

Abstract
Scope:
This work aims at evaluating the effect of dietary ellagic acid (EA) and its microbial metabolite urolithin A (UA) on glucose metabolism and insulin resistance (IR) in mice with diet-induced IR.
Methods and Results:
DBA2J mice are fed a high fat/high sucrose diet (HF/HS) for 8 weeks to induce IR and then 0.1% EA, UA, or EA and UA (EA+UA) are added to the HF/HS-diet for another 8 weeks. UA significantly decreases fasting glucose and increases adiponectin compared with HF/HS-controls. During intraperitoneal insulin tolerance test, EA+UA significantly improve insulin-mediated glucose lowering effects at 15 and 120 min and reduce blood triglycerides compared with HF/HS-controls. Serum free fatty acids are significantly decreased by EA, UA, and EA+UA. Differential expression of genes related to mitochondrial function by EA, UA, and EA+UA in liver and skeletal muscle is observed. Primary hepatocytes from IR-mice have higher proton leak, basal and ATP-linked oxygen consumption rates compared with healthy controls. EA and EA+UA but not UA reduce the proton leak in hepatocytes from IR-mice.
Conclusion:
EA and UA induce different metabolic benefits in IR mice. The effects of EA and UA on mitochondrial function suggest a potentially novel mechanism modulating metabolism.
Keywords: ellagic acid, insulin resistance, mitochondria, obesity, urolithin A
1. Introduction
The effects of pomegranate (Pom) polyphenols on conditions with impaired glucose control have previously been reported.[1,2] The most abundant polyphenols in Pom are ellagitannins (ETs) and ellagic acid (EA). ETs undergo acid hydrolysis to EA in the stomach and then are further metabolized by intestinal gut bacteria into urolithins, including urolithin A (UA). While EA is known to be poorly bioavailable due to its low water solubility,[3,4] UA is the most widely studied microbial metabolite of EA, reaching a micromolar level in circulation. UA also persists in the body far longer than EA.[5] Both EA and UA contribute to the metabolic benefits of Pom, however, the mechanisms of the direct effect of EA and UA alone and in combination remain to be elucidated.[6–9]
Urolithins are microbial metabolites produced after consumption of ETs/EA-containing foods including pomegranates and walnuts. ETs/EA-metabolizing phenotypes (urolithin metabotypes A, B and 0) vary among individuals.[10] EA and UA share similar biological activities, such as antiinflammatory potential, but also has distinct functions individually, such as the regulation of mitochondrial function.[9,11,12] In addition, studies have shown that gut microbial composition vary among different urolithin metabotypes.[13–18] The difference in EA and urolithin profiles resulting from individuals’ heterogeneous ability of urolithin production as well as the potential interaction between EA/urolithins and gut microbiome all likely contribute to the observation that subjects of different urolithin metabotypes responded differently to dietary ETs/EA intake.[10,15] The specific effects of UA and EA are difficult to study in isolation after consumption of Pom with its multiple polyphenols and different urolithin metabotypes. Using a high fat/high sucrose (HF/HS) diet induced insulin resistance (IR) mouse model, we designed a study that examines the effects of EA and UA on glucose metabolism and IR, without endogenous production of UA and UB.
Recent in vitro studies demonstrate that EA, UA and UB differentially regulate adipocyte lipid accumulation and inflammatory response, which are important factors in the induction of IR.[19] We hypothesized that administration of EA and UA in our animal model system might have distinct impacts on glucose homeostasis and IR. We investigated both the separate and combined effects of EA and UA at controlled doses in mice that could not produce UA or UB from EA. This design enabled us to examine the effects of EA and UA alone and in combination.
Several mouse strains have been used to study diet induced obesity and T2DM symptoms,[20–22] and mice of different genetic backgrounds (strains) show distinct susceptibilities to developing diet-induced metabolic abnormalities.[23] The most widely used is C57BL/6J fed with high fat diets (HFD, 60% fat), which develop obesity, IR and mild hyperglycemia in response to HFD.[22] However, DBA2/J mice developed most severe IR in response to HFD feeding compared with C57BL6, 129 × 1, BALB/c and FVB/N.[23] A study evaluating HF/HS induced IR in mice of different genetic backgrounds showed that male DBA/2J mice are sensitive to HF/HS diet induced IR.[24] In addition, in contrast to C57BL/6J mice, DBA2/J mice have been shown to develop pancreatic beta cell failure with chronic hyperglycemia and therefore represent a better model of human pre-diabetes and metabolic syndrome.[25] Therefore, we used DBA2/J mice with HF/HS diet induced IR to evaluate the efficacy of EA and UA in glucose homeostasis and IR.
2. Results
2.1. Glucose Homeostasis and HF/HS-Diet Induced IR
We confirmed that experimental mice used in this study were not able to produce UA and/or UB. At the initial screening, only EA but no UA and/or UB was detected in the stool samples collected during 4 day’s Pom juice (PomJ) feeding (Figure S1A,B, Supporting Information). We also confirmed that chronic EA feeding did not induce UA and/or UB production in EA fed mice, and chronic UA feeding did not induce UB production in UA or EA+UA fed mice by examining UA/UB in the colon contents collected at the end of experiment (Figure S1C, Supporting Information). Dietary EA and UA supplementation did not alter food intake, body weight, total visceral fat, liver weight, liver lipid content as well as histology markers of hepatic steatosis of HF/HS-diet fed mice (Figures S3 and S4, Supporting Information).
Glucose metabolism and IR were assessed by intraperitoneal glucose tolerance test (IPGTT), intraperitoneal insulin tolerance test (IPITT) as well as fasting blood glucose and insulin levels. Glucose metabolism was impaired in mice fed HF/HS- diet compared with LF/LS-diet as indicated by higher fasting blood glucose and area under the curve (AUC) during IPGTT (Figures S2 and S5, Supporting Information). Fasting blood glucose levels were significantly lower in the UA group, and showed a trend to decrease in EA or EA+UA groups compared with HF/HS group while fasting blood insulin was similar among all groups (Figure 1A,B). Blood glucose levels during IPGTT were comparable among mice fed the HF/HS-diet and HF/HS-diets supplemented with EA, UA, and EA+UA (Figure S5, Supporting Information).
Figure 1.
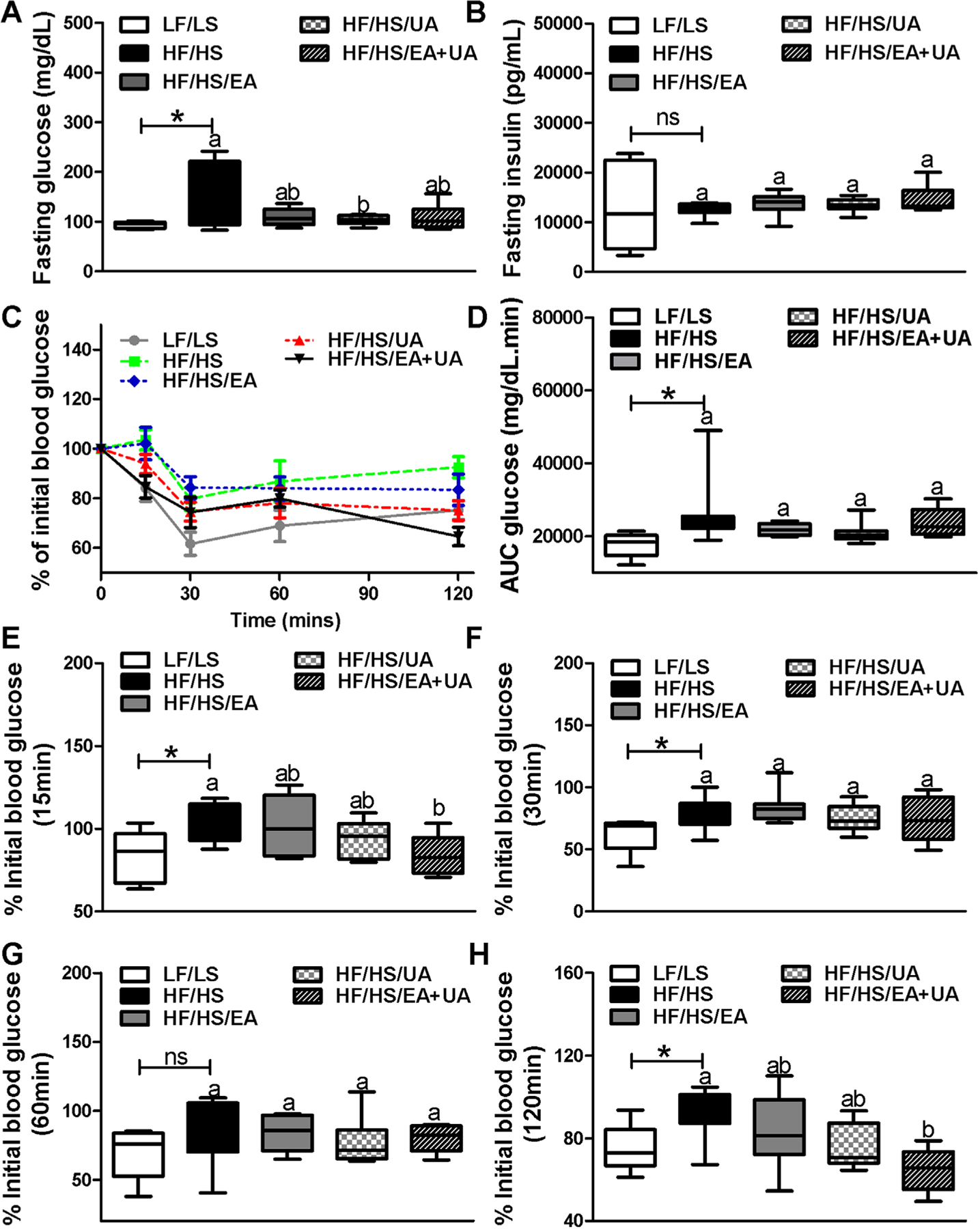
Profiles of glucose control in experimental mice. A–B) Fasting blood glucose and insulin at week 16 (n = 8–12). C–H) Profiles of IPITT in experimental mice at week 16 (n = 8). Data are presented as means ± SEMs. C) Two-way ANOVA analysis showed a significant treatment effects between LF/LS and HF/HS (Treatment p = 0.000; Time p = 0.001; Time x Treatment p = 0.997). D–H) *p < 0.05 LF/LS versus HF/HS mice; for comparisons between HF/HS mice or HF/HS mice with EA, UA or EA+UA supplementation, means in a column without a common letter differ; p < 0.05.
IR assessed by IPITT showed that HF/HS feeding significantly increased IR compared with LF/LS-diet as indicated by higher area under the curve (AUC) as well as a slower decrease of blood glucose levels during the first 30 min of insulin overload (Figure 1C–F). The blood glucose levels were significantly lower at 15 and 120 min during IPITT in the EA+UA group compared with HF/HS group (Figure 1C,E,H).
2.2. Serum Lipids, Adiponectin, and Proinflammatory Cytokines
Levels of serum free fatty acids (FFA) and cholesterol but not triglycerides (TG) were significantly higher in mice fed the HF/HS compared with LF/LS-diet (Figure 2A,B,D). EA, UA, and EA+UA significantly reduced serum FFA, while only EA+UA decreased serum TG (Figure 2A,B). Significant increase of circulating adiponectin was also observed in mice receiving UA (Figure 2C).
Figure 2.
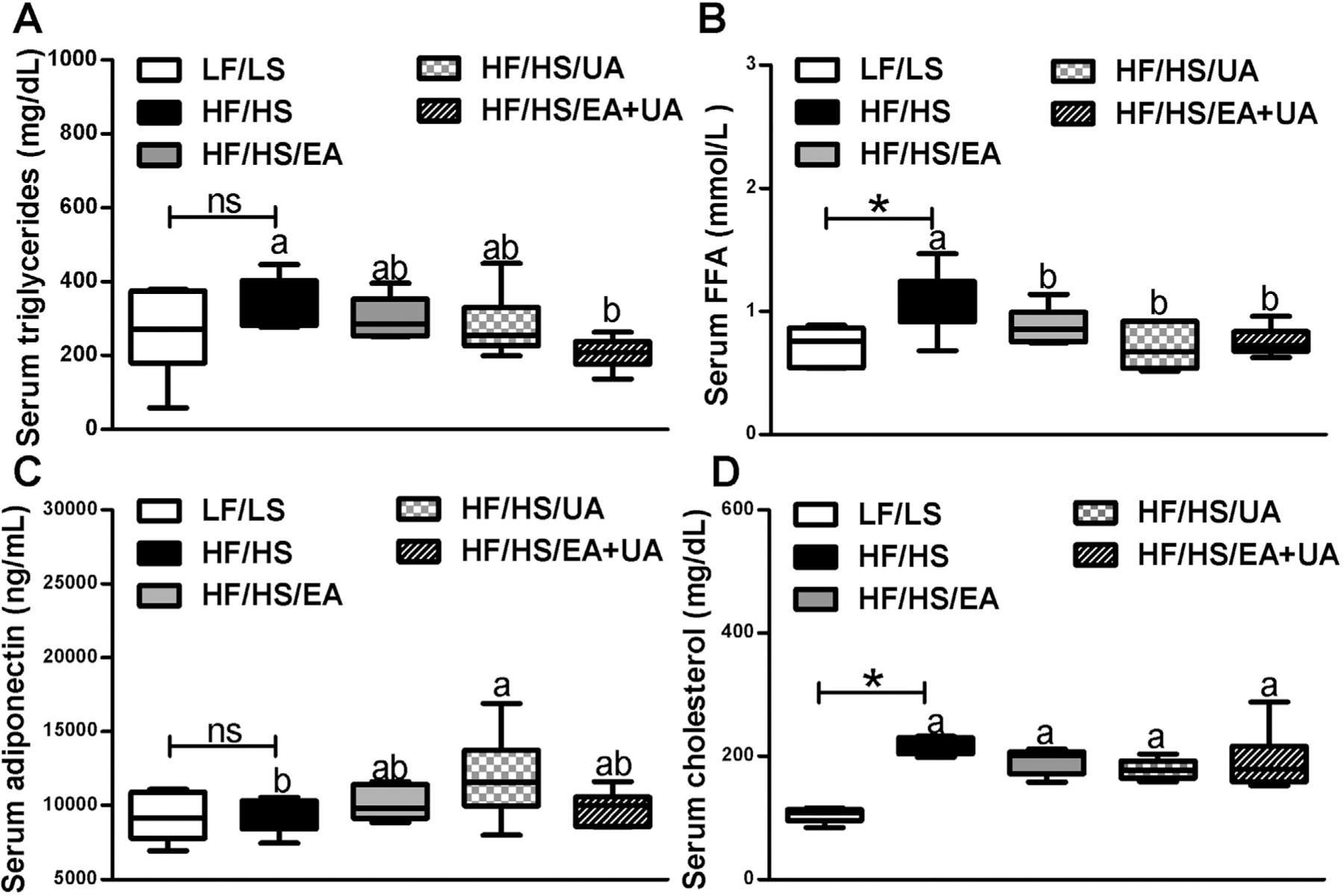
Effects of EA and/or UA on serum A) triglycerides, B) total FFA, C) adiponectin, and D) total cholesterol in experimental mice. Data are presented as means ± SEMs (n = 8–12). *p < 0.05 LF/LS versus HF/HS mice; for comparisons between HF/HS mice or HF/HS mice with EA, UA or EA+UA supplementation, means in a column without a common letter differ; p < 0.05.
Proinflammatory cytokines CXCL1 (KC), MCP1, TNFα, and IL6 were also evaluated (Figure 3). Levels of KC, a cytokine associated with islet function, obesity and T2D,[26] was significantly higher in mice fed the HF/HS compared with LF/LS-diet (Figure 3A). Dietary supplementation of EA, UA, and EA+UA did not change the levels of KC. Proinflammatory cytokines MCP1, TNFα, and IL6 in mice fed HF/HS and LF/LS-diets were at similar levels, and was not altered with dietary EA, UA, and EA+UA supplementation (Figure 3B–D). In addition, expression of Mcp1, Tnfα, and Il6 in liver, skeletal muscle (SM) and epididydimal fat (E-fat) were similar in all experimental groups except that SM Mcp1 had a higher expression level in HF/HS mice compared with LF/LS mice (Figure S6, Supporting Information).
Figure 3.
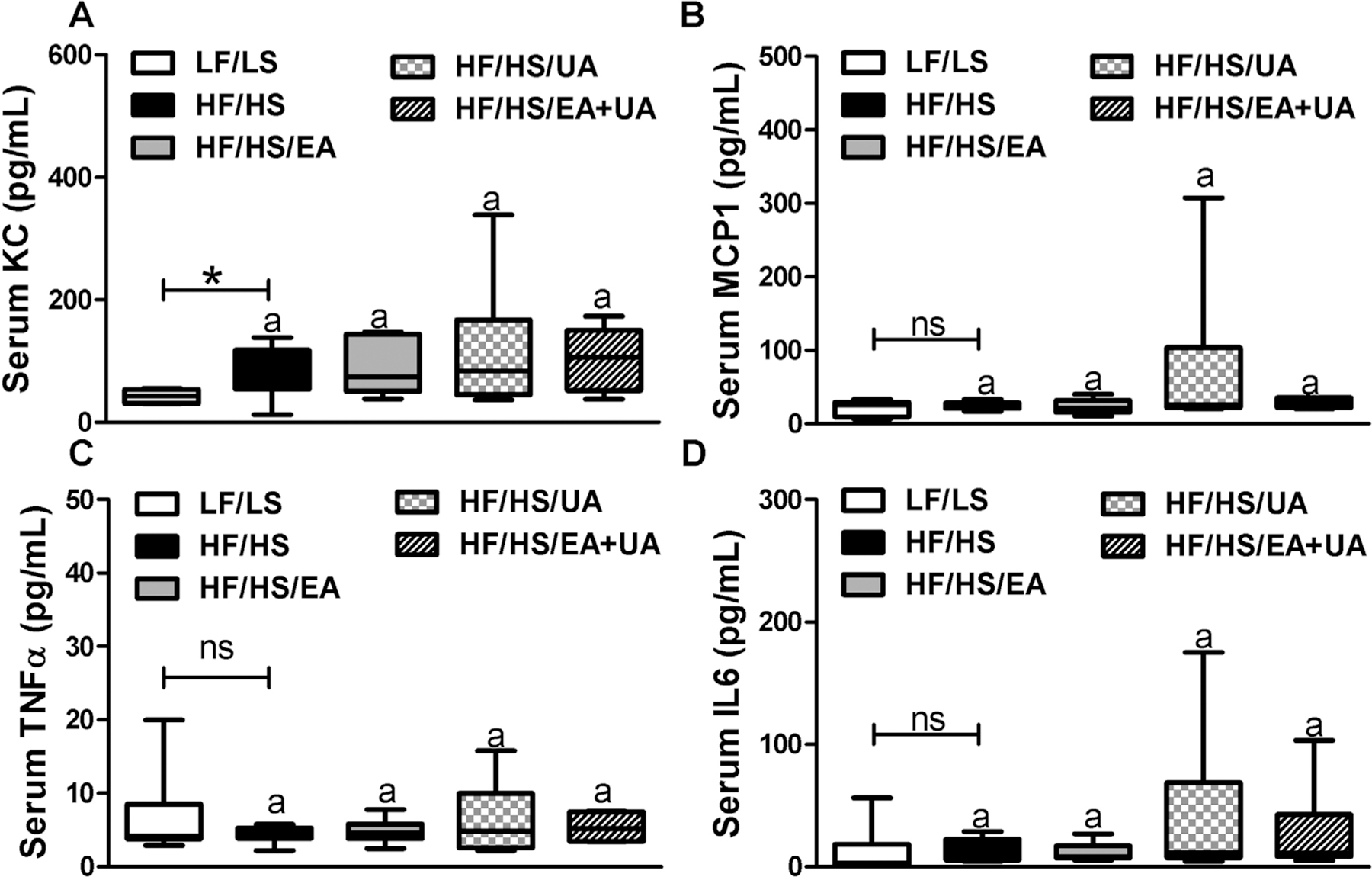
Effects of EA and/or UA on serum A) KC, B) MCP1, C) TNFα, and D) IL6 in experimental mice. Data are presented as means ± SEMs (n = 8–12). *p < 0.05 LF/LS versus HF/HS mice; for comparisons between HF/HS mice or HF/HS mice with EA, UA or EA+UA supplementation, means in a column without a common letter differ; p < 0.05.
2.3. The Expression of Genes Related to Lipid Metabolism in Liver, SM, and E-Fat
The expression of genes involved in regulating lipid metabolism, Cebpa, Pparg, Cpt1a, Cpt1b, Cpt2, Ucp1, Ucp3, Cd36, and Fasn were determined in liver, SM, and E-fat (Figures S7–S9, Supporting Information). Expression levels of Pparg and Cd36 in liver, Cebpa, Cpt1b and Cpt2 in SM, and Cebpa, Cpt1b, and Ucp1 in E-fat were higher in HF/HS mice compared with LF/LS mice. Only Ucp3 gene expression in SM was significantly increased by dietary UA supplementation compared with HF/HS mice. Expression levels of other genes were not altered by EA, UA, and EA+UA. Expression levels of lipolysis marker Lipe and Pnpl2 were higher in HF/HS mice compared with LF/LS mice in E-fat, but were not altered by EA, UA and EA+UA (Figure S9, Supporting Information).
2.4. Markers of Mitochondrial Function
Mitochondrial metabolism was evaluated in liver, SM, and E-fat by determining mitochondrial density (the ratio of mitochondrial DNA (mtDNA) to nuclear DNA (nDNA)) as well as the expression of genes involved in mitochondrial dynamics (Mfn2, Prkn, Pink1, Errα and Pgc1α) (Table 1, Figure S10, Supporting Information). Mitochondrial density was similar in all three tissues of mice fed the HF/HS and LF/LS-diet (Table 1, Figure S10A). In SM, UA supplementation significantly increased mitochondrial density compared with mice fed the HF/HS-diet (Table 1). Mitochondrial fusion marker Mfn2 was decreased in liver but increased in SM in HF/HS mice compare to LF/LS mice. UA and EA+UA supplementation significantly increased Mfn2 gene expression in both liver and SM. Significant decrease of mitophagy markers (Prkn, Pink1) was observed in the liver of HF/HS mice compared with LF/LS mice. UA supplementation significantly increased the gene expression of mitophagy markers (Prkn, Pink1) in both SM and liver. Other mitochondrial biogenesis markers (Errα and Pgc1α) were not changed among experimental groups. In E-fat, all the mitochondrial markers were similar in all experimental groups (Figure S10, Supporting Information).
Table 1.
Relative expression (fold change) of mitochondrial density and genes involved in mitochondrial dynamics in liver and SM of HF/HS, HF/HS/EA, HF/HS/EA, and HF/HS/EA+UA groups compared with LF/LS group.
| LF/LS | HF/HS | HF/HS/EA | HF/HS/UA | HF/HS/EA+UA | |
|---|---|---|---|---|---|
| Liver | |||||
| Err α | 1.06(0.16) | 0.91(0.18)a | 0.90(0.10)a | 1.27(0.13)a | 1.30(0.12)a |
| Pink1 | 1.04(0.12) | 0.64(0.07)*b | 0.74(0.08)ab | 0.89(0.07)a | 0.86(0.13)ab |
| Prkn | 1.00(0.06) | 0.52(0.07)*b | 0.93(0.14)ab | 9.74(6.42)a | 1.38(0.34)ab |
| Pgc1 α | 1.01(0.06) | 1.29(0.2)a | 1.24(0.15)a | 1.38(0.22)a | 1.1(0.08)a |
| Mfn2 | 1.09(0.20) | 0.52(0.07)*b | 0.84(0.15)ab | 1.31(0.23)a | 1.34(0.25)a |
| mtDNA | 1.01(0.20) | 0.52(0.24)a | 0.84(0.40)a | 1.31(0.20)a | 1.34(0.20)a |
| Skeletal muscle | |||||
| Err α | 1.21(0.37) | 1.18(0.18)b | 1.23(0.25)ab | 2.69(0.48)a | 2.55(0.44)ab |
| Pink1 | 1.02(0.1) | 0.94(0.17)b | 1.02(0.25)ab | 2.01(0.42)a | 1.77(0.24)ab |
| Prkn | 1.02(0.11) | 0.75(0.14)b | 0.79(0.16)ab | 1.6(0.33)a | 1.28(0.18)ab |
| Pgc1 α | 1.28(0.36) | 1.29(0.32)a | 1.48(0.25)a | 2.05(0.61)a | 2.16(0.49)a |
| Mfn2 | 1.12(0.26) | 2.33(0.4)*b | 1.92(0.38)b | 4.13(0.3)ab | 4.7(0.96)a |
| mtDNA | 2.42(0.58) | 1.67(0.34)b | 2.15(0.41)ab | 3.95(0.86)a | 2.79(0.57)ab |
Expression data are mean (SEM) of 8–12 animals
p < 0.05 LF/LS versus HF/HS mice
for comparisons between HF/HS mice and HF/HS mice with EA, UA or EA+UA supplementation, means without a common letter (a, b, or ab) differ; p < 0.05.
2.5. Primary Hepatocyte Mitochondrial Respiratory Capacity
We also evaluated the acute effects of EA, UA, and EA+UA on liver mitochondrial respiration by measuring the oxygen consumption rate (OCR) of cells in response to ATP synthase inhibitor (oligomycin), uncoupling agent (FCCP), and complex I and III inhibitor (rotenone and antimycin A) (Figures 4 and S11, Supporting Information). Primary hepatocytes from mice of HF/HS induced IR had higher proton leak as well as higher baseline, maximal and ATP linked OCR (Figure 4). EA and EA+UA significantly decreased proton leak in primary hepatocytes from HF/HS mice. EA, UA, and EA+UA had no effect on basal, maximal and ATP linked OCR of primary hepatocytes from HF/HS mice.
Figure 4.
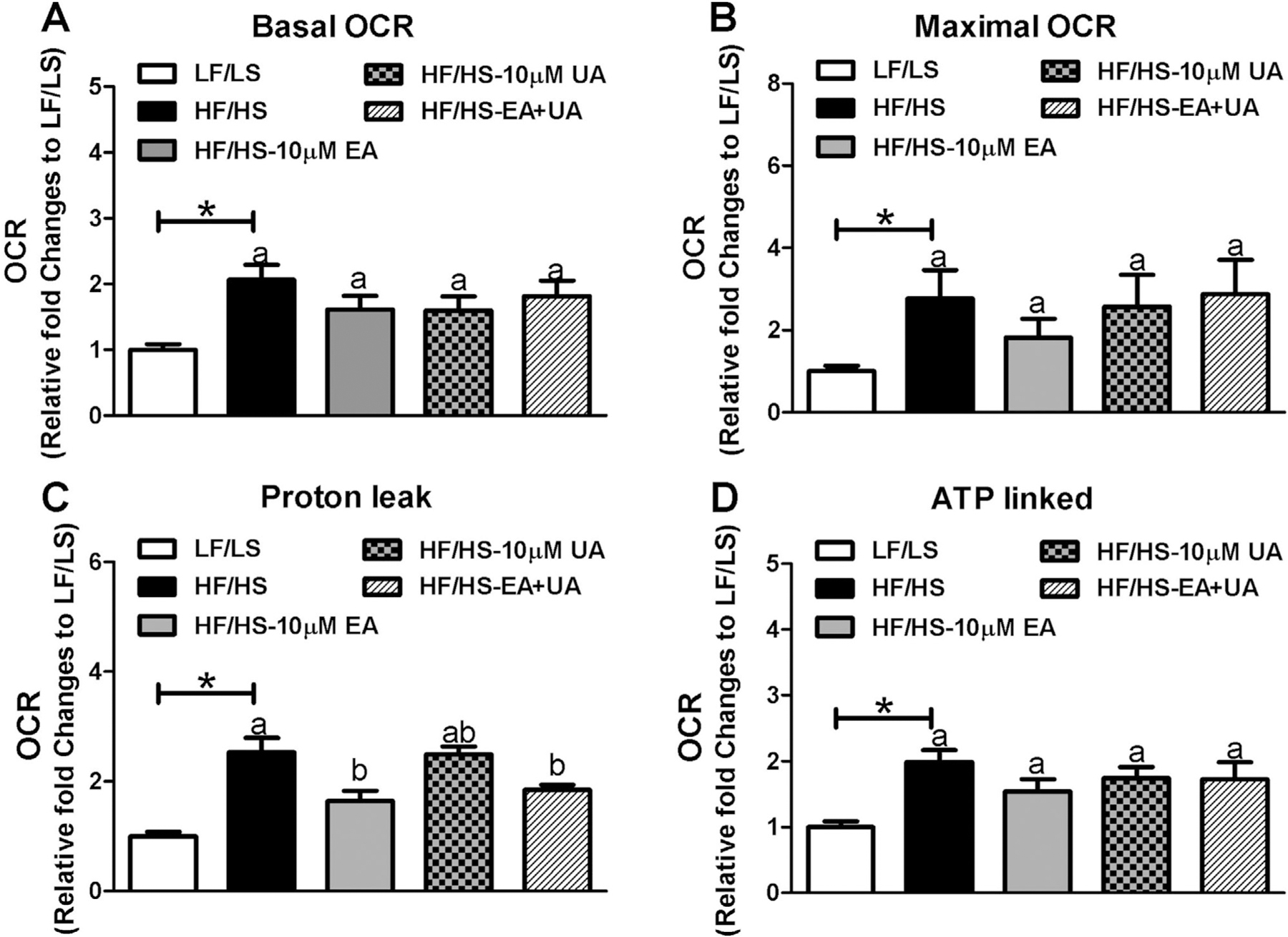
The acute effect of EA and/or UA on primary hepatocytes from mice with HF/HS induced IR. Primary hepatocytes were isolated form LF/LS and HF/HS fed mice. Primary hepatocytes from IR mice were treated with 10 µm EA, 10 µm UA or 10 µm EA and 10 µm UA combined for 18–24 h before seahorse analysis. The experiments were repeated three times. Relative fold changes normalized to mitochondrial respiratory parameters of primary hepatocytes from LF/LS were used. A) Basal OCR, B) Maximal OCR, C) proton leak, and D) ATP linked OCR. *p < 0.05 LF/LS versus HF/HS mice; for comparisons between HF/HS mice or HF/HS mice with EA, UA or EA+UA treatment, means in a column without a common letter differ; p < 0.05.
3. Discussion
Reports of the metabolic benefits of food enriched with ETs and EA from Pom, walnuts, and berries have not considered the effects of EA and UA directly on glucose metabolism.[27] In this study, we investigated the effect of EA, UA alone, or in combination in HF/HS- diet-induced obesity and IR in DBA2J mice that lack the ability to form UA.
HF/HS-diet fed mice showed significantly higher resistance to insulin-induced glucose reduction compared with LF/LS-diet fed mice during the first 30 min of IPITT test, indicating these mice were IR. Dietary supplementation with EA+UA showed potential in improving IR, as indicated by a significant decrease in insulin-mediated glucose concentrations during the first 15min as well as at 2 h of IPITT. Future studies using more specific tests, such as glucose clamp, will be necessary to provide further details of EA and/or EA effects in peripheral insulin sensitivity and beta cell function.[28] Due to low solubility, unabsorbed EA remaining in the GI tract likely acts on intestinal cells as well as gut microbiome, which are known to be important in IR.[29] Future studies are needed to understand the role of intestinal cells and gut microbiome in EA mediated amelioration of obesity and diet-induced IR.
The liver contributes to fasting blood glucose levels via gluconeogenesis. Adiponectin, an important metabolic hormone, inhibits hepatic gluconeogenesis.[30–32] UA resulted in a significant decrease of fasting blood glucose, and increases in adiponectin, suggesting that UA could potentially affect glucose homeostasis via hepatic gluconeogenesis. Expression levels of gluconeogenesis markers G6pc and Pcpk1 in liver were higher in HF/HS mice compared with LF/LS mice (Figure S12, Supporting Information). However, significant reduction of G6pc expression in comparison with HF/HS was observed with EA supplementation. A trend of decrease was observed with UA supplementation. FFAs are formed by lipolysis. This process mainly occurs but is not limited to white adipose tissues providing FFA as energy substrate for other tissues. Increased blood FFA levels have been associated with the pathogenesis of IR.[33,34] EA, UA, and EA+UA in combination significantly decreased fasting blood FFA. Expression levels of lipolysis markers Lipe but not Pnpl2 were higher in HF/HS mice compared with LF/LS mice, but neither was altered by EA, UA, or EA+UA. We also evaluated the acute effect of EA, UA, and EA+UA on lipolytic activity in E-fat from HF/HS-diet fed mice using an ex vivo lipolysis assay. EA, UA, and EA+UA, however, did not induce any acute effects on lipolysis (data not shown). Fasting TG levels were similar between HF/HS and LF/LS mice, however, fasting TG levels were significantly decreased by EA+UA. Our findings suggest a distinct modulating effect of EA, UA, and EA+UA on fasting whole-body energy metabolism. Future studies are needed to examine the effects of EA and UA on specific tissues in reference to overall glucose and lipid metabolism.
Chronic low-grade inflammation has been linked to obesity and related metabolic disorders. Elevated proinflammatory cytokines have frequently been associated with HF feeding in experimental mice.[23] However, in this mouse model, we did not find a difference in blood and tissues (liver, SM, E-fat) levels of proinflammatory cytokines TNFα, IL6, and MCP1 between HF/HS and LF/LS mice. Only the levels of blood KC, a proinflammatory cytokine that affects islet function, and SM Mcp1 gene expression, were significantly higher in HF/HS mice compared with LF/LS mice. In addition, EA, UA, and EA+UA intervention did not alter the levels of the markers of inflammation we evaluated. These observations suggest that inflammation is not essential for the development of IR in this specific DBA/2 J model.
Adipocytes, hepatocytes, and SM cells play a central role in the regulation of nutrient metabolism and energy homeostasis. Mitochondrial function in these tissues is likely involved in the pathogenesis of obesity and related metabolic disorders.[35,36] Mitochondria are highly dynamic and undergo fusion, fission, and mitophagy to maintain function.[37] We therefore evaluated mitochondrial metabolism in these three tissues in response to HF/HS feeding and EA, UA, and EA+UA administration. In agreement with previous publications showing that UA stimulates SM mitophagy,[9,38,39] UA significantly increased gene expression of mitophagy markers (Prkn and Pink1) in SM and Parkin in liver in this study. Other mitochondrial dynamic markers, mtDNA, Mfn2, and Errα, were also increased by UA in SM or liver. EA, UA, and EA+UA did not change any of the mitochondrial markers we evaluated in E-fat.
We also evaluated the acute effects of EA and UA on mitochondrial respiration in primary hepatocytes isolated from mice fed the HF/HS diet. EA and UA undergo phase II metabolism after absorption and the biological activities of EA and UA could differ from their phase II metabolites.[39] We did not include EA and UA phase II metabolites here as primary hepatocytes carry out phase II metabolism.[40] In these experiments, maximal and ATP linked OCR together with proton leak were increased in primary hepatocytes from HF/HS mice compared with LF/LS mice. EA and EA+UA significantly decreased proton leak in primary hepatocytes form HF/HS mice. Mitochondrial proton leak account for about ≈25% resting oxygen consumption in liver and has been implicated in the development of IR.[41–45] However, the physiological relevance of mitochondrial respiratory and proton leak to IR is still controversial due to conflicting results. Increased intracellular lipid accumulation resulting from reduced oxidative capacity of mitochondria in liver was previously reported in IR rats.[46–48] On the other hand, higher resting metabolic rate has also been reported in mice of HFD induced IR compared with chow fed mice.[49] In addition, higher mitochondrial respiration was observed in liver of obese subjects with non-alcoholic fatty liver diseases compared with lean subjects, while significant higher proton leak was detected in obese subjects with non-alcoholic steatohepatitis but not obese subjects with non-alcoholic fatty liver diseases and lean individuals.[50] All these data suggest that mitochondrial phenotypes vary according to physiological status. In this study we observed an increase of mitochondrial respiratory and proton leak in primary hepatocytes as well as higher hepatic lipid content in HF/HS induced IR mice compared with LF/LS mice. Further investigations are needed to determine whether the increased mitochondrial respiratory and proton leak possibly resulted from the adaptation to hepatic lipid accumulation during chronic nutrient overload of HF/HS feeding, and whether this phenotype varies during IR progression. We also found that EA acutely reduced proton leak in primary hepatocytes from IR mice in vitro. The limitations of this experiment included 1) absence of dose-response studies; 2) studies of acute but not chronic effects; 3) lack of testing the responses in different mitochondrial respiratory pathways, such as those for fatty acids; and 4) normalizing our data based on cell number, but not mitochondrial density. Therefore, we do not have enough evidence to conclude that the acute effects of EA on proton leak of isolated hepatocytes is physiologically significant in terms of whole body metabolism and IR status, which need further studies about whole body metabolic parameters, mitochondrial respiratory, and proton leak in vital tissues including SM and liver.
In summary, our results show that both, EA and its microbial metabolite UA, induce differential metabolic benefits on IR. In addition, we demonstrated that UA is a more potent regulator of fasting blood glucose, adiponectin, and mitochondrial dynamic compared with EA. However, significant reduction of proton leak in primary hepatocytes was observed only with EA treatment. Most likely individuals who have the intestinal capacity to convert EA from dietary supplements and foods (walnuts, berries, and Pom) to UA will experience the most benefits in regards to glucose and energy metabolism and mitochondrial health. Since UA production decreases with age, these studies may have implications for the association of aging and IR.[51] Future studies are needed to understand how EA and UA-mediated changes of mitochondrial function contribute to the improvement of IR.
4. Experimental Section
Animal Study:
All mouse procedures were approved by the UCLA Animal Research Committee in compliance with the Association for Assessment and Accreditation of Laboratory Care International. 60 Male DBA/2J mice at 5–6 weeks of age were purchased from the Jackson Laboratory and housed in standard caging systems: four mice per cage on a 12 h light/12 h dark cycle at 22 °C. No special practice was performed to minimize coprophagia. After initial screening of urolithin production capability using pomegranate juice (PomJ), mice without urolithin production capability were switched to regular water and randomly assigned to 2 groups with similar body weight distribution in each group, and fed either a high-fat/high-sucrose (HF/HS: 42% energy from fat, 30% energy from sucrose, n = 48) or low-fat/low-sucrose (LF/LS: 13 energy from fat, 13% energy from sucrose, n = 12) diet (Envigo, WI, USA). Eight weeks later, IPGTT, and IPITT were performed to confirm the development of IR in HF/HS fed mice (Figures S1 and S2, Supporting Information). Mice fed the HF/HS-diet were further assigned to four groups: HF/HS-diet only, HF/HS-diet supplemented with 0.1% EA (94% EA, Ecological Formulas, CA, USA), HF/HS-diet supplemented with 0.1% UA (95% UA, Feitang, Shangdong, China), and HF/HS-diet supplemented with both 0.1% EA and 0.1% UA (n = 12 each), Figure S1, Supporting Information and Table S1, Supporting Information). Body weight and food intake were recorded weekly. After receiving 8 weeks of dietary EA, UA, and EA+UA supplementation, mice were fasted for 5 h and IPGTT and IPITT were performed.[52] Glucose and insulin were given at 1g kg−1 and 1U kg−1 body weight respectively. Mice were then euthanized, blood and tissues were collected, weighed, and stored at −80 °C until analysis.[53]
Dosage Information/Dosage Regimen:
At the beginning of the experiment, mice were given PomJ for 4 days and stool samples were collected every day to determine the urolithin production capability (Figure S1, Supporting Information). In humans, the oral consumption of Pom extract containing 57 mg ETs and 45mg EA for three days (≈0.95 mg ETs and 0.75 mg EA kg−1 body weight) was adequate to determine urolithin production capability in individuals with the necessary microbiota.[10] Mice with an average body weight of 20g drank about 3mL PomJ day−1 containing about 0.3mg EA and 1.1mg ETs, which equals to 1.22 mg ETs and 4.47 mg EA kg−1 body weight in humans, a much higher dose than reported to be an effective dose in humans to induce urolithin production.[10,54] In humans UA was well tolerated and significantly regulated fatty acid metabolism at the dose of 500–1000 mg day−1 (16.7 mg kg−1) via oral administration.[38] It was decided to supplement HF/HS diets with EA and/or UA at 0.1% as mice with average body weight of 25g eat about 4g diets, which was similar to 13.0 mg kg−1 in humans. For the in vitro study, we used EA and UA at the dose of 10 µm to test their acute effect on primary hepatocytes based on previous studies showing that UA dose dependently stimulated skeletal muscle (SM) mitophagy at the range of 10–50 µm. In addition, 1000 mg UA intake in human resulted in peak blood UA levels around 3.5 µm including free UA, UA glucuronide, and UA sulfate.[9,38,39]
Serum Biochemical markers:
Serum TG, total cholesterol and proinflammatory cytokines were measured as previously described.[53] Serum was diluted and FFA were measured using Free Fatty Acid Quantitation Kit (Sigma, Missouri, USA).
Tissue mRNA Extraction and RT-qPCR Assay:
Total RNA from liver and adipose tissues was isolated using RNeasy mini kit (Qiagen, Hilden, Germany). Total RNA from SM were isolated using RNeasy fibrous tissue mini kit (Qiagen). Genomic DNA was removed from RNA samples using TURBO DNA-free Kit (Thermofisher, MA, USA). RNA was then reverse transcripted into cDNA using a first strand cDNA synthesis kit (Clontech, CA, USA). Quantitative RT-PCR was performed using a SYBR green PCR master mix (Clontech) as previously described.[53] The mRNA levels of all genes were normalized using glyceraldehyde 3-phosphate dehydrogenase (Gapdh) as an internal control in liver and adipose tissues, and beta actin was used for SM. The primers used were shown in Table S2, Supporting Information.
Tissue DNA Extraction and mtDNA/nDNA Realtime-PCR:
total DNA was extracted from liver, adipose and SM using QIAamp DNA mini kit (Qiagen). Ratio of mitochondria DNA (mtDNA) to nuclear DNA (nDNA) was evaluated by realtime PCR as previously described.[55]
Primary Hepatocyte Isolation and Seahorse Assay:
Primary hepatocytes were isolated from LF/LS and HF/HS mice at the end of study with modified protocol as previously published.[56] Williams Medium E and CM3000 was used as plating medium, and Williams Medium E and CM4000 was used as cell maintain medium (Thermofisher, MA, USA). 9000 live cells per well were seeded in collagen coated XF96 plates. On the second day, cells from HF/HS mice were treated with 10 µm EA, 10 µm UA or 10 µm EA + 10 µm UA for 18–24 h. Mitochondrial function of treated cells was evaluated using Seahorse XF Cell Mito Stress Test Kit on Seahorse XFe96 Analyzer (Agilent, CA, USA) at Metabolism Core at UCLA. Data were normalized to cell number count.
Statistical Analysis:
Mean values and standard errors were calculated using descriptive statistics. For the IPITT test, two-way ANOVA analysis was performed to determine the effect of the treatment, time and their interactions on the outcome variables. The Greenhouse–Geisser correction was used if the sphericity assumption was violated. To identify the mean difference between EA, UA, EA+UA, and HF/HS fed groups, one-way ANOVA was used when data was normally distributed. Tukey–Kramer multiple comparison procedure was used for Post-hoc comparisons. Kruskal–Wallis test was used when data was not normally distributed. Student t-test or Mann–Whitney Rank test was used to evaluate the difference between LF/LS and HF/HS. p values < 0.05 was considered statistically significant. These statistical analyses were conducted using IBM SPSS Statistics version 23.
Supplementary Material
Acknowledgements
The authors thank Gail Thames for English editing. This project was supported by the Center for Human Nutrition, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles. Some spelling errors and the table 1 layout are corrected on September 29, 2020.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Jieping Yang, Center for Human Nutrition, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095, USA.
Yuanqiang Guo, State Key Laboratory of Medicinal Chemical Biology, College of Pharmacy and Tianjin Key Laboratory of Molecular Drug Research, Nankai University, Tianjin 300350, China.
Susanne M. Henning, Center for Human Nutrition, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095, USA
Brenda Chan, Center for Human Nutrition, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095, USA.
Jianfeng Long, Department of Clinical Nutrition, 2nd XiangYa Hospital, Central South University, Changsha 410011, China.
Jin Zhong, Department of Pathology and Laboratory Medicine, VA Greater Los Angeles Health Care System, Los Angeles CA 90095, USA.
Rebeca Acin-Perez, Division of Endocrinology, Department of Medicine and Department of Molecular and Medical Pharmacology, David Geffen School of Medicine at UCLA, Los Angeles CA 90095, USA.
Anton Petcherski, Division of Endocrinology, Department of Medicine and Department of Molecular and Medical Pharmacology, David Geffen School of Medicine at UCLA, Los Angeles CA 90095, USA.
Orian Shirihai, Division of Endocrinology, Department of Medicine and Department of Molecular and Medical Pharmacology, David Geffen School of Medicine at UCLA, Los Angeles CA 90095, USA.
David Heber, Center for Human Nutrition, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095, USA.
Zhaoping Li, Center for Human Nutrition, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095, USA; Department of Medicine, VA Greater Los Angeles Health Care System, Los Angeles CA 90095, USA.
Data Availability Statement
Data available on request from the authors.
References
- [1].Banihani S, Swedan S, Alguraan Z, Nutr. Res 2013, 33, 341. [DOI] [PubMed] [Google Scholar]
- [2].Belwal T, Nabavi SF, Nabavi SM, Habtemariam S, Nutrients 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Larrosa M, Garcia-Conesa MT, Espin JC, Tomas-Barberan FA, Mol. Aspects Med 2010, 31, 513. [DOI] [PubMed] [Google Scholar]
- [4].Bakkalbasi E, Mentes O, Artik N, Crit. Rev. Food Sci. Nutr 2009, 49, 283. [DOI] [PubMed] [Google Scholar]
- [5].Seeram NP, Aronson WJ, Zhang Y, Henning SM, Moro A, Lee RP, Sartippour M, Harris DM, Rettig M, Suchard MA, Pantuck AJ, Belldegrun A, Heber D, J. Agric. Food Chem 2007, 55, 7732. [DOI] [PubMed] [Google Scholar]
- [6].Adams LS, Seeram NP, Aggarwal BB, Takada Y, Sand D, Heber D, J. Agric. Food Chem 2006, 54, 980. [DOI] [PubMed] [Google Scholar]
- [7].Hollebeeck S, Winand J, Herent MF, During A, Leclercq J, Larondelle Y, Schneider YJ, Food Funct 2012, 3, 875. [DOI] [PubMed] [Google Scholar]
- [8].Bialonska D, Kasimsetty SG, Schrader KK, Ferreira D, J. Agric. Food Chem 2009, 57, 8344. [DOI] [PubMed] [Google Scholar]
- [9].Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, Aebischer P, Sandi C, Rinsch C, Auwerx J, Nat. Med 2016, 22, 879. [DOI] [PubMed] [Google Scholar]
- [10].Gonzalez-Sarrias A, Garcia-Villalba R, Romo-Vaquero M, Alasalvar C, Orem A, Zafrilla P, Tomas-Barberan FA, Selma MV, Es-pin JC, Mol. Nutr. Food Res 2017, 61, 1600830. [DOI] [PubMed] [Google Scholar]
- [11].Boehning AL, Essien SA, Underwood EL, Dash PK, Boehning D, Biomed. Pharmacother 2018, 106, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kang I, Kim Y, Tomas-Barberan FA, Espin JC, Chung S, Mol. Nutr. Food Res 2016, 60, 1129. [DOI] [PubMed] [Google Scholar]
- [13].Garcia-Villalba R, Beltran D, Espin JC, Selma MV, Tomas-Barberan FA, J. Agric. Food Chem 2013, 61, 8797. [DOI] [PubMed] [Google Scholar]
- [14].Selma MV, Beltran D, Luna MC, Romo-Vaquero M, Garcia-Villalba R, Mira A, Espin JC, Tomas-Barberan FA, Front. Microbiol 2017, 8, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Z, Henning SM, Lee RP, Lu QY, Summanen PH, Thames G, Corbett K, Downes J, Tseng CH, Finegold SM, Heber D, Food Funct 2015, 6, 2487. [DOI] [PubMed] [Google Scholar]
- [16].Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI, Nature 2006, 444, 1027. [DOI] [PubMed] [Google Scholar]
- [17].Gonzalez-Barrio R, Edwards CA, Crozier A, Drug Metab. Dispos 2011, 39, 1680. [DOI] [PubMed] [Google Scholar]
- [18].Selma MV, Beltran D, Garcia-Villalba R, Espin JC, Tomas-Barberan FA, Food Funct 2014, 5, 1779. [DOI] [PubMed] [Google Scholar]
- [19].Cisneros-Zevallos L, Bang WY, Delgadillo-Puga C, Int. J. Mol. Sci 2020, 21, 2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Andrikopoulos S, Massa CM, Aston-Mourney K, Funkat A, Fam BC, Hull RL, Kahn SE, Proietto J, J. Endocrinol 2005, 187, 45. [DOI] [PubMed] [Google Scholar]
- [21].Cruciani-Guglielmacci C, Bellini L, Denom J, Oshima M, Fernan-dez N, Normandie-Levi P, Berney XP, Kassis N, Rouch C, Dairou J, Gorman T, Smith DM, Marley A, Liechti R, Kuznetsov D, Wigger L, Burdet F, Lefevre AL, Wehrle I, Uphues I, Hildebrandt T, Rust W, Bernard C, Ktorza A, Rutter GA, Scharfmann R, Xenarios I, Le Stunff H, Thorens B, Magnan C, Ibberson M, Mol. Metab 2017, 6, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schurmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Muller TD, Tschop MH, Nat. Rev. Endocrinol 2018, 14, 140. [DOI] [PubMed] [Google Scholar]
- [23].Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N, Diabetologia 2013, 56, 1129. [DOI] [PubMed] [Google Scholar]
- [24].Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, Castellani LW, Rau CD, Pan C, Phun J, Zhou ZQ, Yang WP, Neuhaus I, Gargalovic PS, Kirchgessner TG, Graham M, Lee R, Tontonoz P, Gerszten RE, Hevener AL, Lusis AJ, Cell Metab 2015, 21, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kooptiwut S, Zraika S, Thorburn AW, Dunlop ME, Darwiche R, Kay TW, Proietto J, Andrikopoulos S, Endocrinology 2002, 143, 2085. [DOI] [PubMed] [Google Scholar]
- [26].Nunemaker CS, Chung HG, Verrilli GM, Corbin KL, Upad-hye A, Sharma PR, J. Endocrinol 2014, 222, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Garcia-Munoz C, Vaillant F, Crit. Rev. Food Sci. Nutr 2014, 54, 1584. [DOI] [PubMed] [Google Scholar]
- [28].Pacini G, Omar B, Ahren B, J. Diabetes Res 2013, 2013, 986906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee CJ, Sears CL, Maruthur N, Ann. N. Y. Acad. Sci 2020, 1461, 37. [DOI] [PubMed] [Google Scholar]
- [30].Lihn AS, Pedersen SB, Richelsen B, Obes. Rev 2005, 6, 13. [DOI] [PubMed] [Google Scholar]
- [31].Combs TP, Marliss EB, Rev. Endocr. Metab. Disord 2014, 15, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim JY, De Wall EV, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE, J. Clin. Invest 2007, 117, 2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Delarue J, Magnan C, Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142. [DOI] [PubMed] [Google Scholar]
- [34].Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS, Annu. Rev. Nutr 2007, 27, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Stepien KM, Heaton R, Rankin S, Murphy A, Bentley J, Sexton D, Hargreaves P, J. Clin. Med 2017, 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bhatti JS, Bhatti GK, Reddy PH, Biochim Biophys Acta Mol. Basis Dis 2017, 1863, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gao AW, Canto C, Houtkooper RH, EMBO Mol. Med 2014, 6, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, Auwerx J, Singh A, Rinsch C, Nat. Metabol 2019, 1, 595. [DOI] [PubMed] [Google Scholar]
- [39].Gonzalez-Sarrias A, Gimenez-Bastida JA, Nunez-Sanchez MA, Larrosa M, Garcia-Conesa MT, Tomas-Barberan FA, Espin JC, Eur. J. Nutr 2014, 53, 853. [DOI] [PubMed] [Google Scholar]
- [40].Gebhardt R, Hengstler JG, Muller D, Glockner R, Buenning P, Laube B, Schmelzer E, Ullrich M, Utesch D, Hewitt N, Ringel M, Hilz BR, Bader A, Langsch A, Koose T, Burger HJ, Maas J, Oesch F, Drug Metab. Rev 2003, 35, 145. [DOI] [PubMed] [Google Scholar]
- [41].Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD, Essays Biochem 2010, 47, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Brand MD, Brindle KM, Buckingham JA, Harper JA, Rolfe DF, Stuart JA, Int. J. Obes. Relat. Metab. Disord 1999, 23, S4. [DOI] [PubMed] [Google Scholar]
- [43].Rolfe DF, Newman JM, Buckingham JA, Clark MG, Brand MD, Am. J. Physiol 1999, 276, C692. [DOI] [PubMed] [Google Scholar]
- [44].Chan CB, Harper ME, Curr. Diabetes Rev 2006, 2, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ramsey JJ, Harper ME, Humble SJ, Koomson EK, Ram JJ, Bevilacqua L, Hagopian K, Comp. Biochem. Physiol. B Biochem. Mol. Biol 2005, 140, 99. [DOI] [PubMed] [Google Scholar]
- [46].Vial G, Dubouchaud H, Leverve XM, Acta Biochim. Pol 2010, 57, 389. [PubMed] [Google Scholar]
- [47].Vial G, Dubouchaud H, Couturier K, Cottet-Rousselle C, Taleux N, Athias A, Galinier A, Casteilla L, Leverve XM, J. Hepatol 2011, 54, 348. [DOI] [PubMed] [Google Scholar]
- [48].Franko A, Kunze A, Bose M, von Kleist-Retzow JC, Paulsson M, Hartmann U, Wiesner RJ, Int. J. Mol. Sci 2017, 18, 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Burnett CM, Grobe JL, Mol. Metab 2014, 3, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, Roden M, Cell Metab 2015, 21, 739. [DOI] [PubMed] [Google Scholar]
- [51].Cortes-Martin A, Garcia-Villalba R, Gonzalez-Sarrias A, Romo-Vaquero M, Loria-Kohen V, Ramirez-de-Molina A, Tomas-Barberan FA, Selma MV, Espin JC, Food Funct 2018, 9, 4100. [DOI] [PubMed] [Google Scholar]
- [52].Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J, Am. J. Physiol. Endocrinol. Metab 2008, 295, E1323. [DOI] [PubMed] [Google Scholar]
- [53].Yang J, Zhang S, Henning SM, Lee R, Hsu M, Grojean E, Pisegna R, Ly A, Heber D, Li Z, J. Nutr. Biochem 2018, 52, 62. [DOI] [PubMed] [Google Scholar]
- [54].Nair AB, Jacob S, J. Basic Clin. Pharm 2016, 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu W, Ruiz-Velasco A, Wang S, Khan S, Zi M, Jungmann A, Camacho-Munoz MD, Guo J, Du GH, Xie LP, Oceandy D, Nicolaou A, Galli G, Muller OJ, Cartwright EJ, Ji Y, Wang X, Nat. Commun 2017, 8, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Akie TE, Cooper MP, J. Vis. Exp 2015, 102, e52982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request from the authors.