

Summary
Dietary fibers act through the microbiome and improve cardiovascular health, metabolic disorders and cancer prevention. To understand health benefits of dietary fiber supplementation we investigated two popular purified fibers, arabinoxylan (AX) and long-chain inulin (LCI), and a mixture of five fibers. We present multi-omic signatures of metabolomics, lipidomics, proteomics, metagenomics, a cytokine panel and clinical measurements on healthy and insulin resistant participants. Each fiber is associated with fiber-dependent biochemical and microbial responses. AX consumption associates with a significant reduction in LDL and an increase in bile acids, contributing to its observed cholesterol reduction. LCI is associated with an increase in Bifidobacterium. However, at the highest LCI dose there is increased inflammation and elevation in the liver enzyme alanine aminotransferase. This study yields insights into the effects of fiber supplementation, it provides insights into mechanisms behind fiber induced cholesterol reduction, and it shows effects of individual, purified fibers on the microbiome.
eTOC
Lancaster et al. directly test the effects of two highly purified fibers on extensive clinical and biochemical profiles. They found that arabinoxylan, a common fiber of Metamucil, reduced cholesterol through bile acid production. Whereas inulin, a common fiber of many vegetables had no effect, while high doses caused inflammation.
Graphical Abstract
Introduction
At the population level, high fiber diets reduce the risk of heart attack, stroke and cardiovascular disease. They act by lowering cholesterol and promoting a healthier lipid profile for people eating a Westernized diet (Chutkan et al., 2012; Mozaffarian et al., 2003; Rimm et al., 1996). High-fiber diets are beneficial, but only 5% of Americans age 19–50 consume the recommended 28g (female)/35g (male) of total fiber per day (Medicine, 2005; Quagliani and Felt-Gunderson, 2017; Trumbo et al., 2002), with 90% of Americans consuming an average of 15 grams of fiber per day (Slavin, 2005). Dietary fiber supplementation has the potential to close this fiber gap, highlighting its broad potential relevance to health-related outcomes.
Dietary fibers are carbohydrates found in plants (i.e. fruits, vegetables, legumes, nuts, seeds, and grains) that are selectively metabolized by gut microbiota but are otherwise indigestible by humans. Once nourished by dietary fiber, the microbiota participates in numerous beneficial functions, including fermentation of carbohydrates into short chain fatty acids (SCFAs)(Marchesi et al., 2016; Scott et al., 2008), synthesis of important vitamins (i.e. K and B vitamins)(LeBlanc et al., 2013) and priming the immune system (Round and Mazmanian, 2009; Sekirov et al., 2010). Understanding how dietary fibers impact the microbiome and in turn human biochemistry and physiology, is critical to effectively using dietary fiber supplementation to improve human health.
Chemically, fibers are diverse molecules in length, branching, solubility, charge and other properties. They are usually studied as complex mixtures from their plant source. We sought to understand the effects of purified individual fiber components and investigated the physiological effects of dietary supplementation of two common and structurally distinct soluble fibers: arabinoxylan (AX), common in whole grains and the hemicellulose of other plants, and long chain inulin (LCI), found in onions, chicory root and Jerusalem artichokes. AX has a xylose backbone and arabinose branching chains. The degree of branching varies between plants and affects its impact on the microbiota (Rumpagaporn et al., 2015; Tuncil et al., 2020). Additionally, AX has bound phenolic compounds like ferulic acid that reduce inflammation by potentially acting as antioxidants (Yin et al., 2019). The other fiber, LCI, consists of a fructose polymer with a glucose cap. Inulin can be a dimer or be polymerized up to 60 or more fructose units, and LCI relative to its shorter-chain oligofructose counterparts, is selected for polymers longer than 25 units. LCI lacks the bound phenolic compounds that are present on AX. Lastly, we also analyzed AX and LCI in the context of five fibers that also included glucomannans, resistant starch, and acacia gum.
Soluble fibers have been individually associated with health benefits, although conflicting results have been reported depending on plant source and population (Armet et al., 2020; Myhrstad et al., 2020). In humans and animals AX is reported to manage hypertension, reduce LDL, reduce total cholesterol, reduce blood glucose levels and improve insulin resistance (Lu et al., 2004; Neyrinck et al., 2011; Ryan et al., 2015; Tong et al., 2014). Several mechanisms have been proposed for the cholesterol lowering effects of fiber (Joyce et al., 2019), including direct binding of cholesterol to fiber preventing absorption. Another proposed mechanism for fiber reduction of cholesterol is to sequester bile acids in the intestines, which inhibits bile acid reabsorption. Free cholesterol is then metabolized to create new primary bile acids, resulting in a systemic reduction of cholesterol (Buhman et al., 1998; Soliman, 2019; Wilson et al., 1998). The breadth of proposed mechanisms also suggests that fibers may have several mechanisms for inducing hypocholesterolemia.
LCI increases abundance of the genus Bifidobacterium – sometimes recognized as a “healthy microbe” (Hidaka et al., 1986; Kolida et al., 2002; Slavin, 2013) – and reduces constipation (Closa-Monasterolo et al., 2017; Collado Yurrita et al., 2014), as well as improves fasting glucose and HbA1C in people with diabetes (Dehghan et al., 2013). It has also been shown to improve heart (Dehghan et al., 2013), colon (Pattananandecha et al., 2016) and bone health (Abrams et al., 2005). However, some studies show little-to-no benefit (Alles et al., 1999; Giacco et al., 2004; Hoving et al., 2018) and others suggest that inulin may be harmful at high doses (Coussement, 1999). For example, high levels of LCI, 20% of chow in mice, has been reported to induce liver damage and ultimately liver cancer in mice after long-term exposure (Singh et al., 2018). Together these mixed findings highlight the personalized nature of fiber supplementation, its dependence on study population, the importance of fiber dose, and the need for a deeper mechanistic understanding of the impacts of fiber.
The current body of literature regarding the effect of dietary fiber on the gut microbiome and host is largely informed by whole-diet interventions (David et al., 2014; Koecher et al., 2019; Walker et al., 2011; Wu et al., 2011) and cross-sectional associations (De Filippo et al., 2010; Schnorr et al., 2014). These investigations provide understanding the effects of dietary fiber; however they can make limited conclusions about the physiological effects of individual fibers (Falony et al., 2016) and of course causal inference is precluded in cross-sectional studies. The impact of purified fibers on the gut microbiome has been tested in a limited number of prospective human intervention studies (Baxter et al., 2019; Venkataraman et al., 2016; Walker et al., 2011), but the resulting impact on host systems biology remains an unresolved question. There is a need for determining the unadulterated effects of individual fibers on the microbiome and for establishing associated health biomarkers (Swanson et al., 2020), ideally by testing different fibers on the same individuals. In this study we examined the effect of AX, LCI and the fiber mixture by analyzing stool metagenomics, plasma proteomics, metabolomics, lipidomics, serum cytokines and clinical values. Importantly, our study design included escalating dosage and washout periods between different fibers revealing both cohort-wide patterns and individual responses to each fiber. These results have significant implications for determining the effects of individual fibers on human health and physiology.
Results
Experimental Design and Cohort
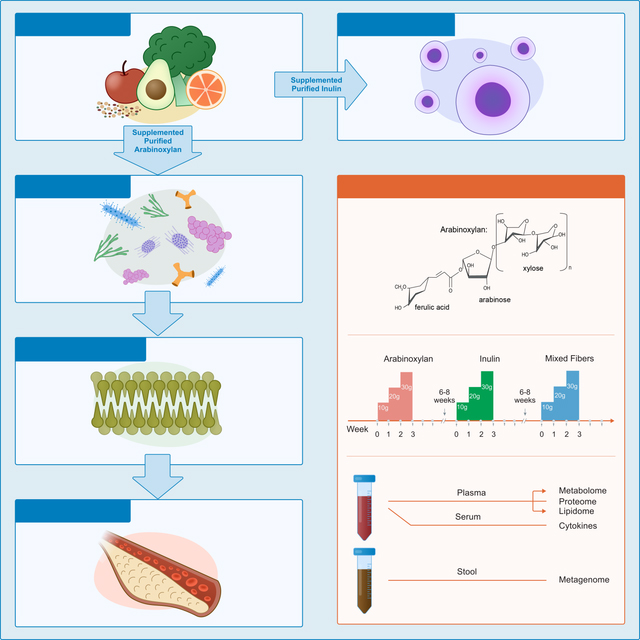
The study is a longitudinal, randomized crossover design (demonstrated in Figure 1) in which 18 IRB (23602) consented participants (8 men and 10 women) had their diets periodically supplemented with two fibers, AX and LCI, and a mixture of fibers consisting of equal parts AX, LCI, acacia gum, glucomannans, and resistant starch. Participants were randomized to consume either AX or LCI first, and the mixed fibers were always administered last. For all fibers, supplementation increased weekly from 10g/day for the first week (at breakfast), to 20g/day the second week (10g at breakfast and 10g at dinner), and lastly to 30g/day the third week (10g with each breakfast, lunch and dinner). A 6–8 week “wash-out” period without fiber supplementation was included between fiber series. We used purified fibers, rather than whole fibrous foods, as this is closest to a standardized intervention, and because a single purified fiber will have effects unadulterated from other phytochemicals. Participants also maintained food logs so their entire diet, including fiber from other sources, was monitored.
Figure 1.

Study design overview. Participants were randomized to two arms, starting with AX (lower structure) or LCI (upper structure) first and second, with mixed fibers last. Participants consumed an increasing dose of fiber daily for three weeks. Samples were collected at baseline, end of every week (Wk1–3), and washout periods day 3 and day 10 (WD3 and WD10). Participants did not take fiber supplements for 6–8 weeks between each cycle to allow a return to baseline. Blood and stool samples were collected at each indicated time point and omics and clinical analyses were performed.
To examine in detail the effects of each fiber we collected plasma, serum, and stool samples along with physiological measures (e.g. HR, blood pressure) at baseline before supplementation, at the end of each week, at washout day 3 (WD3), and at washout day 10 (WD10). The baseline of the subsequent fiber series was used as a final washout (WF). Being last, the mixed fiber does not have a WF. In addition to clinical measures (Table S1A), plasma was stored at −80°C and analyzed by LC-MS for metabolomics, lipidomics, and proteomics; serum was stored at −80°C and analyzed via a 62 cytokine and growth factor panel; and stool was stored at −80°C, sequenced via shotgun metagenomics (35.9 million average unique reads/sample) and analyzed for both species abundance and gene abundance (see methods). The average age of the participants was 56.9 with a standard deviation of 7.7. For most clinical values, our cohort was in a healthy range except LDL and total cholesterol where the average cohort value (113.18 ± 36.46 mg/dL and 193.20 ± 40.40 mg/dL, respectively) was near the high end of the recommended healthy level of <160 mg/dL and <240 mg/dL, respectively (Grundy et al., 2019)(Table S1A; Figure 2B). It is important to note that although the cohort size was small (n=18), the crossover design and deep profiling at different fiber doses increases statistical power and provides a deep assessment of individual responses to the different fibers (e.g. 324 timepoints total are analyzed).
Figure 2.

Baseline statistics and general fiber findings. A) Baseline of selected clinical measurements shown with violin plots. Background colors represent health range of each test: red = above normal range, yellow = borderline, green = normal range. B) Coefficient of variation at baseline for each ome represented as a boxplot with outliers. The number of features is enumerated below each plot. C) Non-metric dimensional scaling for baseline clinical measures. Each color represents a different participant. D) From the metagenomics, Shannon Diversity was calculated for all fibers at all timepoints. Error bars are the standard error of the mean. Change is significant (one-way ANOVA). WD3 = Washout Day 3, WD10 = Washout Day 10, and WF = washout final. E) Scatter plot of change from baseline of microbial gene abundance calculated in AX and LCI when taking 30g. X-axis is AX, y-axis LCI. The bacteria most impacted by fiber supplementation are colored. See also Figure S1; and see Tables S1A and S1B.
General Biological Effects of Fiber
We first examined inter-individual variation for the different readouts at baseline as measured by the coefficient of variation (CV). The most variation occurred in the microbial data, with the CVs in the microbial pathway abundance (median: 1.071, IQR: 1.100) and microbial genes (median: 0.849, IQR: 0.689). Following these, the protein (median: 0.438, IQR: 0.252) and lipid (median: 0.430, IQR: 0.280) data. Among the omes with the lowest variation were the cytokine (median: 0.262, IQR: 0.235) and metabolomic (median: 0.066, IQR: 0.074) data. There were significant differences when comparing the distribution of the CVs across omes (p < 0.05 for all comparisons except for lipids vs proteins, Figure 2B, Table S1B).
Notable fiber-dependent shifts were evident as detailed below. However there are some general observations. First, clinical values and other omics measurements are more similar within an individual as compared to differences between fiber type or fiber dose between individuals (Figure 2C).
Second, across all fibers, we saw a shift in microbiome composition and a decrease in microbial Shannon diversity over the course of fiber supplementation (AX p=1.38*10−7, LCI p=4.91*10−5, Mix p=0.0102, Figure 2D, Table S1C). All fibers, including the fiber mixture, elicited a similar decrease in diversity (Figure 2D). There was no change in richness over the course of fiber supplementation, and a very similar change in Simpson diversity. Although many changes in the microbiome were specific to one fiber (described below), some were shared between AX and LCI (Figure 2E). Specifically, Bacteroides ovatus and Bacteroides xylanisolvens increase for both fibers. Additional pan-fiber changes include a decrease in blood urea nitrogen across all the fibers (AX p=0.00711, LCI p=3.90*10−4, Mix p=0.0102; Figure S1A).
For AX and LCI, 606 of the 128,350 total analytes/microbes from all omes increased in a dose-dependent response in both fibers. These overlapping analytes do not include lipids, cytokines, proteins, or clinical measurements; however they do include the metabolites indolepropionic acid, indole and cinnamoylglycine, among a few others. They contain the microbiome species Bacteroides xylanisolvens and microbial genes from Bacteroides xylanisolvens, Haemophilus parainfluenzae, Veillonella parvula and Veillonella atypica. In the cluster where analytes decreased in a dose dependent fashion, 1,557 of the 128,350 features overlap. Thus, although small, there are more shared analytes/microbes that decreased in a dose-dependent response. This group lacks cytokines, clinical measurements, and proteins, but it does include 4 lipid species (one phosphatidylethanolamine, two phosphatidylethanolamine-ethers, and one phosphatidylethanolamine-plasmalogen), 96 metabolites, and the species Bacteroides stercoris, with the rest of the changes bacterial genes and pathways.
During all fiber supplementations self-reported data was collected regarding participant comfort, stool measurements, and diet (Table S1D; Figure S1). Stool frequency increased over time, with AX and Mix showing the largest effect size (AX p=5.4710−6, LCI p=0.00186, Mix p=3.4710−6; Figure S1E). Stool volume increased across all fibers, with AX and Mix showing the largest change (AX p=1.87*10−4, LCI p=0.0200, Mix p=2.76*10−4; Figure S1G). Stool consistency, evaluated by Bristol stool chart score, increased for AX and Mix (AX p=8.76−6, LCI p=NS, Mix p= 0.00250; Figure S1F). Bloating increased for the LCI and mixed fibers, and decreased after supplementation ended (AX p=NS, LCI p=6.55*10−5, Mix p=0.00454; Figure S1B). Flatulence followed a similar pattern, increasing during supplementation, then decreasing after supplementation ended (AX p=0.00548, LCI p=3.50−7, Mix p=1.19*10−4; Figure S1C).
When correlating the GI symptoms with the multi-omics, interesting trends emerge. Of the lipids that decrease during AX supplementation, bowel movement frequency inversely correlates (all FDR < 0.05) with 8/12 cholesteryl esters (CEs), 2/5 sphingomyelins (SMs) and 5/18 phopshpatidylcholines (PCs). However it does not correlate with clinical cholesterol levels nor bile acids. (Table S1E). Bristol stool scale was inversely correlated with 5/12 CEs. For the metagenome, there were a similar number of correlations with bowel movement frequency in AX and LCI (73 AX and 70 LCI). There were more correlations with bloating change (66 LCI and 3 AX) and other symptoms (25 LCI and 5 AX) in LCI than AX. Lastly there were more associations with stool consistency change (80 in AX and 38 in LCI), Bristol score (29 in AX and 8 in LCI), and stool volume change (60 in AX and 28 in LCI) in AX than in LCI (Tables S2A, S2B).
Diet journals informed about nutrient intake during the course of fiber supplementation. There were few changes, which included an increase in total sugar (66.2g ± 39.1 to 89.3g ± 45.5, p=0.00214) and a slight increase in carbohydrate intake with a modest significance (181.0g ± 62.9 to 224.0g ± 81.1, p=0.0443). However, calories from carbohydrate along with other macronutrient distributions did not change (Table S2C). Given the changes in the time of year between fiber supplementation and the other potential for changes in diet, there were very few changes in participants’ diet.
The Metagenome Changes in Response to Arabinoxylan
We next examined in depth the effect of each individual fiber on clinical and omics measurements. After normalization (see methods), we clustered the analytes into longitudinal patterns of prevalence using C-means clustering. Three major patterns were observed, along with other minor patterns (Tables S2D, S2E, S3A). During AX supplementation, microbes, microbial gene families, and molecular analytes were represented in the Class I cluster pattern (8,016/128,350 analytes) that followed a dose-response increase with fiber (Figure 3A, Supplementary Table 6). Examples include the genus Roseburia (p=0.00286, F=2.33) and species Bacteroides xylanisolvens (p=1.38*10−5; F=3.01) – two taxa known to ferment fibers in the colon. Also in this class is the metagenomic super-pathway of L-citrulline metabolism (p=0.00206, F=2.41) and xylose degradation IV (p=0.0379, F=1.73). This cluster also contains the metagenomic enzymes responsible for metabolizing AX and the phenolic acids bound to it, a xylanase (K01181, p=0.00531, F=2.19) and feruloyl esterase (K09252, p=1.19*10−8, F=5.26). The xylanase is a carbohydrate active enzyme that is a glycoside hydrolase responsible for breaking down the bonds between sugars in arabinoxylan.
Figure 3.

Systems biology changes when taking AX. A) C-means clusters from all omics that follow a dose-response and reverse dose-response. Select pathways and features of interest are displayed for each. Individual analytes tested for significance using multiple linear regression. B) LDL trend for all three fibers over the course of supplementation. Both AX and mix are significant (one-way ANOVA). Data are normalized to baseline and then raised to the mean of the cohort. C) The longitudinal response of the bile acids UDCA, CDCA and LCA over the course of fiber supplementation for all three fibers. Significance determined by one-way ANOVA. D) Significant correlations between bile acids (y-axis) and microbial taxa (x-axis) represented by bubble plot. Bubbles are colored by direction of correlation coefficient and the bubble size is represented by − log10(p-value). P-values determined by Spearman correlations, FDR corrected E) CEs that significantly reduce in a dose-response manner with AX supplementation. Significance determined by one-way ANOVA. F) Correlation network of lipids, bile acids and clinical cholesterol levels, where negative correlations are connected with a red edge and positive correlations are connected with a blue edge. Orange node = bile acid, yellow node = lipid, and blue node = clinical lipid value. All edges are spearman correlations with FDR < 0.05. See also Figure S2 and S3; and see Tables S1C, S2D, 3C, 3D and S4A.
We then performed correlation analyses within the C-means cluster. Within Class I, there are inverse correlations between lipids and the microbiome (all FDR <0.05, Table S3B, Figure S2A). The microbes inversely correlated with the lipid cluster are Haemophilus parainfluenzae, Veillonella parvula, Veillonella dispar, and Megasphaera micronuciformis (all FDR<0.05, Table S3B, Figure S2A). This suggests that those participants with increases in lipids during AX supplementation do not have these microbes (or the microbes are below detection limit), whereas those participants with the weakest lipid response have higher abundances of those microbes.
The Class II cluster (7,149/128,350 analytes) exhibited a decrease in analytes/microbes and revealed gene ontologies associated with arabinose and xylose metabolism (all significant genes FDR < 0.01; K15921, p=0.0111, F=2.02; K18205, p=1.71*10−6, F=4.02; K18206, p=5.34*10−6, F=3.76; and K08138, p=0.00128, F=2.51). These genes encode proteins that metabolize forms of arabinoxylan from other plants, a phenomenon observed previously (Tuncil et al., 2020). Because this class is rich in lipids, including clinical lipids, further in depth discussion is in the section below.
The third major pattern (Class III; 8,128/128,350 analytes; Figure S3A) associated with AX supplementation shows analytes increasing in a non-dose-response manner. These increase at 30g and remain elevated during washout time points D3 and D10 and decrease again at WF to near baseline levels (Table S2D; Figure S3A). This delayed response cluster includes pathways associated with fermentation, such as acetate production, mixed acid fermentation, and several fatty acid pathways (Table S2D). The gene families in this cluster include xylose and arabinose metabolizing genes (all significant genes FDR < 0.05; K02099, p=0.00617, F=2.16; K02100, p=0.00555, F=2.18; K02467, p=0.0113, F=2.02; K06041, p=0.00536, F=2.19; K07806, p=0.00207, F=2.41; K10011, p=0.0123, F=2.00; K10537, p=0.00222, F=2.39; K10538, p=0.0114, F=2.02; K10539, p=0.0125, F=2.00; and K10544, p=0.0219, F=1.86). This delayed class also contains several cytokines (Figure S3A). Thus, these genes took longer to be elevated by fiber supplementation, but also persisted longer after fiber removal. Together these findings show a dynamic and persistent metagenomic response to fiber supplementation. (Figure 3A)
Cholesterol, Bile Acid and Lipid Responses to AX
There are clinically relevant analytes that showed the inverse dose-response with AX (Class II; Figure 3A, Supplementary Table 6). These include LDL and total cholesterol (LDL p=3.12*10−4, Total p=8.79*10−4; Figure 3B). The mixed fiber course also showed a similar inverse dose response to AX, albeit of lesser effect, perhaps because it contains AX at a lower dose. Unlike in AX, LDL did not change in response to LCI supplementation, contradicting some published reports (Figure 3A). HDL cholesterol did not significantly change during AX supplementation (p=0.158). Additionally, a number of CE species detected in the lipidomics data exhibited an inverse dose-response with fiber (Figure 3E, all FDR<0.05). Together these results show the cholesterol pool including LDL is decreased during AX supplementation.
Bile acids are an important class of molecules synthesized from cholesterol and reabsorbed through enterohepatic circulation. In our cohort, the plasma bile acid pool shifts during fiber supplementation (Table S3C). When performing random forest classification on fiber supplementation vs no supplementation, bile acids were among those molecules most significantly predictive of AX – six of the top eight predictors of fiber supplementation are bile acids (Figure S3B). In particular, the bile acids ursodeoxycholic acid (UDCA; p=0.00966) and chenodeoxycholic acid (CDCA; p=0.0439), known modulators of cholesterol (Cohen-Solal et al., 1995; Einarsson, 1994; Poupon et al., 1993; Singhal et al., 1984), undergo dose-dependent increase in stool in response to fiber supplementation (Figure 3C). These bile acids correlate with numerous microbes (all FDR < 0.05, Figure 3D). For example, UDCA positively correlates with Bifidobacterium. These associations highlight the interactions of the microbiome with bile acid abundances. The correlations between bile acids and the microbiome were not shared between fiber types, because there was no correlation between bile acids and the microbes that existed in all three fiber supplementations. Other secondary bile acids like lithocholic acid (LCA; p=0.686), did not significantly increase with AX, and showed a significant decrease in LCI supplementation (p=0.0227). Thus, fiber not only uniquely shapes the microbiome, but also the bile acid pool.
In a follow up cohort of seven individuals, we followed a similar protocol except instead of increasing the dose, we gave the participants 20g of fiber per day for three weeks. The cholesterol trend was the same as in the parent cohort, decreasing significantly during the course of fiber supplementation and returning to baseline during the washout period (p=1.17*10−4, F=7.85; Figure S3C). Additionally we see an increase in the bile acids, particularly chenodeoxycholic acid (p=0.0174, F=2.73; Figure S3D). Thus, these results validate the findings of the original dose response study.
Following a inverse dose-response relationship during AX fiber supplementation was a group of lipids that included PCs, phosphatidylethanolamines (PEs), PE-plasmalogens (PEPs), ceramides (CER), SM, and CEs (Figure S3C; Tables S3D, S3E, S4A; all lipids FDR<0.05). PCs and PEs are secreted in lecithin with bile but do not go through enterohepatic circulation to the same degree as bile acids. These lipids are also enriched members of the myelin sheath. Overall this decrease in lipids demonstrates a generalized hypolipidemic effect of AX. The molecules showing the inverse dose-response correlate together, including the PCs, PEs, CEs, LDL and total cholesterol (all FDR<0.05; Figure 3F; Table S3E). Furthermore, these molecules inversely correlate with the secondary bile acids (all correlations FDR<0.05; Figure 3F; Table S4A). Of the 12 CEs in this cluster, 8 inversely correlate bowel movement frequency after FDR correction. Together these factors show shifts in lipids and bile acids that correlate with the microbiome during AX supplementation.
Cholesterol Responders have Different Systemic Effects
Every participant had a unique cholesterol response to AX fiber supplementation. To understand the different cholesterol responses, we analyzed our cohort in three equal groups of six individuals. Group one had the largest reduction in cholesterol (i.e. strong responders), group two had the intermediate response, and group three were the non-responders. We found a number of significant differences between the strong responders and the non-responders over the three AX doses. There were very few significant differences between the intermediate responders and either other group. From the metagenome, Helicobacter (FDR=0.00864) and hydrogen cyanide synthase (FDR=0.00269) were lower in responders, whereas mixed acid fermentation (FDR<0.0500), taurine dioxygenase (FDR=0.00183), tryptophan permease (FDR=0.00183), p-hydroxybenzoic acid efflux pump (FDR=0.0123), and feruloyl esterase (FDR=0.0123) were higher in responders (Figure 4A; Tables S4B,S4C,S4D).
Figure 4.

Systemic effects associated with strong AX cholesterol responders relative to non-responders. NR = non-responders; R = responders. A) From stool samples relative abundances of the genus Helicobacter and the metagenomic gene feruloyl esterase in the two categories. Y-axis is log2 fold change relative to baseline. P-values determined by Wilcoxon rank sum, FDR corrected. B) In clinical serum samples, comparing total proteins and triglycerides between categories. P-values determined by Wilcoxon rank sum, FDR corrected. C) Volcano plot showing proteins significantly increased in the responder group. X-axis is the value of the protein at 30g AX relative to baseline, and y-axis is log2 fold change. Two sample t-test, permutation based FDR 0.05 (number of randomizations is 250) and S0 correction 0.1 were applied using Perseus. D) The ratio of dietary protein to fiber in responders vs non-responders. P-value determined by Wilcoxon rank sum, FDR corrected. See Tables S4B, S4C, S4d, S5A and S5B.
Strong responders had lower levels of clinically-measured total serum proteins, which are primarily the high abundance proteins albumin, globulin and fibrinogen (Figure 4B; Table S4D). Several key lower abundance proteins, measured by plasma proteomics, were elevated in the responders, including the cardiac ryanodine receptor (RY2R) and apolipoprotein C (APOC4; Figure 4C). Moreover, Ingenuity Pathway Analysis of the proteomics data, revealed that the nuclear receptors regulating the conversion of cholesterol to bile acids, liver X receptor (LXR) and farnesoid X receptor (FXR), are differentially increased in responders vs non. LXR is up regulated during AX supplementation and the expression of FXR-controlled proteins significantly changes (LXR p=1.59*10−33, FXR p=2.49*10−34, Figure S3E–F, Supplementary Table S4E).
The clinical triglycerides, which were not different in the total cohort, are lower during supplementation in responders (p=0.0164; Table S5A). Lastly, we found from diet journals that at baseline the intake ratio of dietary protein to fiber (p=0.00749) as well as total protein intake (p=0.0318) were higher in responders (Figure 4D; Table S5B). Otherwise, the overall nutrient intake did not differ from those of non-responders. Thus, participants with higher dietary protein consumption are likely to have better cholesterol and hypolipidemic responses to AX.
LCI Alters the Metagenome and has a Deleterious Response at 30g
As with AX supplementation, we identified three major longitudinal response patterns to LCI supplementation (Figure 5A). Analytes/microbes from every omic layer followed an increased dose-response (Class I; 5,851/128,350 analytes; Table S2E). The microbiome analysis showed a dose-response of members of butyrate-producing Clostridium cluster XIVa. It also included two Bifidobacterium species, B. bifidum and B. pseudocatenulatum. The bacterial biochemical pathways in Class I include hexitol fermentation to lactate, and microbial gene families in Class I include those involved in the metabolism of fructose, the primary sugar in inulin (all significant genes FDR < 0.05; K01193, p=2.51*10−4, F=2.88; K01624, p=0.0101, F=2.04; K02768, p=0.0356, F=1.75; K02769, p=0.00724, F=2.12; K02770, p=0.0322, F=1.77; K10709, p=0.00857, F=2.08; and K19506, p=0.00174, F=2.45). This group of gene families includes a glycoside hydrolase for breaking apart the ester bonds between the sugars in inulin, K01193. The Class I cluster also contains lipids that are elevated during LCI supplementation but then destabilize during washout after removal of LCI (Figure S4A). For many analytes removing the fiber does not lead to a clean “off” state.
Figure 5.

Systemic changes when taking long-chain inulin. A) The dose-response and reverse dose-response pathways from the multi-omic data, as determined by C-means clustering. Significance for individual analytes determined by multiple linear regression. B) Participants ZOZOW1T, ZVBQY1N and ZL9BTWF all saw an increase in ALT above normal levels at 30g LCI, where most participants (individuals = grey, average of cohort = black) showed little to no change in ALT levels. C) The abundance of Bifidobacterium for each participant can be seen over the course of LCI supplementation, with ALT outliers colored and the average increase for the entire cohort in black. All other participants are gray, and the average of all participants is black with standard error bars. D) Metagenomic gene that decreased in the ALT spikers compared to those who did not spike: bile acid-coenzyme A. Each dot color represents a different participant. P-value determined by Wilcoxon rank sum, FDR corrected. NC ALT = no change in ALT. E) Entericidin B increased in ALT responders. P-value determined by Wilcoxon rank sum, FDR corrected. F) Cytokines decreased when taking 10g and 20g of LCI as compared to baseline; however at 30g there was a significant increase in cytokine abundance. Individual cytokines that significantly increased are denoted (*) using a one-way ANOVA. See Figure S4; and see Tables S2E, S3C, S4C, S5C, S5D
Similarly, there is a inverse dose-relationship with inulin for analytes from every omic layer (Class II; 7,724/128,350 analytes). Within this cluster there are antibiotic genes and a xylose symporter (all significant genes FDR < 0.05; K19061, p=3.06*10−16, F=10.8; K10673, p=6.91*10−4, F=2.65; K08223, p=0.0391, F=1.72; K08218 p=8.41*10−4, F=2.61; K04343, p=1.31*10−5, F=3.55; and K13252, p=0.0447, F=1.69; K08138 p=0.00128, F=2.51). (Figure 5A)
Another prominent longitudinal pattern in LCI supplementation is a spike of analytes/microbes at 30g (Class III; 8,824/128,350 analytes; Figure S4B). From the metagenome, taxa that spike include the phylum Actinobacteria, primarily driven by Bifidobacterium animalis (Figure 5C), and members of the genera Dorea, Lachnospiraceae and Klebsiella. In this group there are genes associated with fructose metabolism, the biosynthesis of antibiotics, antibiotic resistance genes and a quorum sensing gene (all significant genes FDR < 0.05; K16306, p=0.00739, F=2.17; K18333, p=2.03*10−9, F=5.72; K01597, p=2.77*10−4, F=2.86; K18926, p=3.91*10−6, F=3.82; K18900, p=0.00220, F=2.39; K18893, p=4.99*10−4, F=2.72; K18348, p=1.13*10−19, F=14.1; K18214, p=4.91*10−4, F=2.73; K14982, p=1.59*10−5, F=3.50); the latter may indicate denselypacked bacterial species competing in an overcrowded niche. These genes may also indicate the bacteria in this group include strong antibiotic producers, although Streptomyces, the best-known antibiotic producer, was not included in the Actinobacteria that spiked.
Also evident at the 30g LCI spike were analytes associated with poor health. Although not a cohort-wide response, the liver enzyme alanine aminotransferase (ALT) spiked outside the normal range at 30g for three participants (Figure 5B). Neither bilirubin nor primary bile acids were increased in the clinical or metabolomic measurements, indicating that the ALT spikes we observed have etiologies other than cholestasis. Participants were quickly removed from inulin after the ALT response, so they could later develop cholestasis if exposure to LCI persisted. Similar to LDL in AX, we compared the participants with the spike in ALT (responders) with those that did not respond (non-responders). Significant differences include increases in the microbial gene Entericidin B K36148 (Figure 5E; p=0.0394), the genus Klebsiella (p=0.0443), the biofilm formation gene K11907 (p=0.0393) and the protein IGHV3–53 (p=0.00728), whereas lower prevalence was the gene bile acid-coenzyme A ligase K15868 (p=0.0421; Figure 5D) in the ALT-spike group (Tables S5B, S5C).
Importantly, there is a significant cytokine cascade at 30g that includes the cytokines IL-6 (p=0.0484), TGF-b (p=0.0359), and VEGF-A (p=0.0456), among others, suggesting a systemic inflammatory response. At lower doses cytokines were below baseline (Figure 5F), indicating a dose-dependent effect of LCI-associated inflammation. The people with the largest cytokine spikes were not those with the ALT spikes, indicating two different responses to high inulin stress. Correlations within this class indicated the cytokines are inversely correlated to Pediococcus pentosaceus, Subdoligranulum variabile, Clostridium ramosum, Haemophilus parainfluenzae and the genus Streptococcus (all FDR<0.05, Table S5E, Figure S4C). People with the inflammatory response had lower levels of these microbes.
Mixed Fibers Show Smaller, Intermediate Phenotype
Consuming a diversity of different fibers is important for human health (reviewed in (Cockburn and Koropatkin, 2016)). In our cohort, mixed fiber supplementation yielded fewer significant changes compared to either individual fiber, suggesting individual fibers may have the strongest effects at least as administered in our study. The mixed fiber supplementation contained both AX and LCI, and we observed many of the same analytes trend toward similar dose-dependent responses as the pure fibers (Figure S4A; Table S3A). For example, there was a moderate reduction in cholesterol (LDL p=0.00351, total cholesterol p=0.00204, Figure 3B) compared to AX supplementation alone. Additionally, there was an intermediate cytokine response compared to inulin alone (Figure S5B; Table S6A), with fewer significant cytokines and a smaller magnitude of response. Thus, we find intermediate phenotypes with the combination of fibers as compared to the pure fibers.
Personalized Responses to Fiber
Although trends were observed across the cohort, each participant also showed a unique response to fiber supplementation. As described above, a majority of the cohort had a reduction of cholesterol from AX, but there were individuals that did not respond. In the other fiber, LCI, some participants even had increased LDL (Supplementary Figure 6A). As often occurs in clinical studies a range of responses can occur, and fiber studies have been found to have high variability, mostly attributed to the complexity of the microbiome, fiber source and population studied.
To assess personal responses systematically, we used outlier analysis to detect strong individual responses (Figure S5C). Importantly, every individual had some outlying analytes/microbes during fiber supplementation; however, some people had analytes concentrated in particular pathways.
To highlight several interesting responses, participant ZOZOW1T had an inflammatory response during mixed fiber supplementation (Supplementary Figure 5C; Table S5B) during week 2 and week 3. After further investigation into clinical notes and survey responses, no clinical symptoms or noted sickness was found for the participant during this time. These findings suggest that an individual can for several weeks have large changes in their cytokines and immune system without any presentation of symptoms or changes in regular clinical tests. Analysis of the metabolites that changed in ZOZOW1T during the time of the cytokine spike indicated large changes in energy metabolism (Supplementary Figure 6B). Additionally, although inulin causes negative health effects at 30g for several members of our cohort, participant ZQFL1P3 had higher concentrations of citrulline, phenolic acids and plasma bile acids, all generally associated with a healthy fiber response. These health improvements are corroborated by enrichment in healthy metabolic pathways including pantothenate and CoA biosynthesis and arachidonic acid metabolism (Supplementary Figure 6C; Table S5C). Thus, these participants had a unique response to fiber supplementation that may give insights to their personal health and/or the health of their microbiome.
Discussion
Fiber is associated with improved metabolic and cardiovascular health, but understanding effects of individual fibers on microbial and metabolomic response has not been studied using a multi-omics dataset. In the studies investigating clinical markers (e.g. cholesterol and glucose) the reported results have sometimes been inconsistent. Using a crossover study design and deep profiling of thousands of markers and microbial pathways, we have found that individual fibers can have dramatic and distinct responses both at the population and individual level.
Overall, AX improved lipid profiles, reducing LDL, total cholesterol and other lipids in a dose-dependent manner. However, individual responses varied and some participants saw little to no change in cholesterol levels. Those who were strong responders show different systemic responses, including more phenolic acid metabolizing genes in their metagenome and lower clinical triglycerides. During LCI supplementation, low doses showed a modest reduction of cytokines and increase of bacteria generally recognized as beneficial. However, at 30g some participants experienced a spike in cytokines and the liver enzyme ALT, suggesting too much of this fiber may be harmful. Overall, our findings show the benefits of fiber are dependent on fiber type, dose, and participant – a landscape of factors resulting from interactions between fiber, the gut microbiome, and host. These results have important implications in personalized response and interventions.
Heterogenous fibers from whole fruits and vegetables have been suggested to be the most beneficial for health (reviewed in (Cockburn and Koropatkin, 2016), although differences in their fermentability and other chemical properties have not been comprehensively studied (Williams et al., 2017). In our study, mixed fiber had intermediate effects relative to individual fibers, indicating that this particular combination of fibers was not synergistic. For example, the mixture of fibers showed a similar trend of cholesterol reduction as purified AX, but to a lesser extent. Additionally, there is a spike in a subset of cytokines during 30g of supplementation with mixed fibers similar to LCI, but to a lesser degree (Supplementary Figure 5A). Several high-fiber foods have cholesterol-reducing effects (Brown et al., 1999), and our study suggests that these reductions may be driven by individual constituents of the mix of fibers in unrefined plant foods.
It is worth noting some limitations of the current study. We did not include insoluble fibers in our mixed fibers supplementation, which may alter the properties of soluble mixed fibers. Also, in our study design, the mixed fibers always came after the purified fibers, which perhaps introduced a confounding variable. However, we included a lengthy washout period between each fiber supplementation, making this scenario unlikely. Furthermore, our study did not compare the mixed fiber supplements with consumption of mixed fibers from natural foods. Thus, our results may be affected by the modality by which it is consumed, and whole plant foods may include other phytochemicals and nutrients known to drive systems biology in other, possibly healthier directions.
AX from multiple plants reduces cholesterol (Tong et al., 2014), and studies have hypothesized this is due to AX directly binding cholesterol in the intestinal lumen, helping cholesterol pass through the body (Soliman, 2019). Our results suggest AX decreasing cholesterol is related to increased bile acid production or other changes in bile (Everson et al., 1992; Marlett and Fischer, 2002; Turley et al., 1991). In particular, we find bile acids increased during AX supplementation. In addition to draining the cholesterol pool, they also act as signaling molecules for cholesterol metabolism, binding the receptors FXR and LXR, which control cholesterol metabolism and its conversion to bile acids. These receptors are preferentially regulated in the participants in our study with the greatest cholesterol reduction. Consistent with this hypothesis, bile acids are inversely correlated with cholesterol and cholesteryl esters.
PCs, PEs, CEs and other lipids also decrease during AX supplementation. Similar to cholesterol, these lipids are secreted in bile as lecithin but do not participate in enterohepatic circulation to the extent of bile acids. Thus, lipids in bile that go through enterohepatic circulation increase (i.e. bile acids), while those that do not go through it (i.e. PEs, PCs and CEs) decrease. This increased bile flow is likely driven by the microbiome, as many of the bile acids are significantly correlated with the microbiome (the association between bile acids and the microbiome is reviewed in (Di Ciaula et al., 2017)). Hence we find that AX, through altering the microbiome, diversifies and enlarges the bile acid pool, increases bile flow, and changes bile composition - all of which contribute to depleting the cholesterol pool (Figure 6). Furthermore, in AX supplementation, several CEs inversely correlate with bowel movement frequency, although cholesterol itself does not. This indicates that the hypocholesterolemic effects of increased bowel movements may be due to the passing of cholesterol esters. CEs do not decrease during LCI supplementation despite increased bowel movement frequency, indicating this phenomenon is specific to AX. Thus, we suggest that to completely understand the reduction in cholesterol from dietary fiber, one should consider the multiple mechanisms that impact cholesterol levels, including changes in bile and the bile acid pool driven by microbial production of secondary bile acids.
Figure 6.

Observed effects of fiber on host systems biology. Green analytes indicate those that increased and in red those that decreased. Black arrows indicate movement of analytes through the body. The hypothesized mechanism of action for AX.
To further understand the cholesterol response, we compared the strongest cholesterol responders to the weakest responders. There were shifts in microbial communities and gene pools, including increases in the metagenomic enzymes that metabolize two phenolic acids: feruloyl esterase and p-hydroxybenzoic acid efflux pump. The shifts in these enzymes are likely due to phenolic acids bound to AX that become bioavailable after fermentation by gut microbes (Pasinetti et al., 2018). Phenolic acids themselves are hydrophobic molecules known to have antioxidative properties (Medicine, 2005; Piazzon et al.) and along with polyphenols are known to help healthy microbes thrive (Mompeo et al., 2020; Pasinetti et al., 2018). Ferulic acid has also been suggested to lower LDL in both mice and humans (Bumrungpert et al., 2018; Wang et al., 2018). Thus, phenolic acids, not present in inulin, may be specifically selecting microbes that contribute to lowered LDL cholesterol and altered lipid profile.
The reduction of LDL was more pronounced in people who had a higher dietary protein to fiber ratio. Given that both LDL responder and non-responder groups had similar total fiber intake, people with a high protein diet may have greater benefit from dietary fiber supplementation.
Inulin is a commonly used fiber supplement that has been associated with numerous positive health outcomes (Chambers et al., 2019; Hoffman et al., 2019). Since our study began, however, there have also been studies linking inulin consumption in mice with liver damage (Singh et al., 2018). Our study substantiates the adverse effects of high doses of inulin in humans, observing an increase in ALT in a subset of our participants. The increase in ALT, which is a known potential marker of liver damage, occurred without the presence of cholestasis suggesting other mechanisms might contribute to this phenomenon. We also saw a number of inflammatory and cytokine markers induced at 30g of LCI (most strongly IL-6, TGF-b and VEGF-A), showing this fiber causes inflammation at higher doses. The spikes of ALT and cytokines occurred in different people, suggesting differences in personal responses to high inulin stress. It has been proposed that high doses of inulin cause dysregulation of fermentation in the gut (Singh et al., 2018) and daily consumption over 20g may negatively impact health (Schaafsma and Slavin, 2015). However, inulin is believed to be safe in humans at low doses, and consistent with this possibility, at low doses of LCI we saw relatively a modest decrease in inflammation markers. Additionally there was a decrease in lithocholic acid and an increase in Bifidobacterium during LCI supplementation, both associated with long term gut health. These findings show inulin dose is crucial for deriving health benefits and further demonstrate health effects vary among individuals.
Two limitations of our study are the short time period that participants consumed fiber and the small number of participants. These limitations were balanced by the deep molecular and microbial profiling of a large number of samples collected and analyzed (324). The careful doses used and washout analyses enabled us to follow the dose-response effects of the different fibers as well as the kinetics with which their effects lasted. The number of participants was small, but the crossover, longitudinal design enabled us to follow the effects of each fiber on the same participants.
Overall, our study highlights the systemic effects of two popular purified dietary fiber supplements: LCI and AX. Our findings include 1) discovering the arabinoxylan glycoside hydrolase responsible for arabinoxylan metabolism, 2) discovering that in our population, individual fibers have stronger effects than a mix of fibers, 3) an increase in plasma bile acids, which one would not expect if fiber was depleting bile acids, 4) increased risk of liver damage in humans as a result of a high dose of inulin, 5) inflammation in humans from inulin supplementation, 6) the full panel of lipids contributing to the hypolipidemic effect of arabinoxylan, in particular the PCs, PEs, and CEs, 7) a decrease in microbial diversity after purified fiber supplementation, and 8) personalized response to purified fibers which is crucial for understanding personalized nutrition. Our results demonstrate that the physiological, microbial and molecular effects of the two fibers differ substantially. Further, our results demonstrate the tantalizing prospect of using targeted fibers, mediated by the microbiome, to drive health and systems biology in an predictable, personalized direction (reviewed in Lordan, 2020) (Lordan et al., 2020)).
STAR formatted Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael P. Snyder (mpsnyder@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The data sets and code supporting the current study have been deposited in Metabolomics Workbench #ST002112. They will be made available on publication and are available from the corresponding author on request.
All original code has been deposited on GitHub (https://github.com/slancast) and is available up on request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Study
The participants in this study were 10 adult women and 8 adult men, Stanford IRB No. 23602. The IRB approved this study on 10/15/2014. IRB # 23602. First enrolled participant was on 11/22/2014. Subjects were randomized to either arm of the experimental study.
Primary Outcome Measurements:
-
Comprehensive phenotyping of host profile during prebiotic intervention.
Our first primary outcome measurement was to determine the changes in host (human) phenotypic profile during the prebiotic intervention. The phenotypic profile composed of the metagenome, transcriptome, metabolome and cytokines of participants while taking the prebiotic intervention.
-
Mapping of microbiota changes in response to prebiotic intervention.
Our second primary outcome measurement was to determine a detailed map of microbes and microbial gene abundance changes while taking different purified prebiotic fibers. We used shotgun sequencing to determine the microbiota complexity.
-
Biological signatures identified from the integration of microbiome-host omes.
We developed biological signatures associated with purified prebiotic fibers from the integration of the omics data with microbial metabolite levels.
Clinical Trial #: NCT04706858
Title of clinical trial registration: Integrative Personal Omics for Dynamic Molecular Phenotypes Monitoring During Fiber Supplementation.
Participant enrollment/sample size for the study was calculated using a two-group repeated measures design which allows for participant attrition, as well as a number of variance-covariance structures across the repeated measures (Hedeker et al., 1999). The two-level mixed-effects model accounting for the correlation between repeated observations is below:
In this model, the joint distribution of random intercept and random slope of time (random intercept term(i) and random slope term(i)), which also captures the correlation within repeated observations is assumed to be multivariate normal with mean 0 and variance covariance matrix VS.
Here, the primary parameter of interest is the slope of interaction between group and time (β3). This parameter describes the differences in the linear growth trends of each group over time. Under this model, with an N of 54 (3 series of 18), we can attain 80% power with alpha set to 0.05, tau (group by time interaction) set to 0.450 and an attrition rate of 5%.
METHOD DETAILS
General Study Design
18 participants were enrolled from a larger cohort in the iPOP study (integrative Personal Omic Profile, Stanford IRB No. 23602). All participants provided written informed consent and all evaluations and blood/stool samples were obtained at the Clinical and Translational Research Unit (CTRU) after overnight fasting. Participants were recruited from the Stanford University surrounding community via online advertisements. Inclusion criteria for this study include: 1) be 18 years of age or older, 2) not be pregnant 3) be generally healthy (with no apparent symptoms at the time of enrollment) or have type 2 diabetes, 4) be willing to provide written informed consent for all study procedures. Exclusion criteria for this study include: 1) any medical condition that physicians believe would interfere with study participation or evaluation of results, 2) mental incapacity and/or cognitive impairment on the part of the patient that would preclude adequate understanding of, or cooperation with, the study protocol. There were a total of 37 individuals interested in participating in this study. Fifteen people expressed interest but did not join. Four were excluded: one for unstable blood sugar with diabetes, one difficulty getting blood, needing multiple punctures and two having experienced recent significant weight loss. The other eleven decided not to join due to a number of reasons including the time commitment, travel or living far from Stanford University, where the study was conducted.
Participants went through 3 cycles of fiber supplementation, each cycle was three weeks long with weekly increasing doses of 10 g/day during the first week, 20g/day during the second week and 30 g/day during the third week. Randomized for the first two cycles, fibers tested were chicory inulin (99%>5 dp; range 2–60dp; average >23 dp) and arabinoxylan (psyllium husks powder Now Foods) and a mix of 5 fibers during the third cycle. The fiber mix included equal amounts of inulin, arabinoxylan, glucomannan (Now Foods), resistant starch (Hi Maize from Honeyville), and acacia fiber (Now Foods). Washout period between the cycles was from 6–10 weeks. Fiber was provided in 10 g sachets and participants were instructed to resuspend content of the sachet in at least 8 oz of water and drink one with breakfast for the first week, one with breakfast and one with dinner during the second week, and one with each meal (breakfast, lunch and dinner) during the third week.
For each of the fiber cycles, blood, urine and stool samples were collected at seven timepoints: baseline, end of week one, end of week two, end of week three, day 3 after end of supplementation and day 10 after end of supplementation. Comprehensive anthropometric, clinical, and plasma measures of general health (weight, blood pressure, hematocrit, fasting blood sugar, fasting insulin, lipids, liver function tests, and kidney function tests) were performed at each visit.
Blood was fractionated into plasma, serum and peripheral blood mononucleotide cells (PBMCs). We performed a series of multiomic analyses on these samples, targeted cytokine profiling, and a panel of 52 clinicals. Please find the clinical measurements in Supplementary Table 1, including p-values to determine changes over time. From the serum fraction of blood we performed the targeted cytokine profiling. Shotgun metagenomics was performed on stool samples. Proteomics, lipidomics, and metabolomics were performed on the plasma fraction of the blood. Once all samples were in the correct aliquots, they were stored at −80°C. Samples were only thawed when prepared for analysis for each of the respective omics assay which were all performed within 5 years of collecting the samples.
Diet Records
For this study, participants were asked to not change their diet or eating habits for the time they were enrolled in the study. Participants were required to log all food and beverage items consumed for one week at baseline and one week at 30g fiber supplementation (i.e. week 3). Based on the food logs that were collected from the participants, nutrient analysis was conducted by research dietitians using ESHA Food Processor Nutrition Analysis software (ESHA Research, Salem, OR), including the USDA’s Food and Nutrient Database for Dietary Studies (FNDDS).
The nutrients that were assessed in this study were as follows: energy (kcal), carbohydrate (g), protein (g), fat (g), macronutrient distribution (i.e. calories from each macronutrient divided by the total calories), sugar (g), total fiber (g; called ‘fiber’), cholesterol (mg), sodium (mg), ratio of energy to fiber, ratio of carbohydrate to fiber, ratio of protein to fiber, ratio of fat to fiber, ratio of cholesterol to fiber, and ratio of sugar to fiber. To examine changes in the nutrients over time (between baseline and week 3) of the cohort, Wilcoxon Signed-Rank test (i.e. the paired samples Wilcoxon test) was performed. P values were 2-tailed and considered significant at <0.05.
GI symptom was assessed by using the Gastrointestinal Symptom Rating Scale (GSRS)(Svedlund et al., 1988) at baseline and by a modified questionnaire from Winham and Hutchins (Winham and Hutchins, 2011) at each subsequent visit. Assessed GI symptoms included daily bowel movements, stool frequency, stool volume, stool consistency, bloating, and flatulence. To test changes in GI symptoms in each fiber group, Kruskal–Wallis test was performed due to non-normality of the data. P values were 2-tailed and considered significant at <0.05. Statistical analysis was performed using the stats package, and data visualization was performed using ggplot2 in The R Project for Statistical Computing environment, available from the ComprehensiveRArchiveNetwork.
Metagenomics via shotgun sequencing
Stool samples were aliquoted into 0.25g samples. Whole metagenome DNA was isolated using the QIAamp PowerFecal DNA kit (Qiagen cat# 51804). Library preparation performed in a PCR free manner using the NuGEN Ovation Rapid library kit. DNA was fragmented by shearing using the Covaris E210R. The Covaris settings were Intensity: 4; Duty Cycle: 10%; Cycles per Burst: 200; Treatment Time: 80s; Temperature: 7C with the E210 Intensifier (PN 500141). After shearing samples were tested on the bioanalyzer to determine correct fragment length, averaging ~300bp. Samples were then pooled and sequenced 96 at a time to a depth of 30 to 50 million reads per sample on a HiSeq2000. Sequences were analyzed using the HUMAnN2 and MetaPhlAn pipelines to generate pathway coverage values, gene family abundances, and taxa abundances (Franzosa et al., 2018). These were mapped to the Human Microbiome Project database for alignment. These generate relative counts used for future analyses.
Target Cytokine Analysis
Cytokines were profiled using a Luminex xMAP cytokine profiling system at the Stanford Human Immune Monitoring Center. This involves attaching antibodies specific to cytokines to beads using a capture molecule. The fluorescent molecules are attached to the molecule of interest (in this case the cytokine), and fluorescence is used as the measure of cytokine abundance. The fluorescent values were used for downstream analyses.
Metabolomics
Plasma metabolite extraction.
We performed an untargeted metabolomics using a platform, previously described in Contrepois et al., 2015 (Contrepois et al., 2015), to profile plasma extracted metabolites from individuals who completed the study as described above. Prior to sample preparation, aliquots of plasma were frozen and stored at −80°C. Samples were thawed and 100 μL of plasma was transferred into a 2mL Protein Lobind eppendorf tube. In order to precipitate and remove proteins, 400 μl of 1:1:1 methanol:acetonitrile:acetone solution, including 17 internal standards to control for extraction efficiency and evaluate LC-MS performance, was added to each plasma sample while on a cold plate. In the cold room, samples were placed on the vortex shaker for 15 minutes and placed at −20°C for two hours to allow for sufficient protein precipitation. Samples were centrifuged at 10,000 rpm for 10 min at 4°C and the supernatant (metabolite extract) from each sample was carefully removed and placed in a new 2mL Protein Lobind eppendorf tube. Samples were placed in the turbovap, evaporated to dryness under nitrogen and stored at −80°C. Before analysis on the MS, samples were reconstituted in 100 μl 1:1 methanol:water solution and vortexed for 30s, placed in a bath sonicator for 30s and incubated on ice for 30s. This process was repeated 3 times for all samples and were centrifuged at 10,000 rpm for 10 min at 4°C. The supernatant of each of these was then transferred to MS tubes and stored at −20°C until ran on the mass spectrometer.
Multiple quality control measures were performed to ensure data quality. First, all samples were randomized prior to both protein extraction and data acquisition. We regularly review internal standards to qualify mass accuracy, retention time and peak shape. Finally, we created a pooled quality control (QCs) sample by adding 30 μL of each sample in the study. The QCs were injected every 10 injections in the sequence to monitor instrument performance, to correct for MS drift with time and normalization.
Data acquisition.
Plasma samples were analyzed using HILIC and RPLC separation in both positive and negative ionization modes. HILIC data were acquired on a Thermo Q Exactive HF mass spectrometer while RPLC data were acquired using a Thermo Q Exactive mass spectrometer (Thermo Fisher Scientific). Both mass spectrometers were equipped with a HESI-II probe and operated in full MS scan mode. HILIC experiments used a ZIC-HILIC column (2.1 × 100mm, 3.5 mm, 200 Å; EMD Millipore) that used mobile phase solvents (A) 10 mM ammonium acetate in 50:50 acetonitrile:water and (B) 10 mM ammonium acetate in 95:5 acetonitrile:water. RPLC experiments used a Zorbax-SB-aq column (2.1 × 50mm, 1.7mm, 100Å; Agilent) that used mobile phase solvents (A) 0.06% acetic acid in water and (B) 0.06% acetic acid in methanol. Before running any samples, LC-MS systems were calibrated, and the columns were equilibrated by injecting 12 and 6 pooled (QCs) for HILIC and RPLC, respectively. Pooled QCs were also used to acquire MS/MS data at normalized collision energies (NCE) of 25 and 35 for HILIC and 25 and 50 for RPLC.
Data processing.
Raw data were imported into Progenesis QI 2.3 software (Water, Milford, MA, USA) to align and quantify chromatographic peaks. Data from all 4 acquisition modes (HILIC positive, HILIC negative, RPLC positive, RPLC negative) were processed independently. Using in house R code, we 1) removed noise, 2) imputed data and 3) adjusted for MS drift with time using the LOESS normalization method on pooled QCs injected every 10 injections in the sequence. We used MetID and our MS/MS data to identify 12740 metabolites with confidence levels ranging from 1–3, where 1 matches MS/MS, retention time and m/z from standards on our platform (843 metabolites), 2 has MS/MS and m/z matches from a database (395 metabolites), and 3 matches the m/z of a database (11,502). These metabolite names, m/z, retention time and their confidence levels can be found in Table S5D. Bile acids were of specific interest to this study and were further validated using MS/MS from standards we ran on our platform which we could then match retention time and fragmentation Table S5D.
Proteomics
Sample preparation and data acquisition.
In a randomized order, frozen plasma samples were thawed on ice, prepared by in-solution tryptic digestion and analyzed by SWATH-MS (Gillet et al., 2012). Undepleted plasma proteins (8ug, 5uL) were used for in-solution trypsin digestion protocol and liquid chromatography separation on a NanoLC 425 System (SCIEX, Eksigent system). The mobile phase solvent (A) consisted of 92.9% water, 2% acetonitrile, 5% dimethyl sulfoxide, and 0.1% formic acid, solvent (B) 92.9% acetonitrile, 2% water, 5% dimethyl sulfoxide (DMSO), and 0.1% formic acid, and solvent (C) 97.9% water, 2% acetonitrile, and 0.1% formic acid. The LC system setting used a trap and analytical column. A flow of 5uL/min (5min) of solvent (C) was used on the trap column ChromXP C18 0.5 × 10 mm, 5 μm, 120 Å (SCIEX, cat# 5028898). Peptides were eluted from an analytical column ChromXP C18 column 0.3 × 150 mm, 3 μm, 120 Å (SCIEX Cat# 5022436) using a 43-minute gradient from 4–32% B with 1-hour total run. Sciex TripleTOF 6600 System was used for SWATH Acquisition method, and this was assembled with a DuoSpray Source and 25 μm ID electrode (SCIEX). In Analyst TF Software (v1.7) was created a MS method with 100 variable Q1 windows and high sensitivity MS/MS mode. At the beginning and end of each batch, a quality control sample (QC) was injected from a premix of an equimolar pool from samples in this study. Samples were run in four batches and QC data were used to control for batch effects.
Data processing.
Label-free quantification of SWATH-MS data was performed in the OpenSWATH environment. SWATH wiff files were converted into mzML files using ProteoWizard and analyzed using OpenSWATH workflow (Gillet et al., 2012). For the protein identification, we used the SWATH-MS proteomic library from the PRIDE database (Liu et al., 2015; Vizcaino et al., 2016). For this workflow, peak groups were statistically scored with pyProphet tool (v2.0.1) from individual runs, and all injection samples were cross-run aligned using TRIC strategy (Rost et al., 2016). This work utilized computing resources provided by the Stanford Genetics Bioinformatics Service Center (SCG Genomic cluster) at Stanford University.
Statistical analysis.
For the bioinformatics analysis we used SWATH2stats (Blattmann et al., 2016), mapDIA (Teo et al., 2015), Perseus (Tyanova et al., 2016) and R. SWATH2stats transformed SWATH data from the OpenSWATH software into a format readable by other statistics packages as mapDIA while performing filtering, annotation and FDR estimation using protein FDR 5% and peptide FDR 0.5%. The mapDIA software was used for statistical analysis of protein expression using DIA fragment-level intensities considering a minimum of 3 fragments per peptide and 1 peptide per protein. Next using Perseus, data matrix was Log2 transformed, filtered 70% of valid values and imputed missing values drawing from a log normal distribution (‘mean = ‘Sample mean’ − (1.8*sample standard deviation)’ and ‘standard deviation = 0.3 * sample standard deviation’), and data normalization by subtracting median grouping by sample preparation plate number (batch). We produced proteomics plots using R and Perseus.
Metabolomic and Proteomic Enrichment Analysis
Results from proteomics and validated bile acids analysis from baseline were combined and uploaded into Qiagen’s Ingenuity Pathway Analysis (IPA) for core analysis using the reference set Ingenuity pathway knowledge base (genes and endogenous chemicals QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis). IPA identified the canonical pathways enriched between baseline and 30g fiber. The experimental dataset was filtered using p-values (<0.2) and log-ratio in the differential expression (−0.3 down, 0.3 up, Log2 (fold change) The canonical pathway and the differentially expressed features resulting from IPA were plotted in Cytoscape (Shannon et al., 2003). \
Lipidomics
Sample Preparation.
Lipids were extracted using a modified biphasic separation protocol with methyl tertiary-butyl ether (MTBE)(Matyash et al., 2008). Pipetting was performed using Rainin BioClean Ultra pipette tips. In 2 ml Protein LoBind Eppendorf tubes (cat# 022431102), 40 μl of neat plasma were mixed with 260 μl ice-cold MeOH and briefly vortexed to denature proteins. Next, 40 μL of an isotopically labeled standard lipid stock that approximates the blood plasma lipid composition was added to each sample (SCIEX, cat# 5040156, LPISTDKIT-101), which was prepared according to the instruction in Lipidomics Workflow Manager (LWM). The mix was briefly vortexed. Next, 1000 μL ice-cold MTBE was added after which samples were vortexed for 10 seconds followed by shaking at 4°C for 30 min. Phase separation was induced by adding 250 μL of HPLC grade water. Samples were subsequently vortexed for 60 seconds. Next, samples were centrifuged at 16,000 g for 5 min at room temperature. 750 μL of the upper organic (MTBE) phase were transferred to a new 2 mL tube. This MTBE lipid fraction was dried down in a nitrogen blower for about 2 hours. For storage at −20°C, 200 μL of MeOH was added to dried-down samples.
Lipid analysis (Lipidyzer).
On the day of measurement, lipids stored at −20°C in MeOH were dried down in a nitrogen blower. Then, lipids were reconstituted in 300 μL MeOH-toluene with 10 mM ammonium acetate (9:1). Samples were vortexed for 10 seconds, left for equilibration at room temperature for 20 minutes. To remove potentially insoluble material, samples were centrifuged at 16,000 g for 5 min at room temperature. Finally, 250 μL were transferred to 2 mL amber vials (Waters, cat# 186000848) containing a 300 μL glass inlet (Waters, cat# WAT094170DV) and sealed with Teflon lids (Waters, cat# 186000274).
Data Acquisition.
The Lipidyzer (SCIEX), a QTRAP system with SelexION ion mobility, was used for targeted profiling as described previously (Contrepois et al., 2018). In brief, flow injection analysis was performed with a LC-30AD (Shimazdu) operating at 8 μL/min (50 μL injection volume) using a running solution that consisted of 10 mM ammonium acetate in dichloromethane:MeOH (50:50). DMS separates lipids based on the principle that each lipid class has a different head group dipole moment and thus mobility in the DMS aperture (Schneider et al., 2010).
The lipid molecular species were identified and quantified using multiple reaction monitoring (MRM) and positive/negative switching. Two acquisition methods were employed covering 10 lipid classes across positive and negative mode. Method 1 had SelexION voltages turned on while Method 2 had SelexION voltages turned off. Method 1 employed an isocratic flow of 8 μL/min for 7.9 min, followed by a 2 minute wash at 30 μL/min. Method 2 employed an isocratic flow of 8 μL/min for 6 minutes, followed by 2 minute wash at 30 μL/min. Each lipid was acquired throughout 20 cycles. Lipid classes targeted in positive mode: SM, DAG, CE, CER, and TAG. Lipid classes targeted in negative mode: LPE, LPC, PC, PE, and FFA. Lipids were quantified using the LWM software, which compares endogenous lipids to the known concentrations of structurally most similar spiked-in lipid standards and reports all detected lipids in nmol/g.
Data analysis.
Data were downloaded from the Lipidyzer LWM and merged and processed in R. In brief, Excel files (LWM output) were read with the “loadWorkbook” package. From all samples, lipid concentrations determined in a blank control (sample processed in parallel without the addition of cells) were subtracted to correct for background signals. The data set was further filtered accepting only lipid species that detected in at least 25% of all samples. Missing values were imputed by drawing from a random distribution with the following log scale properties: ‘mean = ‘Sample mean’ − (1.8*sample standard deviation)’ and ‘standard deviation = 0.3 * sample standard deviation’.
QUANTIFICATION AND STATISTICAL ANALYSIS
Batch correction
Batch correction was performed using the CoMBAT program in the R SVA package. For the metagenomic analysis there were 5 batches. Additional variables included in the batch correction model were the fiber, the dosage of fiber, and participants to ensure any variation from these data were not removed during batch correction. After batch correction, these datasets were used in downstream processing.
Data integration: data preprocessing
Due to the skewed, non-normal distribution of omic datasets, data was log2 adjusted prior to analyses to make the different datasets more comparable. Imputation of missing data in the lipidomics and clinical measurements were calculated using a k-nearest neighbor analysis from R using the function inpute::impute.knn(). Every omics measured was split into thirds corresponding with each fiber course, generating our dataset 1 for each ome. In order to generate the most stable baseline measurement, an average of the three baseline measurements were taken for every analyte for each person, which was dataset 2. Two additional datasets were created for analyzing the data. First a dataset normalized to the average baseline measurements by subtracting the baseline for every person values from the full dataset (3). Lastly an aggregated dataset was generated for either the average value for each analyte from the baseline normalized data (4). This generated four primary datasets that we worked with for each ome.
C-means clustering
To determine changes in expression over longitudinal data, we used C-means clustering, which allowed us to find which analytes following a dose-response with fiber. This was performed using the R package Mfuzz. The function Mfuzz::standardise2 was used to facilitate comparisons between data with varying distributions. These functions generate a z-score, normalizing each analyte to its average and standard deviation, and then normalizing every initial value to 0. Because the first time point was the average of three baseline measurements subtracted from the single baseline of that fiber series, the analytes did not start at exactly zero without this normalization. The fuzzification factor was determined using the function Mfuzz::mestimate(). From these normalized data, clusters were determined using Mfuzz::mfuzz(), and those clusters following a dose-response with fiber were used to determine which analytes were responding to fiber supplementation. The clusters were plotted using a density function, coloring the most denselypacked lines red, and the outer lines yellow then blue. On metabolites from relevant classes, targeted linear models determine significance. The formula was as follows: analyte = visit + participant.
Correlation networks
One aspect of systems biology is understanding which analytes respond together. To find this we determined correlation networks, both on the entire datasets and on aggregated values in the datasets. Normalized values were used on the entire dataset to subtract out baseline inter-individual variation and only capture individual variation in response to fiber. Correlations were found using the program Hmisc::rcorr(). On these data a Benjamini-Hochberg FDR correction was applied. Every correlation below 0.05 after correction was saved as part of the network. Data was visualized using the R package igraph with the layout algorithm Fruchterman Reingold.
Supervised clustering and machine learning
Supervised statistical methods allow for prediction of known treatment groups from expression data. To this end, we used both supervised clustering and random forest machine learning model. For both methods the data were classified into a binary classifier as either fiber treatment (at 10g, 20g, and 30g) or no fiber treatment (baseline and washout day 10). Washout day 3 was eliminated from these analyses because it might serve as an intermediate value, not clearly classifiable in the binary scale. For the supervised clustering, the R function supclust::pelora() was used, and for the random forest, the Python function sklearn.grid_search.fit() was used. For supervised clustering the number of clusters was determined by using the elbow method on the “crit” value. For machine learning, analytes with the highest weights determined to be most predictive of fiber supplementation were used. During plotting, only significant analytes were used from the supervised clustering and the top predicting analytes from the random forest.
Responders and non-responders
To determine the systemic differences of people responding and not responding to dietary fiber, a combination of approaches was used. For LDL, the responses were broken up in tertiles based on the response normalized to baseline at 10g, 20g, and 30g of fiber. When comparing between responders and non-responders the top and bottom tertiles were used. Since there were only 3 responders for ALT in LCI, only the top 3 were used, and compared to the rest of the cohort. A Wilcoxon rank sum was performed on participants values during fiber treatment for the other log normalized analytes, followed by BH FDR correction. A cutoff of 0.05 was used to determine significance after correction. Plotting of individual responses was done using a jittered boxplot in ggplot2::ggplot(), colored by participant, or by a line graph in the same package.
Additional statistical measurements
For testing individual time points against each other Wilcoxon rank sum tests were used, and for testing the p-value for the trends a one-way ANOVA was used. Both were performed using the R base package, and unless otherwise stated were performed on the dataset that was baseline normalized.
Supplementary Material
Table S1. A) Clinical data for the entire cohort. B) Coefficients of variation at baseline for every analyte in the cohort. C) Gut microbiome taxa data generated using MetaPhlAn with associated metadata. D) Gastrointestinal symptoms during fiber supplementation. E) GI symptoms correlated with lipids and bile acids in AX. Related to Figures 2 and 3.
Table S2. A) GI symptoms correlated with metagenome in AX. S2B) GI symptoms correlated with metagenome in LCI. C) Diet data in responders and non-responders during fiber supplementation. D) Analytes associated with their respective cluster in C-means clustering during AX supplementation. Class I is cluster 11, Class II is cluster 10, and Class III is cluster 16. E) Analytes associated with their respective cluster from C-means clustering during LCI supplementation. Class I is cluster 11, Class II is cluster 7, and Class III is cluster 9. Related to Figures 3 through 5.
Table S3. A) Analytes associated with their respective cluster in C-means clustering during Mix supplementation. Class I is cluster 13 and Class II is cluster 2. B) Correlation within Class I analytes in AX. Only those with FDR<0.05 are included. C) Metabolomics data with associated metadata during fiber supplementation. D) Lipid data with associated metadata during fiber supplementation. E) Lipids that follow the reverse dose-response to fiber, and linear regression p-value/F-statistic. Related to Figures 3 and 5.
Table S4. A) Correlations between lipids, bile acids, and clinical cholesterol. Only correlation with FDR < 0.05 are listed. B) MetaPhlAn analytes in LDL responders vs non-responders from AX data, with Wilcoxon rank sum and FDR adjusted p-values. C) Pathway abundance analytes in LDL responders vs non-responders from the AX data, with Wilcoxon rank sum and FDR adjusted p-values. D) Gene family analytes in LDL responders vs non-responders from the AX data, with Wilcoxon rank sum and FDR adjusted p-values. E) IPA analysis for LXR and FXR pathways. Related to Figures 3 and 4.
Table S5. A) Clinical analytes in LDL responders vs non-responders in the AX data, with Wilcoxon rank sum and FDR adjusted p-values. B) Diet analytes in LDL responders vs non-responders in the AX data. C) MetaPhlAn analytes in ALT responders vs non-responders in the LCI cohort, with Wilcoxon rank sum and FDR adjusted p-values. D) Gene family analytes in ALT responders vs non-responders in the LCI cohort, with Wilcoxon rank sum and FDR adjusted p-values. E) Correlations between analytes in LCI Class I C-means cluster. Only correlations with FDR<0.05 are present. Related to Figures 4 and 5.
Table S6. A) C-means clustering for the cytokines during Mix supplementation. Class I is cluster 3 and Class II is cluster 6. B) Personalized C-means clustering for participant ZOZOW1T. C) Personalized C-means clustering for participant ZQFL1P3. D) Metadata associated with metabolomics. Related to Figures 2, 3 and 5.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| EDTA-plasma and PBMCs prepared from intravenous whole blood collection | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Methanol | Fisher Scientific | A456-4 |
| Methyl tert-butyl ether | Sigma-Aldrich | 650560-1L |
| Water | Fisher Scientific | W6-4 |
| Acetone | Fisher Scientific | A9291 |
| Aeconitrile | Fisher Scientific | A955-4 |
| Toluene | Sigma-Aldrich | 650579-1L |
| Ammonium Acetate | Sigma-Aldrich | 73594-25G-F |
| Acetic acid | Sigma-Aldrich | 73594-25G-F |
| Isopropanol | Fisher Scientific | A461-4 |
| Dichloromethane | Sigma-Aldrich | 34856 |
| Deposited data | ||
| Raw and analyzed data | This paper | Metabolomics Workbench: ST002112 |
| Critical Commercial Assays | ||
| AllPrep DNA/RNA/Protein kit | Qiagen | cat# 80004 |
| Clinical labs | This paper | N/A |
| QIAamp PowerFecal | Qiagen | cat# 51804 |
| Rapid DNA-seq | Nugen | cat# 0567-24 |
| Software and Algorithms | ||
| Impute | Hastie et al., 2021 | https://www.bioconductor.org/packages/release/bioc/html/impute.html |
| Analyst TF | Sciex | https://sciex.com/products/software/analyst-tf-software |
| Progenesis QI | Nonlinear Dynamics | https://www.nonlinear.com/progenesis/qi/ |
| Lipidomics Workflow Manager | Sciex | https://sciex.com/ |
| Mfuzz | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/Mfuzz.html |
| Ingenuity pathway analysis | Qiagen | https://digitalinsights.qiagen.com/productsoverview/discovery-insightsportfolio/analysis-and-visualization/qiagenipa/ |
| igraph | Bioconductor | https://cran.rproject.org/web/packages/igraph/index.html |
| MetaPhlAn | Segata et al., 2012 | https://huttenhower.sph.harvard.edu/metaphlan2/ |
Highlights:
Crossover clinical trial examines how highly purified fibers select a specific microbiome
Arabinoxylan decreases cholesterol in part through increased bile acid synthesis
Cholesterol responders ate more protein and had higher levels of feruloyl esterase
High doses of inulin cause inflammation and elevation of ALT
Acknowledgements
We acknowledge the Genome Sequencing Service Center by Stanford Center for Genomics and Personalized Medicine Sequencing Center, supported by the grant award NIH S10OD025212 and NIH/NIDDK P30DK116074. Further we used services by the Stanford Genetics Bioinformatics Service Center, and the Stanford Human Immune Monitoring Center for their technical assistance during this study. We acknowledge the NIH NCCIH and training grant 5T32AI007290 for funding, and the Stanford iPOP team for their continued support.
Footnotes
Declaration of Interests
The authors declare no competing interests.
ADDITIONAL RESOURCES
Clinical Trial #: NCT04706858
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams SA, Griffin IJ, Hawthorne KM, Liang L, Gunn SK, Darlington G, and Ellis KJ (2005). A combination of prebiotic short- and long-chain inulin-type fructans enhances calcium absorption and bone mineralization in young adolescents. Am J Clin Nutr 82, 471–476. [DOI] [PubMed] [Google Scholar]
- Alles MS, de Roos NM, Bakx JC, van de Lisdonk E, Zock PL, and Hautvast GA (1999). Consumption of fructooligosaccharides does not favorably affect blood glucose and serum lipid concentrations in patients with type 2 diabetes. Am J Clin Nutr 69, 64–69. [DOI] [PubMed] [Google Scholar]
- Armet AM, Deehan EC, Thone JV, Hewko SJ, and Walter J (2020). The Effect of Isolated and Synthetic Dietary Fibers on Markers of Metabolic Diseases in Human Intervention Studies: A Systematic Review. Adv Nutr 11, 420–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter NT, Schmidt AW, Venkataraman A, Kim KS, Waldron C, and Schmidt TM (2019). Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattmann P, Heusel M, and Aebersold R (2016). SWATH2stats: An R/Bioconductor Package to Process and Convert Quantitative SWATH-MS Proteomics Data for Downstream Analysis Tools. PLoS One 11, e0153160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L, Rosner B, Willett WW, and Sacks FM (1999). Cholesterol-lowering effects of dietary fiber: a meta-analysis. Am J Clin Nutr 69, 30–42. [DOI] [PubMed] [Google Scholar]
- Buhman KK, Furumoto EJ, Donkin SS, and Story JA (1998). Dietary psyllium increases fecal bile acid excretion, total steroid excretion and bile acid biosynthesis in rats. J Nutr 128, 1199–1203. [DOI] [PubMed] [Google Scholar]
- Bumrungpert A, Lilitchan S, Tuntipopipat S, Tirawanchai N, and Komindr S (2018). Ferulic Acid Supplementation Improves Lipid Profiles, Oxidative Stress, and Inflammatory Status in Hyperlipidemic Subjects: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers ES, Byrne CS, Morrison DJ, Murphy KG, Preston T, Tedford C, Garcia-Perez I, Fountana S, Serrano-Contreras JI, Holmes E, et al. (2019). Dietary supplementation with inulin-propionate ester or inulin improves insulin sensitivity in adults with overweight and obesity with distinct effects on the gut microbiota, plasma metabolome and systemic inflammatory responses: a randomised cross-over trial. Gut 68, 1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkan R, Fahey G, Wright WL, and McRorie J (2012). Viscous versus nonviscous soluble fiber supplements: mechanisms and evidence for fiber-specific health benefits. J Am Acad Nurse Pract 24, 476–487. [DOI] [PubMed] [Google Scholar]
- Closa-Monasterolo R, Ferre N, Castillejo-DeVillasante G, Luque V, Gispert-Llaurado M, Zaragoza-Jordana M, Theis S, and Escribano J (2017). The use of inulin-type fructans improves stool consistency in constipated children. A randomised clinical trial: pilot study. Int J Food Sci Nutr 68, 587–594. [DOI] [PubMed] [Google Scholar]
- Cockburn DW, and Koropatkin NM (2016). Polysaccharide Degradation by the Intestinal Microbiota and Its Influence on Human Health and Disease. J Mol Biol 428, 3230–3252. [DOI] [PubMed] [Google Scholar]
- Cohen-Solal C, Parquet M, Ferezou J, Serougne C, and Lutton C (1995). Effects of hyodeoxycholic acid and alpha-hyocholic acid, two 6 alpha-hydroxylated bile acids, on cholesterol and bile acid metabolism in the hamster. Biochim Biophys Acta 1257, 189–197. [DOI] [PubMed] [Google Scholar]
- Collado Yurrita L, San Mauro Martin I, Ciudad-Cabanas MJ, Calle-Puron ME, and Hernandez Cabria M (2014). Effectiveness of inulin intake on indicators of chronic constipation; a meta-analysis of controlled randomized clinical trials. Nutr Hosp 30, 244–252. [DOI] [PubMed] [Google Scholar]
- Contrepois K, Jiang L, and Snyder M (2015). Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)-Mass Spectrometry. Mol Cell Proteomics 14, 1684–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrepois K, Mahmoudi S, Ubhi BK, Papsdorf K, Hornburg D, Brunet A, and Snyder M (2018). Cross-Platform Comparison of Untargeted and Targeted Lipidomics Approaches on Aging Mouse Plasma. Sci Rep 8, 17747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussement PA (1999). Inulin and oligofructose: safe intakes and legal status. J Nutr 129, 1412S–1417S. [DOI] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, and Lionetti P (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107, 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghan P, Pourghassem Gargari B, and Asgharijafarabadi M (2013). Effects of high performance inulin supplementation on glycemic status and lipid profile in women with type 2 diabetes: a randomized, placebo-controlled clinical trial. Health Promot Perspect 3, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ciaula A, Garruti G, Lunardi Baccetto R, Molina-Molina E, Bonfrate L, Wang DQ, and Portincasa P (2017). Bile Acid Physiology. Ann Hepatol 16, s4–s14. [DOI] [PubMed] [Google Scholar]
- Einarsson K (1994). Effect of ursodeoxycholic acid on hepatic cholesterol metabolism. Scand J Gastroenterol Suppl 204, 19–23. [DOI] [PubMed] [Google Scholar]
- Everson GT, Daggy BP, McKinley C, and Story JA (1992). Effects of psyllium hydrophilic mucilloid on LDL-cholesterol and bile acid synthesis in hypercholesterolemic men. J Lipid Res 33, 1183–1192. [PubMed] [Google Scholar]
- Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, et al. (2016). Population-level analysis of gut microbiome variation. Science 352, 560–564. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco R, Clemente G, Luongo D, Lasorella G, Fiume I, Brouns F, Bornet F, Patti L, Cipriano P, Rivellese AA, et al. (2004). Effects of short-chain fructo-oligosaccharides on glucose and lipid metabolism in mild hypercholesterolaemic individuals. Clin Nutr 23, 331–340. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11, O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, et al. (2019). 2018AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139, e1082–e1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedeker D, Mermelstein RJ, and Weeks KA (1999). The thresholds of change model: an approach to analyzing stages of change data. Ann Behav Med 21, 61–70. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Eida T, Takizawa T, Tokunaga T, and Tashiro Y (1986). Effects of Fructooligosaccharides on Intestinal Flora and Human Health.
- Hoffman JD, Yanckello LM, Chlipala G, Hammond TC, McCulloch SD, Parikh I, Sun S, Morganti JM, Green SJ, and Lin AL (2019). Dietary inulin alters the gut microbiome, enhances systemic metabolism and reduces neuroinflammation in an APOE4 mouse model. PLoS One 14, e0221828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoving LR, Katiraei S, Pronk A, Heijink M, Vonk KKD, Amghar-El Bouazzaoui F, Vermeulen R, Drinkwaard L, Giera M, van Harmelen V, et al. (2018). The prebiotic inulin modulates gut microbiota but does not ameliorate atherosclerosis in hypercholesterolemic APOE*3-Leiden.CETP mice. Sci Rep 8, 16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce SA, Kamil A, Fleige L, and Gahan CGM (2019). The Cholesterol-Lowering Effect of Oats and Oat Beta Glucan: Modes of Action and Potential Role of Bile Acids and the Microbiome. Front Nutr 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koecher KJ, McKeown NM, Sawicki CM, Menon RS, and Slavin JL (2019). Effect of whole-grain consumption on changes in fecal microbiota: a review of human intervention trials. Nutr Rev 77, 487–497. [DOI] [PubMed] [Google Scholar]
- Kolida S, Tuohy K, and Gibson GR (2002). Prebiotic effects of inulin and oligofructose. Br J Nutr 87 Suppl 2, S193–197. [DOI] [PubMed] [Google Scholar]
- LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, and Ventura M (2013). Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol 24, 160–168. [DOI] [PubMed] [Google Scholar]
- Liu Y, Buil A, Collins BC, Gillet LC, Blum LC, Cheng LY, Vitek O, Mouritsen J, Lachance G, Spector TD, et al. (2015). Quantitative variability of 342 plasma proteins in a human twin population. Mol Syst Biol 11, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lordan C, Thapa D, Ross RP, and Cotter PD (2020). Potential for enriching next-generation health-promoting gut bacteria through prebiotics and other dietary components. Gut Microbes 11, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZX, Walker KZ, Muir JG, and O’Dea K (2004). Arabinoxylan fibre improves metabolic control in people with Type II diabetes. Eur J Clin Nutr 58, 621–628. [DOI] [PubMed] [Google Scholar]
- Marchesi JR, Adams DH, Fava F, Hermes GD, Hirschfield GM, Hold G, Quraishi MN, Kinross J, Smidt H, Tuohy KM, et al. (2016). The gut microbiota and host health: a new clinical frontier. Gut 65, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlett JA, and Fischer MH (2002). A poorly fermented gel from psyllium seed husk increases excreta moisture and bile acid excretion in rats. J Nutr 132, 2638–2643. [DOI] [PubMed] [Google Scholar]
- Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, and Schwudke D (2008). Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res 49, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicine I.o. (2005). Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Washington, DC: The National Academies Press; ). [DOI] [PubMed] [Google Scholar]
- Mompeo O, Spector TD, Matey Hernandez M, Le Roy C, Istas G, Le Sayec M, Mangino M, Jennings A, Rodriguez-Mateos A, Valdes AM, et al. (2020). Consumption of Stilbenes and Flavonoids is Linked to Reduced Risk of Obesity Independently of Fiber Intake. Nutrients 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D, Kumanyika SK, Lemaitre RN, Olson JL, Burke GL, and Siscovick DS (2003). Cereal, fruit, and vegetable fiber intake and the risk of cardiovascular disease in elderly individuals. JAMA 289, 1659–1666. [DOI] [PubMed] [Google Scholar]
- Myhrstad MCW, Tunsjo H, Charnock C, and Telle-Hansen VH (2020). Dietary Fiber, Gut Microbiota, and Metabolic Regulation-Current Status in Human Randomized Trials. Nutrients 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyrinck AM, Possemiers S, Druart C, Van de Wiele T, De Backer F, Cani PD, Larondelle Y, and Delzenne NM (2011). Prebiotic effects of wheat arabinoxylan related to the increase in bifidobacteria, Roseburia and Bacteroides/Prevotella in diet-induced obese mice. PLoS One 6, e20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinetti GM, Singh R, Westfall S, Herman F, Faith J, and Ho L (2018). The Role of the Gut Microbiota in the Metabolism of Polyphenols as Characterized by Gnotobiotic Mice. J Alzheimers Dis 63, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattananandecha T, Sirilun S, Duangjitcharoen Y, Sivamaruthi BS, Suwannalert P, Peerajan S, and Chaiyasut C (2016). Hydrolysed inulin alleviates the azoxymethane-induced preneoplastic aberrant crypt foci by altering selected intestinal microbiota in Sprague-Dawley rats. Pharm Biol 54, 1596–1605. [DOI] [PubMed] [Google Scholar]
- Piazzon A, Vrhovsek U, Masuero D, Mattivi F, Mandoj F, and Nardini M (2012). Antioxidant activity of phenolic acids and their metabolites: synthesis and antioxidant properties of the sulfate derivatives of ferulic and caffeic acids and of the acyl glucuronide of ferulic acid. J Agric Food Chem 60, 12312–12323. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Ouguerram K, Chretien Y, Verneau C, Eschwege E, Magot T, and Poupon R (1993). Cholesterol-lowering effect of ursodeoxycholic acid in patients with primary biliary cirrhosis. Hepatology 17, 577–582. [DOI] [PubMed] [Google Scholar]
- Quagliani D, and Felt-Gunderson P (2017). Closing America’s Fiber Intake Gap: Communication Strategies From a Food and Fiber Summit. Am J Lifestyle Med 11, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimm EB, Ascherio A, Giovannucci E, Spiegelman D, Stampfer MJ, and Willett WC (1996). Vegetable, fruit, and cereal fiber intake and risk of coronary heart disease among men. JAMA 275, 447–451. [DOI] [PubMed] [Google Scholar]
- Rost HL, Liu Y, D’Agostino G, Zanella M, Navarro P, Rosenberger G, Collins BC, Gillet L, Testa G, Malmstrom L, et al. (2016). TRIC: an automated alignment strategy for reproducible protein quantification in targeted proteomics. Nat Methods 13, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, and Mazmanian SK (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9, 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpagaporn P, Reuhs BL, Kaur A, Patterson JA, Keshavarzian A, and Hamaker BR (2015). Structural features of soluble cereal arabinoxylan fibers associated with a slow rate of in vitro fermentation by human fecal microbiota. Carbohydr Polym 130, 191–197. [DOI] [PubMed] [Google Scholar]
- Ryan PM, Ross RP, Fitzgerald GF, Caplice NM, and Stanton C (2015). Functional food addressing heart health: do we have to target the gut microbiota? Curr Opin Clin Nutr Metab Care 18, 566–571. [DOI] [PubMed] [Google Scholar]
- Schaafsma G, and Slavin JL (2015). Significance of Inulin Fructans in the Human Diet. Comprehensive Reviews in Food Science and Food Safety 14, 37–47. [DOI] [PubMed] [Google Scholar]
- Schneider BB, Covey TR, Coy SL, Krylov EV, and Nazarov EG (2010). Planar differential mobility spectrometer as a pre-filter for atmospheric pressure ionization mass spectrometry. Int J Mass Spectrom 298, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, Turroni S, Biagi E, Peano C, Severgnini M, et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat Commun 5, 3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KP, Duncan SH, and Flint HJ (2008). Dietary fibre and the gut microbiota. Nutrition Bulletin 33, 201–211. [Google Scholar]
- Sekirov I, Russell SL, Antunes LC, and Finlay BB (2010). Gut microbiota in health and disease. Physiol Rev 90, 859–904. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, Lapek JD Jr., Zhang L, Wang WB, Hao S, et al. (2018). Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell 175, 679–694 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal AK, Cohen BI, Mosbach EH, Une M, Stenger RJ, McSherry CK, May-Donath P, and Palaia T (1984). Prevention of cholesterol-induced gallstones by hyodeoxycholic acid in the prairie dog. J Lipid Res 25, 539–549. [PubMed] [Google Scholar]
- Slavin J (2013). Fiber and prebiotics: mechanisms and health benefits. Nutrients 5, 1417–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin JL (2005). Dietary fiber and body weight. Nutrition 21, 411–418. [DOI] [PubMed] [Google Scholar]
- Soliman GA (2019). Dietary Fiber, Atherosclerosis, and Cardiovascular Disease. Nutrients 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedlund J, Sjodin I, and Dotevall G (1988). GSRS--a clinical rating scale for gastrointestinal symptoms in patients with irritable bowel syndrome and peptic ulcer disease. Dig Dis Sci 33, 129–134. [DOI] [PubMed] [Google Scholar]
- Swanson KS, de Vos WM, Martens EC, Gilbert JA, Menon RS, Soto-Vaca A, Hautvast J, Meyer PD, Borewicz K, Vaughan EE, et al. (2020). Effect of fructans, prebiotics and fibres on the human gut microbiome assessed by 16S rRNA-based approaches: a review. Benef Microbes 11, 101–129. [DOI] [PubMed] [Google Scholar]
- Teo G, Kim S, Tsou CC, Collins B, Gingras AC, Nesvizhskii AI, and Choi H (2015). mapDIA: Preprocessing and statistical analysis of quantitative proteomics data from data independent acquisition mass spectrometry. J Proteomics 129, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong LT, Zhong K, Liu L, Qiu J, Guo L, Zhou X, Cao L, and Zhou S (2014). Effects of dietary wheat bran arabinoxylans on cholesterol metabolism of hypercholesterolemic hamsters. Carbohydr Polym 112, 1–5. [DOI] [PubMed] [Google Scholar]
- Trumbo P, Schlicker S, Yates AA, Poos M, Food, and Nutrition Board of the Institute of Medicine, T.N.A. (2002). Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein and amino acids. J Am Diet Assoc 102, 1621–1630. [DOI] [PubMed] [Google Scholar]
- Tuncil YE, Thakkar RD, Arioglu-Tuncil S, Hamaker BR, and Lindemann SR (2020). Subtle Variations in Dietary-Fiber Fine Structure Differentially Influence the Composition and Metabolic Function of Gut Microbiota. mSphere 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley SD, Daggy BP, and Dietschy JM (1991). Cholesterol-lowering action of psyllium mucilloid in the hamster: sites and possible mechanisms of action. Metabolism 40, 1063–1073. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Venkataraman A, Sieber JR, Schmidt AW, Waldron C, Theis KR, and Schmidt TM (2016). Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. (2011). Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J 5, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Pan Y, Zhou H, Wang L, Chen X, Song G, Liu J, and Li A (2018). Ferulic acid suppresses obesity and obesity-related metabolic syndromes in high fat diet-induced obese C57BL/6J mice. Food and Agricultural Immunology 29, 1116–1125. [Google Scholar]
- Williams BA, Grant LJ, Gidley MJ, and Mikkelsen D (2017). Gut Fermentation of Dietary Fibres: Physico-Chemistry of Plant Cell Walls and Implications for Health. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TA, Romano C, Liang J, and Nicolosi RJ (1998). The hypocholesterolemic and antiatherogenic effects of Cholazol H, a chemically functionalized insoluble fiber with bile acid sequestrant properties in hamsters. Metabolism 47, 959–964. [DOI] [PubMed] [Google Scholar]
- Winham DM, and Hutchins AM (2011). Perceptions of flatulence from bean consumption among adults in 3 feeding studies. Nutr J 10, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin ZN, Wu WJ, Sun CZ, Liu HF, Chen WB, Zhan QP, Lei ZG, Xin X, Ma JJ, Yao K, et al. (2019). Antioxidant and Anti-inflammatory Capacity of Ferulic Acid Released from Wheat Bran by Solid-state Fermentation of Aspergillus niger. Biomed Environ Sci 32, 11–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A) Clinical data for the entire cohort. B) Coefficients of variation at baseline for every analyte in the cohort. C) Gut microbiome taxa data generated using MetaPhlAn with associated metadata. D) Gastrointestinal symptoms during fiber supplementation. E) GI symptoms correlated with lipids and bile acids in AX. Related to Figures 2 and 3.
Table S2. A) GI symptoms correlated with metagenome in AX. S2B) GI symptoms correlated with metagenome in LCI. C) Diet data in responders and non-responders during fiber supplementation. D) Analytes associated with their respective cluster in C-means clustering during AX supplementation. Class I is cluster 11, Class II is cluster 10, and Class III is cluster 16. E) Analytes associated with their respective cluster from C-means clustering during LCI supplementation. Class I is cluster 11, Class II is cluster 7, and Class III is cluster 9. Related to Figures 3 through 5.
Table S3. A) Analytes associated with their respective cluster in C-means clustering during Mix supplementation. Class I is cluster 13 and Class II is cluster 2. B) Correlation within Class I analytes in AX. Only those with FDR<0.05 are included. C) Metabolomics data with associated metadata during fiber supplementation. D) Lipid data with associated metadata during fiber supplementation. E) Lipids that follow the reverse dose-response to fiber, and linear regression p-value/F-statistic. Related to Figures 3 and 5.
Table S4. A) Correlations between lipids, bile acids, and clinical cholesterol. Only correlation with FDR < 0.05 are listed. B) MetaPhlAn analytes in LDL responders vs non-responders from AX data, with Wilcoxon rank sum and FDR adjusted p-values. C) Pathway abundance analytes in LDL responders vs non-responders from the AX data, with Wilcoxon rank sum and FDR adjusted p-values. D) Gene family analytes in LDL responders vs non-responders from the AX data, with Wilcoxon rank sum and FDR adjusted p-values. E) IPA analysis for LXR and FXR pathways. Related to Figures 3 and 4.
Table S5. A) Clinical analytes in LDL responders vs non-responders in the AX data, with Wilcoxon rank sum and FDR adjusted p-values. B) Diet analytes in LDL responders vs non-responders in the AX data. C) MetaPhlAn analytes in ALT responders vs non-responders in the LCI cohort, with Wilcoxon rank sum and FDR adjusted p-values. D) Gene family analytes in ALT responders vs non-responders in the LCI cohort, with Wilcoxon rank sum and FDR adjusted p-values. E) Correlations between analytes in LCI Class I C-means cluster. Only correlations with FDR<0.05 are present. Related to Figures 4 and 5.
Table S6. A) C-means clustering for the cytokines during Mix supplementation. Class I is cluster 3 and Class II is cluster 6. B) Personalized C-means clustering for participant ZOZOW1T. C) Personalized C-means clustering for participant ZQFL1P3. D) Metadata associated with metabolomics. Related to Figures 2, 3 and 5.
Data Availability Statement
The data sets and code supporting the current study have been deposited in Metabolomics Workbench #ST002112. They will be made available on publication and are available from the corresponding author on request.
All original code has been deposited on GitHub (https://github.com/slancast) and is available up on request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.