

Abstract
Background
Acute myocardial infarction (AMI) is one of the leading causes of mortality worldwide. Undesirable myocardial damage may occur during reperfusion of the ischemic myocardium, and this is known as “ischemic reperfusion injury” (IRI). Currently, there are few effective drugs to alleviate IRI. Dl-3-n-butylphthalide (NBP) is recommended for the treatment of acute ischemic stroke in China. This study investigated the effects of NBP on IRI and its underlying mechanisms.
Methods
The left anterior descending (LAD) coronary arteries of rats were occluded for 30 minutes and reperfused for 6 hours to establish the ischemia/reperfusion (I/R) model. NBP was administered intraperitoneally 2 hours before modeling and immediately after reperfusion. At 6 hours after reperfusion, 2,3,5-triphenyltetrazolium chloride (TTC) staining, enzyme-linked immunosorbent assay (ELISA), oxidative stress index, myocardial injury index, hematoxylin and eosin (HE) staining, transmission electron microscopy (TEM), real-time polymerase chain reaction (PCR), immunofluorescence staining, and Western blot analyses were performed to investigate the protective effects of NBP against IRI.
Results
In the rat I/R model, NBP remarkably reduced the myocardial infarct size, alleviated myocardial injury and oxidative stress, improved the pathological alteration of cardiomyocytes and mitochondria, and upregulated mitophagy. In addition, the study demonstrated that the protective effects of NBP against IRI involved mitophagy mediated by the PTEN-induced putative kinase protein-1 (PINK1)/Parkin signaling pathway.
Conclusions
NBP was able to protect the myocardium from IRI in rats through inhibiting oxidative stress and activating mitophagy, mediated by the PINK1/Parkin pathway.
Keywords: Dl-3-n-butylphthalide (NBP), ischemic reperfusion injury (IRI), mitophagy, oxidative stress
Introduction
Acute myocardial infarction (AMI) is a serious and critical disease, and is one of the leading causes of morbidity and mortality worldwide. Approximately 1.5 million individuals in the United States suffer from AMI every year (1,2). The mortality associated with AMI has decreased significantly over the past decades by virtue of timely reperfusion strategies, such as thrombolysis, percutaneous coronary intervention (PCI), and coronary artery bypass grafting (CABG). However, reperfusion of the ischemic myocardium itself may cause undesirable damage to the myocardium, known as “ischemic reperfusion injury” (IRI). Early studies have shown that about 50% of the final myocardial infarction area is caused by reperfusion injury (3,4). The exact pathophysiological mechanisms of IRI remain to be fully elucidated. According to previous studies, mitochondrial dysfunction plays a crucial role in IRI (5,6). The mitochondria make up more than 30% of the volume of cardiomyocytes (7), and the heart relies heavily on mitochondrial oxidative phosphorylation to promote myocardial contraction (8). The integrity and functional status of the mitochondria are essential for maintaining the structure and function of cardiomyocytes. During myocardial IRI, the mitochondria produce reactive oxygen species (ROS), initiate oxidative stress, and cause further mitochondrial damage. Damaged mitochondria subsequently produce more ROS, directly damage mitochondrial proteins and DNA, aggravate mitochondrial dysfunction, and may activate mitochondrial-related apoptosis pathways, resulting in cell death.
Mitophagy refers to the selective autophagy process of removing dysfunctional or superfluous mitochondria to regulate mitochondrial numbers and maintain cell energy metabolism (9). However, mitophagy is a double-edged sword in IRI. Moderate mitophagy can selectively remove damaged mitochondria, eliminating further damage to cells, while excessive mitophagy can result in insufficient numbers and functional decline of the mitochondria, leading to aggravation of cardiomyocyte damage (10). Increasing evidence indicates that modulation of mitophagy plays a pivotal role in myocardial protection during IRI (11). The most representative mitophagy regulatory pathway is the PTEN-induced putative kinase protein-1 (PINK1)/Parkin pathway. Protective effects of mitophagy mediated by the PINK1/Parkin pathway have been described in several models of ischemia/reperfusion (I/R) or hypoxia/reperfusion (H/R) (12,13).
Dl-3-n-butylphthalide (NBP; C12H14O2; Figure 1A) is a synthetic, racemic compound developed from L-3-n-butylphthalide, which is extracted from the seeds of Apium graveolens. NBP was approved by the State Food and Drug Administration of China for the treatment of acute ischemic stroke in 2002. Previous studies have shown that NBP has various neuroprotective effects, such as improving mitochondrial function and anti-oxidative stress, inhibiting apoptosis and inflammation, regulating autophagy, improving microcirculation, and promoting neuron regeneration (14-16). In recent studies, NBP has been shown to exert cardioprotective effects on cardiac ischemic injury or IRI by inhibiting oxidative stress, affecting mitochondrial apoptosis, and regulating mitochondrial biogenesis (17-19). However, the effects of NBP on mitophagy in IRI has not been elucidated. Ligation of the left anterior descending (LAD) coronary artery in rats is a widely accepted and established method for generating I/R models. Therefore, in this study, the rat I/R model was used to evaluate the effects of NBP on myocardial IRI and its potential molecular mechanisms were investigated. The role of NBP in cardiac mitochondrial quality control was also examined from the perspective of mitophagy. We present the following article in accordance with the ARRIVE reporting checklist (available at https://jtd.amegroups.com/article/view/10.21037/jtd-22-585/rc).
Figure 1.

NBP decreased the myocardial infarct size in rats subjected to I/R injury. (A) The chemical structure of NBP. (B) A representative TTC staining image of rat hearts in each group. (C) Quantitative analysis of the myocardial infarct size (n=5). ##, indicates P<0.01 compared with the sham group; **, indicates P<0.01 compared with the I/R group. I/R, ischemia/reperfusion; NBP, Dl-3-n-butylphthalide; NBP-L, low dose of NBP; NBP-H, high dose of NBP; TTC, 2,3,5-triphenyltetrazolium chloride.
Methods
Experimental animals
A total of 90 specific pathogen-free and healthy adult male Sprague-Dawley rats (aged 7–8 weeks, and weighing 270±20 g), were provided by Huafukang Biological Company [Experimental animal license No. SCXK (Beijing) 2019-0008; Beijing, China]. Animals were housed in a clean environment with a temperature of 22 ℃, a relative humidity of 50%, and a 12-hour light/dark cycle with free access to water and food. All rats were subjected to the experiment after 5 days of adaptive feeding. All animal experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and approved by the Laboratory Animal Ethics Committee of the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (ethics application number: IRM-DWLL-2020076).
Model establishment and experimental protocol
Considering surgery-related mortality, rats were assigned randomly into four groups: sham-operated group (sham, n=15); myocardial I/R group (I/R, n=25); myocardial I/R group treated with a low dose of NBP (100 mg/kg, NBP-L, n=25);myocardial I/R group treated with a high dose of NBP (200 mg/kg, NBP-H, n=25). Rats were anesthetized by inhalation of 2–3% isoflurane (20191204, Ringup, Tianjin, China) using a small animal anesthesia machine (F700, Harvard Apparatus, USA). After successful anesthesia, the LAD coronary artery was subjected to 30 minutes of temporal occlusion followed by 6 hours of reperfusion. In the sham group, the LAD artery was only threaded without ligation. The procedure described above was in accordance with the previous report (20). Except for the sham group, the postoperative survival rate of rats in the other groups was about 80%.
NBP (Enbipu Pharmaceutical, Shijiazhuang, China) was dissolved in olive oil. Rats in the NBP-treated groups were injected intraperitoneally with corresponding doses of NBP 2 hours before ischemia and immediately after reperfusion. Rats in other groups were given an equal volume of olive oil. After 6 hours of reperfusion, the rats were anesthetized with chloral hydrate and sacrificed. Subsequently, 5 hearts from each group were prepared for 2,3,5-triphenyltetrazolium chloride (TTC) staining. Blood samples from the remaining rats were collected from the abdominal aorta, followed by immediately centrifugation (2,500 r/min, 15 minutes) at 4 ℃ to isolate the serum. The hearts were washed in phosphate-buffered saline (PBS). The serum and hearts were stored frozen at −80 ℃ until further analysis.
TTC staining
The myocardial infarct area was assessed by TTC staining. After 6 hours of reperfusion, the rat hearts were frozen and the left ventricular (LV) tissue was sliced into 2 mm-thick transverse slices. The slices were incubated for 20 minutes in 1% TTC (Sigma, Saint Louis, USA) at 37 ℃, followed by fixation in 4% paraformaldehyde overnight. The images were photographed using a digital camera and the areas of infarcted myocardium (white area) and viable tissue (red staining) were evaluated using Adobe Photoshop CS6 software (Adobe Systems Incorporated). The ratio of the infarct size (white area) was calculated as a percentage of the LV area.
Enzyme-linked immunosorbent assay (ELISA)
Myocardial injury was assessed by measuring the serum levels of lactate dehydrogenase (LDH), creatine kinase-myocardial isoenzyme (CK-MB), and cardiac troponin I (cTnI). Oxidative stress was evaluated using malondialdehyde (MDA), superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and myeloperoxidase (MPO) levels. ELISAs (Nanjing Jiancheng Institute of Bioengineering, China) were performed according to manufacturer’s instructions.
Hematoxylin and eosin (HE) staining of myocardial tissue
Cardiac tissues were fixed with 4% paraformaldehyde for 24 hours at room temperature, embedded in paraffin, and then sliced into 4 µm-thick sections. After dewaxing and rehydration, the sections were stained with HE for morphological analysis. The morphological structure of myocardial tissue was observed under light microscopy (DM3000 LED, Leica, Germany).
Transmission electron microscopy (TEM)
Ischemic areas of the LV heart tissue were cut into 1 mm3 sections on ice and placed into 2.5% pre-chilled glutaraldehyde solution immediately, fixed at 4 ℃ for 24 hours, and postfixed in 1% osmium tetroxide for 2 hours. Samples were then dehydrated in alcohols, embedded in epoxy resin, and cut into ultra-thin sections of 50 nm thickness. After double staining with uranyl acetate and lead citrate, ultrathin sections were observed and photographed under a TEM (JEM-1400Plus; JEOL, Tokyo, Japan).
Western blot analysis
Total protein was extracted from rat hearts using radioimmunoprecipitation assay (RIPA) buffer. The protein concentrations were quantified with the Bicinchoninic Acid (BCA) Protein Assay Kit (E-BC-K318-M, Elabscience Biotechnology, Wuhan, China). Equivalent amounts of proteins were separated by 8–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; P1200, Solarbio, Beijing, China) and transferred to polyvinylidene difluoride (PVDF) membranes (YA1701, Solarbio, Beijing, China). Membranes were blocked with 5% nonfat milk for 2 hours at room temperature, followed by incubation with primary antibodies against PINK1 (1:800, 23274-1-AP, Proteintech, Wuhan, China), microtubule-associated protein 1 light chain 3 (LC3; 1:1,000, 14600-1-AP, Proteintech, Wuhan, China), p62 (1:5,000, 18420-1-AP, Proteintech, Wuhan, China), Parkin (1:5,000, 66674-1-lg, Proteintech, Wuhan, China), Beclin1 (1:5,000, 11306-1-AP, Proteintech, Wuhan, China), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:4,000, 10494-1-AP, Proteintech, Wuhan, China) at 4 ℃ overnight. After washing with Tris-buffered saline-Tween 20 (TBST) 3 times, the membranes were incubated with the corresponding horse radish peroxidase (HRP)-conjugated goat anti-mouse antibody (1:4,000, ab205719, Abmart, China) or goat anti-rabbit antibody (1:4,000, ab205718, Abmart, China) at room temperature for 2 hours. The protein bands were detected using the enhanced chemiluminescence (ECL) kit (Advansta, Menlo Park, USA) and the BLT GelView 6000Plus system (Bolu, Guangzhou, China). The band intensities were analyzed quantitatively using Image J software (National Institutes of Health, Bethesda, MD, USA). Protein expression was normalized to GAPDH.
Real-time polymerase chain reaction (PCR)
Total RNA was isolated from myocardial tissues using TRIzol Reagent (Life Technologies). Reverse transcription was performed using a kit purchased from Takara, Japan. The SYBR Premix Ex Tag Kit (TaKaRa, Dalian, China) was used for real-time quantitative PCR (the instrument was purchased from Bio-Rad, USA) to detect the expression levels of PINK1, Beclin1, and Parkin. The primer sequences used are shown in Table 1. Each experiment was performed in triplicate for calibration with GAPDH as the internal reference gene. The Ct values obtained from different samples were compared using the 2−ΔΔCt method.
Table 1. Primer sequences for quantitative real-time PCR.
| Gene | Primer sequences |
|---|---|
| Beclin1 | Forward: TTT TCT GGA CTG TGT GCA GC |
| Reverse: CCA CTT GAG ATT CGT CAG CA | |
| PINK1 | Forward: CCC ACA CCC TAA CAT CAT CC |
| Reverse: CTG CTC CTC AAG GTA CTG GC | |
| Parkin | Forward: TGG AAA GCT CCG AGT TCA GT |
| Reverse: CCT TGT CTG AGG TTG GGT GT | |
| GAPDH | Forward: GGG TGT GAA CCA CGA GAA AT |
| Reverse: CCA CAG TCT TCT GAG TGG CA |
PCR, polymerase chain reaction; PINK1, PTEN-induced putative kinase protein-1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Immunofluorescence staining
Paraffin-embedded heart sections were baked in an oven at 65 ℃ for 2 hours. After deparaffinization, the sections were rinsed 3 times with PBS for 5 minutes each time, and then placed in ethylenediaminetetraacetic acid (EDTA) buffer for microwave repair. Sections were subsequently incubated in 3% H2O2 solution at room temperature for 10 minutes and blocked with 5% bovine serum albumin (BSA) for 20 minutes. Sections were then incubated at 4 ℃ overnight with the following primary antibodies: Parkin (1:200, 66674-1-lg, Proteintech, Wuhan, China), voltage-dependent anion-selective channel protein 1 (VDAC1; 1:200, ab154856, Abcam, UK), and LC3 (1:400, 14600-1-AP, Proteintech, Wuhan, China). The following day, sections were incubated with the corresponding fluorescent secondary antibody for 1 hour at 37 ℃. 4',6-diamidino-2-phenylindole (DAPI) was used to label cardiomyocytes (the total DNA in all cell nuclei was counterstained blue with DAPI). The co-location of Parkin and VDAC1, and the expression level of LC3 were observed under fluorescence microscopy (DM3000 LED, Leica, Germany) and confocal scanning microscopy (TCS SP5, Leica, Germany).
Statistical analysis
All statistical analyses were performed with SPSS 26.0 software (IBM Corp., Chicago, IL, USA). Data were presented as the mean ± standard deviation (SD). Differences among groups were determined by one-way analysis of variance (ANOVA). Tukey post-hoc tests were used to identify significant differences between means. A P value <0.05 was considered statistically significant. All graphs were made using GraphPad Prism 8.0 software (Motulsky, San Diego, CA, USA).
Results
The effects of NBP treatment on rats subjected to I/R injury
A total of 90 rats were included in the study, and 74 rats survived the surgery, completed the specimen collection, and data analyses. There were 16 deaths during or within 6 hours after surgery. There were no significant differences in body weight among the groups. TTC staining was performed to assess the effects of NBP on myocardial infract size. Compared with the sham group, the myocardial infarct size of the I/R group was significantly increased (38.47%±5.67%), while treatment with NBP reduced the myocardial infarct size induced by IRI (100 mg/kg NBP: 29.22%±5.10%; 200 mg/kg NBP: 23.24%±4.41%; P<0.05; Figure 1B,1C). However, there was no significant difference between the 2 doses of NBP (P=0.083). CK-MB, cTnI, and LDH are widely used as markers of acute myocardial injury. The serum levels of these markers were significantly elevated after exposure to IRI, and these effects were attenuated in a dose-dependent manner after administration of NBP (P<0.05; Figure 2A-2C).
Figure 2.

NBP attenuated the I/R-induced myocardial injury and oxidative stress. The levels of myocardial injury and oxidative stress indicators. (A) CK-MB isoenzyme levels; (B) cTnI levels; (C) LDH levels; (D) MDA levels; (E) MPO levels; (F) SOD levels; and (G) GSH-Px levels. n=10; ##, indicates P<0.01 compared with the sham group; **, indicates P<0.01 compared with the I/R group; *, indicates P<0.05 compared with the I/R group. I/R, ischemia/reperfusion; NBP, Dl-3-n-butylphthalide; NBP-L, low dose of NBP; NBP-H, high dose of NBP; CK-MB, creatine kinase-myocardial isoenzyme; cTnI, cardiac troponin I; LDH, lactate dehydrogenase; MDA, malondialdehyde; MPO, myeloperoxidase; SOD, superoxide dismutase; GSH-Px, glutathione peroxidase.
The effects of NBP treatment on oxidative stress in rats subjected to I/R injury
The levels of MDA, MPO, SOD, and GSH-Px were assessed by ELISA to determine the effects of NBP on oxidative stress in vivo. Compared to the sham group, the levels of MDA and MPO were increased, while the levels of SOD and GSH-Px were decreased in the I/R group. However, administration of NBP reduced the levels of MDA and MPO, and improved the levels of SOD and GSH-Px (Figure 2D-2G). These results suggested that NBP attenuated the I/R-induced oxidative stress in vivo.
The effects of NBP on the morphological structure of myocardial tissue and mitochondria in I/R rats
The morphological structure of myocardial tissue was evaluated by HE staining and the ultrastructure of the cardiomyocytes was examined by TEM. In the sham group, cardiomyocytes were arranged in an orderly manner, with nuclei located in the center of muscle fibers, slightly stained, and the mitochondria had complete double-layer structural membranes, dense matrix particles, and tightly packed cristae. In contrast, cardiomyocytes in the I/R group showed severe damage, irregular morphology, interstitial edema, incomplete structure, disordered arrangement, and more significant changes in mitochondrial ultrastructural morphology, including swelling, disintegration, loss of mitochondrial cristae, and more vacuole formation in the matrix. However, NBP treatment significantly ameliorated these changes (Figure 3A,3B), suggesting that NBP exerted a protective effect on cardiomyocytes and the mitochondria.
Figure 3.

The effects of NBP on pathological injury in the heart. (A) The morphology of heart tissues visualized with HE staining (magnification ×200). (B) Representative transmission electron micrograph images of the mitochondria in cardiomyocytes; the white arrow indicates a mitochondrion (magnification ×10,000). I/R, ischemia/reperfusion; NBP, Dl-3-n-butylphthalide; NBP-L, low dose of NBP; NBP-H, high dose of NBP; HE, hematoxylin and eosin.
The effects of NBP on cardiac mitophagy and PINK1/Parkin signaling pathway in I/R rats
The expression of Beclin1, p62, and the ratio of LC3 II/LC3 I was used to assess mitophagy. Compared with the sham group, the I/R group showed significantly decreased expression of Beclin1 and LC3 II/LC3 I ratio (P<0.01), while the expression of p62, a substrate ubiquitin binding protein known to be negatively correlated with autophagy (21), was enhanced. NBP treatment significantly elevated the LC3 II/LC3 I ratio and Beclin1 expression levels, and reduced the expression of p62 (Figure 4A). In addition, reverse transcription PCR (RT-PCR) analysis revealed that Beclin1 mRNA expression was consistent with Beclin1 protein expression observed with Western blots (Figure 4B). Furthermore, immunofluorescence staining revealed that NBP treatment significantly increased LC3-positive punctuated staining compared with the I/R group (Figure 4C).
Figure 4.

NBP promoted autophagy in rats subjected to I/R injury. (A) Western blot analysis of the expression levels of autophagy-related proteins (Beclin1, LC3 II/LC3 I, and p62) in heart tissue. (B) RT-PCR detection of gene expression levels in rat heart tissue. (C) Representative images of immunofluorescence labelling with LC3 in different groups. Scale bar =50 µm. The white arrow indicates the expression of LC3. ##, indicates P<0.01 compared with the sham group; #, indicates P<0.05 compared with the sham group; **, indicates P<0.01 compared with the I/R group; *, indicates P<0.05 compared with the I/R group. I/R, ischemia/reperfusion; NBP, Dl-3-n-butylphthalide; NBP-L, low dose of NBP; NBP-H, high dose of NBP; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; LC3, light chain 3; DAPI, 4',6-diamidino-2-phenylindole; RT-PCR, reverse transcription polymerase chain reaction.
The effect of NBP on the PINK1/Parkin pathway was examined. After administration of NBP, the protein expression of PINK1 and Parkin was distinctly elevated (Figure 5A), which was consistent with the results from RT-PCR (Figure 4B). Immunofluorescence staining with the mitochondrial outer membrane protein VDAC1 was performed to locate the mitochondria. Colocalization of Parkin and VDAC1 staining suggested that the PINK1/Parkin pathway mediated mitophagy. These results indicated that NBP treatment significantly upregulated Parkin and VDAC1 co-expression in a dose-dependent manner (Figure 5B).
Figure 5.
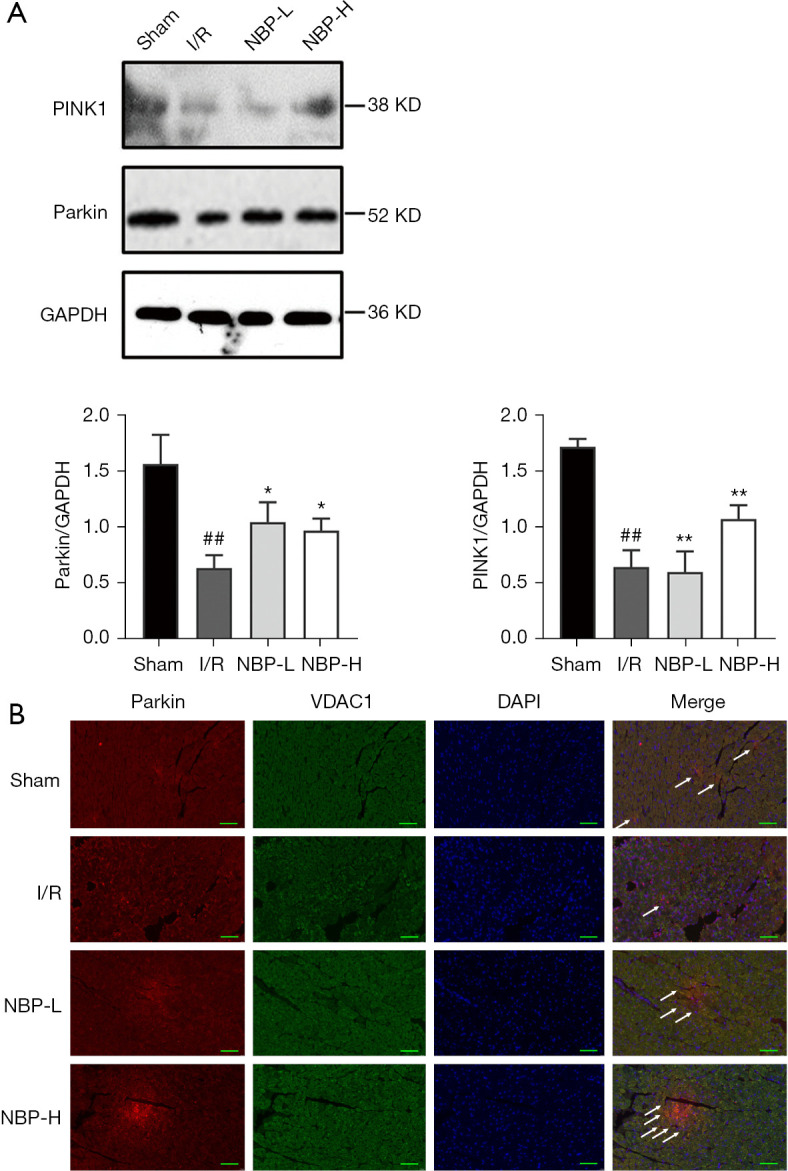
NBP activated PINK1/Parkin-mediated mitophagy in rats subjected to I/R injury. (A) Western blot analysis of the expression levels of PINK1 and Parkin in the heart of rats. ##, indicates P<0.01 compared with the sham group; **, indicates P<0.01 compared with the I/R group; *, indicates P<0.05 compared with the I/R group. (B) PINK1/Parkin-mediated mitophagy as shown by double immunofluorescence labelling with Parkin and VDAC1. Scale bar =50 µm. The white arrow indicates the expression of Parkin in mitochondria. I/R, ischemia/reperfusion; NBP, Dl-3-n-butylphthalide; NBP-L, low dose of NBP; NBP-H, high dose of NBP; PINK1, PTEN-induced putative kinase protein-1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; VDAC1, voltage-dependent anion-selective channel protein 1; DAPI, 4',6-diamidino-2-phenylindole.
Taken together, these results suggested that NBP activated PINK1/Parkin-mediated mitophagy in I/R rats. This may be a potential mechanism for NBP to protect cardiomyocytes from IRI.
Discussion
Increasing evidence indicates that a wide range of pathological processes are involved in the development of myocardial IRI, including oxidative stress, endothelial cell inflammation, calcium overload, mitochondrial dysfunction and various programs of cell death, such as necrosis, apoptosis, autophagy and ferroptosis (22,23). Mitochondria have been recognized as key triggers of myocardial IRI. Mitochondrial quality control mechanisms, a series of adaptive responses after IRI to protect mitochondrial structure and function, ensuring myocardial cell survival and cardiac function, including mitochondrial fission, mitochondrial fusion, mitophagy and mitochondrial-dependent cell death (24). Mitochondrial protection may become a promising target for myocardial protection of IRI.
Previous reports have identified that NBP has extensive pharmacological effects in nervous system diseases (14-16). While a limited number of studies have shown that NBP has cardioprotective effects, the precise mechanisms are not fully understood. This present study explored the effects of NBP on myocardial IRI and the potential mechanisms of action. NBP greatly reduced the myocardial infarct size, alleviated myocardial injury and oxidative stress, improved the pathological alteration of cardiomyocytes and mitochondria, and upregulated autophagy, especially mitophagy, which may be mediated by the PINK1/Parkin pathway. These results demonstrated a novel mechanism by which NBP exerted a cardioprotective effect during IRI. Indeed, NBP may be a potential pharmacological candidate for preventative therapy in myocardial IRI.
CK-MB isoenzyme, cTnI, and LDH leakage from myocardial cells lead to increased serum concentrations, which can reflect the degree of myocardial injury (25). These indexes, together with the myocardial infarction area, were used to evaluate the degree of myocardial injury. The data demonstrated that these serum indexes and the myocardial infarction area were all significantly increased in the I/R group, while significantly decreased in a dose-dependent manner in the NBP-treated groups. Consistent with previous studies, our results verified the cardioprotective effect of NBP in IRI (18,26). This was further confirmed by the pathological alterations observed.
Excessive production of ROS is an important mechanism of IRI, resulting in destruction of the mitochondrial structure, mitochondrial swelling, cell membrane structure damage, and cell energy metabolism disorder (27). Oxidative stress occurs due to excessive production of ROS or insufficient antioxidant capacity of the body. MDA is a presumptive marker of lipid peroxidation and an accepted parameter for determining the increase in free radical formation and tissue damage. MPO is a special enzyme of neutrophils. Its activity can quantitatively reflect the activation and aggregation of neutrophils in tissues and indirectly reflect the degree of oxidative stress. SOD and GSH-Px are main enzymes reflecting the body’s antioxidant activity, which can scavenge ROS and resist oxidative damage. The present study found that the antioxidant capacity of cardiomyocytes decreased during IRI, which was manifested by the decrease in SOD and GSH-Px activities and the increase of MDA and MPO content. However, NBP treatment improved this imbalance between oxidation and antioxidant systems, and this is consistent with the effects of NBP on renal IRI (28). Therefore, the antioxidant stress effects of NBP may be conducive to cardioprotection against IRI.
As mentioned above, increasing evidence suggests that modulation of mitophagy plays a pivotal role during myocardial IRI (11), and it may become a novel target for cardioprotection. The PINK1/Parkin signaling pathway is the most recognized pathway leading to mitophagy in mammalian cells (29). PINK1, a serine/threonine kinase, is imported from the cytoplasm to the inner mitochondrial membrane in healthy mitochondria. The degradation of PINK1 is a signal to maintain mitochondrial membrane potential. The decrease in damaged mitochondrial membrane potential results in aggregation of PINK1 on the mitochondrial surface, resulting in the phosphorylation of various substrates. Among them is the E3 ubiquitin ligase Parkin, which has high affinity for phosphoS65-ubiquitin, translocates from the cytoplasm to the mitochondrial surface, and promotes ubiquitination of the outer mitochondrial membrane (OMM) proteins. Parkin-mediated hyper-ubiquitination of OMM is recognized by ubiquitin-binding receptors, such as p62, OPTN, NBR1, NDP52, which further binds to LC3 and initiates mitophagosome formation (30). Most studies have shown that mitophagy attenuates rather than exacerbates myocardial damage in IRI (31-33). PINK1 or Parkin overexpression protects cardiomyocytes from IRI, while deficiency in PINK1 or Parkin in knockout models makes the heart more vulnerable to IRI, and this may be related to the decrease of mitophagy and the increase of mitochondrial function imbalance (34,35). Several studies have shown the cardioprotective effects from pharmacological activation of PINK1/Parkin-mediated mitophagy after I/R or H/R (12,13,36). For instance, dexpramipexole (DPX) treatment reduced the myocardial infarct size and tissue damage induced by I/R, and protected cells from H/R-induced mitochondrial dysfunction. DPX enhanced mitophagy in vivo and in vitro, accompanied by increased expression of PINK1 and Parkin. Knockout of PINK1 and Parkin by specific small interfering RNA (siRNAs) reversed the DPX-induced inhibition of myocardial I/R injury, suggesting that PINK1/Parkin-mediated mitophagy is a target of myocardial protection in IRI (36).
NBP is a recommended medication for the management of acute ischemic stroke in China, and has been suggested to be involved in maintaining mitochondrial dynamics, including mitochondrial division and fusion, and PINK1/Parkin-mediated mitophagy in a mouse model of Parkinson’s disease (37). NBP has also been shown to play a cardioprotective role against cardiac ischemic injury via the regulation of mitochondrial function and biogenesis (17). The present study demonstrated that NBP treatment could not only reduce myocardial injury caused by IRI, but also improve the morphology, structure, and integrity of the mitochondria, as confirmed by TEM. Meanwhile, NBP inhibited the oxidative stress induced by IRI. Furthermore, Western blot analyses, real-time PCR, and immunofluorescence staining assays confirmed that NBP upregulated the expression of autophagy marker proteins, such as Beclin1 and LC3 II/LC3 I, and activated PINK1/Parkin-mediated mitophagy in the myocardium suffering from IRI. Overall, this study identified a potential pharmacological candidate, NBP, for the prevention and treatment of myocardial IRI.
There are certain limitations to the present report. First, inhibitors of mitophagy or the PINK1/Parkin signaling pathway should be added in future experiments to verify the role of NBP. Second, further clinical trials are warranted to determine whether NBP exerts a myocardial protective effect in patients with IRI.
Conclusions
In summary, this work provided evidence that NBP protects the myocardium from IRI by reducing oxidative stress and activating mitophagy, through activation of the PINK1/Parkin pathway in vivo. The mitochondrial protective effect of NBP may be related to its antioxidative role and activation of mitophagy. Thus, NBP may be a potential cardioprotective drug, and its effects on IRI warrant further investigation.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This study was supported by the Health Department of Hebei Province, China (No. 20220285).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All animal experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and approved by the Laboratory Animal Ethics Committee of the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (ethics application number: IRM-DWLL-2020076).
Reporting Checklist: The authors have completed the ARRIVE reporting checklist. Available at https://jtd.amegroups.com/article/view/10.21037/jtd-22-585/rc
Data Sharing Statement: Available at https://jtd.amegroups.com/article/view/10.21037/jtd-22-585/dss
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://jtd.amegroups.com/article/view/10.21037/jtd-22-585/coif). The authors have no conflicts of interest to declare.
(English Language Editor: J. Teoh)
References
- 1.GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1545-602. 10.1016/S0140-6736(16)31678-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu S, Jiang J, Zhang Y, et al. Discovery of potential plasma protein biomarkers for acute myocardial infarction via proteomics. J Thorac Dis 2019;11:3962-72. 10.21037/jtd.2019.08.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piper HM, García-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res 1998;38:291-300. 10.1016/S0008-6363(98)00033-9 [DOI] [PubMed] [Google Scholar]
- 4.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007;357:1121-35. 10.1056/NEJMra071667 [DOI] [PubMed] [Google Scholar]
- 5.Maneechote C, Palee S, Chattipakorn SC, et al. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J Cell Mol Med 2017;21:2643-53. 10.1111/jcmm.13330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulek AR, Anzell A, Wider JM, et al. Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury. Cells 2020;9:214. 10.3390/cells9010214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Picca A, Mankowski RT, Burman JL, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol 2018;15:543-54. 10.1038/s41569-018-0059-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol 2018;15:457-70. 10.1038/s41569-018-0044-6 [DOI] [PubMed] [Google Scholar]
- 9.Ji H, Wu D, Kimberlee O, et al. Molecular Perspectives of Mitophagy in Myocardial Stress: Pathophysiology and Therapeutic Targets. Front Physiol 2021;12:700585. 10.3389/fphys.2021.700585 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Zhang W, Siraj S, Zhang R, et al. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 2017;13:1080-1. 10.1080/15548627.2017.1300224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.García-Niño WR, Zazueta C, Buelna-Chontal M, et al. Mitochondrial Quality Control in Cardiac-Conditioning Strategies against Ischemia-Reperfusion Injury. Life (Basel) 2021;11:1123. 10.3390/life11111123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andres AM, Hernandez G, Lee P, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal 2014;21:1960-73. 10.1089/ars.2013.5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang Q, Wu M, Zhang L, et al. Gerontoxanthone I and Macluraxanthone Induce Mitophagy and Attenuate Ischemia/Reperfusion Injury. Front Pharmacol 2020;11:452. 10.3389/fphar.2020.00452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niu X, Li M, Gao Y, et al. DL-3-n-butylphthalide suppressed autophagy and promoted angiogenesis in rats with vascular dementia by activating the Shh/Ptch1 signaling pathway. Neurosci Lett 2021;765:136266. 10.1016/j.neulet.2021.136266 [DOI] [PubMed] [Google Scholar]
- 15.Wang BN, Wu CB, Chen ZM, et al. DL-3-n-butylphthalide ameliorates diabetes-associated cognitive decline by enhancing PI3K/Akt signaling and suppressing oxidative stress. Acta Pharmacol Sin 2021;42:347-60. 10.1038/s41401-020-00583-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang C, Cui L, He W, et al. Dl-3-n-butylphthalide promotes neurite outgrowth of primary cortical neurons by Sonic Hedgehog signaling via upregulating Gap43. Exp Cell Res 2021;398:112420. 10.1016/j.yexcr.2020.112420 [DOI] [PubMed] [Google Scholar]
- 17.Wang YG, Li Y, Wang CY, et al. L-3-n-Butylphthalide protects rats' cardiomyocytes from ischaemia/reperfusion-induced apoptosis by affecting the mitochondrial apoptosis pathway. Acta Physiol (Oxf) 2014;210:524-33. 10.1111/apha.12186 [DOI] [PubMed] [Google Scholar]
- 18.Tian X, He W, Yang R, et al. Dl-3-n-butylphthalide protects the heart against ischemic injury and H9c2 cardiomyoblasts against oxidative stress: involvement of mitochondrial function and biogenesis. J Biomed Sci 2017;24:38. 10.1186/s12929-017-0345-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai M, Pan CL, Jiang GX, et al. Effects of butylphthalide on oxidative stress and inflammatory response in rats with myocardial infarction through Akt/Nrf2 signaling pathway. Eur Rev Med Pharmacol Sci 2019;23:9642-50. [DOI] [PubMed] [Google Scholar]
- 20.Wu S, Chang G, Gao L, et al. Trimetazidine protects against myocardial ischemia/reperfusion injury by inhibiting excessive autophagy. J Mol Med (Berl) 2018;96:791-806. 10.1007/s00109-018-1664-3 [DOI] [PubMed] [Google Scholar]
- 21.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012;8:445-544. 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibáñez B, Heusch G, Ovize M, et al. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 2015;65:1454-71. 10.1016/j.jacc.2015.02.032 [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Fan H, Wang S, et al. Ferroptosis: A Novel Therapeutic Target for Ischemia-Reperfusion Injury. Front Cell Dev Biol 2021;9:688605. 10.3389/fcell.2021.688605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B 2020;10:1866-79. 10.1016/j.apsb.2020.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao Z, Ma J, Liu H. Evaluation of the antioxidant potential of Salvia miltiorrhiza ethanol extract in a rat model of ischemia-reperfusion injury. Molecules 2011;16:10002-12. 10.3390/molecules161210002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Hao G, Jiang Y, et al. Influence of dl-3-N-butylphthalide on infarction size in rats with acute myocardial infarction. Int J Clin Exp Pathol 2020;13:1707-11. [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan X, Juan Z, Zhang R, et al. Clemastine Fumarate Protects Against Myocardial Ischemia Reperfusion Injury by Activating the TLR4/PI3K/Akt Signaling Pathway. Front Pharmacol 2020;11:28. 10.3389/fphar.2020.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong Y, Yin J, Chen T, et al. Dl-3-n-butylphthalide pretreatment attenuates renal ischemia/reperfusion injury. Biochem Biophys Res Commun 2021;557:166-73. 10.1016/j.bbrc.2021.04.006 [DOI] [PubMed] [Google Scholar]
- 29.Turkieh A, El Masri Y, Pinet F, et al. Mitophagy Regulation Following Myocardial Infarction. Cells 2022;11:199. 10.3390/cells11020199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdrakhmanov A, Gogvadze V, Zhivotovsky B. To Eat or to Die: Deciphering Selective Forms of Autophagy. Trends Biochem Sci 2020;45:347-64. 10.1016/j.tibs.2019.11.006 [DOI] [PubMed] [Google Scholar]
- 31.Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res 2017;120:1812-24. 10.1161/CIRCRESAHA.117.311082 [DOI] [PubMed] [Google Scholar]
- 32.Lv W, Jiang J, Li Y, et al. MiR-302a-3p aggravates myocardial ischemia-reperfusion injury by suppressing mitophagy via targeting FOXO3. Exp Mol Pathol 2020;117:104522. 10.1016/j.yexmp.2020.104522 [DOI] [PubMed] [Google Scholar]
- 33.Chang X, Zhang T, Meng Q, et al. Quercetin Improves Cardiomyocyte Vulnerability to Hypoxia by Regulating SIRT1/TMBIM6-Related Mitophagy and Endoplasmic Reticulum Stress. Oxid Med Cell Longev 2021;2021:5529913. 10.1155/2021/5529913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubli DA, Zhang X, Lee Y, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 2013;288:915-26. 10.1074/jbc.M112.411363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khuanjing T, Palee S, Kerdphoo S, et al. Donepezil attenuated cardiac ischemia/reperfusion injury through balancing mitochondrial dynamics, mitophagy, and autophagy. Transl Res 2021;230:82-97. 10.1016/j.trsl.2020.10.010 [DOI] [PubMed] [Google Scholar]
- 36.Tang L, Li YP, Hu J, et al. Dexpramipexole attenuates myocardial ischemia/reperfusion injury through upregulation of mitophagy. Eur J Pharmacol 2021;899:173962. 10.1016/j.ejphar.2021.173962 [DOI] [PubMed] [Google Scholar]
- 37.Li H, Wang H, Zhang L, et al. Dl-3-n-Butylphthalide Alleviates Behavioral and Cognitive Symptoms Via Modulating Mitochondrial Dynamics in the A53T-α-Synuclein Mouse Model of Parkinson's Disease. Front Neurosci 2021;15:647266. 10.3389/fnins.2021.647266 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as