

Abstract
The x-ray crystal structure is presented for a nitrogenase MoFe protein where the alpha subunit residue at position 70 (α-70Val) has been substituted by the amino acid isoleucine (α-70Ile). Substitution of ?-70Val by ?-70Ile results in a MoFe protein that is hampered in its ability to reduce a range of substrates including acetylene and N2, yet retains normal proton reduction activity. The 2.3 Å structure of the α-70Ile MoFe protein is compared to the α-70Val wild type MoFe protein, revealing that the δ methyl group of ?-70Val is positioned over Fe 6 within the active site FeMo-cofactor. This work provides strong crystallographic support for the previously proposed model that substrates bind and are reduced at a single 4Fe-4S face of the FeMo-cofactor and that when ?-70Val is substituted by ?-70Ile access of substrates to Fe6 of this face is effectively blocked. Furthermore the detailed examination of the structure provides the basis for understanding the ability to trap and characterize hydrides in the variant, contributing significantly to our understanding of substrate access and substrate reduction at the FeMo-cofactor active site of nitrogenase.
Keywords: Nitrogenase, MoFe protein, FeMo-cofactor, Proton reduction, Hydride intermediate
Introduction
The enzyme nitrogenase catalyzes the reduction of N2 to two ammonia molecules in a reaction having an ideal stoichiometry shown in equation 1.
| Eq 1 |
In the Mo-dependent nitrogenase, this reaction is catalyzed by two component proteins called the Fe protein and the MoFe protein [1]. The Fe protein delivers electrons to the MoFe protein in a reaction dependent on the hydrolysis of MgATP. The MoFe protein contains the site of substrate binding, which occurs at a heterometallic cofactor called FeMo-cofactor [7Fe-9S-Mo-X-homocitrate] [2,3]. FeMo-cofactor contains many possible sites of N2 binding including three identical 4Fe-4S faces and Mo [4,5]
The x-ray structure of MoFe proteins shows FeMo-cofactor is covalently bound to the MoFe protein through Cys ligation to an Fe at one end and through His ligation to the Mo on the other end [6,7]. The MoFe protein environment surrounding FeMo-cofactor is asymmetric, with a wide range of different amino acids defining the first shell of non-covalent interactions. In an effort to define where and how substrates interact with FeMo-cofactor, amino acids within this region have been substituted with different amino acids [8–13]. This approach was used to identify ?-70Val as a residue that controls substrate access to FeMo-cofactor [14,15]. The side chain of ?-70Val is in close contact to one Fe-S face of FeMo-cofactor (Fe atoms 2, 3, 6, and 7 in the numbering system established in 1M1N.PDB) [7], indicating that it could control access of small molecules to the site of substrate interaction on FeMo-cofactor. To test this model, α-70Val was substituted by a variety of different amino acids [16]. Substitution of ?-70Val by residues with smaller side chains resulted in a capacity to reduce compounds that are normally very poor substrates for nitrogenase, such as hydrazine (H2N-NH2), propyne (HC≡C-CH3), propargyl alcohol (HC≡C-CH2OH), and 1-butyne (HC≡C-CH2CH3) [17]. In contrast, when the α-70Val is substituted by α-70Ile, with a larger side chain, the resulting MoFe protein showed a significantly lowered activity for reduction of a number of substrates except protons [16]. In the absence of other substrates, nitrogenase reduces protons to make H2 (eq 1), and thus this reaction is an important control that indicates the overall catalytic function of nitrogenase. In the case of the α-70Ile substituted MoFe protein normal proton reduction activity was observed, confirming that the underlying catalytic functions of the enzyme are undisturbed. The findings with the α-70Ala, α-70Gly, and α-70Ile substituted MoFe proteins were interpreted as evidence that the residue at position α-70 controls access to FeMo-cofactor for binding of both alkyne and nitrogenous substrates, suggesting that substrate binding occurs at a specific FeS cluster face of FeMo-cofactor (Fe atoms 2, 3, 6, 7) [18]. Subsequent studies on a state of the α-70Ala MoFe protein with the substrate propargyl alcohol and propargyl amine trapped suggested that a specific Fe atom (number 6) might be the site of propargyl alcohol binding [19]. The α-70Ile MoFe protein could be freeze trapped during turnover under argon (reducing protons to make H2) in an EPR active state that was assigned to FeMo-cofactor with one or two hydrides bound [20]. It was not clear why the α-70Ile MoFe protein trapped these likely intermediates during the H2 evolution reaction.
Here, we report the x-ray crystal structure of the α-70Ile variant MoFe protein at 2.3 Å resolution. A comparison of the structure of the α-70Ile MoFe protein with the wild type MoFe protein reveals a likely explanation for the earlier observations regarding substrate interactions in the α-70Ile MoFe protein, in turn providing new insights into a specific site on FeMo-cofactor that could constitute the site of substrate interaction and suggests a role for the α-70Val as a gate keeper. Further, the α-70Ile structure suggests an explanation for why the hydride intermediate is trapped in this variant MoFe protein.
Materials and Methods
The α-70Ile MoFe protein with a polyhistidine tag near the carboxyl terminus of the nifD gene was purified from Azotobacter vinalandii strain DJ1373 using a metal affinity chromatography method as described earlier [21]. All manipulations were done in the absence of O2 under an argon atmosphere using gas tight syringes to transfer solutions and gases. The α-70Ile MoFe protein was judged to be > 95% homogeneous from Coomassie blue staining of SDS-gels. The final protein was in Tris-HCl buffer, pH 8.0, with 250 mM NaCl, 2 mM dithionite. The protein had a specific activity for H2 evolution of greater than 2200 nmol/min/mg MoFe protein (95 % of the wild type specific activity).
For crystallization, the protein was diluted to 38 mg/mL in 50 mM Tris buffer, pH 8.0, and 250 mM NaCl before setting up trials under anaerobic conditions in a nitrogen atmosphere glove box (UniLAB, MBRAUN, NH) using a micro-capillary batch diffusion method [22]. The α-70Ile variant of the MoFe protein crystallized in 30% polyethylene glycol (PEG) 4000, 100 mM Tris (pH 8.0), 170–190 mM sodium molybdate and 1mM dithionite over a period of 3–4 weeks to give dark brown crystals of ~100 × 200 × 200 µm. The crystals were flash frozen in liquid nitrogen on rayon loops before data collection on beam line 11–1 at the Stanford Synchrotron Radiation Laboratory under a continuous flow of liquid nitrogen at 100 K. A single wavelength data set was collected λ = 0.89 up to a resolution 2.3 Å and the data was integrated and scaled using the HKL2000 software package [23]. The data statistics are summarized in Table 1. Calculation of the Matthews coefficient [24,25] and consideration of reasonable solvent content of the crystals suggested that each asymmetric unit of the crystal contained one α2β2 heterotetramer.
Table 1:
Data Statistics
| Cell dimensions | a = 77.02 Å |
| b = 129.36 Å | |
| c = 107.09 Å | |
| α = γ = 90.00° | |
| β = 109.01 | |
| space group | P21 |
| Wavelength (Å) | λ1 = 0.88557 |
| Resolution (Å) | 50 – 2.3 |
| Completeness (%) | 99.9 (99.9)a |
| Observed reflections | 361917 |
| Unique reflections | 187881 (18797) |
| Average redundancy | 3.8 (3.5) |
| I/σ | 11.8 (2.7) |
| Rsymb (%) | 11.5 (23.3) |
|
| |
| Refinement Statistics | |
|
| |
| Resolution (Å) | 26.6 – 2.3 |
| Rcyrstc (%) | 22.7 |
| Rfreec (%) | 27.8 |
| Real Space CCd (%) | 91.1 |
| Mean B Value (Å2 ) | 26.6 |
| Coordinate Error (based on maximum likelihood, Å) | 0.22 |
| RMSD from ideality: | |
| Bonds (Å) | 0.007 |
| Angles (°) | 2.812 |
| Ramchandran statisticse: | |
| Most favored (%) | 97.3 |
| Additional allowed (%) | 2.65 |
| Outliers (%) | 0.05 |
Numbers in parenthesis refer to the highest resolution shell.
where Ii(h) is the ith measurement of reflection h and <I(h)> is the average value of the reflection intensity.
where Fo and Fc are the observed and calculated structure factor amplitudes used in refinement. Rfree is calculated as Rcryst, but using the “test” set of structure factor amplitudes that were withheld from refinement.
Correlation coefficient (CC) is agreement between the model and 2mFo-DFc electron density map.
Calculated using Molprobity [32]
AutoMR of CCP4 suite of programs [26] was used to accomplish molecular replacement using the α2β2 MoFe protein heterotetramer (PDB ID: 2MIN) [27] as the starting model. The search resulted in a solution with Rcryst of 30.0% and correlation coefficient of 70%, and the model was further refined in REFMAC5 [28] to improve the quality of the model. The refinement runs made use of medium noncrystallographic symmetry (NCS) restraints (medium for main chains and loose for side chains), B-factor restraints and 2 screw (Translation/Libration/ Screw) tensors (per polypeptide chain). The final model was refined to Rcryst of 22.7% and obeys good stereochemistry with up to 97.3% of all residues in the Ramchandran [29] allowed regions (Table 1). All figures in the manuscript were generated using PyMOL [30].
Results and Discussion
Structure of the α-70Ile MoFe protein
The x-ray structure of α-70Ile MoFe protein from Azotobacter vinelandii refined to 2.3 Å shows a very similar overall structure when compared to the wild-type MoFe protein [7]. The protein was crystallized under strict anaerobic conditions in the presence of 2 mM dithionite to capture the reduced form and has been refined to an Rcryst of 22.7%. The structure exhibits good geometry with 97.3% of all residues in the Ramchandran [29] allowed regions (Table 1). Serine 255 in the β-subunit lies in the Ramchandran disallowed region in the structure reported here and this distortion in the protein stereo chemistry has been previously observed in the wild-type structure as well [7]. The distortion in the protein back bone in this region probably arises from steric hindrance from the surrounding residues in chain B. The overall structure of the α-70Ile MoFe protein is quite similar to the wild type protein with the secondary structure elements overlaying with significant similarities. Most of the differences are seen in the FeMo-cofactor binding region and details of the differences are highlighted in the following sections.
Insights into the location of substrate binding on FeMo-cofactor
The secondary structure of the α-70Ile MoFe protein was compared to the structure of the wild-type MoFe protein (1MIN.PDB) using the protein structure comparison service SSM at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm) [31]. An examination of the root mean square standard deviation (RMSD) among all residues reveals the near identity of the structures, with an average RMSD of 0.2 Å. The close structural agreement between the α-70Ile and wild-type MoFe proteins is evident from the small RMSD values. A short stretch of amino acids in the α-subunit between residues 105–120 shows slightly higher deviations compared to all other residues. These residues are located on the surface of the MoFe protein, and thus some variation in the position of the residues in this region could be expected.
Residues that define the FeMo-cofactor binding pocket between the wild-type and α-70Ile MoFe proteins are shown in Fig. 1. Near identity in the positions of most amino acids in this region is observed, with a few exceptions. The side chains of α-96Arg, α-359Arg, α-195His refine to slightly different positions in the α-70Ile MoFe protein structure relative to the wild-type MoFe protein structure and these differences are supported by the electron density maps in this region. The position of the α-70Val and α-70Ile side chains is seen to be nearly identical, with the methyl group (Cδ1) on the isoleucine approaching Fe 6 of FeMo-cofactor (Fig. 2). The distance between the Cδ1 of α-70Ile and the Fe 6 is found to be 3.9 Å, confirming the close contact between the isoleucine side chain and this Fe atom (Table 2).
Figure 1:
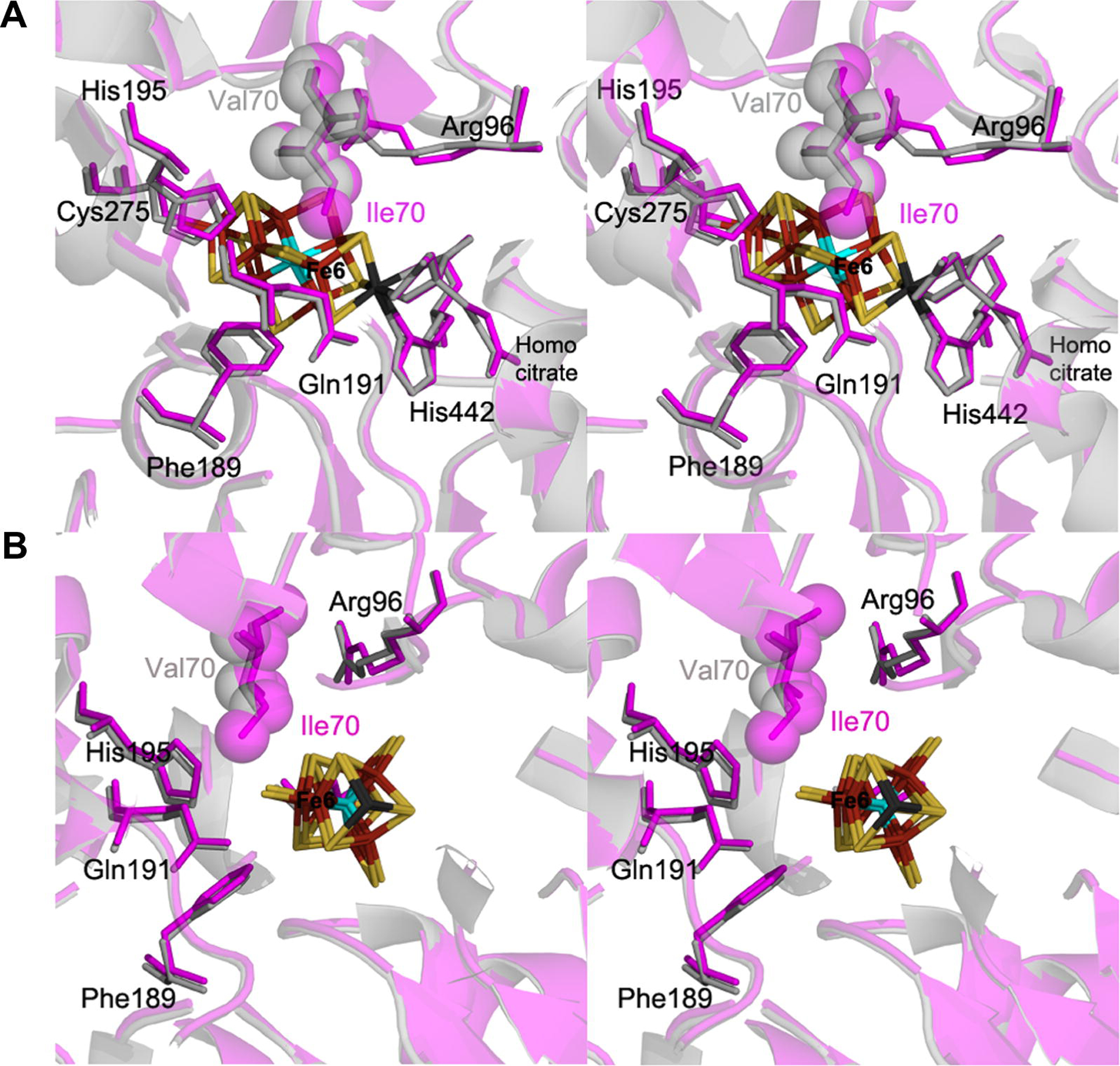
Stereoviews of the protein environment surrounding the FeMo-cofactor. Shown is a portion of α-carbon trace and select amino acid side chains for both the α-70Ile (magenta) and wild type (cyan) MoFe proteins in two orientations (A and B) separated by an ~90? rotation. The protein backbone is shown as ribbons, with the residues immediately surrounding FeMo-cofactor from the α-subunit and the FeMo-cofactor shown in line angle representations with the exception of either the α-70Val (wild-type) side chain and the α-70Ile (variant) side chain shown in space filling / van der Waals representations. Fe atoms are colored in rust, S in yellow, Mo in black,and X in cyan.
Figure 2:
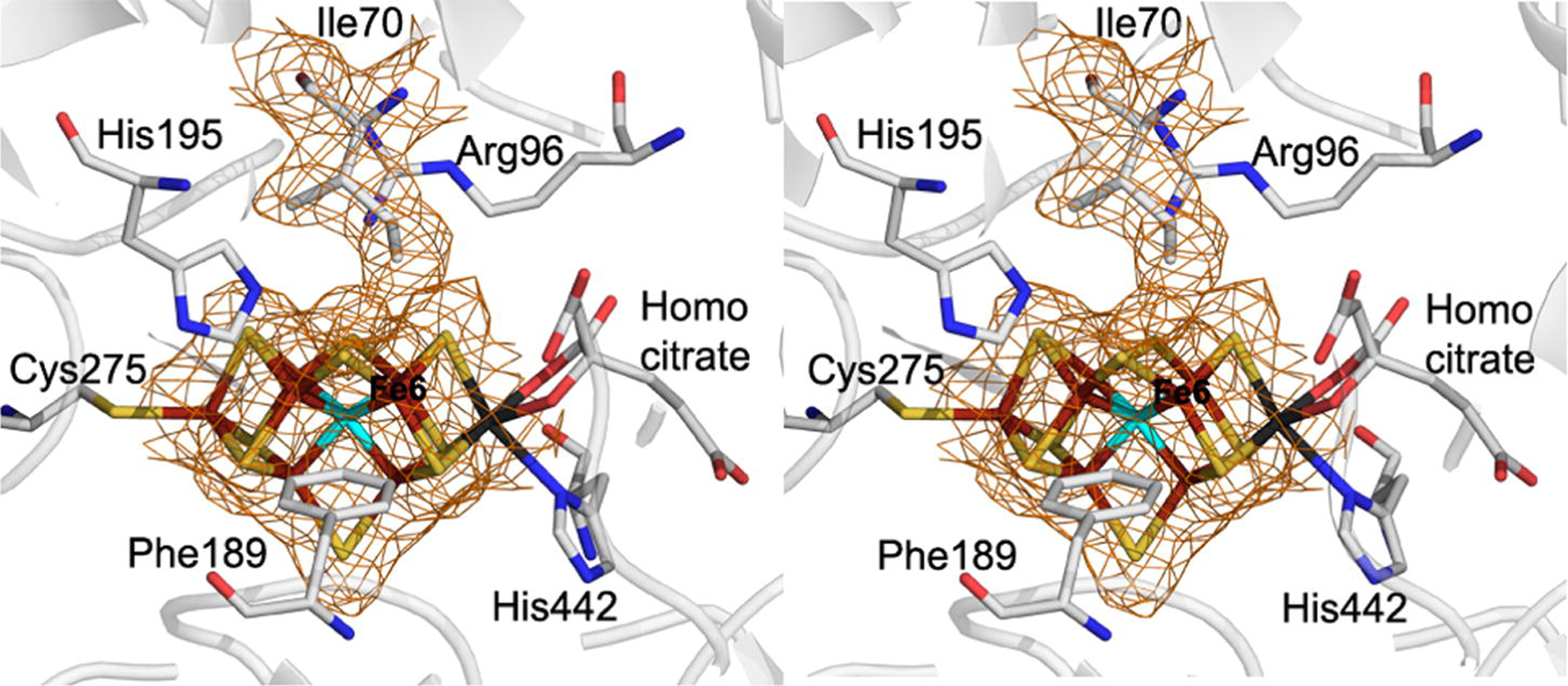
Stereoview of the 2Fo-Fc electron density maps (1.0σ) of the region surrounding the FeMo-cofactor and the adjacent α-70Ile residue. The protein backbone is shown in ribbons with residues in the FeMo-cofactor environment from the α-subunit are shown in line angle representations color coded with Fe in rust, S in yellow, C in grey, N in blue, O in red, and Mo in black.
Table 2 :
Distances between α-70Val and α-70Ile side chain groups and the FeMo-cofactor
| Native MoFe protein interactions |
α-70Ile MoFe protein interactions |
||
|---|---|---|---|
| Interaction | Distance (Å) | Interaction | Distance (Å) |
| α-70Val (Cγ1) – Fe2 | 4.4 | α-70Ile (Cγ2) – Fe2 | 4.6 |
| α-70Val (Cγ1) – Fe3 | 5.2 | α-70Ile (Cγ2) – Fe3 | 5.1 |
| α-70Val (Cγ2) – Fe6 | 4.4 | α-70Ile (Cγ1) – Fe6 | 4.0 |
| α-70Val (Cγ2) – Fe7 | 5.5 | α-70Ile (Cδ1) – Fe6 | 3.9 |
| α-70Ile (Cγ1) – Fe7 | 5.1 | ||
| α-70Ile (Cδ1) – Fe7 | 5.5 | ||
The position of Cδ1 in α-70Ile can be considered in light of studies indicating that the same Fe atom is a site of substrate interactions. When α-70Val was substituted by α-70Ile, the MoFe protein showed much lower activity for reduction of most compounds that are effective substrates for the wild-type MoFe protein. Proton reduction rates are unaffected by the α-70Ile substitution. A simple explanation for this observation is that the Cδ1 on α-70Ile has blocked substrates (except protons) from accessing the binding site on FeMo-cofactor. This model was further supported by the observation that when α-70Val is substituted by amino acids having smaller side chains (e.g., alanine or glycine), then larger compounds that are normally poor substrates for nitrogenase can be actively reduced. For example, the α-70Ala MoFe protein is found to reduce larger alkynes such as propyne (HC≡C-CH3) and propargyl alcohol (HC≡C-CH2-OH), whereas these compounds are only poor substrates in the wild-type MoFe protein [17]. Likewise, the nitrogenous substrate hydrazine (H2N-NH2) can be reduced at much higher rates by the α-70Ala MoFe protein when compared to the wild-type MoFe protein [18]. In the α-70Gly MoFe protein, 1-butyne (HC≡C-CH2-CH3) is found to be a substrate, whereas this compound is only reduced at very low rates by the wild-type MoFe protein. Taken together, these results can be explained if the side chain at α-70 acts as a gate-keeper, controlling the size of substrates that can gain access to a substrate binding site on FeMo-cofactor. Given the location of the side chain of α-70 over one Fe-S face of FeMo-cofactor, the simplest interpretation of the results is that this FeS face constitutes a substrate binding location.
Other studies also pointed to Fe atom 6 of FeMo-cofactor as a possible substrate binding location. For example, it was found that propargyl alcohol under turnover conditions can be freeze trapped on FeMo-cofactor in the α-70Ala MoFe protein [4]. Using isotopically labeled substrate (13C and 1/2H) and electron-nuclear double resonance (ENDOR) spectroscopy, it was concluded that the species bound to FeMo-cofactor was the partially reduced allyl alcohol (H2C=CH-CH2OH) [4]. It was further concluded that allyl alcohol was bound side-on between the first and second carbons to one or two Fe atoms. Considering the wild-type structure of the MoFe protein, molecular mechanics calculations were done to try to further refine where on the FeS face of FeMo-cofactor the allyl alcohol was likely to be bound [19]. A central observation to these calculations was the proposed role of the side chain of α-195His in H bonding to the OH of allyl alcohol in stabilizing the bound state, favoring binding of substrates to Fe 6 compared to the other Fe atoms on this FeS face.
In a separate study, it was found that the α-70Gly substituted MoFe protein can reduce 1-butyne (HC≡C-CH2CH3) but not 2-butyne (H3C-C≡C-CH3). To explain these results, both 1-butyne and 2-butyne were modeled into the wild type structure bound side-on to Fe6 of FeMo-cofactor [18], which indicated 2-butyne cannot bind because of steric overlap with the side chain of α-191Gln. This model predicted that substitution of α-191Gln by α-191Ala might allow 2-butyne to fit into a position to bind side-on to Fe6. The α-191Ala substituted MoFe protein was found to reduce 2-butyne at considerable rates, also locating the substrate binding site to Fe 6 [18]. Finally, a density functional theory study favors binding of alkynes such as propargyl alcohol to Fe 6 [19]. The x-ray structure of the α-70Ile MoFe protein presented here, coupled with the earlier studies, provides strong evidence for Fe 6 acting as a specific binding site for substrate interactions on FeMo-cofactor
Insights into a hydride trapped state on the α-70Ile MoFe protein
A spectroscopic investigation of the α-70Ile MoFe protein revealed that when this protein is freeze quenched during turnover conditions, a unique EPR active state is trapped that has been assigned to one or two hydrides bound to the FeMo-cofactor (modeled as bound to one or more Fe atoms). It is not clear why the α-70Ile amino acid substitution should result in the trapping of hydrides to FeMo-cofactor, although there are several possible explanations. One model is that a proton is bound to FeMo-cofactor (formally as a hydride with electrons coming from FeMo-cofactor) and that the addition of the second proton needed to form H2 is restricted by the additional methyl group of Ile, allowing the hydride bound state to accumulate. The second proton could come either directly from the protein through the protein backbone or via a proton relay through amino acid side chains such as the α-195His residue, or from a large pool of waters located around R-homocitrate (Fig. 3).
Figure 3:
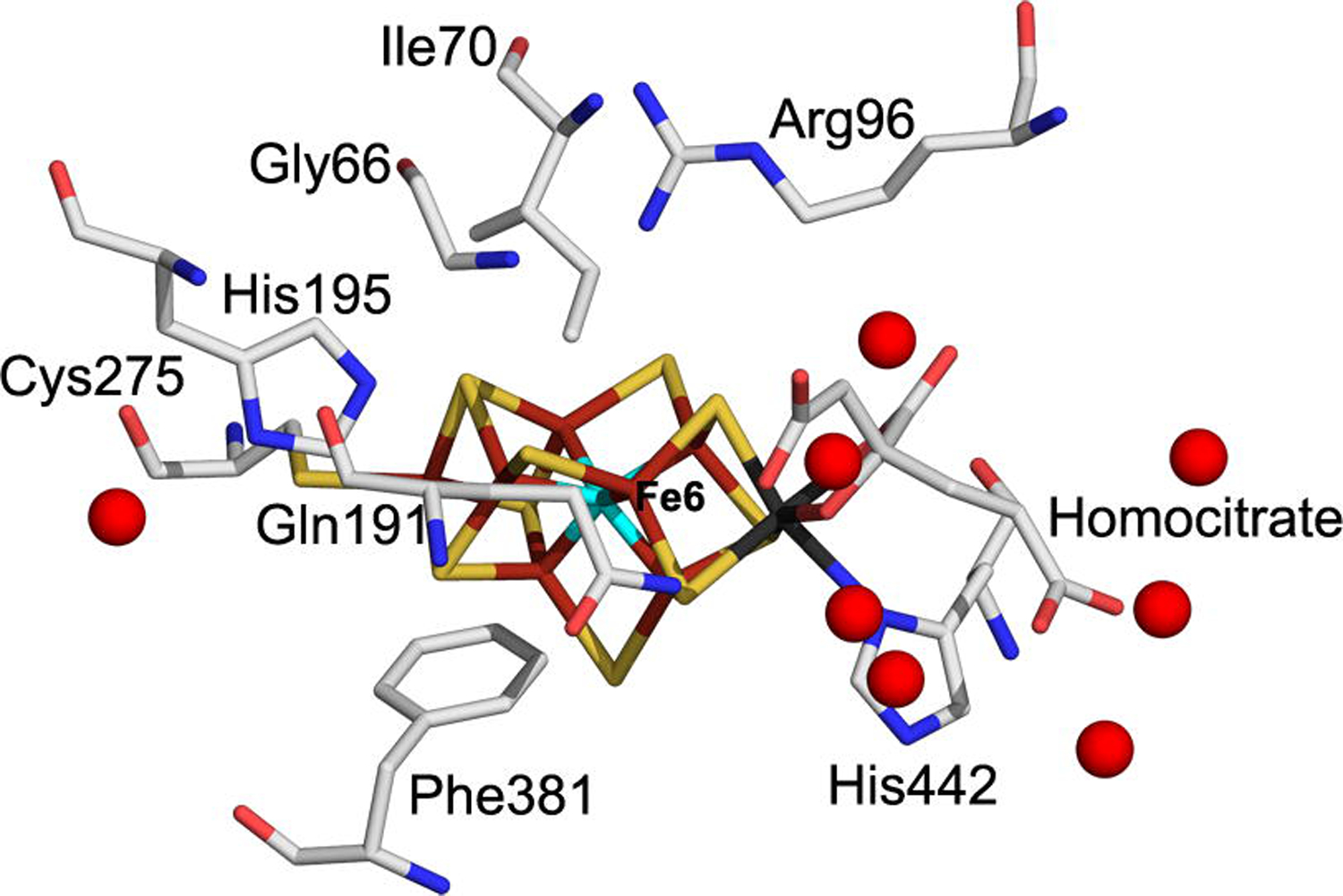
FeMo-cofactor binding site in the the α-70Ile MoFe protein structure showing the location of the surrounding water molecules represented as red spheres. The residues in the FeMo-cofactor and surrounding protein environment shown with line angle representations color coded as in Figure 2..
Here we consider the possibility that the side chain of α-70Ile might obstruct the proton flow from the pool of waters near R-homocitrate. Other protein components that would be predicted to define the flow of protons from the water pool are the α carbon of α-66Gly, the β carbon of α-191Gln, the ε nitrogen of α-195His, and part of the R-homocitrate. Two different models could be considered to explain how the α-70Ile might block the flow of these protons. One model is that the methyl group of isoleucine might block proton transfer from the α-195His residue to a hydride bound to Fe 6. The α-195His residue has been previously implicated as a possible proton donor for the reduction of nitrogenous substrates. Alternatively, if a hydride is bound to Fe2, then the α-70Ile could block the proton flow through waters that reside next to R-homocitrate. Both models would explain how a hydride bound state might populate when the α-70Ile MoFe protein is trapped during turnover.
Summary
The 2.3 Å structure of the α-70Ile MoFe protein variant is presented. The localization of the side chain of isoleucine in the structure explains earlier kinetic studies that indicated that the side chain of the amino acid at position α-70 acts as a gate keeper to control access of substrates to a binding location on FeMo-cofactor. Based on the location of the side chain of α-70Ile directly over Fe6 and previous molecular mechanics studies, a model is constructed for substrate interactions at this specific Fe site. Further, the structure offers insights into the observation of trapping of hydrides on the α-70Ile MoFe protein when it is frozen during H2 evolution.
Acknowledgments
This work was supported by National Institutes of Health Grants R01-GM069938 (JWP) and R01-GM59087 (LCS and DRD). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The NASA Astrobiology Biogeocatalysis Research Center (NNA08CN85A) is supported by the NASA Astrobiology Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The coordinates for this structure have been submitted to the RCSB Protein Data Bank for release upon submission (PDB code 3K1A).
References
- [1].Peters JW, Szilagyi RK, Curr. Opin. Chem. Biol 10 (2006) 101–108. [DOI] [PubMed] [Google Scholar]
- [2].Barney BM, Lee HI, Dos Santos PC, Hoffman BM, Dean DR, Seefeldt LC, Dalton Trans (2006) 2277–2284. [DOI] [PubMed] [Google Scholar]
- [3].Howard JB, Rees DC, Proc. Natl. Acad. Sci. USA 103 (2006) 17088–17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee HI, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM, J. Am. Chem. Soc 126 (2004) 9563–9569. [DOI] [PubMed] [Google Scholar]
- [5].Dance J Am. Chem. Soc 126 (2004) 11852–11863. [DOI] [PubMed] [Google Scholar]
- [6].Peters JW, Fisher K, Newton WE, Dean DR, J. Biol. Chem 270 (1995) 27007–27013. [DOI] [PubMed] [Google Scholar]
- [7].Einsle O, Tezcan FA, Andrade SL, Schmid B, Yoshida M, Howard JB, Rees DC, Science 297 (2002) 1696–1700. [DOI] [PubMed] [Google Scholar]
- [8].Scott DJ, Dean DR, Newton WE, J. Biol. Chem 267 (1992) 20002–20010. [PubMed] [Google Scholar]
- [9].Dean DR, Setterquist RA, Brigle KE, Scott DJ, Laird NF, Newton WE, Mol. Microbiol 4 (1990) 1505–1512. [DOI] [PubMed] [Google Scholar]
- [10].Govezensky D, Zamir A, J. Bacteriol 171 (1989) 5729–5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kent HM, Ioannidis I, Gormal C, Smith BE, Buck M, Biochem. J 264 (1989) 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Scott DJ, May HD, Newton WE, Brigle KE, Dean DR, Nature 343 (1990) 188–190. [DOI] [PubMed] [Google Scholar]
- [13].Kim CH, Newton WE, Dean DR, Biochemistry 34 (1995) 2798–2808. [DOI] [PubMed] [Google Scholar]
- [14].Benton PM, Laryukhin M, Mayer SM, Hoffman BM, Dean DR, Seefeldt LC, Biochemistry 42 (2003) 9102–9109. [DOI] [PubMed] [Google Scholar]
- [15].Seefeldt LC, Hoffman BM, Dean DR, Annu. Rev. Biochem 78 (2009) 701–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC, L.C. J. Biol. Chem 279 (2004) 53621–53624. [DOI] [PubMed] [Google Scholar]
- [17].Mayer SM, Niehaus WG, Dean DR, Dalton Trans (2002) 802–807. [Google Scholar]
- [18].Dos Santos PC, Mayer SM, Barney BM, Seefeldt LC, Dean DR, J. Inorg. Biochem 101 (2007) 1642–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Igarashi RY, Dos Santos PC, Niehaus WG, Dance IG, Dean DR, Seefeldt LC, J. Biol. Chem 279 (2004) 34770–34775. [DOI] [PubMed] [Google Scholar]
- [20].Igarashi RY, Laryukhin M, Dos Santos PC, Lee HI, Dean DR, Seefeldt LC, Hoffman BM, J. Am. Chem. Soc 127 (2005) 6231–6241. [DOI] [PubMed] [Google Scholar]
- [21].Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR, Biochemistry 37 (1998) 12611–12623. [DOI] [PubMed] [Google Scholar]
- [22].Georgiadis MM, Komiya H, Chakrabarti P, Woo D, Kornuc JJ, Rees DC, Science 257 (1992) 1653–1659. [DOI] [PubMed] [Google Scholar]
- [23].Otwinowski Z, Minor W, in: Carter CW, C.W., Jr., Sweet RM (Eds.), Macromolecular Crystallography, vol. 276, Acad. Press Inc., New York, pp. 307–326. [Google Scholar]
- [24].Matthews BW, J. Mol. Biol 33 (1968) 491–497. [DOI] [PubMed] [Google Scholar]
- [25].A Kantardjieff K, Rupp B, Prot. Science 12 (2003) 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].CCP4 Acta Crystallogr. D Biol. Crystallogr 50 (1994) 760–763. [DOI] [PubMed] [Google Scholar]
- [27].Peters JW, Stowell MH, Soltis SM, Finnegan MG, Johnson MK, Rees DC, Biochemistry 36 (1997)1181–1187. [DOI] [PubMed] [Google Scholar]
- [28].Murshudov GN, Vagin AA and Dodson EJ (1997) Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [29].Laskowski RA, Macarthur MW, Moss DS, Thornton JM, J. Appl. Crystallogr 26 (1993) 283–291. [Google Scholar]
- [30].DeLano WL, DeLano Scientific LLC, San Carlos, CA, USA. (2002) http://www.pymol.org [Google Scholar]
- [31].Krissinel E, Henrick K, Acta Crystallogr. D Biol. Crystallogr 60 (2004) 2256–2268. [DOI] [PubMed] [Google Scholar]
- [32].Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, et al. Nucleic Acids Res 35 (2007) W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]