

Abstract
Summary
Distinguishing biologically relevant interfaces from crystallographic ones in biological complexes is fundamental in order to associate cellular functions to the correct macromolecular assemblies. Recently, we described a detailed study reporting the differences in the type of intermolecular residue–residue contacts between biological and crystallographic interfaces. Our findings allowed us to develop a fast predictor of biological interfaces reaching an accuracy of 0.92 and competitive to the current state of the art. Here we present its web-server implementation, PRODIGY-CRYSTAL, aimed at the classification of biological and crystallographic interfaces. PRODIGY-CRYSTAL has the advantage of being fast, accurate and simple. This, together with its user-friendly interface and user support forum, ensures its broad accessibility.
Availability and implementation
PRODIGY-CRYSTAL is freely available without registration requirements at https://haddock.science.uu.nl/services/PRODIGY-CRYSTAL.
1 Introduction
Interactions between proteins are at the origin of essential function in cells. Correct identification of biologically relevant assemblies is therefore fundamental for a proper understanding of their function and mode of action. X-ray crystallography is the most widely used technique to experimentally determine structures of protein–protein complexes. However, distinguishing biologically relevant interfaces from crystallographic ones, which are non-specific and mere artifacts of the crystallization process, remains a non-trivial problem.
We have previously demonstrated the fundamental role of interfacial residue–residue contacts in describing and predicting binding properties. These have led to high-performing binding affinity predictors for protein–protein (Vangone and Bonvin, 2015) and protein–small ligand binding affinities (Kurkcuoglu et al., 2018) implemented into the PRotein binDIng enerGY prediction (PRODIGY) web server available at http://haddock.science.uu.nl/services/PRODIGY (Vangone and Bonvin, 2017; Xue et al., 2016) and PRODIGY-LIGand (PRODIGY-LIG), available at http://haddock.science.uu.nl/services/PRODIGY-LIG (Vangone et al., 2018), respectively.
We have recently further extended the concept of intermolecular contacts into a new methodology for the classification of biological/crystallographic interfaces in protein–protein complexes, reported in Elez et al. (2018). Our predictor is based on simple structural properties of the interface: The residue–residue contacts classified by their polar/apolar/charged character and the type of amino acid involved in the contact. By using the Many dataset (Baskaran et al., 2014), we trained a machine learning predictor which reaches an accuracy of 0.92 (Elez et al., 2018). Our method is competitive method with respect to the current state of the art PISA (Krissinel and Henrick, 2007) and EPPIC (Duarte et al., 2012) which reach accuracies of 0.83 and 0.88 on the same dataset, respectively.
A few web-tools for interface classification have been made available to the scientific community (Bernauer et al., 2008; Duarte et al., 2012; Krissinel and Henrick, 2007; Mitra et al., 2011; Ponstingl et al., 2003). Here we present PRODIGY-CRYSTAL, a fast and user-friendly online tool which implements our contact-based methodology to classify interfaces into biological or crystallographic ones.
2 The web server
The input page
PRODIGY-CRYSTAL has been implemented as a user-friendly web server within our PRODIGY collection of web-tools, which are aimed at predicting various properties in biomolecules complexes. The server is freely available without registration at http://haddock.science.uu.nl/services/PRODIGY-CRYSTAL. It provides a fast prediction of the protein–protein interface nature, classifying it as biological or crystallographic. Users are required to provide the following information:
Structures: coordinates file in PDB or mmCIF format. The file can be uploaded or automatically retrieved from the PDB by specifying a PDB ID. A compressed archive file (tar, tgz, zip, bz2 or tar.gz) containing multiple complexes can also be provided for classification of multiple interfaces in one submission.
Chain IDs: the chain identifiers for the interface to be analyzed. These must be provided in the two boxes named ‘Interactor 1’ and ‘Interactor 2’, respectively. One interface can also be formed by more than one chain (e.g. chains A and B). In that case the chains should be reported as a comma separated list (‘A, B’).
Job ID (Optional): personalized job name.
Email (Optional): in the case an email is provided, a link to the results page will be sent to the user.
Upon successful validation of the input data, users are redirected to the job page, which displays the status of the job during execution and the results upon completion.
The output page
Prediction is usually completed within a few seconds and the results displayed online. If the user closes the page, the calculation will still be carried out and the results will remain accessible for 2 weeks provided the user saved the URL or provided an email address.
The results page displays the following information:
Class of the interface predicted, reported as ‘biological’ or ‘crystallographic’ together with the probability values, which range between 0 and 1 and indicate how likely is the interface to belong to each of the two classes;
Number of residue contacts found at the interface, classified according to their polar/apolar/charged character;
Link Density value (Elez et al., 2018);
List of residue contacts, reported as a downloadable table in .txt format.
If an interface is predicted as ‘biological’, the server will run the PRODIGY module (Xue et al., 2016) in order to provide the binding affinity prediction (reported in kcal mol−1), which is then displayed in the main results page.
Online visualization of the query interfaces is also provided, powered by the NGL Viewer (Rose et al., 2018) JavaScript software. The interacting chains are colored in blue/pink for Interactor 1 and Interactor 2, respectively. The interface, composed of the residues within 5.0 Å distance from the other chain, is highlighted in green for a predicted biological interface or in red for a crystallographic one. Finally, it is possible to download an archive folder of the PRODIGY-CRYSTAL output (in .tgz format).
An overview of PRODIGY-CRYSTAL output page is reported in Figure 1.
Fig. 1.
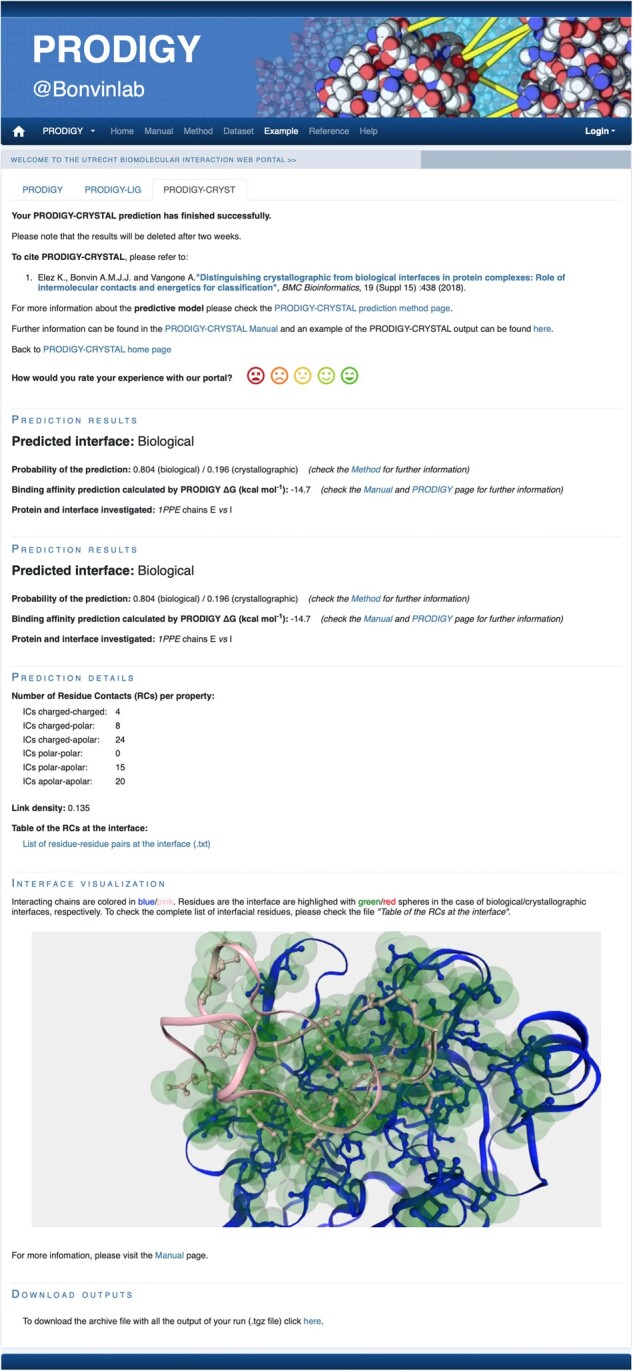
Example of the PRODIGY-CRYSTAL output page for the complex 1PPE, chains E versus I. The four sections of the output pages are shown (Prediction results, Prediction details, Interface visualization and Download outputs). A three-dimensional representation of the complex interface is shown in the gray box, powered by NGL Viewer. The interacting chains will be reported in pink/blue ribbons, while the interface will be marked in green spheres (will be reported in red for interfaces predicted as crystallographic)
The web-tool
As for the other PRODIGY tools, the home page is organized with a top banner displaying the several available sections. Dedicated tabs help the user read the content of each page and easily navigate through them.
Besides the Home page, which reports general information and presents the form for job submission, Manual, Method, Dataset, References and Help pages are also provided. Explanations regarding the usage of the web-tool, the required input and output formats are reported in Manual. The implemented methodology and dataset used for training and testing (also available for download through the SBGRID repository at https://data.sbgrid.org/dataset/566/) can be found in Method and Dataset, respectively. A typical output page can be found by clicking on the Example tab. It is also possible to run a test job from scratch in the Home input page, by clicking on the Load Example Data button. The References page provides the relevant literature related to the PRODIGY services and the Help page provides links to the dedicated user support forum (http://ask.bioexcel.eu/c/prodigy) and the standalone code freely available from GitHub. Indeed, in addition to the web-server we also provide a standalone version of the PRODIGY-CRYSTAL code for local use. Information about use, installation and download are reported in our GitHub repository at https://github.com/haddocking/prodigy-cryst.
The PRODIGY-CRYSTAL web server was written in Python 3.6.5 based on the Flask framework (version 1.0.2). The server is containerized with Docker (1.20.1) and deployed on a dedicated GNU/Linux server.
3 Conclusion
We have presented here an expansion of PRODIGY, namely PRODIGY-CRYSTAL, for the classification of protein–protein interfaces in crystallographic complexes. The web-tool takes a PDB or mmCIF file as input and applies our contact-based prediction method to classify interfaces as biological or crystallographic, not requiring any additional data. Its user-friendly interface, free availability and extensive online documentation and support will target a large audience, reaching researchers with different background and limited computational resources.
Funding
This work was supported by the European Union’s Horizon 2020 e-Infrastructure grants West-Life [grant number 675858]; BioExcel [grant numbers 675728, 823830] and EOSC-Hub [grant number 777536]; and by the Dutch Foundation for Scientific Research (NWO) [TOP-PUNT grant 718.015.001]. A.V. was supported by the Marie Skłodowska-Curie Individual Fellowship H2020 MSCA-IF-2015 [BAP-659025].
Conflict of Interest: none declared.
Contributor Information
Brian Jiménez-García, Computational Structural Biology Group, Bijvoet Center for Biomolecular Research, Faculty of Science - Chemistry, Utrecht University, Utrecht 3512 JE, The Netherlands.
Katarina Elez, Computational Structural Biology Group, Bijvoet Center for Biomolecular Research, Faculty of Science - Chemistry, Utrecht University, Utrecht 3512 JE, The Netherlands.
Panagiotis I Koukos, Computational Structural Biology Group, Bijvoet Center for Biomolecular Research, Faculty of Science - Chemistry, Utrecht University, Utrecht 3512 JE, The Netherlands.
Alexandre Mjj Bonvin, Computational Structural Biology Group, Bijvoet Center for Biomolecular Research, Faculty of Science - Chemistry, Utrecht University, Utrecht 3512 JE, The Netherlands.
Anna Vangone, Computational Structural Biology Group, Bijvoet Center for Biomolecular Research, Faculty of Science - Chemistry, Utrecht University, Utrecht 3512 JE, The Netherlands.
References
- Baskaran K. et al. (2014) A PDB-wide, evolution- based assessment of protein-protein interfaces. BMC Struct. Biol., 14, 22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernauer J. et al. (2008) DiMoVo: a Voronoi tessellation-based method for discriminating crystallographic and biological protein–protein interactions. Bioinformatics, 24, 652–658. [DOI] [PubMed] [Google Scholar]
- Duarte J.M. et al. (2012) Protein interface classification by evolutionary analysis. BMC Bioinformatics, 13, 334.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elez K. et al. (2018) Distinguishing crystallographic from biological interfaces in protein complexes: role of intermolecular contacts and energetics for classification. BMC Bioinformatics, 19 (Suppl. 15), 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol., 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Kurkcuoglu Z. et al. (2018) Performance of HADDOCK and a simple contact-based protein-ligand binding affinity predictor in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des., 32, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P. et al. (2011) Combining Bayes classification and point group symmetry under Boolean framework for enhanced protein quaternary structure inference. Structure, 19, 304–312. [DOI] [PubMed] [Google Scholar]
- Ponstingl H. et al. (2003) Automatic inference of protein quaternary structure from crystals. J. Appl. Cryst., 36, 1116–1122. [Google Scholar]
- Rose A.S. et al. (2018) NGL Viewer: web-based molecular graphic for large complexes. Bioinformatics, 34, 3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A., Bonvin A.M.J.J. (2015) Contacts-based prediction of binding affinity in protein-protein complexes. Elife, 4, e07454.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A., Bonvin A.M.J.J. (2017) PRODIGY: a contact-based predictor of binding affinity in protein-protein complexes. Bio Protoc., 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A. et al. (2018) Large scale prediction of binding affinity in protein-small ligand complexes: the PRODIGY-LIG web server. Bioinformatics, 35, 1585–1587. [DOI] [PubMed] [Google Scholar]
- Xue L.C. et al. (2016) PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics, 32, 3676–3678. [DOI] [PubMed] [Google Scholar]