a sensitivity analysis excluding trials at high risk of bias (Figure 4). Overall, the evidence for the estimated MD in IOP reduction from baseline was low certainty after downgrading two levels for high risk of bias due to deviation from the intended intervention and missing outcome data.
2.1. Analysis.

Comparison 2: Rho kinase inhibitor versus beta‐blocker, Outcome 1: Mean IOP changes from baseline
4.

Glaucoma progression using valid anatomic (structural) criteria
No trials comparing ROKi versus BB reported glaucoma progression using valid anatomic (structural) criteria.
Participant‐reported outcomes
No trials comparing ROKi versus BB reported participant‐reported outcomes.
Mean change in the number of glaucoma medications
No trials comparing ROKi versus BB reported mean change in the number of glaucoma medications.
Need for intraocular pressure‐lowering medications
No trials comparing ROKi versus BB reported need for IOP‐lowering medications.
Adverse effects
Four trials (1678 participants) examined the AEs of netarsudil 0.02% (once per day, p.m.) compared with timolol 0.5% (twice per day, a.m./p.m.) (Kahook 2019 (ROCKET‐2); Khouri 2019 (ROCKET‐4); NCT02246764 (ROCKET‐3); Serle 2018 (ROCKET‐1); Table 2).
Total ocular adverse events: treatment with netarsudil probably resulted in a higher rate of ocular AEs compared with timolol, with 21 more ocular AEs per 100 person‐months (95% CI 14 to 27; I2 = 93%; Analysis 2.2). Excluding trials with reporting thresholds of 5% or greater (Khouri 2019 (ROCKET‐4); NCT02246764 (ROCKET‐3)) did not affect this difference in incidence rates (22 more events per 100 person‐months, 95% CI 3 to 41; I2 = 97%; Figure 5). Additionally, there was no evidence of a difference between subgroups defined by reporting threshold of AEs (Analysis 2.3). Overall, the evidence for the estimated MD in ocular AEs was moderate certainty after downgrading one level for risk of bias due to incomplete outcome reporting or selective outcome reporting.
2.2. Analysis.

Comparison 2: Rho kinase inhibitor versus beta‐blocker, Outcome 2: Total ocular adverse events (per person‐month) – incidence risk difference
5.

2.3. Analysis.

Comparison 2: Rho kinase inhibitor versus beta‐blocker, Outcome 3: Total ocular adverse events (per person‐month) – subgroup analysis by levels of reporting threshold
In general, treatment with ROKi resulted in more events of conjunctival hyperemia (Analysis 2.4) and cornea verticillata compared with timolol. There was no evidence of a difference in terms of ocular pain and irritation (Analysis 2.5). Kahook 2019 (ROCKET‐2) reported more than 5% of AEs were serious for both treatments. For a detailed description, see Appendix 9.
2.4. Analysis.

Comparison 2: Rho kinase inhibitor versus beta‐blocker, Outcome 4: Conjunctival hyperemia as adverse event (per person‐month)*
2.5. Analysis.

Comparison 2: Rho kinase inhibitor versus beta‐blocker, Outcome 5: Ocular pain or irritation as adverse event (per person‐month)*
Rho kinase inhibitor versus prostaglandin analog
Primary outcome
Glaucoma progression
None of the trials comparing ROKi versus PA reported quantifiable data on glaucoma progression such as visual field defects, thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer or thinning of the macular ganglion cell layer.
Secondary outcomes
Difference in mean intraocular pressure
Four trials (1283 participants) compared netarsudil 0.02% (once per day, p.m.) and latanoprost 0.005% (once per day, p.m.) (Asrani 2019 (MERCURY‐1); Bacharach 2015; Lewis 2016; Walters 2019 (MERCURY‐2); Figure 6).
6.

After one to three months, latanoprost likely reduced IOP more than netarsudil (MD 0.97 mmHg, 95% CI 0.67 to 1.27; I2 = 4%; Analysis 3.1). Overall, the evidence for the estimated MD in IOP reduction from baseline was moderate certainty after downgrading one level for risk of bias (Table 3).
3.1. Analysis.

Comparison 3: Rho kinase inhibitor versus prostaglandin analog, Outcome 1: Mean IOP changes from baseline (mmHg)
Glaucoma progression using valid anatomic (structural) criteria
No trials comparing ROKi versus PA reported glaucoma progression using valid anatomic (structural) criteria.
Participant‐reported outcomes
No trials comparing ROKi versus PA reported participant‐reported outcomes
Mean change in the number of glaucoma medications
No trials comparing ROKi versus PA reported mean change in the number of glaucoma medications.
Need for intraocular pressure‐lowering medications
No trials comparing ROKi versus PA reported need for IOP‐lowering medications.
Adverse effects
Four trials (1286 participants) examined the AEs of netarsudil 0.02% (once per day, p.m.) and latanoprost 0.005% (once per day, p.m.) (Asrani 2019 (MERCURY‐1); Bacharach 2015; Lewis 2016; Walters 2019 (MERCURY‐2)).
Total ocular adverse events: netarsudil may lead to more ocular AEs than latanoprost with 29 more ocular AEs per 100 person‐months (95% CI 17 to 42; I2 = 95%; Analysis 3.2). As all trials were at high risk of bias overall, we did not conduct a sensitivity analysis based on the risk of bias judgment (Figure 7). However, the evidence was of low certainty after downgrading it one level for high risk of bias in selective outcome reporting and one level for unclear bias in outcome measurement.
3.2. Analysis.

Comparison 3: Rho kinase inhibitor versus prostaglandin analog, Outcome 2: Total ocular adverse events (per person‐month) – incidence rate difference
7.

In general, compared with latanoprost, treatment with ROKi resulted in slightly more events of conjunctival hyperemia (Analysis 3.3), cornea verticillata and ocular pain and irritation (Analysis 3.4). Asrani 2019 (MERCURY‐1) and Lewis 2016 reported that more than 5% of AEs were serious with latanoprost. For a detailed description, see Appendix 9.
3.3. Analysis.

Comparison 3: Rho kinase inhibitor versus prostaglandin analog, Outcome 3: Conjunctival hyperemia as adverse event (per person‐month)*
3.4. Analysis.

Comparison 3: Rho kinase inhibitor versus prostaglandin analog, Outcome 4: Ocular pain or irritation as adverse event (per person‐month)*
Rho kinase inhibitor plus prostaglandin analog versus prostaglandin analog
Primary outcome
Glaucoma progression
None of the trials comparing ROKi plus PA versus PA reported quantifiable data on glaucoma progression such as visual field defects, thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer or thinning of the macular ganglion cell layer.
Secondary outcomes
Difference in mean intraocular pressure
Four trials (1319 participants) examined the efficacy of combination therapy with ROKi and PA as either netarsudil 0.02% and latanoprost 0.005% (PG‐324) administered once per day (p.m.), or as ripasudil 0.4% (twice per day, a.m. and p.m.) and latanoprost 0.005% (once per day, p.m.) compared with monotherapy with latanoprost 0.005% (once per day, p.m.) (Asrani 2019 (MERCURY‐1); Lewis 2016; Tanihara 2015c; Walters 2019 (MERCURY‐2)).
The trials were too heterogeneous to draw conclusions on combination therapy with pooled ROKi and latanoprost versus latanoprost (I2 = 91.5%; Figure 8). Thus, we examined findings from two subgroups based on the type of ROKi (Analysis 4.1). Latanoprost and netarsudil may decrease IOP more than combination therapy with ripasudil and latanoprost. After one to three months of treatment with netarsudil and latanoprost, IOP decreased by an MD of 1.64 mmHg (95% CI 1.11 to 2.16; I2 = 67%) more than with latanoprost monotherapy. Based on findings from one trial (205 participants), there was no evidence that adding ripasudil to baseline therapy of latanoprost decreased IOP further (MD 0.29 mmHg, 95% CI −0.28 to 0.86).
8.

4.1. Analysis.

Comparison 4: Rho kinase inhibitor + prostaglandin analog versus prostaglandin analog, Outcome 1: Mean IOP changes from baseline
Overall, the evidence for the estimated MD in IOP reduction from baseline was of moderate certainty after downgrading one level for risk of bias in selective outcome reporting as not all studies reported uncorrected mean changes in diurnal IOP from baseline (Table 4).
Glaucoma progression using valid anatomic (structural) criteria
No trials comparing ROKi plus PA versus PA reported glaucoma progression using valid anatomic (structural) criteria.
Participant‐reported outcomes
No trials comparing ROKi plus PA versus PA reported participant‐reported outcomes
Mean change in the number of glaucoma medications
No trials comparing ROKi plus PA versus PA reported mean change in the number of glaucoma medications.
Need for intraocular pressure‐lowering medications
No trials comparing ROKi plus PA versus PA reported need for IOP‐lowering medications.
Adverse effects
Four trials (1321 participants) examined the AEs of combination therapy with ROKi and PA as PG‐324 0.02% (once per day, p.m.) (fixed‐dose compound [FDC] of netarsudil 0.02% and latanoprost 0.005%) or as ripasudil 0.4% (twice per day, a.m./p.m.) combined with latanoprost 0.005% (once per day, p.m.) compared with latanoprost 0.005% (once per day, p.m.) (Asrani 2019 (MERCURY‐1); Lewis 2016; Tanihara 2015c; Walters 2019 (MERCURY‐2)) (Table 4).
Total ocular adverse events: the studies were too heterogeneous to combine outcome data. Thus, we evaluated the subgroups based on type of ROKi. Combination of netarsudil and latanoprost may lead to more ocular AEs than latanoprost monotherapy with 29 more events per 100 person‐months (95% CI 11 to 47; I2 = 96%; Analysis 4.2). All trials investigating netarsudil were at high risk of bias, and thus we did not perform a sensitivity analysis. Combination therapy of ripasudil and latanoprost may lead to 21 more ocular AEs per 100 person‐months (95% CI 11 to 31) than the latanoprost monotherapy (Figure 9).
4.2. Analysis.

Comparison 4: Rho kinase inhibitor + prostaglandin analog versus prostaglandin analog, Outcome 2: Total ocular adverse events (per person‐month) – incidence rate difference
9.

The evidence was uncertain comparing combination therapy of ROKi and latanoprost to latanoprost. Overall, the evidence was of low certainty after downgrading two levels for high risk of bias in selective reporting of adverse outcomes.
In general, treatment with ROKi resulted in more reported events of conjunctival hyperemia (Analysis 4.3) and cornea verticillata. There was no evidence of a difference in terms of ocular pain and irritation (Analysis 4.4). Asrani 2019 (MERCURY‐1) and Lewis 2016 reported that more than 5% of AEs were serious with latanoprost. For a detailed description, see Appendix 9.
4.3. Analysis.

Comparison 4: Rho kinase inhibitor + prostaglandin analog versus prostaglandin analog, Outcome 3: Conjunctival hyperemia as adverse event (per person‐month)*
4.4. Analysis.

Comparison 4: Rho kinase inhibitor + prostaglandin analog versus prostaglandin analog, Outcome 4: Ocular pain or irritation as adverse event (per person‐month)*
Rho kinase inhibitor plus prostaglandin analog versus rho kinase inhibitor
Primary outcome
Glaucoma progression
None of the trials comparing ROKi plus PA versus ROKi reported quantifiable data on glaucoma progression such as visual field defects, thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer or thinning of the macular ganglion cell layer.
Secondary outcomes
Difference in mean intraocular pressure
Three trials (1132 participants) investigated the efficacy of combination therapy with netarsudil and latanoprost (PG‐324 0.02% once per day, p.m.) compared with monotherapy with netarsudil 0.02% (once per day, p.m.) (Asrani 2019 (MERCURY‐1); Lewis 2016; Walters 2019 (MERCURY‐2); Table 5). Treatment with netarsudil and latanoprost likely decreased IOP more compared with netarsudil monotherapy (MD 2.66 mmHg, 95% CI 2.35 to 2.98; I2 = 0%; Analysis 5.1; Figure 10). Overall, the evidence was of moderate certainty after downgrading one level for risk of bias in selective outcome reporting.
5.1. Analysis.

Comparison 5: Rho kinase inhibitor + prostaglandin analog versus Rho kinase inhibitor, Outcome 1: Mean IOP changes from baseline
10.

Glaucoma progression using valid anatomic (structural) criteria
No trials comparing ROKi plus PA versus ROKi reported glaucoma progression using valid anatomic (structural) criteria.
Participant‐reported outcomes
No trials comparing ROKi plus PA versus ROKi reported participant‐reported outcomes.
Mean change in the number of glaucoma medications
No trials comparing ROKi plus PA versus ROKi reported mean change in the number of glaucoma medications.
Need for intraocular pressure‐lowering medications
No trials comparing ROKi plus PA versus ROKi reported need for IOP‐lowering medications.
Adverse effects
Three trials (1131 participants) investigated the AEs of combination therapy with ROKi and PA as PG‐324 0.02% (once per day, p.m.) (FDC of netarsudil 0.02% and latanoprost 0.005%) compared with monotherapy with netarsudil 0.02% (once per day, p.m.) (Asrani 2019 (MERCURY‐1); Lewis 2016; Walters 2019 (MERCURY‐2)).
Total ocular adverse events: combination therapy may not lead to more ocular AEs than netarsudil monotherapy, with one more ocular AEs per 100 person‐months (95% CI 0 to 3; I2 = 50%) associated with the combination therapy than with the monotherapy (Analysis 5.2). The evidence was low certainty after downgrading two levels for high risk of bias for selective outcome reporting in all included studies.
5.2. Analysis.

Comparison 5: Rho kinase inhibitor + prostaglandin analog versus Rho kinase inhibitor, Outcome 2: Total ocular adverse events (per person‐month) – incidence rate difference
In general, we found weak evidence of differences between treatments in terms of conjunctival hyperemia (Analysis 5.3) and ocular pain and irritation (Analysis 5.4). Cornea verticillata was reported as an AE of both treatments. Less than 5% of participants reported SAEs. For a detailed description, see Appendix 9.
5.3. Analysis.

Comparison 5: Rho kinase inhibitor + prostaglandin analog versus Rho kinase inhibitor, Outcome 3: Conjunctival hyperemia as adverse event (per person‐month)*
5.4. Analysis.

Comparison 5: Rho kinase inhibitor + prostaglandin analog versus Rho kinase inhibitor, Outcome 4: Ocular pain or irritation as adverse event (per person‐month)*
Rho kinase inhibitor plus beta‐blocker versus beta‐blocker
Primary outcome
Glaucoma progression
None of the trials comparing ROKi plus BB versus BB reported quantifiable data on glaucoma progression such as visual field defects, thinning of the neuroretinal rim at the optic disk, thinning of the peripapillary retinal nerve fiber layer or thinning of the macular ganglion cell layer.
Secondary outcomes
Difference in mean intraocular pressure
One trial (208 participants) examined the efficacy of combination therapy with ripasudil 0.4% and timolol (twice per day, a.m./p.m.) compared with monotherapy with timolol (twice per day, a.m./p.m.) (Tanihara 2015b).
After two months, treatment with combination therapy of ripasudil and timolol may decrease IOP more than timolol monotherapy (MD 0.75 mmHg, 95% CI 0.21 to 1.29; Analysis 6.1). Overall, the evidence for the estimated MD in IOP reduction from baseline was of moderate certainty after downgrading one level for imprecision due to small sample size (Table 6).
6.1. Analysis.

Comparison 6: Rho kinase inhibitor + beta‐blocker versus beta‐blocker, Outcome 1: Mean IOP changes from baseline
Glaucoma progression using valid anatomic (structural) criteria
No trials comparing ROKi plus BB versus BB reported glaucoma progression using valid anatomic (structural) criteria.
Participant‐reported outcomes
No trials comparing ROKi plus BB versus BB reported participant‐reported outcomes.
Mean change in the number of glaucoma medications
No trials comparing ROKi plus BB versus BB reported mean change in the number of glaucoma medications.
Need for intraocular pressure‐lowering medications
No trials comparing ROKi plus BB versus BB reported need for IOP‐lowering medications.
Adverse effects
One trial (208 participants) examined the AEs of combination therapy with ripasudil 0.4% (twice per day, a.m./p.m.) and timolol 0.5% twice per day (a.m./p.m.) compared with monotherapy with timolol 0.5% (twice per day, a.m./p.m.) (Tanihara 2015b).
Total ocular adverse events: combination therapy with ripasudil and timolol resulted in 35 additional events per 100 person‐months (95% CI 25 to 45) compared with timolol monotherapy (Analysis 6.2). The level of certainty for the single‐study estimate was moderate, downgraded one level for imprecision due to small sample size.
6.2. Analysis.

Comparison 6: Rho kinase inhibitor + beta‐blocker versus beta‐blocker, Outcome 2: Total ocular adverse events (per person‐month) – incidence rate difference
Treatment with ripasudil resulted in more events of conjunctival hyperemia compared with timolol (Analysis 6.3). There was weak evidence of RDs between treatments in terms of ocular pain and irritation (Analysis 6.4). Cornea verticillata was not reported. Less than 5% of participants reported SAEs. For a detailed description, see Appendix 9.
6.3. Analysis.

Comparison 6: Rho kinase inhibitor + beta‐blocker versus beta‐blocker, Outcome 3: Conjunctival hyperemia as adverse event (per person‐month)
6.4. Analysis.

Comparison 6: Rho kinase inhibitor + beta‐blocker versus beta‐blocker, Outcome 4: Ocular pain or irritation as adverse event (per person‐month)
Discussion
Summary of main results
In this review, we analyzed findings from 17 trials that evaluated the efficacy and safety of the three marketed ROKi‐based drugs; netarsudil 0.02% (once per day, p.m.), ripasudil 0.04% (twice per day, a.m./p.m.) and PG‐324 (once per day, p.m.) (FDC of netarsudil 0.02% and latanoprost 0.005%) compared with either placebo, timolol 0.5% (twice per day, a.m./p.m.), latanoprost 0.005% (once per day, p.m.) or netarsudil 0.2% (once per day, p.m.) in people with (P)OAG or OHT.
ROKi was evaluated as mono‐ and combination therapy, showing that combination therapy with netarsudil and latanoprost may be superior to monotherapy. Furthermore, we found that timolol and latanoprost may be slightly better in reducing IOP in people diagnosed with OHT or POAG. The review focused on commercially available formulations of ROKi.
People treated with ROKi experienced more ocular AEs than those treated with placebo, latanoprost or timolol. In general, AEs were mild, transient and reversible upon treatment discontinuation. ROKi was not associated with any systematically reported SAEs, which were AEs reported to be the primary cause of treatment discontinuation.
Two pharmaceutical companies funded 16 trials: Aerie Pharmaceuticals and Kowa Company. Aerie Pharmaceuticals sponsored trials investigating netarsudil‐based interventions, whereas Kowa Company sponsored trials investigating ripasudil‐based interventions. Trial findings of industry‐controlled and sponsored trials may be influenced by the agenda of the pharmaceutical companies. However, all included trials except Inoue 2018 published the study protocol on either ClinicalTrials.gov or ClinicalTrials.jp before study initiation, which may reduce the influence of funding sources on the analysis and reporting of results. One trial provided no information about funding sources (Inoue 2018).
Overall completeness and applicability of evidence
This review found several trials that investigated the IOP‐reducing effect and adverse events of ROKi in people with OHT or POAG, but data on glaucoma progression such as changes in visual field defects are lacking. The included trials reported only two of the prespecified secondary outcomes (IOP reduction and AEs). IOP outcomes were incompletely and variably reported among the trials. Thus, we included both single time point IOP and mean diurnal IOP measurements in the quantitative analyses. The circadian IOP regulation as well as differences in the pharmacodynamic/pharmacokinetic properties of the different interventions may have introduced bias. Thus, single time point IOP may not adequately reflect the actual effect of the different types of ROKi on the mean diurnal IOP. All trials that had investigated ripasudil reported time‐matched IOP, whereas trials investigating netarsudil differed between time‐matched and mean diurnal IOP. Results from both clinical trials (Peace 2021; Tanihara 2015a) and in vivo experiments (Kaneko 2017; Sturdivant 2016) suggest that netarsudil has a prolonged duration of effect compared with ripasudil, which is reflected in the different recommended dosing frequencies. Therefore, it may be inappropriate to draw conclusions about the efficacy of ripasudil compared with netarsudil in terms of reducing the mean diurnal IOP based on an indirect comparison. Nevertheless, our meta‐analyses reflect the variations in drug efficacies at 10 to 12 hours after administration. As ripasudil has a relatively short duration of IOP‐lowering effect, the early efficacy of ripasudil may be underestimated.
Cornea verticillate is a well‐established AE to treatment with cationic, amphiphilic drugs such as amiodarone or aminoquinolines caused by drug‐induced phospholipidosis (Raizman 2017). In most cases, the condition is asymptomatic and reversible. Netarsudil is a cationic and amphiphilic drug (Lin 2018b), and some included trials reported cornea verticillate as an asymptomatic and reversible AE to treatment (Appendix 9).
We addressed treatment safety in the meta‐analysis of total ocular AEs; the most frequently reported ocular AEs were conjunctival hyperemia and ocular pain or irritation. There was considerable statistical heterogeneity among the trials, which reflected the clinical and methodological heterogeneity. To compare AEs across trials of different durations, the review team decided to employ incidence rates of reported AEs. This method did not account for temporal associations of occurrence of AE. The incidence rate of short‐term AEs thus may underestimate the effects in trials of longer duration.
Most trials reported the number of participants with at least one ocular AE instead of the mean number of ocular AEs per person. The latter was approximated as the sum of each reported ocular AE divided by the number of analyzed participants. This approach may underestimate the actual mean depending on reporting threshold or degree of details when reporting AE. Thus, our estimate reflected the occurrence of AEs among all treated participants and not among participants experiencing AEs.
AEs were inconsistently reported across trials and differed substantially regarding terminology, degree of detail and reporting threshold. To address this problem, we categorized clinically similar or identical AEs to allow the comparison of AEs among trials (Appendix 8). The limitation of this approach was a risk of overestimating the number of specific AEs, especially relevant for categories composed of numerous AEs such as ocular pain and irritation. Furthermore, trials reporting safety outcomes with a low degree of detail and a high reporting threshold would be at risk of being under‐represented and vice versa. The AEs categorized as ocular pain or irritation included both acute AEs upon instillation and more long‐term AEs that could lead to discontinuation of medication.
When addressing SAEs, some trials applied a standardized terminology (i.e. Medical Dictionary for Regulatory Activities, MedDRA) with strict definitions, whereas others provided limited or no information about the definition of SAEs. The review team decided to apply the FDA's definition of SAE (FDA 2016). Thus, AEs that met the FDA criteria were classified as SAEs independently of the classification used in trial reports. Furthermore, when no degree of severity or qualitative description of a given AE was provided, the AE evaluation was classified based on the potential severity (e.g. unspecified asthma would be considered as an SAE). This approach may have overestimated the number of SAEs reported.
Quality of the evidence
We evaluated the confidence in the estimates of the effects using the GRADE approach (GRADE Handbook).
None of the studies investigated our primary outcome of disease progression. We were able to assess only two of the six prespecified secondary outcomes. Most trials evaluated the short‐term effects of ROKi (less than six months of treatment), and thus we were unable to evaluate the long‐term effects of treatment with ROKi. The assessment and reporting of AEs varied substantially among the trials. The trials included in the analyses were heterogeneous and thus difficult to compare. As the quantitative analyses included fewer than 10 studies, the risk of publication bias was not assessed employing funnel plots. Our judgments of certainty of evidence ranged from very low to moderate, depending upon the outcome and comparison of intervention arms.
Limitations to the trials were mainly insufficient reporting on study design, differences in methods of measuring and reporting the outcomes, and differences in descriptive terminology. The short duration of the included trials did not permit meaningful analysis of the comparative effectiveness of ROKi on glaucoma progression. Thus, advocates of ROKi must design and conduct trials with longer follow‐up of treatment and observation, and measurement of outcomes that focus on changes in visual field and morphology associated with glaucoma in order to provide convincing data regarding their effectiveness and safety.
Potential biases in the review process
We followed the Cochrane Handbook for Systematic Reviews of Interventions and guidelines to conduct a systematic review. We applied a broad search strategy to ensure that all relevant papers were included in this review. Two review authors independently extracted data and assessed risk of bias; we also followed the prespecified methods of meta‐analysis and investigations of subgroup differences. To our knowledge, there should be no bias in the review process with respect to trial selection and analysis of available data. The primary bias is the short follow‐up of participants in individual trials so that intermediate‐ and long‐term effects of ROKi on IOP and glaucoma progression could not be examined, a factor beyond the control of the review authors. Thus, those effects remain unknown.
Agreements and disagreements with other studies or reviews
Since treatment with ROKi is newly introduced in the USA and Europe (USA in 2017 [FDA 2017] and Europe in 2019 [Aerie 2019]), the number of reviews that have examined this treatment is limited. Other reviews have included trials and data for the same trials that we did. To our knowledge, this is the first systematic review to assess both the efficacy and safety of ROKi.
Tanna 2018 and Mehran 2020 concluded that monotherapy with ROKi was not superior to the traditional antiglaucomatous eye drops. However, they suggested that people could benefit from combination therapy with ROKi, as ROKi primarily acts on the outflow resistance of the AH. The two reviews found that the incidence of AEs was higher for ROKi compared with other treatments. Moura‐Coelho 2019 agreed that the efficacy and safety of ROKis were convincing and could be used to gain an additive IOP‐reducing effect from combination therapy. This conclusion supports our finding that the greatest reduction of IOP was in the trials of ROKi and latanoprost combination therapy.
The IOP‐lowering effect of ROKi depends on the responsiveness/functionality of the TM. It may be hypothesized that ROKi would be more effective in early disease stages of POAG or other subtypes of glaucoma in the absence of, perhaps, irreversible TM/SC cellular dysfunction or structural damage. Likewise, the efficacy of ROKi may to some extent depend on the anatomy of the iridocorneal angle. If overactivity of rho GTPase/ROCK signaling pathway is a causal factor of OHT/increased TM resistance, ROKis may prove to be the preferred drugs compared with other glaucoma medications in some subtypes of glaucoma/OHT such as glucocorticoid‐induced glaucoma/OHT (Li 2021b). However, the amount of data from interventional trials is limited, and the current evidence is inconclusive regarding the preventive effect of ROKi on topical corticosteroid‐induced OHT (Price 2021).
Preclinical data from in vivo animal models indicate that ROKi expresses neuroprotective properties (promoting axonal outgrow, survival of retinal ganglia cells and increasing blood flow to the optic disk) (Rao 2017). Whether these effects can be translated into humans is unknown. Furthermore, it is questionable whether persistent topical administration will lead to sufficient intravitreal concentrations of ROKi to promote meaningful clinical effects in the glaucomatous retina.
Authors' conclusions
Implications for practice.
Rho kinase inhibitors (ROKis) are currently prescribed to reduce intraocular pressure (IOP) in people with open‐angle glaucoma (OAG) or ocular hypertension (OHT). With reservations because of the limited amount and quality of clinical evidence, the results from our review suggest that ROKi monotherapy (netarsudil 0.02% once per day or ripasudil 0.4% twice per day) reduces IOP in people diagnosed with OAG or OHT, but treatment with ROKi may be inferior compared with monotherapy with timolol 0.5% twice per day or latanoprost 0.005% once per day. Furthermore, combining ROKi (netarsudil 0.02% once per day or ripasudil 0.4%) with either latanoprost 0.005% or timolol 0.5% twice per day probably results in additional IOP reduction compared with monotherapy (netarsudil 0.02% once per day, latanoprost 0.005% once per day or timolol 0.5% twice per day). In terms of adverse events (AEs), ROKi monotherapy and combination therapy may increase the rate of ocular AEs compared with latanoprost and timolol monotherapy. However, the certainty of evidence was low or very low for all comparisons except with timolol (moderate‐certainty evidence).
Implications for research.
This review highlights the need for additional comparable trials investigating the efficacy and safety of ROKi in people with OAG and OHT in order to evaluate their effectiveness and safety in routine clinical care. Even though the review included 17 trials, the trials were too heterogeneous to include in a reasonable, collective meta‐analysis. Future trials should be sufficiently large and follow participants for a sufficiently long time to provide reliable information about glaucoma progression, relevant outcome measurements of IOP and a detailed description of AEs to treatment using similar terminology in order to ensure the robustness and confidence of the found results.
History
Protocol first published: Issue 12, 2020
Risk of bias
Risk of bias for analysis 1.1 Mean intraocular pressure (IOP) changes from baseline.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.1.1 Netarsudil 0.02% once per day | ||||||||||||
| Araie 2021 | Low risk of bias | All the eligible patients were randomized (1:1:1:1) by a computer‐generated randomization list using an interactive web response system (...). There was no statistically significant difference in demographics and other baseline characteristics across the treatment groups (Table 1). | Low risk of bias | Patients and designated study site personnel (investigators, sponsor, monitor, data manager, statistician, and personnel involved in study management) were fully masked to treatment assignments. | Low risk of bias | All randomized persons were included in the ITT. No lost to follow up. | Low risk of bias | The IOP was measured at screening visit, qualification visit and all study visits using a calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma). | Low risk of bias | Protocol published on clinicaltrials.gov. Primary efficacy endpoint (IOP) reported as a number, which is the only way to report that outcome. All IOP measurements were listed in NCT whereas IOP measurements in the published paper were modified ITT results although well described. The mITT population may affect the results in the following manners: The ANCOVA model explains additional variability (i.e., reduces standard error) through inclusion of baseline covariates and therefore improves statistical power. The LS means calculated from this model are the means adjusted for the covariate. LS means are better estimates of true population means compared to arithmetic means. |
Low risk of bias | Based on the low risk of bias judgement in all domains, the overall judgement is low risk of bias. |
| Peace 2021 | Low risk of bias | Randomization schedule prepared by the sponsor representative. The study centre was provided with a predefined order sequence (i.e., kit number) for the dispensation of the study drug to patients. Investigator, subjects, sponsor, sponsor representative, and clinical monitors were not allowed to know the treatment assigned to each subject. Active and vehicle study medications packaged in identical bottles with identical labels. . No obvious differences in appearance(e.g., colour or viscosity) between the netarsudil and vehicle solutions. No apparent differences in baseline characteristics between treatment groups. | Low risk of bias | Double‐masked. The intent to treat (ITT) population served as the primary efficacy population. All randomized subjects who received at least one dose of the study drug were included in the ITT population. Subjects in the ITT population were analyzed in accordance with their assigned randomized treatment, even if the actual treatment the subject received was different from the planned treatment (...). Of the 12 patients randomized, 100% received their assigned study medication and therefore comprised the ITT population. | Low risk of bias | All 12 patients enrolled completed the study (8:4). | Low risk of bias | IOP was measured at each time point by study staff masked to treatment using a Perkins tonometer. The method of IOP measurements are well described, e.g. head position, and number of measurements. | Low risk of bias | Study registered at ClinicalTrials.gov before study initiation with a priori analysis described and outcome reported in the publication as planned. Primary efficacy endpoint (IOP) reported as a number which is the only way to report that outcome. | Low risk of bias | Judged as low risk of bias in all domains. Thus, the overall risk of bias is judged as low. |
| Sit 2021 | Low risk of bias | Pre‐published analysis plan: A randomization schedule will be prepared by an independent individual (unmasked personnel) who is not involved in the day‐to‐day conduct of the study. Randomization will be stratified by site using permuted blocks (...). Randomization numbers will be assigned sequentially to subjects in the order in which they become eligible for randomization. The site staff will dispense to the subject the study kit labelled with the corresponding randomization number. Baseline differences are not relevant as each patient will receive placebo in one eye and treatment in the other eye. | Some concerns | Double‐masked (article and NCT). Patients included in mITT if 1) received at least one dose of medication 2) had all baseline and post‐baseline measurements for outflow facility 3) responder, defined as a decrease of at least 2 mmHg in diurnal IOP (post‐baseline). | Low risk of bias | 18/20 (90%) patients included in the analysis ‐ all responders. Two patients discontinued the study; 1 for an adverse event of conjunctival hyperemia and the other for a protocol violation. | Low risk of bias | IOP was measured with a pneumotonometer (Model 30 Classic, Reichert Inc., Depew, NY, USA). Minimum 2 IOP measurements. |
Low risk of bias | Priori statistical analysis plan published. Primary efficacy endpoint (IOP) reported as a number, which is the only way to report that outcome. |
Some concerns | Because there were some concerns in Domain 2, the overall risk of bias was judged as having some concerns. |
| Subgroup 1.1.2 Ripasudil 0.4% twice per day | ||||||||||||
| Tanihara 2013 | Low risk of bias | Permuted blocks method with allocation ratio of 1:1:1:1. Site investigators were not informed about the block size throughout the study period. Allocation concealment not described. Even though statistical comparisons of baseline characteristics are not performed, the intervention groups seem alike (table 1). | Some concerns | Double‐masked. Site investigators not informed about block size throughout the study period. A total of 7 patients were excluded from analyses because of adverse events in 6 and protocol deviations in 1. In the remaining 203 patients, 52 in the placebo group, 50 in the 0.1% group, 52 in the 0.2% group, and 49 in the 0.4% group, subsequent statistical analyses were conducted for the estimation of efficacy in IOP reduction. | Low risk of bias | A total of 7 patients were excluded from analyses because of adverse events in 6 and protocol deviations in 1. In the remaining 203 patients, 52 in the placebo group, 50 in the 0.1% group, 52 in the 0.2% group, and 49 in the 0.4% group, subsequent statistical analyses were conducted for the estimation of efficacy in IOP reduction. | Low risk of bias | A calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma [3]) was used for IOP measurements. Double‐masked. Site investigators were not informed about the block site throughout the study period. |
Low risk of bias | Protocol JAPIC 101015 and a priori analysis plan were conducted. Primary efficacy endpoint (IOP) reported as a number, which is the only way to report that outcome. Reports covariance adjusted timepoint‐specific means pre‐and post‐administration. Pre‐administration values extracted. |
Some concerns | Based on the judgment of some concerns in Domain 2, the overall risk of bias was judged as having some concerns. |
Risk of bias for analysis 1.2 Mean IOP changes from baseline: sensitivity analysis.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.2.1 Netarsudil 0.02% once per day | ||||||||||||
| Araie 2021 | Low risk of bias | All the eligible patients were randomized (1:1:1:1) by a computer‐generated randomization list using an interactive web response system (...). There was no statistically significant difference in demographics and other baseline characteristics across the treatment groups (Table 1). | Low risk of bias | Patients and designated study site personnel (investigators, sponsor, monitor, data manager, statistician, and personnel involved in study management) were fully masked to treatment assignments. | Low risk of bias | All randomized persons were included in the ITT. No lost to follow up. | Low risk of bias | The IOP was measured at screening visit, qualification visit and all study visits using a calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma). | Low risk of bias | Protocol published on clinicaltrials.gov. Primary efficacy endpoint (IOP) reported as a number, which is the only way to report that outcome. All IOP measurements were listed in NCT whereas IOP measurements in the published paper were modified ITT results although well described. The mITT population may affect the results in the following manners: The ANCOVA model explains additional variability (i.e., reduces standard error) through inclusion of baseline covariates and therefore improves statistical power. The LS means calculated from this model are the means adjusted for the covariate. LS means are better estimates of true population means compared to arithmetic means. |
Low risk of bias | Based on the low risk of bias judgement in all domains, the overall judgement is low risk of bias. |
| Peace 2021 | Low risk of bias | Randomization schedule prepared by the sponsor representative. The study centre was provided with a predefined order sequence (i.e., kit number) for the dispensation of the study drug to patients. Investigator, subjects, sponsor, sponsor representative, and clinical monitors were not allowed to know the treatment assigned to each subject. Active and vehicle study medications packaged in identical bottles with identical labels. . No obvious differences in appearance(e.g., colour or viscosity) between the netarsudil and vehicle solutions. No apparent differences in baseline characteristics between treatment groups. | Low risk of bias | Double‐masked. The intent to treat (ITT) population served as the primary efficacy population. All randomized subjects who received at least one dose of the study drug were included in the ITT population. Subjects in the ITT population were analyzed in accordance with their assigned randomized treatment, even if the actual treatment the subject received was different from the planned treatment (...). Of the 12 patients randomized, 100% received their assigned study medication and therefore comprised the ITT population. | Low risk of bias | All 12 patients enrolled completed the study (8:4). | Low risk of bias | IOP was measured at each time point by study staff masked to treatment using a Perkins tonometer. The method of IOP measurements are well described, e.g. head position, and number of measurements. | Low risk of bias | Study registered at ClinicalTrials.gov before study initiation with a priori analysis described and outcome reported in the publication as planned. Primary efficacy endpoint (IOP) reported as a number which is the only way to report that outcome. | Low risk of bias | Judged as low risk of bias in all domains. Thus, the overall risk of bias is judged as low. |
| Subgroup 1.2.2 Ripasudil 0.4% twice per day | ||||||||||||
| Tanihara 2013 | Low risk of bias | Permuted blocks method with allocation ratio of 1:1:1:1. Site investigators were not informed about the block size throughout the study period. Allocation concealment not described. Even though statistical comparisons of baseline characteristics are not performed, the intervention groups seem alike (table 1). | Some concerns | Double‐masked. Site investigators not informed about block size throughout the study period. A total of 7 patients were excluded from analyses because of adverse events in 6 and protocol deviations in 1. In the remaining 203 patients, 52 in the placebo group, 50 in the 0.1% group, 52 in the 0.2% group, and 49 in the 0.4% group, subsequent statistical analyses were conducted for the estimation of efficacy in IOP reduction. | Low risk of bias | A total of 7 patients were excluded from analyses because of adverse events in 6 and protocol deviations in 1. In the remaining 203 patients, 52 in the placebo group, 50 in the 0.1% group, 52 in the 0.2% group, and 49 in the 0.4% group, subsequent statistical analyses were conducted for the estimation of efficacy in IOP reduction. | Low risk of bias | A calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma [3]) was used for IOP measurements. Double‐masked. Site investigators were not informed about the block site throughout the study period. |
Low risk of bias | Protocol JAPIC 101015 and a priori analysis plan were conducted. Primary efficacy endpoint (IOP) reported as a number, which is the only way to report that outcome. Reports covariance adjusted timepoint‐specific means pre‐and post‐administration. Pre‐administration values extracted. |
Some concerns | Based on the judgment of some concerns in Domain 2, the overall risk of bias was judged as having some concerns. |
Risk of bias for analysis 2.1 Mean IOP changes from baseline.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Kahook 2019 (ROCKET‐2) | Low risk of bias | Randomized by a computer‐generated method. No statistical baseline differences between groups (table 1, Serle et al 2018). | High risk of bias | Double‐masked:Patients and designated study site personnel were fully masked to treatment assignments. A vehicle bottle was provided for AM dosing in the netarsudil q.d. PM treatment groups to maintain masking. In order to include the results from the most patients, we used the results from the NCT (PP with no IOP‐restrictions; Netarsudil (QD) 206/251, timolol (BID) 217/251 = All subjects who did not have a major protocol violation likely to seriously affect the primary outcome of the study). | High risk of bias | Article reports results from PP (PP population with maximum baseline IOP < 25 mm Hg, 3 months). In order to include the results from the most patients, we used the results from the NCT (PP with IOP‐restrictions; Netarsudil (QD) 206/251, timolol (BID) 217/251 = All subjects who did not have a major protocol violation likely to seriously affect the primary outcome of the study). In the original report at 3 months, 82% (205/251), 60% (153/254), and 94% (237/251) of patients in the netarsudil q.d., netarsudil b.i.d., and timolol b.i.d. groups, respectively, completed the first 3 months. | Low risk of bias | IOP was measured using a calibrated Goldmann applanation tonometer. Two consecutive IOP measurements of each eye were obtained. If the 2 measurements differed by more than 2 mm Hg, a third measurement was obtained. IOP was to be recorded as the mean of 2 measurements or as the median of 3 measurements. | Low risk of bias | Statistical and trial plan published before the study initiation and followed. Primary efficacy endpoint (IOP) reportet as a number ‐ only way to report that outcome. | High risk of bias | Judged as high risk of bias in Domain 2 and 3. Thus, the overall risk of bias was judged as high. |
| Khouri 2019 (ROCKET‐4) | Low risk of bias | Patients were randomized (1:1) by a computer‐generated method. Randomization was stratified by study site and maximum baseline IOP (< 25 mm Hg vs ≥ 25 mmHg). No description of allocation concealment. No significant differences in baseline values (table 1). | High risk of bias | Double‐masked. 708 patients were randomized, but only 186 in each treatment arm were included in the per‐protocol analysis ( = 1) no serious protocol violations 2) baseline IOP < 25 mmHg). Data is extracted from NCT (PP for overall study population = 306 (netarsudil) and 316 (timolol)). According to the NCT, 243 netarsudil and 314 timolol patients completed the study, respectively. Thus, a substantial amount of patients that per protocol definition didn't complete the study. |
High risk of bias | Data is extracted from NCT ( PP for overall study population = . 306 (netarsudil) and 316 (timolol)). According to the NCT 243 (Netarsudil) and 314 timolol patients completed the study, respectively. Thus, a substantial amount of patients that per protocol definition didn't complete the study have been used in the analysis of the netarsudil‐group. | Low risk of bias | IOP measured with a calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma [3]). |
Low risk of bias | A priori analysis was described and followed. IOP is reported as a number, which is the only way to report that outcome. Detailed description of measurements e.g. timeslots and number of measurements. |
High risk of bias | Based on the judgment of high risk of bias in domains 2 and 3, the overall risk of bias is judged as high. |
| Serle 2018 (ROCKET‐1) | Low risk of bias | Eligible patients were randomized by a computer‐generated method (...). No information on allocation concelament. Significant differences in "iris colour" between intervention groups (P=0,0085). However, no clinical influence. | High risk of bias | Double‐masked: Patients and study site personnel were fully masked to treatment assignments. A vehicle bottle was provided for AM dosing in the netarsudil to maintain masking. In ROCKET‐1, the primary efficacy population was the per‐protocol population with maximum baseline IOP < 27 mm Hg. PP is defined as subjects without major protocol violations likely to seriously affect the primary outcome of the study. Netarsudil‐group: 182/202 analyzed (netarsudil 171 completed the study according to NCT), timolol‐group: 188/209 (timolol 196 completed the study according to NCT). Thus, per study definition several non‐completing subjects have been added to the analysis in the netarsudil‐group. |
High risk of bias | In ROCKET‐1, the primary efficacy population was the per‐protocol population with maximum baseline IOP < 27 mm Hg. PP is defined as subjects without major protocol violations likely to seriously affect the primary outcome of the study. Netarsudil‐group: 182/202 analyzed (netarsudil 171 completed the study according to NCT), timolol‐group: 188/209 (timolol 196 completed the study according to NCT). Thus, per study definition several non‐completing subjects have been added to the analysis in the netarsudil‐group. | Low risk of bias | IOP was measured using a calibrated Goldmann applanation tonometer. Two consecutive IOP measurements of each eye were obtained. If the 2 measurements differed by more than 2 mm Hg, a third measurement was to be obtained. IOP was to be recorded as the mean of 2 measurements or as the median of 3 measurements Double‐masked: Patients and study site personnel were fully masked to treatment assignments. | Low risk of bias | Statistical plan are published and followed. Primary efficacy endpoint (IOP) reportet as a number ‐ only way to report that outcome. | High risk of bias | Judged as high risk of bias in Domain 2 and 3. Thus, the overall risk of bias was judged as high. |
Risk of bias for analysis 3.1 Mean IOP changes from baseline (mmHg).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Asrani 2019 (MERCURY‐1) | Low risk of bias | Patients randomized (..) in a 1:1:1 ratio (...). Randomization was determined by a computer‐generated randomization code and was stratified by site and maximum IOP(<25 vs. ≥ 25 mmHg). The randomization code was prepared by an independent biostatistician not involved in day‐to‐day study conduct (reference: Asrani et al 2019). Baseline demographic characteristics were similar across all 3 treatment groups (Table 1). | Low risk of bias | Treatment assignments were masked to the investigator, clinical study team, and patients. An independent person at the investigative site not responsible for performing any study procedure was assigned to dispense, collect, and store study treatment. The primary efficacy analysis was performed on the ITT, which included all randomized patients who received ≥ 1 dose of study medication. | Low risk of bias | ITT: all randomized participants were included in the analysis. Missing data were imputed using Markov Chain Monte Carlo multiple imputation techniques. | Low risk of bias | A calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma [3]) was used for IOP measurement (stated in the ClinicalTrials.gov). | Low risk of bias | Statistical plan detailed described in the paper and followed. IOP is reported as a number, which is the only way to report that. The plan of analysis was well described in the paper. Timepoint specific IOP with SD at 10 AM (baseline and week 12) day have been extracted from ClinicalTrials.gov. | Low risk of bias | Judged as low risk of bias in all domains. Thus, the overall risk of bias was judged as low. |
| Bacharach 2015 | Some concerns | No information about the randomization process. The allocation sequence is not described. Concealment not described. However, no significant baseline differences between intervention groups. | Low risk of bias | Double masked (NCT: Quadruple). Patients were dispensed masked study medication. Three subjects were excluded from the modified intent‐to‐treat (mITT) population (1 each in the 0.01% AR‐13324, 0.02% AR‐13324, and latanoprost groups, respectively) because they did not have time‐specific posttreatment IOP measures, leaving 221 patients. Ten subjects were excluded from the per‐protocol population for major violations of entry criteria (2, 3, and 5 each, respectively). leaving 211 patients. | Low risk of bias | AR‐13324 ophthalmic solution 0.01 % (74/75) AR‐13324 ophthalmic solution 0.02 % (72/71) Latanoprost 0.005% (76/77) | Low risk of bias | IOP was measured using a Calibrated Goldmann tonometer. Two consecutive measurements of IOP in each eye were obtained at each time point. If the 2 measurements differed by >2 mmHg, a third measurement was obtained. We analyzed IOP as the mean of 2 measurements or as the median of 3 measurements. Double masked (NCT: Quadruple) | Low risk of bias | Protocol published before study initiation and followed. Primary efficacy endpoint (IOP) reported as a number ‐ only way to report that outcome. Reports mean diurnal IOP. | Some concerns | Some concerns in Domain 1. Thus, the overall risk of bias was judged as having some concern. |
| Lewis 2016 | Low risk of bias | Qualified individuals were randomised according to a computer‐generated schedule. Use of masked medication. Double‐masked. No significant differences between intervention groups. | Low risk of bias | Double‐masked. Use of masked medication. The primary population for efficacy was a modified intent‐to‐treat, defined as all randomised patients who received at least one dose of study medication, had all three baseline IOP measurements (ie, visit 3, day 1 at 08:00, 10:00 and 16:00 h), and had at least one scheduled post‐treatment time‐specific IOP measurement. | Low risk of bias | 2 (0.7 %) patients were not included in the mITT, as they did not match the required criteria for the mITT. | Low risk of bias | A calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma) was used for IOP measurements. Double‐masked. | Low risk of bias | A priori analysis is described and outcomes reported as planned. Primary efficacy endpoint (IOP) reported as a number, which is the only way to report that outcome. Mean diurnal IOP‐reported. Paper refers to wrong NCT (Rocket 1 study) with a later corrigendum (NCT02207491). |
Low risk of bias | Based on the judgment of low risk of bias in all five domains, the overall risk of bias was judged to be low. |
| Walters 2019 (MERCURY‐2) | Low risk of bias | Randomized (1:1:1) through an interactive web‐based response system. Randomization was stratified by study site and maximum baseline IOP (<25 vs. ≥25 mmHg), and the randomization code was prepared by an independent biostatistician not involved in the study’s day‐to‐day conduct. Baseline demographics of randomized patients were similar across the 3 treatment arms (Table 1). | Low risk of bias | Treatment assignments were masked to the investigator, clinical study team, and patients. The primary efficacy analysis was performed on the ITT population. | Low risk of bias | All persons randomized included in ITT. Missing data were imputed using Markov Chain Monte Carlo multiple imputation techniques. | Low risk of bias | A calibrated Goldmann applanation tonometer (the most clinically accurate and the standard tonometer used in the diagnosis and treatment of glaucoma [3]) was used for measurements of IOP. Detailed description of time slots for IOP measurements. Treatment assignments were masked to the investigator, clinical study team, and patients. |
Low risk of bias | A priori analysis plan and outcomes analysed as planned. Primary efficacy endpoint (IOP) is reported as a number, which is the only way to report that outcome. Timepoint specific IOP have been extracted from ClinicalTrials.gov. |
Low risk of bias | Judged as low risk of bias in all domains. Thus, the overall risk of bias was judged as low. |
Acknowledgements
We thank Lori Rosman, Information Specialist for Cochrane Eyes and Vision (CEV), who created and executed the electronic search strategies.
We also thank Alison Su‐Hsun Liu, Sueko Ng, and Louis Leslie of CEV@US; Renee Wilson, Assistant Managing Editor for CEV@US; and Anupa Shah, Managing Editor for CEV, for support and guidance in the preparation of this review. We also thank Naira Khachatryan for her prior contribution to the protocol.
We would also like to thank the following peer reviewers for their comments: Renee Bovelle (Howard University) and Anthony King (Spire Nottingham Hospital) for the review manuscript.
This review was managed by CEV@US and was signed off for publication by Tianjing Li.
Appendices
Appendix 1. CENTRAL search strategy
#1 MeSH descriptor: [Glaucoma] explode all trees #2 MeSH descriptor: [Ocular Hypertension] explode all trees #3 MeSH descriptor: [Intraocular Pressure] explode all trees #4 glaucom* #5 (POAG or OHT) #6 (ocular or intra*ocular) near/3 (hypertension* or tension* or pressur*) #7 IOP #8 {or #1‐#7} #9 MeSH descriptor: [rho‐Associated Kinases] explode all trees #10 ROCK OR ROK OR ROKalpha OR "rho associated" OR p160ROCK #11 (rho near/3 kinase*) OR (rhoA near/3 kinase*) #12 (protein* near/3 kinase*) #13 {or #9‐#12} #14 #8 AND #13
Appendix 2. MEDLINE Ovid search strategy
1. Randomized Controlled Trial.pt. 2. Controlled Clinical Trial.pt. 3. (randomized or randomised).ab,ti. 4. placebo.ab,ti. 5. drug therapy.fs. 6. randomly.ab,ti. 7. trial.ab,ti. 8. groups.ab,ti. 9. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 10. exp animals/ not humans.sh. 11. 9 not 10 12. exp Glaucoma/ 13. exp Ocular Hypertension/ 14. exp Intraocular Pressure/ 15. glaucom*.tw. 16. (POAG or OHT).tw. 17. ((ocular* or intra*ocular) adj3 (hypertension* or tension* or pressur*)).tw. 18. IOP.tw. 19. or/12‐18 20. exp rho‐Associated Kinases/ 21. (ROCK or ROK or ROKalpha or "rho associated" OR p160ROCK).tw. 22. ((rho adj3 kinase*) or (rhoA adj3 kinase*)).tw. 23. (protein* adj3 kinase*).tw. 24. or/20‐23 25. 19 and 24 26. 11 and 25
The search filter for trials at the beginning of the MEDLINE strategy is from the published paper by Glanville 2006.
Appendix 3. Embase.com search strategy
#1 'randomized controlled trial'/exp #2 'randomization'/exp #3 'double blind procedure'/exp #4 'single blind procedure'/exp #5 random*:ab,ti #6 #1 OR #2 OR #3 OR #4 OR #5 #7 'animal'/exp OR 'animal experiment'/exp #8 'human'/exp #9 #7 AND #8 #10 #7 NOT #9 #11 #6 NOT #10 #12 'clinical trial'/exp #13 (clin* NEAR/3 trial*):ab,ti #14 ((singl* OR doubl* OR trebl* OR tripl*) NEAR/3 (blind* OR mask*)):ab,ti #15 'placebo'/exp #16 placebo*:ab,ti #17 random*:ab,ti #18 'experimental design'/exp #19 'crossover procedure'/exp #20 'control group'/exp #21 'latin square design'/exp #22 #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 #23 #22 NOT #10 #24 #23 NOT #11 #25 'comparative study'/exp #26 'evaluation'/exp #27 'prospective study'/exp #28 control*:ab,ti OR prospectiv*:ab,ti OR volunteer*:ab,ti #29 #25 OR #26 OR #27 OR #28 #30 #29 NOT #10 #31 #30 NOT (#11 OR #23) #32 #11 OR #24 OR #31 #33 'glaucoma'/exp #34 'intraocular pressure'/exp #35 'intraocular pressure abnormality'/de #36 'intraocular hypertension'/exp #37 glaucom*:ab,ti,kw #38 (POAG OR OHT):ab,ti,kw #39 ((intra*ocular OR ocular*) NEAR/3 (hypertension* OR tension* OR pressur*)):ab,ti,kw #40 iop:ab,ti,kw #41 #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 #42 'rho kinase'/exp #43 rock:ab,ti,kw OR rok:ab,ti,kw OR rokalpha:ab,ti,kw OR 'rho associated':ab,ti,kw OR 'rhoa associated':ab,ti,kw OR p160ROCK:ab,ti,kw #44 ((rho NEAR/3 kinase*):ab,ti,kw) OR ((rhoa NEAR/3 kinase*):ab,ti,kw) #45 (protein* NEAR/3 kinase*):ab,ti,kw #46 #42 OR #43 OR #44 OR #45 #47 #41 AND #46 #48 #32 AND #47
Appendix 4. PubMed search strategy
1. ((randomized controlled trial[pt]) OR (controlled clinical trial[pt]) OR (randomised[tiab] OR randomized[tiab]) OR (placebo[tiab]) OR (drug therapy[sh]) OR (randomly[tiab]) OR (trial[tiab]) OR (groups[tiab])) NOT (animals[mh] NOT humans[mh]) 2. glaucom*[tw] 3. POAG[tw] OR OHT[tw] 4. ((ocular*[tw] OR "intra ocular"[tw]) AND (hypertension*[tw] OR tension*[tw] OR pressur*[tw])) 5. IOP[tw] 6. #2 OR #3 OR #4 OR #5 7. (ROCK[tw] OR ROK[tw] OR ROKalpha[tw] OR "rho associated"[tw] OR p160ROCK[tw]) 8. ((rho[tw] AND kinase*[tw]) OR (rhoA[tw] AND kinase*[tw])) 9. (protein*[tw] AND kinase*[tw]) 10. #7 OR #8 OR #9 11. #6 AND #10 12. #1 AND #11 13. Medline[sb] 14. #12 NOT #13
Appendix 5. LILACS search strategy
(MH:C11.525$ OR glaucom$ OR "Ocular Hypertension" OR "Hipertensión Ocular" OR "Hipertensão Ocular" OR MH:G14.440$ OR ((intraocular OR "intra‐ocular" OR ocular$) AND (hypertension$ OR tension$ OR pressur$)) OR "Presión Intraocular" OR "Pressão Intraocular" OR IOP OR MH:E04.540.450$ OR MH:E04.540.825.249$ OR POAG OR OHT) AND (MH:D08.811.913.696.620.682.700.814$ OR MH:D12.644.360.590$ OR MH:D12.776.476.595$ OR ROCK OR ROK OR ROKalpha OR "rho associated" OR p160ROCK OR RhoA OR (rho AND kinase$) OR (protein$ AND kinase$) OR "Quinasas Asociadas a rho" OR "Quinases Associadas a rho")
Appendix 6. ClinicalTrials.gov search strategy
(glaucoma OR hypertension OR intraocular pressure OR POAG OR IOP) AND ("rho associated" OR "Rho kinase" OR ROCK OR ROK OR ROKalpha OR "rhoA associated" OR p160ROCK OR "Protein Kinase")
Appendix 7. WHO ICTRP search strategy
glaucoma AND rho OR glaucoma AND rhoA OR glaucoma AND ROCK OR glaucoma AND ROK OR glaucoma AND RokAlpha OR glaucoma AND "protein kinase" OR glaucoma AND p160ROCK OR hypertension AND rho OR hypertension AND rhoA OR hypertension AND ROCK OR hypertension AND ROK OR hypertension AND RokAlpha OR hypertension AND "protein kinase" OR hypertension AND p160ROCK OR "intraocular pressure" AND rho OR "intraocular pressure" AND rhoA OR "intraocular pressure" AND ROCK OR "intraocular pressure" AND ROK OR "intraocular pressure" AND RokAlpha OR "intraocular pressure" AND "protein kinase" OR "intraocular pressure" AND p160ROCK
Appendix 8. Pooling of adverse events
| Category | Event 1 | Event 2 | Event 3 | Event 4 | Event 5 | Event 6 | Event 7 | Event 8 |
| Conjunctival hyperemia | Conjunctival hyperemia | — | — | — | — | — | — | — |
| Ocular hyperemia | Ocular hyperemia | — | — | — | — | — | — | — |
| Ocular pain and irritation | Instillation foreign body sensation | Instillation pain | Instillation discomfort | Foreign body sensation | Ophthalmalgia | Ocular irritation | Eye pain | Eye pruritus |
| Cornea verticillata | Cornea verticillata | Corneal deposits | Corneal opacity | — | — | — | — | — |
| Increased lacrimation | Increased lacrimation | Eye discharge | — | — | — | — | — | — |
| Punctate keratitis | Punctate keratitis | Corneal erosion | Vital dye staining of cornea | — | — | — | — | — |
| Visual AE | Blurred vision | Visual acuity reduced | Eyesight deterioration | — | — | — | — | — |
| Allergy | Conjunctivitis allergic | Conjunctival follicles | — | — | — | — | — | — |
| Edema | Eye swelling | Conjunctival edema | Eyelid edema | — | — | — | — | — |
| Iritis | Photophobia | Iritis | — | — | — | — | — | — |
Appendix 9. Detailed overview of specific adverse events
Adverse effects
ROKi versus placebo
Conjunctival hyperemia: after two months, treatment with ripasudil led to an increased rate of conjunctival hyperemia, with an incidence rate difference (IRD) of 26 more events per 100 person‐months (95% confidence interval [CI] 14 to 38). Treatment with netarsudil yielded 79 more events per 100 person‐months (95% CI 16 to 142) than placebo. In combination, treatment with ROKi resulted in 46 more events per 100 person‐months (95% CI 19 to 73; I2 = 62.4%). We found no evidence of a difference between subgroups defined by the type of ROKi (P = 0.10; Analysis 1.5).
Araie 2021 found a mean conjunctival hyperemia score (0 to 3: 0 = none, 1 = mild, 2 = moderate, 3 = severe) evaluated by biomicroscopic examination of 0.2 for netarsudil and 0 for placebo at 4‐week follow‐up compared to a mean baseline score of 0 for both interventions. The mean conjunctival hyperemia score remained relatively constant across week 1, 2 and 4. Sit 2021 evaluated 9/13 events as mild and 4/13 events as moderate in the netarsudil group. Tanihara 2013 evaluated all events of conjunctival hyperemia in the ripasudil group (32/32) and the placebo group (7/7) as mild. Thirty events in the ripasudil group and five events in the placebo group resolved spontaneously within 12 hours or less after application.
Ocular pain and irritation: we found no evidence of a difference between treatments in terms of ocular pain and irritation (IRD 4 more events per 100 person‐months, 95% CI −7 to 15; I2 = 61%). Neither did we find any evidence of differences between subgroups based on the type of ROKi (Analysis 1.6). Araie 2021 evaluated all events as mild, whereas Sit 2021 reported 1/3 events as mild and 2/3 events as moderate in the netarsudil group.
Cornea verticillata: none of the studies reported any events of cornea verticillata.
Serious adverse events (SAE): Araie 2021 reported one SAE (1.9%) (corneal abrasion) in the netarsudil group and no SAEs in the placebo group. Peace 2021, Aerie 2017, and Sit 2021 reported no SAEs in all treatment groups. Tanihara 2013 reported no SAEs with ripasudil and two SAEs (3.7%; iron‐deficiency anemia and retinal tear) with placebo. None of the SAEs were attributed to treatment with ROKi.
ROKi versus beta‐blocker
Conjunctival hyperemia: after three to 12 months, treatment with netarsudil led to 7 more events of conjunctival hyperemia per 100 person‐months (95% CI 4 to 11; I2 = 93%) compared to timolol (Analysis 2.4).
Kahook 2019 (ROCKET‐2) reported a mean conjunctival hyperemia score (0 to 3 scale) of 0.5 to 0.7 for netarsudil compared to 0.2 for timolol (baseline values of 0.2 for both interventions). Similarly, Khouri 2019 (ROCKET‐4) reported a mean conjunctival hyperemia score (0 to 3 scale) of 0.7 for netarsudil and 0.2 for timolol (baseline of 0.2 for both interventions) at six months' follow‐up. The mean hyperemia score remained constant across the six months of the study for both treatments. Serle 2018 (ROCKET‐1) described all events of conjunctival hyperemia as mild. The hyperemia was described as primarily transient/intermittent by Kahook 2019 (ROCKET‐2) and Khouri 2019 (ROCKET‐4).
Ocular pain and irritation: we found no evidence of a difference between treatments in terms of ocular pain and irritation (IRD 1 more events per 100 person‐months (95% CI −1 to 2; I2 = 46%; Analysis 2.5).
Cornea verticillata: at 12 months' follow‐up, Kahook 2019 (ROCKET‐2) detected 65 events (25.49%) of cornea verticillata with netarsudil compared to two events (0.8%) with timolol. At 6 months' follow‐up, Khouri 2019 (ROCKET‐4) reported 86 (24.5%) events in the netarsudil group and 0 events in the timolol group. At 12 months' follow‐up, NCT02246764 (ROCKET‐3) reported 18 (52.94%) events in the netarsudil group and 0 events in the timolol group. At 3 months' follow‐up, Serle 2018 (ROCKET‐1) detected 11 (5.42%) events in the netarsudil group and no events in the timolol group. Kahook 2019 (ROCKET‐2) reported a mean time to onset of 172.9 days (range 40 to 396 days) and a mean time to resolution of 341.2 days after treatment discontinuation. All but one participant had complete resolution, in which cornea verticillata improved and stabilized. Khouri 2019 (ROCKET‐4) reported a mean time to onset of 109.2 days (range 30 to 183) and a mean time to resolution of 87.3 days (range 0 to 264). Serle 2018 (ROCKET‐1) reported a range of six to 13 weeks until onset, and complete resolution typically within 13 weeks. The severity was predominantly described as mild without influencing the visual function (i.e. visual acuity, contrast sensitivity).
SAEs: Kahook 2019 (ROCKET‐2) reported 22 SAE (8.76%; 8 cardiac disorders, 1 cholelithiasis, 1 cellulitis, 1 postoperative Ileus, 1 fluid overload, 1 back pain, 1 osteoarthritis, 4 neoplasms, 2 central nervous system disorders, 1 pulmonary artery stenosis and 1 accelerated hypertension) with netarsudil and 18 SAEs (7.17%; 1 cardiac disorder, 1 cataract, 1 gastric ulcer perforation, 1 cholecystitis, 3 infections/infestations, 4 injuries, 1 prostate‐specific antigen increased, 1 synovial cyst, 1 carotid artery stenosis, 2 renal and urinary disorder, 1 pulmonary embolism and 1 peripheral artery occlusion) with timolol. Khouri 2019 (ROCKET‐4) reported 11 SAE (3. 13%; 2 cardiac disorder, 2 gastrointestinal (GI) disorders, 3 neoplasms, 1 central nervous system disorder, 1 renal and urinary disorder, 1 cervical dysplasia, and 1 pneumonia aspiration) with netarsudil and 12 SAE (3.36%; 3 cardiac disorders, 1 GI disorder, 1 pneumonia, 2 injuries, 2 neoplasms, 2 central nervous system disorders and 1 metal status change) with timolol. NCT02246764 (ROCKET‐3) reported no SAE with netarsudil compared to one SAE (4.35%; breast cancer) with timolol. Serle 2018 (ROCKET‐1) reported three SAE (1.48%; 1 cardiac disorder, 1 hypertension, 1 prostate cancer) with netarsudil compared to six SAE (2.88%; 1 cardiac disorder, 1 pneumonia, 2 nervous system disorders, 1 reproductive and breast disorder, and 1 acute respiratory failure) with timolol.
ROKi versus prostaglandin analog
Conjunctival hyperemia: treatment with netarsudil yielded a pooled mean excess of 11 events of conjunctival hyperemia per 100 person‐months (95% CI 3 to 19; I2 = 88%) compared to latanoprost (Analysis 3.3). At 12 months' follow‐up, Asrani 2019 (MERCURY‐1) reported a mean conjunctival hyperemia score of 0.6 for netarsudil compared to 0.3 for latanoprost (0 to 3 scale, baseline 0.2 for both interventions). Walters 2019 (MERCURY‐2) reported a mean conjunctival hyperemia score of 0.5 for netarsudil and 0.2 for latanoprost (0 to 3 scale, baseline of 0.1 for both outcomes). The mean hyperemia score remained relatively constant during study conduct (Asrani 2019 (MERCURY‐1); Walters 2019 (MERCURY‐2)).
Ocular pain and irritation: treatment with netarsudil resulted in a pooled mean excess of 2 events of ocular pain and irritation per 100 person‐months (95% CI 1 to 3; I2 = 0%) compared to treatment with latanoprost (Analysis 3.4).
Cornea verticillata: after 12 months of treatment, Asrani 2019 (MERCURY‐1) reported 33 events (13;58%) of cornea verticillata in the netarsudil group and no events in the latanoprost group. All cases were asymptomatic and predominantly judged as mild. The mean time to onset was 216.6 days, whereas the mean time to resolution was 86.4 days after treatment discontinuation. After three months of treatment, Walters 2019 (MERCURY‐2) found 25 events (9.8%) with netarsudil compared to no events with latanoprost. All cases were asymptomatic and predominantly judged as mild.
SAEs: Asrani 2019 (MERCURY‐1) reported 11 SAEs (4.52%; 1 cardiac disorder, 2 GI disorders, 1 cellulitis, 1 hypoglycemia, 2 neoplasms, 1 spontaneous abortion, 1 bronchitis chronic and 2 vascular disorders) with netarsudil monotherapy compared to 14 SAEs (5.91%; 2 cardiac disorder, 1 GI disorders, 1 sepsis, 2 injuries, 1 osteoarthritis, 3 neoplasms, 2 nervous system disorders, 1 pulmonary embolism and 1 deep vein thrombosis) with latanoprost. Bacharach 2015 reported no SAEs with netarsudil compared to two SAEs (2.6%; 1 pneumonia and 1 fall) with latanoprost. Lewis 2016 reported no SAEs with netarsudil compared to four SAEs (5.48%; 1 ulcerative keratitis, and 3 gastrointestinal/hepatobiliary disorders) with latanoprost. Walters 2019 (MERCURY‐2) reported seven SAE (2.75%; 4 cardiac disorder, 1 bronchitis, 1 central nervous system disorder and 1 pulmonary embolism) with netarsudil compared to five SAEs (1.99%; 1 cardiac disorder, 1 retinal detachment, 1 cholecystitis acute and 2 infections) with latanoprost.
ROKi plus prostaglandin analog versus prostaglandin analog
Conjunctival hyperemia: combination therapy with ripasudil and latanoprost caused on average an additional 24 events per 100 person‐months (95% CI 16 to 32) compared to latanoprost monotherapy, whereas combination therapy with netarsudil and latanoprost resulted in a pooled mean excess of 10 events per 100 person‐months (95% CI 0 to 20; I2 = 81%) compared to latanoprost monotherapy (Analysis 4.3). Overall, combination therapy with ROKi and latanoprost led to more events compared to latanoprost monotherapy, with an IRD of 15 more events per 100 person‐months (95% CI 3 to 27; I2 = 92%). We found no evidence of a difference between subgroups defined by the type of ROKi (I2 = 78%).
Asrani 2019 (MERCURY‐1) found a mean conjunctival hyperemia score of 0.6 for combination therapy with netarsudil and latanoprost compared to 0.3 for latanoprost (0 to 3 scale, baseline 0.2 for both interventions). Walters 2019 (MERCURY‐2) reported a mean conjunctival hyperemia score of 0.7 for combination therapy with netarsudil and latanoprost compared to 0.2 for latanoprost (baseline values of 0.2 for netarsudil/latanoprost and 0.1 for latanoprost). The mean hyperemia score remained relatively constant during study conducts (Asrani 2019 (MERCURY‐1); Walters 2019 (MERCURY‐2)).
Ocular pain and irritation: we found weak evidence of a minor difference between treatments in terms of ocular pain and irritation, with an IRD of 3 events per 100 person‐months (95% CI 0 to 7; I2 = 86%), with the combination of netarsudil and latanoprost associated with more excess AEs than the latanoprost monotherapy group (Analysis 4.4).
Cornea verticillata: after 12 months of treatment, Asrani 2019 (MERCURY‐1) reported 42 events (17.65%) of cornea verticillata with combination therapy of netarsudil and latanoprost compared to no events with latanoprost, whereas Walters 2019 (MERCURY‐2) reported 32 events (13.11%) with combination therapy of netarsudil and latanoprost compared to no events with latanoprost at three months' follow‐up. Cases were asymptomatic and predominantly judged as mild (Asrani 2019 (MERCURY‐1); Walters 2019 (MERCURY‐2)). Mean onset was 180.6 days after initiation, while mean time to resolution after treatment discontinuation was 48.7 days (Asrani 2019 (MERCURY‐1)).
SAEs: Asrani 2019 (MERCURY‐1) reported six SAEs (2.52%; 1 anemia, 1 GI disorder, 1 cholecystitis, 1 pneumonia, 1 neoplasm and 1 central nervous system disorder) with combination therapy compared to 14 SAEs (5.91%) with latanoprost (see detailed description above). Lewis 2016 reported no SAEs with combination therapy and four SAEs (5.48%) with latanoprost (see detailed description above). Tanihara 2015c reported three SAEs (2.94%) (1 otitis media and 2 visual field defects) with ripasudil plus latanoprost and five SAEs (1.99%) (2 cataract, 2 otitis media and 1 visual field defect) with latanoprost. Walters 2019 (MERCURY‐2) reported three SAEs (2.94%) (2 cardiac disorders and 1 mental status change) with combination therapy compared to five SAEs (4.85%) with latanoprost (see detailed description above).
ROKi plus prostaglandin analog versus rho kinase inhibitor
Conjunctival hyperemia: we found no evidence of a difference between treatments in terms of conjunctival hyperemia (IRD 1 more event per 100 person‐months, 95% CI 0 to 2; I2 = 0%; Analysis 5.3). Asrani 2019 (MERCURY‐1) found a mean conjunctival hyperemia score of 0.6 for both combination therapy with netarsudil and latanoprost and netarsudil monotherapy (0 to 3 scale, baseline 0.2 for both interventions).
Walters 2019 (MERCURY‐2) reported a mean conjunctival hyperemia score of 0.7 for combination therapy with netarsudil and latanoprost compared to 0.5 for netarsudil monotherapy (0 to 3 scale, baseline values of 0.2 for combination therapy and 0.1 for netarsudil monotherapy). The mean hyperemia score remained relatively constant during study conducts (Asrani 2019 (MERCURY‐1); Walters 2019 (MERCURY‐2)).
Ocular pain and irritation: we found no evidence of a difference between treatments in terms of ocular pain and irritation (IRD 0 per 100 person‐months (95% CI 0 to 1; I2 = 65%; Analysis 5.4).
Cornea verticillata: Asrani 2019 (MERCURY‐1) reported 42 events (17.65%) of cornea verticillata with combination therapy of netarsudil and latanoprost compared to 33 events (13.58%) with netarsudil, whereas Walters 2019 (MERCURY‐2) reported 32 events (13.11%) with combination therapy of netarsudil and latanoprost compared to 25 events (9.8%) with netarsudil. Asrani 2019 (MERCURY‐1) reported a mean onset of 180.6 days after initiation and mean time to resolution after discontinuation of 48.7 days for combination therapy. Considering netarsudil monotherapy, the mean time until onset was 216.6 days, whereas the mean time until resolution was 86.4 days after treatment discontinuation.
SAEs: Asrani 2019 (MERCURY‐1) reported six SAEs (2.52%) with combination therapy compared to 11 SAEs (4.53%) with netarsudil monotherapy. Lewis 2016 reported no SAE for both combination therapy and monotherapy with netarsudil. Walters 2019 (MERCURY‐2) reported three SAEs (1.23%) with combination therapy compared to seven SAEs (2.75%) with monotherapy (see detailed description above).
ROKi plus beta‐blocker versus beta‐blocker
Conjunctival hyperemia: on average combination therapy with ripasudil and timolol resulted in 30 more events of conjunctival hyperemia per 100 person‐months (95% CI 22 to 38) compared to timolol monotherapy (Analysis 6.3). The severity of conjunctival hyperemia was not examined.
Ocular pain and irritation: we found no evidence of a difference between treatments in terms of ocular pain and irritation (IRD 4 more events per 100 person‐months, 95% CI 0 to 8; Analysis 6.4).
Cornea verticillata: the trial did not investigate events of cornea verticillata.
SAEs: Tanihara 2015b reported one SAE (0.96%; visual field defect) with combination therapy and four SAEs (4.81%; 2 spinal osteoarthritis, 1 asthma and 1 cholelithiasis) with timolol.
Data and analyses
Comparison 1. Rho kinase inhibitor versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Mean intraocular pressure (IOP) changes from baseline | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1.1 Netarsudil 0.02% once per day | 3 | 155 | Mean Difference (IV, Random, 95% CI) | ‐3.11 [‐3.62, ‐2.59] |
| 1.1.2 Ripasudil 0.4% twice per day | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐2.15, ‐0.45] |
| 1.2 Mean IOP changes from baseline: sensitivity analysis | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 Netarsudil 0.02% once per day | 2 | 119 | Mean Difference (IV, Fixed, 95% CI) | ‐2.96 [‐3.56, ‐2.37] |
| 1.2.2 Ripasudil 0.4% twice per day | 1 | 103 | Mean Difference (IV, Fixed, 95% CI) | ‐1.30 [‐2.15, ‐0.45] |
| 1.3 Total ocular adverse events (per person‐month) – incidence risk difference | 5 | Risk Difference (IV, Random, 95% CI) | 0.66 [0.28, 1.03] | |
| 1.3.1 Netarsudil 0.02% once per day | 4 | Risk Difference (IV, Random, 95% CI) | 0.86 [0.38, 1.34] | |
| 1.3.2 Ripasudil 0.4% twice per day | 1 | Risk Difference (IV, Random, 95% CI) | 0.27 [0.13, 0.41] | |
| 1.4 Total ocular adverse events (per person‐month): sensitivity analysis | 2 | Risk Difference (IV, Random, 95% CI) | 0.67 [0.55, 0.79] | |
| 1.5 Conjunctival hyperemia as adverse event (per person‐month)* | 5 | Risk Difference (IV, Random, 95% CI) | 0.46 [0.19, 0.73] | |
| 1.5.1 Ripasudil 0.4% twice per day | 1 | Risk Difference (IV, Random, 95% CI) | 0.26 [0.14, 0.38] | |
| 1.5.2 Netarsudil 0.02% once per day | 4 | Risk Difference (IV, Random, 95% CI) | 0.79 [0.16, 1.42] | |
| 1.6 Ocular pain or irritation as adverse event (per person‐month)* | 5 | Risk Difference (IV, Random, 95% CI) | 0.04 [‐0.07, 0.15] | |
| 1.6.1 Ripasudil 0.4% twice per day | 1 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.05, 0.07] | |
| 1.6.2 Netarsudil 0.02% once per day | 4 | Risk Difference (IV, Random, 95% CI) | 0.08 [‐0.14, 0.30] |
Comparison 2. Rho kinase inhibitor versus beta‐blocker.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Mean IOP changes from baseline | 3 | 1415 | Mean Difference (IV, Fixed, 95% CI) | 0.66 [0.41, 0.91] |
| 2.2 Total ocular adverse events (per person‐month) – incidence risk difference | 4 | Risk Difference (IV, Random, 95% CI) | 0.21 [0.14, 0.27] | |
| 2.3 Total ocular adverse events (per person‐month) – subgroup analysis by levels of reporting threshold | 4 | Risk Difference (IV, Random, 95% CI) | 0.21 [0.14, 0.27] | |
| 2.3.1 Reporting threshold ≥ 3% | 2 | Risk Difference (IV, Random, 95% CI) | 0.22 [0.03, 0.41] | |
| 2.3.2 Reporting threshold ≥ 5% | 2 | Risk Difference (IV, Random, 95% CI) | 0.20 [0.14, 0.26] | |
| 2.4 Conjunctival hyperemia as adverse event (per person‐month)* | 4 | Risk Difference (IV, Random, 95% CI) | 0.07 [0.04, 0.11] | |
| 2.5 Ocular pain or irritation as adverse event (per person‐month)* | 4 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.01, 0.02] |
Comparison 3. Rho kinase inhibitor versus prostaglandin analog.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Mean IOP changes from baseline (mmHg) | 4 | 1283 | Mean Difference (IV, Random, 95% CI) | 0.97 [0.67, 1.27] |
| 3.2 Total ocular adverse events (per person‐month) – incidence rate difference | 4 | Risk Difference (IV, Random, 95% CI) | 0.29 [0.17, 0.42] | |
| 3.3 Conjunctival hyperemia as adverse event (per person‐month)* | 4 | Risk Difference (IV, Random, 95% CI) | 0.11 [0.03, 0.19] | |
| 3.4 Ocular pain or irritation as adverse event (per person‐month)* | 4 | Risk Difference (IV, Random, 95% CI) | 0.02 [0.01, 0.03] |
Comparison 4. Rho kinase inhibitor + prostaglandin analog versus prostaglandin analog.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Mean IOP changes from baseline | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1.1 Netarsudil 0.02% + latanoprost 0.005% (FDC) once per day vs latanoprost 0.005% once per day | 3 | 1114 | Mean Difference (IV, Random, 95% CI) | ‐1.64 [‐2.16, ‐1.11] |
| 4.1.2 Ripasudil 0.4% twice per day + latanoprost 0.005% once per day vs latanoprost 0.005% once per day | 1 | 205 | Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.86, 0.28] |
| 4.2 Total ocular adverse events (per person‐month) – incidence rate difference | 4 | Risk Difference (IV, Random, 95% CI) | 0.26 [0.13, 0.40] | |
| 4.2.1 Netarsudil 0.02% + latanoprost 0.005% (FDC) once per day vs latanoprost 0.005% once per day | 3 | Risk Difference (IV, Random, 95% CI) | 0.29 [0.11, 0.47] | |
| 4.2.2 Ripasudil 0.4% twice per day + latanoprost 0.005% once per day vs latanoprost 0.005% once per day | 1 | Risk Difference (IV, Random, 95% CI) | 0.21 [0.11, 0.31] | |
| 4.3 Conjunctival hyperemia as adverse event (per person‐month)* | 4 | Risk Difference (IV, Random, 95% CI) | 0.15 [0.03, 0.27] | |
| 4.3.1 Netarsudil 0.02% + latanoprost 0.005% (FDC) once per day vs latanoprost 0.005% once per day | 3 | Risk Difference (IV, Random, 95% CI) | 0.10 [0.00, 0.20] | |
| 4.3.2 Ripasudil 0.4% twice per day + latanoprost 0.005% once per day vs latanoprost 0.005% once per day | 1 | Risk Difference (IV, Random, 95% CI) | 0.24 [0.16, 0.32] | |
| 4.4 Ocular pain or irritation as adverse event (per person‐month)* | 4 | Risk Difference (IV, Random, 95% CI) | 0.03 [‐0.00, 0.07] | |
| 4.4.1 Netarsudil 0.02% + latanoprost 0.005% (FDC) once per day vs latanoprost 0.005% once per day | 3 | Risk Difference (IV, Random, 95% CI) | 0.05 [0.01, 0.09] | |
| 4.4.2 Ripasudil 0.4% twice per day + latanoprost 0.005% once per day vs latanoprost 0.005% once per day | 1 | Risk Difference (IV, Random, 95% CI) | ‐0.02 [‐0.06, 0.02] |
Comparison 5. Rho kinase inhibitor + prostaglandin analog versus Rho kinase inhibitor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Mean IOP changes from baseline | 3 | 1132 | Mean Difference (IV, Fixed, 95% CI) | ‐2.66 [‐2.98, ‐2.35] |
| 5.2 Total ocular adverse events (per person‐month) – incidence rate difference | 3 | Risk Difference (IV, Fixed, 95% CI) | 0.01 [‐0.00, 0.03] | |
| 5.3 Conjunctival hyperemia as adverse event (per person‐month)* | 3 | Risk Difference (IV, Fixed, 95% CI) | 0.01 [0.00, 0.02] | |
| 5.4 Ocular pain or irritation as adverse event (per person‐month)* | 3 | Risk Difference (IV, Fixed, 95% CI) | 0.00 [‐0.00, 0.01] |
Comparison 6. Rho kinase inhibitor + beta‐blocker versus beta‐blocker.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Mean IOP changes from baseline | 1 | 208 | Mean Difference (IV, Fixed, 95% CI) | ‐0.75 [‐1.29, ‐0.21] |
| 6.2 Total ocular adverse events (per person‐month) – incidence rate difference | 1 | Risk Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.3 Conjunctival hyperemia as adverse event (per person‐month) | 1 | Risk Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.4 Ocular pain or irritation as adverse event (per person‐month) | 1 | Risk Difference (IV, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aerie 2017.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 42 participants Number of participants (eyes) randomized per group: placebo: 15, netarsudil 0.02%: 15, netarsudil 0.04%: 12 participants Total number of participants (eyes) lost to follow‐up: 1 participant lost to follow‐up, 2 participants excluded from analysis Number of participants (eyes) lost to follow‐up per group: placebo: 1, netarsudil 0.02%: 1, netarsudil 0.04%: 0 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 28 days Actual length of follow‐up: 28 days How missing outcome data were handled: Monte Carlo Markov Chain multiple imputation techniques Was the trial single/double/triple‐masked: triple‐masked (NCT), double‐masked (protocol) Was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: mITT Duration of washout for each drug class before interventions began: 4 weeks (prostaglandins and BBs); 2 weeks (adrenergic agonists); 5 days (muscarinic agonists, carbon anhydrase inhibitors) |
|
| Participants |
Baseline characteristics Placebo
Netarsudil 0.02%, once per day (p.m.)
Netarsudil 0.04%, once per day (p.m.)
Overall
Inclusion criteria: aged ≥ 18 years; of Japanese ethnicity within the second generation defined as (a) first generation born in Japan, immigrated to US and (b) second generation – parents are first generation and the patient was born in US as an American citizen; diagnosis of OAG or OHT in both eyes; medicated IOP ≥ 15 mmHg and < 30 mmHg in both eyes at screening OAG eyes, unmedicated IOP ≥ 15 mmHg and < 35 mmHg at 2 qualification visits at 8 a.m., 10 a.m. and 4 p.m.; OHT eyes, unmedicated IOP ≥ 22 mmHg and < 35 mmHg at 8 a.m., 10 a.m. and 4 p.m.; best corrected visual acuity + 1.0 logMAR or better by ETDRS in each eye; able to give signed informed consent and follow instructions Exclusion criteria: clinically significant ocular disease; pseudoexfoliation or pigment dispersion component glaucoma, history of angle closure glaucoma or narrow angles; IOP ≥ 35 mmHg in either eye; ocular hyperemia score of moderate (+2) at qualification visit #2; previous glaucoma intraocular surgery; refractive surgery in either eye; ocular injury within 6 months prior to screening or ocular surgery or non‐refractive laser treatment within 3 months prior to screening; recent or current ocular infection or inflammation in either eye; use of ocular medication in either eye of any type within 30 days of screening and throughout the study; mean central corneal thickness > 620 µm in either eye; any abnormality preventing reliable applanation tonometry of either eye; known hypersensitivity to benzalkonium chloride or excipients of netarsudil ophthalmic solution; clinically significant abnormalities in screening laboratory tests; clinically significant systemic disease that might interfere with the study; participated in any investigational study within 30 days prior to screening; systemic medication that could have a substantial effect on IOP within 30 days prior to screening or anticipated during the study; women of child‐bearing potential who are pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control Pretreatment: no formal comparison results reported |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: 40 locations Online trial registration site: ClinicalTrials.gov Trial registration #: NCT03310580 Phase of trial: phase 2 Current publication reported findings from > 1 trial: no Year publication accepted: 2019 (results posted) Year study initiation (participants screening, enrollment and treatment): 2017 |
|
| Notes | Other study ID: AR‐13324‐CS205 | |
Araie 2021.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 215 participants Number of participants (eyes) randomized per group: placebo: 55, netarsudil 0.01%: 54, netarsudil 0.02%: 51, netarsudil 0.04%: 55 participants Total number of participants (eyes) lost to follow‐up: 8 participants Number of participants (eyes) lost to follow‐up per group: placebo: 1, netarsudil 0.01%: 1, netarsudil 0.02%: 5, netarsudil 0.04%: 1 participant Power calculation and sample size consideration reported: yes Planned length of follow‐up: 4 weeks Actual length of follow‐up: 4 weeks How missing outcome data were handled: Monte Carlo Markov chain multiple imputation techniques Was the trial single/double/triple‐masked: double‐masked Based on the study hypothesis, was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: 5 days for muscarinic agonists or carbonic anhydrase inhibitors; 2 weeks for adrenergic agonists; 4 weeks for prostaglandins or BBs; 6 weeks for ROKi |
|
| Participants |
Baseline characteristics Placebo, once per day (8–10 p.m.)
AR‐13324 (netarsudil), 0.01%, once per day (8–10 p.m.)
AR‐13324 (netarsudil), 0.02%, once per day (8–10 p.m.)
AR‐13324 (netarsudil), 0.04%, once per day (8–10 p.m.)
Overall
Inclusion criteria: aged ≥ 20 years; diagnosis of OAG or OHT in both eyes (OAG in 1 eye and OHT in the fellow eye was acceptable); BCVA ≥ 0.1 in decimal unit using Landolt‐C chart or its equivalent; willing to give signed informed consent and following study instructions Exclusion criteria: clinically significant ocular diseases; pseudoexfoliation or pigment dispersion component glaucoma, history of narrow angle closure glaucoma or narrow angles; previous glaucoma intraocular surgery; refractive surgery in either eye; ocular trauma; ocular infection or inflammation; known hypersensitivity to benzalkonium chloride or excipient of netarsudil ophthalmic solution; cannot demonstrate proper delivery of the eye drop; clinically significant abnormalities in screen laboratory tests; clinically significant systemic disease; participation in any investigational study within 30 days of screening; women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control Pretreatment characteristics between groups: no significant differences |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: Japan Setting: multicenter, 25 locations Online trial registration site: ClinicalTrials.gov Trial registration #: NCT03844945 Phase of the trial (phase 2/phase 3/unclear): phase 2 Current publication reported findings from > 1 trial: no Year of publication accepted: 2021 Year of study initiation (participants screening, enrollment and treatment): 2019 |
|
| Notes | A netarsudil dose‐finding trial (phase 2) | |
Asrani 2019 (MERCURY‐1).
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 718 participants Number of participants (eyes) randomized per group: netarsudil: 244, FDC: 238, latanoprost: 236 participants Number of participants (eyes) lost to follow‐up per group: netarsudil: 43, FDC: 37, latanoprost: 13 participants Total number of participants (eyes) lost to follow‐up: 93 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 3 months for efficacy; 12 months for safety Actual length of follow‐up: 3 months for efficacy; safety outcomes at 12 months were reported separately (Brubaker 2020) How missing outcome data were handled: Markov Chain Monte Carlo multiple imputation techniques Was the trial single/double/triple‐masked: double‐masked Based on the study hypothesis, was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: 4 weeks for PAs and BBs, 2 weeks for adrenergic agonists, and 5 days for muscarinic agonists and carbonic anhydrase inhibitors |
|
| Participants |
Baseline characteristics Netarsudil 0.02%, once per day (p.m.)
Latanoprost 0.005%, once per day (p.m.)
Netarsudil 0.02%/latanoprost 0.005% (FDC), once per day (p.m.)
Overall
Inclusion criteria: bilateral OAG or OHT and were aged ≥ 18 years with unmedicated IOP > 20 and < 36 mmHg in both eyes at 8 a.m. at 2 qualification visits (2–7 days apart) and > 17 and < 36 mmHg in both eyes at 10 a.m. and 4 p.m. at the second qualification visit. Patients using ocular hypotensive medications were required to undergo washout before study entry: 4 weeks for PAs and BBs, 2 weeks for adrenergic agonists, and 5 days for muscarinic agonists and carbonic anhydrase inhibitors. BCVA in each eye was +1.0 logMAR or better by ETDRS measurement Exclusion criteria: treated with > 2 ocular hypotensive medications within 30 days of screening; pseudoexfoliation or pigment dispersion glaucoma; history of iridocorneal angle closure or narrow angles (including previous peripheral iridotomy); previous glaucoma incisional or laser surgery; previous refractive surgery; central corneal thickness > 620 mm; known hypersensitivity or contraindications to netarsudil or latanoprost (or their excipients); with clinically significant ocular disease other than glaucoma in either eye or systemic disease that might interfere with the study; women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy, or not using a medically acceptable form of birth control Pretreatment differences between groups: baseline demographics were similar across treatment groups Other description of the overall study population at baseline: in total, 55.3% (397/718) receiving prostaglandin monotherapy, 7.0% (50/718) other monotherapy and 11.6% (83/718) combination therapy |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: funding/support: the MERCURY‐1 study was sponsored by Aerie Pharmaceuticals, Inc., who participated in the design and conduct of the study; the collection, management, analysis and interpretation of data; and the preparation, review and approval of the manuscript. Country: USA Setting: 56 active sites in 21 states Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02558400 Current publication reported findings from > 1 trial: no Phase of the trial (phase 2/phase 3/unclear): phase 3 Year of study initiation (participants screening, enrollment and treatment): 2017 Year of publication accepted: 2019 |
|
| Notes | ||
Bacharach 2015.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 224 participants Number of participants (eyes) randomized per group: netarsudil 0.01%: 75, netarsudil 0.02%: 72, latanoprost: 77 participants Total number of participants (eyes) lost to follow‐up: 1 participant Number of participants (eyes) lost to follow‐up per group: netarsudil 0.01%: 1, netarsudil 0.02%: 0, latanoprost: 0 participant Power calculation and sample size consideration reported: yes Planned length of follow‐up: 28 days (+ day 29 and day 30) Actual length of follow‐up: 28 days (+ day 29 and day 30) How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: quadruple‐masked (NCT), double‐masked (article) Was the trial an equivalence/superiority/non‐inferiority study: non‐inferiority study Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: mITT Duration of washout for each drug class before interventions began: prostaglandins: 4 weeks; BBs: 4 weeks; adrenergic agonists (including alfa‐agonists such as brimonidine and apraclonidine): 2 weeks; muscarinic agonists (e.g. pilocarpine), carbonic anhydrase inhibitors (topical or oral): 5 days |
|
| Participants |
Baseline characteristics Netarsudil 0.01%, once per day (10–11 p.m.)
Netarsudil 0.02%, once per day (10–11 p.m.)
Latanoprost 0.005%, once per day (10–11 p.m.)
Overall
Inclusion criteria: aged ≥ 18 years; diagnosis of OAG or OHT; unmedicated (post‐washout) IOP ≥ 24 mmHg at 2 eligibility visits (8 hours), 2–7 days apart, and ≥ 22 mmHg at 10 a.m. and 4 p.m. at second qualification visit; if only 1 eye met the IOP criteria it must have been the same eye that met the criteria at all the qualification time points; corrected visual acuity in each eye +1.0 logMAR or better by ETDRS in each eye (equivalent to 20/200); able and willing to give signed informed consent and follow study instructions Exclusion criteria: glaucoma: pseudoexfoliation or pigment dispersion component, history of angle closure or narrow angles. Note: previous laser peripheral iridotomy was NOT acceptable; IOP > 36 mmHg known hypersensitivity to any component of the formulation (benzalkonium chloride, etc.), or to topical anesthetics; previous glaucoma intraocular surgery or glaucoma laser procedures in study eye(s) (e.g. laser trabeculoplasty); refractive surgery in study eye(s) (e.g. radial keratotomy, photorefractive keratectomy, laser eye surgery, etc.); ocular trauma within past 6 months, or ocular surgery or laser treatment within the past 3 months prior to screening; evidence of ocular infection, inflammation, clinically significant blepharitis or conjunctivitis at screening (visit 0), or history of herpes simplex keratitis; ocular medication of any type within 30 days of visit 0, except for (a) ocular hypotensive medications (which must have been washed out according to the provided schedule), (b) lid scrubs (which may have been used prior to, but not after visit 0) or (c) lubricating drops for dry eye (which may have been used throughout the study); clinically significant ocular disease (e.g. uveitis, severe keratoconjunctivitis sicca) which might interfere with the study, including glaucomatous damage so severe that washout of ocular hypotensive medications for 1 month is not judged safe (i.e. cup–disk ratio > 0.8); central corneal thickness > 600 µm; any abnormality preventing reliable applanation tonometry of either eye. Systemic: clinically significant abnormalities (as determined by the treating physician) in laboratory tests at screening; known hypersensitivity or contraindication to latanoprost; clinically significant systemic disease (e.g. myasthenia gravis, hepatic, renal, endocrine or cardiovascular disorders) which might interfere with the study; participation in any investigational study within 30 days prior to screening; changes of systemic medication that could have a substantial effect on IOP within 30 days prior to screening, or anticipated during the study; due to the current status of the preclinical safety program, women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy, or not using a medically acceptable form of birth control. An adult woman was considered of child‐bearing potential unless she was 1 year postmenopausal or three months postsurgical sterilization. All females of child‐bearing potential must have had a negative urine pregnancy test result at the screening examination and must not have intended to become pregnant during the study Pretreatment differences between groups: no significant differences (Table 3) Other description of the overall study population at baseline: none |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals, Inc. Country: USA Setting: 22 private practice ophthalmology clinics Online trial registration site: ClinicalTrials.gov Trial registration #: NCT01731002 Phase of the trial: phase 2b Current publication reported findings from > 1 trial: no Year of publication accepted: 2014 Year of study initiation (participants screening, enrollment and treatment): 2012 |
|
| Notes | 3 severe adverse events were judged not to be related to treatment: 1 pneumonia (latanoprost), 1 influenza and syncope (latanoprost), 1 death from leukemia (netarsudil 0.01%) | |
Inoue 2018.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 51 participants Number of participants (eyes) randomized per group: added group: 25, switched group: 26 participants Total number of participants (eyes) lost to follow‐up: 5 participants Number of participants (eyes) lost to follow‐up per group: added group: 4, switched group: 1 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 3 months Actual length of follow‐up: 3 months How missing outcome data were handled: exclusion from the analysis Was the trial single/double/triple‐masked: open‐label Was the trial an equivalence/superiority/non‐inferiority study: unclear Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: mITT Duration of washout for each drug class before interventions began: not reported |
|
| Participants |
Baseline characteristics Ripasudil 0.4% twice per day + latanoprost 0.005%/travoprost 0.004%/tafluprost 0.005% once per day (added group)
Timolol 0.5% + latanoprost 0.005%/travoprost 0.004%/tafluprost 0.005% FDC, once per day (switched group)
Overall
Inclusion criteria: POAG with insufficient IOP control while taking PA monotherapy insufficient IOP reduction after > 3 months of treatment (mean treatment duration, 96.9 [SD 52.5] months; range 3–198 months) with latanoprost (40 eyes), travoprost (7 eyes), or tafluprost (4 eyes) Exclusion criteria: not reported Pretreatment: at baseline, no significant between‐group differences were found Other description of the overall study population at baseline: not reported |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: no sponsorship source reported. Conflicts of interest: K Inoue, none; K Ishida, none; G Tomita, none. Country: Japan Setting: Inouye Eye Hospital (Tokyo, Japan) Online trial registration site: not reported Trial registration #: not reported Phase of the trial (phase 2/phase 3/unclear): unclear Current publication reported findings from > 1 trial: no Year of publication accepted: 2018 Year of study initiation (participants screening, enrollment and treatment): 2015 |
|
| Notes | ||
Kahook 2019 (ROCKET‐2).
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 756 participants Number of participants (eyes) randomized per group: netarsudil 0.02% once per day: 251, netarsudil 0.02% twice per day: 254, timolol: 251 participants Total number of participants (eyes) lost to follow‐up: 124 participants Number of participants (eyes) lost to follow‐up per group: netarsudil 0.02% once per day: 46, netarsudil 0.02% twice per day: 101, timolol: 14 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: primary outcome at 3 months; safety outcomes at 12 months Actual length of follow‐up: primary outcome at 3 months; safety outcomes at 12 months How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: double‐masked (article), quadruple‐masked (NCT) Was the trial an equivalence/superiority/non‐inferiority study: non‐inferiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: NCT: the PP population included all participants who did not have a major protocol violation likely to seriously affect the primary outcome of the study Duration of washout for each drug class before interventions began: 28 days for BBs, prostaglandins and the dorzolamide‐timolol fixed combination; 14 days for alfa‐ and alfa/beta‐agonists, 5 days for miotics and oral or topical carbonic anhydrase inhibitors, and 3 days if participants were receiving no IOP‐lowering therapy |
|
| Participants |
Baseline characteristics Netarsudil 0.02%, once per day (p.m.)
Netarsudil 0.02%, twice per day
Timolol 0.5%, twice per day
Overall
Inclusion criteria: 0–2 years of age and ≥ 18 years; diagnosis of OAG or OHT; unmedicated (post‐washout) IOP > 20 mmHg and < 27 mmHg in the study eye at 2 qualification visits; corrected visual acuity in each eye equivalent to 20/200; able and willing to give signed informed consent (parent or guardian consent for children) and follow study instructions Exclusion criteria: glaucoma: pseudoexfoliation or pigment dispersion component, history of angle closure, or narrow angles. Note: previous laser peripheral iridotomy is NOT acceptable; IOP ≥ 27 mmHg (unmedicated) in both eyes or use of > 2 ocular hypotensive medications within 30 days of screening. Note: fixed dose combinations count as 2 medications; known hypersensitivity to any component of the formulations to be used (benzalkonium chloride, etc.), to topical anesthetics or BBs; previous glaucoma intraocular surgery or glaucoma laser procedures in either eye; refractive surgery in either eye; ocular trauma in either eye within the 6 months prior to screening, or ocular surgery or non‐refractive laser treatment within the 3 months prior to screening; recent or current evidence of ocular infection or inflammation in either eye; current evidence of clinically significant blepharitis, conjunctivitis or a history of herpes simplex or zoster keratitis at screening in either eye; ocular medication in either eye of any type within 30 days of screening; clinically significant ocular disease in either eye (e.g. corneal edema, uveitis, severe keratoconjunctivitis sicca) which might interfere with the study, including glaucomatous damage so severe that washout of ocular hypotensive medications for 1 month is not judged safe; central corneal thickness in either eye > 600 µm at screening; any abnormality in either eye preventing reliable applanation tonometry of either eye. Systemic: clinically relevant abnormalities (as determined by the investigator) in laboratory tests at screening which may impact the study; known hypersensitivity or contraindication to BBs (e.g. chronic obstructive pulmonary disease or bronchial asthma; abnormally low blood pressure or heart rate; second or third‐degree heart block or congestive heart failure; severe diabetes); clinically significant systemic disease (e.g. uncontrolled diabetes, myasthenia gravis, hepatic, renal, endocrine or cardiovascular disorders) which might interfere with the study; participation in any investigational study within 30 days prior to screening; changes of systemic medication that could have an effect on IOP within 30 days prior to screening, or anticipated during the study; women of child‐bearing potential who are pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control. An adult woman was considered of child‐bearing potential unless she was 1 year postmenopausal or 3 months postsurgical sterilization. All females of child‐bearing potential must have had a negative urine pregnancy test result at the screening examination and must not have intended to become pregnant during the study Pretreatment characteristics between groups: no statistically significant differences in demographics between treatment groups Other description of the overall study population at baseline: not reported |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: multicenter (62 clinical trial sites) Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02207621 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: no Year of publication accepted: 2019 Year of study initiation (participants screening, enrollment and treatment): 2014 |
|
| Notes | Primary efficacy data of ROCKET‐2 at 3 months were reported in Serle 2018; safety data of ROCKET‐2 at 12 months in Kahook 2019. | |
Khouri 2019 (ROCKET‐4).
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 708 participants Number of participants (eyes) randomized per group: netarsudil: 351, timolol: 357 participants Total number of participants (eyes) lost to follow‐up: 86 participants Number of participants (eyes) lost to follow‐up per group: netarsudil: 45, timolol: 41 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 6 months Actual length of follow‐up: 6 months How missing outcome data were handled: exclusion of participants Was the trial single/double/triple‐masked: double‐masked (article), quadruple‐masked (NCT) Was the trial an equivalence/superiority/non‐inferiority study: non‐inferiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: PP Duration of washout for each drug class before interventions began: 4 weeks for participants using PAs or BBs prior to study entry, 2 weeks for those using adrenergic agonists, and 5 days for those using muscarinic agonists or topical carbonic anhydrase inhibitors |
|
| Participants |
Baseline characteristics Netarsudil 0.02%, once per day (p.m.)
Timolol 0.5%, twice per day
Overall
Inclusion criteria: aged ≥ 18 years; diagnosis of OAG or OHT in both eyes; post‐washout IOP > 20 mmHg and < 30 mmHg in 1 or both eyes at 2 qualification visits; corrected visual acuity equivalent to 20/200; able to give informed consent and follow study instructions Exclusion criteria: clinically significant ocular disease; pseudoexfoliation or pigment dispersion component glaucoma, history of angle closure or narrow angles; unmedicated IOP ≥ 30 mmHg; use of > 2 ocular hypotensive medications within 30 days of screening; known hypersensitivity to any component of the formulation; previous glaucoma surgery or refractive surgery; ocular trauma within 6 months prior to screening; any ocular surgery or non‐refractive laser treatment within 3 months prior to screening; recent or current ocular infection or inflammation in either eye; used ocular medication in either eye of any type within 30 days of screening; mean central corneal thickness > 620 µm at screening; any abnormality preventing reliable applanation tonometry of either eye; clinically significant abnormalities in laboratory tests at screening; known hypersensitivity or contraindication to BBs; clinically significant systemic disease; participation in any investigational study within 60 days prior to screening; used any systemic medication that could have a substantial effect in IOP within 30 days prior to screening; women who are pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control Pretreatment differences between groups: baseline demographics of randomized participants were similar between the 2 treatment groups Other description of the overall study population at baseline: not reported |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: 52 activity sites Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02558374 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: no Year of publication accepted: 2019 Year of study initiation (participants screening, enrollment and treatment): 2015 |
|
| Notes | ROCKET‐4; the citation links to a prior conference abstract whereas the full text was published in Khouri 2019 with a different title. | |
Lewis 2016.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 298 participants Number of participants (eyes) randomized per group: netarsudil 0.01%/latanoprost 0.005%: 74; netarsudil 0.02%/latanoprost 0.005%: 73; latanoprost 0.005%: 73; netarsudil 0.02%: 78 participants Total number of participants (eyes) lost to follow‐up: 2 participants Number of participants (eyes) lost to follow‐up per group: netarsudil 0.01%/latanoprost 0.005%: 1; netarsudil 0.02%/latanoprost 0.005%: 1; latanoprost 0.005%: 0; netarsudil 0.02%: 0 participant Power calculation and sample size consideration reported: yes Planned length of follow‐up: 28 days Actual length of follow‐up: 28 days How missing outcome data were handled: exclusion Was the trial single/double/triple‐masked: double‐masked Was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: mITT Duration of washout for each drug class before interventions began: 28 days for BB, prostaglandins and the dorzolamide‐timolol fixed combination; 14 days for alfa‐ and alfa/beta‐agonists, 5 days for miotics and oral or topical carbonic anhydrase inhibitors, and 3 days if participants were receiving no IOP‐lowering therapy (based on Lewis 2016). |
|
| Participants |
Baseline characteristics Netarsudil 0.01%/latanoprost 0.005% (PG‐324), once per day
Netarsudil 0.02%/latanoprost 0.005% (PG‐324), once per day
Latanoprost 0.005%, once per day
Netarsudil 0.02%, once per day
Overall
Inclusion criteria: aged ≥ 18 years; diagnosis of OAG or OHT; corrected visual acuity in each eye equivalent to 20/200 or better; able and willing to give signed informed consent and follow study instructions Exclusion criteria: ophthalmic: glaucoma: pseudoexfoliation or pigment dispersion component, history of angle closure, or narrow angles; IOP >36 mmHg; known hypersensitivity to any component of the formulation, latanoprost or to topical anesthetics; previous glaucoma intraocular surgery or glaucoma laser procedures in study eye(s); refractive surgery in study eye(s); ocular trauma within the 6 months prior to screening, or ocular surgery or laser treatment within the 3 months prior to screening; evidence of ocular infection and inflammation; clinically significant ocular disease, which might interfere with the study, including glaucomatous damage so severe that washout of ocular hypotensive medications for 1 month is not judged safe; central corneal thickness > 600 μm; any abnormality preventing reliable applanation tonometry of either eye. Systemic: clinically significant abnormalities (as determined by the investigator) in laboratory tests at screening; clinically significant systemic disease; participation in any investigational study within 30 days prior to screening; changes in systemic medication; women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control Pretreatment characteristics between groups: no clinically or statistically significant differences among treatment groups (Table 1, Lewis 2016) Other description of the overall study population at baseline: the population was 79% Caucasian (236/298), 18% African–American (54/298), 2% Asian (7/298) and 0.3% Native American (1/298). 22% (64/298) of participants self‐identified as Hispanic. The most frequent iris color was brown/black (62%, 184/298), followed by blue/grey/green (27%, 79/298) and hazel (12%, 35/298) |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: multicenter, 23 study locations Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02057575 Phase of the trial: phase 2 Current publication reported findings from > 1 trial: no Year of publication accepted: 2015 Year of study initiation (participants screening, enrollment and treatment): 2014 |
|
| Notes | ||
NCT02246764 (ROCKET‐3).
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 93 participants Number of participants (eyes) randomized per group: netarsudil 0.02% once per day: 34; netarsudil 0.02% twice per day: 36; timolol: 23 participants Total number of participants (eyes) lost to follow‐up: 0 Number of participants (eyes) lost to follow‐up per group: netarsudil 0.02% once per day: 0; netarsudil 0.02% twice per day: 0; timolol: 0 participants Power calculation and sample size consideration reported: no Planned length of follow‐up: 12 months Actual length of follow‐up: 12 months How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: quadruple‐masked (NCT) Was the trial an equivalence/superiority/non‐inferiority study: unclear Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: ≥ 4 weeks for participants using PAs or BBs before study entry, ≥ 2 weeks for those using α‐agonists, and ≥ 5 days for those using muscarinic agonists or carbonic anhydrase inhibitors |
|
| Participants |
Baseline characteristics Netarsudil 0.02%, once per day
Netarsudil 0.02%, twice per day
Timolol 0.5%, twice per day
Overall
Inclusion criteria: aged ≥ 19 years; diagnosis of OAG or OHT; unmedicated (post‐washout) IOP > 20 mmHg and < 27 mmHg in the study eye at 2 qualification visits (8 a.m.), 2–7 days apart. At second qualification visit, IOP > 17 mmHg and < 27 mmHg at 10 a.m. and 4 p.m.(in the same eye); corrected visual acuity in each eye +1.0 logMAR or better by ETDRS in each eye (equivalent to 20/200); able and willing to give signed informed consent and follow study instructions Excluded criteria: glaucoma: pseudoexfoliation or pigment dispersion component, history of angle closure, or narrow angles. Note: previous laser peripheral iridotomy is NOT acceptable; IOP ≥ 27 mmHg (unmedicated) in both eyes or use of > 2 ocular hypotensive medications within 30 days of screening. Note: fixed dose combinations count as 2 medications; known hypersensitivity to any component of the formulations to be used (benzalkonium chloride, etc.), to topical anesthetics or BBs; previous glaucoma intraocular surgery or glaucoma laser procedures in either eye; refractive surgery in either eye; ocular trauma in either eye within the 6 months prior to screening, or ocular surgery or non‐refractive laser treatment within the 3 months prior to screening; recent or current evidence of ocular infection or inflammation in either eye; current evidence of clinically significant blepharitis, conjunctivitis or a history of herpes simplex or zoster keratitis at screening in either eye; ocular medication in either eye of any type within 30 days of screening; clinically significant ocular disease in either eye (e.g. corneal edema, uveitis, severe keratoconjunctivitis sicca) which might interfere with the study, including glaucomatous damage so severe that washout of ocular hypotensive medications for 1 month is not judged safe; central corneal thickness in either eye > 600 µm at screening; any abnormality in either eye preventing reliable applanation tonometry of either eye. Systemic: clinically relevant abnormalities (as determined by the investigator) in laboratory tests at screening which may impact the study; known hypersensitivity or contraindication to BBs (e.g. chronic obstructive pulmonary disease or bronchial asthma; abnormally low blood pressure or heart rate; second or third‐degree heart block or congestive heart failure; severe diabetes); clinically significant systemic disease (e.g. uncontrolled diabetes, myasthenia gravis, hepatic, renal, endocrine or cardiovascular disorders) which might interfere with the study; participation in any investigational study within 30 days prior to screening; changes of systemic medication that could have an effect on IOP within 30 days prior to screening, or anticipated during the study; women of child‐bearing potential who are pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control. An adult woman was considered of child‐bearing potential unless she was 1 year postmenopausal or 3 months postsurgical sterilization. All females of child‐bearing potential must have had a negative urine pregnancy test result at the screening examination and must not have intended to become pregnant during the study Pretreatment characteristics between groups: no formal comparison results were shown Other description of the overall study population at baseline: not reported |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: multicenter Comments: ROCKET‐3 (a safety trial); data extracted based on the NCT record; early termination due to slow recruitment Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02246764 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: yes Year of publication accepted: 2018 (reported) Year of study initiation (participants screening, enrollment and treatment): 2014 |
|
| Notes | ||
Peace 2021.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 12 participants Number of participants (eyes) randomized per group: placebo: 8, netarsudil 0.02%: 4 participants Total number of participants (eyes) lost to follow‐up: 0 participants Number of participants (eyes) lost to follow‐up per group: placebo: 0, netarsudil 0.02%: 0 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 7 days Actual length of follow‐up: 7 days How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: double‐masked (article), quadruple‐masked (NCT) Was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: 4 weeks for PAs and BBs; 2 weeks for adrenergic agonists (including alfa‐agonists); or 5 days for muscarinic agonists and carbonic anhydrase inhibitors (topical or oral formulations) |
|
| Participants |
Baseline characteristics Placebo
Netarsudil 0.02%, once per day (8‐10 p.m.)
Overall
Inclusion criteria: aged ≥ 18 years; ocular hypertension or OAG in both eyes; unmedicated IOP > 17 mmHg in 1 or both eyes and < 30 mmHg in both eyes; corrected visual acuity in each eye equivalent to 20/200 or better; able and willing to give signed informed consent and follow study instructions Exclusion criteria: glaucoma with pseudoexfoliation or pigment dispersion component, history of angle closure, narrow angles; IOP ≥ 30 mmHg; use of ocular medications within 30 days; known hypersensitivity to any component of the test formulations or to medications used routinely during a clinical eye examination; previous eye surgery (other than cataract); ocular trauma within 6 months; clinically significant ocular disease that might interfere with the study; central corneal thickness > 620 µm Pretreatment characteristics between groups: no statistical comparisons reported (Table 2) Other description of the overall study population at baseline: women and men were equally distributed in each treatment group, there were more participants aged ≥ 65 years (66.7%) than < 65 years (33.3%), and a higher percentage of participants were black or African American (75.0%) than any other race (25.0%) |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie pharmaceuticals Country: USA Setting: single‐center Comments: IOP data: mean nocturnal IOP from NCT report; cup–disc ratio Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02874846 Phase of the trial (phase 2/phase 3/unclear): phase 2 Current publication reported findings from > 1 trial: no Year of publication accepted: 2020 Year of study initiation (participants screening, enrollment and treatment): 2016 |
|
| Notes | ||
Serle 2018 (ROCKET‐1).
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 411 participants Number of participants (eyes) randomized per group: netarsudil: 202, timolol: 209 participants Total number of participants (eyes) lost to follow‐up: 1 participant Number of participants (eyes) lost to follow‐up per group: netarsudil: 0, timolol: 1 participant Power calculation and sample size consideration reported: yes Planned length of follow‐up: 3 months Actual length of follow‐up: 3 months How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: double‐masked (article), quadruple‐masked (NCT) Was the trial an equivalence/superiority/non‐inferiority study: non‐inferiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: PP Duration of washout for each drug class before interventions began: 28 days for BBs, prostaglandins and the dorzolamide‐timolol fixed combination; 14 days for alfa‐ and alfa/beta‐agonists, 5 days for miotics and oral or topical carbonic anhydrase inhibitors, and 3 days if participants were receiving no IOP‐lowering therapy (based on Serle 2018). |
|
| Participants |
Baseline characteristics Netarsudil 0.02%, once per day (p.m.)
Timolol 0.5%, twice per day
Overall
Inclusion criteria: aged 0–2 years or ≥ 18 years; diagnosis of OAG or OHT; unmedicated (post‐washout) IOP > 20 mmHg and < 27 mmHg in the study eye at 2 qualification visits; corrected visual acuity in each eye equivalent to 20/200; able and willing to give signed informed consent (parent or guardian consent for children) and follow study instructions Exclusion criteria: glaucoma: pseudoexfoliation or pigment dispersion component, history of angle closure, or narrow angles. Note: previous laser peripheral iridotomy was NOT acceptable; IOP ≥ 27 mmHg (unmedicated) in both eyes (individuals who are excluded for this criterion were not allowed to attempt requalification), or use of > 2 ocular hypotensive medications within 30 days of screening. Note: fixed‐dose combinations count as 2 medications; known hypersensitivity to any component of the formulations to be used (benzalkonium chloride, etc.), to topical anesthetics or BBs; previous glaucoma intraocular surgery or glaucoma laser procedures in either eye; refractive surgery in either eye; ocular trauma in either eye within the 6 months prior to screening, or ocular surgery or non‐refractive laser treatment within the 3 months prior to screening; recent or current evidence of ocular infection or inflammation in either eye; current evidence of clinically significant blepharitis, conjunctivitis or history of herpes simplex or zoster keratitis at screening in either eye; ocular medication in either eye of any type within 30 days of screening; clinically significant ocular disease in either eye (e.g. corneal edema, uveitis, severe keratoconjunctivitis sicca) which might interfere with the study, including glaucomatous damage so severe that washout of ocular hypotensive medications for 1 month is not judged safe; central corneal thickness in either eye > 600 µm at screening; any abnormality in either eye preventing reliable applanation tonometry of either eye. Systemic: clinically relevant abnormalities (as determined by the investigator) in laboratory tests at screening which may impact the study; known hypersensitivity or contraindication to BBs (e.g. chronic obstructive pulmonary disease or bronchial asthma; abnormally low blood pressure or heart rate; second‐ or third‐degree heart block or congestive heart failure; severe diabetes); clinically significant systemic disease (e.g. uncontrolled diabetes, myasthenia gravis, hepatic, renal, endocrine or cardiovascular disorders) which might interfere with the study; participation in any investigational study within 30 days prior to screening; changes of systemic medication that could have an effect on IOP within 30 days prior to screening, or anticipated during the study; women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control. An adult woman was considered of child‐bearing potential unless she was 1 year postmenopausal or 3 months postsurgical sterilization. All females of child‐bearing potential must have had a negative urine pregnancy test result at the screening examination and must not have intended to become pregnant during the study Pretreatment characteristics between groups: no statistical differences (Table 2, Serle 2018) Other description of the overall study population at baseline: the only characteristic that was statistically significantly different between treatment groups was iris color |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: multicenter Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02207491 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: yes Year of publication accepted: 2017 Year of study initiation (participants screening, enrollment and treatment): 2014 |
|
| Notes | ROCKET‐1 trial results; primary outcome in participants whose baseline IOP < 27 mmHg (PP analysis) | |
Sit 2021.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: eyes Total number of participants (eyes) randomized: 40 eyes Number of participants (eyes) randomized per group: placebo: 20, netarsudil: 20 eyes Total number of participants (eyes) lost to follow‐up: 4 eyes Number of participants (eyes) lost to follow‐up per group: placebo: 2, netarsudil: 2 eyes Power calculation and sample size consideration reported: yes (analysis plan and published study protocol) Planned length of follow‐up: 7 days Actual length of follow‐up: 7 days How missing outcome data were handled: missing data were not imputed Was the trial single/double/triple‐masked: double‐masked Was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: mITT Duration of washout for each drug class before interventions began: ≥ 4 weeks for prostaglandins and BBs, 2 weeks for alfa‐agonists and 5 days for muscarinic agonists and carbonic anhydrase inhibitors |
|
| Participants |
Baseline characteristics Overall
Inclusion criteria: aged ≥ 18 years; diagnosis of POAG or OHT in both eyes; unmedicated IOP > 20 mmHg and < 30 mmHg in both eyes at first qualification visit; BCVA equivalent to 20/200 Snellen or better; able to give informed consent and follow study instructions Excluded criteria: clinically significant ocular disease; pseudoexfoliation or pigment dispersion component glaucoma, history of angle closure glaucoma, or narrow angles; IOP ≥ 30 mmHg in either eye; difference in IOP between eyes > 4 mmHg at qualification visit; use of > 2 ocular hypotensive medications within 30 days of screening; known hypersensitivity to any component of the formulation; previous glaucoma surgery or refractive surgery; keratorefractive surgery in either eye; report of ocular injury in either eye within the 6 months prior to screening or ocular or non‐refractive surgery within 3 months prior to screening; recent or current ocular infection or inflammation in either eye; use of ocular medication in either eye of any type within 30 days of screening; mean central corneal thickness > 620 μm in either eye; any abnormality preventing reliable applanation tonometry of either eye; lack of suitable episcleral vein prior to performing episcleral venus pressure measurement (applicable to 1 site only). Systemic: clinically significant abnormalities within 6 weeks prior to screening; clinically significant systemic disease; participation in any investigational study within 60 days prior to screening; use of systemic medication that could have an effect on IOP within 30 days prior to screening; women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control Pretreatment differences between groups: irrelevant for within‐person study |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA Setting: multicenter, 2 clinical sites Online trial registration site: ClinicalTrials.gov Trial registration #: NCT03233308 Phase of the trial (phase 2/phase 3/unclear): phase 2 Current publication reported findings from > 1 trial: no Year of publication accepted: 2021 Year of study initiation (participants screening, enrollment and treatment): 2017 |
|
| Notes | Within‐person study | |
Tanihara 2013.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 2010 participants Number of participants (eyes) randomized per group: placebo: 54; ripasudil 0.1%: 53; ripasudil 0.2%: 54; ripasudil 0.4%: 49 participants Total number of participants (eyes) lost to follow‐up: 7 participants Number of participants (eyes) lost to follow‐up per group: placebo: 2; ripasudil 0.1%: 3; ripasudil 0.2%: 2; ripasudil 0.4%: 0 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 8 weeks Actual length of follow‐up: 8 weeks How missing outcome data were handled: exclusion Was the trial single/double/triple‐masked: double‐masked Was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: mITT Duration of washout for each drug class before interventions began: 4 weeks for prostaglandin and sympatholytic, 2 weeks for other antiglaucoma medication |
|
| Participants |
Baseline characteristics Placebo, twice per day (9 a.m., 9 p.m.)
Ripasudil 0.1%, twice per day (9 a.m., 9 p.m.)
Ripasudil 0.2%, twice per day (9 a.m., 9 p.m.)
Ripasudil 0.4%, twice per day (9 a.m., 9 p.m.)
Overall
Inclusion criteria: men or women (excluding women of child‐bearing potential who were pregnant, breast‐feeding or planning a pregnancy) with POAG or OHT; aged ≥ 20 years; untreated IOP (after washout) ≥ 21 mmHg and IOP differences were within 3 mmHg in ≥ 1 eye at 2 eligibility visits (9 a.m.), 2–14 days apart; untreated IOP was < 35 mmHg in both eyes Exclusion criteria: people with narrow angles defined as grade 2 or less of the Shaffer classification by gonioscopy or who had undergone ocular surgery (other than cataract surgery > 1 year ago, retinal laser treatment and yttrium–aluminum–garnet laser posterior capsulotomy > 90 days ago, and eyelid surgery > 120 days ago); severe visual field defects or with a corrected visual acuity of worse than 0.3 (decimal fraction) in either eye During the trial, participants were prohibited from receiving other IOP‐lowering agents, receiving any ophthalmic agents (excluding artificial tears) or steroids, wearing contact lenses and changing dosages of any systemic medications that may affect IOP Pretreatment characteristics between groups: no statistical comparisons are provided in Table 1 of Tanihara 2013 Other description of the overall study population at baseline: none |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Kowa company, Ltd Country: Japan Setting: multicenter, 29 clinical centers Online trial registration site: www.clinicaltrials.jp Trial registration #: JAPIC101015 Phase of the trial (phase 2/phase 3/unclear): phase 2a Current publication reported findings from > 1 trial: no Year of publication accepted: 2013 Year of study initiation (participants screening, enrollment and treatment): 2010 |
|
| Notes | Adverse events that lead to withdrawal from the trial were iron‐deficiency anemia (1 in placebo group), retinal tear (1 in placebo group), fracture of the femoral neck (1 in 0.1% group), asthma (in 0.2% and 0.4% groups), photophobia (in 0.1% group) | |
Tanihara 2015a.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: cross‐over Unit of randomization: participant Total number of participants (eyes) randomized: 28 participants Number of participants (eyes) randomized per group: 28 participants Total number of participants (eyes) lost to follow‐up: 0 participants Number of participants (eyes) lost to follow‐up per group: 0 participants Power calculation and sample size consideration reported: no Planned length of follow‐up: 24 hours Actual length of follow‐up: 24 hours How missing outcome data were handled: NA Was the trial single/double/triple‐masked: open‐label Was the trial an equivalence/superiority/non‐inferiority study: equivalence Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: 4 weeks for prostaglandin and BBs; 2 weeks for other IOP‐lowering agents; 5–30 days between cross‐over periods |
|
| Participants |
Baseline characteristics Group C
Inclusion criteria: Japanese men or non‐pregnant women diagnosed with POAG or OHT, aged 20–64 years; untreated or post‐washout IOP levels ≥ 21 mmHg in 1 or both eyes at the screening visit Exclusion criteria: people with ocular disease (other than POAG or OHT) or ocular surgery (other than eyelid surgery performed < 120 days prior to screening); severe visual field defects or with a corrected visual acuity of worse than decimal visual acuity 0.3 (commensurate with 0.5 logMAR); people with IOP ≥ 30 mmHg excluded in terms of safety concerns During the study, participants were prohibited from receiving other IOP‐lowering medications, any ophthalmic medications or steroids, wearing contact lenses and changing dosages of any systemic medications Pretreatment characteristics between groups: not relevant |
|
| Interventions |
Intervention characteristics
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: sponsored by Kowa Company, Ltd, Nagoya, Japan. Sponsor participated in study design, conducting the study, data collection, data management, data analysis, review and approval of the manuscript Country: Japan Setting: 3 clinical pharmacology facilities Online trial registration site: www.clinicaltrials.jp Trial registration #: study no. 090708 Phase of the trial (phase 2/phase 3/unclear): phase 2 Current publication reported findings from > 1 trial: no Year of publication accepted: 2014 Year of study initiation (participants screening, enrollment and treatment): 2009 |
|
| Notes | A phase 2, cross‐over, 3‐arm trial of ripasudil 0.2%, 0.4% vs placebo. Only data from group C in dosing period 1 (placebo) and 2 (0.2%) are eligible for inclusion. | |
Tanihara 2015b.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 208 participants Number of participants (eyes) randomized per group: timolol: 104, ripasudil + timolol: 104 participants Total number of participants (eyes) lost to follow‐up: 6 participants Number of participants (eyes) lost to follow‐up per group: timolol: 4, ripasudil + timolol: 2 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 8 weeks Actual length of follow‐up: 8 weeks How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: double‐masked Was the trial an equivalence/superiority/non‐inferiority study: equivalence Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: none but a 4‐week of run‐in period with the intervention medication was applied |
|
| Participants |
Baseline characteristics Timolol 0.5%, twice per day (9 a.m. and 9 p.m.)
Ripasudil 0.4% + timolol 0.5% (FDC), twice per day (9 a.m. and 9 p.m.)
Overall
Inclusion criteria: men or women with POAG or OHT; aged ≥ 20 years; IOP after run‐in periods (treated with timolol, 0.5%, twice per day or latanoprost, 0.005%, once per day for ≥ 4 weeks) ≥ 18 mmHg, IOP difference within 3 mmHg in ≥ 1 eye at 2 eligibility visits (9 a.m.) 2–14 days apart, and treated IOP < 35 mmHg in both eyes Exclusion criteria: people with narrow angles defined as grade 2 or less by the Shaffer classification by gonioscopy or who had undergone ocular surgery (other than cataract surgery > 1 year ago, retinal laser treatment or neodymium‐doped yttrium aluminum garnet laser posterior capsulotomy > 90 days ago, and eyelid surgery > 120 days ago) in either eye; severe visual field defects or a corrected visual acuity of worse than 20/70 in either eye Pretreatment characteristics between groups: no significant differences in table 1 of Tanihara 2015b |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Kowa Company Country: Japan Setting: multicenter (29 Japanese clinical centers) Online trial registration site: www.clinicaltrials.jp Trial registration #: JAPIC111701 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: yes Year of publication accepted: 2015 Year of study initiation (participants screening, enrollment and treatment): 2011 |
|
| Notes | Data extracted for K‐115 (ripasudil) + timolol vs placebo + timolol | |
Tanihara 2015c.
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 205 participants Number of participants (eyes) randomized per group: placebo + latanoprost: 103, ripasudil + latanoprost: 102 participants Total number of participants (eyes) lost to follow‐up: 9 participants Number of participants (eyes) lost to follow‐up per group: placebo + latanoprost: 6, ripasudil + latanoprost: 3 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 8 weeks Actual length of follow‐up: 8 weeks How missing outcome data were handled: unclear Was the trial single/double/triple‐masked: double‐masked Was the trial an equivalence/superiority/non‐inferiority study: equivalence Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: none but a 4‐week run‐in period for latanoprost was applied |
|
| Participants |
Baseline characteristics Placebo, twice per day (9 a.m. and 9 p.m.) + latanoprost 0.005%, once per day
Ripasudil 0.4%, twice per day (9 a.m. and 9 p.m.) + latanoprost 0.005%, once per day
Overall
Inclusion criteria: men or women with POAG or OHT; aged ≥ 20 years; IOP after run‐in periods (treated with timolol, 0.5% twice per day or latanoprost, 0.005%, once per day for ≥ 4 weeks) of ≥ 18 mmHg; IOP difference within 3 mmHg in ≥ 1 eye at 2 eligibility visits (9 a.m.) 2–14 days apart, and treated IOP < 35 mmHg in both eyes Exclusion criteria: people with narrow angles defined as grade 2 or less by the Shaffer classification by gonioscopy or who had undergone ocular surgery (other than cataract surgery > 1 year ago, retinal laser treatment or neodymium‐doped yttrium aluminum garnet laser posterior capsulotomy > 90 days ago, and eyelid surgery > 120 days ago) in either eye; severe visual field defects or a corrected visual acuity of worse than 20/70 in either eye Pretreatment characteristics between groups: no significant differences in Table 1 of Tanihara 2015c Other description of the overall study population at baseline: NA |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Kowa Company Country: Japan Setting: multicenter (29 Japanese clinical centers) Online trial registration site: www.clinicaltrials.jp Trial registration #: JAPIC111700 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: yes Year of publication accepted: 2015 Year of study initiation (participants screening, enrollment and treatment): 2011 |
|
| Notes | Data extracted for K‐115 (ripasudil) + latanoprost vs placebo + latanoprost | |
Walters 2019 (MERCURY‐2).
| Study characteristics | ||
| Methods |
Study design: randomized controlled trial Study grouping: parallel group Unit of randomization: participant Total number of participants (eyes) randomized: 750 participants Number of participants (eyes) randomized per group: netarsudil: 245, latanoprost: 255, netarsudil/latanoprost: 250 participants Total number of participants (eyes) lost to follow‐up: 3 participants Number of participants (eyes) lost to follow‐up per group: netarsudil: 1, latanoprost: 0, netarsudil/latanoprost: 2 participants Power calculation and sample size consideration reported: yes Planned length of follow‐up: 3 months Actual length of follow‐up: 3 months How missing outcome data were handled: missing data were imputed using Markov Chain Monte Carlo multiple imputation techniques Was the trial single/double/triple‐masked: double‐masked Was the trial an equivalence/superiority/non‐inferiority study: superiority Extracted outcome results were based on ITT/mITT/CC/PP/PT analysis: ITT Duration of washout for each drug class before interventions began: 4 weeks for PAs or BBs, 2 weeks for adrenergic agonists and 5 days for muscarinic agonists or carbonic anhydrase inhibitors |
|
| Participants |
Baseline characteristics Netarsudil 0.02%, once per day (p.m.)
Latanoprost 0.005%, once per day (p.m.)
Netarsudil 0.02%/latanoprost 0.005% (FDC), once per day (p.m.)
Overall
Inclusion criteria: ≥ 18 years (≥ 19 years in Canada); diagnosis of OAG or OHT in both eyes; unmedicated IOP > 20 mmHg and < 36 mmHg in both eyes at 2 qualification visits; BCVA equivalent to 20/200 Snellen or better; able to give informed consent and follow study instructions Excluded criteria: clinically significant ocular disease; pseudoexfoliation or pigment dispersion component glaucoma, history of angle closure or narrow angles; unmedicated IOP ≥ 36 mmHg in either eye or use of > 2 ocular hypotensive medications within 30 days of screening; known hypersensitivity to any component of the formulation or latanoprost; previous glaucoma surgery or refractive surgery; ocular trauma within 6 months prior to screening; any ocular surgery or non‐refractive laser treatment within 3 months prior to screening; recent or current ocular infection or inflammation in either eye; use of ocular medication in either eye of any type within 30 days of screening and throughout of the study; mean central corneal thickness > 620 µm at screening in either eye; any abnormality preventing reliable applanation tonometry of either eye. Systemic: clinically significant abnormalities in laboratory tests at screening; clinically significant systemic diseases; participation in any investigational study within 60 days prior to screening; systemic medication that could have had a substantial effect on IOP within 30 days prior to screening, or anticipated to be used during the study; women of child‐bearing potential who were pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable form of birth control Pretreatment characteristics between groups: baseline demographics similar across the 3 treatment arms (Table 1 of Walters 2019 (MERCURY‐2)) Other description of the overall study population at baseline: 91.3% of participants completed 3 months of treatment (netarsudil: 90.2%, latanoprost: 89.4%, netarsudil/latanoprost: 94.4%) |
|
| Interventions |
|
|
| Outcomes |
Primary outcome reported (time points assessed and reported)
Other outcomes reported (time points assessed and reported)
|
|
| Identification |
Sponsorship source: Aerie Pharmaceuticals Country: USA and Canada Setting: 60 active sites Online trial registration site: ClinicalTrials.gov Trial registration #: NCT02674854 Phase of the trial (phase 2/phase 3/unclear): phase 3 Current publication reported findings from > 1 trial: no Year of publication accepted: 2019 Year of study initiation (participants screening, enrollment and treatment): 2016 |
|
| Notes | IOP data were extracted from ClinicalTrials.gov | |
BB: beta‐blocker; BCVA: best‐corrected visual acuity; CC: complete case; ETDRS: Early Treatment Diabetic Retinopathy Study; FDC: fixed‐dose compound; IOP: intraocular pressure; ITT: intention to treat; mITT: modified intention to treat; OAG: open‐angle glaucoma; OHT: ocular hypertension; PA: prostaglandin analog; POAG: primary open‐angle glaucoma; PP: per protocol; PT: per treatment; ROKi: Rho kinase inhibitor; SD: standard deviation.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Dubiner 2014 | Ineligible study design: dose‐finding study |
| Inazaki 2017 | Ineligible study design: observational study |
| Komizo 2019 | Ineligible study design: observational study |
| Kopczynski 2018 | Ineligible study design: review article |
| NCT02173223 | Ineligible study population: people with uncontrolled advanced glaucoma with prior failed trabeculectomy or tube shunt |
| NCT03808688 | Ineligible study design: observational study |
| Sakamoto 2019 | Ineligible study design: observational study |
| Tanihara 2016 | Ineligible study design: observational study |
| UMIN000026228 | Ineligible study design: observational study |
| Weiss 2013 | Ineligible intervention: not the dosage of interest |
Characteristics of studies awaiting classification [ordered by study ID]
CTRI/2018/04/013091.
| Methods | Randomized, parallel group, active controlled trial Method of generating randomization sequence: computer‐generated randomization Method of allocation concealment: sequentially numbered, sealed, opaque envelopes Masking: participant and investigator (double‐masked) |
| Participants |
Inclusion criteria: people with IOP 22–30 mmHg at time of screening in any 1 eye. In case both eyes are affected then the eye fulfilling the criteria will be considered for the evaluations. If both eyes fulfill the criteria, then the right eye will be considered for the study; voluntary willingness of person to give written informed consent prior to participation in trial Exclusion criteria: women who are not using an effective means of birth control or who are pregnant or breast‐feeding; any severe or advanced cases of glaucoma; Shaffer angle grade < 2 in either eye (range, 0 [complete or partial closure] to 3 [wide open angle, > 20]), as measured by gonioscopy; people who are blind or have a single eye; severe central visual field loss in either eye measured by perimetry; cup–disk ratio > 0.80; scheduled to undergo eye surgery during the study; history of/current chronic, recurrent or severe inflammatory eye disease (e.g. scleritis, uveitis, herpes keratitis) or current other severe ocular pathology (including severe dry eye) that would affect the conduct of the study; history of ocular trauma or any other intraocular surgery within the past 6 months in either eye; current/history of ocular infection or inflammation within the past 3 months as determined by patient history or eye examination, or both |
| Interventions |
|
| Outcomes |
Primary outcome
Secondary outcome
|
| Notes | Phase 3; date completed: 12 June 2018 |
CTRI/2020/01/022619.
| Methods | Randomized, parallel group, active controlled trial Method of generating randomization sequence: computer‐generated randomization Method of allocation concealment: sequentially numbered, sealed, opaque envelopes Masking: participant and investigator (double‐masked) |
| Participants |
Inclusion criteria: men and women aged 18–65 years; newly diagnosed primary open‐angle glaucoma/ocular hypertension in 1 or both eyes confirmed by Goldmann applanation tonometry; IOP 22–27 mmHg at time of screening in any 1 eye. In case both the eyes of a single subject are affected then the eye fulfilling the criteria will be considered for the evaluations. If both eyes are fulfilling the criteria, then the right eye will be considered for the study; visual acuity ≥ 6/60; willingness to give written informed consent prior to participation in trial Exclusion criteria: any of the following. Ophthalmic: any severe or advanced cases of glaucoma; Shaffer angle grade ≤ 2 in either eye, as measured by gonioscopy; with pseudoexfoliation or pigment dispersion component glaucoma; blind or have 1 eye; severe central visual field loss in either eye measured by perimetry; cup–disk ratio > 0.80; current or history within 3 months prior to baseline of significant ocular disease, e.g. corneal edema, uveitis, ocular infection or ocular trauma in either eye; current corneal abnormalities that would prevent accurate IOP readings with the Goldmann applanation tonometer; history of any ocular surgery in either eye (e.g. peripheral iridotomy, glaucoma incisional or laser surgery, refractive surgery) or are planning to undergo any ocular surgery during the study period; central corneal thickness > 600 mm in either eye; history of clinically relevant or progressive retinal disease such as retinal degeneration, diabetic retinopathy or retinal detachment; contact lens user; contraindication or hypersensitivity to any component of study medication; history of chronic use of any ocular medication; local administration of corticosteroids injections in the eye. Others: use within 1 month prior to baseline of systemic corticosteroid or high‐dose salicylate therapy or oral carbonic anhydrase inhibitor; pregnant, breast‐feeding or planning a pregnancy; of child‐bearing potential who do not agree to utilize an adequate form of birth control; history of CVS, hepatic, psychiatric, cancer or renal diseases that could be considered significant for enrollment in the study; history of bronchial asthma, bronchial hyper‐reactivity or severe COPD that would preclude the safe administration of a topical beta‐blocker; people receiving high‐dose (> 1 g per day) salicylate, topical as well as oral beta‐blockers, alpha agonists and blockers, angiotensin‐converting enzyme inhibitors and calcium channel blockers, systemic administration of corticosteroids or immunosuppressive agents; participation in any clinical study within 30 days prior to entry into the study; unwilling to comply with the study protocol and does not provide written informed consent to participation; with type 1 and uncontrolled type 2 diabetes mellitus (i.e. defined as glycosylated hemoglobin > 7%) |
| Interventions |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Phase 3 |
NCT03284853.
| Methods | Randomized parallel trial Masking: participant, care provider, investigator (triple‐masked) |
| Participants |
Inclusion criteria: aged ≥ 18 years; diagnosis of open‐angle glaucoma or ocular hypertension; participants insufficiently controlled or considered in need for combination therapy; medicated IOP ≥ 17 mmHg and < 28 mmHg in both eyes at screening visit; unmedicated (post‐washout) IOP > 20 mmHg in 1 or both eyes and < 36 mmHg in both eyes at 2 qualification visits. At second qualification visit to have IOP > 17 mmHg in 1 or both eyes and < 36 mmHg in both eyes. If only 1 eye qualifies at second qualification visit it MUST be the same eye qualified on the first visit (this will be known as the study eye); best corrected visual acuity +1.0 logMAR or better; willing and able to give informed consent and follow instructions; women must be either of non‐child‐bearing potential, or women with child‐bearing potential and men with reproductive potential must be willing to practice acceptable methods of birth control during study; women of child‐bearing potential must have a negative urine pregnancy test within 7 days of first dose of study treatment and agree to use highly effective birth control from time of randomization and for 3 months after last dose of study medication; men with a female partner of child‐bearing potential must have either had a prior vasectomy or agree to use effective birth control from time of randomization and for 3 months following the last dose of study medication; in France, a person will be eligible for inclusion only if either affiliated to or as a beneficiary of a social security number Exclusion criteria: ophthalmic: clinically significant ocular disease which might interfere with interpretation of the study results, including people with glaucomatous damage so severe that washout of ocular hypotensive medication for ≥ 4 weeks would not be judged as safe; pseudoexfoliation or pigment dispersion glaucoma, history of narrow angles or angle closure glaucoma. Previous laser peripheral iridotomy is not permitted; IOP (note: fixed‐dose combinations, for the purpose of this exclusion criterion count as 1 medication); however, people currently taking 2 fixed‐dose combination products are excluded; treatment‐naïve people; prior treatment with GANFORT topical eye drops, where person's IOP did not achieve target IOP and was considered a failure or an insufficient response; currently (prior to screening visit) being treated with GANFORT; known hypersensitivity to any component of the investigational formulations to be used; previous glaucoma intraocular surgery; refractive surgery in either eye; ocular trauma within 6 months prior to screening, or ocular surgery or non‐refractive laser treatment within 3 months prior to screening; recent or current evidence of ocular infection or inflammation in either eye; use of ocular medication in either eye within 30 days of screening and throughout the study except for permitted ocular medication (which must be the same medication for 30 days prior to screening) as prescribed by the investigator; mean central corneal thickness > 620 μm at screening; any abnormality preventing reliable Goldmann applanation tonometry of either eye. Systemic: clinically significant abnormalities in laboratory tests at screening; known hypersensitivity or contraindication to GANFORT and to beta‐blockers; clinically significant systemic disease that might interfere with the study; participation in any investigational study within 30 days prior to screening; systemic medication (including corticosteroids) that could have a substantial effect on IOP which HAVE NOT been maintained at a consistent dose and regimen within 30 days prior to screening, and are anticipated to change in dose or regimen (or both) during the study; use of topical steroids containing medications on the face or in or around the eyes; women of child‐bearing potential who are pregnant, breast‐feeding, planning a pregnancy or not using a medically acceptable and highly effective form of birth control; vulnerable people such as minors, adults under legal protection or unable to express their consent; people deprived of liberty or people subject to psychiatric care |
| Interventions |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Phase 3; date completed: 6 November 2020; results submitted to ClinicalTrials.gov on 5 November 2021 |
COPD: chronic obstructive pulmonary disease; CVS: cardiovascular surgical; IOP: intraocular pressure.
Characteristics of ongoing studies [ordered by study ID]
JapicCTI‐194920.
| Study name | A comparison study on WP‐1303 and ripasudil ophthalmic solution in patients with primary open‐angle glaucoma (POAG) or ocular hypertension (OH) |
| Methods | Study type: interventional Study design: multicenter, randomized, single‐masked, parallel‐group |
| Participants |
Inclusion criteria: men and women with POAG or OHT; IOP < 35 mmHg for both eyes; 1 eye is ≥ 21 mmHg; aged > 20 years Exclusion criteria: visual acuity < 0.3; presence of active eye disease (e.g. uveitis, ocular infection, severe dry eye); pregnant or lactating women or women who desire to be pregnant; people judged inappropriate to participate in the study by investigator or subinvestigator |
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcome
|
| Starting date | 6 September 2019 |
| Contact information | Wakamoto Pharmaceutical Co., Ltd. Clinical Development. Address: 2‐2, Nihonnbashi Honcho 2‐chome, Chuo‐ku Tokyo. Telephone: +81‐3‐3279‐0370 |
| Notes | Study terminated on 25 December 2019. |
UMIN000019017.
| Study name | Efficacy of Rho‐kinase inhibitor ophthalmic solution on bleb formation after trabeculectomy, a randomized parallel study |
| Methods | Basic design: parallel Randomization: randomized Randomization unit: participants Masking: open‐label |
| Participants |
Inclusion criteria: men and women who have trabeculectomy as the therapy of open‐angle glaucoma which is not sufficiently controlled; age ≥ 20 years Key exclusion criteria: people who had intraocular surgeries within 6 months; IOP unable to be measured by Goldmann applanation tonometer; conjunctival surgeries (including glaucoma surgeries); allergy to the ingredients of Rho‐kinase inhibitor ophthalmic solution; used Rho‐kinase inhibitor ophthalmic solution before surgery; pregnant or breast‐feeding; people unsuitable for study according to doctor. |
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Starting date | 1 November 2015 (no longer recruiting); last follow‐up date: 30 September 2022 |
| Contact information | okumic@hiroshima‐u.ac.jp |
| Notes | Study enrolled 112 participants; results of the study were unpublished. |
IOP: intraocular pressure; OHT: ocular hypertension; POAG: primary open‐angle glaucoma.
Differences between protocol and review
Types of participants
Normal‐tension glaucoma was considered as a subgroup of POAG. As some studies had open‐angle glaucoma (OAG) and not primary open‐angle glaucoma (POAG) as inclusion criteria, the review team decided to include studies both investigating OAG and POAG. This was not clearly stated in the protocol.
Types of interventions
Various trials evaluated different non‐clinically prescribed doses of marketed ROKis as well as non‐marketed ROKis such as AR‐12286, SNJ‐1656, AMA0076 and WP‐1303. However, due to the modest amount and heterogeneous nature of the clinical evidence, the review team decided not to evaluate the effects of non‐therapeutic doses of commercial as well as non‐commercial ROKis.
Outcomes
We specified in the protocol that we would collect adverse effects (AEs) as measured in dichotomous measures at the longest follow‐up. However, with the variable lengths of follow‐up across studies, we decided to calculate incidence rates of AEs by averaging the numbers of adverse events reported over the relevant person‐periods exposed to the treatment (in 100 person‐months). The protocol also specified that we would collect ocular AEs related to the intervention drug. Yet some studies did not clarify whether the reported ocular AEs were considered related or unrelated to the intervention drug. Thus, the numbers extracted for analysis were for all ocular AEs and for studies that did not report whether it was directly related to the intervention drug. Furthermore, if a given AE was classified as an ocular AE or serious AE by the review team, the AE would be regarded as so independently of the statement of the trial investigators.
Measures of treatment effect
For categorical data with dichotomous measures, the protocol specified that the treatment effect would be estimated using risk ratios (RR) with 95% CIs. This was only relevant for AE data. As the studies varied in follow‐up period, we calculated the AE incidence RDs with 95% CIs instead. None of the included studies reported continuous measures obtained from different measurement tools.
Unit of analysis issues
None of the eligible studies reported data for both eyes of participants treated with the same intervention. Hence, no estimates that accounted for the between‐eye correlation were made. Of the eligible studies, only one reported data from participants receiving different interventions in each eye (Sit 2021). Therefore, the review team decided not to access the within‐person effect as otherwise stated in the protocol. Rather, we performed a sensitivity analysis to evaluate the single study effect (Analysis 1.2).
Subgroup analysis and investigation of heterogeneity
We did not perform a subgroup analysis based on population, as the included studies either did not include both subgroups or did not clearly distinguish between OAG and OHT. We performed a subgroup analysis based on the different types of ROKis.
We did not evaluate responses of the highest and lowest dose of non‐therapeutic doses of ROKi due to limited data.
Reporting of outcomes
For the secondary outcomes 'change in IOP from baseline', the protocol stated that data from the longest available follow‐up would be reported. However, it should be specified that 'the longest available follow‐up' is the follow‐up period prespecified for the primary and secondary outcomes in the included studies, since the studies were specifically designed for these time points. This was not clearly described in the protocol.
In the protocol, we stated that the outcomes 'change in IOP from baseline' and 'number of adverse events' should be reported at short (less than six months) or medium‐to‐long term (six months or more). However, very few of the included studies reported medium‐to‐long term outcomes. Thus, the review team decided not to report these outcomes based on short‐ and medium‐ to long‐term considerations.
Contributions of authors
JCF, AVS, NK, MK, AA and GV designed, drafted, and wrote the protocol. All authors approved the final protocol.
JCF and AVS carried out the searches, assessed inclusion of publications and extracted data.
JCF and AVS drafted the review.
MK, AA and GV commented on the draft, and JCF adjusted the final draft accordingly.
JVF, AVS, MK, AA and GV approved the published version of the review.
Sources of support
Internal sources
No sources of support provided
External sources
-
National Eye Institute, National Institutes of Health, USA
Cochrane Eyes and Vision US Project, supported by grant UG1EY020522 (PI: Tianjing Li, MD, MHS, PhD)
-
Queen's University Belfast, UK
Gianni Virgili, Co‐ordinating Editor for Cochrane Eyes and Vision’s work is funded by the Centre for Public Health, Queen’s University of Belfast, Northern Ireland.
Declarations of interest
JCF: none.
AVS: none.
MK is on the advisory board or is a consultant for AbbVie, Thea Pharmaceuticals and Santen. Collaborations are not related to this current review.
AA: none.
GV: none known. Dr Virgili is a member of the editorial team and was not involved in the editorial processes.
New
References
References to studies included in this review
Aerie 2017 {published data only}
- NCT03310580. Study of netarsudil ophthalmic solution in Japanese/Japanese-American subjects with open-angle glaucoma or ocular hypertension. clinicaltrials.gov/ct2/show/results/NCT03310580 (first received 16 October 2016).
Araie 2021 {published data only}
- Araie M, Sugiyama K, Aso K, Kanemoto K, Kothapalli K, Kopczynski C, et al. Phase 2 randomized clinical study of netarsudil ophthalmic solution in Japanese patients with primary open-angle glaucoma or ocular hypertension. Advanced Therapies 2021;38(4):1757-75. [DOI: 10.1007/s12325-021-01634-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03844945. Efficacy and systemic safety of netarsudil 0.01%, 0.02%, 0.04% relative to placebo in subjects with open-angle glaucoma or ocular hypertension in Japan. clinicaltrials.gov/ct2/show/NCT03844945 (first received 19 February 2019).
Asrani 2019 (MERCURY‐1) {published data only}
- Asrani S, Bacharach J, Holland E, McKee H, Sheng H, Lewis RA, et al. Fixed-dose combination of netarsudil and latanoprost in ocular hypertension and open-angle glaucoma: pooled efficacy/safety analysis of phase 3 MERCURY-1 and -2. Advanced Therapeutics 2020;37(4):1620-31. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrani S, Robin Al, Serle JB, Lewis RA, Usner DW, Kopczynski CC, et al. Netarsudil/latanoprost fixed-dose combination for elevated intraocular pressure: three-month data from a randomized phase 3 trial. American Journal of Ophthalmology 2019;207:248-57. [DOI: 10.1016/j.ajo.2019.06.016] [DOI] [PubMed] [Google Scholar]
- Brubaker JW, Teymoorian S, Lewis RA, Usner D, McKee HJ, Ramirez N, et al. One year of netarsudil and latanoprost fixed-dose combination for elevated intraocular pressure: Phase 3, randomized MERCURY-1 study. Ophthalmology Glaucoma 2020;3(5):327-38. [DOI: 10.1016/j.ogla.2020.05.008] [DOI] [PubMed] [Google Scholar]
- FDA Regulatory Document. Drug approval package: rocklatan ophthalmic solution. www.accessdata.fda.gov/drugsatfda_docs/nda/2019/208259Orig1s000TOC.cfm (accessed 18 February 2022).
Bacharach 2015 {published data only}
- Bacharach J, Dubiner HB, Levy B, Kopczynski CC, Novack GD. Double-masked, randomized, dose-response study of AR-13324 versus latanoprost in patients with elevated intraocular pressure. Ophthalmology 2015;122(2):302-7. [DOI: 10.1016/j.ophtha.2014.08.022] [DOI] [PubMed] [Google Scholar]
Inoue 2018 {published data only}
- Inoue K, Ishida K, Tomita G. Effectiveness and safety of switching from prostaglandin analog monotherapy to prostaglandin/timolol fixed combination therapy or adding ripasudil. Japanese Journal of Ophthalmology 2018;62(4):508-16. [DOI: ] [DOI] [PubMed] [Google Scholar]
Kahook 2019 (ROCKET‐2) {published data only}
- Kahook MY, Serle JB, Mah FS, Kim T, Raizman MB, Heah T, et al. Long-term safety and ocular hypotensive efficacy evaluation of netarsudil ophthalmic solution: rho kinase elevated IOP treatment trial (ROCKET-2). American Journal of Ophthalmology 2019;200:130-7. [DOI: 10.1016/j.ajo.2019.01.003] [DOI] [PubMed] [Google Scholar]
- Serle JB, Kahook M, Lewis RA, Heah T, Ramirez-Davis N, Kopczynski CC, et al. Long-term safety and ocular hypotensive efficacy of netarsudil ophthalmic solution: the ROCKET-2 study. Investigative Ophthalmology and Visual Science 2018;59:9. [Google Scholar]
- Serle JB, Katz LJ, McLaurin E, Heah T, Ramirez-Davis N, Usner DW, et al. Two phase 3 clinical trials comparing the safety and efficacy of netarsudil to timolol in patients with elevated intraocular pressure: rho kinase elevated IOP treatment trial 1 and 2 (ROCKET-1 and ROCKET-2). American Journal of Ophthalmology 2018;186:116-27. [DOI: 10.1016/j.ajo.2017.11.019] [DOI] [PubMed] [Google Scholar]
Khouri 2019 (ROCKET‐4) {published data only}
- FDA Regulatory Document. Rhopressa (netarsudil) ophthalmic solution. www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208254Orig1s000TOC.cfm (accessed 18 February 2022).
- Khouri AS, Serle JB, Bacharach J, Usner DW, Lewis RA, Braswell P, et al. Once-daily netarsudil versus twice-daily timolol in patients with elevated intraocular pressure: the randomized phase 3 ROCKET-4 study. American Journal of Ophthalmology 2019;204:97-104. [DOI: 10.1016/j.ajo.2019.03.002.] [DOI] [PubMed] [Google Scholar]
- Khouri A, Heah T, Kopczynski C, Novack GD. A double-masked, randomized, parallel study of netarsudil ophthalmic solution, 0.02% QD compared to timolol maleate ophthalmic solution, 0.5% BID in patients with elevated intraocular pressure (ROCKET-4). Investigative Ophthalmology and Visual Science 2017;58(8):ARVO E-abstract 2461. [Google Scholar]
Lewis 2016 {published data only}
- Dubiner H, Levy B, Ramirez N, Kopczynski C, Novack GD. Fixed dose combination of AR-13324 and latanoprost (PG324): a double-masked, randomized, controlled study in patients with open-angle glaucoma (OAG) or ocular hypertension (OHT). Investigative Ophthalmology and Visual Science 2015;56(7):ARVO E-abstract 5704. [Google Scholar]
- Lewis RA, Levy B, Ramirez N, Kopczynski CC, Usner DW, Novack GD. Fixed-dose combination of AR-13324 and latanoprost: a double-masked, 28-day, randomised, controlled study in patients with open-angle glaucoma or ocular hypertension. British Journal of Ophthalmology 2016;100(3):339-44. [DOI: 10.1136/bjophthalmol-2015-306778] [DOI] [PubMed] [Google Scholar]
- NCT02057575. Study assessing safety and efficacy of PG324 ophthalmic solution in patients with elevated intraocular pressure. clinicaltrials.gov/ct2/show/NCT02057575 (first received 7 February 2014).
NCT02246764 (ROCKET‐3) {published data only (unpublished sought but not used)}
- NCT02246764. Study of netarsudil (AR-13324) ophthalmic solution in patients with glaucoma or ocular hypertension. clinicaltrials.gov/ct2/show/NCT02246764 (first received 23 September 2014).
Peace 2021 {published data only}
- Peace JH, Kopczynski CC, Heah T. Ocular hypotensive efficacy of netarsudil ophthalmic solution 0.02% over a 24-hour period: a pilot study. Investigative Ophthalmology & Visual Science 2017;58(8):ARVO E-abstract 2460. [Google Scholar]
- Peace JH, McKee HJ, Kopczynski CC. A randomized, phase 2 study of 24-h efficacy and tolerability of netarsudil in ocular hypertension and open-angle glaucoma. Ophthalmology Therapy 2021;10(1):89-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Serle 2018 (ROCKET‐1) {published data only}
- FDA Regulatory Document. Rhopressa (netarsudil) ophthalmic solution. www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208254Orig1s000TOC.cfm (accessed 18 February 2022).
- Serle JB, Katz LJ, McLaurin E, Heah T, Ramirez-Davis N, Usner DW, et al. Two phase 3 clinical trials comparing the safety and efficacy of netarsudil to timolol in patients with elevated intraocular pressure: rho kinase elevated IOP treatment trial 1 and 2 (ROCKET-1 and ROCKET-2). American Journal of Ophthalmology 2018;186:116-27. [DOI: 10.1016/j.ajo.2017.11.019] [DOI] [PubMed] [Google Scholar]
Sit 2021 {published data only}
- Sit AJ, Gupta D, Kazemi A, McKee H, Challa P, Liu KC, et al. Netarsudil improves trabecular outflow facility in patients with primary open angle glaucoma or ocular hypertension: a phase 2 study. American Journal of Ophthalmology 2021;226:262-9. [DOI: 10.1016/j.ajo.2021.01.019] [DOI] [PubMed] [Google Scholar]
- Sit AJ, Gupta D, Kazemi A, McKee H, Lopez J, Kopczynski C, et al. Improvement of trabecular outflow facility by netarsudil ophthalmic solution in patients with primary open angle glaucoma or ocular hypertension. Investigative Ophthalmology and Visual Science 2019;60(9):ARVO E-abstract 5164. [Google Scholar]
Tanihara 2013 {published data only}
- Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Araie M. Phase 2 randomized clinical study of a Rho kinase inhibitor, K-115, in primary open-angle glaucoma and ocular hypertension. American Journal of Ophthalmology 2013;156(4):731-6. [DOI: 10.1016/j.ajo.2013.05.016] [DOI] [PubMed] [Google Scholar]
Tanihara 2015a {published data only}
- Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Suganami H, et al. Intra-ocular pressure-lowering effects of a Rho kinase inhibitor, ripasudil (K-115), over 24 hours in primary open-angle glaucoma and ocular hypertension: a randomized, open-label, crossover study. Acta Ophthalmologica 2015;93(4):e254-60. [DOI: 10.1111/aos.12599] [DOI] [PubMed] [Google Scholar]
Tanihara 2015b {published data only}
- Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Suganami H, et al. Additive intraocular pressure-lowering effects of the rho kinase inhibitor ripasudil (K-115) combined with timolol or latanoprost: a report of 2 randomized clinical trials. JAMA Ophthalmology 2015;133(7):755-61. [DOI: 10.1001/jamaophthalmol.2015.0525] [DOI] [PubMed] [Google Scholar]
Tanihara 2015c {published data only}
- Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Suganami H, et al. Additive intraocular pressure-lowering effects of the rho kinase inhibitor ripasudil (K-115) combined with timolol or latanoprost: a report of 2 randomized clinical trials. JAMA Ophthalmology 2015;133(7):755-61. [DOI: 10.1001/jamaophthalmol.2015.0525] [DOI] [PubMed] [Google Scholar]
Walters 2019 (MERCURY‐2) {published data only}
- Walters TR, Ahmed LI, Lewis RA, Usner DW, Lopez J, Kopczynski CC, et al. Once-daily netarsudil/latanoprost fixed-dose combination for elevated intraocular pressure in the randomized phase 3 MERCURY-2 study. Ophthalmology Glaucoma 2019;2(5):280-9. [DOI: 10.1016/j.ogla.2019.03.007] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Dubiner 2014 {published data only}
- Dubiner H, Levy B, Kopczynski C, Novack GD. Double-masked, randomized, dose response study of AR-13324 ophthalmic solution compared to latanoprost in patients with elevated intraocular pressure. Investigative Ophthalmology and Visual Science 2014;55(13):545. [DOI] [PubMed] [Google Scholar]
Inazaki 2017 {published data only}
- Inazaki H, Kobayashi S, Anzai Y, Satoh H, Sato S, Inoue M, et al. One-year efficacy of adjunctive use of ripasudil, a rho-kinase inhibitor, in patients with glaucoma inadequately controlled with maximum medical therapy. Graefe's Archive for Clinical and Experimental Ophthalmology 2017;255(10):2009-15. [DOI] [PubMed] [Google Scholar]
Komizo 2019 {published data only}
- Komizo T, Ono T, Yagi A, Miyata K, Aihara M. Additive intraocular pressure-lowering effects of the Rho kinase inhibitor ripasudil in Japanese patients with various subtypes of glaucoma. Japanese Journal of Ophthalmology 2019;63(1):40-5. [DOI] [PubMed] [Google Scholar]
Kopczynski 2018 {published data only}
- Kopczynski CC, Heah T. Netarsudil ophthalmic solution 0.02% for the treatment of patients with open-angle glaucoma or ocular hypertension. Drugs Today 2018;54(8):467-78. [DOI] [PubMed] [Google Scholar]
NCT02173223 {published data only}
- NCT02173223. A study to assess the effect of rho-kinase inhibitor AR-12286 ophthalmic solution 0.5% and 0.7% in patients with uncontrolled advanced glaucoma with prior failed trabeculectomy or tube shunt. clinicaltrials.gov/show/NCT02173223 (first received 24 June 2014).
NCT03808688 {published data only}
- NCT03808688. Study of Rhopressa® for the reduction of elevated intraocular pressure in patients with glaucoma or ocular hypertension in a real-world setting. clinicaltrials.gov/ct2/show/NCT03808688 (first received 17 January 2019).
Sakamoto 2019 {published data only}
- Sakamoto E, Ishida W, Sumi T, Kishimoto T, Tada K, Fukuda K, et al. Evaluation of offset of conjunctival hyperemia induced by a Rho-kinase inhibitor; 0.4% ripasudil ophthalmic solution clinical trial. Scientific Reports 2019;9:3755. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanihara 2016 {published data only}
- Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Fukushima A, et al. One-year clinical evaluation of 0.4% ripasudil (K-115) in patients with open-angle glaucoma and ocular hypertension. Acta Ophthalmologica 2016;94(1):e26-e34. [DOI: 10.1111/aos.12829] [DOI] [PubMed] [Google Scholar]
- Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Fukushima A, et al. Specific clinical features of intraocular pressure-lowering effect in ripasudil (K-115): insights from 52-week phase 3 study. Investigative Ophthalmology and Visual Science 2016;57(12):ARVO E-abstract 3033. [Google Scholar]
UMIN000026228 {published data only}
- UMIN000026228. One-year efficacy of adjunctive use of ripasudil, a rho-kinase inhibitor, in patients with glaucoma inadequately controlled under maximum medical therapy. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000030126 (first received 20 February 2017).
Weiss 2013 {published data only}
- Aerie Pharmaceuticals Ireland Ltd. Assessment report: Rhokiinsa (netarsudil). www.ema.europa.eu/en/documents/assessment-report/rhokiinsa-epar-public-assessment-report_en.pdf (accessed prior to 18 March 2022).
- NCT01528787. Study of AR-13324 in patients with elevated intraocular pressure. clinicaltrials.gov/ct2/show/study/NCT01528787 (first received 8 February 2012).
- Weiss M, Levy B, Kopczynski C, Haarlem T, Novack G, AR-13324-CS201 Study Group. Evaluation of AR-13324, a novel dual mechanism agent, in lowering of IOP in glaucoma and ocular hypertension. Investigative Ophthalmology and Visual Science 2013;54(15):ARVO E-abstract 754. [Google Scholar]
References to studies awaiting assessment
CTRI/2018/04/013091 {published data only (unpublished sought but not used)}
- CTRI/2018/04/013091. A clinical trial to check the effect, safety and tolerability of ripasudil hydrochloride hydrate eye drops 0.4% w/v vs timolol maleate eye drops 0.5% w/v in patients with ocular hypertension/glaucoma. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=21731&EncHid=&userName=ripasudil (first received 6 April 2018).
CTRI/2020/01/022619 {published data only (unpublished sought but not used)}
- CTRI/2020/01/022619. A study of netarsudil ophthalmic solution 0.02% w/v vs timolol maleate eye drops 0.5% w/v in treatment of elevated intraocular pressure in patient with open angle glaucoma or ocular hypertension. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=38394&EncHid=&userName=timolol maleate (first received 6 January 2020).
NCT03284853 {published data only (unpublished sought but not used)}
- EUCTR2015-001528-41-AT. A prospective, double-masked, randomized, multicenter, active-controlled, parallel-group, 6-month study assessing the safety and ocular hypotensive efficacy of PG324 ophthalmic solution compared to GANFORT® (bimatoprost 0.03%/timolol 0.5%) ophthalmic solution in subjects with elevated intraocular pressure (MERCURY 3). www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2015-001528-41-AT (first received 15 February 2018).
- NCT03284853. Safety and efficacy study of PG324 (netarsudil/latanoprost 0.02%/0.005%) ophthalmic solution compared to GANFORT® ophthalmic solution in open angle glaucoma or ocular hypertension. clinicaltrials.gov/show/NCT03284853 (first received 15 September 2017).
- UCTR2015-001528-41-ES. A prospective, double-masked, randomized, multicenter, active-controlled, parallel-group, 6-month study assessing the safety and ocular hypotensive efficacy of PG324 ophthalmic solution compared to GANFORT® (bimatoprost 0.03%/timolol 0.5% ophthalmic solution) in subjects with elevated intraocular pressure (MERCURY 3). www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2015-001528-41-ES (first received 3 July 2017).
References to ongoing studies
JapicCTI‐194920 {published data only (unpublished sought but not used)}
- JapicCTI-194920. WP-1303 phase 3 comparison study. www.clinicaltrials.jp/cti-user/trial/ShowDirect.jsp?japicId=JapicCTI-194920 (first received 20 August 2019).
UMIN000019017 {published data only (unpublished sought but not used)}
- UMIN000019017. Efficacy of Rho-kinase inhibitor ophthalmic solution on bleb formation after trabeculectomy, a randomized parallel study. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000021996 (first received 24 September 2015).
Additional references
Aerie 2019
- Aerie Pharmaceuticals Ireland Ltd. Rhokiinsa – EMEA/H/C/004583. www.ema.europa.eu/en/medicines/human/EPAR/rhokiinsa (accessed 14 September 2021).
Braunger 2015
- Braunger BM, Fuchshofer R, Tamm ER. The aqueous humor outflow pathways in glaucoma: a unifying concept of disease mechanisms and causative treatment. European Journal of Pharmaceutics and Biopharmaceutics 2015;95:173-81. [DOI] [PubMed] [Google Scholar]
Cedrone 2008
- Cedrone C, Mancino R, Cerulli A, Cesareo M, Nucci C. Epidemiology of primary glaucoma: prevalence, incidence, and blinding effects. In: Nucci C, Cerulli L, Osborne NN, Bagetta G, editors(s). Progress in Brain Research. Vol. 173. Amsterdam (the Netherlands): Elsevier, 2008:3-14. [DOI] [PubMed] [Google Scholar]
Chang 2012
- Chang EE, Goldberg JL. Glaucoma 2.0: neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology 2012;119(5):979-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Conlon 2017
- Conlon R, Saheb H, Ahmed II. Glaucoma treatment trends: a review. Canadian Journal of Ophthalmology 2017;52(1):114-24. [DOI] [PubMed] [Google Scholar]
Covidence [Computer program]
- Covidence. Melbourne, Australia: Veritas Health Innovation, accessed 14 September 2021. Available at covidence.org.
FDA 2016
- Food and Drug Administration (FDA). What is a serious adverse event? www.fda.gov/safety/reporting-serious-problems-fda/what-serious-adverse-event (accessed 14 September 2021).
FDA 2017
- Food and Drug Administration (FDA). Novel drug approvals for 2017. www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2017 (accessed 14 September 2021).
Gabelt 2005
- Gabelt BT, Kaufman PL. Changes in aqueous humor dynamics with age and glaucoma. Progress in Retinal and Eye Research 2005;24(5):612-37. [DOI] [PubMed] [Google Scholar]
Garnock‐Jones 2014
- Garnock-Jones KP. Ripasudil: first global approval. Drugs 2014;74(18):2211-5. [DOI] [PubMed] [Google Scholar]
Gazzard 2019
- Gazzard G, Konstantakopoulou E, Garway-Heath D, Garg A, Vickerstaff V, Hunter R, et al. Selective laser trabeculoplasty versus eye drops for first-line treatment of ocular hypertension and glaucoma (LiGHT): a multicentre randomised controlled trial. Lancet 2019;393(10180):1505-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glanville 2006
- Glanville JM, Lefebvre C, Miles JN, Camosso-Stefinovic J. How to identify randomized controlled trials in MEDLINE: ten years on. Journal of the Medical Library Association 2006;94(2):130-6. [PMC free article] [PubMed] [Google Scholar]
GRADE Handbook
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
Heijl 2002
- Heijl A, Leske MC, Bengtsson B, Hyman L, Bengtsson B, Hussein M. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma trial. Archives of Ophthalmology 2002;120(10):1268-79. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S. Chapter 16: Missing data. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Higgins 2021a
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Higgins 2021b
- Higgins JP, Li T, Deeks JJ. Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Isobe 2014
- Isobe T, Mizuno K, Kaneko Y, Ohta M, Koide T, Tanabe S. Effects of K-115, a rho-kinase inhibitor, on aqueous humor dynamics in rabbits. Current Eye Research 2014;39(8):813-22. [DOI] [PubMed] [Google Scholar]
Jonas 2017
- Jonas JB, Aung T, Bourne RR, Bron AM, Ritch R, Panda-Jonas S. Glaucoma. Lancet 2017;390(10108):2183-93. [DOI] [PubMed] [Google Scholar]
Kaneko 2017
- Kaneko Y, Ohta M, Isobe T, Nakamura Y, Mizuno K. Additive intraocular pressure-lowering effects of ripasudil with glaucoma therapeutic agents in rabbits and monkeys. Journal of Ophthalmology 2017;2017:Article ID 7079645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kazemi 2018
- Kazemi A, McLaren JW, Kopczynski CC, Heah TG, Novack GD, Sit AJ. The effects of netarsudil ophthalmic solution on aqueous humor dynamics in a randomized study in humans. Journal of Ocular Pharmacology and Therapeutics 2018;34(5):380-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kolko 2015
- Kolko M. Present and new treatment strategies in the management of glaucoma. Open Ophthalmology Journal 2015;9:89-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kreuger 1980
- Kreuger DE, Milton RC, Maunder LR. The Framingham Eye study: introduction to the monograph. Survey of Ophthalmology 1980;24(6):614-20. [DOI] [PubMed] [Google Scholar]
Kwon 2009
- Kwon YH, Fingert JH, Kuehn MH, Alward WL. Primary open-angle glaucoma. New England Journal of Medicine 2009;360(11):1113-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2016
- Li T, Lindsley K, Rouse B, Hong H, Shi Q, Friedman DS, et al. Comparative effectiveness of first-line medications for primary open-angle glaucoma: a systematic review and network meta-analysis. Ophthalmology 2016;123(1):129-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2021a
- Li T, Higgins JP, Deeks JJ. Chapter 5: Collecting data. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Li 2021b
- Li G, Lee C, Read AT, Wang K, Ha J, Kuhn M, et al. Anti-fibrotic activity of a rho-kinase inhibitor restores outflow function and intraocular pressure homeostasis. Elife 2021;10:e60831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2018a
- Lin CW, Sherman B, Moore LA, Laethem CL, Lu DW, Pattabiraman PP, et al. Discovery and preclinical development of netarsudil, a novel ocular hypotensive agent for the treatment of glaucoma. Journal of Ocular Pharmacology and Therapeutics 2018;34(1-2):40-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2018b
- Lin CW, Foley BE, Carbajal K, Kopczynski C. Evaluation of the potential of AR-13324 and AR-13503 to Induce phospholipidosis. Investigative Ophthalmology and Visual Science 2018;59(9):6125. [Google Scholar]
Lütjen‐Drecoll 1999
- Lütjen-Drecoll E. Functional morphology of the trabecular meshwork in primate eyes. Progress in Retinal and Eye Research 1999;18(1):91-119. [DOI] [PubMed] [Google Scholar]
McKenzie 2021
- McKenzie JE, Brennan SE, Ryan RE, Thomson HJ, Johnston RV. Chapter 9: Summarizing study characteristics and preparing for synthesis. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Mehran 2020
- Mehran NA, Sinha S, Razeghinejad R. New glaucoma medications: latanoprostene bunod, netarsudil, and fixed combination netarsudil-latanoprost. Eye 2020;34(1):72-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analysis: the PRISMA statement. PLoS Medicine 2009;6(7):e1000097. [DOI] [PMC free article] [PubMed]
Moura‐Coelho 2019
- Moura-Coelho N, Ferreira JT, Bruxelas CP, Dutra-Medeiros M, Cunha JP, Proença RP. Rho kinase inhibitors – a review on the physiology and clinical use in ophthalmology. Graefe's Archive for Clinical and Experimental Ophthalmology 2019;257:1101-17. [DOI: 10.1007/s00417-019-04283-5] [DOI] [PubMed] [Google Scholar]
Okumura 2017
- Okumura N, Kinoshita S, Koizumi N. Application of Rho kinase inhibitors for the treatment of corneal endothelial diseases. Journal of Ophthalmology 2017;2017:Article ID 2646904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Price 2021
- Price MO, Feng MT, Price FW Jr. Randomized, double-masked trial of netarsudil 0.02% ophthalmic solution for prevention of corticosteroid-Induced ocular hypertension. American Journal of Ophthalmology 2021;222:382-7. [DOI] [PubMed] [Google Scholar]
Quigley 2006
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. British Journal of Ophthalmology 2006;90(3):262-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Quigley 2011
- Quigley HA. Glaucoma. Lancet 2011;377(9774):1367-77. [DOI] [PubMed] [Google Scholar]
Raizman 2017
- Raizman MB, Hamrah P, Holland EJ, Kim T, Mah FS, Rapuano CJ, et al. Drug-induced corneal epithelial changes. Survey of Ophthalmology 2017;62(3):286-301. [DOI] [PubMed] [Google Scholar]
Rao 2017
- Rao PV, Pattabiraman PP, Kopczynski C. Role of the Rho GTPase/Rho kinase signaling pathway in pathogenesis and treatment of glaucoma: bench to bedside research. Experimental Eye Research 2017;158:23-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan Web 2022 [Computer program]
- Review Manager Web (RevMan Web). Version 4.1.0. The Cochrane Collaboration, 2022. Available at revman.cochrane.org.
Rohatgi 2021 [Computer program]
- WebPlotDigitizer. Ankit Rohatgi, August 2021. automeris.io/WebPlotDigitizer.
Schünemann 2019
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing 'Summary of findings' tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from training.cochrane.org/handbook/archive/v6.
Stamer 2012
- Stamer WD, Acott TS. Current understanding of conventional outflow dysfunction in glaucoma. Current Opinion in Ophthalmology 2012;23(2):135-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:I4898. [DOI] [PubMed] [Google Scholar]
Sturdivant 2016
- Sturdivant JM, Royalty SM, Lin CW, Moore LA, Yingling JD, Laethem CL, et al. Discovery of the ROCK inhibitor netarsudil for the treatment of open-angle glaucoma. Bioorganic and Medicinal Chemistry Letters 2016;26(10):2475-80. [DOI] [PubMed] [Google Scholar]
Tanna 2018
- Tanna AP, Johnson M. Rho kinase inhibitors as a novel treatment for glaucoma and ocular hypertension. Ophthalmology 2018;125(11):1741-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tham 2014
- Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 2014;121(11):2081-90. [DOI] [PubMed] [Google Scholar]
Varma 2004
- Varma R, Ying-Lai M, Francis BA, Nguyen BB, Deneen J, Wilson MR, et al. Prevalence of open-angle glaucoma and ocular hypertension in Latinos: the Los Angeles Latino Eye study. Ophthalmology 2004;11(8):1439-48. [DOI] [PubMed] [Google Scholar]
Wang 2015
- Wang RF, Williamson JE, Kopczynski C, Serle JB. Effect of 0.04% AR-13324, a ROCK, and norepinephrine transporter inhibitor, on aqueous humor dynamics in normotensive monkey eyes. Journal of Glaucoma 2015;24(1):51-4. [DOI] [PubMed] [Google Scholar]
Weinreb 2014
- Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA 2014;311(18):1901-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolfram 2019
- Wolfram C, Stahlberg E, Pfeiffer N. Patient-reported nonadherence with glaucoma therapy. Journal of Ocular Pharmacology and Therapeutics 2019;35(4):223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Freiberg 2020
- Freiberg JC, Spreckelsen A, Khachatryan N, Kolko M, Azuara-Blanco A, Virgili G. Rho kinase inhibitor for primary open-angle glaucoma and ocular hypertension [ ]. Cochrane Database of Systematic Reviews 2020, Issue 12. Art. No: CD013817. [DOI: 10.1002/14651858.CD013817] [DOI] [PMC free article] [PubMed] [Google Scholar]