

Abstract
Salivary gland neoplasms are uncommon, and most exhibit epithelial differentiation. Mesenchymal neoplasms of the salivary gland are rare, and the incidence ranges from 1.9% to 5%. The aim of this study is to identify the types and clinical-pathological features of mesenchymal salivary neoplasm and review their differential diagnosis. A retrospective search for mesenchymal neoplasms of salivary glands from our institution's pathology archives from the 2004–2021 period and consultation files of one of the authors (AER) was performed. The clinical data were obtained from available medical records, and the histological slides and ancillary studies were retrieved and reviewed. We identified a total of 68 cases that form the study cohort. Thirty-five patients were male, and thirty-three patients were female, with a mean age of 48 years (range, 7 months–79 years), and the male to female ratio was 1:.94. Sixty-three (92.6%) of sixty-eight tumors were benign and included: 38 (56%) lipomas, 9 (13%) hemangiomas, 7 (10.3%) schwannomas, 3 (4.4%) neurofibromas, 3 (4.4%) lymphangioma, 2 (3%) solitary fibrous tumors, 1 (1.5%) myofibroma. Five of sixty-eight (7.4%) were malignant and included: 3 (4.4%) Adamantinoma-like Ewing sarcomas, 1 (1.5%) malignant peripheral nerve sheath tumor (MPNST), and 1 (1.5%) malignant solitary fibrous tumor. The involved sites included: parotid (55), submandibular gland (5), parapharyngeal space (5), buccal mucosa minor salivary gland (2), and sublingual gland (1). Sixty-seven patients underwent surgical resection. One patient with lymphangioma manifested a recurrence/persistence a week post-surgery. One patient with a parotid hemangioma developed post-operative numbness, and another patient developed chronic postauricular pain after surgery. Two patients with MPNST and one patient with adamantinoma-like Ewing sarcoma underwent neoadjuvant chemoradiation and were disease-free after treatment. The remaining 37 patients with available follow-up ranging from 7 days to 96 months (mean, 18 months) had a favorable outcome and were disease-free after treatment. Mesenchymal neoplasms of salivary gland are rare; most are benign and demonstrate adipocytic, endothelial, and schwannian differentiation; awareness of their development is important for adequate diagnosis. The mainstay of treatment is surgical excision, with the extent determined by tumor type. Adjuvant therapy is reserved for high-grade sarcomas and may be given in a neoadjuvant or adjuvant setting.
Keywords: Salivary gland, Lipoma, Hemangioma, Lymphangioma, Solitary fibrous tumor, Adamantinoma like Ewing sarcoma, Malignant peripheral nerve sheath tumor
Introduction
Salivary gland neoplasms are uncommon. They account for approximately 3% of head and neck tumors, and approximately 95% are epithelial in phenotype [1]. Mesenchymal tumors of the salivary gland are unusual, and they represent 1.9%–5% of salivary gland neoplasms [1–4]. They may arise from the structures that compose the salivary gland, traverse it, extend from a tumor in neighboring tissues, or maybe a manifestation of metastatic disease. The vast majority are benign, and the most common are lipomas, hemangiomas, and lymphangiomas [2, 3, 5]. However, a wide variety of soft tissue tumors arise in these structures. The aim of this study is to identify the spectrum and clinical-pathological features of mesenchymal salivary gland neoplasms and review their differential diagnosis.
Materials and methods
With prior approval from our Institutional Review Board, a retrospective search for mesenchymal tumors of salivary glands from the pathology archives of the University of Miami and Jackson Memorial Hospital from 2004 to 2021 and the consultation files of one of the authors (AER) was performed. Metastasis and tumors that involved the salivary gland by direct extension were excluded from our cohort. The clinical data were obtained from available medical records, and the histological slides and ancillary studies were retrieved and reviewed.
Results
We identified a total of 68 cases, and they form the study cohort. Six cases were obtained from the consultation service of AER, and the remaining cases were retrieved from the department's surgical pathology archives. Thirty-five patients were male, and thirty-three patients were female (M: F = 1:0.94), with a mean age of 48 years (range: 7 months -79 years). The tumors included 38 (56%) lipomas, 9 (13%) hemangiomas, 7 (10.3%) schwannomas, 3 (4.4%) neurofibromas, 3 (4.4%) lymphangioma, 2 (3%) solitary fibrous tumors, 1 (1.5%) myofibroma, 3 (4.4%) Adamantinoma-like Ewing sarcomas, 1 (1.5%) malignant peripheral nerve sheath tumor (MPNST), and 1 (1.5%) malignant solitary fibrous tumor. The involved sites included: parotid gland 55 (81%), submandibular gland 5 (7.3%), parapharyngeal space 5 (7.3%), minor salivary gland 2 (3%), and sublingual gland 1 (1.4%). The clinicopathologic data are summarized in Table 1.
Table 1.
Clinicopathologic features of mesenchymal tumors of the salivary gland
| Case | Gender | Age (years) | Location | Diagnosis | Size (cm) | Follow up (months) |
|---|---|---|---|---|---|---|
| 1 | M | 18 | Right parotid | Conventional lipoma | 3.0 | 84, NED |
| 2 | M | 62 | Right parotid | Spindle cell lipoma | 4.0 | 5, NED |
| 3 | M | 55 | Parapharyngeal space mass | Conventional lipoma | 10.0 | 26, NED |
| 4 | F | 42 | Right parotid | Conventional lipoma | 3.0 | 28, NED |
| 5 | F | 66 | Left parotid | Conventional lipoma | 6.5 | 7, NED |
| 6 | F | 48 | Left parotid | Conventional lipoma | 2.0 | 11, NED |
| 7 | F | 65 | Right parotid | Conventional lipoma | 4.0 | 30, NED |
| 8 | M | 55 | Right parapharyngeal space | Conventional lipoma | 3.0 | 4, NED |
| 9 | F | 47 | Deep parotid | Conventional lipoma | 4.5 | 31, NED |
| 10 | M | 26 | Left parotid | Conventional lipoma | 5.4 | 0.23, NED |
| 11 | M | 63 | Right parotid | Fibrolipoma | 4.0 | 14, NED |
| 12 | M | 44 | Right parotid | Conventional lipoma | 1.2 | 7, NED |
| 13 | M | 56 | Left parotid | Conventional lipoma | 3.5 | 2, NED |
| 14 | F | 68 | Right parotid | Conventional lipoma | 3.5 | 11, NED |
| 15 | F | 56 | Right parotid | Conventional lipoma | 6.0 | 7, NED |
| 16 | M | 70 | Right parotid | Conventional lipoma | 6.5 | 2, NED |
| 17 | M | 64 | left parotid | Conventional lipoma | 3.5 | 12, NED |
| 18 | F | 36 | Right parotid | Conventional lipoma | 6.5 | 6, NED |
| 19 | F | 19 | Left parapharyngeal space | Fibrolipoma | 7.9 | 7, NED |
| 20 | M | 64 | Parotid | Conventional lipoma | 5.2 | 0.46, NED |
| 21 | F | 46 | Right parotid | Conventional lipoma | 4.0 | 1, NED |
| 22 | M | 55 | Right parotid | Conventional lipoma | 3.0 | 0.26, NED |
| 23 | M | 65 | Left parotid | Fibrolipoma | 1.4 | NA |
| 24 | M | 48 | Right parotid | Conventional lipoma | 3.0 | NA |
| 25 | F | 51 | Left anterior parotid | Spindle cell lipoma | 1.2 | NA |
| 26 | F | 51 | Right parotid | Conventional lipoma | 2.0 | 96, NED |
| 27 | M | 63 | Left parotid | Conventional lipoma | 5.7 | 0.03, NED |
| 28 | M | 63 | Lip minor salivary gland | Pleomorphic lipoma (AER consult) | 1.0 | NA |
| 29 | M | 64 | Right parotid | Conventional lipoma | NA | NA |
| 30 | M | 59 | Right parotid | Conventional lipoma | 2.0 | NA |
| 31 | F | 67 | Left submandibular gland | Conventional lipoma | 8.9 | 25, NED |
| 32 | M | 59 | Right parotid | Conventional lipoma | 3.0 | 96, NED |
| 33 | M | 50 | Right parotid | Conventional lipoma | 4.0 | NA |
| 34 | M | 46 | Left parotid | Conventional lipoma | 3.0 | 11, NED |
| 35 | M | 53 | Left parotid | Conventional lipoma | 3.5 | 0.63, NED |
| 36 | M | 43 | Right parotid | Conventional lipoma | 6.0 | 0.1, NED |
| 37 | M | 55 | Left parotid | Conventional lipoma (AER consult) | NA | NA |
| 38 | M | 55 | Left tail of parotid | Spindle cell lipoma | 2.0 | NA |
| 39 | M | 53 | Right Parotid | Schwannoma | 6.4 | 32, NED |
| 40 | F | 40 | Parapharyngeal space mass | Plexiform neurofibroma | 3.4 | 6, NED |
| 41 | M | 54 | Right parotid | Schwannoma | 0.9 | 21, NED |
| 42 | F | 45 | Parapharyngeal space mass | Schwannoma | NA | NA |
| 43 | F | 15 | Left parapharyngeal space | MPNST | 6.7 | 9, NED |
| 44 | F | 43 | Right parotid | Schwannoma | NA | NA |
| 45 | F | 29 | Left neck mass within parotid sheath | Schwannoma | 2.5 | NA |
| 46 | F | 53 | Parotid mass | Schwannoma | 3.9 | NA |
| 47 | F | 36 | Right Parotid | Schwannoma | 8 | 10, NED |
| 48 | M | 28 | Left parotid | Neurofibroma (AER consult) | NA | NA |
| 49 | F | 30 | Left parotid | Neurofibroma | 2.6 | NA |
| 50 | F | 40 | Left parotid | Adamantinoma-like Ewing Sarcoma | 2.4 | 96, NED |
| 51 | F | 21 | Submandibular gland | Adamantinoma-like Ewing Sarcoma | 5 | NA |
| 52 | F | 79 | Left parotid | Adamantinoma-like Ewing Sarcoma | 2 | 16, NED |
| 53 | M | 59 | Tail of parotid | Hemangioma | 2.9 | 1, NED |
| 54 | F | 64 | Right parotid | Hemangioma | 3 | 11, NED |
| 55 | F | 46 | Right parotid | Venous hemangioma | 1.6 | 2, NED |
| 56 | F | 66 | Right parotid | Cavernous hemangioma | NA | 20, NED |
| 57 | M | 14 | Left sublingual gland | Cavernous and capillary hemangioma | 3.5 | NA |
| 58 | F | 41 | Left submandibular gland | Hemangioma | 1.5 | 36, NED |
| 59 | F | 63 | Right parotid | Venous hemangioma | 2.5 | NA |
| 60 | M | 31 | Submandibular gland | Capillary hemangioma | NA | NA |
| 61 | M | 70 | Right parotid | Hemangioma | 3 | 9, NED |
| 62 | F | 12 | Submandibular gland | Lymphangioma | 1 | NA |
| 63 | M | 1 | Right parotid | Lymphangioma | 5.2 | 12, NED |
| 64 | M | 60 | Buccal mucosa (minor salivary gland) | Solitary fibrous tumor (AER consult) | 1.5 | NA |
| 65 | F | 57 | Left parotid | Solitary fibrous tumor | 3.2 | NA |
| 66 | F | 72 | Left parotid | Malignant solitary fibrous tumor (AER consult) | 7.0 | NA |
| 67 | M | 7 months | Left parotid | Myofibroma (AER consult) | 3.8 | NA |
| 68 | F | 37 | Right tail of parotid | Lymphangioma | 3.0 | 2, NED |
NA Not available, MPNST Malignant peripheral nerve sheath tumor, NED no evidence of disease
The 38 lipomas included 31 (81.6%) of the conventional type, 3 (7.9%) fibrolipomas, and 4 (10.5%) spindle cell/pleomorphic lipomas. The patients typically presented with an asymptomatic, slow-growing mass. Twenty-five patients were male, and thirteen were female; their mean age was 53 years, with a range of 18–70 years. The most common site of origin was the parotid gland/parapharyngeal space—36 (94.7%), followed by the submandibular gland—1 (2.7%) and lip/minor salivary gland 1 (2.7%) (Fig. 1A). Tumor size known in 36 cases ranged from 1.2 cm to 10 cm with an average of 4.1 cm. Twelve patients had pre-operative fine-needle aspiration (FNA). Three cases contained fragments of mature adipose tissue admixed with non-neoplastic salivary gland elements, suggesting the possibility of lipoma. Nine cases were considered non-diagnostic: three contained non-neoplastic salivary gland elements only, and six were acellular. All tumors were well circumscribed and encapsulated with a yellow-tan lobulated cut surface. Histologically, conventional lipomas were circumscribed and composed of lobules of adipocytes separated by thin fibrous septa (Fig. 1B). The fibrolipomas contained prominent fibrous septa with hypocellular bundles of collagen scattered throughout the mass. The spindle cell/pleomorphic lipomas were composed of adipocytes with various amounts of bland spindle cells, ropy collagen, mast cells, and a prominent myxoid background. All patients had surgical resection of the tumor; none of the 29 patients with available follow-up information (range 1 day- 96 months, average 16 months) developed clinical evidence of recurrence.
Fig. 1.

A Reformatted Axial CT scan with contrast shows low attenuation intraparotid fatty tumor measuring 3.6 × 3.2 × 2.3 cm. B Intraparotid lipoma: mature adipocytes separated from major salivary gland tissue by fibrous septae (HES × 10)
The 9 benign vascular tumors included 6 (67%) venous hemangiomas, 2 (22%) cavernous hemangiomas (one was mixed cavernous and capillary), and 1 (11%) capillary hemangioma. Five patients were female, and 4 were male; their mean age was 50 years, with a range of 14–70 years. The most common site of origin was the parotid gland 6 (67%), followed by the submandibular gland 2 (22%) and the sublingual gland 1 (11%). The patients presented with swelling, local sensitivity, and tenderness. The tumor size known in 6 cases was 1.5 cm–3.5 cm, average of 2.5 cm. Five patients had pre-operative FNA, and all cases were considered non-diagnostic due to absent cellularity, presence of non-neoplastic salivary gland elements, and blood only. Histologically venous hemangiomas were composed of aggregates of small and large venous channels lined by flat endothelial cells with irregular, smooth muscle walls. The cavernous and capillary hemangiomas were composed of compact aggregates of dilated, thin-walled blood vessels lined by bland endothelium, and the walls lacked smooth muscle. Follow-ups were available on 6 patients and ranged from 1–36 months, average 13 months. One patient developed numbness in the area post-surgery, and one patient developed chronic postauricular pain after surgery. The numbness improved over time, and the patient otherwise did well. None of the patients developed recurrence.
Three cases of lymphangioma were identified. Two patients had pre-operative FNA. One was interpreted as a lymphoepithelial cyst, and the other contained histiocytes and blood only. One lymphangioma occurred in a 1-year-old male with a 5.3 cm right parotid mass that was resected by parotidectomy. A week after the surgery, the patient developed a recollection of fluid in the surgical bed. The fluid was drained on three occasions, and one month later, the patient was taken to the operative room to remove a possible seroma. During surgery, a cystic structure overlying the entire superficial parotidectomy defect was identified and resected, revealing recurrent/persistent lymphangioma. After 12 months of follow-up, there was no local recurrence. The second lymphangioma occurred in a 37-year female that presented with right neck pain and tail of parotid mass. The patient underwent right nerve-sparing parotidectomy, and after 2 months of follow-up, the patient is doing well without evidence of local recurrence. The third lymphangioma was a 1.0 cm mass in the submandibular gland in a 12-year-old female. It was resected, and no follow-up information is available. Histologically, the lymphangiomas were composed of variably sized anastomosing sinusoidal-like spaces lined by small endothelial cells, and small collections of lymphocytes were within the lumina admixed with proteinaceous lymphatic fluid and erythrocytes.
The 7 schwannomas affected 5 females and 2 males; their mean age was 45 years, with a range of 29–54 years. The most common site of origin was the parotid 6 (86%), followed by the parapharyngeal space 1 (14%). Five patients presented with symptoms including headache (1), facial paralysis (1), and a palpable mass (3). Three patients had pre-operative FNA. Two cases were interpreted as benign spindle cell neoplasms, and one case was acellular. Six patients had surgical resection; information regarding one patient was not available. The tumor size known in 5 cases was 1.0 cm—8.0 cm, average 4.3 cm. Histologically the schwannomas were encapsulated, contained Antoni A and B regions, and were composed of spindle cells with elongate wavy nuclei arranged in short interconnecting fascicles enmeshed in a variably myxocollagenous stroma within which were dilated blood vessels that had focally hyalinized walls. Follow-up available on three patients ranged from 10 to 32 months, average 21 months. None developed local recurrence. The 3 neurofibromas developed in a 40-year-old female with a history of neurofibromatosis type 1, a 28-year-old healthy male, and a 30-year-old female with prior medical history of papillary thyroid carcinoma. The tumor in the patient with neurofibromatosis type 1 was 3.4 cm, located in a parapharyngeal space, and was a plexiform neurofibroma. The tumor was excised, and after six months of follow-up, there has been no recurrence. The tumor present in the male patient was in the left parotid, and a biopsy revealed a neurofibroma; no follow-up information is available. The tumor in the 30-year-old female was 2.6 cm, arose from the facial nerve traversing through the left parotid gland, and biopsy revealed a neurofibroma. Immunohistochemistry showed that the tumor cells were positive for S100.
One case of malignant peripheral nerve sheath tumor was identified. The patient was a 15-year-old female with a history of neurofibromatosis type 1 status post resection of a left parapharyngeal plexiform neurofibroma 5 years previously. She presented with a recurrent 6.7 cm mass in the left parapharyngeal space in the area of prior surgery (Fig. 2A). The recurrent tumor arose in a plexiform neurofibroma (Fig. 2B, C), and immunohistochemistry showed that the tumor cells were positive for CD34 and negative for Sox10, S100, desmin, myogenin, MYOD1, and STAT6. Expression of H3k27me3 was lost in a minority of tumor cells. Rereview of the original mass revealed the presence of sarcoma that was not appreciated at the time of the original diagnosis. The patient underwent adjuvant chemoradiation, and after 9 months follow-up, she is doing well with no evidence of disease.
Fig. 2.

A Reformatted Axial T1 MRI shows an avidly enhancing left infratemporal fossa mass. B Plexiform neurofibroma: multiple expanded nerve bundles within major salivary gland (HES × 10). C Atypical neurofibroma-MPNST: Loosely arranged spindle cells with varying degrees of cellularity and nuclear atypia adjacent to hypercellular fascicles of hyperchromatic spindled cells with nuclear pleomorphism (HES × 20)
The 3 cases of adamantinoma-like Ewing sarcoma occurred in females 21, 40, and 79 years old. The tumors presented as an enlarging painless face/neck mass. Two tumors arose in the parotid gland and 1 in the submandibular gland. The tumor size was 2.0 cm–5.0 cm, average 3.1 cm. All patients underwent surgical resection. Histologically, the tumors were centered within the salivary parenchyma and were composed of sheets of small round blue round cells that had round nuclei and fine chromatin. Two cases exhibited scattered squamous cell nests with keratinization, including squamous pearls (Fig. 3A). Immunohistochemistry showed that synaptophysin was positive in all cases; two cases tested were positive for p40 and CD99, and chromogranin was negative in all cases. Polymerase chain reaction performed in one case demonstrated EWSR1-Fli1 fusion, and the other two cases had EWSR1 rearrangement by fluorescence in-situ hybridization (Fig. 3B). Follow-up information was available for two patients. One patient was treated with surgery and adjuvant chemoradiation, and no local recurrence or distant metastases were observed after 96 months. The other patient was treated with surgery only and is currently under close surveillance without evidence of disease after 16 months. No follow-up information was available for the other patient.
Fig. 3.

A Adamantinoma like Ewing Sarcoma: sheets of small round blue round cells with area of focal abrupt keratinization (HES × 40). B FISH for EWSR1 rearrangement, dual-color break-apart probe for EWSR1 gene shows the separation of green and red signals, consistent with positive EWSR1 rearrangement
The three cases of solitary fibrous tumors included 2 benign and 1 malignant variant. Two tumors arose in the parotid and 1 in the minor salivary gland of the buccal mucosa. Both benign tumors occurred in males, and their ages were 57 and 60 years. One patient with a benign solitary fibrous tumor had pre-operative FNA that was interpreted as spindle cell proliferation. The tumors were 3.2 cm and 1.5 cm, respectively, and composed of short uniform bland spindle to ovoid cells with scant cytoplasm and small, uniform, vesicular nuclei arranged in a patternless pattern; the stroma was collagenous with wire-like collagen fibers and harbored a prominent staghorn vasculature. The malignant solitary fibrous tumor occurred in a 72-year-old female with a 5-year history of a slowly enlarging left parotid mass that resulted in facial paralysis. The 7.0 cm resected tumor was a lobulated mass with a yellow-white cut surface. The tumor was histologically cellular and composed of pleomorphic spindle cells arranged in fascicles with a staghorn vasculature. Necrosis and increased mitotic activity (16 mitoses per 20 HPF) were present. Immunohistochemically, the tumor cells were positive for CD34, actin and were negative for desmin, keratin, EMA, and p63. None of the patients have follow-up information available.
The myofibroma occurred in a 7-month-old boy with a 3.8 cm left parotid mass. Histologically, the hypercellular tumor was composed of plump spindle cells with eosinophilic cytoplasm arranged in short fascicles and whorls with a staghorn vascular tree (Fig. 4). No necrosis or increased mitotic activity was identified. The tumor was resected with positive margins, and no follow-up information is available.
Fig. 4.
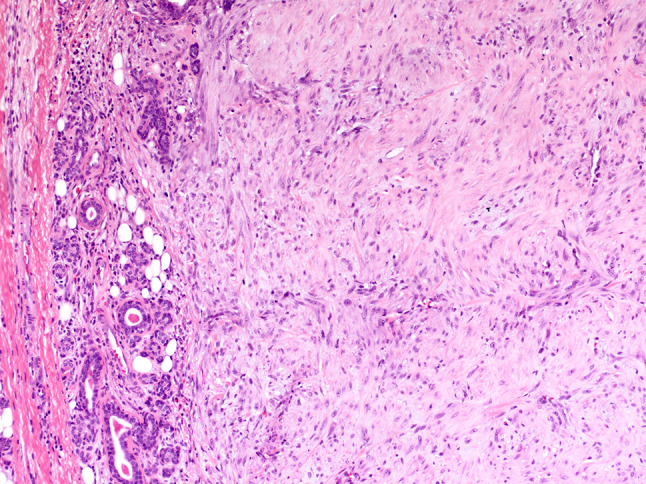
Myofibroma: hypercellular tumor composed of plump spindle cells with eosinophilic cytoplasm arranged in short fascicles and whorls involving salivary gland parenchyma (HES × 20)
Discussion
Mesenchymal tumors of the salivary glands are uncommon and constitute a heterogeneous group of lesions. The majority are benign, arise in the major salivary glands, exhibit adipocytic, endothelial, or schwannian differentiation, and have a good prognosis. The differential diagnosis is broad and ancillary studies may be necessary to render a precise diagnosis (Table 2). Fine needle aspiration cytology (FNAC) can be used as a diagnostic tool in the pre-operative setting of patients, although inadequate specimens are not uncommon [6]. This procedure can distinguish neoplastic versus non-neoplastic salivary gland lesions, and in some cases, a definitive diagnosis can be rendered and help guide appropriate patient management.
Table 2.
Ancillary studies in mesenchymal tumors of salivary gland and select differential diagnosis
| Tumor | Immunohistochemistry | FISH | NGS |
|---|---|---|---|
| Conventional lipoma | HMGA2 rearrangement | ||
| Spindle cell lipoma | Loss of nuclear Rb expression | ||
| Atypical lipomatous tumor | Overexpression of nuclear MDM2 and CDK4 | MDM2 amplification | |
| Lymphangioma | D2-40 ( +) and CD-31 ( +) | ||
| Hemangioma | CD-31 ( +), ERG ( +) and D2-40 (-) | ||
| Schwannoma | S100 ( +) and Sox-10 ( +) | ||
| Neurofibroma | Mixture of S100( +), CD34( +), and EMA( +) cells | ||
| Perineurioma | EMA( +), claudin-1 ( +), GLUT-1( +), S100 (-) and Sox-10 (-) | ||
| MPNST | Focal S100 protein ( +) and SOX10( +) |
NF1 mutation CDKN2A mutation TP53 mutation |
|
| Synovial sarcoma |
EMA ( +) Nuclear TLE1( +) Nuclear SS18-SSX ( +) |
SS18 rearrangement | SS18-SSX gene fusion |
| ALES | Cytokeratin ( +), p40 ( +), CD99 ( +), and NKX 2.2 ( +) | EWSR1 rearrangement | EWSR1-Fli-1 gene fusion |
| SFT | CD34( +) and nuclear STAT6 ( +) | NAB2-STAT6 gene fusion | |
| Myofibroma | SMA( +) | ||
| Fibromatosis | Nuclear β-catenin ( +) | Somatic mutations in β-catenin CTNNB1 gene on 3p21 (sporadic tumors) | |
| Nodular fasciitis | SMA ( +), nuclear β-catenin (-) | USP6 gene rearrangement | USP6-MYH9 gene fusion |
FISH Fluorescent in situ hybridization, NGS Next-generation sequencing, MPNST malignant peripheral nerve sheath tumor, ALES Adamantinoma-like Ewing sarcoma, SFT solitary fibrous tumor
Lipoma is a benign neoplasm of white adipocytes. It is the most common soft tissue neoplasm in adults and is usually diagnosed in the fifth to sixth decade of life [7, 8]. It has a predilection for the trunk, extremities, and head/neck [9]. Approximately 13% of lipomas occur in the head and neck region, and the posterior neck is the most common site [10–12]. Lipoma rarely arises in the salivary gland, and when it does, most develop in the parotid gland, where it accounts for 0.6–4.4% of benign salivary gland neoplasms [7, 13]. Lipoma of the parotid gland presents as a slow-growing, painless mass, most commonly located in the superficial lobe [4, 14, 15]. It can be classified based on location (intraparotid or periparotid) and histologic subtype [15]. Magnetic resonance imaging is the preferred imaging modality, given that it can accurately identify adipocytic neoplasms and determine the size, location, and anatomic extent [16]. In our series, lipoma was the most common mesenchymal tumor of the salivary gland and was more common in males than females (M:F ratio-1.9:1). All patients underwent surgical resection with no clinical evidence of recurrences. The important differential diagnosis for the conventional and spindle cell/ pleomorphic variants is atypical lipomatous tumor which can be identified by the presence of thick fibrous bands and the predominance of mononuclear cells with large hyperchromatic nuclei. For problematic cases, chromogenic in-situ hybridization or fluorescence hybridization is-situ for amplification of MDM2 can help distinguish between these neoplasms.
Hemangiomas are common in the head and neck but infrequently develop in salivary glands. Over 90% of salivary gland hemangiomas involve the parotid gland, where they account for 0.4%–0.6% of all parotid tumors [17]. The remainder usually occur in the submandibular gland [18, 19]. The tumor usually presents in the parotid as a swelling involving the superficial lobe, and less commonly, the deep lobe. Treatment is surgical removal, with facial nerve preservation when possible. The recurrence rate following surgery is reported to be low (4%) [19–23]. Other treatment modalities include watchful observation, endovascular sclerotherapy, and medical management (systemic corticosteroid, vincristine, and beta-blockers). Many patients require a combination of therapies to achieve resolution [24]. If observation is chosen, it is important to know that capillary hemangiomas of the parotid gland in the pediatric population tend to involute; however, cavernous hemangiomas in adults may not involute [21, 23]. The differential diagnosis includes lymphangioma and angiosarcoma. Lymphangiomas are characterized by variably sized inter-anastomosing vascular spaces lined by bland-appearing endothelial cells that often contain abundant lymph, scattered lymphocytes, and erythrocytes [25]. Angiosarcomas are composed of a proliferation of malignant epithelioid or spindle endothelial cells that line vascular lumens and have an infiltrative growth pattern [26].
Lymphangiomas are composed of lymphatic vessels of variable caliber; up to 90% occur in the head and neck region and are most commonly observed in the pediatric population [27–30]. Among those found in the head and neck region, the posterior triangle of the neck is most frequently involved, with occasional cases involving the salivary glands (parotid and submandibular gland) [31]. They usually present as a painless, progressively enlarging mass. These tumors do not involute, and the treatment of choice includes parotidectomy with preservation of the facial nerve [32]. Other treatment forms are observation, sclerotherapy, and medical management (systemic sildenafil and sirolimus) [27]. The recurrence rate following surgery can be high (10%–38%) and may be due to incomplete tumor removal, as seen in one of our cases [33, 34]. The differential diagnosis includes hemangioma, which is composed of aggregates of round to oval thin-walled blood-filled capillaries.
Approximately 25% of schwannomas originate in the neural structures of the head and neck region, and up to 9% occur in the extratemporal part of the facial nerve and can mimic a primary parotid tumor [35, 36]. Parotid schwannoma usually presents as an asymptomatic slow-growing firm mass that arises from the intraparotid segment of the facial nerve [37]. The current treatment of choice is surgical excision with facial nerve preservation when possible; the recurrence rate is reported to be low (2%) [38, 39]. Observation or external beam irradiation can be considered as alternatives to surgical resection so that facial nerve function can be preserved [40]. The differential diagnosis includes neurofibroma, solitary fibrous tumor, and schwannoma-like pleomorphic adenoma Neurofibromas usually lack encapsulation and do not contain verocay bodies [41]. Solitary fibrous tumors have a more collagenous stroma and a staghorn vascular tree. Schwannoma-like pleomorphic adenoma has predominant nuclear palisading reminiscent of Antoni A areas with verocay bodies and can show cystic degeneration and hemorrhage [42]. In problematic cases, immunohistochemistry is helpful because schwannoma is diffusely positive for S100 and SOX 10, unlike neurofibroma (which has a background population of negative cells) and solitary fibrous tumor [43]. Solitary fibrous tumor is positive for CD34 and STAT6, whereas schwannoma is negative for these markers. P63 is strongly positive in the spindle myoepithelial cells of schwannoma-like pleomorphic adenoma and is negative in schwannoma [42, 44].
Neurofibroma is a benign peripheral nerve sheath tumor composed of a variable mixture of Schwann, perineurial, and fibroblastic cells. Neurofibromas can arise from the extratemporal part of the facial nerve, which traverses in between the superficial and deep lobe of the parotid [45]. Approximately 25% of all neurofibromas are found in the head and neck region, and only up to 0.4% occur in the salivary glands [46, 47]. Observation is an alternative to definitive resection if morbidity secondary to the sacrifice of the associated nerve is high. Given that neurofibromas are usually intimately attached to nerves, en bloc surgical excision with the involved nerve is the curative treatment of choice [48]. A benign course characterizes the clinical behavior of neurofibroma with a low frequency of recurrence after surgical excision [46, 49, 50]. Infrequently, neurofibroma can undergo malignant transformation into malignant peripheral nerve sheath tumor (10%–15%), especially in the setting of neurofibromatosis type 1 [45]. The differential diagnosis includes schwannoma (discussed previously) and perineurioma. Perineurioma is composed exclusively of perineural cells and is composed of bland spindle cells with long, delicate cytoplasmic processes arranged in various growth patterns (storiform, whorling, lamellar, fascicular), and the stroma is collagenous and myxoid. Immunohistochemistry can help distinguish between these tumors as perineurioma is negative for S100 and SOX 10 and positive for EMA, claudin1, and GLUT-1.
Approximately 9% of malignant peripheral nerve sheath tumors arise in the soft tissues of the head and neck, and a minority can arise within the salivary gland with an incidence of about 0.01% in the parotid gland [51–55]. It occurs more commonly in males than in females (ratio of 2.2:1), and patients range in age from 3.5 to 82 years, average 48.4 years. Over 93% of salivary gland MPNST involve the parotid gland; one case has been reported in the submandibular gland [56]. The tumors usually present as a rapidly enlarging mass associated with pain and facial paralysis. The average size of the tumor at presentation is 5.3 cm, range 1.0–10 cm. They can develop sporadically or in association with neurofibromatosis type 1. Next-generation sequencing studies have shown recurrent co-occurring alterations in NF1, CDKN2A, SUZ12, and TP53 genes in NF1-associated and sporadic MPNST [57, 58]. Of the reported cases of salivary gland MPNST, 33% of patients died of disease, at an average of 10.75 months from the time of diagnosis [54, 55, 59–70]. Thirty-three percent of patients developed metastasis to the lungs (26%), followed by lymph nodes (15%) and temporal bone (6%). The local recurrence rate is high (~ 31%) [54, 55, 59, 71]. The recommended treatment is wide en bloc resection for small and early-stage tumors [71]. Larger tumors may require systemic therapy and radiation [71–73]. The differential diagnosis includes spindle cell melanoma and synovial sarcoma. Synovial sarcoma is composed of uniform spindle cells arranged in short fascicles associated with hyalinized areas, calcifications, variable epithelial differentiation, and specific SS18-SSX fusion gene [74]. Differentiating MPNST from spindle cell melanoma can be challenging. Immunohistochemistry can aid with this distinction -S100 and Sox-10 are often diffusely positive in spindle cell melanoma and negative or only focally positive in MPNST. Loss of H3K27me3 nuclear expression is not helpful as it may occur in both MPNST and spindle cell melanoma [75].
Adamantinoma-like Ewing sarcoma is a rare round cell sarcoma with epithelial differentiation that harbors the EWSR1-Fli-1 gene fusion [76]. Approximately 5% of adamantinoma-like Ewing sarcomas occur in the head and neck and appear to affect a wide variety of anatomic subsites, including periorbital soft tissues, thyroid gland, parotid gland, and the sinonasal tract [77]. Salivary gland adamantinoma-like Ewing sarcoma is rare; the majority of reported cases involve the parotid gland, followed by the submandibular gland, and occur in older patients (mean 52 years) than reported in other head and neck subsites [76]. This tumor's prognosis is not entirely clear because of limited follow-up data; however, tumors that arise in the salivary gland appear to have a good outcome. Of the reported cases, only one patient died due to treatment complications, one had persistent disease, and none died of disease with a follow-up time of 0–96 months, average 20 months including our cases [76]. The current recommended treatment is surgery, neoadjuvant radiation, and systemic chemotherapy [76]. The differential diagnosis in this location includes basal cell adenocarcinoma, solid adenoid cystic carcinoma, Merkel cell carcinoma, and basaloid squamous cell carcinoma. Unlike these tumors, adamantinoma like Ewing sarcoma shows expression of cytokeratin, p40, CD99, and NKX 2.2.
Head and neck solitary fibrous tumors account for 6% of all solitary fibrous tumors, and the most commonly affected regions are the sinonasal tract (30%) and orbit (25%), followed by the oral cavity (15%) and salivary glands (14%). Solitary fibrous tumor of the salivary glands is rare, and the majority of reported cases involve the parotid gland, followed by the submandibular and sublingual glands [78, 79]. It occurs in males and females equally and is diagnosed in patients ranging in age from 11 to 79 years, average 48.5 years [78–80]. The tumors usually present as a well-defined, slow-growing, painless mass that has often been present for a significant period of time (ranging from 3 to 120 months; mean, 24.7 months) [78]. Features associated with an increased rate of local recurrence in the head and neck include cellular pleomorphism, size (> 5.0 cm), increased mitotic rate (> 4 mitoses per 10 HPF), epithelioid morphology, and necrosis [80]. It is uncertain whether the risk assessment criteria developed for soft tissue and orbital tumors apply to those originating in the salivary glands [81, 82]. A high rate of local recurrence (up to 36%) in other head and neck sites has been reported; however, the recurrence rate in the parotid gland appears to be lower (5%), with an average follow-up time of 45.6 months [78]. Given that the overall follow-up time in the reported cases is short, additional studies are needed to better characterize clinically significant parameters. Therefore, long-term follow-up is recommended to exclude recurrence or metastatic disease [78, 80, 83]. The most common treatment for both benign and malignant solitary fibrous tumor is complete surgical excision, if feasible. Tumors that cannot be excised entirely or show malignant histologic features may respond to radiation and/or chemotherapy [84, 85]. Despite the wide histological spectrum, these tumors all share the NAB2-STAT6 gene fusion and staining for STAT6. The differential diagnosis is broad and includes several benign and malignant soft tissue tumors, including fibrous histiocytoma, giant cell angiofibroma, synovial sarcoma, and myopericytoma. The distinction can be made after histopathologic evaluation in conjunction with immunohistochemical and molecular findings.
Myofibroma is a rare mesenchymal neoplasm most commonly found in the head and neck region (up to 22% of solitary lesions), including the scalp, forehead, orbit, parotid area, and oral cavity [86–89]. Most head and neck myofibromas are solitary and present as a firm, well-circumscribed submucosal or subcutaneous mass in children [87, 90]. Myofibroma of the salivary gland is rare. There is only one case reported of myofibroma in the submandibular gland and our case in the parotid gland. The prognosis of this tumor in the salivary gland is uncertain because of limited cases and follow-up data. However, it has been reported that the biological behavior of myofibroma depends on the location and extent of the disease. Usually, solitary lesions have an excellent clinical course with cure after complete surgical excision; however, multicentric disease with involvement of viscera has a worse prognosis [91, 92]. The differential diagnosis includes leiomyoma, nodular fasciitis, and fibromatosis. Leiomyomas have perpendicularly oriented fascicles of spindle cells with fibrillar eosinophilic cytoplasm and lack a staghorn-like vascular tree. Fibromatosis is composed of broad fascicles of uniform spindle cells with elongate nuclei with tapered ends and indistinct eosinophilic cytoplasm. The tumors cells are associated intimately with abundant undulating collagen fibers, have an infiltrative growth pattern, and lack a staghorn-like vascular tree. Nodular fasciitis is composed of plump spindle and stellate fibroblasts and myofibroblasts in a myxoid stroma with extravasated red blood cells and scattered mononuclear cells that has a tissue culture-like appearance.
Limitations of this study are the incomplete and short follow-up on many patients; this limits the information regarding the clinical course of the tumors.
Conclusion
Mesenchymal tumors of salivary gland are uncommon. The benign variants greatly outnumber their malignant counterparts. The majority are lipomas followed by vascular tumors (hemangioma and lymphangioma), peripheral nerve sheath tumors (schwannoma and neurofibroma), solitary fibrous tumor, and myofibroma. Sarcomas are rare and include adamantinoma like Ewing sarcoma, malignant peripheral nerve sheath tumor, and malignant solitary fibrous tumor. The mainstay of treatment is surgical excision, and sarcomas may require adjuvant chemotherapy and radiation. Although minor areas of numbness are a common outcome of parotid surgery, they generally have no functional implications. The management of vascular tumors may include surgery, observation, endovascular sclerotherapy, and medical management (systemic corticosteroid, vincristine, and beta-blockers). Immunohistochemical and molecular studies are useful to precisely classify some of these tumors. A multidisciplinary team should manage these patients because treatment and outcome depend on their combined expertise (pathologist, radiologist, radiation, and medical oncologist).
Author Contributions
JVT, AER, JADP, and EMD contributed to the study conception and design. Material preparation, data collection, and analysis were performed by JVT, AER, EAM, EMD, JL, DW, GT, FC, DA, CGF, and JADP. JVT and AER wrote and edited the manuscript. JVT and AER provided radiographs and micrographs.
Funding
No funding was obtained.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article.
Declarations
Conflict of interest
The authors have no conflicts of interest to disclose.
Ethical Approval
All procedures performed in this retrospective data analysis involving human participants were in accordance with the ethical standards of the institutional review board, which did not require informed consent.
Consent for publication
All authors have approved the final manuscript and agree with submission to Head and Neck Pathology.
Consent to participate
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
11/17/2022
Family name of author Jaylou M. Velez Torres is corrected to Velez Torres.
References
- 1.Takahama A, Jr, Leon JE, de Almeida OP, Kowalski LP. Nonlymphoid mesenchymal tumors of the parotid gland. Oral Oncol. 2008;44(10):970–974. doi: 10.1016/j.oraloncology.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Cho KJ, Ro JY, Choi J, Choi SH, Nam SY, Kim SY. Mesenchymal neoplasms of the major salivary glands: clinicopathological features of 18 cases. Eur Arch Otorhinolaryngol. 2008;265(Suppl 1):S47–56. doi: 10.1007/s00405-007-0488-5. [DOI] [PubMed] [Google Scholar]
- 3.Ito FA, Ito K, Vargas PA, de Almeida OP, Lopes MA. Salivary gland tumors in a Brazilian population: a retrospective study of 496 cases. Int J Oral Maxillofac Surg. 2005;34(5):533–536. doi: 10.1016/j.ijom.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Lee YW, Chung J. Intraglandular ordinary lipoma of the submandibular gland. Ear Nose Throat J. 2019 doi: 10.1177/0145561319895603. [DOI] [PubMed] [Google Scholar]
- 5.Seifert G, Oehne H. Mesenchymal (non-epithelial) salivary gland tumors Analysis of 167 tumor cases of the salivary gland register. Laryngol Rhinol Otol (Stuttg). 1986;65(9):485–91. doi: 10.1055/s-2007-1008020. [DOI] [PubMed] [Google Scholar]
- 6.Chhieng DC, Cohen JM, Cangiarella JF. Fine-needle aspiration of spindle cell and mesenchymal lesions of the salivary glands. Diagn Cytopathol. 2000;23(4):253–259. doi: 10.1002/1097-0339(200010)23:4<253::aid-dc8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Chikui T, Yonetsu K, Yoshiura K, Miwa K, Kanda S, Ozeki S, et al. Imaging findings of lipomas in the orofacial region with CT, US, and MRI. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84(1):88–95. doi: 10.1016/s1079-2104(97)90302-4. [DOI] [PubMed] [Google Scholar]
- 8.Johnson CN, Ha AS, Chen E, Davidson D. Lipomatous soft-tissue tumors. J Am Acad Orthop Surg. 2018;26(22):779–788. doi: 10.5435/JAAOS-D-17-00045. [DOI] [PubMed] [Google Scholar]
- 9.Goldblum JR, Folpe AL, Weiss SW. Benign lipomatous tumors. Enzinger & Weiss's Soft Tissue Tumors. 7. Philadelphia, Pennsylvania: Elsevier; 2020. pp. 476–81. [Google Scholar]
- 10.Baykul T, Aydin MA, Findik Y, Yildirim D. Huge lipoma of the right parotid gland: Case report and review of 42 cases. Ear Nose Throat J. 2016;95(1):E8–E13. doi: 10.1177/014556131609500103. [DOI] [PubMed] [Google Scholar]
- 11.El-Monem MH, Gaafar AH, Magdy EA. Lipomas of the head and neck: presentation variability and diagnostic work-up. J Laryngol Otol. 2006;120(1):47–55. doi: 10.1017/S0022215105004597. [DOI] [PubMed] [Google Scholar]
- 12.Gooskens I, Manni JJ. Lipoma of the deep lobe of the parotid gland: report of 3 cases. ORL J Otorhinolaryngol Relat Spec. 2006;68(5):290–295. doi: 10.1159/000093404. [DOI] [PubMed] [Google Scholar]
- 13.Som PM, Scherl MP, Rao VM, Biller HF. Rare presentations of ordinary lipomas of the head and neck: a review. AJNR Am J Neuroradiol. 1986;7(4):657–664. [PMC free article] [PubMed] [Google Scholar]
- 14.Houle A, Mandel L. Diagnosing the parotid lipoma. Case report. N Y State Dent J. 2015;81(3):48–50. [PubMed] [Google Scholar]
- 15.Tong KN, Seltzer S, Castle JT. Lipoma of the parotid gland. Head Neck Pathol. 2020;14(1):220–223. doi: 10.1007/s12105-019-01023-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starkman SJ, Olsen SM, Lewis JE, Olsen KD, Sabri A. Lipomatous lesions of the parotid gland: analysis of 70 cases. Laryngoscope. 2013;123(3):651–656. doi: 10.1002/lary.23723. [DOI] [PubMed] [Google Scholar]
- 17.Choi HJ, Lee JC, Kim JH, Lee YM, Lee HJ. Cavernous hemangioma with large phlebolith of the parotid gland. J Craniofac Surg. 2013;24(6):e621–e623. doi: 10.1097/SCS.0b013e3182a2d87b. [DOI] [PubMed] [Google Scholar]
- 18.Batsakis JG. Vascular tumors of the salivary glands. Ann Otol Rhinol Laryngol. 1986;95(6 Pt 1):649–650. doi: 10.1177/000348948609500622. [DOI] [PubMed] [Google Scholar]
- 19.Mantravadi J, Roth LM, Kafrawy AH. Vascular neoplasms of the parotid gland. Parotid vascular tumors. Oral Surg Oral Med Oral Pathol. 1993;75(1):70–5. doi: 10.1016/0030-4220(93)90409-w. [DOI] [PubMed] [Google Scholar]
- 20.Achache M, Fakhry N, Varoquaux A, Coulibaly B, Michel J, Lagier A, et al. Management of vascular malformations of the parotid area. Eur Ann Otorhinolaryngol Head Neck Dis. 2013;130(2):55–60. doi: 10.1016/j.anorl.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Nussbaum M, Tan S, Som ML. Hemangiomas of the salivary glands. Laryngoscope. 1976;86(7):1015–1019. doi: 10.1288/00005537-197607000-00017. [DOI] [PubMed] [Google Scholar]
- 22.Beahrs OH, Woolner LB, Carveth SW, Devine KD. Surgical management of parotid lesions. Review of seven hundred sixty cases. Arch Surg. 1960;80:890–904. doi: 10.1001/archsurg.1960.01290230008002. [DOI] [PubMed] [Google Scholar]
- 23.Lara-Sanchez H, Peral-Cagigal B, Madrigal-Rubiales B, Verrier-Hernandez A. Cavernous hemangioma of the parotid gland in adults. J Clin Exp Dent. 2014;6(5):e592–e594. doi: 10.4317/jced.51750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss I, Lipari BA, Meyer L, Berenstein A, Waner M, O TM. Current treatment of parotid hemangiomas. Laryngoscope. 2011;121(8):1642–50. doi: 10.1002/lary.21358. [DOI] [PubMed] [Google Scholar]
- 25.Lymphangioma TL. Lymphangioma. Ear Nose Throat J. 2006;85(1):18–19. doi: 10.1177/014556130608500106. [DOI] [PubMed] [Google Scholar]
- 26.Shustef E, Kazlouskaya V, Prieto VG, Ivan D, Aung PP. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70(11):917–25. doi: 10.1136/jclinpath-2017-204601. [DOI] [PubMed] [Google Scholar]
- 27.Aluffi Valletti P, Brucoli M, Boffano P, Benech A, Toso A, Dell'Era V, et al. A single-center experience in the management of head and neck lymphangiomas. Oral Maxillofac Surg. 2020;24(1):109–115. doi: 10.1007/s10006-020-00832-z. [DOI] [PubMed] [Google Scholar]
- 28.Chinnakkulam Kandhasamy S, Ramasamy Raju T, Sahoo AK, Gunasekaran G. Adult cystic lymphangioma of the parotid gland: an unwonted presentation. Cureus. 2018;10(5):e2644. doi: 10.7759/cureus.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waner M, O TM. Multidisciplinary approach to the management of lymphatic malformations of the head and neck. Otolaryngol Clin North Am. 2018;51(1):159–72. doi: 10.1016/j.otc.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy TL, Whitaker M, Pellitteri P, Wood WE. Cystic hygroma/lymphangioma: a rational approach to management. Laryngoscope. 2001;111(11 Pt 1):1929–1937. doi: 10.1097/00005537-200111000-00011. [DOI] [PubMed] [Google Scholar]
- 31.Tsui SC, Huang JL. Parotid lymphangioma. A case report. Int J Pediatr Otorhinolaryngol. 1996;34(3):273–8. doi: 10.1016/0165-5876(95)01274-5. [DOI] [PubMed] [Google Scholar]
- 32.Stenson KM, Mishelle J, Toriumi DM. Cystic hygroma of the parotid gland. Ann Otol Rhinol Laryngol. 1991;100(6):518–520. doi: 10.1177/000348949110000617. [DOI] [PubMed] [Google Scholar]
- 33.Ricciardelli EJ, Richardson MA. Cervicofacial cystic hygroma. Patterns of recurrence and management of the difficult case. Arch Otolaryngol Head Neck Surg. 1991;117(5):546–53. doi: 10.1001/archotol.1991.01870170092021. [DOI] [PubMed] [Google Scholar]
- 34.Mandel L. Parotid area lymphangioma in an adult: case report. J Oral Maxillofac Surg. 2004;62(10):1320–1323. doi: 10.1016/j.joms.2003.12.040. [DOI] [PubMed] [Google Scholar]
- 35.Gross BC, Carlson ML, Moore EJ, Driscoll CL, Olsen KD. The intraparotid facial nerve schwannoma: a diagnostic and management conundrum. Am J Otolaryngol. 2012;33(5):497–504. doi: 10.1016/j.amjoto.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Forton GE, Moeneclaey LL, Offeciers FE. Facial nerve neuroma. Report of two cases including histological and radiological imaging studies. Eur Arch Otorhinolaryngol. 1994;251(1):17–22. doi: 10.1007/BF00175952. [DOI] [PubMed] [Google Scholar]
- 37.Seo BF, Choi HJ, Seo KJ, Jung SN. Intraparotid facial nerve schwannomas. Arch Craniofac Surg. 2019;20(1):71–74. doi: 10.7181/acfs.2018.02250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Lu X, Xie S, Li Z, Shan X, Cai Z. Intraparotid facial nerve schwannoma: a 17-year, single-institution experience of diagnosis and management. Acta Otolaryngol. 2019;139(5):444–450. doi: 10.1080/00016489.2019.1574983. [DOI] [PubMed] [Google Scholar]
- 39.Zhang GZ, Su T, Xu JM, Cheng ZQ. Clinical retrospective analysis of 9 cases of intraparotid facial nerve schwannoma. J Oral Maxillofac Surg. 2016;74(8):1695–1705. doi: 10.1016/j.joms.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Ingrosso G, Ponti E, di Cristino D, Terenzi S, Cicchetti S, Morelli P, et al. Intra-parotid facial nerve schwannoma with intra-temporal extension; a case report. Is there a role for stereotactic radiotherapy? Am J Otolaryngol. 2013;34(3):258–61. doi: 10.1016/j.amjoto.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Plotkin SR, Wick A. Neurofibromatosis and schwannomatosis. Semin Neurol. 2018;38(1):73–85. doi: 10.1055/s-0038-1627471. [DOI] [PubMed] [Google Scholar]
- 42.Chandra SR, Karim F, Rawal YB. Divergent schwannoma-like phenotype in a pleomorphic adenoma. Head Neck Pathol. 2017;11(4):567–574. doi: 10.1007/s12105-017-0817-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bianchi G, Sambri A, Pedrini E, Pazzaglia L, Sangiorgi L, Ruengwanichayakun P, et al. Histological and molecular features of solitary fibrous tumor of the extremities: clinical correlation. Virchows Arch. 2020;476(3):445–454. doi: 10.1007/s00428-019-02650-5. [DOI] [PubMed] [Google Scholar]
- 44.Zhu S, Schuerch C, Hunt J. Review and updates of immunohistochemistry in selected salivary gland and head and neck tumors. Arch Pathol Lab Med. 2015;139(1):55–66. doi: 10.5858/arpa.2014-0167-RA. [DOI] [PubMed] [Google Scholar]
- 45.McGuirt WF, Sr, Johnson PE, McGuirt WT. Intraparotid facial nerve neurofibromas. Laryngoscope. 2003;113(1):82–84. doi: 10.1097/00005537-200301000-00015. [DOI] [PubMed] [Google Scholar]
- 46.Marocchio LS, Oliveira DT, Pereira MC, Soares CT, Fleury RN. Sporadic and multiple neurofibromas in the head and neck region: a retrospective study of 33 years. Clin Oral Investig. 2007;11(2):165–169. doi: 10.1007/s00784-006-0096-6. [DOI] [PubMed] [Google Scholar]
- 47.Rai A, Kumar A. Neurofibroma of facial nerve presenting as parotid mass. J Maxillofac Oral Surg. 2015;14(Suppl 1):465–468. doi: 10.1007/s12663-014-0681-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kavanagh KT, Panje WR. Neurogenic neoplasms of the seventh cranial nerve presenting as a parotid mass. Am J Otolaryngol. 1982;3(1):53–56. doi: 10.1016/s0196-0709(82)80033-1. [DOI] [PubMed] [Google Scholar]
- 49.Needle MN, Cnaan A, Dattilo J, Chatten J, Phillips PC, Shochat S, et al. Prognostic signs in the surgical management of plexiform neurofibroma: the Children's Hospital of Philadelphia experience, 1974–1994. J Pediatr. 1997;131(5):678–682. doi: 10.1016/s0022-3476(97)70092-1. [DOI] [PubMed] [Google Scholar]
- 50.Katz AD, Passy V, Kaplan L. Neurogenous neoplasms of major nerves of face and neck. Arch Surg. 1971;103(1):51–56. doi: 10.1001/archsurg.1971.01350070077018. [DOI] [PubMed] [Google Scholar]
- 51.Sordillo PP, Helson L, Hajdu SI, Magill GB, Kosloff C, Golbey RB, et al. Malignant schwannoma–clinical characteristics, survival, and response to therapy. Cancer. 1981;47(10):2503–2509. doi: 10.1002/1097-0142(19810515)47:10<2503::aid-cncr2820471033>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 52.Minovi A, Basten O, Hunter B, Draf W, Bockmuhl U. Malignant peripheral nerve sheath tumors of the head and neck: management of 10 cases and literature review. Head Neck. 2007;29(5):439–445. doi: 10.1002/hed.20537. [DOI] [PubMed] [Google Scholar]
- 53.Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006–21. doi: 10.1002/1097-0142(19860515)57:10<2006::AID-CNCR2820571022>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 54.Guraya SS, Prayson RA. Peripheral nerve sheath tumors arising in salivary glands: A clinicopathologic study. Ann Diagn Pathol. 2016;23:38–42. doi: 10.1016/j.anndiagpath.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Imamura S, Suzuki H, Koda E, Usami S, Yoshizawa A. Malignant peripheral nerve sheath tumor of the parotid gland. Ann Otol Rhinol Laryngol. 2003;112(7):637–643. doi: 10.1177/000348940311200711. [DOI] [PubMed] [Google Scholar]
- 56.Alvi M, Pilkington R, Sahota RS, Adams J. Malignant peripheral nerve sheath tumour arising in the submandibular gland. BMJ Case Rep. 2020 doi: 10.1136/bcr-2020-238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pemov A, Li H, Presley W, Wallace MR, Miller DT. Genetics of human malignant peripheral nerve sheath tumors. Neurooncol Adv. 2020;2(Suppl 1):i50–i61. doi: 10.1093/noajnl/vdz049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gogate BP, Anand M, Deshmukh SD, Purandare SN. Malignant peripheral nerve sheath tumor of facial nerve: Presenting as parotid mass. J Oral Maxillofac Pathol. 2013;17(1):129–131. doi: 10.4103/0973-029X.110708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chis O, Albu S. Malignant peripheral nerve sheath tumor of the parotid gland. J Craniofac Surg. 2014;25(5):e424–e426. doi: 10.1097/SCS.0000000000000938. [DOI] [PubMed] [Google Scholar]
- 61.Biswa P, Kar A, Mohanty L, Pattnaik K, Nayak M. Malignant peripheral nerve sheath tumor in parotid gland-a rare and challenging case. J Clin Case Rep. 2013 doi: 10.4172/2165-7920.1000243. [DOI] [Google Scholar]
- 62.Athow AC, Kirkham N. Malignant parotid salivary gland peripheral nerve sheath tumour in a twelve-year-old girl. J Laryngol Otol. 1992;106(8):748–750. doi: 10.1017/s002221510012078x. [DOI] [PubMed] [Google Scholar]
- 63.Punjabi AP, Haug RH, Chung-Park MJ, Likavek M. Malignant peripheral nerve sheath tumor of the parotid gland: report of case. J Oral Maxillofac Surg. 1996;54(6):765–769. doi: 10.1016/s0278-2391(96)90700-4. [DOI] [PubMed] [Google Scholar]
- 64.Nepka C, Karadana M, Karasavvidou F, Barbanis S, Kalodimos G, Koukoulis G. Fine needle aspiration cytology of a primary malignant peripheral nerve sheath tumor arising in the parotid gland: a case report. Acta Cytol. 2009;53(4):423–426. doi: 10.1159/000325344. [DOI] [PubMed] [Google Scholar]
- 65.Laurian N, Zohar Y. Malignant neurilemmoma of parotid gland. J Laryngol Otol. 1970;84(12):1267–1271. doi: 10.1017/s0022215100073011. [DOI] [PubMed] [Google Scholar]
- 66.De Stefano A, Kulamarva G, Citraro L, Borgia L, Croce A. Malignant peripheral nerve sheath tumour (malignant epithelioid Schwannoma) of the parotid gland. Bratisl Lek Listy. 2012;113(10):628–631. doi: 10.4149/bll_2012_143. [DOI] [PubMed] [Google Scholar]
- 67.Colmenero C, Rivers T, Patron M, Sierra I, Gamallo C. Maxillofacial malignant peripheral nerve sheath tumours. J Craniomaxillofac Surg. 1991;19(1):40–46. doi: 10.1016/s1010-5182(05)80270-7. [DOI] [PubMed] [Google Scholar]
- 68.Aslan I, Oysu C, Bilgic B, Basaran B, Yazicioglu E. Malignant peripheral nerve sheath tumor of the parotid gland. Kulak Burun Bogaz Ihtis Derg. 2007;17(1):53–57. [PubMed] [Google Scholar]
- 69.Daga G, Paul R, Mandal G, Kumar R. Malignant peripheral nerve sheath tumor of the parotid gland. Indian J Surg Oncol. 2018;9(4):629–632. doi: 10.1007/s13193-018-0779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shindel J, Markowicz H. Surgery of the parotid gland. Review and follow up of 173 cases. Proc Staff Meet Pethah Tiqva Isr Beilinson Hosp. 1961;10:86–102. [PubMed] [Google Scholar]
- 71.Arshi A, Tajudeen BA, St JM. Malignant peripheral nerve sheath tumors of the head and neck: demographics, clinicopathologic features, management, and treatment outcomes. Oral Oncol. 2015;51(12):1088–1094. doi: 10.1016/j.oraloncology.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 72.Bishop AJ, Zagars GK, Torres KE, Bird JE, Feig BW, Guadagnolo BA. Malignant peripheral nerve sheath tumors: a single institution's experience using combined surgery and radiation therapy. Am J Clin Oncol. 2018;41(5):465–470. doi: 10.1097/COC.0000000000000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailet JW, Abemayor E, Andrews JC, Rowland JP, Fu YS, Dawson DE. Malignant nerve sheath tumors of the head and neck: a combined experience from two university hospitals. Laryngoscope. 1991;101(10):1044–1049. doi: 10.1288/00005537-199110000-00003. [DOI] [PubMed] [Google Scholar]
- 74.Thway K, Fisher C. Synovial sarcoma: defining features and diagnostic evolution. Ann Diagn Pathol. 2014;18(6):369–380. doi: 10.1016/j.anndiagpath.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 75.Le Guellec S, Macagno N, Velasco V, Lamant L, Lae M, Filleron T, et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: a study of 387 cases including mimicking lesions. Mod Pathol. 2017;30(12):1677–1687. doi: 10.1038/modpathol.2017.91. [DOI] [PubMed] [Google Scholar]
- 76.Rooper LM, Jo VY, Antonescu CR, Nose V, Westra WH, Seethala RR, et al. Adamantinoma-like ewing sarcoma of the salivary glands: a newly recognized mimicker of basaloid salivary carcinomas. Am J Surg Pathol. 2019;43(2):187–194. doi: 10.1097/PAS.0000000000001171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bishop JA, Alaggio R, Zhang L, Seethala RR, Antonescu CR. Adamantinoma-like Ewing family tumors of the head and neck: a pitfall in the differential diagnosis of basaloid and myoepithelial carcinomas. Am J Surg Pathol. 2015;39(9):1267–1274. doi: 10.1097/PAS.0000000000000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bauer JL, Miklos AZ, Thompson LD. Parotid gland solitary fibrous tumor: a case report and clinicopathologic review of 22 cases from the literature. Head Neck Pathol. 2012;6(1):21–31. doi: 10.1007/s12105-011-0305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lim DWJ, Tan TSH, Tan JL, Venkateswaran K. Solitary fibrous tumour of the parotid gland: a case report and a 15-year literature review. AME Case Rep. 2019;3:14. doi: 10.21037/acr.2019.04.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith SC, Gooding WE, Elkins M, Patel RM, Harms PW, McDaniel AS, et al. Solitary fibrous tumors of the head and neck: a multi-institutional clinicopathologic study. Am J Surg Pathol. 2017;41(12):1642–1656. doi: 10.1097/PAS.0000000000000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thompson LDR, Liou SS, Feldman KA. Orbit solitary fibrous tumor: a proposed risk prediction model based on a case series and comprehensive literature review. Head Neck Pathol. 2020 doi: 10.1007/s12105-020-01184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Demicco EG, Wagner MJ, Maki RG, Gupta V, Iofin I, Lazar AJ, et al. Risk assessment in solitary fibrous tumors: validation and refinement of a risk stratification model. Mod Pathol. 2017;30(10):1433–1442. doi: 10.1038/modpathol.2017.54. [DOI] [PubMed] [Google Scholar]
- 83.Demicco EG, Park MS, Araujo DM, Fox PS, Bassett RL, Pollock RE, et al. Solitary fibrous tumor: a clinicopathological study of 110 cases and proposed risk assessment model. Mod Pathol. 2012;25(9):1298–1306. doi: 10.1038/modpathol.2012.83. [DOI] [PubMed] [Google Scholar]
- 84.Yang XJ, Zheng JW, Ye WM, Wang YA, Zhu HG, Wang LZ, et al. Malignant solitary fibrous tumors of the head and neck: a clinicopathological study of nine consecutive patients. Oral Oncol. 2009;45(8):678–682. doi: 10.1016/j.oraloncology.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 85.Goodlad JR, Fletcher CD. Solitary fibrous tumour arising at unusual sites: analysis of a series. Histopathology. 1991;19(6):515–522. doi: 10.1111/j.1365-2559.1991.tb01499.x. [DOI] [PubMed] [Google Scholar]
- 86.Coffin CM, Alaggio R. Fibroblastic and myofibroblastic tumors in children and adolescents. Pediatr Dev Pathol. 2012;15(1 Suppl):127–180. doi: 10.2350/10-12-0944-PB.1. [DOI] [PubMed] [Google Scholar]
- 87.Lopes RN, Alves Fde A, Rocha AC, Suassuna TM, Kowalski LP, de Castro JF, et al. Head and neck solitary infantile myofibroma: Clinicopathological and immunohistochemical features of a case series. Acta Histochem. 2015;117(4–5):431–436. doi: 10.1016/j.acthis.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 88.Calsina M, Philipone E, Patwardhan M, Eisig S, Prat J, Kazim M. Solitary orbital myofibroma: clinical, radiographic, and histopathologic findings. A report of two cases Orbit. 2011;30(4):180–182. doi: 10.3109/01676830.2011.574773. [DOI] [PubMed] [Google Scholar]
- 89.Vered M, Allon I, Buchner A, Dayan D. Clinico-pathologic correlations of myofibroblastic tumors of the oral cavity. II. Myofibroma and myofibromatosis of the oral soft tissues. J Oral Pathol Med. 2007;36(5):304–14. doi: 10.1111/j.1600-0714.2007.00528.x. [DOI] [PubMed] [Google Scholar]
- 90.Sugatani T, Inui M, Tagawa T, Seki Y, Mori A, Yoneda J. Myofibroma of the mandible. Clinicopathologic study and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;80(3):303–9. doi: 10.1016/s1079-2104(05)80388-9. [DOI] [PubMed] [Google Scholar]
- 91.Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer. 1981;48(8):1807–1818. doi: 10.1002/1097-0142(19811015)48:8<1807::aid-cncr2820480818>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 92.Miwa T, Oi S, Nonaka Y, Tamogami R, Sasaki H, Akiyama M, et al. Rapid spontaneous regression of multicentric infantile myofibromatosis in the posterior fossa and lumbar vertebra. Childs Nerv Syst. 2011;27(3):491–496. doi: 10.1007/s00381-010-1306-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.