

Abstract
Myoglobin based biosynthetic models of perturbed cytochrome c oxidase (CcO) active site are reconstituted, in situ, on electrodes where glutamate residues are systematically introduced in the distal site of the heme/Cu active site instead of a tyrosine residue. These biochemical electrodes show efficient 4e−/4H+ reduction with turnover rates and numbers more than 107 M−1 s−1 and 104, respectively. The H2O/D2O isotope effects of these series of crystallographically characterized mutants bearing zero, one, and two glutamate residues near the heme Cu active site of these perturbed CcO mimics are 16, 4, and 2, respectively. In situ SERRS-RDE data indicate complete change in the rate-determining step as proton transfer residues are introduced near the active site. The high selectivity for 4e−/4H+ O2 reduction and systematic variation of KSIE demonstrate the dominant role of proton transfer residues on the isotope effect on rate and rate-determining step of O2 reduction.
Keywords: biosynthetic model, cytochrome c oxidase, O2 reduction reaction, proton transfer residues, kinetic rate constant, kinetic isotope effect
Graphical Abstract
INTRODUCTION
Cytochrome c oxidase (CcO) and nitric oxide reductase (NOR) are two very important members of the Heme-copper oxidases (HCOs) superfamily.1–6 Crystal structure of the CcO and NOR indicate the similar organization of their active sites which consist of a heme coordinated to the protein by a histidine (His) ligand at the proximal side and a nonheme metal binding site comprising on three His ligands on the distal side.3,7,8 In the case of CcO, apart from the three His residues required to bind the distal Cu (CuB), a conserved, post-translationally modified tyrosine residue (Tyr244), covalently bound to one of the His ligand in the distal metal binding site, is present.9–11 Alternatively, in NOR a FeB ion replaces the CuB and a few conserved glutamate residues replace the tyrosine residue. In spite of similarities in the organization of their active sites, NOR primarily catalyzes the 2e− reduction of nitric oxide (NO) to nitrous oxide (N2O) and CcO primarily catalyzes the 4e−/4H+ reduction of oxygen (~90% of molecular oxygen reduction in the biosphere) to water and drives the synthesis of ATP by using the free energy generated from the chemical reduction of oxygenase.2,12–15 The very distinct reactivity of these two active sites likely originate from the differences in distal metal and/or the key second sphere distal residues.16–19 O2 reduction to H2O is a multistep process which requires 4e−/4H+. In the absence of requisite numbers of electrons, O2 may be partially reduced to H2O2. Recent investigations using in situ spectro-electrochemistry has unearthed the importance of the preorganized proton transfer channels in determining both rate and selectivity of the O2 reduction.20 The role of these proton transfer residues is in itself paramount. In particular, the rates of steps involving coupled proton and electron transfer (PCET)21 or proton transfer could be strongly affected by the presence of preorganized proton channel.22 However, these effects are difficult to investigate in native CcO as it is a membrane-bound protein. Synthetic complexes, however, are amenable to judicious modifications of the chemical functionalities in the second sphere and may help to understand the roles played by these proton transfer residues.
Several groups have been involved in modeling the structure and reactivity of HCOs using synthetic chemical models.23–25 Some of these models show 4e−/4H+ reduction of O2 to H2O, and the selectivity was enhanced with the incorporation of a distal CuB and a redox active tyrosine residue.23,26 In particular, some of these complexes have incorporated carboxylic acid groups (e.g., Hangman porphyrins) in their design to mimic the proton donor residues in HCOs.27,28 Some of these models have been used to assemble O2 reducing electrodes.23,29 These elegant complexes are easily accessible to proton sources from the solvent and, thus, they mostly provide a preorganized hydrogen bonding mediated proton transfer pathway. However, rarely very high selectivity (>90%) has been observed.27 Lu and co-workers have successfully installed the heme Cu site of CcO enzyme inside the active site of myoglobin (CuBMb) and further enhanced the selectivity of this mutant for O2 reduction by mutation of the Gly65 to Tyr (G65YCuBMb) in the distal site.16–18,30–32 Apart from modeling the active site of CcO, Lu et.al. has successfully designed and engineered the active site of NOR where instead of the CuB, FeB is introduced in the distal site and a glutamate residue is introduced mutating the Val68 residue in near the active site to Glu (Figure 1A, V68ECuBMb).19 Introduction of another glutamate residue mutating the Ile107 (Figure 1B, V68E/I107ECuBMb) showed greater NOR reactivity.16 The fact that one can easily switch the distal metal between Fe and Cu in these mutants empowers one to gain insight into the roles played by these metals and residues in distinguishing the selectivity of CcO and NOR; two members of the heme/Cu family. These artificial CcO mimics, while not very appealing as ORR catalysts, provide a great opportunity for understanding the key factors that control the rate and selectivity of this 4e−/4H+ process.
Figure 1.
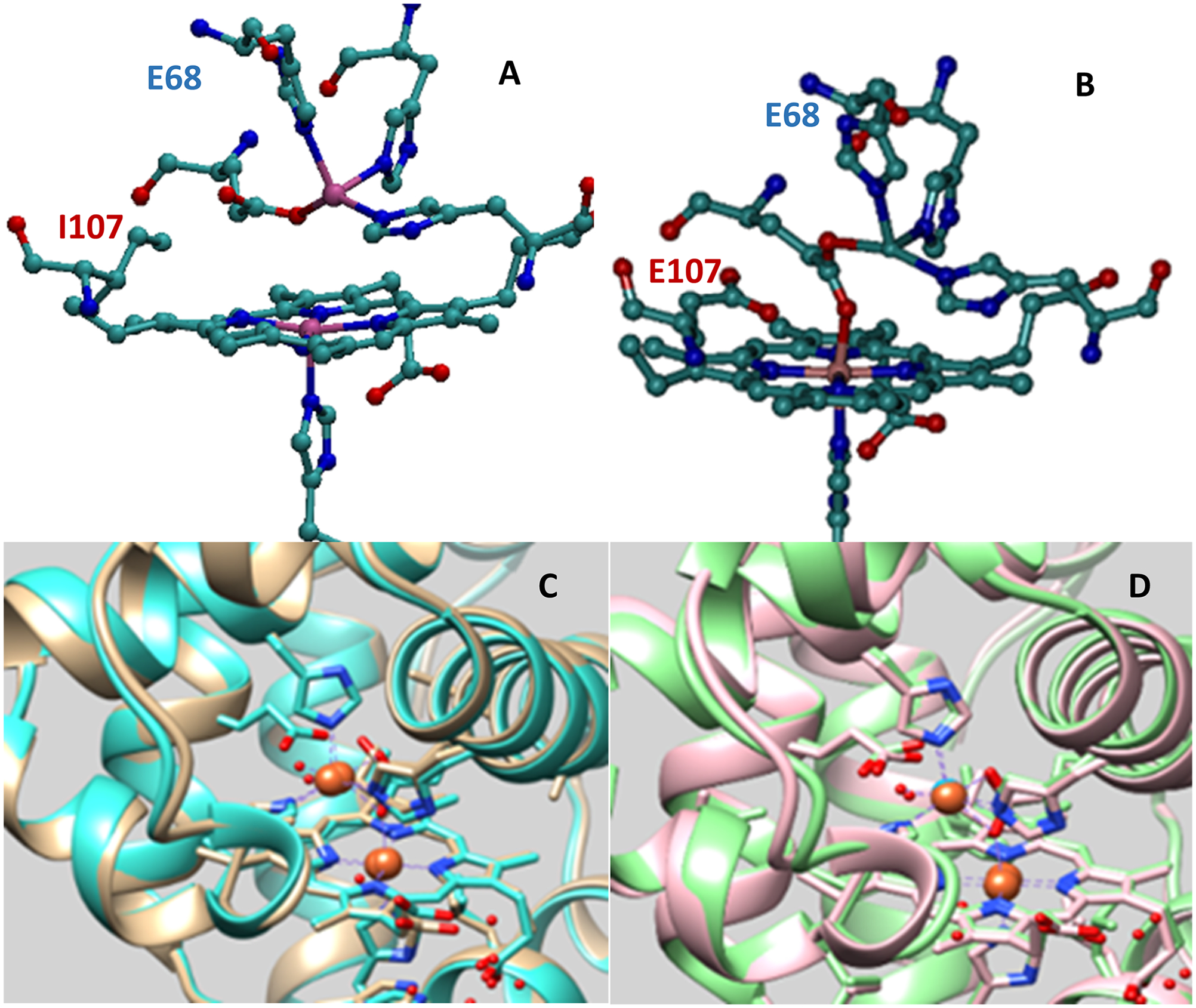
Crystal structure of (A) FeB(II)-V68ECuBMb (PDB: 3K9Z, tan); (B) FeB(II)-V68E/I107ECuBMb (PDB: 3M39, turquoise); (C) overlay of the crystal structure of FeB(II)-V68ECuBMb (tan) and FeB(II)-V68E/I107ECuBMb (turquoise); and (D) overlay of the crystal structure of FeB(II)-V68E/I107ECuBMb (light green) and CuB(II)-V68E/I107ECuBMb (PDB: 3M3A, pink), water molecules, and Fe(II) are represented by red and brown, respectively.
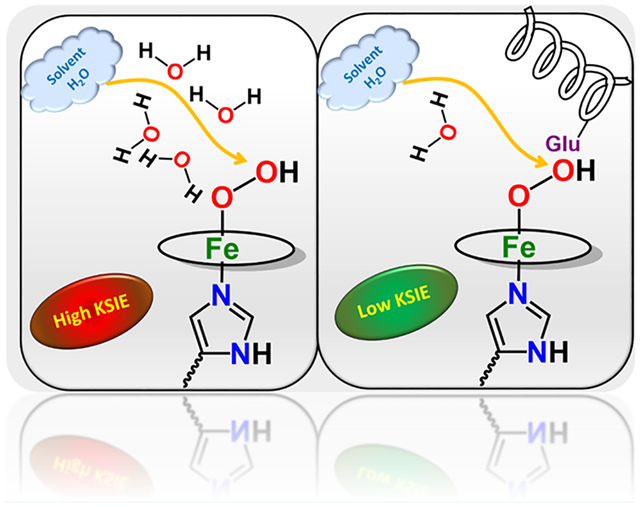
The key step in the selective and facile reduction of O2 to water is the heterolysis of O–O bond of the Fe(III)-hydroperoxide species. Selective 4e−/4H+ reduction of O2 can be achieved if protonation of a ferric hydroperoxide intermediate occurs at the distal oxygen atom, alternatively protonation of the proximal oxygen leads to the production of H2O2; the 2e−/2H+ reduction product. The axial ligand as well as the second sphere play a major role in selective and facile heterolytic cleavage of this O–O bond (Figure 2). A generous “push” effect from the axial ligand and an effective “pull” effect from the second sphere geometry result in a facile protonation to the distal oxygen atom and a facile oxygen reduction rate.20,33,34 Both the “push” and “pull” effects, which facilitate protonation, also increase the possibility of H2O2 release, which requires, specifically, protonation of the proximal oxygen atom. Thus, a preorganized distal pocket in a biosynthetic model used here may allow making this discrimination in reactivity.
Figure 2.
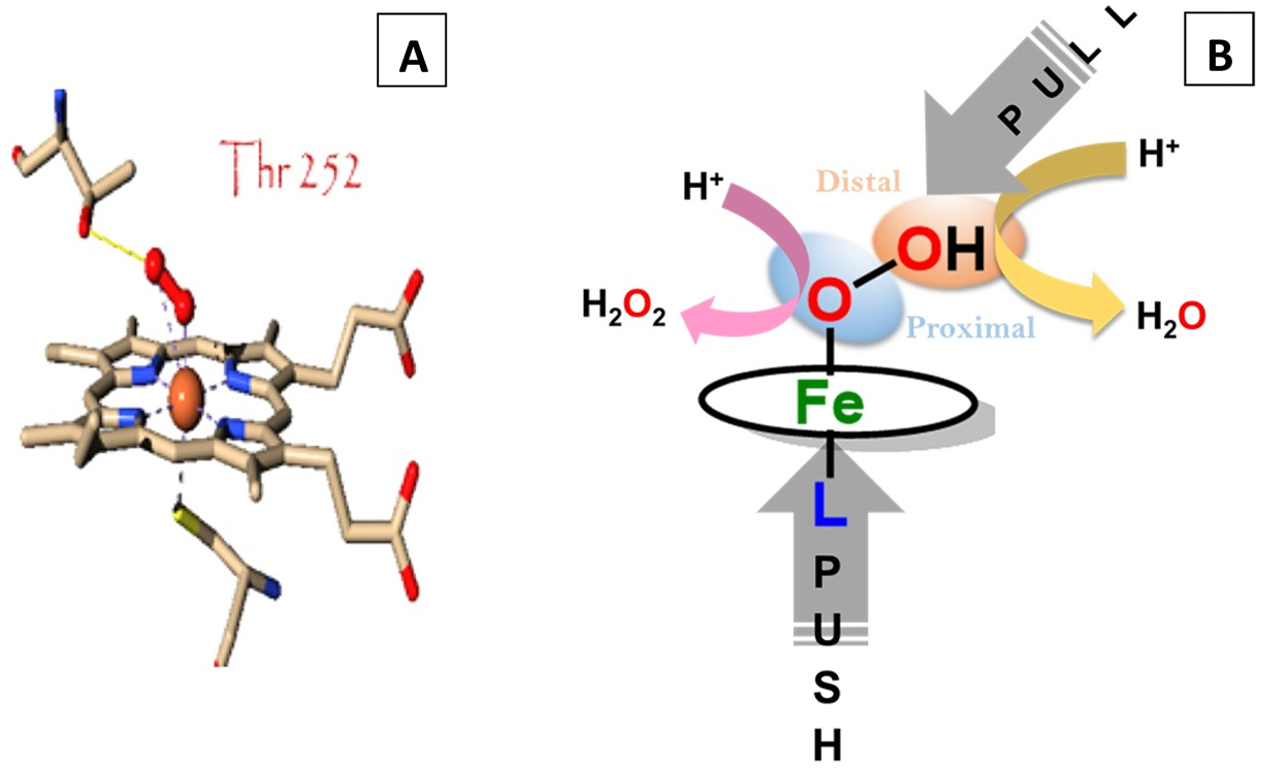
Crystal structure of (A) cytochrome P450 active site having a “pull” effect from Threonine 252 and a “push” effect from axial cysteine ligation. (B) Graphical representation of the impact of the “push “ and the “pull” effect in the selective and facile O2 reduction.
In this paper the FeB in the distal site of V68ECuBMb and V68E/I107ECuBMb biosynthetic models of NOR are replaced with Cu to create perturbed models of CcO which does not include the redox active tyrosine residue but contains proton transfer glutamate residues instead; mimicking the proton donor acidic residues in HCOs. These glutamate residues are at the protein–solvent interface (Figure 1C) and, thus, are ideally suited to transfer protons from bulk solvent to the active site. The overlay of the crystal structure of FeB(II)-V68E/I107ECuBMb and CuB(II)-V68E/I107ECuBMb (Figure 1D), shows that the nature of the distal metal neither alters the active site geometry nor changes the protein conformation substantially. These mutants are immobilized on modified Au electrodes and their electrochemical O2 reduction properties are investigated. A combination of H2O/D2O solvent isotope effect and in situ surface enhanced resonance Raman spectroscopy (SERRS) reveals the predominant role played by the proton transfer residues in modulating the rate-determining steps in 4e−/4H+ O2 reduction.
EXPERIMENTAL SECTION
Materials and Methods.
1-Azidoundecane-11-thiol and Hemin-yne were synthesized following the reported procedure.15,16 6-Mercaptohexanoic acid was purchased from Sigma-Aldrich. Disodium hydrogen phosphate dihydrate (Na2HPO4.2H2O) was purchased from Merck. 2,6-Lutidine was purchased from Avra Synthesis Pvt., Ltd. These chemicals were used without further purification. Au wafers were purchased from Platypus Technologies (1000 Å of Au on 50 Å of Ti adhesion layer on top of a Si(III) surface). Au and Ag discs for the Rotating Ring Disc Electrochemistry (RRDE) and Surface Enhanced Resonance Raman Spectroscopy (SERRS) experiments, respectively, were purchased from Pine Instruments, U.S.A. The Mb mutants were prepared as reported in the literature.17,18 Analysis of the components of the rR spectrum were done by using Lorenztian line shape of peak fit software.
Formation of Mixed SAM and Covalent Attachment of Hemin-yne onto the SAM.
A mixed self-assembled monolayer of 1-azidoundecan-11-thiol and 6-mercaptohexanoic acid was formed on immersing the properly cleaned Au wafers or disks into the deposition solution containing 1-azidoundecan-11-thiol and 6-mercaptohexanoic acid in 10 mL of ethanol in the desired ratio (typically 1:49). The total thiol concentration of these deposition solutions was always maintained at 1 mM. On this SAM Hemin-yne was covalently attached using “Click” reaction (Figure S1 of the Supporting Information, SI). The covalent attachment does not affect the nature of the SAM (Figure S2).35
Reconstitution of Apo Mutants to CuBMb/V68ECuBMb/V68E/I107ECuBMb with CuB on Heterogeneous SAM Surface.
For all the experiments on heterogeneous SAM surfaces the Hemin-yne modified–COOH SAM surfaces were incubated with a 20 μM apo mutant solution for 2 h. The supernatant solution was drained and the surface was thoroughly cleaned with DI water. In the case of the Cu bound mutants, the Cu is loaded to the apoprotein prior to in situ reconstitution on the electrode by adding 2 equiv of 1 mM CuSO4 to the apoprotein solution and stirred for 2–3 h.
Characterization of SAM Modified Electrodes.
The SAM modified biosynthetic model bearing electrodes were characterized by using AFM, SERRS, XPS, CV, and UV–Vis spectroscopy (Figures S3–S7, Tables S1 and S2).
Cyclic Voltammetry.
The CV was performed using Au wafers sandwiched between two Teflon blocks of the Plate material evaluating cell from ALS Japan (http://www.als-japan.com/1398.html). All electrochemical experiments were done in pH 7 phosphate buffer containing Potassium hexafluorophosphate. Anaerobic cyclic voltammetry experiments were done by using a degassed buffer (three cycles of freeze–pump–thaw). Ag/AgCl satd(KCl) reference electrode and Pt counter electrode were used throughout all the electrochemical experiments. The capacitive current is monitored at a potential where there is no Faradaic process (generally between −0.5 V to 0.1 V vs Ag/AgCl satd(KCl)) after every step to ensure that the SAM is intact.
Surface Enhanced Resonance Raman Spectroscopy (SERRS) and SERRS-RDE.
The excitation wavelength used in the Resonance Raman experiments was 413.1 nm and the power applied to the sample was 10 mW. The spectrograph was calibrated against naphthalene. The Ag surfaces were roughened before SERRS experiments following literature protocols.36 The Surface Enhanced Resonance Raman Spectroscopy coupled to Rotating Disc Electrochemistry (SERRSRDE) set up has been previously described.37 The data for the oxidized state was obtained by holding the potential of the disc at 0 mV vs NHE and the data during steady state ORR was obtained by holding the disc at −300 mV vs NHE and the disc was rotated at 300 rpm. The data becomes independent of rotation rate above 100 rpm(Figure S8).37 Data were acquired over a period of 300 s. The components of ν3 are fit in Peakfit software using Lorentzian peaks with an fwhm of 5–7 cm−1. The fwhm are kept the same between all the components in the same fit.
Atomic Force Microscopy.
The AFM data were obtained at room temperature in a Veecodicp II (Model no: AP-0100) instrument bearing a phosphate doped Si cantilever (1–10 ohm.cm, thickness 3.5–4.5 μm, length 115–135 μm, width 30–40 μm, resonance frequency 245–287 kHz, and elasticity 20–80 N/m).
X-ray Photoelectron Spectroscopy.
The XPS data were collected in an Omicron (model: 1712-62-11) spectrometer using a high-resolution monochromatic Al–Kα source at 1486.7 eV under 15 kV voltage and 10 mA current maintaining a base pressure of 5 × 10−10 mbar. The binding energies were calibrated to the Ag 3d5/2 peak at 368.2 eV.
RESULTS AND ANALYSIS
The O2 reduction by the perturbed CcO mutants are investigated by covalently attaching the modified heme (Hemin-yne, Scheme 1) reconstituted CuB-V68E and CuB-V68E/I107ECuBMb mutants to the mixed self-assembled monolayer of 1-azidoundecan-11-thiol and 6-mercaptohexanoic acid formed on cleaned Au electrodes using “Click” reaction. Note that due to the direct attachment of heme to the electrode, the electron transfer rate from the electrode to the heme active site is facile.32,35 The number of glutamate near the active site is systematically increased from zero in CuBMb (L29H, F43H, H64) to one in V68ECuBMb (L29H, F43H, H64, V68E) and to two in V68E/I107ECuBMb (L29H, F43H, H64, V68E and I107E), respectively, allowing investigating the role of local acidic residues in determining the selectivity and kinetics of O2 reduction in these active sites which are not very solvent accessible. The CuBMb, V68ECuBMb and V68E/I107ECuBMb constructs are structurally characterized and these mutations do not cause any change in the overall structure of the protein (Figure S9). The E68 residue binds the distal FeB in V68ECuBMb (Figure 2A) and moves into a bridging position in the I107E/V68ECuBMb (Figure 2B). While it is not possible to ascertain that the protein structure, in solution, is retained upon its attachment to the electrode, several analytical techniques (Figures S4 and S6) like AFM (size), SERRS (heme vibrations), CV (redox activity), and XPS (metal and cofactor ionizations) are all consistent with attachment of these intact proteins on the modified electrode.
Scheme 1.
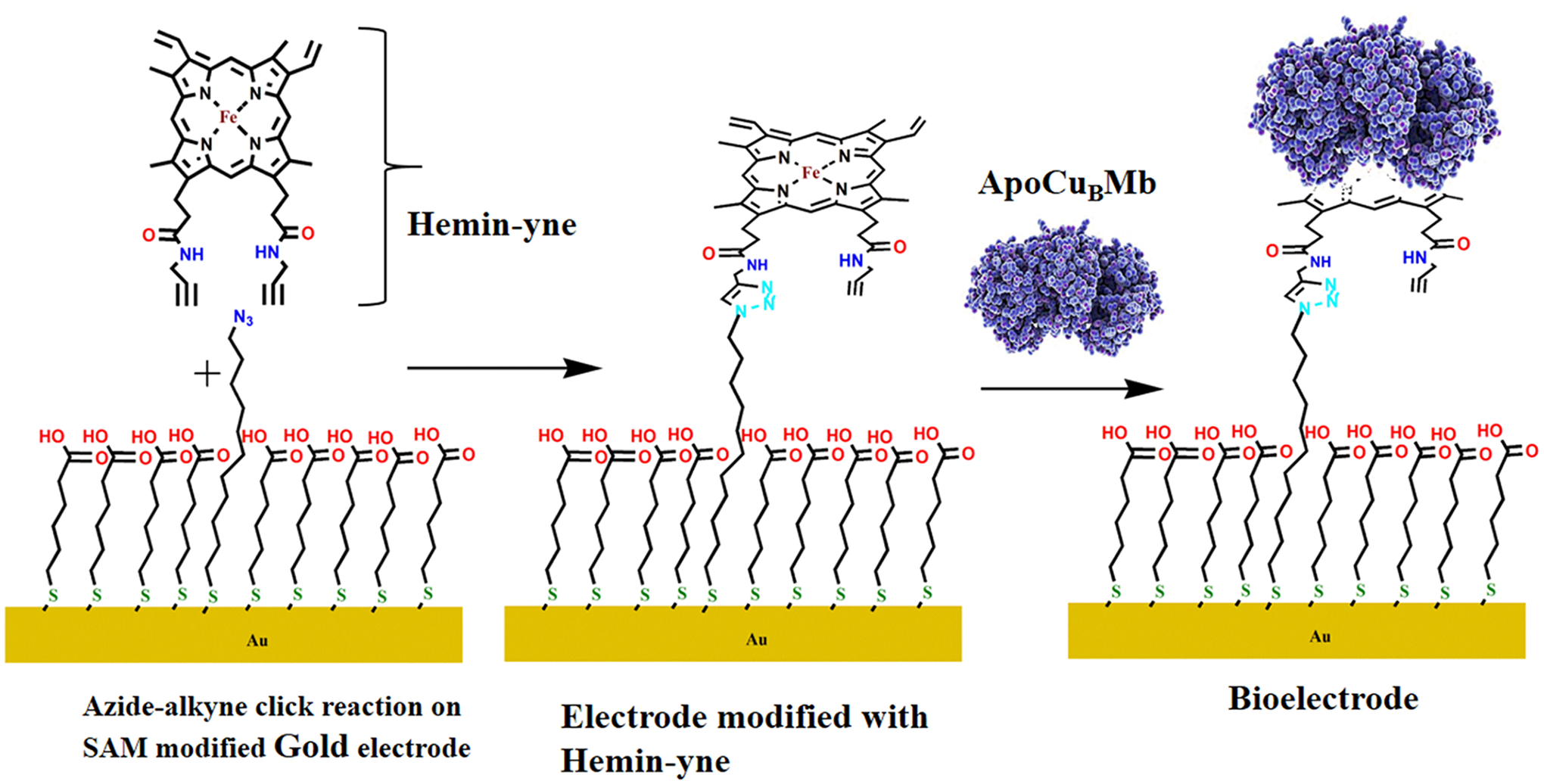
Construction of Electrode Bearing Bio-Synthetic Models
Linear sweep voltammograms (LSV) of electrodes, bearing CuBMb or V68ECuBMb or V68E/I107ECuBMb, in aerated buffers at pH 7 at room temperature show large electrocatalytic O2 reduction currents (icat) at potentials where the heme gets reduced (Figure S6, Table S2). At this potential the SAM-coated electrode did not show any background O2 reduction (Figure S10). In the active site of the O2 reducing enzymes, O2 may be reduced by 4e− and 4H+ to H2O or by 1e− or 2e− to produce reactive oxygen species (ROS) like O2− or H2O2. The number of electrons involved and the second order rate constant of the O2 reduction reaction (ORR), kORR can be determined using rotating disc electrochemistry (RDE) where the catalytic O2 reduction current (icat) is O2 diffusion limited and increases with increasing rotation rates (Figure 3A, C) following the Kouteky-Levich (K-L) equation.38 Plots of icat−1 at multiple rotation rates with the inverse square root of the angular rotation rate (ω−1/2) for V68ECuBMb (Figure 3B) are linear. Furthermore, the number of electrons (n) donated to the substrate, i.e., O2 by the electrocatalyst can be experimentally estimated from the slope of the K-L plot which is expressed as 1/[n{0.62FA(Do2)2/3ν−1/6}]. The slope obtained from the experimental data for V68ECuBMb (Figure 3B), V68E/I107ECuBMb (Figure 3D) are close to the slope expected for a 4e− process (Figure 3B and D, dotted purple line) and very different from the slope for a 2e− process (Figure 3B and D, dotted green line). Thus, like G65YCuBMb analog,32 both V68ECuBMb and V68E/I107ECuBMb proteins predominantly catalyze a 4e−/4H+ O2 reduction with the distal CuB. The selectivity for O2 reduction is more accurately reflected in the very limited production of H2O2 detected in situ (Table 1, fourth column) using rotating ring disc electrochemistry (RRDE) (Figure S12). The second order rate of O2 reduction by the G65YCuBMb, V68ECuBMb, and V68E/I107ECuBMb is determined to be 1.98 ± 0.1 × 107 M−1 s−1, 2.2 ± 0.3 × 107 M−1 s−1 and 0.86 ± 0.17 × 107 M−1 s−1, respectively (Table 1, second column). The corresponding pseudo first order rate constants (assuming [O2] = 0.26 × 10−3 M) are 5148 ± 260 s−1, 5720 ± 780 s−1, and 2243 ± 338 s−1. The highest second order O2 reduction rate reported for any synthetic heme Cu system is ~105 M−1 s−1; that too on a multilayer.39,40 The biosynthetic model (V68ECuBMb) examined here has a second order rate constant of >107 M−1 s−1 which is 2 orders of magnitude higher than the value reported for any synthetic heme/Cu O2 reduction catalyst. The turn over number (TON = moles of O2 reduced per mole of catalyst determined from the electrochemical ORR currents normalized with respect to catalyst degradation) of the catalyst V68ECuBMb, are estimated to be at least 2.8 × 104.
Figure 3.
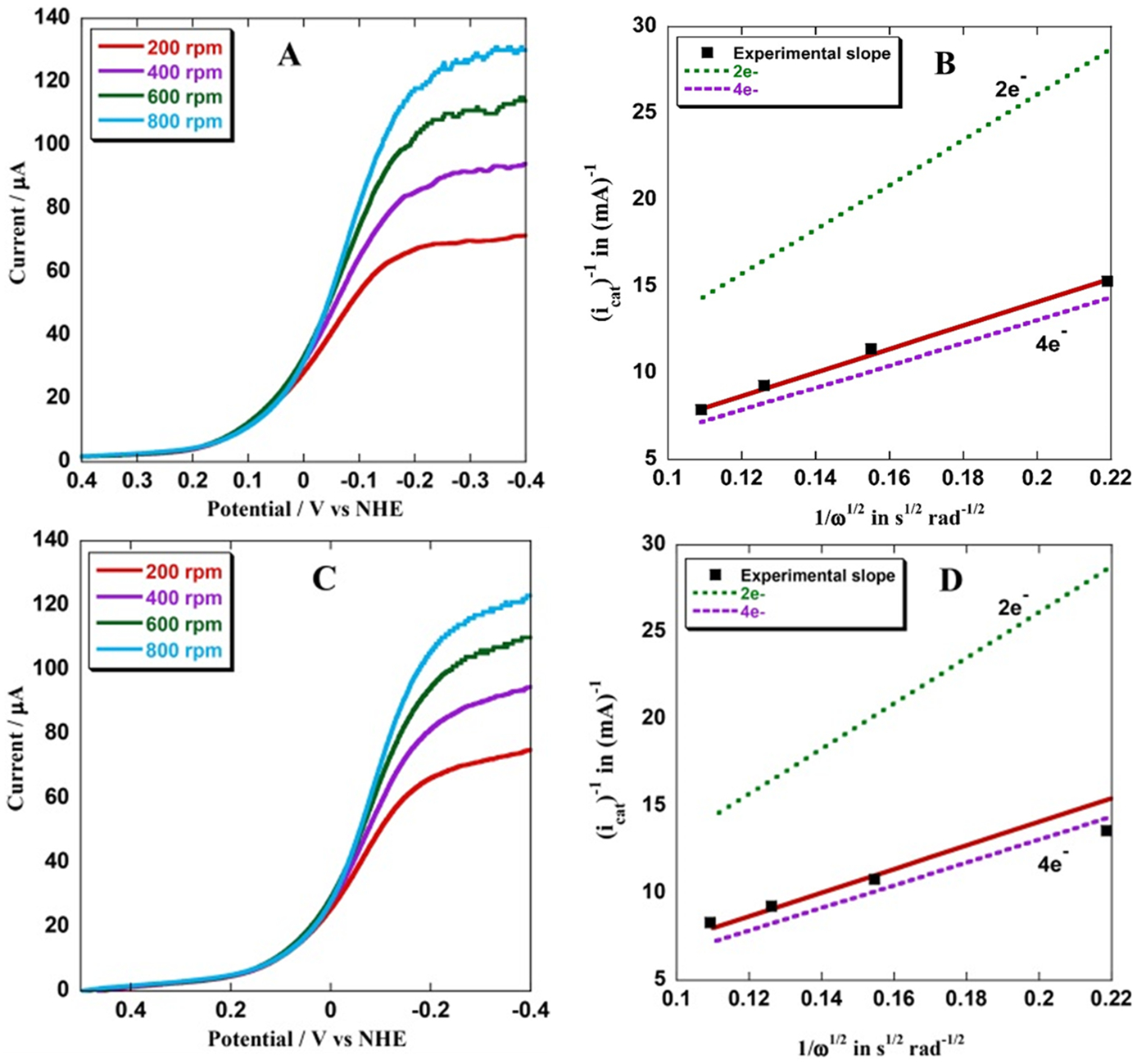
(A) RDE data of V68ECuBMb in air saturated pH 7 100 mM phosphate buffer solution at 100 mV/s scan rate. (B) K-L plot of icat−1 for V68ECuBMb at −300 mV vs the inverse square root of the angular rotation rate ω−1/2). (C) RDE data of V68E/I107ECuBMb in air saturated pH7 100 mM phosphate buffer solution at 100 mV/s scan rate. (D) K-L plot of V68E/I107ECuBMb at −300 mV vs ω−1/2.
Table 1.
Rate Constants (kORR), Turn over Number (TON), ROS Generated and SKIE of Different CuB/Heme Bound Biosynthetic Models for O2 Reduction Reaction
| mutants (contains both CuB and heme) | rate (kORR) 2nd order in 107 M−1 s−1, 1st order in s−1 | TON (104) | ROS (%) (pD 7) | SKIE |
|---|---|---|---|---|
| CuBMb | ND | ND | 6±1 | |
| G65YCuBMb | 1.98 ± 0.10 | >0.65 | 6 ± 1 (5.3 ± 1) | 16.00 ± 1.0 |
| 5148 ± 260 | ||||
| V68ECuBMb | 2.20 ± 0.30 | >2.8 | 4 ± 1 (4 ± 1) | 4.30 ± 0.5 |
| 5720 ± 780 | ||||
| V68E/I107E CuBMb | 0.86 ± 0.17 | >0.5 | 7±1 | 2.43 ± 0.07 |
| 2243 ± 338 | ||||
| *(~3560 ± 850) |
The presence of the additional proton donor residues near the active site do not compromise the selectivity of 4e−/4H+ as these V68ECuBMb and V68E/I107ECuBMb mutants generate very limited partially reduced oxygen species (Table 1). Note that the rate of O2 reduction by the V68E/I107ECuBMb mutant is lower than the others likely due to the presence of an inactive LS FeIII species which could not be reduced at −300 mV (Figure S13). The amount of this species can be approximately estimated to be ~37 ± 1% (SI Figure S14). Thus, the corrected rate of the active component in V68E/I107ECuBMb mutant is ~3560 s−1 (Figure S14). Due to the presence of a majority of LS species, the rate obtained is only approximate and hence is not included in the further discussion. However, the inactive LS species, which does not get reduced at −300 mV vs NHE, will not contribute to the ORR current and only the active HS species will. Thus, the ROS produced (ratio of the disc and ring current) and the H2O/D2O effect (ratio of the catalytic current in H2O and D2O) on the ORR rate only represents those emanating from the active component and is discussed here.
The kORR obtained in deuterated buffer (Figure S15) allow estimation of H2O/D2O isotope effects which, in turn, reflects the involvement of proton transfer in the rate-determining step (rds). An H2O/D2O solvent kinetic isotopic effect (SKIE, Table 1) on O2 reduction for G65YCuBMb is 16.0 which is reduces to 4.3 in V68ECuBMb (Figure S15). Thus, the introduction of H+ transfer residue “Glu” in V68ECuBMb reduces the SKIE. In the V68E/I107ECuBMb mutant introduction of an additional H+ transfer “Glu” residue further reduces the SKIE to ~2.4. Large SKIE are observed on the mutation of the residues involved in the proton transfer in heme enzymes like cytochrome P450 and peroxidases.41,42 For example, mutating the proton transfer residues Asp251 in cytochrome P450 causes a marked enhancement of SKIE from ~2.4 in WT to 10.0 in the mutant. The loss of preorganized proton transfer channel due to mutation was proposed to be responsible for high SKIE in these cases. Similarly SKIE > 18 has been observed on the rate-limiting protonation of a FeIII–OOH species involved in O2 reduction catalyzed by a thiolate bound iron porphyrin complex43 Native CcO, where there is a proton channel containing aspartate residues, the SKIE is only ~1.4.2 Thus, the large H/D isotope effect, 16, in G65YCuBMb indicates a rate determining protonation step and the lack of an efficient proton transfer channel to the heme/Cu active site. Note that a rate determining protonation step in the catalytic cycle of G65YCuBMb, suggested by the large SKIE obtained here, is consistent with previous proposals on the same mutant made based on in situ SERRS-RDE data.32 Introducing proton transfer residues, glutamates, near the active site results in increased efficiency of proton transfer for this 4e−/4H+ reduction process result in similar oxygen reduction reaction rates and lower H2O/D2O SKIE suggesting a change in the rate-determining step.
Normally, SERRS data on these proteins would show oxidation and spin state marker vibrations typical for resting ferric (Figure 4A, red) and ferrous (Figure 4A, cyan) when oxidizing and reducing potentials are applied in the absence of oxygen. SERRS-RDE is used to investigate the mechanism of O2 reduction by these biosynthetic CcO models in-operando.32,37 In this technique, the resonance Raman (rR) spectra of the electrocatalyst is collected while the system is involved in steady-state O2 reduction at a potential where catalysis is mass transfer limited. A combination of oxidation and spin state marker bands are used to gain insight into the nature of intermediates involved in the catalytic cycle.37 The species preceding the rds will accumulate where the system is at steady state and can be identified by fitting the different components in the ν3 region (Figure 4B).
Figure 4.
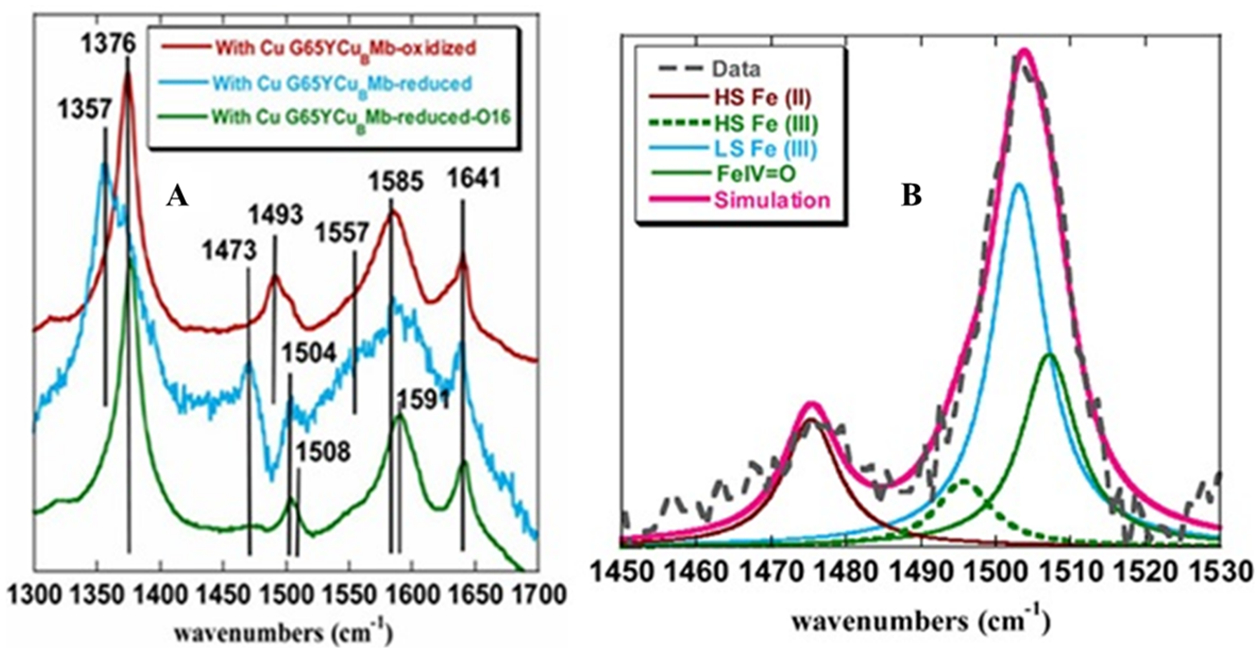
(A) SERRS-RDE spectra of oxidized (red), reduced (under N2 atm; sky blue) and oxygen bound (molecular oxygen:16O2; green) G65YCuBMb bearing electrode in 100 mM pH 7 phosphate buffer. (B) Components of the rR spectrum determined by simulating the spectra of the G65YCuBMb-bearing electrode in the presence of O2 (O16) saturated 100 mM pH 7 phosphate buffer.32
SERRS-RDE of G65YCuBMb shows the presence of H.S FeII, resting H.S FeIII, LS FeIII and ferryl (FeIV=O) species with ν3 at 1473, 1493, 1504, and 1508 cm−1 (Figure 4).32 Similarly the ν2 shows a shift at higher energy to 1591 cm−1 under steady state indicating the presence of a FeIV=O species (Figure 4A, green). Thus, the rates of formation of these species are greater than rates of decay and the rate of decay of the species with highest population (LS FeIII) is the rds of the O2 reduction. The 18O2/16O2 difference spectra show isotope sensitive vibrations between 700 and 900 cm−1 (Figure 5A). A band at 701 cm−1 which shifts to 677 cm−1 with 18O2 could represent the Fe–O vibration of an LS FeIII–peroxide. Similarly two vibrations at 760 and 745 cm−1 shifts to 753 and 734 cm−1, respectively, in 18O2 which could originate from the Fe–O vibration from a FeIV=O species Fermi coupling to another vibration (likely a heme band). The overwhelming intensity of the LS. FeIII species (Figure S16) and the 18O2 sensitive vibrations (Figure 5A) in conjunction with the large H2O/D2O SKIE suggests the protonation of LS ferric peroxide species is likely to be the rds.
Figure 5.
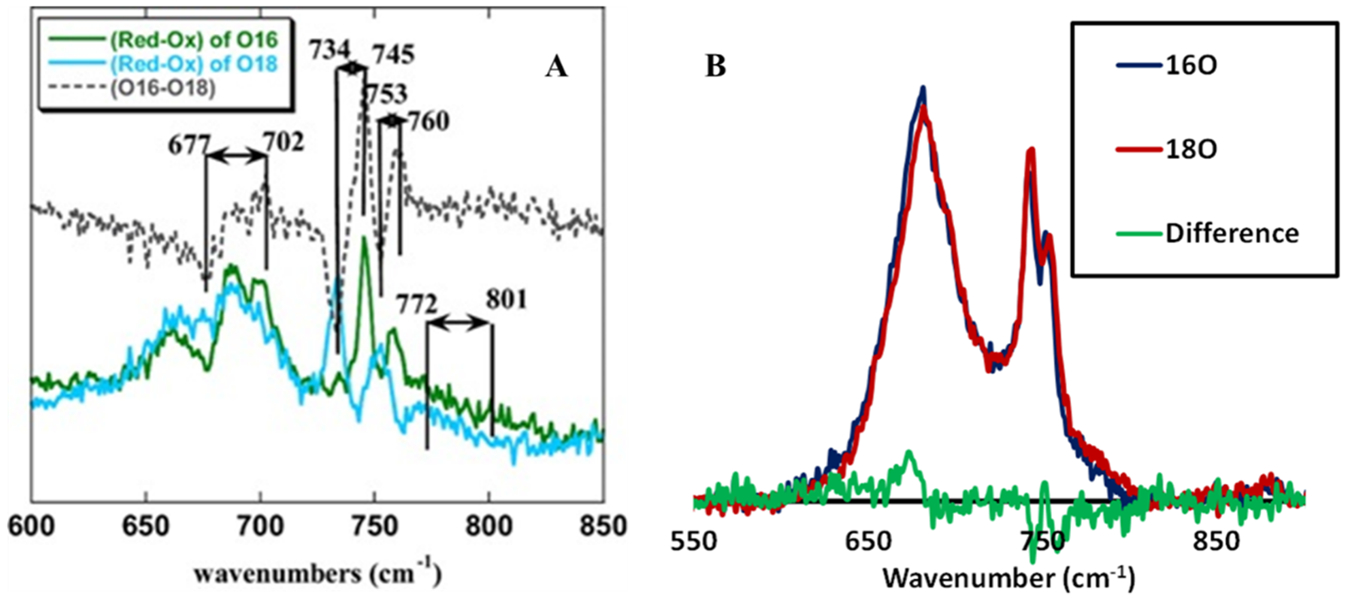
(A) Difference spectra of reduced and oxidized state in 16O2 and 18O2 saturated pH 7 phosphate buffer at high range of wavenumbers for G65YCuBMb, showing the increase in intensity of the peaks from 18O2 to 16O2. (B) The SERRS-RDE 16O2/18O2 data of V68ECuBMb obtained under reducing potentials where the system is under steady state.
The SERRS-RRDE data of V68ECuBMb, the construct with the highest rate, shows only HS FeIII species under steady state (Figure 6A) and unlike in G65YCuBMb no LS FeIII peroxide species and FeIV=O species are observed (Figure 6B). The 18O2 and 16O2 SERRS-RDE data do not show any differences in the 500–900 cm−1 region as well (Figure 5B) consistent with the analysis of the marker bands accumulated under a steady state in G65YCuBMb which did not show any LS FeIII (6C heme peroxide species are LS) or FeIV=O species. Thus, neither an LS FeIII–peroxide nor a FeIV=O species are observed in the V68ECuBMb mutant. Since decay of both of these species requires protons, the fact that these species do not accumulate during steady state is consistent with faster protonation steps in V68ECuBMb relative to G65YCuBMb. The efficient proton transfer pathway in V68ECuBMb is likely manifested in its low H2O/D2O isotope effect relative to G65YCuBMb. SERRS-RDE of the V68E/I107ECuBMb double glutamate mutant could not be explored due to the presence of the LS FeIII species at −300 mV even in the absence of O2 (Figure S13).
Figure 6.
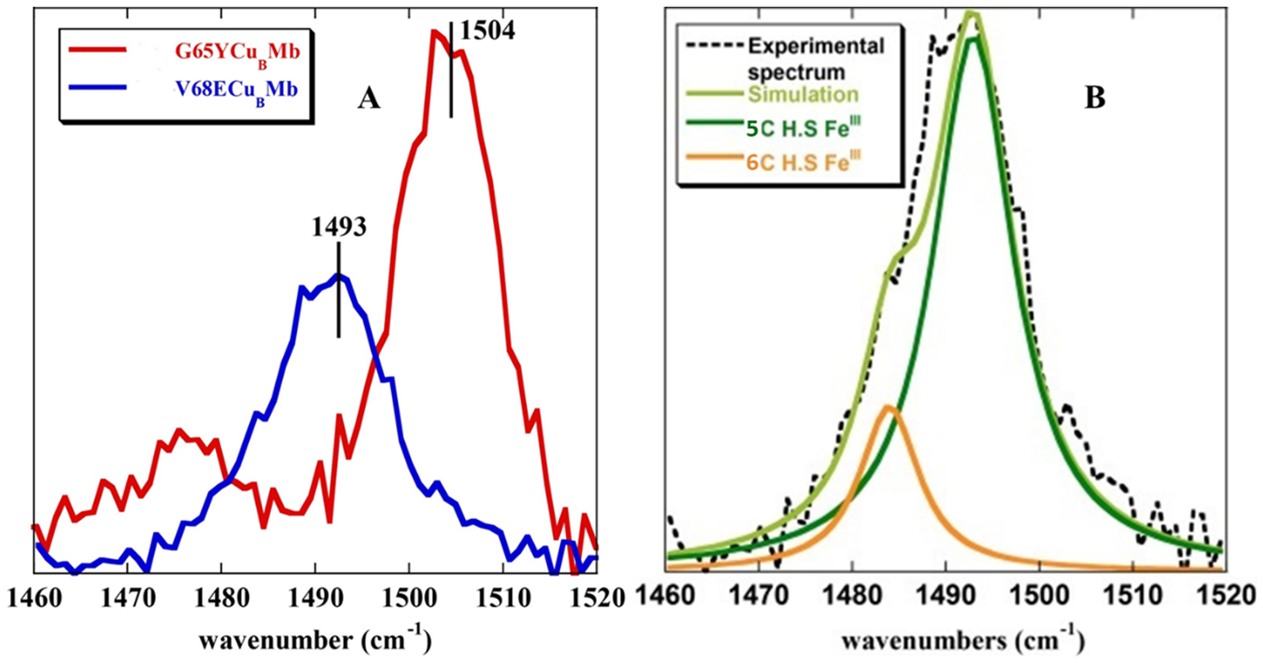
(A) Overlay of the SERRS-RDE spectra of V68ECuBMb and G65YCuBMb, bearing electrode in aerated 100 mM pH 7 phosphate buffer when the electrode is held at −0.3 V w.r.t NHE. (B) Components of the rR spectra determined by simulating ν3 band of the spectra of V68ECuBMb.
DISCUSSION
Biosynthetic models that mimic the active site of CcO are immobilized on SAM covered electrodes using a modular approach. In this approach, a heme cofactor is first immobilized on the SAM bearing azide-terminal groups using “click” reaction and these heme groups are then inserted in apo-proteins using in situ reconstitution. These biochemical models have been shown to mimic the O2 reduction activity of CcO in solution deriving electrons from a reductant like sodium dithionite.44 The electrodes bearing a site isolated dilute monolayer of these biosynthetic models of CcO, bioinspired electrodes, can reduce O2 to H2O with exceptionally high rates(>107 M−1 s−1) and selectivity >90% for the 4e−/4H+ process. These findings also add to a building body of literature that stresses the advantages of protein environment in catalysis.45–47
The ability to design the distal site by site-directed mutagenesis allows creating of “perturbed” CcO active site models where the tyrosine residue is replaced by glutamate(s). The motivation for such replacement, of course, is derived from the key differences in the active site structure of CcO and NOR; the two members of the heme/Cu oxidase family which varies dramatically in their substrate selectivity.1,4 The efficient O2 reduction by the heme/Cu sites with glutamate observed here clearly indicates that the presence of glutamates is not at all detrimental to O2 reduction, both rates, and selectivity, under the conditions where the electron transfer to the active site is not rate limiting. However, a conclusive statement on the issue can only be made when the O2 reduction by these species are evaluated under rate limiting electron transfer fluxes where the role of the tyrosine is well documented.23
These mutants, G65YCuBMb, V68ECuBMb, and V68E/I107ECuBMb, with very high overpotentials for ORR and low electrode coverage, are not practicable as ORR catalysts. However, understanding the factors that enhance rates and efficiencies of O2 reduction in these systems will herald the development of artificial catalysts for the same purpose. The detailed electrochemical study of oxygen reduction reaction for all the mutants, G65YCuBMb, V68ECuBMb, and V68E/I107ECuBMb, shows that all these mutants reduce oxygen in more or less comparable rate but the main variation lies in their H2O/D2O SKIE value (2.0–16.0) during the course of reaction. Generally large H2O/D2O SKIE indicates the involvement of proton transfer in the rds. The large variation in the H2O/D2O SKIE on kORR observed in these mutants (2.0–16.0) appears to be diagnostic of a preorganized proton transfer channel to the active site. But the same rds for G65YCuBMb, V68ECuBMb, and V68E/I107ECuBMb, having 0, 1, and 2 preorganized proton transfer channels, respectively, would result in a variation in the rate of oxygen reduction which is not the case suggesting that the rate-determining steps are different depending on the nature of the second sphere residue of these mutants. For V68E/I107ECuBMb the order of the rate of O2 reduction is as high as for the other mutants. The slight lower value of the kORR is due to the presence of 37% LS Fe(III) component unable to take part in ORR mechanism. Thus, the increase in the number of the proton transfer channel alters the rds for ORR and this reflects on the change in the SKIE value.
On the basis of the electrocatalytic and SERRS-RDE data, the mechanism of ORR by the G65YCuBMb and V68EcuBMb mutants can be proposed. The data obtained for the G65YCuBMb is consistent with the mechanism proposed for the active site of CcO (Figure 7, lower panel) including the direct reduction of the 6C HS FeIII–OH (intermediate VIA, Figure 7) species to the active 5C FeII state (intermediate I, Figure 7).32 A low spin Ferric peroxide (intermediate IIIA, Figure 7) and a Ferryl (intermediate VA, Figure 7) intermediate observed under steady state in the G65YCuBMb are consistent with the relative rates of formation and decay reported for these species in the active site of CcO. Although the involvement of the Tyr 65 residue in the G65YCuBMb is not explicitly shown, it must be mentioned that this mutant shows substantially lower PROS than the WT CuBMb. The SERRS-RDE data on the V68ECuBMb, however, does not show either a low spin ferric hydroperoxide or a ferryl intermediate (intermediate IVB, Figure 7) under steady state suggesting that the ORR mechanism has been perturbed significantly upon the introduction of a proton transfer residue in the distal site. Because both the decay of ferric peroxide and ferryl species require protons, not seeing them accumulate under steady state in the V68ECuBMb construct is reasonable. Similarly, the rate of hydrolysis of a 6C HS FeIII–OH (intermediate VB, Figure 7) species resulting from the reduction of the ferryl intermediate may be expected to be enhanced in the V68ECuBMb mutant explaining the observation of the 5C HS FeIII (intermediate VIB, Figure 7) in this mutant under steady state. Recently it has been suggested that the mechanism of oxygen reduction in solution for G65YCuBMb and V68ECuBMb are different. The nonheme Cu in V68ECuBMb does not only donate an electron to the oxygen bound to heme iron but also activates O–O bond leading to a facile O–O bond cleavage by forming a bridging peroxide.48 A bridging peroxide has not yet been established for G65YCuBMb. It is likely there exists a bifurcation in the ORR mechanistic pathway for G65YCuBMb (orange arrow, lower panel, Figure 7) and for V68ECuBMb (blue arrow, upper panel, Figure 7) resulting from the differences in the nature of the peroxide intermediate. The SERRS-RDE data suggest that for G65YCuBMb the slowest steps are the protonation of a peroxide (intermediate IIIA, Figure 7) and ferryl intermediate (intermediate VA, Figure 7). This proposal is based on the accumulation of LS FeIII species during steady state which the 16O2/18O2 tentatively supports. The V68E mutant, however, is likely to form a bridging peroxide intermediate which will result in the oxidation of the distal Cu(I) earlier in the catalytic cycle than in that of CcO and G65YCuBMb. Currently, with the available electrochemical and SERRS-RDE data, it is not immediately clear what the rds is in the V68E mutant. As the KSIE implies that the step involves a proton transfer as well. The lack of direct spectroscopic data on the distal CuB site (SERRS-RDE only probes the heme) is a major deterrent in deducing its role (or the lack of) in determining the rds.
Figure 7.
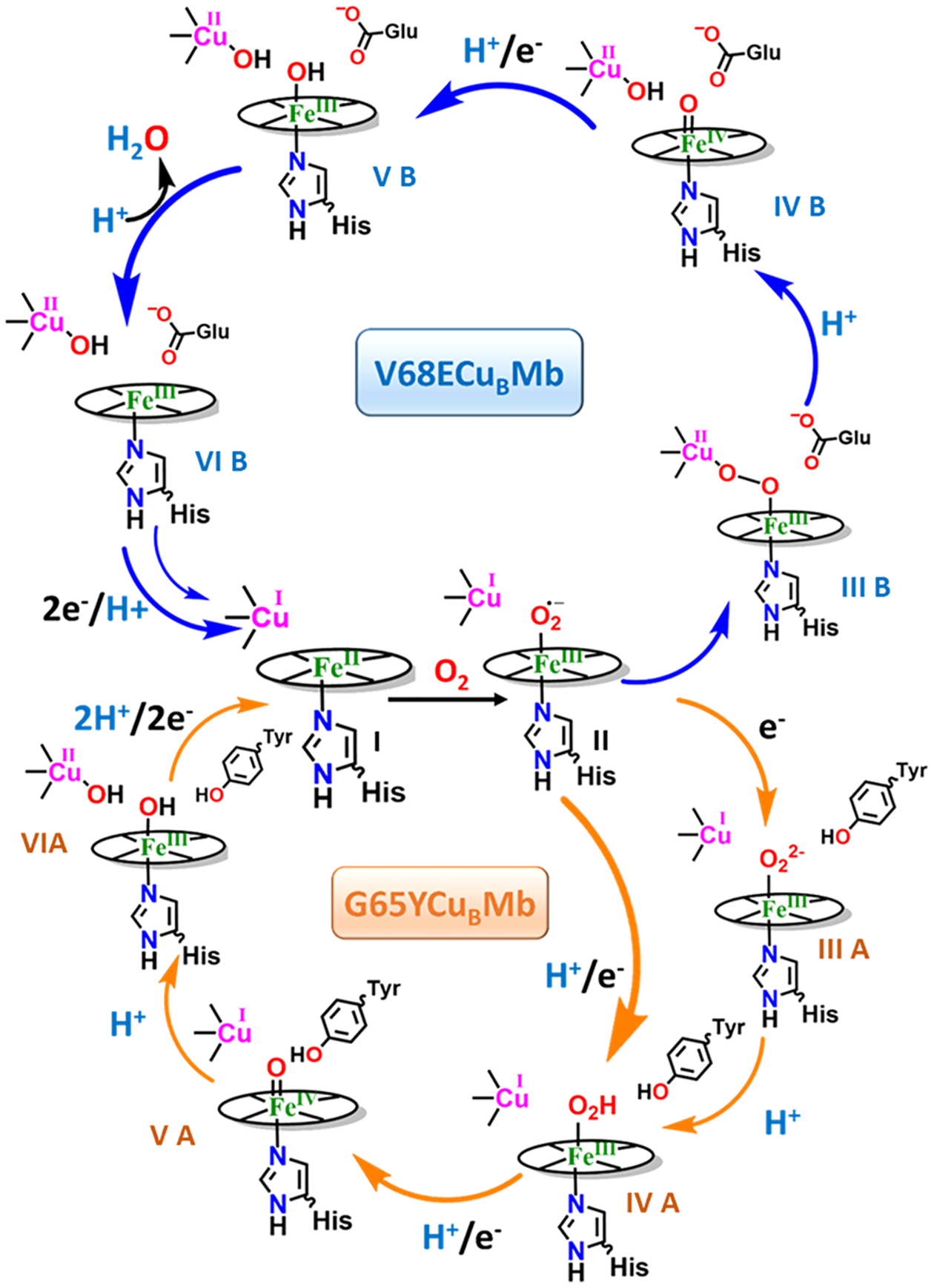
Comparison between the mechanisms of oxygen reduction of the two biosynthetic models while attached on top of the electrode.
In membrane-bound native CcO the H+ required for catalytic O2 reduction are transferred through two proton channels— the D channel and the K Channel via specific residues to the active site. The distal glutamate residues in the V68ECuBMb and V68EI107ECuBMb biosynthetic mimics, which are present at the protein–water interface, act as H+ transfer residues akin to those present in natural enzyme resulting in facile and selective 4e−/4H+ reduction of O2 and also the significant lowering in the value of H2O/D2O SKIE on kORR.
CONCLUSIONS
Oxygen reduction is a multistep (4e−/4H+) process involving several intermediates. A combination of electrochemical and in situ SERRS-RDE data on the site-directed mutants, where the number of proton translocating glutamate residues are systematically varied, clearly indicate the important role a well-defined proton transfer channel plays in dictating the rate-determining step. The G65YCuBMb and the V68ECuBMb variants, which are 16 kDa proteins (more than 140 residues) that differ only by one or two residues, have very similar rates of ORR and selectivity, yet they behave different mechanistically. The study further reinstates the importance of in-operando spectroscopic investigations of electrocatalysts to realize factors that control rates and selectivity.
Supplementary Material
ACKNOWLEDGMENTS
This research is sponsored by Department of Science and Technology, India (SB/S1/IC-25/2013) and U.S. National Institute of Health (GM062211).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b02240.
The characterization data (AFM, XPS, CV, etc.) and control experiments (CV and SERRS) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Averill BA Dissimilatory nitrite and nitric oxide reductases. Chem. Rev 1996, 96, 2951–2964. [DOI] [PubMed] [Google Scholar]
- (2).Ferguson-Miller S; Babcock GT Heme/Copper Terminal Oxidases. Chem. Rev 1996, 96, 2889–2908. [DOI] [PubMed] [Google Scholar]
- (3).Hino T; Matsumoto Y; Nagano S; Sugimoto H; Fukumori Y; Murata T; Iwata S; Shiro Y Structural Basis of Biological N2O Generation by Bacterial Nitric Oxide Reductase. Science 2010, 330, 1666–1670. [DOI] [PubMed] [Google Scholar]
- (4).Zumft WG Nitric oxide reductases of prokaryotes with emphasis on the respiratory, heme-copper oxidase type. J. Inorg. Biochem 2005, 99, 194–215. [DOI] [PubMed] [Google Scholar]
- (5).Cheesman MR; Zumft WG; Thomson AJ The MCD and EPR of the Heme Centers of Nitric Oxide Reductase from Pseudomonas stutzeri: Evidence That the Enzyme Is Structurally Related to the Heme-Copper Oxidases. Biochemistry 1998, 37, 3994–4000. [DOI] [PubMed] [Google Scholar]
- (6).van der Oost J; de Boer APN; de Gier J-WL; Zumft WG; Stouthamer AH; van Spanning RJM The heme-copper oxidase family consists of three distinct types of terminal oxidases and is related to nitric oxide reductase. FEMS Microbiol. Lett 1994, 121, 1–10. [DOI] [PubMed] [Google Scholar]
- (7).Shiro Y; Sugimoto H; Tosha T; Nagano S; Hino T Structural basis for nitrous oxide generation by bacterial nitric oxide reductases. Philos. Trans. R. Soc., B 2012, 367, 1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kim E; Chufán EE; Kamaraj K; Karlin KD Synthetic Models for Heme–Copper Oxidases. Chem. Rev 2004, 104, 1077–1134. [DOI] [PubMed] [Google Scholar]
- (9).Tsukihara T; Aoyama H; Yamashita E; Tomizaki T; Yamaguchi H; Shinzawa-Itoh K; Nakashima R; Yaono R; Yoshikawa S Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Ã. Science 1995, 269, 1069–1074. [DOI] [PubMed] [Google Scholar]
- (10).Yoshikawa S; Shinzawa-itoh K; Nakashima R; Yaono R; Yamashita E; Inoue N; Yao M; Fei MJ; Libeu CP; Mizushima T; Yamaguchi H; Tomizaki T; Tsukihara T Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 1998, 280, 1723–1729. [DOI] [PubMed] [Google Scholar]
- (11).Karlin S; Zhu Z-Y; Karlin KD Extended Metal Environments of Cytochrome c Oxidase Structures. Biochemistry 1998, 37, 17726–17734. [DOI] [PubMed] [Google Scholar]
- (12).Sakurai N; Sakurai T Genomic DNA Cloning of the Region Encoding Nitric Oxide Reductase inParacoccus halodenitrificansand a Structure Model Relevant to Cytochrome Oxidase. Biochem. Biophys. Res. Commun 1998, 243, 400–406. [DOI] [PubMed] [Google Scholar]
- (13).Wasser IM; de Vries S; Moënne-Loccoz P; Schröder I; Karlin KD Nitric Oxide in Biological Denitrification: Fe/Cu Metalloenzyme and Metal Complex NOx Redox Chemistry. Chem. Rev 2002, 102, 1201–1234. [DOI] [PubMed] [Google Scholar]
- (14).Watmough NJ; Field SJ; Hughes RJL; Richardson DJ The bacterial respiratory nitric oxide reductase. Biochem. Soc. Trans 2009, 37, 392–399. [DOI] [PubMed] [Google Scholar]
- (15).Yeung N; Lu Y One Heme, Diverse Functions: Using Biosynthetic Myoglobin Models to Gain Insights into Heme-Copper Oxidases and Nitric Oxide Reductases. Chem. Biodiversity 2008, 5, 1437–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lin Y-W; Yeung N; Gao Y-G; Miner KD; Tian S; Robinson H; Lu Y Roles of glutamates and metal ions in a rationally designed nitric oxide reductase based on myoglobin. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 8581–8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Miner KD; Mukherjee A; Gao Y-G; Null EL; Petrik ID; Zhao X; Yeung N; Robinson H; Lu Y A Designed Functional Metalloenzyme that Reduces O2 to H2O with Over One Thousand Turnovers. Angew. Chem., Int. Ed 2012, 51, 5589–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sigman JA; Kwok BC; Lu Y From Myoglobin to Heme-Copper Oxidase: Design and Engineering of a CuB Center into Sperm Whale Myoglobin. J. Am. Chem. Soc 2000, 122, 8192–8196. [Google Scholar]
- (19).Yeung N; Lin Y-W; Gao Y-G; Zhao X; Russell BS; Lei L; Miner KD; Robinson H; Lu Y Rational design of a structural and functional nitric oxide reductase. Nature 2009, 462, 1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chatterjee S; Sengupta K; Mondal B; Dey S; Dey A Factors Determining the Rate and Selectivity of 4e−/4H+ Electrocatalytic Reduction of Dioxygen by Iron Porphyrin Complexes. Acc. Chem. Res 2017, 50, 1744–1753. [DOI] [PubMed] [Google Scholar]
- (21).Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- (22).Dey A; Jenney FE; Adams MWW; Babini E; Takahashi Y; Fukuyama K; Hodgson KO; Hedman B; Solomon EI Solvent Tuning of Electrochemical Potentials in the Active Sites of HiPIP Versus Ferredoxin. Science 2007, 318, 1464–1468. [DOI] [PubMed] [Google Scholar]
- (23).Collman JP; Devaraj NK; Decréau RA; Yang Y; Yan Y-L; Ebina W; Eberspacher TA; Chidsey CED A Cytochrome c Oxidase Model Catalyzes Oxygen to Water Reduction Under Rate-Limiting Electron Flux. Science 2007, 315, 1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Collman JP; Ghosh S Recent Applications of a Synthetic Model of Cytochrome c Oxidase: Beyond Functional Modeling. Inorg. Chem 2010, 49, 5798–5810. [DOI] [PubMed] [Google Scholar]
- (25).Collman JP; Yang Y; Dey A; Decreau RA; Ghosh S; Ohta T; Solomon EI A functional nitric oxide reductase model. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 15660–15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Boulatov R; Collman JP; Shiryaeva IM; Sunderland CJ Functional Analogues of the Dioxygen Reduction Site in Cytochrome Oxidase:Mechanistic Aspects and Possible Effects of CuB. J. Am. Chem. Soc 2002, 124, 11923–11935. [DOI] [PubMed] [Google Scholar]
- (27).McGuire R Jr.; Dogutan DK; Teets TS; Suntivich J; Shao-Horn Y; Nocera DG Oxygen reduction reactivity of cobalt(ii) hangman porphyrins. Chem. Sci 2010, 1, 411–414. [Google Scholar]
- (28).Rigsby ML; Wasylenko DJ; Pegis ML; Mayer JM Medium Effects Are as Important as Catalyst Design for Selectivity in Electrocatalytic Oxygen Reduction by Iron–Porphyrin Complexes. J. Am. Chem. Soc 2015, 137, 4296–4299. [DOI] [PubMed] [Google Scholar]
- (29).Collman JP; Dey A; Yang Y; Ghosh S; Decreau RA O2 reduction by a functional heme/nonheme bis-iron NOR model complex. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 10528–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lu C; Zhao X; Lu Y; Rousseau DL; Yeh S-R Role of Copper Ion in Regulating Ligand Binding in a Myoglobin-Based Cytochrome c Oxidase Model. J. Am. Chem. Soc 2010, 132, 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sigman JA; Kim HK; Zhao X; Carey JR; Lu Y The role of copper and protons in heme-copper oxidases: Kinetic study of an engineered heme-copper center in myoglobin. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mukherjee S; Mukherjee A; Bhagi-Damodaran A; Mukherjee M; Lu Y; Dey A A biosynthetic model of cytochrome c oxidase as an electrocatalyst for oxygen reduction. Nat. Commun 2015, 6, 8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dey S; Mondal B; Chatterjee S; Rana A; Amanullah S; Dey A Molecular electrocatalysts for the oxygen reduction reaction. Nature Reviews Chemistry 2017, 1, 0098. [Google Scholar]
- (34).Carver CT; Matson BD; Mayer JM Electrocatalytic Oxygen Reduction by Iron Tetra-arylporphyrins Bearing Pendant Proton Relays. J. Am. Chem. Soc 2012, 134, 5444–5447. [DOI] [PubMed] [Google Scholar]
- (35).Mukherjee S; Bandyopadhyay S; Dey A Tuning the apparent formal potential of covalently attached ferrocene using SAM bearing ionizable COOH groups. Electrochim. Acta 2013, 108, 624–633. [Google Scholar]
- (36).Bulova A; Talaikytė Z; Niaura G; Kažemėkaitė M; Marcinkevičienė, L.; Bachmatova, I.; Meškys, R.; Razumas, V. Double-layered Ag/Au electrode for SERS spectroscopy: preparation and application for adsorption studies of chromophoric compounds. CHEMIJA 2007, 18, 9–15. [Google Scholar]
- (37).Sengupta K; Chatterjee S; Samanta S; Dey A Direct observation of intermediates formed during steady-state electrocatalytic O2 reduction by iron porphyrins. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 8431–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bard AJ; Faulkner LR: Electrochem. Methods; J. Wiley: New York, 1980. [Google Scholar]
- (39).Boulatov R; Collman JP; Shiryaeva IM; Sunderland CJ Functional Analogues of the Dioxygen Reduction Site in Cytochrome Oxidase:Mechanistic Aspects and Possible Effects of CuB. J. Am. Chem. Soc 2002, 124, 11923–11935. [DOI] [PubMed] [Google Scholar]
- (40).Chatterjee S; Sengupta K; Hematian S; Karlin KD; Dey A Electrocatalytic O2-Reduction by Synthetic Cytochrome c Oxidase Mimics: Identification of a ″Bridging Peroxo″ Intermediate Involved in Facile 4e-/4H+ O2-Reduction. J. Am. Chem. Soc 2015, 137, 12897–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vidakovic M; Sligar SG; Li H; Poulos TL Understanding the Role of the Essential Asp251 in Cytochrome P450cam Using Site-Directed Mutagenesis, Crystallography, and Kinetic Solvent Isotope Effect. Biochemistry 1998, 37, 9211–9219. [DOI] [PubMed] [Google Scholar]
- (42).Efimov I; Badyal SK; Metcalfe CL; Macdonald I; Gumiero A; Raven EL; Moody PCE Proton Delivery to Ferryl Heme in a Heme Peroxidase: Enzymatic Use of the Grotthuss Mechanism. J. Am. Chem. Soc 2011, 133, 15376–15383. [DOI] [PubMed] [Google Scholar]
- (43).Chatterjee S; Sengupta K; Samanta S; Das PK; Dey A Concerted Proton-Electron Transfer in Electrocatalytic O2 Reduction by Iron Porphyrin Complexes: Axial Ligands Tuning H/D Isotope Effect. Inorg. Chem 2015, 54, 2383–2392. [DOI] [PubMed] [Google Scholar]
- (44).Miner KD; Mukherjee A; Gao Y-G; Null EL; Petrik ID; Zhao X; Yeung N; Robinson H; Lu Y A Designed Functional Metalloenzyme that Reduces O2 to H2O with Over One Thousand Turnovers. Angew. Chem., Int. Ed 2012, 51, 5589–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kleingardner JG; Kandemir B; Bren KL Hydrogen Evolution from Neutral Water under Aerobic Conditions Catalyzed by Cobalt Microperoxidase-11. J. Am. Chem. Soc 2014, 136, 4–7. [DOI] [PubMed] [Google Scholar]
- (46).Cavazza C; Bochot C; Rousselot-Pailley P; Carpentier P; Cherrier MV; Martin L; Marchi-Delapierre C; Fontecilla-Camps JC; Ménage S Crystallographic snapshots of the reaction of aromatic C–H with O2 catalysed by a protein-bound iron complex. Nat. Chem 2010, 2, 1069–1076. [DOI] [PubMed] [Google Scholar]
- (47).Grönberg KLC; Roldán MD; Prior L; Butland G; Cheesman MR; Richardson DJ; Spiro S; Thomson AJ; Watmough NJ A Low-Redox Potential Heme in the Dinuclear Center of Bacterial Nitric Oxide Reductase: Implications for the Evolution of Energy-Conserving Heme–Copper Oxidases. Biochemistry 1999, 38, 13780–13786. [DOI] [PubMed] [Google Scholar]
- (48).Bhagi-Damodaran A; Michael MA; Zhu Q; Reed J; Sandoval BA; Mirts EN; Chakraborty S; Moënne-Loccoz P; Zhang Y; Lu Y Why copper is preferred over iron for oxygen activation and reduction in haem-copper oxidases. Nat. Chem 2017, 9, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.