

Abstract
Novel drug compound hunting was carried out for SARS-CoV-2 proteins with low mutation susceptibility. The probability of escape mutation and drug resistance is lower if conserved microbial proteins are targeted by therapeutic drugs. Mutation rate of all SARS-CoV-2 proteins were analyzed via multiple sequence alignment Non-Structural Protein 13 and Non-Structural Protein 16 were selected for the current study due to low mutation rate among viral strains and significant functionality. Cross-species mutation rate analysis for NSP13 and NSP16 showed these are well-conserved proteins among four coronaviral species. Viral helicase inhibitors, identified using literature-mining, were docked against NSP13. Pharmacophore-based screening of 11,375 natural compounds was conducted for NSP16. Stabilities of top compounds inside human body were confirmed via molecular dynamic simulation. ADME properties and LD50 values of the helicase inhibitors and Ambinter natural compounds were analyzed. Compounds against NSP13 showed binding affinities between −10 and −5.9 kcal/mol whereby ivermectin and scutellarein showed highest binding energies of −10 and −9.9 kcal/mol. Docking of 18 hit compounds against NSP16 yielded binding affinities between −8.9 and −4.1 kcal/mol. Hamamelitannin and deacyltunicamycin were the top compounds with binding affinities of −8.9 kcal/mol and −8.4 kcal/mol. The top compounds showed stable ligand–protein interactions in molecular dynamics simulation. The analyses revealed two hit compounds against each targeted protein displaying stable behavior, high binding affinity and molecular interactions. Conversion of these compounds into drugs after in vitro experimentation can become better treatment options to elevate COVID management.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s11030-022-10468-8.
Keywords: COVID-19, SARS-CoV-2, Non-structural protein 16, Non-structural protein 13, Molecular docking, Pharmacophore, Virtual screening, Molecular dynamics simulations
Introduction
The causative specimen for COVID-19 is the seventh coronavirus called Severe Acute Respiratory Syndrome CoronaVirus-2 which belongs to family Coronaviridae, order Nidovirales [1]. SARS-CoV-2 strain is composed of peplomers that take up typical crown-shape [2, 3]. Since December 2019 till recent times (as on November 2021), the fast-transmitting virus has affected more than 251,788,329 people in 223 countries worldwide (Situation reports, WHO). In the meanwhile the death number has reached to 5,077,907. The most common symptoms of SARS-CoV-2 infection are dry cough, fever and fatigue [4]. The infection can also lead to pneumonia and death in severe cases [5, 6]
The viral genetic material is composed of a single-stranded positive RNA genome containing 5’ cap and 3’ poly-A tail [7]. SARS-CoV-2 is composed of twenty-six proteins which include four structural proteins, sixteen non-structural proteins and six accessory proteins [8]. The translation of two overlapping open reading frames, ORF1a and ORF1b produces pp1a and pp1ab [9]. Then cleavage of pp1a and pp1ab is responsible for the formation of 16 non-structural protein [10].
Identification of new compounds against significant viral proteins can be an effective approach to combat COVID-19 [11]. Firstly, mutation rate of all SARS-CoV-2 proteins were analyzed in this study. The probability of escape mutation and drug resistance is lower if conserved microbial proteins are targeted by therapeutic drugs [9] Non-Structural Protein 13 (NSP13) and Non-Structural Protein 16 (NSP16) were selected due to low mutation rate among viral strains and significant functionality in the viral replication and survival mechanism.
NSP16 is an m7GpppA-specific 2ʹ-O-MTase, which depends on SAM molecules to make the viral RNA cap methylated at ribose 2ʹ-O positions and produce a cap-1 structure [12]. The ortho-methylation of NSP16 avoids host immunological response [13] by imitating host mRNA so that the viral strain is not recognized by host cells [14]. NSP16 of SARS-2-CoV showcases high percentage similarity with SARS-CoV, Bat-CoV-RaTG13 and Bat -SL-CoV Rs4247 [15].
In SARS-CoV, NSP13 (Helicase) catalyze the formation of single strands from the uncoupling of duplex oligonucleotides (either RNA or DNA) in 5’-3’ direction via ATP dependent reaction [16]. ATP-binding site in helicase is almost conserved in all 3 classes of helicases: SF1, SF2 and SF3 [17]. SARS-CoV-2 NSP13 helicase shares a sequence similarity index of 99.8% with SARS-CoV NSP13 differing in only one amino acid molecule (I570V). Thus, it is likely that all the functions are conserved in SARS-CoV-2 helicase. Targeting helicase to interfere with viral replication was successful in flaviviruses, picornaviruses, dengue virus and hepatitis C Virus [18–20].
NSP16 and NSP13 are essential components for viral replication and could therefore be good target for inhibition of SARS-CoV-2. Molecular docking studies with viral helicase inhibitors against SARS-COV-2 NSP13 helicase were conducted in this study. Structure-based pharmacophore screening was performed with natural compound library of 11,325 compounds to identify hit compounds for NSP16. Docking with 18 hit compounds and NSP16 showed that certain natural chemical entities exhibited high binding affinity with the target macromolecule. ADMET profiles of these chemical entities were checked. Furthermore, molecular dynamics simulation was conducted for top compounds.
Materials and method
Workflow
Method Overview provide in a flowchart (Fig. 1).
Fig. 1.

Graphical illustration of the workflow. The workflow entails the step-wise methods followed in case of non-structural protein 16 (NSP16) and non-structural protein 13 (NSP13). The workflow is a detailed depiction of the methodology followed in the present study
SARS-CoV-2 proteome alignment and conservancy analysis
The probability of escape mutation and drug resistance is lower if conserved microbial proteins are targeted by therapeutic drugs. NCBI (https://www.ncbi.nlm.nih.gov/) and GISAID website (https://www.gisaid.org/) were used to understand whether the proteins of SARS-Coronovirus-2 are conserved or not. NCBI web-server contains repository of SARS-CoV-2 strains from all the continents around the globe. There was a total of 11,634 SARS-CoV-2 Coronaviral strains in the repository of NCBI database by 1st September 2020. Multiple sequence alignment of a total of 439 strains for all proteins of SARS-2-CoV was completed using tools in GISAID (list of IDs in Supplementary Table S1). Thus, stratified sampling technique was used to determine sample size of SARS-2-coronaviral strains whereby each continent was considered as a stratum. The equation, n = N/(1 + Ne2) was used to calculate sample size of each Strata (continent) with 10% margin of error and 95% Confidence interval [21, 22]. The detailed sample size for each stratum (continent) is depicted in Supplementary Figure S1.
Comparison of NSP13 and NSP16 among various coronavirus species and among various strains of SARS-CoV-2
In order to evaluate whether NSP16 and NSP13 proteins are well-conserved among various species of coronavirus or not, multiple sequence alignment via Clustal Omega website (https://www.ebi.ac.uk/Tools/msa/clustalo/) and Mega6 [23] was performed. Clustal Omega catches the phylogenetic trees of input sequences and aligns them to represent their similarity. Mega6 displays the aligned sequences with different color codes. The FASTA sequence of reference strain of each species (SARS-CoV, SARS-CoV-2, SARS BJ01 and Bat SARS-like CoV) were collected from NCBI database (https://www.ncbi.nlm.nih.gov/sars-cov-2/). The sequences were aligned (Supplementary Figure S2) using Clustal Omega website (https://www.ebi.ac.uk/Tools/msa/clustalo/) and the result was viewed in Mega6.
Identification of compounds that inhibit viral helicases in vitro for NSP13
Experimentally solved three-dimensional structure of NSP13 in Protein Data Bank (PDB) (http://www.rcsb.org) was searched. Protein structure with PDB ID 6XEZ was selected for the present study. There were no ligands associated with the X-Ray crystallography structure of the protein (PDB ID 6XEZ). There were no small molecules bound to the chain of NSP13 in the three-dimensional structure of protein submitted to PDB. Bound small molecules are used to conduct ligand-based pharmacophore by comparing the chemical properties with a given database was generated. Hence, extensive literature-mining was carried to find out molecules that were successful in inhibiting NSP13 of other viruses in vitro.
Published scientific articles on Google Scholar using the key words, “in vitro,” “viral,” “helicase” and “inhibitor.” Published articles on helicase protein complexes were thoroughly searched for laboratory-based inhibitors of NSP13. In total, 30 helicase inhibitors that work on viral helicase were identified and have proved to inhibit viral helicase activity. Among these, structure data files (SDFs) of 19 compounds were retrieved from PubChem database (Table 1). However, the 3D structure of other 11 chemical compounds were not found in the repository of PubChem, ZINC, DrugBank, ChEMBL and Bind-DB databases (Supplementary Table S2).
Table 1.
Identified helicase inhibitors via literature review selected for molecular docking studies
| Drug | Virus against which the helicase inhibitor worked | References |
|---|---|---|
| Ivermectin | Yellow Fever Virus, Tick Borne Virus, Japanese Encephalitis virus, Dengue virus | [52] |
| Scutellarein, | Severe Acute Respiratory Syndrome | [53] |
| Myricetin | Severe Acute Respiratory Syndrome | [53] |
| Suramin | Dengue Virus | [54] |
| Ribavirin 5'-triphosphate | Hepatitis Virus | [18, 20] |
| Ribavirin 5'-diphosphate | Hepatitis Virus | [18, 20] |
| ST-193 | Dengue Virus | [55] |
| SSYA100-001 | Severe Acute Respiratory Syndrome | [31] |
| SSYA100-002 | Severe Acute Respiratory Syndrome | [31] |
| GTPL10881 | Middle East Respiratory Syndrome | [56] |
| Nogalamycin | Japanese Encephalitis virus | [57] |
| Doxorubicin | Hepatitis Virus | [57] |
| Paclitaxel | Hepatitis Virus | [58] |
| DRBT | Japanese Encephalitis virus, Hepatitis Virus | [59, 60] |
| TBBT | Japanese Encephalitis virus, Hepatitis Virus | [59, 60] |
| Amenamevir | Varicella zoster virus | [31] |
| Pritelivir | Herpes Simplex Virus | [31] |
| Famciclovir | Herpes simplex virus 1, Herpes simplex virus 2 | [19] |
| Valaciclovir | Herpes simplex virus 1, Herpes simplex virus 2 | [19] |
Molecular docking of compounds with NSP13
To prepare macromolecular structures into a more suitable form, molecular protein preparation is needed. Refinement process such as removing attached water molecules, hydrogen bonds addition and optimization, atomic clashes removal, and operations are needed for docking investigations. For this study experimentally validated electron microscopy structure having a resolution 3.50 Å of NSP13 protein was obtained from the protein data bank (PDB ID: 6XEZ) [15]. This electron microscopy structure was then prepared by using Biovia Discovery Studio 2020 Client (BIOVIA Dassault System 2020) by performing the following actions—(i) water molecules were removed (ii) Cofactors, mineral ions and other heteroatoms were removed and (iii) hydrogen bonds were added and optimized.
The Structure Data Files of 19 drug compounds (found via literature-mining) were collected from PubChem database. Each structure was opened separately in PyRx® (https://pyrx.sourceforge.io/) interlinked with Autodock Vina® for molecular Docking purposes [24]. The ligands were minimized in terms of energy using default options in Autodock Vina®. Universal Force Field (UFF) was used for energy minimization and bond angle optimization for all the ligands [24]. Then these were converted to pdbqt format for further steps. The prepared protein files in PDB format were opened in Autodock Vina to carry out docking of NSP13 with each of these drugs separately. Molecular docking was then carried out in virtual screening software: PyRx. PyRx is a extensively used virtual screening software that has identified many potential drug candidates [25–28]. Lamarckian genetic algorithm (LGA) scoring-based AutoDock and AutoDock Vina both are included in PyRx [29]. Binding affinity (kcal/mol) were retrieved of the resultant docked compounds with the proteins and visualized by using BIOVIA Discovery Studio Visualizer Tool.
Structure-Based pharmacophore mapping and virtual screening against natural compounds for NSP16
LigandScout® software [30] was used to generate structure-based pharmacophore using default parameters. For the generation of structure-based pharmacophore models, experimentally solved three-dimensional structure of NSP16 was retrieved from RCSB Protein Data Bank (http://www.rcsb.org). Till May 2021, there was a total of 18 PDB structures available for NSP16. The PDB IDs of these structures are as follows: 7JYY, 7JZ0, 6WVN, 6WQ3, 6W4H, 6WKQ, 7C2I, 6W75, 6W61, 6WJT, 7JPE, 7JHE, 7C2J, 7JIB, 7BQ7, 6YZ1, 6XKM and 6WRZ. From these solved protein structures three ligands: s-adenosyl-l-methionine (SAM), s-adenosyl-homocysteine (SAH) and sinefungin were found to be bound to NSP16 chain. SAM analogs were identified as potential inhibitor of 2ʹ-0-methyltransferase [31]. SAM and its analogs, SAH and sinefungin were found to be potential inhibitors of methyltransferase activity [32]. So chemical interactive features of these ligands were further used for the generation of the pharmacophore.
For the preparation of a structure-based pharmacophore model LigandScout 4.3 software was used. Ligand scout works by retrieving meaningful interactions between amino acids of the active sites of the target protein and inhibitors. Vital ligand-receptor interactions then can be interpreted as pharmacophore features such as hydrogen bond acceptors, hydrogen bond donor, hydrophilic and hydrophobic regions electronic charges by using LigandScout. The common interaction features of the three ligands with NSP16 in 18 solved proteins structure were then used to generate a structure-based pharmacophore by merging them using default parameters. Virtual screening using pharmacophore-based models are extensively used for the identification of structurally novel and effective compounds [33]
The pharmacophore of NSP16 complex was screened against 11,325 natural compounds available in Ambinter web-server (February 2020) (http://www.ambinter.com) in search of similar pharmacophore generating compounds. Ambinter (http://www.ambinter.com/) is an advanced chemical supplier worldwide with a huge freely available library of 36 million online chemical structures, which is being utilized to identify the potential lead compounds. For the study, Ambinter natural product library containing 11,325 natural compounds was chosen as the screening target for pharmacophore-based virtual screening. SDF data format in Ambinter was converted to Idb database format that was used in LigandScout as input for the Pharmacophore‑based virtual screening. The generated database containing all the natural compounds was then subjected to screening against the validated structural-based pharmacophore according to their features. The following parameters were maintained during the screening: (i)maximum number of omitted features-3, (ii)scoring function-pharmacophore fit and (iii)screening mode-match all query features. According to pharmacophore score, hit compounds were identified and retrieved and subjected to further investigation.
Molecular docking of good pharmacophore generating compounds with NSP16
After carrying out structure-based pharmacophore screening of NSP16 complex, 18 natural compounds based on pharmacophore-fit score were docked with NSP16. For the purpose of molecular docking of NSP16 with selected compounds we used X-Ray diffraction structure with a resolution 2.37 Å of which was retrieved from protein data bank (PDB ID:7BQ7). Protein was then prepared accordingly.
The protein was cleaned by discarding water molecules and heteroatoms in Biovia Discovery Studio and converted to pdbqt format. The SDF files of 18 natural compounds were energy minimized by using UFF force field and converted to AutoDock ligand pdbqt format. Then Molecular docking was performed using the Vina wizard in PyRx and binding affinities were retrieved.
ADME and toxicity (ADMET) analysis
For developing a molecule into a drug, the evaluation of absorption, distribution, metabolism and excretion (ADME) properties is one of the utmost priorities [34]. Nowadays this characterizations can be done via computer-based analysis and can act as an early stage prediction of these properties [35]. For this study, Swiss-ADME (http://www.swissadme.ch/) server was used to evaluate the ADME properties such as solubility profile, GIT absorption, bioavailability profile for all the selected compounds of NSP-13 and NSP-16. The various ADME properties of the helicase inhibitors and Ambinter natural compounds (hit molecules) were collected from Swiss-ADME web-server. The Swiss-ADME, is a free web tool used in evaluating the chemistry pharmacokinetics and drug-likeness (ADME and physicochemical properties) of small molecules widely used in computational biology [36].
Toxicity profile evaluation is necessary to determine the possibility of harmful effects of compounds on human and animals. In silico computational-based approaches can be used to measure the toxicity and safety profile the desired compounds [34]. For this study, LD50 values were determined using Toxicity Estimation Software Tool (TEST), a freely available software to estimate toxicity of the compounds [37]. TEST tools employ Quantitative Structure–Activity Relationships (QSARs) algorithms for estimation of the toxicity and gives a primary reliable prediction [38, 39].
Oral bioavailability analysis
Bioavailability radar enables a first glance at the drug-likeness of a molecule and provides a graphical snapshot of the drug-likeness parameters for an orally available bioactive drug. For analyzing the oral bioavailability capabilities of our compounds the bioavailability radar for all of our compounds were retrieved from Swiss-ADME. After inputting information on the smile IDs of the compounds, the relevant ADME information for all the compounds were shown. From the bioavailability section of the given ADME information, bioavailability radar images were collected. The radar is presented as a hexagon (Fig. 4) with each of the vertices representing a defining parameter for the bioavailable drug. The bioavailability radar profiles of top 2 compounds (hamamelitannin and deacyltunicamycin) of NSP16 and top 2 compounds (ivermectin and scutellarein) of NSP13 are showcased in (Fig. 4).
Fig. 4.

The bioavailability radar for the hit molecules. These bioavailability radars have been retrieved from Swiss-ADME server for hamamelitannin, deacyltunicamycin, ivermectin and scutellarein
Molecular dynamics (MD) simulation
Molecular dynamics simulation of the protein–ligand (drug) complex was performed by following method described by Zinnia et al. [40]. GROMACS (v2021) [41] and the CHARMM36 all-atom additive force field (version 2021) [42] were used for simulation study. In brief, the protein topology for NSP13 and NSP16 were generated by using GROMACS pdb2gmx function following addition of hydrogen. Next, the complex was immersed at the center of a cubic box of a simple point charge extended (SPC/E) water model [43]. The solvated system was neutralized by incorporating an aqueous solution having 0.15 M of Na+ (sodium) and Cl− (chloride). Equilibration was done for 100 picoseconds (ps) by using the NVT and NPT ensemble. For NVT simulation, the system was gradually heated to desired temperatures after equilibrating under an isothermal ensemble, for next 100 ps, by soft coupling with the Berendsen thermostat [44]. In the NPT simulation, all chemical bonds were restrained using the LINCS (LINear Constraint Solver) algorithm [45]. Periodic boundary conditions (PBC) were used with a constant number of particles in the system, constant pressure, and constant temperature simulation criteria (NPT) in order to eliminate the boundary effects. NPT ensemble was performed at 300 K. The system was coupled with Parrinello–Rahman barostat [46] to equilibrate at 1 bar for 100 ps. The long-range electrostatic interactions were processed using the Particle Mesh Ewald (PME) protocol and the short-range van der Waals cutoff (rvdW) interactions were calculated using a cutoff of 1.0 nm. MD simulations were done for 50 ns (ns). Trajectory plots of solvent accessible surface area (SASA), root mean square fluctuation (RMSF), root mean square deviation (RMSD), the radius of gyration (Rg) and hydrogen bond (H-bond) were generated using in-house python script using matplotlib module [47] along with the NumPy [48] library, as well as using R (version 3.6.3) [49] program with Peptides library [50].
Results
NSP13 and NSP16 show high sequence similarity with other SARS-CoV species
Both NSP13 and NSP16 are well-conserved between SARS-CoV, SARS-CoV-2, SARS BJ01 and Bat SARS-like-CoV (Supplementary Figure S1). Multiple Sequence alignment for NSP16 showed > 90% sequence similarity of SARS-CoV-2 with other beta coronaviruses (93% with SARS-CoV, 94% with SARS BJ01 and 93.3% similarity with Bat SARS-like coronavirus). The percentage of sequence similarity for NSP13 between SARS-CoV-2 NSP13 and NSP13 of other beta coronaviruses are even higher: 99% with SARS, 99.3% with SARS BJ01 and 99.5% similarity with Bat SARS-like coronavirus (Fig. 2). Both NSP13 and NSP16 are well-conserved between SARS-CoV, SARS-CoV-2, SARS BJ01 and Bat SARS-like-CoV.
Fig. 2.
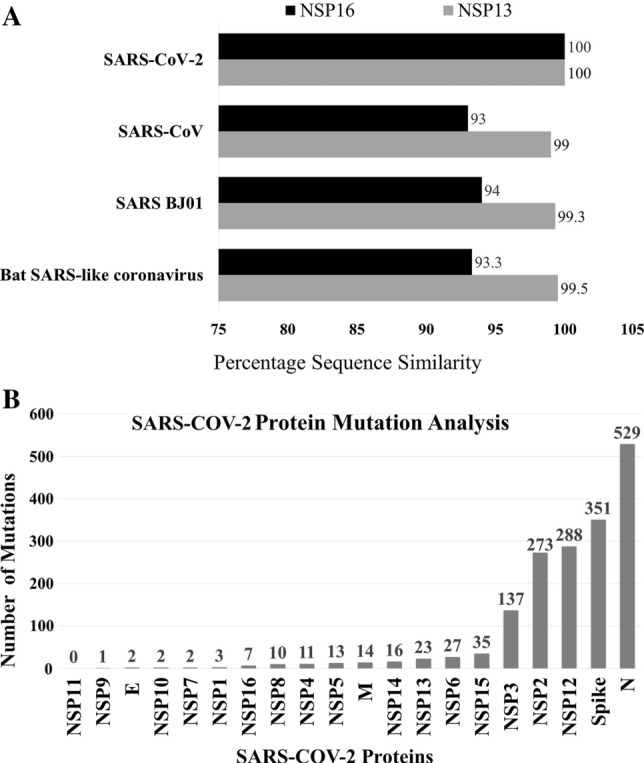
The highly conserved NSP13 and NSP16 proteins show lowest mutation rate. A Bar chart representing sequence similarity of SARS-CoV-2 NSP16 and NSP13 with other beta coronaviruses: Sequence similarity of NSP16 (93% with SARS, 94% with SARS BJ01 and 93.3% similarity with Bat SARS-like coronavirus); Sequence similarity of NSP13 (99% with SARS, 99.3% with SARS BJ01 and 99.5% similarity with Bat SARS-like coronavirus). The high sequence similarity can be considered as a mark of conservancy of both NSP13 and NSP16 among beta lineage of coronaviruses. B Bar chart showing number of mutations for the sample size of 439 SARS-CoV-2 viral strains (retrieved from NCBI virus database) with which multiple sequence alignment was performed via GISAID. Mutation rate among the 439 viral strains studied is low for both NSP13 and NSP16 proteins with rate of 23 mutations and 7 mutations, respectively. The nucleocapsid protein (529 mutations), spike protein (351 mutations) and non-structural protein 12 (288 mutations) exhibited highest mutation rate
Mutation rate analysis show low mutation rate of NSP13 and NSP16 as compared to other SARS-CoV-2 proteins
With the help of NCBI virus database and GISAID web-server, mutation rate of all proteins of SARS-CoV-2 were studied with 439 strains. Mutation rate among the 439 strains studied is low for both NSP13 and NSP16 proteins. The nucleocapsid protein (529 mutations), spike protein (351 mutations) and non-structural protein 12 (288 mutations) exhibited highest mutation rate. Both NSP13 and NSP16 showed lower mutation rate of 23 mutations and 7 mutations, respectively, as compared to other proteins (Fig. 2B).
Helicase inhibitors can be promising in inhibiting NSP13
Helicase inhibitors with reported helicase inhibitory activities against viruses like Severe Acute Respiratory Syndrome, Dengue Virus, Hepatitis Virus, Middle East Respiratory Syndrome, Japanese Encephalitis virus, Hepatitis Virus, Varicella zoster virus, Herpes Simplex Virus were identified using literature-mining. In this study, the activities of 19 helicase inhibitors as identified by literature-mining against SARS-CoV-2 NSP13 were checked (Table 1). Then docking [51] with NSP13 and chemical entities with previous history of helicase inhibitory activity was performed. In our analysis, ivermectin, scutellarein and myricetin showcases the highest binding affinity among the selected inhibitors with binding affinity of −10, −9.9 and −9.7 kcal/mol, respectively, (Table 2). Among the nineteen compounds, seven chemical entities had binding affinihes greater than or equal to −9 kcal/mol. These chemicals are ivermectin, scutellarein, myricelin, suramin, nogalamycin, doxorubrun and paclitaxel as shown by Table 2. Five of the compounds showcased binding affinities greater than or equal to—8 kcal/mol. Ribavirin 5-triphosphate had a binding affinity of −7.8 kcal/mol while DRBT showed binding energy of −7.4 kcal/mol Rest of the five compounds (famciclovir, compound 16, valayclovir, tetrabrobenzotriazde and SSYA 10) showed binding energy in the range −6.7 kcal/mol to −5.9 kcal/mol.
Table 2.
The binding affinities and Oral LD50 (rat) values of selected helicase inhibitors against NSP13
| Ligand (Pubchem ID) | Binding affinity (kcal/mol) | Oral LD50 (rat) (mg/kg) |
|---|---|---|
| Ivermectin (CID6321424) | −10 | 29.69 |
| Scutellarein (CID5281697) | −9.9 | 1854.96 |
| Myricetin (CID5281672) | −9.7 | 1251.16 |
| Suramin (CID5361) | −9.6 | 11,671.01 |
| Nogalamycin (CID5289019) | −9.4 | 171.21 |
| Doxorubicin (CID31703) | −9 | 859.5 |
| Paclitaxel (CID36314) | −9 | 316.76 |
| SSYA10-002 (CID2826467) | −8.8 | 715.56 |
| Amenamevir (CID11397521) | −8.6 | 291.04 |
| ST-193 (CID349985067) | −8.3 | 2790.18 |
| Ribavirin 5'-diphosphate (CID124970) | −8 | 4289.2 |
| Pritelivir (CID491941) | −8 | 115.04 |
| Ribavirin 5'-triphosphate (CID12210) | −7.8 | 1531.84 |
| DRBT (CID506146) | −7.4 | 3023.69 |
| Famciclovir (CID3324) | −6.7 | 3391.22 |
| GTPL10881-Compound-16 (CID146170994) | −6.7 | 722.23 |
| Valacyclovir (CID135398742) | −6.2 | 288.98 |
| 4,5,6,7-Tetrabromobenzotriazole (CID1694) | −6.1 | 48.65 |
| SSYA10-001 (CID2807230) | −5.9 | 1473.61 |
Pharmacophore mapping and virtual screening reveals potential inhibitors for targeting NSP16
Structure-based pharmacophore 3D mapping method is among one of the quintessential techniques to aid in drug development. Virtual screening of large databases can prove to be an excellent way to identify the compounds which have highest probability to interact and subsequently bind to the protein of interest [21, 61, 62].
We generated structure-based target-ligand complex pharmacophore of NSP16 in LigandScout® [30]. After performing screening operations, a total of 18 out of 11,325 molecules with similar pharmacophore generating properties were found. The hit rate was approximately 0.1589%. The Pharmacophore-fit score of the hit compounds ranged from 80 to 90%.
Molecular docking of NSP16 with screened natural compounds
After performing pharmacophore-based screening, there were 18 hit compounds. In order to explore whether our selected 18 compounds can potentially interact with NSP16, we performed molecular docking using Autodock Vina® (Table 3). In our analysis, we found that hamamelitannin (Amb21855910), deacyltunicamycin (Amb23438471) and desferrichrome (Amb8397892) showcase the highest binding affinity among the docked compounds with binding affinity of −8.9, −8.4 and −8.4 kcal/mol, respectively (Table 3). Among the 18 compounds, ten chemical entities had binding affinities greater than or equal to −7 kcal/mol. These chemicals are hamamelitannin (Amb21855910), deacyltunicamycin (Amb23438471), desferrichrome (Amb8397892), validamycin A (Amb22731536), cyanidin 3-xylosyl (feruloylglucosyl)galactoside (Amb24326030), pulchinenoside E1 (Amb29085853), MCULE-3415537068 (Amb15770173), sinefungin (Amb23438712), s-(5'-Adenosyl)-L-homocysteine (Amb6364853), hygrovetine (Amb10845248) and kukoamine A (Amb22584539) as shown by Table 3.
Table 3.
The binding affinities and LD50 values of selected helicase inhibitors against Oral LD50 (rat) 16
| Ligand (Ambinter* ID) | Binding affinity (kcal/mol) | Oral LD50 (rat) mg/kg |
|---|---|---|
| Hamamelitannin(Amb21855910) | −8.9 | 5475.87 |
| Deacyltunicamycin(Amb23438471) | −8.4 | 5891.48 |
| Desferrichrome (Amb8397892) | −8.4 | 17,228.07 |
| Validamycin A (Amb22731536) | −7.9 | 17,385.06 |
| Cyanidin 3-xylosyl(feruloylglucosyl)galactoside (Amb24326030) | −7.9 | N/A |
| Pulchinenoside E1 (Amb29085853) | −7.6 | 127.08 |
| MCULE-3415537068 (Amb15770173) | −7.5 | 5132.52 |
| Sinefungin (Amb23438712) | −7.5 | 1165.1 |
| S-(5'-Adenosyl)-L-homocysteine (Amb6364853) | −7.4 | 612.84 |
| Hygrovetine (Amb10845248) | −7 | 17,143.69 |
| Kukoamine A (Amb22584539) | −6.9 | 797.19 |
| S-Lactoylglutathione (Amb28974356) | −6.9 | 3790.26 |
| MCULE-8740758758 (Amb24326044) | −5.9 | 3356.94 |
| QUISQUALIC ACID (Amb19133767) | −5.8 | 5326.58 |
| DL-Methyldopa (Amb3940868) | −5.6 | 4141.93 |
| L-Homoarginine hydrochloride (Amb19132515) | −4.7 | 1192.18 |
| L-Citrulline (Amb2718751) | −4.6 | 4462.25 |
| L-Cystine-dimethyl Ester Dihydrochloride (Amb8514731) | −4.1 | N/A |
The database is used for high-throughput screening and as building blocks for combinatorial chemistry
N/A not applicable
*Ambinter database includes a library of over 36 million advanced chemicals for drug discovery applications and purposes.
Intermolecular interaction study of docked complexes for both NSP13 and NSP16
Intermolecular interaction of protein–ligand docked complexes were studied using Discovery studio software. In case of both NSP13 and NSP16, ligand protein–ligand interactions were analyzed for all the selected compounds. The result of the molecular interaction study for NSP13 with the nineteen helicase inhibitors are shown in Table 4. The NTPase activity of the helicase enzyme in SARS-CoV is coordinated in a cleft at the base situated between 1 and 2A domains. It consists of six important residues such as Lys288, Ser289, Asp374, Glu375, Gln404 and Arg567 [63]. Some of our selected inhibitors interact with the residues important for NTPase activity (Table 3). For example, Ribavirin di-phosphode interacts with K288, S289 Q404 and R567 of these six important amino acid residues. Ribavivin triphosphate interacts with the residues K288, E375 and R567. Doxorubicin interacts with S289 while valaciclovir forms bond with S289 and D374. The chemical entity GTPL 10,881- Compound 16 showcases interaction with D374. Also, SSYA 10,881-001 forms bond with the amino acid residues D374. Ivermectin forms three hydrogen bonds at the position A135, D383 and H230 with NSP13 (Fig. 3A). The distance of these hydrogen bonds between drug and protein are as follows 2.1369 Å, 3.6768 Å and 3.5106 Å, respectively. It also forms 4 alkyl and 2 π-alkyl bonds. Scutellarein forms three hydrogen bonds at the position Y120, Y421 and T380. The distance of these hydrogen bonds between drug and protein are as follows 2.3066 Å, 2.0207 Å and 3.40944 Å, respectively. Alongside, it forms two π-alkyl, one π-σ and one π–π T-shaped bond (Fig. 3B). Myricetin forms three hydrogen bonds at the position F422 Y421, Y120 and T380 with the target macromolecule (Table 4).
Table 4.
Interaction of ligand molecules with NSP13 macromolecule
| Ligand (Pubchem ID) | Hydrogen bond | Hydrophobic interaction | ||||||
|---|---|---|---|---|---|---|---|---|
| Alkyl | π-alkyl | π-σ | π-πT-shaped | π-Cation | π-anion | π-sulfur | ||
| Ivermectin (CID6321424) | A135, D383, H230 | A338, V181, K139, L138 | H311, Y382 | |||||
| Scutellarein (CID5281697) | Y120, Y420, T380 | L138, K139 | L138 | Y120 | ||||
| Myricetin (CID5281672) | F422, Y421, Y120, T380 | L138, T380 | Y120 | |||||
| Suramin (CID5361) | A407, T410, L412, T413, | L405 | P408 | Y515 | R560 | D534 | Y515 | |
| S486, N516, H554, R560, P175 | ||||||||
| Nogalamycin (CID5289019) | A407, N516, R560, D534 | P175 | P408, Y515, H554 | |||||
| Doxorubicin (CID31703) | Y324, S289, G285 | H290 | G538 | |||||
| Paclitaxel (CID36314) | N179, R409, T380 | K139 | A407, L139 | T380 | Y120 | D383 | ||
| SSYA10-002 (CID2826467) | T380, T410, P172 | R409 | E142 | |||||
| Amenamevir (CID11397521) | T410, E142, F145 | A407, K146, P408, Y180 | ||||||
| ST-193 (CID349985067) | T380 | L138, R409, L411 | K139, R409, A135, Y120 | L138 | E143 | |||
| Ribavirin 5'-diphosphate (CID124970) | P283, T286, S289, G285, Q404, T566, R567, K288, P284, H290 | |||||||
| Pritelivir (CID491941 | AR339, N361, T228, N179 | R409, V181 | K146 | V181 | ||||
| Ribavirin 5'-triphosphate (CID122108) | K288, E375, G282, P283, G285, R567 | |||||||
| DRBT (CID506146) | T286, E540, E375 | L317, K320 | R443 | R567 | ||||
| Famciclovir (CID3324) | N179, R409, T410, E143, T228, E142 | |||||||
| GTPL10881-Compound-16 (CID146170994) | T380, M380, D383 | A407, R409 | T380 | D374 | M378 | |||
| Valaciclovir (CID135398742) | K320, D374, G282, S289, G400 | A316, L317 | ||||||
| 4,5,6,7-Tetrabromobenzotriazole (CID1694) | Q531, T530, D204, S523 | L526, P529 | P529 | E201, D204 | ||||
| SSYA10-001 (CID2807230) | G538, R567 | A313, A316, L317, P284 | D374 | |||||
Fig. 3.
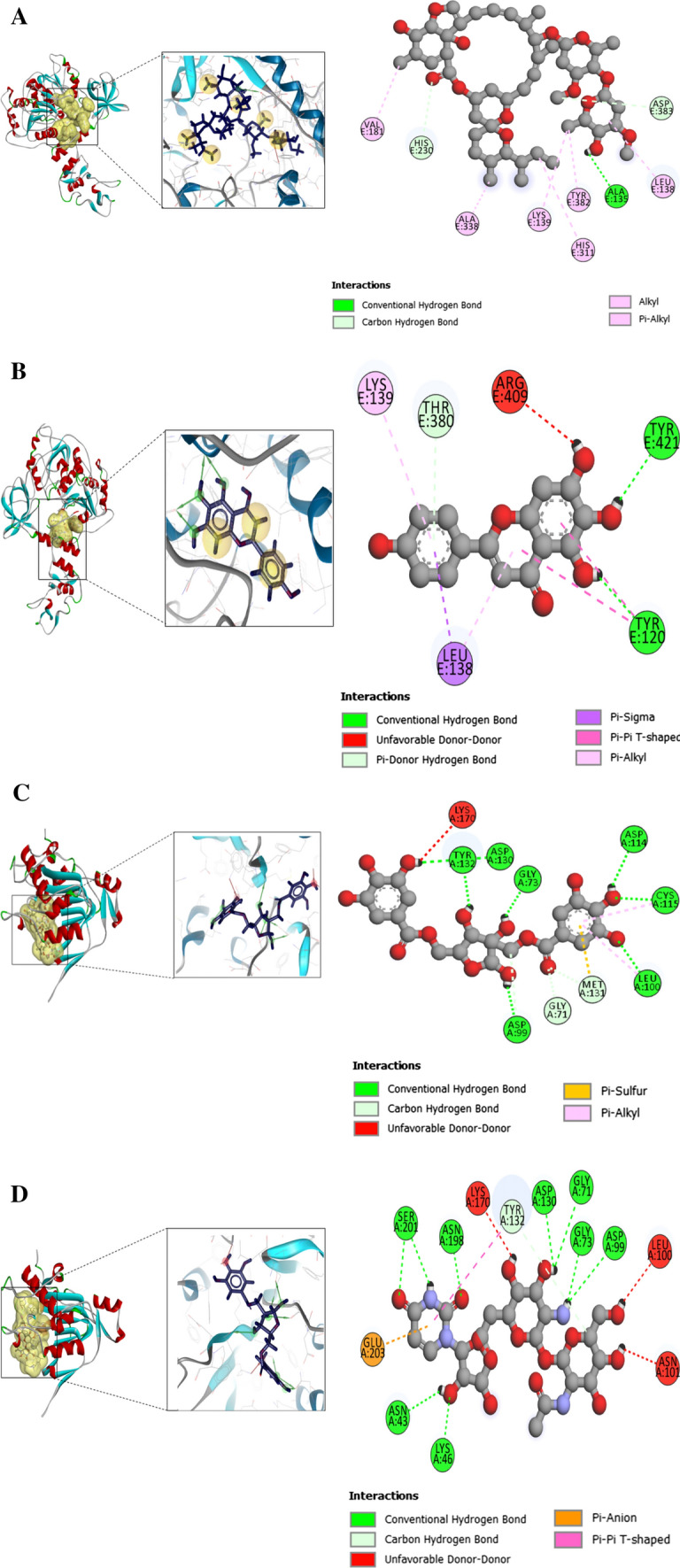
Protein–ligand interaction for the SARS-CoV-2 NSP13 and NSP16. A Protein–ligand interaction of the SARS-CoV-2 NSP13 and ivermectin after completion of docking operation in Autodock Vina. The interaction in the docked complex was visualized using Discovery Studio. B Protein–ligand interaction of the SARS-CoV-2 NSP13 and scutellarein after completion of docking operation in Autodock Vina. C Protein–ligand interaction of the SARS-CoV-2 NSP16 and hamamelitannin after completion of docking operation in Autodock Vina. The interaction in the docked complex was visualized using Discovery Studio. D Protein–ligand interaction of the SARS-CoV-2 NSP16 and deacyltunicamycin after completion of docking operation in Autodock Vina. The interaction in the docked complex was visualized using Discovery Studio
The four amino acid residues which are particularly important for the enzyme (NSP16) to exhibit its MTase activity are K46, D130, K170, E203, together known as the KDKE motif in SARS coronavirus. The motif is highly conserved between SARS coronavirus and SARS-CoV-2. In our analysis as viewed in Discovery Studio® for NSP16 and the eighteen hit compounds, hamamelitannin forms hydrogen bond with D130 residue of the KDKE motif in NSP16 protein (Fig. 3C). The distance of these hydrogen bonds between drug and protein is as follows 2.65567 Å. Deacyltunicamycin forms hydrogen bond with D130, K46 and E203(Fig. 3D). The distance of these hydrogen bonds between drug and protein are as follows 2.194 Å, 3.194 Å and 3.7528 Å, respectively. Like the above two compounds, we also found strong interactions between NSP16 and our selected molecules. For example, desferrichrome forms hydrogen bond with D130 and K170 while validomycin A forms hydrogen bond with D130 and K70 (Table 5).
Table 5.
Interaction of ligand molecules with NSP13 macromolecule
| Ligand(Ambinter ID) | Hydrogen bond | Hydrophobic interaction | ||||||
|---|---|---|---|---|---|---|---|---|
| Alkyl | π-alkyl | π-σ | π-π T-shaped | π-Cation | π-anion | π-sulfur | ||
| Hamamelitannin (Amb21855910) | G73, D114, Y132, D99, D130, L100, C115, G71, M131 | L100, C115 | M131 | |||||
| Deacyltunicamycin (Amb23438471) | N43, S201, G71, D130, G73, D99, K46, N198, Y132 | Y132 | E203 | |||||
| Desferrichrome (Amb8397892) | D130, Y30, D32, N43, K170, N198, S201, S202 | |||||||
| Validamycin A (Amb22731536) | D130, G71, N43, Y132, D99, K170 | |||||||
| Cyanidin 3-xylosyl(feruloylglucosyl)galactoside (Amb24326030) | Q18, E217, Y53, N13, W190, W58 | M189 | W189 | |||||
| Pulchinenoside E1 (Amb29085853) | K123, N286, A121 | L163, K263 | Y211 | |||||
| MCULE-3415537068 (Amb15770173) | D130, G73, G71, Y47, N101, Y132 | L100 | M131 | |||||
| Sinefungin (Amb23438712) | N43, G73, D99, D114, G113, D130, L100, G71 | |||||||
| S-(5'-Adenosyl)-L-homocysteine (Amb6364853) | D130, G71, D99, D114, L100 | |||||||
| Hygrovetine (Amb10845248) | Y132, D114, G71, L100, S98, G113 | |||||||
| Kukoamine A (Amb22584539) | D114, G73, D133, E142, L100 | K146 | K146 | D99 | ||||
| S-Lactoylglutathione (Amb28974356) | G73, D130, Y47, D99, G71, L100, N43, K46, K170 | |||||||
| MCULE-8740758758 (Amb24326044) | Y211, G213, L161 | I267 | ||||||
| QUISQUALIC ACID (Amb19133767) | W5, S243, K249, Q52 | |||||||
| DL-Methyldopa (Amb3940868) | A121, E284, R66, K123 | K123 | E284 | |||||
| L-Homoarginine hydrochloride (Amb19132515) | A121, E284, R66, L262, K123 | |||||||
| L-Citrulline (Amb2718751) | M20, N143, D144 | |||||||
| L-Cystine-dimethyl Ester Dihydrochloride (Amb8514731) | V118, Y152, T151, H119, Q159, T120 | A116 | H119,Y152 | F156 | ||||
Selected compounds showed favorable ADME and toxicity (ADMET) profiles
Every drug substance undergoes absorption, distribution, metabolism, excretion after administration into human body. Desirable or undesirable physiological or pharmacological effect can result from interactions with the target biological macromolecules with the drug. Thus safety and efficacy of the drug greatly depends on its ADME properties. So in this study the ADME properties of the selected compounds were evaluated by using in silico Swiss-ADME [36] (www.swissadme.ch/index.php) tool to see the important ADME parameters such as molecular weight, number of H-bond acceptors, number of H-bond donors, number of rotatable bonds, ESOL Class, iLOGP, TPSA, GI absorption, bioavailability score, number of Lipinski violations, BBB permeant, number of lead-likeness violations and synthetic accessibility (Supplementary Table S3 and S4).
We evaluated toxicity of these potential inhibitors of NSP13 and NSP16. The LD50 values of all of our studied compound were calculated using T.E.S.T software (Tables 2, 3). Usually, the smaller the value of LD50, the more toxic is the chemical. Acute toxicity threshold, which is for Oral LD50 (rat), is considered below 56 mg/kg. Most of the compound shows low toxicity in our analysis.
Selected molecules possess better drug-likeness properties
The bioavailability radar gives graphical interpretation including lipophilicity, compound size, insolubility, polarity, insaturation and flexibility in its six hexagonal vertices [36]. A compound may be considered to have drug-like properties if these parameters are balanced. The balance of these parameters can be understood by studying the graphical illustration of the bioavailability radar. The vertices represent drug-likeness parameters of a chemical compound when these remain within the first tier of the hexagonal structure [36]. For a comprehensive review we looked at the bioavailability radar from the Swiss-ADME for our top two compounds of NSP13 and NSP16.
The bioavailability radar (Fig. 4) shows a good balance of the parameters for scutellarein and hemamelitannin. In case of deacyltunicamycin, the size and polarity is over the edge of acceptable level. Ivermectin too is a high molecular weight compound with low solubility. However, drugs with high polarity and high molecular weight can be developed into drug candidates after formulation amendments and indicating good ADME and bioavailability properties.
Molecular dynamics simulation reveals stable complex formed by NSP13 and NSP16 with respective helicase inhibitors
To understand the stability of our top hit drugs (scutellarein, hamamelitannin and deacyltunicamycin) with NSPs, we performed molecular dynamics (MD) simulation (Fig. 5). RMSD plots shows that the system mostly became stabilized after 2 ns of simulation and tended to remain in the plateau phase thereafter for the rest of the period. Growing up swiftly, the RMSD value of protein was remained stable in the range from 0.2–0.3 nm. The average RMSF value was around 0.15 nm in all cases. We found that the SASA of the complex is less than that of protein alone—which indicates that the protein is connected to the ligand. The radius of gyration (Rg) plots explains structure compactness. The protein can be assumed to have a stably folded structure as evident from a relatively steady Rg value. The average Rg of the protein and ligand complexes was around 2.0 nm and quite stable over the time. All these trajectories pointing stable interaction with target molecules and can be said as direct target of these drugs. The potential, temperature, pressure, density, total energy, hydrogen bonds, hydrogen bond distribution and radial distribution from MD analyses for the three drugs were also calculated in this study (Supplementary figures S3, S4 and S5). The parameters utilized in the simulated environment during molecular dynamics simulation are represented in Supplementary Tables (S4, S5 and S6) All these results indicate stability of the complex.
Fig. 5.

Trajectory plots of Molecular Dynamics (MD) of target protein-drug complex. Plot of radius of gyration (Rg) (1st column panel), root mean square deviation (RMSD) (2nd column panel), root mean square fluctuation (RMSF) (3rd column panel), solvent accessible surface area (SASA) with respect to time (nanoseconds) (4th column panel) during MD simulation of NSP13 complexed with scutellarein (1st row) and NSP16 complex with deacyltunicamycin (2nd row) and hamamelitannin drugs. Green color line indicates protein, blue indicates drug and red color indicates protein-drug complex together
Discussion
Identification of chemical entities that bind to and inhibit the viral proteins involved in viral replication mechanism can be considered as one of the most effective strategy in antiviral drug discovery [64, 65]. Some researchers have adopted computational drug discovery methodology to hunt for molecules that can inhibit other SARS-2-Coronaviral proteins’ function [66–70]. Recent work by researchers showed that some drugs like Ritonavir, Lopinavir and Favipiravir were repurposed to treat COVID-19 and are under clinical trials at present [71]. Romeo et al. collected natural products which can be considered as potential candidates for developing drugs that can target SARS-CoV-2 proteins and boost immunity of host body. Several natural compounds such as Lactoferrin, Berbrine, Quercetin, Hanfangchin, Artemisinin, Glycyrrhizin, Cholchicine, Resveratrol and Vitamin C and Vitamin D showed effectivity against SARS-CoV-2 as per their review work and these compounds are in clinical trials at present [72]. In another review work, natural products and their derivatives such as silvestrol, homoharringtonine, lycorine, tylophorine, ouabain and 7-methoxycryptopleurine were reported to inhibit SARS-Coronavirus and its significant proteins [40]. However, till recent times, none of the chemical compounds have been identified as specific inhibitors for COVID-19 viral infection. Significant research is ongoing in this field to explore novel compounds as well as exploit scope to repurpose currently marketed drugs against SARS-CoV-2 to manage and treat the viral infection.
In this pandemic situation, computer-aided drug design can prove to be an effective and time-saving alternative to cumbersome screening of huge number of compounds using laboratory assays. Computational drug discovery methodology can be utilized to identify potential inhibitors that can bind and inhibit the viral proteins involved in viral replication mechanism [73]. Molecular docking, pharmacophore-based virtual screening, target identification and molecular dynamics simulation are essential to study and analyze potential compounds against druggable target proteins. Molecular docking is used to study and predict binding patterns and affinities of the ligands investigated for proposed target receptor proteins [74]. Structure-based molecular interaction evaluation is needed to get a better perception of the interaction of bioactive compounds with target receptors [75].
Viral proteins with high mutation rate among viral strains are poor choice as druggable targets. The reason behind this is the high probability of escape mutation shown by druggable target protein if the protein has a tendency to mutate constantly. Thus, drug resistance to the chemical entities targeting viral proteins with high mutation rate can be a serious problem. Certain proteins like the spike protein of SARS-2-Coronavirus is known to exhibit high variability among coronavirus species. However, functionally vital protein such as NSP13 is highly conserved which opens doors for investigating the effect of helicase inhibitors in closely related species of coronavirus on SARS-2 NSP13. Previous work by scientists on viral helicase enzymes can prove to be an excellent source for searching effective chemical species against COVID-19.
Non-structural protein 13 is a polypeptide chain consisting of 601 amino acid residues [63]. In SARS-Coronavirus, the five domains including zinc-binding domain, 1A domain, 1B domain, stalk domain and 2A domain are shown to function by coordinating with each other in order to accomplish the final unwinding process [76]. Many pathogenic viruses code for RNA helicases which are known to be essential for viral replication and pathogenesis [77, 78].
Yua et.al performed chemical assays on 66 natural compounds to analyze the effect of these compounds on inhibition of SARS NSP13 ATP hydrolysis activity. They found that at a concentration level of 10 μM, myricetin and scutellarein stopped the ATPase activity of NSP13 by more than 90% In further laboratory assessment, they exposed normal breast epithelial MCF 10A cell line to scutellarein or myricetin and showed that neither scutellarein nor myricetin affect the growth of the MCF 10A cells at concentrations close to their IC50 values. Thus, both myricetin and scutellarein can be used safely at concentrations required for their efficacy [79]. Our findings also suggest strong affinity between these two inhibitors and SARS-CoV-2 helicase and we suggest further investigation of ivermectin and these two compounds. Ivermectin is a broadly used anthelminthic drug that was discovered using molecular modeling to be a potent helicase inhibitor of YFV replication (sub-nanomolar EC50 values), as well as other flaviviruses such as DENV (sub-micromolar values), JEV and TBEV. Recently, ivermectin was shown to reduce SARS-CoV-2 RNA by approximately 5000 folds at 48 h [52].
NSP16 of SARS-CoV-2 lies on the range of 6799–7096 on orf1ab residues, encoding 298 amino acids The NSP16 protein structure as observed in SARS Coronavirus, showcases the typical folding of Type I methyltransferase family viruses In NSP16, the range of amino acid molecules, 30-209, form the main MTase domain [15, 80].
Previous research on SARS and MERS Coronaviral NSP16 showed that by mutating the KDKE motif, the viral strains can be weakened both in vivo and in vitro [81]. The viruses devoid of MTase activity were more sensitive to IFN-treatment than fully active viruses. The mutant viral strains could be recognized by host Mda5 (cytoplasmic RNA sensor) [81]. Thus, any change of amino acid sequence on the junction of NSP16 and NSP10 can be predicted to affect binding of substrates and alter the MTase activity of NSP16 [15]. The experimentally solved X-ray Crystallography structures of NSP10-16 complex (PDB ID: 6W4H, 6W61 and 6W75) revealed that the KDKE (K46, D130, K170, E203) motif, involved in methyl transfer by 2’-O-MTases, is highly conserved in SARS-CoV-2 strain [76, 82]. The residues N43, Y47, G71, A72, S74, G81, D99, N101, L100, D114 and M131, that contribute to SAM substrate binding in SARS-CoV-1 through water-mediated interactions and by the formation of hydrogen bonds are also fully conserved in SARS-CoV-2 [82]. The substrate m7GpppA-RNA and s-adenosyl-l-methionine can be bound together by NSP10/16 complex in order to initiate and implement 2'-O-MTase activity [15]. Therefore, these residues are important targets to stop NSP16 MTase activity. Any ligand showing interaction with these residues can be predicted to stop the activity of NSP16 protein. In our analysis as viewed in Discovery Studio®, hamamelitannin forms hydrogen bond with D130 residue of NSP16 protein (Fig. 3C). Previously, it inhibited neuraminidase activity of human papillomavirus and influenza A virus [83].
Deacyltunicamycin is a derivative of tunicamycin produced by strains of Streptomyces chartreusis [84]. Tunicamycin previously showed strong antiviral activity against enveloped RNA viruses, e.g., vesicular stomatitis and Sindbis viruses by halting multiplication and glycosylation of the proteins intended for the viral envelope [85]. Tunicamycin (TM) is a glucosamine-containing antibiotic with potent antiviral activity. In studies with Newcastle disease virus (NDV), it has been found that tunicamycin interferes with glycoprotein biosynthesis. We found that it forms hydrogen bond with D130, K46 and E203 [86]. Hydrogen bond plays an important role in stability of enzyme-inhibitor complex [87, 88]. Since deacyltunicamycin forms hydrogen bonds with some of the residues of KDKE (K46, D130, K170, E203) motif, it can be assumed that the compound will be effective in stopping the NSP16 MTase activity.
The absorption, distribution, metabolism and excretion (ADME) profile of a chemical entity provides integral information on its use inside animal models and subsequently inside human bodies to render effective and safe therapeutic efficacy. After administration of drug component through any route inside animal model or human body, interaction with the target macromolecules might produce desirable or in some cases undesirable pharmacological effect [89]. The bioavailability of an administered drug depends on a number of factors which can be evaluated by analyzing the ADME properties of the potential drug candidate. The Lipinski rule of five is considered as a useful parameter to understand the “drug-likeness” of a candidate compound [90]. Alongside this, safety and toxicity profiles of any chemical entity is equally important to understand their utilization as drug molecules [89]. In silico models are nowadays used to predict the toxicity profile of novel chemical compounds [39]. Usually, the smaller the value of LD50, the more toxic is the chemical. Acute toxicity threshold, which is for Oral LD50 (rat) is considered below 56 mg/kg [89]. Most of the compound shows low toxicity in our analysis. Although ivermectin has hepatotoxicity and low LD50 value, it is counterbalanced by the dosage regimen and its high absorbance. Additionally, myricetin, scutellarein, hamamelitannin and deacyltunicamycin show low toxicity as per our toxicity analysis.
Molecular dynamics (MD) simulation is eventually becoming an important tool for analyzing biomolecular configuration and dynamics, the relationship between which is crucial for understanding significant cellular processes [41]. Molecular dynamics simulation studies were performed to evaluate the stability, molecular interactions and binding mode of potential inhibitors of NSP13 and NSP16 that were selected from molecular docking analysis. MD simulations were performed over a timeframe of 50 ns (ns). From these analyses, radius of gyration (Rg), root mean square deviation (RMSD), root mean square fluctuation (RMSF) and solvent accessible surface area (SASA) with respect to time (in nanoseconds) of NSP13 complexed with scutellarein and NSP16 complexed with deacyltunicamycin and hamamelitannin drugs were generated. The MD simulation trajectory showed that the compound scutellarein displayed excellent stability with the protein complex of NSP13 in the simulated environment. Hamamelitannin and deacyltunicamycin with NSP16 complex was checked out for stability via Molecular Dynamic simulation and these compounds showed subsequent stability in the simulated environment.
Conclusion
In the present study, computational approaches have been utilized to identify potential compounds that have the capacity to inhibit functionally vital NSP13 and NSP16 proteins of SARS-CoV-2. These two proteins are highly conserved among beta lineage of coronaviruses as shown in this study. Moreover, these proteins exhibit lowest mutation rate among other viral proteins of SARS-CoV-2. Thus, NSP13 and NSP16 of SARS-2-CoV can be considered as good druggable targets. In this research work, chemical entities have been identified that can bind to and restrict the functionality of these proteins. The binding affinities and toxicity analyses done in this study indicate that ivermectin, scutellarein, hamamelitannin and deacyltunicamycin have very good potentials as antiviral medicines against SARS-CoV-2, the culprit of COVID-19 pandemic. The outcome of molecular dynamics simulation indicates stable interaction of the aforementioned molecules with respective viral protein targets inside human bodies. The results of this study may be beneficial for future pre-clinical and clinical evaluations. The overall findings of the study will help in the development of potential inhibitors that will inhibit SARS-CoV-2 reproduction.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
We acknowledge high performance computing support from Centre for Bioinformatics Learning Advancement and Systematics Training (cBLAST), University of Dhaka for MD analysis. We also acknowledge support of Biomolecular Research Foundation (BMRF), Dhaka, Bangladesh.
Author contribution
MNS, NM, RR and MA contributed to idea generation and manuscript writing. MNS, NM, RR performed the docking analyses and generated the data. ABMMKI curated MD simulation data. MA supervised the work, and MA and ABMMKI reviewed the manuscript, edited the draft manuscript and ABMMKI curated molecular dynamic simulation data.
Funding
This research did not receive any grant from any public, commercial, or not-for-profit funding agencies.
Declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Md. Nazmus Samdani, Niaz Morshed, and Rumman Reza have contributed equally to this work.
Contributor Information
Muhammad Asaduzzaman, Email: asaduzzaman@du.ac.bd.
Abul Bashar Mir Md. Khademul Islam, Email: khademul@du.ac.bd.
References
- 1.Cucinotta D, Vanelli M (2020) WHO declares COVID-19 a pandemic. Acta Biomed [DOI] [PMC free article] [PubMed]
- 2.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/nejmoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan JFW, Yuan S, Kok KH, To KKW, Chu H, Yang J, Xing F, Liu J, Yip CCY, Poon RWS, Tsoi HW, Lo SKF, Chan KH, Poon VKM, Chan WM, Ip JD, Cai JP, Cheng VCC, Chen H, Hui CKM, Yuen KY. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sohrabi C, Alsafi Z, O’Neill N, Khan M, Kerwan A, Al-Jabir A, Iosifidis C, Agha R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19) Int J Surg. 2020;76:71–76. doi: 10.1016/j.ijsu.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Malone B, Llewellyn E, Grasso M, Shelton PMM, Olinares PDB, Maruthi K, Eng ET, Vatandaslar H, Chait BT, Kapoor TM, Darst SA, Campbell EA. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020;182:1560–1573.e13. doi: 10.1016/j.cell.2020.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Wang Y, Ye D, Liu Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int J Antimicrob Agents. 2020;55:105948. doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshimoto FK. The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. 2020;39:198–216. doi: 10.1007/s10930-020-09901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Astuti I, Ysrafil (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab Syndr 14:407–412. 10.1016/j.dsx.2020.04.020 [DOI] [PMC free article] [PubMed]
- 10.Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene reports. 2020;19:100682. doi: 10.1016/j.genrep.2020.100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang J, Wang MX, Ang IYH, Tan SHX, Lewis RF, Chen JI-P, Gutierrez RA, Gwee SXW, Chua PEY, Yang Q, Ng XY, Yap RKS, Tan HY, Teo YY, Tan CC, Cook AR, Yap JC-H, Hsu LY. Potential rapid diagnostics, vaccine and therapeutics for 2019 novel coronavirus (2019-nCoV): a systematic review. J Clin Med. 2020;9:623. doi: 10.3390/jcm9030623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Grotthuss M, Wyrwicz LS, Rychlewski L. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viswanathan T, Arya S, Chan SH, Qi S, Dai N, Misra A, Park JG, Oladunni F, Kovalskyy D, Hromas RA, Martinez-Sobrido L, Gupta YK. Structural basis of RNA cap modification by SARS-CoV-2. Nat Commun. 2020;11:4–10. doi: 10.1038/s41467-020-17496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morales P, Curtis NL, Zárate SG, Bastida A, Bolanos-Garcia VM. Interfering with mrna methylation by the 2′ o-methyltransferase (NSP16) from sars-cov-2 to tackle the covid-19 disease. Catalysts. 2020;10:1–13. [Google Scholar]
- 15.Lin S, Chen H, Ye F, Chen Z, Yang F, Zheng Y, Cao Y, Qiao J, Yang S, Lu G. Crystal structure of SARS-CoV-2NSP10/NSP16 2′-O-methylase and its implication on antiviral drug design. Signal Transduct Target Ther. 2020;5:5–8. doi: 10.1038/s41392-020-00241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang KJ, Jeong S, Kang DY, Sp N, Yang YM, Kim DE. A high ATP concentration enhances the cooperative translocation of the SARS coronavirus helicase NSP13 in the unwinding of duplex RNA. Sci Rep. 2020;10:1–13. doi: 10.1038/s41598-020-61432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sargsyan K, Chen T, Grauffel C, Lim C (2020) Identifying COVID-19 Drug-sites susceptible to clinically safe Zn-ejector drugs using evolutionary/physical principles. 1–10
- 18.Borowski P, Mueller O, Niebuhr A, Kalitzky M, Hwang LH, Schmitz H, Siwecka MA, Kulikowski T (2000) ATP-binding domain of NTPase/helicase as a target for hepatitis C antiviral therapy. Acta Biochim Pol 47:173–180. 10.18388/abp.2000_4075 [PubMed]
- 19.De Clercq E, Field HJ. Antiviral prodrugs - the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. 2006;147:1–11. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon CP, Keller PA. Control of hepatitis C: a medicinal chemistry perspective. J Med Chem. 2005;48:1–20. doi: 10.1021/jm0400101. [DOI] [PubMed] [Google Scholar]
- 21.Rafi JH, Jafar T, Pathan MT, Reza R, Islam S, Sourna IJ, Alam R, Samad A, Ahammad F (2021) High expression of bone morphogenetic protein 1 (BMP1) is associated with a poor survival rate in human gastric cancer, a dataset approaches. Genomics 113:1141–1154. 10.1016/j.ygeno.2020.11.012 [DOI] [PubMed]
- 22.Morse JM. Determining sample size. Qual Health Res. 2000;10:3–5. doi: 10.1177/104973200129118183. [DOI] [Google Scholar]
- 23.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6 . 0. 30:2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed]
- 24.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahammad F, Alam R, Mahmud R, Akhter S, Talukder EK, Tonmoy AM, Fahim S, Al-Ghamdi K, Samad A, Qadri I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief Bioinform. 2021 doi: 10.1093/bib/bbab098. [DOI] [PubMed] [Google Scholar]
- 26.Lu S-H, Wu JW, Liu H-L, Zhao J-H, Liu K-T, Chuang C-K, Lin H-Y, Tsai W-B, Ho Y. The discovery of potential acetylcholinesterase inhibitors: a combination of pharmacophore modeling, virtual screening, and molecular docking studies. J Biomed Sci. 2011;18:8. doi: 10.1186/1423-0127-18-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaker B, Yu M-S, Lee J, Lee Y, Jung C, Na D. User guide for the discovery of potential drugs via protein structure prediction and ligand docking simulation. J Microbiol. 2020;58:235–244. doi: 10.1007/s12275-020-9563-z. [DOI] [PubMed] [Google Scholar]
- 28.Tallei TE, Tumilaar SG, Niode NJ, Fatimawali KBJ, Idroes R, Effendi Y, Sakib SA, Bin ET. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica (Cairo) 2020 doi: 10.1155/2020/6307457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. In: Chemical biology. Springer, pp 243–250 [DOI] [PubMed]
- 30.Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 31.Adedeji AO, Singh K, Calcaterra NE, DeDiego ML, Enjuanes L, Weiss S, Sarafianos SG. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob Agents Chemother. 2012;56:4718–4728. doi: 10.1128/AAC.00957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Zheng YG. SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chem Biol. 2016;11:583–597. doi: 10.1021/acschembio.5b00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 34.van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 35.Pradeepkiran JA, Kumar KK, Kumar YN, Bhaskar M. Modeling, molecular dynamics, and docking assessment of transcription factor rho: a potential drug target in Brucella melitensis 16M. Drug Des Devel Ther. 2015;9:1897–1912. doi: 10.2147/DDDT.S77020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin TM (2020) User’s Guide for T. E. S. T. (Toxicity Estimation Software Tool)
- 38.Hadni H, Elhallaoui M. 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J Chem. 2020;44:6553–6565. doi: 10.1039/C9NJ05767F. [DOI] [Google Scholar]
- 39.Raies AB, Bajic VB. In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci. 2016;6:147–172. doi: 10.1002/wcms.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zinnia MA, Islam ABMMK. Fenugreek steroidal saponins hinder osteoclastogenic bone resorption by targeting CSF-1R which diminishes the RANKL/OPG ratio. Int J Biol Macromol. 2021;186:351–364. doi: 10.1016/j.ijbiomac.2021.06.197. [DOI] [PubMed] [Google Scholar]
- 41.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 42.Huang J, MacKerell AD., Jr CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269–6271. doi: 10.1021/j100308a038. [DOI] [Google Scholar]
- 44.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 45.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 46.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 47.Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 48.Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, Kern R, Picus M, Hoyer S, van Kerkwijk MH, Brett M, Haldane A, del Río JF, Wiebe M, Peterson P, Gérard-Marchant P, Sheppard K, Reddy T, Weckesser W, Abbasi H, Gohlke C, Oliphant TE. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.R Core Team (2020)
- 50.Osorio D, Rondón-Villarreal P, Torres R (2015) Peptides: a package for data mining of antimicrobial peptides. R J 7:4. 10.32614/RJ-2015-001
- 51.Salmaso V, Moro S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: an overview. Front Pharmacol. 2018;9:923. doi: 10.3389/fphar.2018.00923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res. 2020;178:3–6. doi: 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subissi L, Imbert I, Ferron F, Collet A, Coutard B, Decroly E, Canard B (2020) Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID- 19 . The COVID-19 resource centre is hosted on Elsevier Connect, the company’s public news and information
- 54.Byrd CM, Grosenbach DW, Berhanu A, Dai D, Jones KF, Cardwell KB, Schneider C, Yang G, Tyavanagimatt S, Harver C, Wineinger KA, Page J, Stavale E, Stone MA, Fuller KP, Lovejoy C, Leeds JM, Hruby DE, Jordan R. Novel benzoxazole inhibitor of dengue virus replication that targets the NS3 helicase. Antimicrob Agents Chemother. 2013;57:1902–1912. doi: 10.1128/AAC.02251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davies RJ, Freeman A, Morris LS, Bingham S, Dilworth S, Scott I, Laskey RA, Miller R, Coleman N. Analysis of minichromosome maintenance proteins as a novel method for detection of colorectal cancer in stool. Lancet (London, England) 2002;359:1917–1919. doi: 10.1016/S0140-6736(02)08739-1. [DOI] [PubMed] [Google Scholar]
- 56.Zaher NH, Mostafa MI, Altaher AY. Design, synthesis and molecular docking of novel triazole derivatives as potential CoV helicase inhibitors. Acta Pharm. 2020;70:145–159. doi: 10.2478/acph-2020-0024. [DOI] [PubMed] [Google Scholar]
- 57.Borowski P, Schalinski S, Schmitz H. Nucleotide triphosphatase/helicase of hepatitis C virus as a target for antiviral therapy. Antiviral Res. 2002;55:397–412. doi: 10.1016/S0166-3542(02)00096-7. [DOI] [PubMed] [Google Scholar]
- 58.Borowski P, Niebuhr A, Schmitz H, Hosmane RS, Bretner M, Siwecka MA, Kulikowski T (2002) NTPase/helicase of Flaviviridae: Inhibitors and inhibition of the enzyme. Acta Biochim Pol 49:597–614. 10.18388/abp.2002_3769 [PubMed]
- 59.Bretner M, Najda A, Podwińska R, Baier A, Paruch K, Lipniacki A, Piasek A, Borowski P, Kulikowski T. Inhibitors of the NTPase/helicases of hepatitis C and related Flaviviridae viruses. Acta Pol Pharm. 2004;61(Suppl):26–28. [PubMed] [Google Scholar]
- 60.Bretner M, Baier A, Kopańska K, Najda A, Schoof A, Reinholz M, Lipniacki A, Piasek A, Kulikowski T, Borowski P. Synthesis and biological activity of 1H-benzotriazole and 1H-benzimidazole analogues - Inhibitors of the NTPase/helicase of HCV and of some related Flaviviridae. Antivir Chem Chemother. 2005;16:315–326. doi: 10.1177/095632020501600504. [DOI] [PubMed] [Google Scholar]
- 61.Gupta MK, Vemula S, Donde R, Gouda G, Behera L, Vadde R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1751300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tabassum A, Samdani MN, Dhali TC, Alam R, Ahammad F, Samad A, Karpiński TM. Transporter associated with antigen processing 1 (TAP1) expression and prognostic analysis in breast, lung, liver, and ovarian cancer. J Mol Med. 2021;1:1293–1309. doi: 10.1007/s00109-021-02088-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jia Z, Yan L, Ren Z, Wu L, Wang J, Guo J, Zheng L, Ming Z, Zhang L, Lou Z, Rao Z. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus NSP13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iftikhar H, Ali HN, Farooq S, Naveed H, Shahzad-Ul-Hussan S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput Biol Med. 2020;122:103848. doi: 10.1016/j.compbiomed.2020.103848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Hu T, Hua T, Zhang B, Yang X, Li J, Yang H, Liu Z, Xu W, Guddat LW, Wang Q, Lou Z, Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 68.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ton A-T, Gentile F, Hsing M, Ban F, Cherkasov A (2020) Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 Billion Compounds. Mol Inform 39:e2000028. 10.1002/minf.202000028 [DOI] [PMC free article] [PubMed]
- 70.Li J-Y, You Z, Wang Q, Zhou Z-J, Qiu Y, Luo R, Ge X-Y. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 2020;22:80–85. doi: 10.1016/j.micinf.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar V, Parate S, Yoon S, Lee G, Lee KW. Computational simulations identified marine-derived natural bioactive compounds as replication inhibitors of SARS-CoV-2. Front Microbiol. 2021;12:647295. doi: 10.3389/fmicb.2021.647295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Romeo I, Mesiti F, Lupia A, Alcaro S (2021) Current updates on naturally occurring compounds recognizing SARS-CoV-2 druggable targets. Mol. 26 [DOI] [PMC free article] [PubMed]
- 73.Beck BR, Shin B, Choi Y, Park S, Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adedeji AO, Marchand B, Te Velthuis AJW, Snijder EJ, Weiss S, Eoff RL, Singh K, Sarafianos SG. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PLoS ONE. 2012;7:e36521. doi: 10.1371/journal.pone.0036521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kleymann G, Fischer R, Betz UAK, Hendrix M, Bender W, Schneider U, Handke G, Eckenberg P, Hewlett G, Pevzner V, Baumeister J, Weber O, Henninger K, Keldenich J, Jensen A, Kolb J, Bach U, Popp A, Mäben J, Frappa I, Haebich D, Lockhoff O, Rübsamen-Waigmann H. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat Med. 2002;8:392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 78.Frick DN. Helicases as antiviral drug targets. Drug News Perspect. 2003;16:355. doi: 10.1358/dnp.2003.16.6.829307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao Y, Yuan Q, Torres J, Tam JP, Liu DX. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology. 2006;349:264–275. doi: 10.1016/j.virol.2006.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ashour HM, Elkhatib WF, Rahman MM, Elshabrawy HA. Insights into the recent 2019 novel coronavirus (Sars-coV-2) in light of past human coronavirus outbreaks. Pathogens. 2020;9:1–15. doi: 10.3390/pathogens9030186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Menachery VD, Yount BLJ, Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2’-o-methyltransferase activity. J Virol. 2014;88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bollati M, Milani M, Mastrangelo E, Ricagno S, Tedeschi G, Nonnis S, Decroly E, Selisko B, de Lamballerie X, Coutard B. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: implications for RNA-capping mechanisms in Flavivirus. J Mol Biol. 2009;385:140–152. doi: 10.1016/j.jmb.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 83.Theisen LL, Erdelmeier CAJ, Spoden GA, Boukhallouk F, Sausy A, Florin L, Muller CP. Tannins from Hamamelis virginiana bark extract: characterization and improvement of the antiviral efficacy against influenza A virus and human papillomavirus. PLoS ONE. 2014;9:e88062–e88062. doi: 10.1371/journal.pone.0088062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doroghazi JR, Ju K-S, Brown DW, Labeda DP, Deng Z, Metcalf WW, Chen W, Price NPJ. Genome sequences of three tunicamycin-producing Streptomyces Strains, S. chartreusis NRRL 12338, S. chartreusis NRRL 3882, and S. lysosuperificus ATCC 31396. J Bacteriol. 2011;193:7021–7022. doi: 10.1128/JB.06262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leavitt R, Schlesinger S, Kornfeld S. Tunicamycin inhibits glycosylation and multiplication of Sindbis and vesicular stomatitis viruses. J Virol. 1977;21:375–385. doi: 10.1128/jvi.21.1.375-385.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takatsuki A, Tamura G (1971) Effect of tunicamycin on the synthesis of macromolecules in cultures of chick embryo fibroblasts infected with Newcastle disease virus. J Antibiot (Tokyo) 24:785–794 [DOI] [PubMed]
- 87.Itoh Y, Nakashima Y, Tsukamoto S, Kurohara T, Suzuki M, Sakae Y, Oda M, Okamoto Y, Suzuki T. N+-C-H···O Hydrogen bonds in protein-ligand complexes. Sci Rep. 2019;9:1–5. doi: 10.1038/s41598-018-36987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du X, Li Y, Xia Y-L, Ai S-M, Liang J, Sang P, Ji X-L, Liu S-Q. Insights into protein–ligand interactions: mechanisms, models, and methods. Int J Mol Sci. 2016;17:144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin TM, Harten P, Venkatapathy R, Das S, Young DM. A hierarchical clustering methodology for the estimation of toxicity. Toxicol Mech Methods. 2008;18:251–266. doi: 10.1080/15376510701857353. [DOI] [PubMed] [Google Scholar]
- 90.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.