

Abstract
Background and Objective
Trofinetide, a synthetic analog of tripeptide glycine-proline-glutamate, is an investigational agent for the treatment of Rett syndrome, a neurodevelopmental disorder with affected individuals requiring lifelong support. Food can affect the pharmacokinetic profile of a drug, and this phase 1 study assessed the potential effect of food on the pharmacokinetics of trofinetide. The study also evaluated the potential effect of evening dosing on trofinetide bioavailability and characterized the pharmacokinetic profile of trofinetide in urine.
Methods
A 60 mL oral solution of trofinetide (12 g) was administered in three dosing periods: morning fasted (A; reference), morning fed (B), and evening fasted (C). Healthy adult subjects (18−45 years) were randomized to sequence ABC (n = 19) or BAC (n = 22). Blood and urine samples were collected at scheduled timepoints for trofinetide pharmacokinetic analysis. Bioequivalence was confirmed if 90% confidence intervals for geometric mean ratio between B/A or C/A fell within 80–125% equivalence limits for area under the concentration-time curve (AUC) and maximum concentration (Cmax) in whole blood.
Results
Bioequivalence criteria were met for all conditions (i.e., morning fed vs. morning fasted and evening fasted vs. morning fasted) except Cmax in the fed versus fasted condition, which was just below the bioequivalence limit (75.49%), suggesting a negligible food effect and lack of diurnal variation on bioavailability. Trofinetide was primarily excreted unchanged in urine. Trofinetide was well tolerated, and there were no significant changes in vital signs or laboratory parameters.
Conclusion
This study supports dosing of trofinetide without regard to food.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40261-022-01156-4.
Key Points
| This phase 1 study in healthy adults suggests there is a negligible food effect and no diurnal variation on the bioavailability of trofinetide, an investigational agent for the treatment of Rett syndrome. |
| Trofinetide was primarily excreted unchanged in urine. |
Introduction
Rett syndrome (RTT) is a neurodevelopmental disorder caused in most cases by loss of function mutations in the gene encoding methyl-CpG-binding protein 2 (MECP2) [1, 2], which results in neurological dysfunction due to ineffective or defective synaptic connections [3–5]. RTT primarily affects females, occurring in approximately 1 in 10,000–15,000 female births worldwide [6–9]. While survival of individuals with RTT has improved over time, with many living into adulthood, affected individuals require lifelong support and medical care from caregivers and healthcare systems [9–11].
Trofinetide (glycyl-l-2-methylprolyl-l-glutamic acid, also known as ACP-2566 or NNZ-2566) is a novel synthetic analog of glycine-proline-glutamate (GPE), which is a naturally occurring protein in the brain [12]. GPE has been shown to partially reverse core symptoms in Mecp2-deficient mice, improving motor and respiratory function and heart rate, increasing brain weight, and extending life span [12]. In a rat model of hypoxic insult (acute focal stroke), both GPE and trofinetide, which has a longer half-life than GPE, attenuated apoptosis and reduced infarct size, with trofinetide exhibiting a dose-dependent reduction in infarct size [13].
In a phase 2 study of 82 children/adolescents with RTT who received a twice-daily oral solution of trofinetide according to three weight-based doses (50 mg/kg, 100 mg/kg, or 200 mg/kg) for 6 weeks, the 200 mg/kg dose significantly improved clinician- and caregiver-assessed efficacy measures [1].
In initial clinical pharmacokinetic studies (including population pharmacokinetic modeling in healthy adult subjects), oral trofinetide administration led to linear kinetics with no time- or dose-dependent effect on pharmacokinetic parameters [1, 14]. Systemic exposure to trofinetide was dose-proportional across the studied dose range (up to 200 mg/kg), with no metabolic auto-inhibition or auto-induction. There was minimal to no accumulation following multiple-dose administration [1, 14]. In these early analyses, there was an observed difference in the bioavailability of trofinetide between morning and evening dosing, which could potentially be attributed to a food effect or circadian fluctuations in absorption or metabolism. Pharmacokinetic parameters of trofinetide were best described by a two-compartment model in the population pharmacokinetic modeling study [14]; however, a noncompartmental analysis is typically favored for characterizing pharmacokinetics in a single study.
The concomitant intake of food may affect the pharmacokinetic profile of orally administered drugs through several mechanisms, including delaying gastric emptying, changing the pH of the gastrointestinal tract, or changing the luminal metabolism of a drug [15, 16]. It is also well recognized that a drug’s pharmacokinetics can be modified according to the time of administration due to the impact of circadian rhythms on physiological functions (e.g., changes in hepatic blood flow, absorption from the gastrointestinal tract) [17]. This could have implications for oral medications administered more than once daily and could require changes to the dosing regimen or route of administration to optimize systemic exposure. The primary purpose of the current study was to characterize the potential effects of food on pharmacokinetic parameters of trofinetide following oral administration at the highest proposed clinical dose (12 g) to individuals with RTT. Potential diurnal variation in bioavailability was assessed as an exploratory endpoint by comparing the effect of evening and morning dosing on pharmacokinetic parameters of trofinetide. Another exploratory objective of this study was to characterize the pharmacokinetic profile of trofinetide in urine.
Methods
Study Design
This was a phase 1, open-label, single-dose study that enrolled healthy adult subjects to investigate the effect of food on the pharmacokinetics of a single 12-g dose of trofinetide oral solution. The effect of evening dosing on trofinetide pharmacokinetics was also investigated to determine potential diurnal variation. In addition, the pharmacokinetics of trofinetide in urine were characterized. A schema is provided in Fig. 1. The study was designed and performed in line with the US Food and Drug Administration’s (FDA) Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies [18], and was conducted at a specialist phase 1 unit (Worldwide Clinical Trials in the United States) between November 2019 and February 2020.
Fig. 1.

Study design. aUrine samples were not collected during the evening fasted dosing period. A fasted in the morning (reference; morning fasted state), B fed in the morning with a high-fat meal (test; morning fed state), C fasted in the evening (test; evening fasted state), D day, EOS end of study
Potential subjects were screened to assess eligibility within 28 days prior to trofinetide administration. The study included three dosing periods: fasted in the morning (A; morning fasted state [reference]), fed in the morning with a high-fat meal (B; morning fed state [test]), and fasted in the evening (C; evening fasted state [test]). Subjects were randomized to treatment conditions A and B for the first and second dosing periods, and all subjects were to receive treatment condition C in the third dosing period (i.e., subjects were randomized 1:1 to receive either treatment condition sequence ABC or BAC). The washout period after the first and second dosing periods was between 5 and 10 days.
For treatment condition A (morning fasted state), subjects were fasted overnight for approximately 10 h prior to trofinetide administration at around 08:00 (± 2 h). Subjects continued to fast for 4 h after administration of trofinetide. For treatment condition B (morning fed state), subjects were fasted overnight for approximately 10 h, then received a standardized high-fat breakfast consisting of two eggs fried in butter, two strips of bacon, two slices of toast with butter, 4 ounces of hash brown potatoes, and 8 ounces of whole milk. Subjects were administered trofinetide 30 min after completing the high-fat meal, after which they fasted for 4 h. For treatment condition C (evening fasted state), subjects fasted for approximately 6 h prior to trofinetide administration in the evening (at approximately 20:00 h), after which subjects continued to fast for a further 4 h.
Prior to trofinetide administration in each dosing period, an electrocardiogram (ECG) was performed in triplicate, and whole blood and urine samples were collected (urine samples were not collected in dosing period C). Following these procedures, a single 12-g dose of trofinetide was administered as a 60 mL solution. Subjects were allowed up to 250 mL of water following trofinetide administration. There were two follow-up visits to assess safety: (1) in the clinic approximately 1 week after the end of treatment procedures; (2) subjects received a telephone call 30 days after the last trofinetide dose in the third dosing period.
Study Population
Healthy male and female subjects aged 18–45 years, with a body mass index between 18 and 30 kg/m2 and body weight > 50 kg and < 100 kg, were eligible for this study. In the opinion of the investigator, subjects were in good health (defined by the absence of evidence of any active or chronic disease), as determined based on screening medical history, physical examination, laboratory test profile, vital signs, and ECG.
Subjects were excluded if they had a history or presence at screening of a cardiac conduction abnormality, or tested positive for alcohol, illicit drug, or cannabis at screening or day −1. Subjects were also excluded if they had received any prescription or non-prescription medications or supplements during the 14 days or five half-lives (whichever was longer) preceding day −1; an exception was made for acetaminophen or ibuprofen. Subjects who consumed > 500 mg of caffeine or xanthine-containing products per day were also excluded.
Female subjects were non-pregnant and those of childbearing potential or with a partner of childbearing potential agreed to use highly effective non-hormonal contraception for at least 28 days prior to trofinetide administration and for 28 days after the last dose of trofinetide. Male subjects with a female partner of childbearing potential agreed to use highly effective contraception from screening and for 90 days after the last dose of trofinetide.
The use of caffeine and xanthine-containing products was not permitted during confinement periods. Subjects were also required to abstain from alcohol, grapefruit, or Seville orange-containing foods (e.g., orange marmalade) or beverages from 48 h prior to check-in on day −1 through to the end of the study. Prescription or over-the-counter medications (with the exception of acetaminophen and ibuprofen) and herbal or nutritional supplements were not allowed during the study.
Study Assessments
Materials
Acadia Pharmaceuticals Inc. supplied trofinetide oral solution as an aqueous, ready-to-use, strawberry-flavored liquid in 500-mL high-density polyethylene plastic bottles with a child-resistant closure. Reference and internal standards for trofinetide were supplied by Acadia Pharmaceuticals Inc. (San Diego, CA, USA).
Dose Selection
Based on exposure-response data from a previous phase 2 study with trofinetide in female children/adolescents with RTT [1], a 12-g dose was expected to achieve target exposure (area under the blood concentration-time curve [AUC]: 800–1000 µg × h/mL) in the weight range of included subjects (50–100 kg) and thus was selected as the maximum dose for evaluation. As no accumulation was expected with trofinetide, a single dose was representative of the pharmacokinetic profile following multiple-dose administration. The highest dose in the recently completed phase 3 study of trofinetide for the treatment of RTT is 12 g twice a day.
Whole Blood Sample Preparation
Blood samples (each 4 mL) for pharmacokinetic analysis of trofinetide were taken via indwelling intravenous (IV) catheter or by direct venipuncture and were taken during each dosing period within 1 h pre-dose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 18, 24, 36, and 48 h post-dose. A 5-min window was permitted around the nominal sampling timepoints. A 15-min window was permitted for the 18-h timepoint and beyond.
Pharmacokinetic parameters in blood for trofinetide included maximum observed drug concentration (Cmax), time to maximum drug concentration (Tmax), area under the blood concentration-time curve from time 0 to time t (AUC0–t), area under the blood concentration-time curve from time 0 to infinity (AUC0–∞), apparent terminal elimination half-life (t½), terminal phase elimination rate constant (λz), apparent systemic clearance following non-intravenous (e.g., oral) administration (CL/F), and apparent volume of distribution following non-intravenous administration (Vz/F). Calculation methods and equations for each pharmacokinetic parameter in blood are shown Table 1.
Table 1.
Calculation methods for pharmacokinetic parameters in blood and urine
| Parameter | Definition | Calculation method/equation |
|---|---|---|
| Blood | ||
| Cmax | Maximum observed blood drug concentration, determined directly from individual concentration-time data | Observed value |
| Tmax | Time to maximum blood concentration, determined directly from individual concentration-time data | Occurrence of observed Cmax |
| Clast | Last quantifiable drug concentration, determined directly from individual concentration-time data | Observed value |
| Tlast | Time of last quantifiable drug concentration, determined directly from individual concentration-time data | Occurrence of observed Clast |
| AUC0–t | Area under the blood concentration-time curves from time 0 to the time of the last quantifiable concentration | Linear trapezoidal rule AUC0–t = δt × (C1 + C2)/2 |
| AUC0–∞ | Area under the blood concentration-time curve from time 0 to infinity | AUC0–∞ = AUC0–t + Clast/λz |
| %AUCext | Percentage of area under the curve extrapolated from time t to infinity | 100 × (1 − [AUC0–t/AUC0–∞]) |
| λz | Terminal phase elimination rate constant | Linear regression of the terminal linear portion of the log concentration versus time curve |
| t½ | Apparent terminal elimination half-life | t½ = ln(2)/λz |
| CL/F | Apparent systemic clearance following nonintravenous (e.g., oral) administration, where F is the bioavailability | CL/F = Dose/AUC0–∞ |
| Vz/F | Apparent volume of distribution following nonintravenous (e.g., oral) administration, where F is the bioavailability | Vz/F = Dose/(AUC0–∞ × λz) |
| Urine | ||
| Ae | Amount of drug excreted unchanged into urine per collection interval | Ae = urine concentration × volume |
| Total Ae | Cumulative amount of drug excreted unchanged into urine over the entire collection interval | Sum of Ae |
| Fe% | Percentage of dose excreted in urine per collection interval | Fe% = Ae/Dose × 100 |
| Total Fe% | Total percentage of dose excreted in urine over the entire collection interval | Fe% = Total Ae/Dose × 100 |
| CLr | Renal clearance | CLr = Total Ae/AUC0–∞ |
| CLnr | Nonrenal clearance of drug from blood | CLnr = CL/F − CLr |
| CLratio | Ratio of renal clearance to systemic clearance | CLratio = CLr/(CL/F) |
| Tmax_Rate | Midpoint of the collection interval associated with the maximum observed excretion rate | Observed value |
| Max_Rate | Maximum observed excretion rate, at time Tmax_Rate | Observed value |
Urine Sample Preparation
For the first and second dosing periods (morning fasted and morning fed states), a single urine sample was obtained on day 1 prior to dosing. For the first 12 h after dosing on day 1, urine was collected (and pooled) over 2-h intervals. During the 12- to 48-h period after dosing, urine was collected (and pooled) over 6-h intervals. Urine volumes were measured and recorded, and 2 × 2-mL aliquots were obtained from the pooled samples at each specified time interval. The collected samples were used for measurement of trofinetide concentration in the urine specimen.
Pharmacokinetic parameters included maximum urinary excretion rate, renal clearance (CLr), ratio of renal clearance to systemic clearance (CLr/CL), non-renal clearance (CLnr), and percentage of dose excreted renally as unchanged drug. Calculation methods and equations for pharmacokinetic parameters in urine are shown in Table 1.
Bioanalysis and Pharmacokinetic Assessment
Bioanalytical samples (blood and urine) were received frozen in good condition, logged in and stored at −80 °C until analysis. Details of the bioanalytical and pharmacokinetic assessment of blood and urine samples are summarized in the Online Supplemental Materials (OSM), Resource 1.
Safety Assessments
Safety and tolerability were evaluated by treatment-emergent adverse event (TEAE) review, physical examinations, clinical laboratory evaluations, vital sign measurements, and ECGs.
Statistical Analysis
The safety analysis set included all subjects who received at least one dose of trofinetide. The pharmacokinetic analysis set included all subjects who received at least one dose of trofinetide and provided sufficient data to calculate the pharmacokinetic parameters for administration periods to be used for the planned comparisons and who did not experience any protocol deviations that impacted pharmacokinetic measures.
Concentration-time data for trofinetide were analyzed using non-compartmental methods in Phoenix® WinNonlin® (Version 8.1 or higher [Certara L.P., NJ, USA]) in conjunction with the internet-accessible implementation of Pharsight® Knowledgebase ServerTM (PKSO; Version 4.0.4 or higher [Certara L.P.]) using the extravascular model (Model 200 for blood and Model 210 for urine).
The effect of food and evening dosing on the pharmacokinetics of trofinetide was assessed by comparing Cmax, AUC0–t, and AUC0–∞ in the morning fed versus morning fasted states (treatment condition B vs. A [reference]) and in the evening fasted versus morning fasted states (treatment condition C vs. A [reference]). Each pharmacokinetic parameter was analyzed using a mixed effects general linear model. The model included treatment and sequence as fixed effects and subject nested within sequence as a random effect. The two-sided 90% confidence interval (CI) for the geometric mean ratio (GMR) was obtained from the model for the morning fed and evening fasted states versus morning fasted state, and bioequivalence was confirmed if the 90% CIs fell within the limits of 0.8–1.25 for the three pharmacokinetic parameters. All safety measurements were summarized using descriptive statistics.
Thirty-six male or female subjects (18 subjects per treatment sequence) were to be enrolled in the study. The sample size was based on an equivalence test of means using two one-sided tests on data from a two-period crossover design. A total sample size of 36 would provide at least 90% power at a 5% significance level, assuming the true ratio of the means was 1.05, the coefficient of variation on the original unlogged scale was 0.25, and the equivalence limits of the mean ratio were 0.8 and 1.25.
Results
Subject Demographics
A total of 41 subjects were randomized, and 35 (85%) subjects completed the study (Fig. 2). Six (15%) subjects discontinued treatment before the end of the study and were excluded from the pharmacokinetic analysis due to missed dosing periods. One subject discontinued in the ‘ABC’ sequence (the reason was withdrawal by subject). Five subjects discontinued in the ‘BAC’ sequence (the reasons were protocol violation [n = 2], TEAE of appendicitis [n = 1], lost to follow-up [n = 1], and withdrawal by subject [n = 1]). Demographics information is presented in Table 2.
Fig. 2.

Subject disposition. A fasted in the morning (reference; morning fasted state), B fed in the morning with a high-fat meal (test; morning fed state), C fasted in the evening (test; evening fasted state), TEAE treatment-emergent adverse event
Table 2.
Subject baseline characteristics: safety analysis set
| Parameter | Sequence ABC (N = 19) | Sequence BAC (N = 22) | Overall (N = 41) |
|---|---|---|---|
| Age at informed consent (years) | |||
| Mean (SE) | 34.3 (1.73) | 31.6 (1.38) | 32.9 (1.10) |
| Median (min, max) | 37.0 (23, 45) | 32.0 (19, 45) | 32.0 (19, 45) |
| Sex, n (%) | |||
| Male | 13 (68.4) | 15 (68.2) | 28 (68.3) |
| Female | 6 (31.6) | 7 (31.8) | 13 (31.7) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 7 (36.8) | 9 (40.9) | 16 (39.0) |
| Not Hispanic or Latino | 12 (63.2) | 13 (59.1) | 25 (61.0) |
| Race, n (%) | |||
| White | 9 (47.4) | 15 (68.2) | 24 (58.5) |
| Black or African American | 9 (47.4) | 6 (27.3) | 15 (36.6) |
| Asian | 1 (5.3) | 1 (4.5) | 2 (4.9) |
| Weight at screening (kg) | |||
| Mean (SE) | 75.7 (2.14) | 72.8 (2.39) | 74.1 (1.62) |
| Median (min, max) | 74.7 (61, 99) | 73.6 (56, 90) | 73.7 (56, 99) |
| BMI at screening, (kg/m2) | |||
| Mean (SE) | 26.1 (0.66) | 24.9 (0.60) | 25.5 (0.45) |
| Median (min, max) | 26.6 (21, 30) | 24.9 (21, 29) | 25.8 (21, 30) |
A fasted in the morning (reference; morning fasted state), B fed in the morning with a high-fat meal (test; morning fed state), BMI body mass index, C fasted in the evening (test; evening fasted state), SE standard error of the mean
Two (5%) subjects received concomitant medications to treat TEAEs. One subject received hydrocortisone for contact dermatitis at ECG pad sites and prednisone and amoxicillin for a viral illness. The other subject received ondansetron for vomiting and iopamidol, piperacillin-tazobactam, morphine, and dextrose for appendicitis, and was discontinued from the study.
Pharmacokinetics
Whole Blood Pharmacokinetics
A total of 1,692 whole blood samples were collected for analysis. Blood concentration-time curves for trofinetide following administration of trofinetide in all states are presented in Fig. 3a, b. Pharmacokinetic parameters are presented in Table 3, and the analysis of pharmacokinetic parameters and the number of subjects falling below, within, or above bioequivalence are presented in Table 4. GMRs and 90% CIs showing trofinetide pharmacokinetics in the evening fasted and morning fed states compared with the morning fasted (reference) state are presented in Fig. 4.
Fig. 3.

Arithmetic mean (± SD) concentrations of trofinetide in a whole blood, b whole blood (log scale), and c urine following administration of a single oral dose of trofinetide in the morning fasted (treatment A: reference), fed (treatment B: test), and evening fasted (treatment C: test [whole blood only]) states: Pharmacokinetic analysis set. A fasted in the morning (reference; morning fasted state), B fed in the morning with a high-fat meal (test; morning fed state), C fasted in the evening (test; evening fasted state), h hours, SD standard deviation
Table 3.
Summary of the whole blood and urine pharmacokinetic parameters of trofinetide after a single trofinetide dose administered during different treatment conditions: Pharmacokinetic analysis set
| Parameter Mean (SD) unless otherwise noted |
A: morning fasted (reference) (N = 38) | B: morning fed (N = 40) | C: evening fasted (N = 35) |
|---|---|---|---|
| Whole blood | |||
| Cmax, µg/mL | 150 (31.9) | 118 (23.1) | 149 (32.7) |
| Tmax, h (min, max)a | 2.00 (1.50, 4.00) | 2.50 (1.50, 4.08) | 2.00 (1.50, 3.00) |
| AUC0–t, µg × h/mL | 791 (134) | 733 (117) | 872 (177) |
| AUC0–∞, µg × h/mL | 806 (137) | 748 (118) | 883 (178) |
| AUCext, % | 1.77 (0.68) | 2.05 (0.69) | 1.37 (0.56) |
| t½, h | 13.5 (2.76) | 14.7 (2.21) | 9.15 (2.08) |
| λz, 1/h | 0.06 (0.02) | 0.05 (0.01) | 0.08 (0.02) |
| CL/F, L/h | 15.3 (2.81) | 16.5 (3.15) | 14.1 (2.98) |
| Vz/F, L | 296 (69.5) | 350 (88.2) | 187 (57.9) |
| Urine | |||
| Max_Rate, g/h | 1.32 (0.37) | 1.26 (0.30) | – |
| Tmax_Rate, h | 3.00 (1.23) | 3.65 (1.46) | – |
| Total Ae, g | 8.33 (1.45) | 8.14 (1.32) | – |
| Total %Fe | 69.4 (12.0) | 67.8 (11.0) | – |
| CLr, L/h | 10.7 (2.59) | 11.2 (2.69) | – |
| CLnr, L/h | 4.69 (2.08) | 5.33 (2.30) | – |
| CLratio | 0.69 (0.12) | 0.68 (0.11) | – |
A fasted in the morning (reference; morning fasted state), AUC0–t area under the blood concentration-time curve from time 0 to time t, AUC0–∞ area under the blood concentration-time curve from time 0 to infinity, AUCext percentage of area under the curve extrapolated from time t to infinity, B fed in the morning with a high-fat meal (test; morning fed state), C fasted in the evening (test; evening fasted state), CL/F apparent systemic clearance following non-intravenous (e.g., oral) administration, CLr renal clearance, CLnr non-renal clearance of drug from blood, CLratio ratio of renal clearance to systemic clearance, Cmax maximum observed drug concentration, %fe total percentage of dose excreted in urine over the entire collection interval, Max_Rate maximum observed excretion rate at time Tmax_Rate, SD standard deviation, t½ apparent terminal elimination half-life, Tmax time to maximum drug concentration, Tmax_Rate midpoint of the collection interval associated with the maximum observed excretion rate, Total Ae cumulative amount of drug excreted unchanged into urine over the entire collection interval, λz terminal phase elimination rate constant, Vz/F apparent volume of distribution following non-intravenous (e.g., oral) administration, where F is the bioavailability
aMedian (minimum, maximum)
Table 4.
Bioequivalence analysis of Cmax, AUC0–t, and AUC0–∞ of trofinetide following administration in the morning fasted (treatment A [reference]), morning fed (treatment B [test]), and evening fasted (treatment C [test]) states: whole blood pharmacokinetic analysis set
| PK parameter | N test | N reference | GMR (%) (test/reference) | 90% CI (lower–upper) | Number of subjects falling below, within, or above bioequivalence, n (%) | ||
|---|---|---|---|---|---|---|---|
| < 80% | 80–125% | > 125% | |||||
| Morning fed (test) vs. morning fasted (reference) (B vs. A) | |||||||
| Cmax | 40 | 38 | 79.0 | 75.4–82.8 | 20 (54.1) | 16 (43.2) | 1 (2.7) |
| AUC0–t | 40 | 38 | 93.5 | 90.2–97.0 | 3 (8.1) | 33 (89.2) | 1 (2.7) |
| AUC0–∞ | 40 | 38 | 93.8 | 90.5–97.2 | 3 (8.1) | 33 (89.2) | 1 (2.7) |
| Evening fasted (test) vs. morning fasted (reference) (C vs. A) | |||||||
| Cmax | 35 | 38 | 99.8 | 95.1–104.6 | 1 (2.9) | 33 (94.3) | 1 (2.9) |
| AUC0–t | 35 | 38 | 110.2 | 106.2–114.3 | 0 | 31 (88.6) | 4 (11.4) |
| AUC0–∞ | 35 | 38 | 109.8 | 105.9–113.9 | 0 | 31 (88.6) | 4 (11.4) |
A fasted in the morning (reference; morning fasted state), AUC0–t area under the blood concentration-time curve from time 0 to time t, AUC0–∞ area under the blood concentration-time curve from time 0 to infinity, B fed in the morning with a high-fat meal (test; morning fed state), C fasted in the evening (test; evening fasted state), CI confidence interval, Cmax maximum observed drug concentration, GMR geometric mean ratio
Fig. 4.
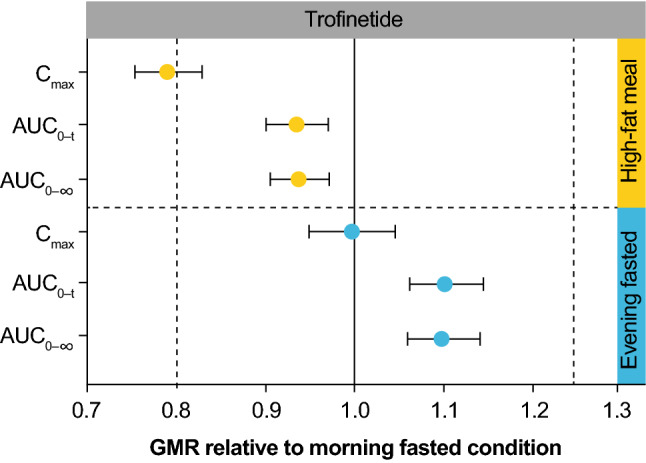
Geometric mean ratio and 90% confidence interval showing the effect of food and evening fasting on exposure to trofinetide: whole blood pharmacokinetic analysis set. AUC0–t area under the blood concentration-time curve from time 0 to time t, AUC0–∞ area under the blood concentration-time curve from time 0 to infinity, CI confidence interval, Cmax maximum observed drug concentration, GMR geometric mean ratio
Following a single 12-g dose, trofinetide pharmacokinetic profiles were similar under morning fasted (reference) and morning fed and evening fasted conditions. Trofinetide was rapidly absorbed into the circulation, with mean Tmax between 2.00 and 2.50 h. Mean Tmax was delayed by 0.50 h in the morning fed state compared with both the morning fasted (reference) and evening fasted states.
The criterion for bioequivalence for the morning fed versus morning fasted states (treatment condition B vs. A [reference]) was met for AUC0–t (GMR 93.5%; 90% CI 90.2–97.0) and AUC0–∞ (GMR 93.8%; 90% CI 90.5–97.2); however, for Cmax it was slightly outside the bioequivalence boundary (GMR 79.0%; 90% CI 75.4–82.8), with exposure around 20% lower in the morning fed versus morning fasted state (Table 4).
In the evening fasted versus morning fasted states (treatment condition C vs. A [reference]), the criterion for bioequivalence was met for all exposure parameters. The respective GMRs for AUC0–t, AUC0–∞, and Cmax were 110.2% (90% CI 106.2–114.3), 109.8% (90% CI 105.9–113.9), and 99.8% (90% CI 95.1–104.6) (Table 4).
The first 12 and 24 h following trofinetide administration accounted for around 85% and 95% of total exposure, respectively, in all three treatment conditions. Inter-subject variability (CV%) for trofinetide systemic exposure parameters (Cmax and AUC) was approximately 20% across all treatment conditions.
Urine Pharmacokinetics
Overall, 893 urine samples were collected during the morning fed and morning fasted dosing periods. Urine concentration-time curves and urine pharmacokinetic parameters were comparable following trofinetide administration in the morning fed versus morning fasted states (Fig. 3c and Table 3). The rate of trofinetide excretion in urine mirrored the clearance in blood with a Tmax (median midpoint) of 3.00 h post-dose. The rate of urinary excretion decreased rapidly over the first 12 h and was significantly reduced thereafter until the end of sample collection. Approximately 70% of the drug was excreted unchanged in urine.
Safety
Of the 41 subjects randomized, 35 subjects received all three single oral doses of 12 g trofinetide, two subjects received two doses, and four subjects received only one dose.
A summary of TEAEs experienced by two or more subjects for any Medical Dictionary for Regulatory Activities system organ class is presented in Table 5. Overall, a single dose of 12 g trofinetide administered as an oral solution was well tolerated under all three treatment conditions. TEAEs were reported in 14 subjects overall. Of these, five subjects experienced TEAEs in the morning fasted state, five in the morning fed state, and seven in the evening fasted state.
Table 5.
TEAEs reported in two or more subjects for any system organ class: safety analysis set
| System organ class MedDRA preferred term | Number of subjects (%) | |||
|---|---|---|---|---|
| A: morning fasted (reference) (N = 38) | B: morning fed (N = 40) | C: evening fasted (N = 35) | Overall (N = 41) | |
| Subjects experiencing any TEAE | 5 (13.2) | 5 (12.5) | 7 (20.0) | 14 (34.1) |
| Cardiac disorder | 0 | 2 (5.0) | 2 (5.7) | 3 (7.3) |
| Palpitations | 0 | 1 (2.5) | 0 | 1 (2.4) |
| Postural orthostatic tachycardia syndrome | 0 | 1 (2.5) | 2 (5.7) | 3 (7.3) |
| Gastrointestinal disorders | 0 | 0 | 3 (8.6) | 3 (7.3) |
| Gastroesophageal reflux disease | 0 | 0 | 1 (2.9) | 1 (2.4) |
| Nausea | 0 | 0 | 1 (2.9) | 1 (2.4) |
| Salivary hypersecretion | 0 | 0 | 1 (2.9) | 1 (2.4) |
| Vomiting | 0 | 0 | 1 (2.9) | 1 (2.4) |
| Infections and infestations | 2 (5.3) | 1 (2.5) | 1 (2.9) | 4 (9.8) |
| Appendicitis | 0 | 1 (2.5) | 0 | 1 (2.4) |
| Viral infection | 0 | 0 | 1 (2.9) | 1 (2.4) |
| Viral upper respiratory tract infection | 2 (5.3) | 0 | 0 | 2 (4.9) |
| Nervous system disorders | 1 (2.6) | 1 (2.5) | 3 (8.6) | 4 (9.8) |
| Dizziness | 1 (2.6) | 0 | 3 (8.6) | 3 (7.3) |
| Headache | 0 | 1 (2.5) | 0 | 1 (2.4) |
| Somnolence | 0 | 1 (2.5) | 0 | 1 (2.4) |
MedDRA Medical Dictionary for Regulatory Activities, TEAE treatment-emergent adverse event
The most common TEAEs (reported in two or more subjects for any preferred term) were postural orthostatic tachycardia syndrome (n = 3 [7.3%]), viral upper respiratory tract infection (n = 2 [4.9%]), and dizziness (n = 3 [7.3%]) (Table 5). All other TEAEs were reported in one subject each. Most TEAEs were mild in severity. Moderate TEAEs of nausea, vomiting, somnolence, and dermatitis contact were reported by one subject each.
Ten treatment-related TEAEs were reported in eight subjects overall. Of these, one subject experienced a treatment-related TEAE in the morning fasted state, two in the morning fed state, and five in the evening fasted state. Treatment-related TEAEs reported in more than one subject for any preferred term were postural orthostatic tachycardia syndrome (n = 3 [7.3%]; morning fed n = 1 and evening fasted n = 2) and dizziness (n = 2 [4.9%]; morning fasted n = 1 and evening fasted n = 1). The incidence of treatment-related TEAEs did not differ meaningfully between treatment conditions.
One subject in the BAC sequence experienced a serious and severe TEAE of acute appendicitis during the first dosing period (morning fed state; treatment condition B). The event was not considered to be treatment related, led to withdrawal of the subject from the study, and later resolved. There were no other serious or severe TEAEs, TEAEs leading to withdrawal, or deaths during the study. There were no clinically meaningful changes in individual laboratory parameters, vital signs, physical findings, or ECG results.
Discussion
Given the potential for food to affect gastrointestinal absorption and alter the pharmacokinetic profile of drugs [19], the present study was primarily designed to assess a potential effect of food on the pharmacokinetic profile of trofinetide, with an exploratory objective to confirm potential diurnal variation in bioavailability as suggested in the early population pharmacokinetic analyses [14, 20] of the previous phase 2 study in female children/adolescents with RTT [1] and phase 1 studies in healthy adult subjects [14]. In these studies, which were conducted under fed conditions, there was an apparent decrease in oral bioavailability following the afternoon/evening dosing (FPM = 0.55) compared with the morning dosing (FAM = 0.70).
In this study, the rate and extent of trofinetide absorption was comparable under morning fasted and evening fasted conditions. Bioequivalence criteria were met for all treatment conditions except Cmax in the morning fed state compared with the morning fasted state, for which the lower 90% CI (75.4%) was just outside the lower equivalence limit of 80%. This small change is unlikely to be clinically meaningful and suggests that trofinetide can be taken with or without food.
This study also demonstrated that trofinetide does not exhibit pharmacokinetic diurnal variation, as indicated by a lack of difference in pharmacokinetic profiles following morning or evening dosing.
It is feasible that other factors were responsible for the observed differences in bioavailability in the previous studies. For instance, lower body weights were associated with lower systemic exposures in the phase 2 study [1], which included individuals with RTT with a mean weight < 30 kg. In contrast, the healthy subjects in this study were required to have body weights > 50 kg. Furthermore, the crossover design in this study accounted for intrapatient variability since each patient acted as a control (morning fasted), thus a more accurate assessment of the effects of food and the timing of dosing was permissible in each subject.
Under morning fasted and fed conditions, the excretion of trofinetide in urine mirrored the clearance in blood. The pharmacokinetics of trofinetide in human urine had not been previously characterized but the findings in this study are consistent with nonclinical data (unpublished; rat ADME study report) and show that approximately 70% of the dose was excreted unchanged in urine, indicating renal excretion as the primary mechanism of drug clearance. Pharmacokinetic analysis of urine samples was not performed for the third dosing period (evening fasted) due to the inconvenience for the subjects, who would have been required to provide urine samples throughout the night. The similar pharmacokinetic profiles in urine under morning fed and morning fasted conditions suggest that renal elimination is unaffected by food and corroborates the findings in blood.
Single doses of 12 g trofinetide were well tolerated under all treatment conditions. Treatment-related TEAEs reported in more than one subject were dizziness (morning fasted n = 1, evening fasted n = 1) and postural orthostatic tachycardia syndrome (morning fed n = 1, evening fasted n = 2). The incidence and severity of TEAEs did not differ meaningfully between treatment conditions. One serious TEAE, acute appendicitis, led to study discontinuation and was considered unrelated to treatment. The safety profile was similar to those reported in clinical trials that investigated trofinetide treatment in individuals with RTT and Fragile X syndrome, during which no sentinel safety events occurred and the few serious TEAEs that did occur were not considered related to trofinetide treatment [1, 21, 22]. The safety profile was also similar to those reported in phase 1 studies conducted in healthy subjects who received IV (loading dose [20 mg/kg] and/or extended infusion [1, 3, and 6 mg/kg/h]) or oral trofinetide at doses up to 100 mg/kg, in which there were no serious TEAEs reported, and the few treatment-related TEAEs included headache and nausea (unpublished data, studies Neu-2566-HV-001, -002, -003, -004, -005).
Limitations
The inclusion of healthy subjects in food effect studies is recommended per FDA guidance [18]; nevertheless, the population studied in this analysis does not represent the target population, which is individuals with RTT, including children/adolescents, who may have gastrointestinal confounders and are typically of a very low weight.
Conclusion
Following a single dose of 12 g trofinetide administered as an oral solution to healthy adult subjects, pharmacokinetic profiles were qualitatively similar under morning fasted, morning fed, and evening fasted conditions. Bioequivalence criteria were met for all conditions except Cmax in morning fed versus morning fasted condition, suggesting a negligible food effect and lack of diurnal variation in the bioavailability of trofinetide. Trofinetide is primarily excreted in the urine, and its pharmacokinetic profile in urine reflected the systemic observations in whole blood.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank the subjects who took part in the study, the staff who assisted at the study site, Mark Forman as medical monitor, and the manuscript writing group.
Declarations
Funding
This study was sponsored by Acadia Pharmaceuticals Inc. Medical writing and editorial support was provided by Lesley Taylor, PhD, and Stuart Murray, MSc, of Evidence Scientific Solutions, Inc. (Philadelphia, PA) and funded by Acadia Pharmaceuticals Inc.
Data availability
Acadia Pharmaceuticals Inc. is adhering to current US and EU requirements so will not make individual de-identified participant data available; however, the protocol and statistical analysis plan will be made available upon request to the corresponding author.
Code availability
Not applicable.
Compliance with ethical standards
All requisite study-related material, including the protocol, were reviewed by an institutional review board/ethics committee (IntegReview, USA). All subjects provided written informed consent for participation in the study. The study was performed in full conformity with the current Declaration of Helsinki, the International Council for Harmonisation Guideline for Good Clinical Practice, and all other applicable regulations.
Conflicts of interest
DD, JH, JMY, MD, and SS are employees of Acadia Pharmaceuticals Inc.
Consent to participate
All subjects provided written informed consent for participation in the study.
Consent to publish
Not applicable.
Author contributions
All authors met ICMJE criteria by contributing to the conception, design, analysis, and/or interpretation of data; drafting the work or revising it critically for important intellectual content; approving the final version to be published; and agreeing to be accountable for all aspects of the work by ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
References
- 1.Glaze DG, Neul JL, Kaufmann WE, Berry-Kravis E, Condon S, Stoms G, Oosterholt S, Della Pasqua O, Glass L, Jones NE, Percy AK, Rett 002 Study Group Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019;92:e1912–e1925. doi: 10.1212/wnl.0000000000007316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kyle SM, Vashi N, Justice MJ. Rett syndrome: a neurological disorder with metabolic components. Open Biol. 2018;8:170216. doi: 10.1098/rsob.170216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, Chou S, Li Y, Ziats M, Ernst C, Jaenisch R, Haggarty SJ, Sur M. Human cerebral organoids reveal deficits in neurogenesis and neuronal migration in MeCP2-deficient neural progenitors. Mol Psychiatry. 2018;23:791. doi: 10.1038/mp.2018.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olson CO, Zachariah RM, Ezeonwuka CD, Liyanage VR, Rastegar M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS One. 2014;9:e90645. doi: 10.1371/journal.pone.0090645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet. 2002;71:1259–1272. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fehr S, Bebbington A, Nassar N, Downs J, Ronen GM, De Klerk N, Leonard H. Trends in the diagnosis of Rett syndrome in Australia. Pediatr Res. 2011;70:313–319. doi: 10.1203/PDR.0b013e3182242461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagberg B. Rett’s syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr Scand. 1985;74:405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- 8.Leonard H, Bower C, English D. The prevalence and incidence of Rett syndrome in Australia. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):8–10. [PubMed] [Google Scholar]
- 9.Kirby RS, Lane JB, Childers J, Skinner SA, Annese F, Barrish JO, Glaze DG, MacLeod P, Percy AK. Longevity in Rett syndrome: analysis of the North American Database. J Pediatr. 2010;156:135–8.e1. doi: 10.1016/j.jpeds.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarquinio DC, Hou W, Neul JL, Kaufmann WE, Glaze DG, Motil KJ, Skinner SA, Lee HS, Percy AK. The changing face of survival in Rett syndrome and MECP2-related disorders. Pediatr Neurol. 2015;53:402–411. doi: 10.1016/j.pediatrneurol.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane JB, Salter AR, Jones NE, Cutter G, Horrigan J, Skinner SA, Kaufmann WE, Glaze DG, Neul JL, Percy AK. Assessment of caregiver inventory for Rett syndrome. J Autism Dev Disord. 2017;47:1102–1112. doi: 10.1007/s10803-017-3034-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, Sieg F, Wen J, Brimble MA, Harris PW, Gluckman PD. NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke. J Neurol Sci. 2009;278:85–90. doi: 10.1016/j.jns.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Oosterholt SP, Horrigan J, Jones N, Glass L, Della PO. Population pharmacokinetics of NNZ-2566 in healthy subjects. Eur J Pharm Sci. 2017;109(Suppl):S98–107. doi: 10.1016/j.ejps.2017.05.032. [DOI] [PubMed] [Google Scholar]
- 15.Welling PG. Influence of food and diet on gastrointestinal drug absorption: a review. J Pharmacokinet Biopharm. 1977;5:291–334. doi: 10.1007/bf01061694. [DOI] [PubMed] [Google Scholar]
- 16.Welling PG. Effects of food on drug absorption. Pharmacol Ther. 1989;43:425–441. doi: 10.1016/0163-7258(89)90019-3. [DOI] [PubMed] [Google Scholar]
- 17.Baraldo M. The influence of circadian rhythms on the kinetics of drugs in humans. Expert Opin Drug Metab Toxicol. 2008;4:175–192. doi: 10.1517/17425255.4.2.175. [DOI] [PubMed] [Google Scholar]
- 18.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry: food-effect bioavailability and fed bioequivalence studies. December 2002. https://www.fda.gov/media/70945/download. Accessed Oct 2021.
- 19.Singh B. Effects of food on clinical pharmacokinetics. Clin Pharmacokinet. 1999;37:213–255. doi: 10.2165/00003088-199937030-00003. [DOI] [PubMed] [Google Scholar]
- 20.Oosterholt S, Jones N, Glass L, Della Pasqua O. Population pharmacokinetics of trofinetide in paediatric Rett syndrome patients. In: pA2 online. E-journal of the British Pharmacological Society. 2017. http://www.pa2online.org/abstract/abstract.jsp?abid=33628&author=Oosterholt&cat=-1&period=67. Accessed Oct 2021.
- 21.Glaze DG, Neul JL, Percy A, Feyma T, Beisang A, Yaroshinsky A, Stoms G, Zuchero D, Horrigan J, Glass L, Jones NE. A double-blind, randomized, placebo-controlled clinical study of trofinetide in the treatment of Rett syndrome. Pediatr Neurol. 2017;76:37–46. doi: 10.1016/j.pediatrneurol.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Berry-Kravis E, Horrigan JP, Tartaglia N, Hagerman R, Kolevzon A, Erickson CA, Hatti S, Snape M, Yaroshinsky A, Stoms G, Glass L, Jones NE, FXS-001 Investigators A double-blind, randomized, placebo-controlled clinical study of trofinetide in the treatment of fragile X syndrome. Pediatr Neurol. 2020;110:30–41. doi: 10.1016/j.pediatrneurol.2020.04.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Acadia Pharmaceuticals Inc. is adhering to current US and EU requirements so will not make individual de-identified participant data available; however, the protocol and statistical analysis plan will be made available upon request to the corresponding author.