

Abstract
Objective
To examine the correlation between verbal and visual memory function and correlation with brain metabolites (lactate and N‐Acetylaspartate, NAA) in individuals with mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS).
Methods
Memory performance and brain metabolites (ventricular lactate, occipital lactate, and occipital NAA) were examined in 18 MELAS, 58 m.3243A > G carriers, and 20 familial controls. Measures included the Selective Reminding Test (verbal memory), Benton Visuospatial Retention Test (visual memory), and MR Spectroscopy (NAA, Lactate). ANOVA, chi‐squared/Fisher’s exact tests, paired t‐tests, Pearson correlations, and Spearman correlations were used.
Results
When compared to carriers and controls, MELAS patients had the: (1) most impaired memory functions (Visual: p = 0.0003; Verbal: p = 0.02), (2) greatest visual than verbal memory impairment, (3) highest brain lactate levels (p < 0.0001), and (4) lowest brain NAA levels (p = 0.0003). Occipital and ventricular lactate to NAA ratios correlated significantly with visual memory performance (p ≤ 0.001). Higher lactate levels (p ≤ 0.01) and lower NAA levels (p = 0.0009) correlated specifically with greater visual memory dysfunction in MELAS. There was little or no correlation with verbal memory.
Interpretation
Individuals with MELAS are at increased risk for impaired memory. Although verbal and visual memory are both affected, visual memory is preferentially affected and more clearly associated with brain metabolite levels. Preferential involvement of posterior brain regions is a distinctive clinical signature of MELAS. We now report a distinctive cognitive phenotype that targets visual memory more prominently and earlier than verbal memory. We speculate that this finding in carriers presages a conversion to the MELAS phenotype.
Introduction
Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) is a maternally inherited progressive multisystemic genetic disorder, originally identified in 1984. 1 Prevalence estimates range from 0.95 to 236 per 100,000. 2 , 3 , 4 , 5 , 6 , 7 , 8 MELAS has been characterized by onset prior to age 40 years, stroke‐like episodes, seizures, exercise intolerance, ragged‐red fibers, and lactic acidosis. 9
Mitochondrial disorders are associated with defects in oxidative phosphorylation (OXPHOS), causing cellular energy failure, NADH reductive stress, and increased reactive oxygen species (ROS). 10 , 11 , 12 , 13 , 14 In MELAS, 80% of cases are caused by an MT‐TL1 gene mutation, where an adenine nucleotide transitions to a guanine at nucleotide position 3243 (mt3243A > G). 15 , 16 , 17 Carriers of this mtDNA mutation range clinically from asymptomatic to fully symptomatic. It is well known that this variability is due to multiple factors, not the least of which is tissue heteroplasmy. Conversion to the fully symptomatic MELAS phenotype is heralded by stroke‐like episodes, often triggered by focal seizures. Seizures and stroke‐like episodes, particularly early in the clinical course, are localized to the posterior brain regions. 18 , 19 , 20 , 21 , 22
Lactic acidosis is a classic biomarker of the m.3243A > G mutation resulting from the OXPHOS defect and NADH reductive stress. 1 , 10 , 23 , 24 , 25 , 26 , 27 , 28 Likewise, changes in N‐acetylaspartate (NAA) levels are associated with the m.3243A > G mutation and reflect tissue damage. Increased lactate and decreased NAA levels in the brain are now recognized as distinctive MELAS biomarkers and have been linked with overall neuropsychological deficits. 26 , 29
Memory impairment specifically has been observed in symptomatic MELAS patients, 26 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 but not in carriers. 26 , 29 , 30 , 31 , 32 However, the relationships between verbal and visual memory, and between brain metabolites and memory, have not been fully explored. Given that focal seizures and stroke‐like episodes are localized to the posterior brain regions early in the clinical course of MELAS, 18 , 19 , 20 , 21 , 22 we have theorized that the cognitive skills subserved by these regions (generally involving visual input) likely will be more impaired than functions localized to other brain regions. Moreover, given that the clinical course of MELAS is associated with both changes in brain metabolites, 26 , 29 and clinical memory impairment, 26 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 we have speculated that these concurrent changes are associated. Working with data collected from a large natural history study, 30 , 31 we selected specific measures for this analysis and have taken a systematic approach to evaluating two types of memory, three brain metabolites, two metabolite ratio scores, across three groups via multiple analyses. We hypothesized that: (1) visual memory will be more impaired than verbal memory, and (2) memory defects will correlate with increased lactate and decreased NAA in the brain.
Methods
A natural history study of individuals with MELAS associated with the m.3243A > G mutation has continued at the Columbia University Medical Center (CUMC) from 1995 to the present. The patient cohort in this report includes participants enrolled in the study from December 1995 through January 2008, and only data collected during this time frame were used in the analysis. 30 , 31 The study has been approved by the CUIMC Institutional Review Board (IRB# AAAB1425) and supported by a Program Project Grant from the National Institutes of Health/National Institute of Child Health and Development (grant numbers P01‐HD080642 and P01‐HD32062) to Drs. Darryl C. De Vivo and Salvatore DiMauro.
Participants
As part of the natural history study, 30 , 31 123 matrilineal relatives from 45 families were enrolled. Ninety‐six participants who completed measures of verbal and visual memory were included in the current analysis and were separated into three groups: (1) 18 fully symptomatic individuals with MELAS (MELAS group), (2) 59 carriers of the m.3243A > G mutation (carrier group), and (3) 20 controls without the m.3243A > G mutation who were patrilineal relatives or married‐in relatives (control group). Patients in the MELAS group were defined as fully symptomatic individuals with the m.3243A > G mutation, lactic acidosis, and stroke‐like episodes with or without focal seizures prior to enrolling in this study. Specific clinical (i.e., neurological, cognitive, and functional), laboratory, and neuroimaging features of this subgroup have been published previously. 30 , 31 Patients in the carrier group were defined as asymptomatic or symptomatic individuals with the m.3243A > G mutation who have not had stroke‐like episodes. Individuals in the control group generally were healthy and did not carry the m.3243A > G mutation.
Procedures
The full prospective cohort study procedures have been described elsewhere. 30 , 31 The study was conducted at CUIMC in New York City. All participants gave written informed consent prior to participation in the study.
Each participant underwent a comprehensive neuropsychological evaluation to assess cognitive functioning across domains, including attention, processing speed, executive function, language, visual–spatial ability, and memory. For the purposes of this selective analysis, verbal, and visual memory performances were examined as the primary outcomes.
Measures
Demographics
Each participant was classified by age (in years), sex (male, female), and education (total years).
Verbal memory
Verbal learning and memory were assessed using the selective reminding test (SRT). 40 This is a rote list‐learning task of 12 words with six learning trials, a delayed recall trial administered 15 min following the last learning trial, and a multiple‐choice recognition trial administered immediately following the delayed recall trial.
Visual memory
Visual learning and memory were assessed using the Benton Visuospatial Retention Test (BVRT). 41 This task involves an initial copy trial of 10 designs, a 10‐second immediate recall trial of 10 different designs, and a multiple‐choice recognition trial of all 10 designs from the immediate recall trial.
Brain Metabolites
Brain Metabolites were measured using multi‐slice proton magnetic resonance spectroscopic imaging (MRS). For the purposes of this study, lactate and NAA were analyzed based on prior association with decreased neuropsychological and neurological functioning. 26 , 29 Biochemical values estimated within the lateral ventricles and occipital lobe gray matter were measured based upon two 15‐mm axial oblique brain segments; full method is described elsewhere. 26 , 30 , 31 , 42 Tissue lactate and NAA values were measured in the gray matter tissue of the occipital lobe. Ventricular lactate was measured within the fluid of the lateral ventricle. All values were measured in institutional units (i.u.). Values were characterized based upon comparison with the amount (mean and standard deviation) of each metabolite measured in the healthy control sample.
Statistical analysis
Descriptive statistics were examined using frequencies and percentages for demographic characteristics, metabolite levels, and verbal/visual memory scores among the three clinical groups – (1) MELAS, (2) carriers, and (3) controls. To compare these variables among groups, ANOVA was used for continuous variables and chi‐squared or Fisher’s exact (for cell counts <5) tests were used for categorical variables. Commonly used cutoffs were identified for the two primary cognitive measures and the three biochemical variables to clearly represent the data. Lactate levels were separately represented by three categories: “within normal limits” (ventricular: ≤4.8 i.u.; occipital: ≤4.5 i.u.), “high” (ventricular: 4.8–5.7 i.u.; occipital: 4.5–5.4 i.u.), and “very high” (ventricular: ≥5.7 i.u.; occipital: ≥5.4 i.u.). Occipital NAA was categorized as “low” (≤14.0 i.u.), “within normal limits” (14.0 i.u.‐17.6 i.u.), and “high” (≥17.6 i.u.). Two ratios were created, (1) occipital lactate to occipital NAA and (2) ventricular lactate to occipital NAA, to examine the relationships between biochemical values and memory performance. For both verbal and visual memory, test scores were converted to percentiles and categorized as “within normal limits” for scores above the 16th percentile, “mildly impaired”: for scores between 16th and 2nd percentile (or between one and two standard deviations from the population mean), and “severely impaired” for scores below the 2nd percentile (more than two standard deviations below the population mean). Chi‐square or Fisher’s exact (for cell counts <5) tests were used to determine the difference in the amount of occipital and ventricular lactate, occipital NAA, verbal memory performance, and visual memory performance among the three clinical groups and separately between MELAS and carriers, MELAS and controls, and carriers and controls. Verbal and visual memory performances were compared within each group using Pearson correlations and paired t‐tests.
The relationships between verbal and visual memory and biochemical variables (occipital lactate, ventricular lactate, and occipital NAA) were examined in several ways. First, six separate Spearman correlations were run to examine the relationship between (1) verbal memory and each biochemical variable separately, and (2) visual memory and each biochemical variable separately. Second, using the pre‐identified cutoffs indicated above, chi‐square or Fisher’s exact (for cell counts <5) tests were used to categorically examine the relationship between the biochemical values and memory, separately for verbal and visual memory. Third, four separate Spearman correlations were run to examine the relationship between each memory variable (verbal, visual) and each ratio variable (occipital lactate / occipital NAA, ventricular lactate / occipital NAA).
Alpha was set at 0.05. All analyses were conducted via SAS 9.4 (SAS Institute, Cary, NC, USA).
Results
Demographic data are presented in Table 1. The mean age of the overall sample was 42.5 (SD = 14.7) years, ranging from age 16 to 76 years. Education ranged from age 9 to 20 years with an average of 14.7 (SD = 2.7) years. The sample was 61.5% female and 88.5% Caucasian. Although groups differed by age (p = 0.002) and sex (p < 0.0001), no differences were identified with respect to ethnicity or total years of education.
Table 1.
Participant characteristics.
| MELAS | Carriers | Controls | Between group comparisons | |
|---|---|---|---|---|
| n = 18 | n = 58 | n = 20 | ||
| Age (in years) | ||||
| Range | 16.0–61.0 | 17.0–76.0 | 32.0–73.0 |
F = 6.84 p = 0.002 |
| Mean (SD) | 35.2 (13.5) | 41.6 (14.7) | 51.6 (11.5) | |
| Sex: N (%) | ||||
| Male | 7 (38.9) | 14 (24.1) | 16 (80.0) | p < 0.0001 |
| Female | 11 (61.1) | 44 (75.9) | 4 (20.0) | |
| Education (in years) | ||||
| Range | 9.0–18.0 | 10.0–20.0 | 10.0–18.0 |
F = 0.43 p = 0.65 |
| Mean (SD) | 14.3 (2.9) | 14.9 (2.8) | 14.5 (2.4) | |
| Ethnicity: N (%) | ||||
| White | 15 (83.3) | 54 (91.4) | 17 (85.0) | p = 0.44 |
| Black | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Hispanic | 1 (5.6) | 3 (5.2) | 1 (5.0) | |
| Asian | 0 (0.0) | 0 (0.0) | 1 (5.0) | |
| American Indian | 2 (11.1) | 2 (3.4) | 1 (5.0) | |
Bold indicates Alpha was set at 0.05.
Ventricular lactate
Overall, the ventricular lactate levels ranged from 2.0 to 14.4 i.u., with a mean of 5.3 i.u. (SD = 2.4) and differed across the groups (p < 0.0001), ranging from 2.4 to 14.4 i.u. in MELAS, 2.0 to 8.6 i.u. in carriers, and 2.4 to 5.6 i.u. in controls (Fig. 1). Ventricular lactate differed between MELAS and carriers (p < 0.0001), MELAS and controls (p < 0.0001), and carriers and controls (p = 0.02). Categorization of ventricular lactate levels by group is summarized in Table 2.
Figure 1.
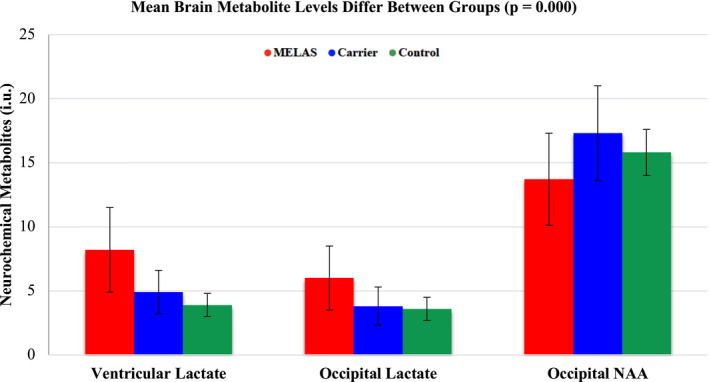
Brain metabolites by group.
Table 2.
Brain metabolites and memory performance across clinical groups.
| MELAS | Carriers | Controls | Between group comparisons | |
|---|---|---|---|---|
| n = 18 | n = 58 | n = 20 | ||
| Verbal memory: N (%)* | ||||
| Within normal limits (>16th %ile) | 8 (44.4) | 44 (75.8) | 13 (65.0) | p = 0.02 |
| Mildly impaired (16th–2nd %ile) | 5 (27.8) | 12 (20.7) | 6 (30.0) | |
| Severely impaired (<2nd %ile) | 5 (27.8) | 2 (3.5) | 1 (5.0) | |
| Visual memory: N (%) § | ||||
| Within normal limits (>16th %ile) | 3 (16.7) | 31 (53.5) | 16 (80.0) | p < 0.0001 |
| Mildly impaired (16th–2nd %ile) | 1 (5.6) | 16 (27.6) | 2 (10.0) | |
| Severely impaired (<2nd %ile) | 14 (77.8) | 11 (18.9) | 2 (10.0) | |
| Occipital NAA levels: N (%)° | ||||
| Low (≤14.0 i.u.) | 11 (61.1) | 9 (15.5) | 5 (25.0) | p = 0.0007 |
| Within normal limits (14.0–17.6 i.u.) | 4 (22.2) | 22 (37.9) | 12 (60.0) | |
| High (≥17.6 i.u.) | 3 (16.7) | 27 (46.6) | 3 (15.0) | |
| Occipital lactate levels: N (%) § | ||||
| Within normal limits (≤4.5 i.u.) | 4 (22.2) | 45 (77.6) | 17 (85.0) | p < 0.0001 |
| High (4.5–5.4 i.u.) | 5 (27.8) | 8 (13.8) | 3 (15.0) | |
| Very high (≥5.4 i.u.) | 9 (50.0) | 5 (8.6) | 0 (0.0) | |
| Ventricular lactate levels: N (%)° | ||||
| Within normal limits (≤4.8 i.u.) | 3 (16.7) | 28 (48.2) | 16 (80.0) | p < 0.0001 |
| High (4.8–5.7 i.u.) | 1 (5.5) | 15 (25.9) | 4 (20.0) | |
| Very high (≥ 5.7 i.u.) | 14 (77.8) | 15 (25.9) | 0 (0.0) | |
Note: Differed between MELAS and carriers, MELAS and controls, and carriers and controls (all p < 0.05). Bold indicated Alpha was set at 0.05.
Differed between MELAS and carriers only (p = 0.0008).
Differed between MELAS and carriers (p < 0.0001) and MELAS and controls (p = 0.000) only.
Differed between MELAS and carriers, MELAS and controls, and carriers and controls (all p = 0.05).
Occipital lactate
Overall, the occipital lactate levels ranged from 1.2 to 10.3 i.u., with a mean of 4.2 i.u. (SD = 1.8). Occipital lactate differed across the three groups (p < 0.0001), ranging from 2.5 to 10.3 i.u. in MELAS, 1.2 to 9.8 i.u. in carriers, and 2.2 to 5.3 i.u. in controls (Fig. 1). Occipital lactate levels differed between MELAS and carriers (p < 0.0001) and MELAS and controls (p = 0.0002), but not between carriers and controls (p = 0.49). Categorization of occipital lactate levels by group is summarized in Table 2.
Occipital NAA
Overall, the occipital NAA levels ranged from 8.4 to 26.4 i.u., with a mean of 16.4 i.u. (SD = 3.7). Occipital NAA differed across the three groups (p = 0.0003), ranging from 8.4 to 20.6 i.u. in MELAS, 9.5 to 26.4 i.u. in carriers, and 12.5 to 18.5 i.u. in controls (Fig. 1). Occipital NAA levels differed between MELAS and carriers (p = 0.0003), MELAS and controls (p = 0.03), and carriers and controls (p = 0.05). Categorization of occipital NAA levels by group is summarized in Table 2.
Verbal memory versus visual memory
A moderate positive correlation was found between verbal and visual memory in the full sample (r = 0.37; p = 0.0002). When separated by group, a strong positive correlation was found between verbal and visual memory in the MELAS group (r = 0.68; p = 0.002), but not in carriers (r = 0.22; p = 0.09) or controls (r = 0.16; p = 0.50). Worse visual memory performance compared with verbal memory was identified in MELAS (p = 0.01) and carriers (p = 0.03); however, this difference was not observed in controls (p = 0.09).
Visual memory
Overall, visual memory performance ranged from 0.05%ile to 98.0%ile, with a mean of 31.3%ile (SD = 32.1%ile). Visual memory performance differed across the groups (p = 0.0003) (Table 3); specifically, differences were identified between MELAS and carriers (p = 0.01), MELAS and controls (p < 0.0001), and carriers and controls (p = 0.01). Categorization of visual memory by group is summarized in Table 2.
Table 3.
Comparison of visual and verbal memory performance within and between clinical groups.
| MELAS | Carriers | Controls | |
|---|---|---|---|
| n = 18 | n = 58 | n = 20 | |
| Visual memory: mean (SD)* | 11.33 (23.48) | 31.27 (31.20) | 52.25 (32.44) |
| Verbal memory: mean (SD) § | 26.31 (31.52) | 42.69 (32.06) | 36.75 (28.07) |
| Visual – verbal: mean (SD) | −14.97 (23.19) | −11.42 (39.48) | 15.50 (39.36) |
| Within group comparison | t = −2.74 (p = 0.01) | t = −2.20 (p = 0.03) | t = 1.76 (p = 0.09) |
Bold indicates Alpha was set at 0.05.
Different among groups (p = 0.0003).
No difference among groups (p = 0.15).
Visual memory & brain metabolites
Occipital Lactate: A weak negative correlation between occipital lactate and visual memory was found (r = −0.28; p = 0.005). Visual memory differed by the amount of occipital lactate (p = 0.01), such that the higher the lactate signal, the worse the observed visual memory performance (Table 4).
Table 4.
Relationship between brain metabolites and memory performance.
| Within normal limits (>16th %ile) | Mildly impaired (16th–2nd %ile) | Severely impaired (<2nd %ile) | Total (%) | |
|---|---|---|---|---|
| Ventricular lactate and verbal memory performance: p = 0.40 | ||||
| Within normal limits (≤ 4.8 i.u.) | 34 | 11 | 2 | 47 (49.0) |
| High (4.8–5.7 i.u.) | 14 | 5 | 1 | 20 (20.8) |
| Very high (≥ 5.7 i.u.) | 17 | 7 | 5 | 29 (30.2) |
| Total (%) | 65 (67.7) | 23 (24.0) | 8 (8.3) | n = 96 |
| Ventricular Lactate and Visual Memory Performance: p < 0.0001 | ||||
| Within normal limits (≤ 4.8 i.u.) | 29 | 10 | 8 | 47 (49.0) |
| High (4.8–5.7 i.u.) | 16 | 2 | 2 | 20 (20.8) |
| Very high (≥ 5.7 i.u.) | 5 | 7 | 17 | 29 (30.2) |
| Total (%) | 50 (52.1) | 19 (19.8) | 27 (28.1) | n = 96 |
| Occipital lactate and verbal memory performance: p = 0.45 | ||||
| Within normal limits (≤ 4.5 i.u.) | 45 | 17 | 4 | 66 (68.7) |
| High (4.5–5.4 i.u.) | 11 | 4 | 1 | 16 (16.7) |
| Very high (≥ 5.4 i.u.) | 9 | 2 | 3 | 14 (14.6) |
| Total (%) | 65 (67.7) | 23 (24.0) | 8 (8.3) | n = 96 |
| Occipital lactate and visual memory performance: p = 0.01 | ||||
| within normal limits (≤ 4.5 i.u.) | 40 | 14 | 12 | 66 (68.7) |
| High (4.5–5.4 i.u.) | 7 | 3 | 6 | 16 (16.7) |
| Very high (≥ 5.4 i.u.) | 3 | 2 | 9 | 14 (14.6) |
| Total (%) | 50 (52.1) | 19 (19.8) | 27 (28.1) | n = 96 |
| Occipital NAA and verbal memory performance: p = 0.44 | ||||
| Low (≤14.0 i.u.) | 15 | 6 | 4 | 25 (26.0) |
| Within normal limits (14.0–17.6 i.u.) | 27 | 10 | 1 | 38 (39.6) |
| High (≥ 17.6 i.u.) | 23 | 7 | 3 | 33 (34.4) |
| Total (%) | 65 (67.7) | 23 (24.0) | 8 (8.3) | n = 96 |
| Occipital NAA and visual memory performance: p = 0.002 | ||||
| Low (≤14.0 i.u.) | 10 | 1 | 14 | 25 (26.0) |
| Within normal limits (14.0–17.6 i.u.) | 19 | 13 | 6 | 38 (39.6) |
| High (≥ 17.6 i.u.) | 21 | 5 | 7 | 33 (34.4) |
| Total (%) | 50 (52.1) | 19 (19.8) | 27 (28.1) | n = 96 |
Ventricular Lactate: A moderate negative correlation between ventricular lactate and visual memory was found (r = −0.39; p < 0.0001). Visual memory differed by the amount of ventricular lactate (p < 0.0001), such that the higher the lactate signal, the worse the observed visual memory performance (Table 4).
Occipital NAA: A weak positive correlation between occipital NAA and visual memory (r = 0.29; p = 0.004) (Fig. 2). Visual memory performance differed by the amount of occipital NAA (p = 0.0009), such that the lower the NAA signal, the worse the observed visual memory performance (Table 4).
Figure 2.
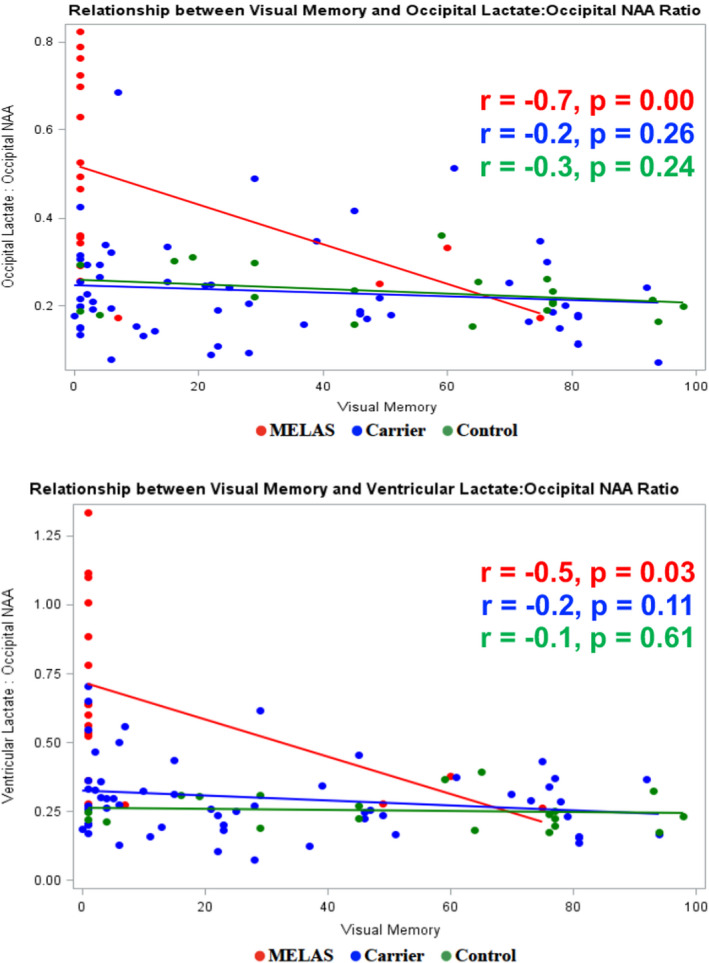
Relationship between visual memory and brain metabolite ratios.
In the full sample, a moderate negative correlation was found between visual memory and 1) the ratio of occipital lactate to occipital NAA (r = −0.37; p = 0.0002) and 2) the ratio of ventricular lactate to occipital NAA (r = −0.42; p < 0.0001). When the three subgroups were analyzed separately, visual memory performance was moderate to highly correlated with the biochemical ratio variables in the MELAS subgroup only (occipital lactate to occipital NAA r = −0.7, p = 0.00; ventricular lactate to occipital NAA r = −0.5, p = 0.03) (Fig. 3).
Figure 3.
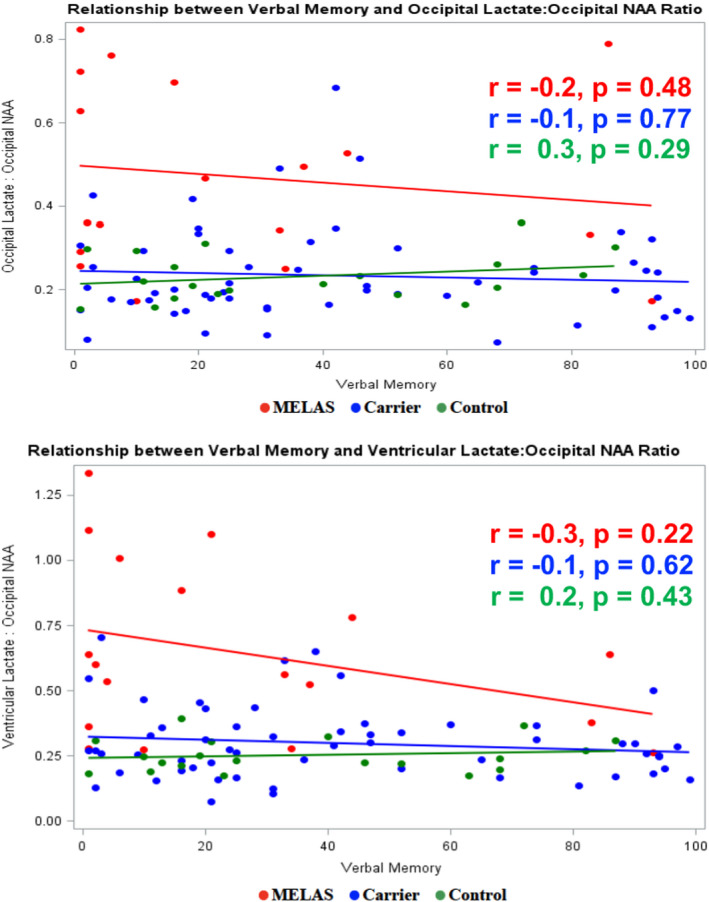
Relationship between verbal memory and brain metabolite ratios.
Verbal memory
Overall, verbal memory performance ranged from 0.9%ile to 99.0%ile, with a mean of 38.4%ile (SD = 31.5%ile). Verbal memory performance differed across the three clinical groups (p = 0.02) (Table 3), but there were no differences between pairs: MELAS and carriers (p = 0.06), MELAS and controls (p = 0.29), or carriers and controls (p = 0.46). Categorization of verbal memory by group is summarized in Table 2.
Verbal memory & brain metabolites
Occipital Lactate: Occipital lactate did not correlate with verbal memory performance (r = −0.15; p = 0.14) (Fig. 1), nor did verbal memory performance differ by the amount (i.e., within normal limits, high, very high i.u) of occipital lactate (p = 0.40) (Table 4).
Ventricular Lactate: Ventricular lactate did not correlate with verbal memory performance (r = −0.19; p = 0.07) (Fig. 1), nor did verbal memory performance differ by the amount (i.e., within normal limits, high, very high i.u) of ventricular lactate (p = 0.34) (Table 4).
Occipital NAA: Occipital NAA level did not correlate with verbal memory performance (r = −0.04; p = 0.66), nor did verbal memory performance differ by the amount (i.e., within normal limits, high, very high i.u) of occipital NAA (p = 0.44) (Table 4).
In the full sample and when the three subgroups were analyzed separately, no correlation was found between verbal memory and (1) the ratio of occipital lactate to occipital NAA (r = −0.14; p = 0.18), or (2) the ratio of ventricular lactate to occipital NAA (r = −0.16; p = 0.12) (Fig. 3).
Discussion
This study examined the relationship between verbal and visual memory function and brain metabolites (i.e., ventricular lactate, occipital lactate, and occipital NAA) in a cohort of 18 MELAS patients, 58 carrier relatives of the m.3243A > G mutation, and 20 control participants. It has previously been shown that in the clinical course of MELAS focal seizures and stroke‐like episodes are primarily localized to occipital brain regions 18 , 19 , 20 , 21 , 22 and there are changes in both brain metabolites 26 , 29 and memory function. 26 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 Building on that work, we theorized that the visual cognitive skills subserved by posterior brain regions would be more impaired than verbal cognitive skills that are subserved by more anteriorly localized brain regions, and that progressive changes in brain metabolites and declining memory are correlated. Thus, we chose to run a specific analysis examining performance only on memory from data collected in our natural history study. 30 , 31 By taking a systematic approach that evaluates the association of three brain metabolites and two metabolite ratio scores to two types of memory performance across three groups, multiple analyses were performed to tease out specific associations. The results support both hypotheses and show that among individuals with MELAS: (1) visual memory is preferentially impaired and (2) brain metabolites do correlate with memory functions.
The occipital regions of the cerebral cortex have a high metabolic demand making these regions more vulnerable to mitochondrial dysfunction in MELAS. It is not surprising, therefore, that brain functions localized to these vulnerable regions may be affected earlier in the clinical course and disproportionately. 43 , 44 We reasoned that memory dependent on visual sensory input would be preferentially affected. Consistent with this reasoning, we found that visual memory was more affected than verbal memory in the MELAS group and, to a lesser degree, in the carrier group. Memory impairment was present in a greater proportion of individuals with the m.3243A > G mutation (MELAS and carriers) compared to controls. A higher percentage of individuals with the m.3243A > G mutation had impaired visual memory (83.4% MELAS, 47.4% carriers) compared with verbal memory (55.6% MELAS, 25.4% carriers). Verbal memory differed across the three clinical groups, with more than a quarter of MELAS patients performing in the severely impaired range compared with less than 10 percent of the carrier and control groups combined. Even more striking, nearly three‐quarters of MELAS patients performed in the severely impaired range on the visual memory task compared with less than 20 percent of the carrier and control groups combined.
In addition, and as expected, 29 MELAS patients were found to have the highest brain lactate levels, lowest brain NAA levels, and most impaired memory functions. Specifically, 83.3% of MELAS patients fell in the high and very high ventricular lactate ranges compared with 51.8% of carriers and 20.0% of controls. Occipital lactate was similarly distributed with 77.8% of MELAS patients in the high or very high categories compared with 22.4% of carriers and 15.0% of controls. Occipital NAA was lowest in 61.1% of MELAS patients compared with 15.5% of carriers and 25.0% of controls. This distinct pattern of neurochemical abnormalities correlated closely with the cognitive profile in the MELAS population. We have always considered brain lactate values to be a surrogate for brain heteroplasmy with the highest brain lactate values being measured in the MELAS group. 29
Our results show that both occipital and ventricular lactate to NAA ratios are moderately correlated with visual memory performance. Furthermore, individuals with higher lactate levels and lower NAA levels demonstrated greater visual memory dysfunction; and most of these individuals were in the MELAS clinical group. In contrast, there was no correlation between verbal memory performance and either occipital lactate to NAA ratio or ventricular lactate to NAA ratio. Our previous work indicates that NAA decreases in the MELAS patients as cerebral lactate continues to increase. 45 The current findings documenting more severely affected visual memory tasks in MELAS correlates function with structure in this vulnerable brain region; visually mediated tasks are more dependent on visual cortex in the occipital lobe, whereas verbally mediated tasks are primarily associated with more anterior brain regions.
Our findings show that most individuals with high lactate and low NAA perform poorly on the visual memory test. Surprisingly perhaps, some of these individuals are still able to perform within normal limits on the verbal memory test. Of particular interest, visual memory performance was poor even within the carrier group before there is any clinical or radiological indication of stroke‐like episodes. This neuropsychological profile is rather distinctive and should raise suspicion for MELAS in the proper clinical setting. Currently, we are following this carrier subgroup to determine if these carriers are at higher risk to convert to the fully symptomatic MELAS group. It is our speculation that early detection of visual memory impairment will serve as a harbinger of this phenotypic conversion. In essence, brain metabolic biomarkers and functional outcome measures likely presage structural damage in the posterior cerebral cortex. Almost half of the carrier group (48%) scored within the impaired range on the visual memory test, and 19% scored within the severely impaired range. These findings strongly suggest that neuropsychological measures of brain function and brain metabolic biomarkers will be more sensitive indicators of cognitive change than the current clinical or radiological characterization of regional brain damage.
MELAS is a multisystemic disease, and as such there may be other disease factors that potentially influence cognitive performance in the study participants. For the current study, the contribution of overall disease burden was not directly assessed in the analysis. The lack of direct measurements of visual acuity and fine motor coordination (which may confound the individual's performance on cognitive measures) is a limitation of the study, yet prior work with the MELAS population has not highlighted these variables as areas of major concern. 29 Similarly, the MRS data collected in the temporal lobe did not clearly focus on the brain regions (i.e., mesial temporal lobe) that subserve verbal memory and therefore could not be analyzed specifically as a comparison to the occipital lobe voxel that subserves aspects of visual memory. Recent studies have also demonstrated that high resolution 7 T imaging shows that carriers of the m.3243A > G mutation have widespread cortical thinning and gray matter volume loss in areas such as the temporal lobes. 46 , 47 Given that the temporal lobes subserve aspects of memory, the relationships between the biomarkers we studied (i.e., visual memory performance and brain metabolites) and possible subtle changes identified on 7 T imaging should be explored in future studies. Another limitation of our study is the significant demographic differences between our control group and the MELAS and carrier groups. We used spouses and patrilinear relatives as controls to best control for environment and familial characteristics. Because of the maternal inheritance pattern of mitochondrial diseases, our control group is both older and more likely to be male than the other two groups. We reason that older age might increase risk for functional decline, so there is little chance of false‐positive conclusions. Moreover, the years of education did not differ between the groups, suggesting the between group findings are likely valid. And while sex may contribute to differences in verbal memory performance, 48 , 49 there was no statistically significant difference between the carrier and control group on verbal memory performance in our sample. This may reflect the inherent differences between males and females and the impact of the mitochondrial dysfunction on the female carrier population.
This study examined the association between brain metabolites and memory performance in this population. Despite limitations such as possible alternative contributions to cognitive changes (e.g., multisystemic disease, physical/emotional/environmental factors) and small proband sample size acknowledging that MELAS is a rare disease, our findings clearly describe a distinct cognitive phenotype in MELAS that specifically reveals visual memory impairment as the surrogate for vulnerability of the posterior brain structures. As discussed previously, the precise mechanism that places the posterior regions at undue risk in MELAS is unknown. We have speculated that the higher basal metabolic rate of the posterior cerebral hemisphere contributes to this regional vulnerability. What is clear, however, from our study is the implication of the regional functional disturbance in anticipating regional structural damage. The disproportionate loss of visual memory, the relative sparing of verbal memory and the changes in regional lactate, and NAA presage the stroke‐like episodes and accompanying radiological signal abnormalities.
Conflict of Interest
Dr. Leaffer reports no financial and nonfinancial disclosure or conflict of interest. Dr. De Vivo reports no financial and nonfinancial disclosure or conflict of interest. Ms. Engelstad reports no financial and nonfinancial disclosure or conflict of interest. Dr. Fryer reports no financial and nonfinancial disclosure or conflict of interest. Dr. Gu reports no financial and nonfinancial disclosure or conflict of interest. Dr. Shungu reports no financial and nonfinancial disclosure or conflict of interest. Dr. Hirano reports no financial and nonfinancial disclosure of conflict of interests. Dr. DiMauro reports no financial and nonfinancial disclosure or conflict of interest. Dr. Hinton report no financial and nonfinancial disclosure or conflict of interest.
Author Contributions
Emily B. Leaffer, PhD, MPH is a manuscript author who analyzed and interpreted the data, designed and conceptualized study, and drafted the manuscript for intellectual content. Darryl C. De Vivo, MD is the study's principal investigator and manuscript author who designed and conceptualized study and revised the manuscript for intellectual content. Kristin Engelstad, MS is a manuscript author who had a major role in the acquisition of all data. Robert H. Fryer, MD, PhD is a manuscript author who analyzed and interpreted the data. Yian Gu, PhD, MD is a manuscript author who analyzed and interpreted the data. Dikoma C. Shungu, PhD is a manuscript author who had a major role in the acquisition of MRS data. Michio Hirano, MD is a manuscript author who analyzed and interpreted the data. Salvatore Di Mauro, MD is a manuscript author who designed and conceptualized study and revised the manuscript for intellectual content. Veronica J. Hinton, PhD is a manuscript author who analyzed and interpreted the data, designed and conceptualized study, and revised the manuscript for intellectual content.
Acknowledgement
Supported by NIH and NICHD (P01‐HD080642, P01‐HD32062).
Funding InformationSupported by NIH and NICHD (P01‐HD080642, P01‐HD32062).
Funding Statement
This work was funded by National Institutes of Health / National Institute of Child Health and Development grants P01‐HD080642 and P01‐HD32062.
Data availability statement
Data not published within the article are available in a public repository and include digital object identifiers (doi) or accession numbers to the datasets or to state that anonymized data will be shared by request from any qualified investigator.
References
- 1. Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984;16(4):481‐488. [DOI] [PubMed] [Google Scholar]
- 2. Majamaa K, Moilanen JS, Uimonen S, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet. 1998;63(2):447‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chinnery P, Johnson M, Wardell T, et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48(2):188‐193. [PubMed] [Google Scholar]
- 4. Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001;49(3):377‐383. [PubMed] [Google Scholar]
- 5. Uusimaa J, Moilanen JS, Vainionpää L, et al. Prevalence, segregation, and phenotype of the mitochondrial DNA 3243A> G mutation in children. Ann Neurol. 2007;62(3):278‐287. [DOI] [PubMed] [Google Scholar]
- 6. Manwaring N, Jones MM, Wang JJ, et al. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7(3):230‐233. [DOI] [PubMed] [Google Scholar]
- 7. Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63(1):35‐39. [DOI] [PubMed] [Google Scholar]
- 8. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77(5):753‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirano M, Pavlakis SG. Topical review: mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4‐13. [DOI] [PubMed] [Google Scholar]
- 10. Sharma R, Reinstadler B, Engelstad K, et al. Circulating markers of NADH‐reductive stress correlate with mitochondrial disease severity. J Clin Invest. 2021;131(2):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taanman JW, Williams SL. Structure and function of the mitochondrial oxidative phosphorylation system. In: Schapira AHV, DiMauro S, eds. Blue Books of Practical Neurology. Academic Press Elsevier; 2002:1‐34. [Google Scholar]
- 12. DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;2008(31):91‐123. [DOI] [PubMed] [Google Scholar]
- 13. Davis RL, Sue CM. The genetics of mitochondrial disease. Semin Neurol. 2011;31(5):519‐530. doi: 10.1055/s-0031-1299790 [DOI] [PubMed] [Google Scholar]
- 14. DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9(8):429‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong LJC. Pathogenic mitochondrial DNA mutations in protein‐coding genes. Muscle Nerve. 2007;36(3):279‐293. [DOI] [PubMed] [Google Scholar]
- 16. Scaglia F. Nuclear gene defects in mitochondrial disorders. Mitochondrial Disorders. 2012;837:17‐34. [DOI] [PubMed] [Google Scholar]
- 17. DiMauro S, Hirano M. MELAS. In: Adams MP, Ardinger HH, eds. Pagon RA. GeneReviews. University of Washington; 2013:1993‐2018. [Google Scholar]
- 18. Veggiotti P, Colamaria V, Dalla Bernardina B, Martelli A, Mangione D, Lanzi G. Epilepsia partialis continua in a case of MELAS: clinical and neurophysiological study. Clin Neurophysiol. 1995;25(3):158‐166. [DOI] [PubMed] [Google Scholar]
- 19. Canifoglia I, Franceschetti S, Antozzi C, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;56:1340‐1346. [DOI] [PubMed] [Google Scholar]
- 20. Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke‐like lesions in MELAS. Neurology. 2003;61(9):1238‐1244. [DOI] [PubMed] [Google Scholar]
- 21. Ito H, Mori K, Harada M, et al. Serial brain imaging analysis of stroke‐like episodes in MELAS. Brain Dev. 2008;30:483‐488. [DOI] [PubMed] [Google Scholar]
- 22. Fryer RH, Bain J, De Vivo DC. Mitochondrial Encephalomyopathy lactic acidosis and stroke‐like episodes (MELAS): a case report and critical reappraisal of treatment options. Pediatr Neurol. 2016;56:59‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Castillo M, Kwock L, Green C. MELAS syndrome: imaging and proton MR spectroscopic findings. Am J Neuroradiol. 1995;16:233‐239. [PMC free article] [PubMed] [Google Scholar]
- 24. Mathews P, Andermann F, Silver K, et al. Proton MR spectroscopic characterization of differences in regional brain metabolic abnormalities in mitochondrial encephalomyopathies. Neurology. 1993;43(12):484‐490. [DOI] [PubMed] [Google Scholar]
- 25. Yatsuga S, Povalko N, Nishioka J, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619‐624. [DOI] [PubMed] [Google Scholar]
- 26. Kaufmann P, Shungu DC, Sano M, et al. Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology. 2004;62(8):1297‐1302. [DOI] [PubMed] [Google Scholar]
- 27. Wilichowski E, Pouwels P, Frahm J, Hanefeld F. Quantitative proton magnetic resonance spectroscopy of cerebral metabolic disturbances in patients with MELAS. Neuropediatrics. 1999;30(5):256‐263. [DOI] [PubMed] [Google Scholar]
- 28. Moller H, Kurlemann G, Putzler M, et al. Magnetic resonance spectroscopy in patients with MELAS. J Neurol Sci. 2005;229:131‐139. [DOI] [PubMed] [Google Scholar]
- 29. Weiduschat N, Kaufmann P, Mao X, et al. Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers. Neurology. 2014;82(9):798‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaufmann P, Engelstad K, Wei Y, et al. Protean phenotypic features of the A3243G mitochondrial DNA mutation. Arch Neurol. 2009;66(1):85‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaufmann P, Engelstad K, Wei Y, et al. Natural history of MELAS associated with mitochondrial DNA m. 3243A> G genotype. Neurology. 2011;77(22):1965‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hinton VJ, Engelstad K, DiMauro S, De Vivo DC. No evidence of cognitive decline among carrier relatives of MELAS patients. Mitochondrion. 2012;12(5):565. doi: 10.1016/j.mito.2012.07.039 [DOI] [Google Scholar]
- 33. Finsterer J. Cognitive decline as a manifestation of mitochondrial disorders (mitochondrial dementia). J Neurol Sci. 2008;272(1):20‐33. [DOI] [PubMed] [Google Scholar]
- 34. Kartsounis L, Troung D, Morgan‐Hughes J, Harding A. The neuropsychological features of mitochondrial myopathies and encephalomyopathies. Arch Neurol. 1992;49(2):158‐160. [DOI] [PubMed] [Google Scholar]
- 35. Pachalska M, DiMauro S, Formińska‐Kapuścik M, et al. The course of vision disturbances in a patient with the MELAS syndrome. Med Sci Monit. 2002;8(2):CS11‐CS20. [PubMed] [Google Scholar]
- 36. Kiejna A, DiMauro S, Adamowski T, et al. Psychiatric symptoms in a patient with the clinical features of MELAS. Signature. 2002;8(7):72. [PubMed] [Google Scholar]
- 37. Sartor H, Loose R, Tucha O, Klein HE, Lange KW. MELAS: a neuropsychological and radiological follow‐up study. Acta Neurol Scand. 2002;106(5):309‐313. [DOI] [PubMed] [Google Scholar]
- 38. Salsano E, Giovagnoli A, Morandi L, et al. Mitochondrial dementia: a sporadic case of progressive cognitive and behavioral decline with hearing loss due to the rare m.3291t>C MELAS mutation. J Neurol Sci. 2011;300:165‐168. [DOI] [PubMed] [Google Scholar]
- 39. Neargarder SA, Murtagh MP, Wong B, Hill EK. The neuropsychologic deficits of MELAS: evidence of global impairment. Cogn Behav Neurol. 2007;20(2):83‐92. [DOI] [PubMed] [Google Scholar]
- 40. Bushke H, Fuld PA. Evaluating storage, retention, and retrieval in disordered memory and learning. Neurology. 1974;24(11):1019‐1025. [DOI] [PubMed] [Google Scholar]
- 41. Benton AL, Hamsher K, Sivan AB. Manual for the Multilingual Aphasia Examination. 3rd ed. AJA Associates; 1994. [Google Scholar]
- 42. Duyn J, Gillen J, Sobering G, et al. Multisection proton MR spectroscopic imaging of the brain. Radiology. 1993;188:277‐282. [DOI] [PubMed] [Google Scholar]
- 43. Turnbull H, Lax N, Diodato D, et al. The mitochondrial brain: from mitochondrial genome to neurodegeneration. Biochim Biophys Acta. 2010;1802:111‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berti V. Brain: normal variations and benign findings in FGD PET/CT imaging. PET Clin. 2014;9(2):129‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dubeau F, De Stefano N, Zifkin BG, et al. Oxidative phosphorylation defect in the brains of carriers of the tRNAleu (UUR) A3243G mutation in a MELAS pedigree. Ann Neurol. 2000;47(2):179‐185. [PubMed] [Google Scholar]
- 46. Haast RAM, Ivanov D, IJsselstein RJT, et al. Anatomic & metabolic brain markers of the m.3243A>G mutation: a multi‐parametric 7T MRI study. Neuroimage Clin. 2018;31(18):231‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haast RAM, De Coo IFM, Ivanov D, et al. Neurodegenerative and functional signatures of the cerebellar cortex in m.3243A > G patients. Brain Commun. 2022;4(1):fcac024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loprinzi PD, Frith E. The role of sex in memory function: considerations and recommendations in the context of exercise. J Clin Med. 2018;31;7(6):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sundermann EE, Maki PM, Rubin LH, et al. Alzheimer's disease neuroimaging initiative. Female advantage in verbal memory: evidence of sex‐specific cognitive reserve. Neurology. 2016;87(18):1916‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not published within the article are available in a public repository and include digital object identifiers (doi) or accession numbers to the datasets or to state that anonymized data will be shared by request from any qualified investigator.