

Abstract
Epigenetic alterations, such as changes in DNA methylation, histones/chromatin structure, nucleosome positioning, and expression of non-coding RNAs, are recognized among key characteristics of carcinogens; they may occur independently or concomitantly with genotoxic effects. While data on genotoxicity are collected through standardized guideline tests, data collected on epigenetic effects is far less uniform. In 2016, we conducted a systematic review of published studies of genotoxic carcinogens that reported epigenetic endpoints to better understand the evidence for epigenetic alterations of human carcinogens, and the potential association with genotoxic endpoints. Since then, the number of studies of epigenetic effects of chemicals has nearly doubled. This review stands as an update on epigenetic alterations induced by occupational and environmental human chemical carcinogens that were previously and recently classified as Group 1 by the International Agency for Research on Cancer. We found that the evidence of epigenetic effects remains uneven across agents. Studies of DNA methylation are most abundant, while reports concerning effects on non-coding RNA have increased over the past 5 years. By contrast, mechanistic toxicology studies of histone modifications and chromatin state alterations remain few. We found that most publications of epigenetic effects of carcinogens were studies in exposed humans or human cells. Studies in rodents represent the second most common species used for epigenetic studies in toxicology; in vivo exposures being the most predominant. Future studies should incorporate dose- and time-dependent study designs and also investigate the persistence of effects following cessation of exposure, considering the dynamic nature of most epigenetic alterations.
Keywords: Epigenetics, Toxicology, Cancer, Genotoxicity, Hazard assessment
1. Introduction
Evaluation of the causative linkages between exposures to environmental agents and cancer includes considerations of the mechanistic evidence; the process of cancer hazard evaluation is now increasingly relying on the evidence from studies other than epidemiology or rodent cancer bioassays [1]. The practice of cancer hazard assessments and incorporation of mechanistic evidence has been streamlined and systematized through the introduction of the key characteristics of carcinogens organizational approach [2, 3]. The key characteristics approach provides guidance and structure for the consistent utilization of the mechanistic evidence and has been developed for many environmental agent-associated toxicities beyond cancer [4–9]. Included among the ten key characteristics of carcinogens is “induces epigenetic effects” and “is genotoxic.” These two key characteristics represent one of the least and one of the most data-rich key characteristics across the peer-reviewed literature related to human carcinogens, respectively, and are the focus of the review presented herein.
While many known carcinogens are genotoxic, they also cause epigenetic alterations [10]; however, most studies consider one or the other even though the linkages between these key characteristics are many [11]. Epigenetic modifications are heritable non-mutational events. They can operate independently or interdependently with genetic alterations to cause dysregulation of gene expression and subsequent molecular changes [12, 13]. Epigenetic events include stable changes in gene expression that are not attributed to the modification of the genomic sequence. These include changes in DNA methylation patterns, histone modifications, alteration of chromatin structure, and dysregulation of non-coding RNA expression. Environmental factors can impact these epigenetic marks, causing phenotypic variation and may potentiate human disease including cancer [14–16].
Studies of environmental agents suggest that epigenetic alterations play an important role in chemically-induced carcinogenesis [16, 17]. As compared to the decades of studies of genotoxicity of chemical agents and exposure-related gene expression responses, studies of epigenetics have been more recently enabled by the advances in sequencing technology. The evidence for epigenetic alterations caused by carcinogens, and the potential association with genotoxic endpoints, is rapidly growing [11]. As recently as five years ago, a systematic literature review of epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens showed that the database was relatively sparse for environmental chemicals [11]. Only three known human chemical carcinogens (aflatoxins, benzene, and benzo[a]pyrene), had ten or more studies with reported epigenetic effects. Other chemical carcinogens had very few studies of this key characteristic. As epigenetics continues to be an area of focus for mechanistic studies in toxicology, the evidentiary basis for the role of these non-genetic changes to DNA and transcription accumulates. This review summarizes the current status of the evidence on the role epigenetic alterations play in the induction of carcinogenesis following exposure to chemical agents (and related occupations) that have strong or moderate evidence of genotoxicity to enable a better understanding of the interplay of epigenetics and DNA damage.
2. Methodology
A multi-step, systematic literature review was conducted to analyze published scientific evidence concerning epigenetic mechanisms of action of human carcinogens that are genotoxic. An update of the recent (since 2016) publications on environmental or occupational hazards classified as Group 1 (“carcinogenic to humans”) by the International Agency for Research on Cancer (IARC) in Monograph Volume 100F was conducted. In addition, carcinogens classified as Group 1 in IARC Monograph Volumes 105-118 were evaluated. The list of substances included in this analysis and their associated IARC monograph reference is provided in Table 1. In summary, substances included in this analysis were (i) classified as IARC Group 1 “human carcinogen”, (ii) identified by IARC Monographs working groups having strong or moderate evidence for a genotoxic mechanism of carcinogenesis, and (iii) had published reports of epigenetic alterations.
Table 1.
Carcinogens classified as Group 1 in IARC Monographs Volume 100F-118 that were included in the systematic literature review.
| Group 1 Carcinogen | Most recent Monograph Number | Evidence of Genotoxicity: Animals | Evidence of Genotoxicity: Humans | Epigenetic data in last monograph | Epigenetic data in this review |
|---|---|---|---|---|---|
| 1,3-Butadiene | 100F | Yes | Yes | Yes | Yes |
| 4,4′-Methylenebis (2-Chlorobensenamine) | 100F | Yes | Yes | Yes | Yes |
| 4-Aminobiphenyl | 100F | Yes | Yes | Yes | Yes |
| Aflatoxins (naturally occurring mixtures) | 100F | Yes | Yes | Yes | Yes |
| Benzene | 100F | Yes | Yes | Yes | Yes |
| Benzidine | 100F | Yes | Yes | Yes | Yes |
| Benzo[a]pyrene | 100F | Yes | Yes | Yes | Yes |
| Coal tar pitch | 100F | Yes | Yes | No | Yes |
| Coke production, occupational exposure | 100F | Yes | Yes | Yes | Yes |
| Engine exhaust, diesel | 105 | Yes | Yes | No | Yes |
| Formaldehyde | 100F | Yes | Yes | Yes | Yes |
| Lindane | 113 | Yes | Yes | No | Yes |
| Occupational exposure as a painter | 100F | No | Yes | No | Yes |
| Outdoor air pollution | 109 | Yes | Yes | Yes | Yes |
| Pentachlorophenol | 117 | No | Yes | Yes | Yes |
| Polychlorinated biphenyls | 107 | Yes | Yes | Yes | Yes |
| Polychlorinated biphenyls, dioxin-like | 107 | Yes | Yes | Yes | Yes |
| Sulfur mustard | 100F | Yes | Yes | Yes | Yes |
| Trichloroethylene | 106 | Yes | Yes | Yes | Yes |
| Vinyl chloride | 100F | Yes | Yes | Yes | Yes |
| Welding fumes | 118 | Yes | Yes | Yes | Yes |
| 1,2-Dichloropropane | 110 | Yes | Yes | No | No |
| 2-Napthylamine | 100F | Yes | Yes | No | No |
| Bis(chloromethyl)ether and chloromethyl methyl ether | 100F | No | Yes | No | No |
| Coal gasification | 100F | Yes | No | No | No |
| Ethylene oxide | 100F | Yes | Yes | No | No |
| Isopropyl alcohol manufacture by the strong-acid process | 100F | No | Yes | No | No |
| Mineral oils, untreated or mildly treated | 100F | No | Yes | No | No |
| Mists from strong inorganic acids | 100F | No | Yes | No | No |
| Occupation exposures during coal-tar distillation | 100F | Yes | Yes | No | No |
| Occupational exposure during aluminum production | 100F | No | Yes | No | No |
| Occupational exposures during iron and steel founding | 100F | No | Yes | No | No |
| Occupational exposures in the rubber manufacturing industry | 100F | No | Yes | No | No |
| Ortho-toluidine | 100F | Yes | Yes | No | No |
| Shale oils | 100F | Yes | No | No | No |
| Soot, as found in the occupation exposure of chimney-sweeps | 100F | No | Yes | No | No |
The systematic literature review was conducted using the Health Assessment Workspace Collaborative (HAWC) web-based tool [18] and was based exclusively on the publications indexed in PubMed. The search terms published in the previous literature review of epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens [11] were used; a list of the search terms used for this assessment is listed in Supplementary Table 1. Inclusion and exclusion criteria are summarized in Supplementary Table 2. Included studies were classified by, in order: chemical, species (human, mouse, rat, or other), and study type (in vitro or in vivo). The final organization of included studies was based on the type of epigenetic alterations in DNA methylation, histone modifications, or non-coding RNA.
Studies that were excluded from the final analyses were grouped by topics or areas of knowledge that were not related to the focus of this review, or not clearly defined. In silico studies were excluded even if they reported epigenetic effects of a chemical of interest. Studies were classified as “not chemical of interest” if the studied agents or substances were not listed in Table 1. Publications that were not available in full text, or were a review/commentary, were excluded. Publications that did report on a chemical of interest but did not report epigenomic alterations were placed within the “No epigenetics” category. A sub-category within this category included “γ-H2AX”, which is an initial cellular response to DNA double-strand break induction that is technically an epigenetic alteration (histone phosphorylation), if this endpoint was used solely as an indicator of DNA damage and the study did not otherwise report on epigenetic alterations.
Dates of the literature search for the IARC Monograph 100F carcinogens ranged from 12/2015 to 12/2020, to capture literature published since the previous literature review of epigenetic alterations related to genotoxic Group 1 agents [11]. Of the 526 articles identified in the search, 391 (74%) were excluded because they did not report epigenetic effects. Other reasons for exclusion included, but were not limited to, the following: research did not pertain to the chemicals of interest, unavailability of full text in English, or no reporting of the primary data. The assessment concerning the Group 1 genotoxic carcinogens included in IARC Monographs 105-118 identified studies published from 01/1968 to 12/2020. A total of 903 scientific reports were identified in the search, with 300 meeting inclusion standards and included in this review (Table 2).
Table 2.
A total number of publications (until December 2020) with data related to epigenetic alterations for each Group 1 carcinogen included in the systematic literature review.
| Group 1 carcinogen | Most Recent Monogr. Listed | DNA Methylation | Histone Modifications | Non-Coding RNAs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Humans | Rodents (mouse or rat) | Other (See footnotes) | Humans | Rodents (mouse or rat) | Other (See footnotes) | Humans | Rodents (mouse or rat) | Other (See footnotes) | Total | |||
| 1,3-Butadiene | 100F | in vitro | 0 | |||||||||
| in vivo | 5 | 5 | 1 | 11 | ||||||||
| 4-Aminobiphenyl | 100F | in vitro | 1 | 2 | 3 | |||||||
| in vivo | 0 | |||||||||||
| Aflatoxins (naturally occurring mixtures) | 100F | in vitro | 8 | 1n, 2o | 1 | 1o | 8 | 1 | 22 | |||
| in vivo | 11 | 4 | 1r | 1 | 1r | 4 | 8 | 1c, 1p | 32 | |||
| Benzene | 100F | in vitro | 11 | 2 | 3 | 1 | 10 | 1 | 28 | |||
| in vivo | 23 | 2 | 5 | 1 | 12 | 2 | 45 | |||||
| Benzidine | 100F | in vitro | 0 | |||||||||
| in vivo | 1 | 1 | ||||||||||
| Benzo[a]pyrene | 100F | in vitro | 18 | 6 | 3n, 2i | 15 | 2 | 1h | 26 | 4 | 77 | |
| in vivo | 11 | 12 | 6w, 2m, 1g, 1r,1s | 3 | 2 | 13 | 1g, 1j, 1t, 1k, 1f | 57 | ||||
| Coal-tar pitch | 100F | in vitro | 1 | 2 | 3 | |||||||
| in vivo | 0 | |||||||||||
| Coke production, occupational exposure | 100F | in vitro | 1 | 1 | ||||||||
| in vivo | 10 | 3 | 4 | 14 | ||||||||
| 1,2-Dichloropropane | 110 | in vitro | 0 | |||||||||
| in vivo | 0 | |||||||||||
| Engine exhaust, diesel | 105 | in vitro | 2 | 2 | 5 | 9 | ||||||
| in vivo | 6 | 5 | 3 | 14 | ||||||||
| Formaldehyde | 100F | in vitro | 1 | 3 | 1v | 1 | 7 | |||||
| in vivo | 1 | 3 | 1d | 5 | ||||||||
| Lindane | 113 | in vitro | 0 | |||||||||
| in vivo | 1 | 1 | ||||||||||
| 4-4′-Methylenebis (2-chlorobenzenamine) | 100F | in vitro | 1 | 1 | ||||||||
| in vivo | 0 | |||||||||||
| Occupational exposure as a painter | 100F | in vitro | 0 | |||||||||
| in vivo | 1 | 1 | 2 | |||||||||
| Outdoor air pollution | 109 | in vitro | 7 | 3 | 16 | 26 | ||||||
| in vivo | 83 | 12 | 1b, 1e | 4 | 3 | 26 | 12 | 1w | 143 | |||
| Pentachlorophenol | 117 | in vitro | 0 | |||||||||
| in vivo | 1 | 1 | ||||||||||
| Polychlorinated biphenyls (non-dioxin-like) | 107 | in vitro | 3 | 1 | 1u | 1 | 1 | 2 | 1 | 2c | 12 | |
| in vivo | 19 | 11 | 1l, 1q, 1a, 1m | 1 | 2 | 3 | 2w | 42 | ||||
| Polychlorinated biphenyls (dioxin-like) | 107 | in vitro | 1 | 2 | 1 | 1 | 6 | |||||
| in vivo | 3 | 3 | 1w | 2 | 8 | |||||||
| Sulfur mustard | 100F | in vitro | 2 | 1 | 2 | 5 | ||||||
| in vivo | 6 | 2 | 8 | |||||||||
| Trichloroethylene | 106 | in vitro | 8 | 2 | 4 | 2 | 2 | 2e | 20 | |||
| in vivo | 1 | 18 | 2 | 1 | 4 | 1w | 27 | |||||
| Vinyl chloride | 100F | in vitro | 0 | |||||||||
| in vivo | 3 | 1 | 1 | 5 | ||||||||
| Welding fumes | 118 | in vitro | 1 | 1 | 2 | |||||||
| in vivo | 3 | 2 | 1 | 6 | ||||||||
Alaska blackfish (Dallia pectoralis)
Barn swallow (Hirundo rustica)
Chicken (Gallus gallus domesticus)
Cynomulgus macaques (Macaca fascicularis)
Dog (Canis lupis familiaris)
Firefly (Coleoptera lampyridea)
Fish (Oeyzias melastigma)
Frog (Xenopus borealis)
Hamster (Mesocricetus auratus)
Japanese Rice Fish (Oryzias latipes)
Lancelet (Brachiostoma belcheri)
Loggerhead sea turtle (Caretta caretta)
Mumichog fish (Fundulus heteroclitus)
Oligodeozynucleotides
Pig (Sus domesticus)
Pig (Sus scrofa domesticus)
Pond Slider (Trachemys scripta)
Rainbow trout (Oncorhynchus mykiss)
Rock dove (Columba livia)
Roundworm (Caenorhabditis elegans)
Sheep (Ovis aries)
Synthetic peptide
Zebrafish (Danio rerio)
Visualizations of the literature tree demonstrating the previously described inclusion and exclusion criteria for Monograph 100F and Monographs 105-118 Group 1 carcinogens with evidence of genotoxicity are shown in Figures 1 and 2, respectively. The systematic literature review information is public and freely accessible. The assessment for the update to Monograph 100F carcinogens is available at: https://hawcproject.org/assessment/1046/. The assessment for Monographs 105-118 carcinogens is available at: https://hawcproject.org/assessment/1063/. While this review aimed to cite most of the studies identified in the systematic literature review, not all of them are referenced and the reader is welcome to explore the literature trees at the links above.
Figure 1.
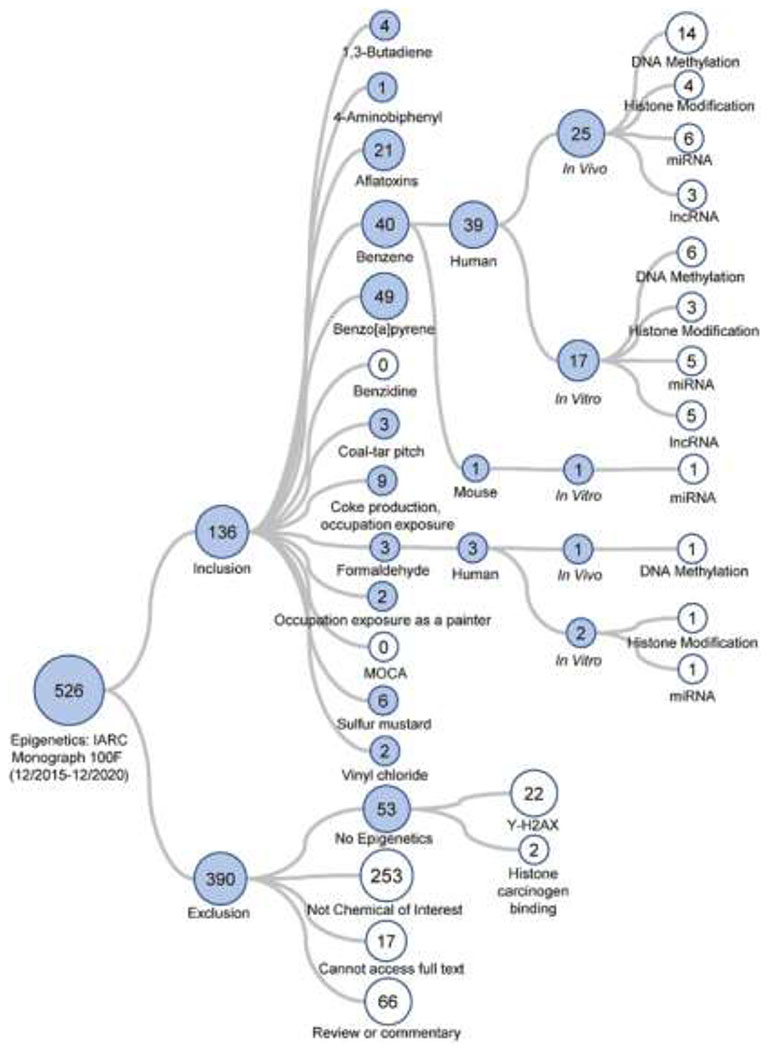
Literature tree of the 526 studies returned from the PubMed search concerning epigenetic endpoints for the human carcinogens included in IARC Monograph 100F (all classified as “Group 1”). The studies were categorized based on inclusion or exclusion criteria as shown. Included studies were further sorted, in order, by cancer hazard, species studied (human, rat, mouse, or other), study design (in vivo or in vitro), and epigenetic endpoint. The numbers within each circle denote how many studies were allocated to that specific category. Blue circles denote that they contain additional sub-categories. White circles indicate a terminal category. The literature tree branches for included studies for carcinogens “Benzene” and “Formaldehyde” are expanded for illustration purposes. The branch for “No Epigenetics” category is shown as an example of an exclusion category. The searches covered literature published between 12/2015 and 12/2020.
Figure 2.
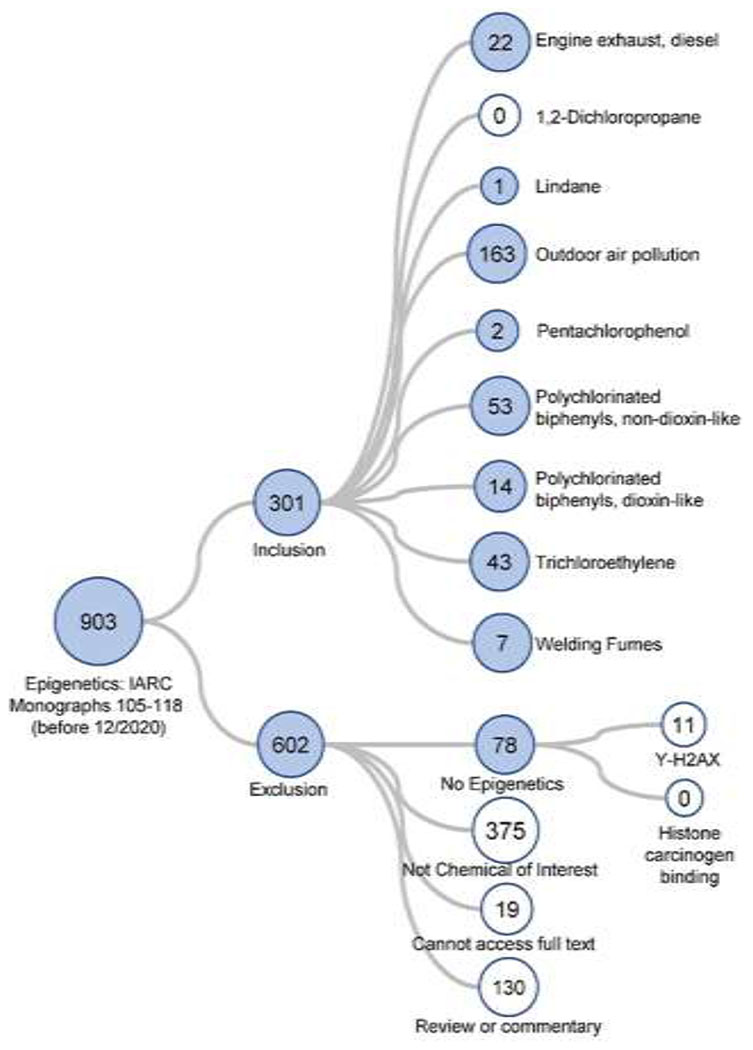
Literature tree of the 903 studies returned from the PubMed search concerning the human carcinogens included in IARC Monographs 105-118 (all classified as “Group 1”) with evaluation of epigenetic alterations. The studies were categorized based on inclusion or exclusion criteria as shown. See Figure 1 legend for further explanation of the figure structure. The searches covered literature published before 12/2020.
3. Categories of epigenetic alterations induced by the chemicals and associated occupations included in the systematic literature review
3.1. DNA Methylation.
DNA methylation (Figure 3) is the covalent addition of a methyl group (CH3) at the carbon-5 position of cytosine, resulting in 5-methylcytosine (5mC). DNA methylation is typically found at cytosine-guanine dinucleotides (CpG sites) and is established by de novo DNA methyltransferases (DNMT3A and DNMT3B) which transfer a methyl group from S-adenosyl methionine (SAM) to the carbon-5 location of the cytosine [19]. DNMT1, a maintenance DNMT, which in cooperation with ubiquitin-like, containing PHD and RING finger domains (UHRF1) is responsible for maintaining the DNA methylation patterns following replication. DNA methylation is a dynamic, multi-step process that is counter-balanced by DNA demethylation. DNA demethylation, or the removal of methyl groups from cytosines, can occur through passive (spontaneous demethylation) or active mechanisms. The latter involves the oxidation of 5mC by the ten-eleven-translocation (TET) pathway, creating 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxycytosine (5caC) through hydroxylase activity [20]. Thymine DNA glycosylase via the base excision repair pathway can excise 5hmC, 5fC, and 5caC which will be replaced with an unaltered cytosine base [21].
Figure 3.
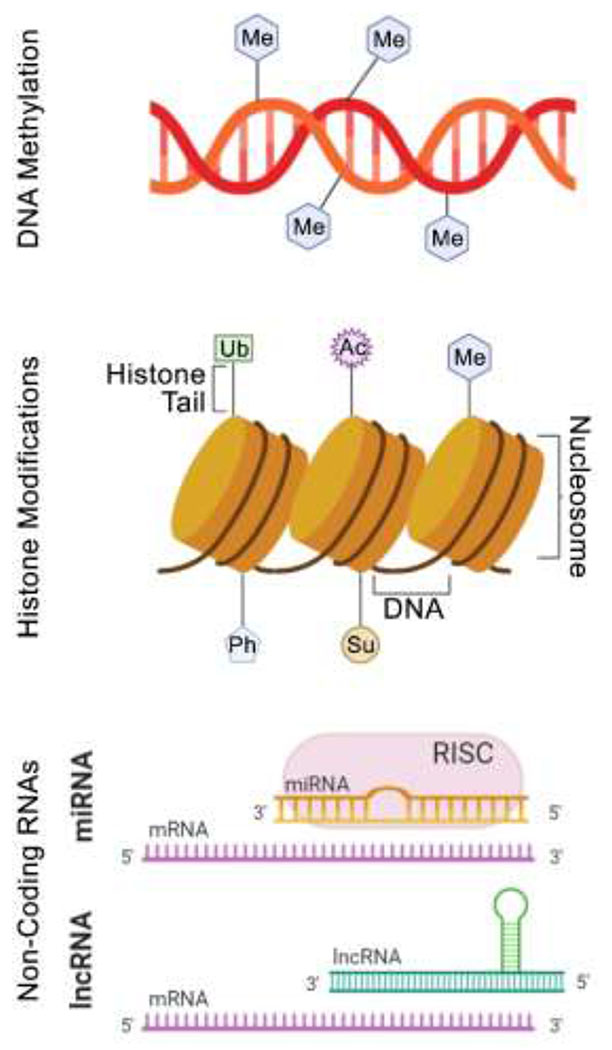
A general graphic summary of the epigenetic alterations that were searched for in this review. Abbreviations: Ac – acetylation; Ph – phosphorylation; Me – methylation; Ub – ubiquitination; Su – sumoylation; mRNA – messenger RNA; miRNA – micro RNA; RISC – RNA induced silencing complex; lncRNA – long non-coding RNA.
DNA methylation is a major and well-studied epigenetic phenomena and has been implicated in the modulation of cell biology [22]; gene expression regulation, genomic stability, overall chromosomal integrity [23, 24]; and silencing of imprinted genes [25]. Aberrant DNA methylation results in malignant tumors through DNA hyper- or hypo-methylation [26], with hypermethylation indicating increased methylation with subsequent transcriptional suppression and decreased tumor suppressor gene expression [27, 28] and hypomethylation indicating decreased methylation with influence over chromosomal stability and activation of oncogenes [29]. Global DNA hypomethylation is normally utilized as a biomarker of cancer induction as it causes genomic instability [26], a hallmark of carcinogens [2].
Previous research in the field of cancer epigenetics, focused mainly on hypermethylation of promoter CpG islands within tumor-suppressor genes as cancer diagnostic biomarkers, identified broad variety of tumor suppressor genes that are hypermethylated and silenced in cancer [30, 31]. Recent genome-wide discoveries of aberrant methylation, both hypermethylation and hypomethylation, of CpG island shores, and even individual CpG sites located in the promoter, gene body, or intergenic regions, in addition to promoter CpG island hypermethylation, substantially enhanced our knowledge on cancer DNA methylation landscape resulting in discoveries of unique CpG loci as potential biomarkers. For example, methylation of specific CpG sites within a promoter has been found to control transcriptional silencing, in contrast to indiscriminate methylation throughout the entire promoter region [32–36]. Furthermore, a recent epigenome-wide association study demonstrated a strong association between methylation changes at unique individual CpG sites in blood and pancreatic cancer that occurred prior cancer diagnostic [37]. Quantification of DNA methylation can be achieved through various techniques, most recent techniques are based on advancements in nucleotide sequencing [38]. Non-sequencing methods include mass spectrometry for global DNA methylation levels or combined bisulfite restriction analysis for gene-specific DNA methylation, warrant consideration [39–41].
3.2. Histone Modifications.
Post-translational histone modifications (Figure 3) comprise several epigenetic marks that have been associated with transcription, DNA repair, and the maintenance of chromatin structure [42]. Nucleosomes, the fundamental building blocks of chromatin, are composed of octamers of the four core histone proteins (H3, H4, H2A, and H2B) around which is wrapped with a 147 base-pair segment of DNA. Each histone protein possesses an N-terminal tail (a side chain) populated with cationic lysine and arginine residues; these side chains may be post-translationally modified by acetylation, citrullination, methylation, phosphorylation, or ubiquitination. Histone modification “writers” (e.g., histone acetyltransferases; HATs) enzymatically add alterations while “erasers” (e.g. histone deacetylases, HDACs) enzymatically remove alterations [43], representing a dynamic system modulating these alterations on the histone side chains. These histone modifications are then recognized by “reader” proteins (e.g., bromodomains and chromodomains) which affect transcription.
Most studies of histone modifications covered in this review focus on acetylation and methylation of lysine resides. Histone acetylation modifies the ε-amino group of lysine residues, neutralizing the positive charge. This alteration has been suggested to disrupt the interaction between the histone side chain and DNA nucleosome, resulting in increased chromatin availability and the promotion of active transcription. Acetylated lysine further promotes chromatin accessibility by attaching to bromodomain-containing transcription factors within chromatin remodeling complexes [44, 45]. Histone methylation has been identified as a key mechanism in development and differentiation [46, 47]. Histone methylation occurs on both lysine and arginine residues with preferential occurrence on histone H3 and H4 [48]. Various methylation states exist (mono-, di-, and trimethylation) and all can elicit different transcriptional outcomes. Recent studies investigate histone modifications through ADP-ribosylation, butyrylation, citrullination, crotonylation, deamination, formylation, O-GlcNAcylation, propionylation, and proline isomerization [42]. Dysregulation of histone modifications can alter chromatin structure, gene expression, and DNA repair mechanisms and therefore has been associated with carcinogenesis [49–53]. For, instance, a recent study [54] showed that exposure to perfluorooctane sulfonate and perfluorooctanoic acid, two ubiquitous environmental contaminants, may promote breast carcinogenesis via inducing persistent cancer-specific alterations in histone lysine acetylation and methylation. Alterations of histone post-translational modification patterns are most frequently studied either globally or at specific loci using ChIP (chromatin immunoprecipitation) followed by sequencing [42].
3.3. Non-Coding RNAs.
Non-coding RNAs (Figure 3) are a class of RNA molecules that are not translated into a protein. Non-coding RNAs that possess a regulatory role in transcription are typically sub-divided into two categories: short-chain non-coding RNAs (miRNAs) and long non-coding RNAs (lncRNAs). These non-encoding RNAs have been shown to have significant roles in epigenetic modifications and regulation on both the gene and chromosomal level [55–57]. Quantitative RT-PCR, RNA sequencing, and microarrays are the most commonly utilized methods for quantitation of non-coding RNAs.
miRNAs are short, single-stranded RNA (~19-25 nt) that originate from the DNA regions prone to structural changes (“fragile sites”); these loci are frequently amplified or deleted in tumor cells [58, 59]. miRNAs play important roles in cell proliferation and apoptosis by controlling the expression of tumor suppressor genes and oncogenes. This regulation is achieved primarily through the binding of miRNA to the 3’ untranslated region of mRNA inducing their degradation or blocking translation [60, 61]. Biogenesis of miRNAs is a complex process [62]; mutations in proteins involved in miRNA synthesis may result in alterations of miRNA processing, stability, and targeting, causing subsequent biological insults, including carcinogenesis [59].
LncRNA are transcripts longer than 200 nt and represent one of the largest and most diverse RNA families [63]. LncRNAs are involved in transcriptional regulation of gene expression [64, 65] and aberrant expression of lncRNAs has been significantly linked to cancer carcinogenesis and progression [66, 67]. LncRNAs may act as scaffolds for various epigenetic endpoints, inducing a cascade of events. For example, lncRNAs may induce histone modifications by linking proteins together to form ribonucleoprotein complexes [65] and interact with DNA methyltransferase enzymes to affect DNA methylation [68].
4. Epigenetic effects associated with IARC Group 1 human carcinogens included in IARC Monograph 100F
A total of 11 Group 1 human carcinogens that met the inclusion criteria for this systematic literature review are discussed herein. A description of the common routes of exposure, associated cancers, and genotoxic potential of each of these, except for coal tar pitch (see section 4.10), was also evaluated previously [11].
4.1. Benzo[a]pyrene.
4.1.1. DNA Methylation.
As detailed in Chappell et al [11], changes in DNA methylation induced by benzo[a]pyrene exposure were primarily studied using human and rodent in vitro models. Recent studies provide additional evidence for gene-specific methylation (e.g. tumor suppressor genes) and identified biomarkers to aid in the diagnosis and prognosis of benzo[a]pyrene-associated cancer. For example, exposure to benzo[a]pyrene via cigarette smoke was significantly associated with increased promoter hypermethylation of p16 and DAPK (genes involved in carcinogenesis pathways), as well as with DNA damage [69]. Furthermore, benzo[a]pyrene increased DNA methyltransferase 1 in smokers diagnosed with bladder cancer [70]. Several studies have identified additional aberrantly methylated genes, e.g., hypermethylated DKK2, EN1, and TRIM36, and hypomethylated LPAR2, associated with the development of benzo[a]pyrene-mediated cancers [71, 72]. Additionally, it has been demonstrated that benzo[a]pyrene exposure of human bronchial epithelial (HBE) cells and lymphocytes induced hypomethylation of nuclear and mitochondrial DNA [71, 73, 74].
4.1.2. Histone Modifications.
In publications reviewed by Chappell et al [11], most studies of histone modifications by benzo[a]pyrene were conducted in human cell lines. The most frequent type of the reported effects was changes in global post-translational modifications of various histones (e.g., levels of H3K4me3 and H3K9ac) as a result of treatment. Since 2016, most of the studies of benzo[a]pyrene and histone modifications were similarly conducted in human cells; however, the emphasis is more on the mechanisms of the benzo[a]pyrene effects on the histones and chromatin, and the utility of histone modifications as biomarkers of exposure to benzo[a]pyrene. For example, decreased acetylation of histones H3 and H4 and upregulation of HDAC2 and HDAC3 histone deacetylases were found to be associated with global loss of DNA methylation following benzo[a]pyrene-exposure of HBE cells ; these effects were concentration-dependent [73]. The effect of benzo[a]pyrene on the chromatin states indicative of reactivation of long interspersed nuclear element-1 (LINE-1) retrotransposon was shown to involve functional modulation of nucleosomal and remodeling deacetylase NuRD, leading to oncogenic transformation of HBE cells [75]. Several studies showed that poly(ADP-ribose) glycohydrolase (PARG) may also play an important role in benzo[a]pyrene-associated changes in histone H2 expression; these studies were conducted in both human lung cancer cell lines and a mouse model of benzo[a]pyrene-induced carcinogenesis [76, 77]. Downregulation of PARG and histone modifications similar to those in human lung cancers were found in benzo[a]pyrene-treated human lung cancer cell lines, suggesting that PARG silencing may be a biomarker for benzo[a]pyrene exposure-related lung carcinogenesis [77].
4.1.3. Non-Coding RNAs.
Formerly reviewed [11] publications that investigated effects of benzo[a]pyrene on the expression of non-coding RNAs utilized human in vitro models; these studies demonstrated the role of aberrant miRNA expression in the dysregulation of DNA repair mechanisms and tumor suppressor genes. Since 2016, the majority of studies also used human cells, with focus on the role of miRNAs in non-genotoxic mechanisms of benzo[a]pyrene-associated carcinogenesis, such as inflammation. For example, benzo[a]pyrene-treated HBE cells demonstrated differential expression of specific miRNAs was significantly associated with the upregulation of activated leukocyte cell adhesion molecule (ACLAM) [78]. In vitro and in vivo (tumor model within nude mice) studies using ALCAM-knockout HBE cells showed inhibition of colony formation and increased cell migration and decreased tumor growth and enhanced metastasis, respectively. Benzo[a]pyrene exposure within human mammary cells resulted in the upregulation of inflammation pathways and dysregulation of tumorigenic miRNAs, progressing benzo[a]pyrene-induced tumorigenesis through non-mutagenic pathways [79]. The influence of benzo[a]pyrene-induced inflammation was further explored within human adenocarcinoma mammary cells cotreated with benzo[a]pyrene and a pro-inflammatory cytokine IL-6 [80]; genotoxicity was enhanced due to the downregulation of miR-27b, facilitating increased CYP1B1-mediated metabolism of benzo[a]pyrene to reactive intermediates. Several studies have shown in vitro benzo[a]pyrene exposure caused prominent changes in the expression of miRNAs and their respective target genes, providing mechanistic insight into benzo[a]pyrene-associated toxicity [74, 81].
In addition to the focus on miRNA, recent studies investigated the role of lncRNA in mediating benzo[a]pyrene-associated effects. For example, the significant upregulation of linc00673 following benzo[a]pyrene exposure within human non-small cell lung cancer cells (A549) increased cell migration, invasion, and epithelial-mesenchymal transition [82]. Human lung-derived cell lines treated with benzo[a]pyrene showed increased lncRNA H19 expression, initiating the downregulation of the S-adenosylhomocysteine hydrolase (SAHH) pathway, and overall methylation of LINE-1 [83].
Overall, the recent evidence demonstrates the ability of benzo[a]pyrene to alter gene-specific methylation patterns that may be related to DNA damage response, chromatin remodeling that is associated with oncogenic transformation, and miRNA expression changes related to inflammation. The majority of these alterations were measured and observed in in vitro studies.
4.2. Benzene.
4.2.1. DNA Methylation.
As detailed in Chappell et al. [11], earlier studies of DNA methylation effects of benzene exposure used human in vivo and in vitro models. The research focused on gene-specific aberrant methylation, explicitly genes associated with tumor suppression and apoptosis inhibition, following benzene exposure. Since 2016, evidence concerning benzene-induced DNA methylation has been similarly gathered from human in vivo and in vitro studies. The majority of new human in vivo studies focused on gene-specific methylation, the identification of potential biomarkers for early detection of benzene toxicity, and the influence of benzene on DNA methyltransferase expression. For example, peripheral blood cells derived from benzene-exposed workers demonstrated hypomethylation of STAT3, which was significantly correlated with oxidative stress variables [84], and MT-COI, a gene associated with oxidative phosphorylation [85]. Also, it was found that low-level occupational benzene exposure was associated with hypermethylation of tumor suppressor genes p15INK4b [86], p14ARF, and p16INK4A [87]; aberrant methylation of p16INK4A was significantly correlated with genomic instability caused by benzene-induced chromosomal abnormalities. Furthermore, occupational benzene exposure led to aberrant promoter methylation of CSF3R that is essential for neutrophil production [88], and ERCC3 that is involved in nucleotide excision repair [89]; these effects were significantly associated with a reduction in neutrophils and CpG methylation-induced hematotoxicity, respectively. Prolonged benzene exposure in workers was associated with chromosomal damage and increased frequency of micronuclei; in vitro validation of these results showed even low-level benzene concentrations cause hypermethylation of DNA repair genes and hypomethylation of LINE-1 within acute myeloid leukemia (AML-5) cells [90].
Single nucleotide polymorphisms (SNPs) in de novo DNMT3 A and DNMT3B were positively associated with hypomethylation following benzene exposure in a study of exposed workers; this effect was hypothesized by the authors to have an influential effect on micronucleus frequency in the same individuals [91]. Increased expression of DNMT3B following benzene exposure found in human in vivo and in vitro studies was correlated with increased promoter hypermethylation, silencing HOXA transcript antisense RNA myeloid-specific 1 (HOTAIRM1) expression, a putative tumor suppressor gene [92]. The DNA methylation status within erythroleukemic cells (K562) exposed to various benzene metabolites determined erythroid gene expression and cell differentiation [93–95]; for example, phenol exposure modulated the methylome to suppress erythroid-specific genes [95] while catechol upregulated gene expression [94].
4.2.2. Histone Modifications.
Previously summarized reports [11] investigating the effect of benzene exposure on histones and/or chromatin structure were conducted within human and mouse in vivo and rat in vitro models. These studies focused on species specificity of benzene-induced histone modifications and the role histone alterations play in topoisomerase IIα (Topo IIα) activity, expression. Since 2016, reports of histone modifications have utilized human in vivo and in vitro models to investigate gene-specific modifications due to aberrant histone expression and the effects of histone alterations on Topo IIα. For example, human bone marrow mononuclear cells treated with benzene increased HDAC activity and decreased Topo IIα activity, subsequently inducing apoptosis [96]. A follow-up full-body inhalation exposure within mice confirmed decreased Topo IIα expression mediated by impaired acetylation of histone H4 and H3. Human bone marrow samples from chronic benzene poisoning cases also showed significant decrease in histone H4 and H3 acetylation levels and lowered mRNA expression of TOPO IIα promoter regulation factors [96]. Recent studies of benzene exposure in workers showed that increased trimethylation of histone 3 lysine 4 (H3K4me3) was positively correlated with DNA damage [97]. A follow-up study within primary human lymphocytes revealed enrichment of H3K4me3 occurred within DNA damage responsive genes, potentially highlighting the mechanism in which histone modifications mediate benzene-induced hematotoxicity.
4.2.3. Non-Coding RNAs.
Studies originally summarized in Chappell et al. [11] concerning the aberrant expression of non-coding RNAs following benzene exposure used human and mouse in vivo data. The most common endpoints covered within these reports included differentially expressed miRNA- and lncRNA-associated immune response pathways, the effects of in utero exposure on immune regulation, and aberrant miRNA expression-associated hematotoxicity. Since 2016, perturbed non-coding RNA expression induced by benzene exposure has been studied within human biological samples and cells. For example, levels of circulating miR-221 following benzene exposure can be utilized as a reliable, minimally invasive biomarker of benzene-induced disease [98]; this finding was corroborated by several recent human in vivo and in vitro studies [99–101]. Downregulation of miR-451a and miR-486-5p was identified as being responsible for the regulation of erythroid differentiation in human cells [102, 103]. Alteration of miRNA-486-5p expression effected expression of MAGI1 (protein scaffolding gene) and RASSF5 (tumor suppressor gene), suggesting miRNA-486-5p may be involved in Ras-associated protein-1 signaling pathway-associated genes to inhibit benzene-induced erythroid differentiation. Aberrant expression of miRNAs induced by poly(ADP-ribose)polymerase-1 (PARP-1) expression has been investigated using TK6 lymphoblastoid [104] and TK6 with miR-7-5p mimic (TK6-miR-7-5p) lines [105]. PARP-1 was found to decrease miR-155 expression through acetylation by modulating MBD2 protein levels [104]. An increase in miR-7-5p expression, following hydroquinone exposure, further aggravated PARP-1 and BRCA1 downregulation in TK6-miR-7-5p cells, subsequently enhancing cell apoptosis and inhibiting proliferation [105]. Investigation into the benzene metabolite 1,4-benzoquinone showed exposure-induced genotoxicity and significant upregulation of miR-222 within human lymphoblast cells [106]; this epigenetic modification inhibited MDMR-p53 expression and subsequently caused inactivation of p53 and abnormal DNA repair capacity.
Expression of lncRNAVNN3 was upregulated within exposed workers and treated human lymphocytes; a positive correlation was noted between this lncRNA and serum autophagy- and apoptosis-associated proteins [107]. Furthermore, LncRNAVNN3 was found to mediate benzene-induced autophagy and apoptosis through the mediation of beclin1 and Bcl-2 phosphorylation. Overexpression of lncRNA-OBFC2A decreased cell proliferation by interacting with Smad3 (a key protein in TGFb signaling cascade) to control the cell cycle through altered Cyclin D1 expression [108, 109]. A human in vivo and in vitro study demonstrated crosstalk between lncRNA FAS-AS1 and DNMT3b with both being dynamically expressed via a mutual inhibition loop; the expression of FAS was downregulated through histone acetylation [110].
Overall, there is a wealth of data on epigenetics alterations related to benzene exposure, much of which has been collected in exposed workers. The epigenetic alterations, including DNA methylation, histone modifications, and miRNA expression, are related to DNA damage and repair, demonstrating that epigenetics plays an important role in the response to the DNA damaging effects of benzene.
4.3. Aflatoxins.
4.3.1. DNA Methylation.
Studies previously reviewed elsewhere [11] predominately focused on modification of the methylome following aflatoxin exposure in humans in vivo. The endpoints most frequently investigated included gene-specific methylation and resulting expression alterations (e.g., RASSF1A), the effect of DNA methylation on telomere length, and global methylation alterations. Since 2016, extensive research has been conducted concerning gene-specific methylation following aflatoxin exposure using human cell lines. For example, chronic aflatoxin B1 (AFB1) exposure was associated with persistent DNA hypomethylation within six identified cancer-related genes: TXNRD1, PCNA, CCNK, DIAPH3, RAB27A, and HIST1H2BF [111]. This study was the first to report persistent epigenetic effects induced following AFB1 exposure within the transcriptome and may provide key insight into potential biomarkers of hepatocellular carcinoma (HCC) development (a disease strongly associated with AFB1 exposure). Minimally cytotoxic in vitro exposures of AFB1 and aflatoxin B2 (AFB2) (a structurally-related non-carcinogenic analog) produced distinct gene-expression profiles with significant DNA methylation alterations [112]; however, no correlation between gene-specific DNA methylation and gene expression changes was noted. In contrast, non-toxic, low-dose AFB2 exposure resulted in a large number of differentially expressed genes but few effects on the global DNA methylome, implying a transcriptomic response independent of DNA methylation modifications [113].
Additional human in vitro studies have shown the interplay of multiple epigenetic mechanisms pivotal to the aflatoxin-induced carcinogenesis pathway. S phase-arrested L02 cells exhibited aflatoxin-induced apoptosis, decreased mitochondrial membrane potential, increased reactive oxygen species generation, and upregulation of global DNA methylation at all exposure concentrations [114]. Results concluded DNA methylation induced by AFB1 exposure played a key role in regulating multiple hormonal and cell fate pathways. AFB1 treatment within immortalized human cells caused p21 promoter (a tumor suppressor gene) hypermethylation via increased DNMT3a expression, a pathway initiated by the upregulation of H3K26me3 and H2AK119Ub [115]; this cascade of epigenetic events resulted in the deregulation of cell cycle regulatory molecules. Another study of AFB1 treatment within human epithelial cells showed the induction of of DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B), histone modifiers (HDAC1, HDAC2, HDAC4, and HDAC6), and histone marks (H2K27me3 and H2AK119Ub) [116]. Together these studies provide strong mechanistic evidence for epigenetic regulation of AFB1 effects.
4.3.2. Histone Modifications.
Previously reviewed [11] studies of aflatoxin exposure-associated histone modifications were conducted in vivo in mice and in vitro in porcine oocytes. Both studies investigated the correlation between histone repression and DNA hypermethylation within oocytes. Since 2016, the effects of aflatoxins on histone marks have only been investigated in human cell lines. As summarized above, crosstalk was identified between histone modifications and genome methylation following AFB1 exposure, specifically the association between repressive histone marks (H3K27me3 and H2AK119Ub) and DNMTs [115, 116]. Chromatin immunoprecipitation analysis showed increased enrichment of H3K27me3 following AFB1 exposure. These epigenetic modifications ultimately caused the deregulation of cell cycle regulatory molecules.
4.3.3. Non-Coding RNAs.
As summarized in Chappell et al. [11], previously reviewed studies investigated the effects of aflatoxin on non-coding RNA expression in human and rodent models. These studies focused on upregulated miRNA within liver tumors, identification of sensitive biomarkers of aflatoxin exposure, and miRNA-mRNA interactions associated with repression of DNA damage repair. Since 2016, human, rodent, and alternative animal models continue to be used and focused on the identification of potential biomarkers of aflatoxin exposure, differentially expressed miRNAs associated with carcinogenesis pathways, and non-coding pathways that enhance immune response pathways. For example, two miRNAs have been identified as biomarkers for the early detection of aflatoxin-associated hepatocellular carcinoma. MiR-4651 was identified as a potential biomarker due to its increased accuracy and sensitivity when compared to α-fetoprotein (a serum marker to identify patients at high risk of HCC) [117]. Upregulation of miR-182 was detectable months before clinical HCC symptoms were present and represent a non-invasive, serum biomarker for disease detection [118]. Utilization of miR-4651 and miR-182 in identifying small-size and early-stage HCC may improve AFB1-induced HCC disease diagnosis and prognosis.
MiR-155 expression in isolated spleen T cells from AFB1-exposed mice was found to be significantly reduced [119]. In the same tissue, significant upregulation of phosphatidylinositol-3, 4, 5-trisphosphate 5-phosphatase 1 (Ship1), and suppressor of cytokine signaling 1 (Socs1) was observed within exposed T cells. These outcomes suggest miR-155 and targeted proteins (Ship1 and Socs1) are involved within AFB1-induced immunotoxicity. Following AFB1 exposure, differentially expressed miRNAs were identified, all being significantly associated with cancer development [120]. Subsequent bioinformatic analyses established miRNA- mRNA regulatory networks related to the onset of AFB1-induced hepatocellular carcinoma.
A long-term exposure study in Roman hens fed AFB1-contaminated food indicated the expression levels of protein-coding genes, miRNAs, and lncRNAs within the liver were altered [121]; there was a strong association between these epigenetic effects and upregulation of PPARG (fatty acid storage regulator) and downregulation of Bcl-6 (autoimmune response regulator).
Overall, the recent evidence demonstrates that AFB1 can alter gene-specific methylation of genes that have a demonstrated relationship to cancer, and miRNA expression changes that are related to immune response pathways. The validation of miRNAs as biomarkers of exposure or aflatoxin-induced toxicity requires additional research. The data on histone modifications is very limited, and suggests alterations to the expression of genes that are involved in cell cycle. The evidence demonstrates that epigenetic alterations likely play a role aflatoxin-induced carcinogenicity, in concert with other mechanisms related to the carcinogenic response.
4.4. Coke Production.
4.4.1. DNA Methylation.
As summarized in Chappell et al. [11], DNA methylation following coke production exposure was only investigated in human exposure scenarios. The endpoints studied within these publications were global and gene-specific promoter methylation, chromosomal abnormalities, and identification of urinary biomarkers for coke production exposure. Since 2016, studies have continued to primarily use human samples from in vivo exposures, reporting retrotransposon methylation (e.g. LINE-1) and gene-specific methylation (e.g. CYP1A1 and FLT1). For example, coke oven workers with reported heavy smoking habits showed significant associations between CYP1A1 hypomethylation and increased levels of 8-OHdG (a biomarker of oxidative DNA damage) [122]; these results indicate the co-exposure of smoking and coke oven work increases the risk for oxidative DNA damage, a key player in disease pathogenesis, through epigenetic alterations of CYP1A1. Urinary samples collected from coke oven workers demonstrated high levels of urinary 1-hydroxypyrene (1-OHP; a PAH metabolite) that were significantly associated with LINE-1 hypomethylation [123]; the additional exposure of tobacco use was found to increase aryl-hydrocarbon receptor repressor (AhRR) hypomethylation. Peripheral blood lymphocytes collected from coke oven workers possessed hypermethylation of fms-related tyrosine kinase 1 (FLT1), a gene previously found to be hypermethylated within several human cancers [124]. The aberrant FLTI methylation status was positively correlated with 1-OHP urinary levels and DNA damage, indicating FLT1 may act as a tumor suppressor and its anomalous modulation may contribute to coke-production-associated carcinogenesis. Coke oven emission-transformed HBE cells demonstrated TRIM36 hypermethylation and overall gene suppression [72]; this gene-specific methylation was significantly associated with 1-OHP levels and DNA damage, indicating aberrant methylation of TRIM36 may contribute to coke oven-associated malignancy.
4.4.2. Histone Modifications.
Trimethylation levels of H3K27 and H3K36 were elevated in peripheral blood lymphocytes collected from coke oven workers [125, 126]. Notably, a positive correlation was identified between H3K36me3 expression and the level of internal PAH-exposure in all subjects (exposed and control), indicating PAH-induced DNA damage may be mediated by H3K36me3 modifications and could be an indicator of PAH exposure. Additionally, lower global H3K79me2 presence, a histone mark that plays a critical role in DNA damage regulation, was found in peripheral blood lymphocytes of coke oven workers [127].
4.4.3. Non-Coding RNAs.
Studies previously reviewed elsewhere [11] that investigated the effects of coke oven emission exposure on non-coding RNA expression used human occupational study data. These reports focused on miRNA expression-mediation of coke production-associated oxidative DNA damage and micronuclei frequency. Since 2016, studies have continued to samples from human exposure scenarios for both miRNA and lncRNA investigations. For example, several plasma miRNAs with significant dysregulation were inversely associated with heart rate variability, representing potential biomarkers for coke production-associated cardiovascular insults [128]. Peripheral blood lymphocytes of coke oven workers demonstrated upregulation of HOTAIR and MALAT1 lncRNA expression; these epigenetic modifications were correlated to DNA damage in an exposure level-dependent manner [129]. Furthermore, elevated HOTAIR expression was significantly associated with H3K27me3 modifications (a biomarker of DNA damage), indicating lncRNA expression may regulate DNA damage through interaction with histone modifications.
Overall, the epigenetic alterations associated with occupations in coke production are related to DNA damage response and gene-specific expression. There is also evidence that epigenetic alterations mediate and/or regulate other cancer-relevant mechanisms and pathways. Because coke oven workers are exposed to complex mixtures of PAH, studies of epigenetic and other effects may be difficult to attribute to the effects of a specific chemical.
4.5. Sulfur Mustard.
4.5.1. DNA Methylation.
One publication reviewed by Chappell et al. [11] demonstrated sulfur mustard exposure induced global hypermethylation in human in vivo and in vitro models. Since 2016, the effects of sulfur mustard on the methylome have been investigated in human endothelial cells and skin samples collected from humans who experienced an accidental exposure [130]. The in vitro study showed significant DNA hypermethylation in late cell passages, suggesting these sulfur mustard-induced epigenetic effects are either specific to these late stages, or persist over time [131]. Human skin samples collected one year following the accidental sulfur mustard exposure exhibited significantly increased global DNA methylation when compared to control skin, supporting the in vitro results.
4.5.2. Histone Modifications.
Human early endothelial cells exposed to low sulfur mustard concentrations [130] were tested for histone acetylation (H3K9, H3K27, H4K8) or di-methylation (H3K9, H3K27, H3K36) at three passage timepoints. No clear generational variation or dose-dependent effects could be discerned within the cells, indicating post-translational histone alterations do not play a key role in sulfur-mustard-induced carcinogenesis.
4.5.3. Non-Coding RNAs.
Previously summarized reports [11] predominately investigated the modulation of miRNA expression following sulfur mustard exposure within human occupational exposure study data; additional studies occurred in human and murine cells. The most common endpoints reported within these studies include diagnostic methods and therapeutic intervention discoveries for sulfur mustard exposure, mechanisms of sulfur mustard-associated cellular function impairment, and identification of miRNA involved in exposure-induced pathogenesis. Since 2016, modulation of miRNA expression following sulfur mustard exposure has continued to be collected from human in vivo and in vitro and rodent in vivo studies. These studies report proposed miRNA-mediated pathways of carcinogenesis and potential biomarkers for sulfur mustard exposure. For example, comparative evaluation of miR-143 and miR-9 (miRNAs associated with key regulatory pathways such as NF-kB signaling) within urine samples collected from occupationally exposed humans showed significantly decreased expression levels [131]. An additional study reported four significantly upregulated miRNAs in serum samples collected from sulfur mustard-exposed veterans [132]. These studies identify specific miRNAs that may represent non-invasive biomarkers of sulfur mustard exposure. Expression of miR-15b-5p and miR-21-5p, and SMAD7 were upregulated within sulfur mustard exposed lung tissue samples, identifying a potential mechanism by which sulfur mustard exposure regulates inflammatory/fibrotic modifications within the lung [133]. Human keratinocyte cells that were made resistant to sulfur mustard (HaCat/SM) displayed significant differences in miRNA expression patterns when compared to controls [134]. The authors postulated that miR-125 and miR-181 upregulation was the mechanism for transcriptional effects and sulfur-mustard resistance in HaCat/SM cells.
A study of male Sprague-Dawley rats exposed to sulfur mustard by inhalation and immortalized rat lung-derived airway epithelial cells (CRL-10354) identified miR-140 expression as a mediator of sulfur mustard effects [135]. Additionally, it was proposed that levels of miR-140 can be used as a predictor of the severity of sulfur mustard-induced health insults.
Overall, there is only limited data on epigenetic alterations related to exposure to sulfur mustard. Most recent evidence suggests that changes in the expression of specific miRNAs may represent potential biomarkers of exposure to sulfur mustard, and be related to inflammatory responses in the lung. The evidence of sulfur mustard exposure-induced changes in DNA methylation is not indicative of pathogenesis. Additional research is necessary to understand the potential of sulfur mustard exposure to alter the epigenome, and how such alterations may be related to disease outcome.
4.6. Formaldehyde.
4.6.1. DNA Methylation.
A study previously reviewed within Chappell et al. [11] showed long-term, low-dose formaldehyde treatment within human bronchial epidermal cells (16HBE) induced global DNA hypomethylation. Since 2016, one publication concerning global methylome modifications following occupational formaldehyde exposure has been published. Beauty salon workers exposed to low levels (up to 0.24 ppm) of formaldehyde demonstrated DNA hypermethylation in whole blood samples [136].
4.6.2. Histone Modifications.
Studies published before 2016 [11] investigating histone modifications following formaldehyde exposure were conducted within human cell lines and histone 4 isolated from calf thymus. The publications focused on the phosphorylation of H3 through JNK and PI3K/Akt pathways (responsible for cellular process regulation) and the inhibition of post-translational modifications by formaldehyde-induced Schiff bases. Since 2016, one publication investigating the epigenetic effects of formaldehyde exposure within human cells has been published. Human bronchial epithelial cells (BEAS-2B) demonstrated the inhibition of covalent modifications of histones H3 and H4 following formaldehyde treatment [137]. This inhibition resulted in aberrant chromatin assembly, altering the expression of multiple cancer-related genes (e.g. JUN, JUNB, CDKN1A, and SERPINB5).
4.6.3. Non-Coding RNAs.
Previously summarized studies [11] concerning the dysregulation of non-coding RNA expression due to formaldehyde exposure utilized human in vitro and animal in vivo models (rodent and alternative species). These publications focused on the identification of potential miRNAs biomarkers for formaldehyde exposure and the role of inflammatory response pathways in formaldehyde-associated carcinogenesis. No studies concerning the effect of formaldehyde on non-coding RNAs were published between 2016 and 2020.
Overall, the evidence of epigenetic alterations related to formaldehyde exposure is limited. Only two studies have been published on such effects since the 2016 review; these studies provide supportive data of previous publications that indicated global DNA hypomethylation and chromatin dysregulation. For DNA hypomethylation, a single study published since the 2016 review reported human in vivo evidence of global DNA hypomethylation, supporting the in vitro evidence reported in a single study included in the 2016 review. The histone modifications were all exclusively studied in cell lines. Additional research is necessary to understand the potential of formaldehyde exposure to alter the epigenome, and how such alterations may be related to disease outcome.
4.7. 1,3-Butadiene.
4.7.1. DNA Methylation.
Previously summarized studies in Chappell et al. [11] reported modification of the methylome following 1,3-butadiene exposure utilized mouse in vivo models; these reports focused on strain- and tissue-specificity of DNA methylation levels. Since 2016, mouse in vivo studies have continued to be the model of choice; scientific interest has focused on variation within damage responses (e.g. sex-specificity) as well as the influence of genetic variation on epigenomic modifications. For example, sex-specific variation in cytosine DNA hypomethylation patterns within the target (liver and lung) and non-target (kidney) tissues was identified in a mouse population-wide model [138]. Furthermore, previous findings of strain-specific effects on DNA methylation were corroborated with respect to the levels of methylation of LINE-1 and short interspersed nuclear element-B1 (SINE B1) retrotransposons detected following an in vivo exposure within a murine model [139]. The liver showed a significant correlation between 1,3-butadiene-induced DNA damage and DNA methylation at the population level; a negative correlation between DNA adduct levels and the extent of SINE B1 methylation was reported. Inter-strain variability in 1,3-butadiene-associated global DNA methylation, as measured by methylation of LINE-1, was far greater than that for DNA adducts or histone methylation.
4.7.2. Histone Modifications.
As stated above, all prior publications concerning histone modifications following 1,3-butadiene exposure were reported within mouse in vivo models. Since 2016, all new research has been collected via mouse in vivo inhalation studies to highlight how population variability may influence susceptibility to epigenetic modifications following 1,3-butadiene exposure. For example, inter-strain variability of global histone modifications following 1,3-butadiene inhalation exposure was demonstrated within 20 Collaborative Cross mouse strains [139]. In the lung, trimethylation of H3K9 and acetylation of H3K27 were significantly decreased after exposure in all 20 strains. Conversely, trimethylation of H4K20 in the liver and H3K6 in the kidney showed significant increases. Interestingly, butadiene-associated effects on variability in chromatin accessibility across tissues in exposed mouse strains only partially explained the observed variability in gene transcription effects, indicating that effects on histones and chromatin are not exclusive regulators of the gene expression effects of butadiene [140].
4.7.3. Non-Coding RNAs.
In a study of two inbred mouse strains (C57BL/6J and CAST/EiJ) exposed to 1,3-butadiene via inhalation, strain-specific differences in miRNA expression in response to treatment were observed [141]: no effects were observed in CAST/EiJ mice, while exposure resulted in changes in the expression of 109 miRNAs in C57BL/6J mice. Notably, miR-326-2p, a key mitigator of cell proliferation and migration in lung carcinogenesis, and miR-150-5p, a regulatory miRNA for glucuronosyltransferases Ugt1a1, Ugt1a2, and Ugt1a5, were affected. These results suggest that butadiene-associated alterations to miRNA expression are strain-specific.
Overall, the recent evidence demonstrates the ability of 1,3-butadiene to alter DNA methylation, chromatin organization, histone modifications, and miRNAs in mice, with variation in such changes between strains and tissues. These data provide support for the involvement of epigenetic alterations in DNA damage response and cell proliferation and migration, as well as genome integrity; however, further research is necessary in human in vitro models or from human samples from in vivo exposures to understand the potential of 1,3-butadiene to alter the epigenome in humans.
4.8. Vinyl Chloride.
4.8.1. DNA Methylation.
As shown within Chappell et al [11], publications concerning epigenetic modifications induced by vinyl chloride exposure were exclusive to DNA methylation in humans . Specifically, these studies focused on the role genetic mutations (e.g. p53 abnormalities) play within vinyl chloride-associated carcinogenesis and the specific methylation of DNA repair genes (e.g. MGMT). Since 2016, one additional report concerning vinyl chloride exposure causing alterations to the methylome has been published, when reported that hepatocytes collected from rats intraperitoneally injected with vinyl chloride showed a positive correlation between DNA damage and promoter methylation of RASSF1A (a gene associated with cell cycle regulation) and MGMT [142].
4.8.2. Histone Modifications.
No studies of histone modifications were identified.
4.8.3. Non-Coding RNAs.
A recent study within human peripheral blood lymphocytes collected from vinyl chloride-exposed workers possessed four significantly dysregulated miRNAs (miR-222-3p, miR-146a-5p, miR-151a-5p, miR-22-3p). Interestingly, miR-22-3p upregulation was positively correlated with high micronuclei frequency, a sign of genotoxicity [143]. These sites of modified expression could act as potential biomarkers of vinyl chloride monomer exposure and resulting carcinogenesis.
Overall, there is very limited evidence of epigenetic alterations related to vinyl chloride. The recent evidence suggests that vinyl chloride exposure can lead to alterations to epigenetic regulation of genes related to DNA damage and/or repair. Additional research is necessary to understand the potential of vinyl chloride exposure to alter the epigenome, and how such alterations may be related to disease outcome.
4.9. Occupational Exposure as a Painter.
4.9.1. DNA Methylation.
No studies of DNA methylation were identified.
4.9.2. Histone Modifications.
No studies of histone modifications were identified.
4.9.3. Non-Coding RNAs.
A recent study of four professional painters within the naval industry aimed to better understand the role of blood miRNAs as biomarkers of exposure to common volatile organic solvents (VOCs) (ethylbenzene, toluene, xylene) used within shipyard painting [144]. Significant upregulation of miR-6819-5p and miR-6778-5p was noted within exposed workers when compared to controls. Evaluated urinary metabolites of exposed workers showed significant associations between miRNAs and organic solvents’ absorbed dose. The authors concluded a more robust study could provide key insight into specific miRNAs that may be used as biomarkers of exposure to match with specific VOCs at defined concentrations.
Overall, there is very limited evidence of epigenetic alterations related to occupational exposure as a painter. The recent evidence suggests that changes in the expression of specific miRNAs may represent potential biomarkers of exposure to VOCs. Additional research is necessary to understand the potential of occupational exposure as a painter to alter the epigenome, and how such alterations may be related to disease outcome.
4.10. Coal Tar Pitch.
4.10.1. DNA Methylation.
Coal-tar pitch extract (CTPE) induced down-regulation of genomic DNA methylation in BEAS-2B cells (lung epithelial cells) [145]. CTPE-induced BEAS-2B cells in culture possessed upregulated DNMT1, DNMT3A DNMT3B, HDAC1, let-7a, and miR-21 levels within early passages, representing potential biomarkers for coal tar pitch-induced carcinogenesis.
4.10.2. Histone Modifications.
No studies of histone modifications were identified.
4.10.3. Non-Coding RNAs.
CTPE-induced BEAS-2B cells, when compared to controls, showed 707 differentially expressed lncRNA [146]. Specifically, lncRNA ENST00000501520 expression increased in malignant-transformed cells exposed to coal tar pitch [146, 147]. When ENST00000501520 was knocked down, cell arrested in G1 phase and increased apoptosis was observed.
Overall, there is very limited evidence of epigenetic alterations related to coal tar pitch. Recent research in vitro has demonstrated that coal tar pitch can alter the expression of genes that encode DNA methyltransferases and histone deacetylate 1, as well as several non-coding RNAs. Additional research is necessary to understand the potential of coal tar pitch exposure to alter the epigenome, and how such alterations may be related to disease outcome.
4.11. 4-Aminobiphenyl.
4.11.1. DNA Methylation.
No studies of DNA methylation were identified.
4.11.2. Histone modifications.
No studies of histone modifications were identified.
4.11.3. Non-Coding RNAs.
As summarized elsewhere [11], previous studies reporting epigenetic alterations due to 4-aminobiphenyl exposure utilized human cells. The reports focused on histone modifications and the role of miRNAs in DNA damage response following exposure. Since 2016, one additional study has been published further investigating the role of miRNAs in 4-aminobiphenyl genotoxicity. Human liver carcinoma cells (HepG2) exposed to 4-aminobiphenyl demonstrated increased reactive oxidative species generation, leading to the increased expression of cAMP-response element-binding protein (CREB; a transcription factor) [148]. The upregulation of CREB increased the expression of miR-630 which modulates RAD18 and MCM8 (genes associated with homologous recombination repair pathway) expression. These findings may increase understanding of molecular mechanisms concerning the role of miR-630 in reducing DNA damage repair within 4-aminobiphenyl-treated hepatic cells.
Overall, there is very limited evidence of epigenetic alterations related to exposure to 4-aminobiphenyl, all of which was collected in vitro. The recent evidence suggests that 4-aminobiphenyl alters the expression of miR-630, which modulates some DNA repair genes. Additional research is necessary to understand the potential of 4-aminobiphenyl exposure to alter the epigenome.
5. Epigenetic effects associated with IARC Group 1 human carcinogens and associated occupations included in Monographs 105-118.
Eight additional human genotoxic carcinogens that were evaluated by IARC after Monograph Volume 100 also met inclusion criteria for this systematic literature review. A description of the scientific evidence concerning DNA methylation (global, gene-specific, and DNMT associated), histone modifications (global, gene-specific, histone enzyme associated, or chromatin structure), and non-coding RNAs (miRNA and lncRNA targeted and untargeted testing) for each carcinogen is summarized herein. Additionally, a brief description of the common routes of exposure, associated cancers, and evidence of genotoxicity is provided. Further background information for each carcinogen is available in the corresponding IARC monograph (Table 1).
5.1. Outdoor Air Pollution.
5.1.1. Routes of exposure, associated cancers, and genotoxicity.
Outdoor air pollution is a multi-source, complex exposure of one or more pollutants that occurs continuously. As summarized within IARC Monograph 109, the main exposure source categories include vehicle emissions; stationary power generation; agricultural and manufacturing emissions; residential sources; terrestrial and aquatic re-emissions; chemical production, distribution, and use; and natural processes. Outdoor air pollution includes photochemical oxidants, particulate matter, sulfur dioxide, carbon monoxide, nitrogen oxide, toxic and/or volatile metals, polycyclic aromatic hydrocarbons (PAHs), organic compounds (VOCs, SVOCs, and particulate organic matter), mineral dust/fibers, and bioaerosols. Target organs for carcinogenesis following outdoor air pollution exposure include the lung, bladder, and breast; hematological malignancies have also been associated with these exposures. Human in vitro and in vivo data significantly indicates outdoor air pollution induces pulmonary cancers through genotoxic mechanisms and promotes tumorigenesis through oxidative stress, responses to oxidative stress, and persistent inflammation. Further exposure and other information are detailed elsewhere [149].
5.1.2. Epigenetic effects.
Scientific evidence of outdoor air pollution-associated epigenetic modifications is voluminous. A total of 163 studies were included in this review; of these, 140 publications reported effects in vivo. DNA methylation is the most studied epigenetic effect of outdoor air pollution (64%), followed by non-coding RNAs (34%) and histone modifications (7%). A total of 135 publications (83% of all included) reported data from studies in humans (112 in vivo and 24 in vitro studies). As most outdoor air pollution exposure publications concern human in vivo investigations into epigenetic modifications, the most recently published studies per endpoint (DNA methylation, histone modifications, and non-coding RNAs) have been identified below to provide insight into current research areas. All other included studies can be reviewed within the HAWC assessment.
5.1.2.1. DNA Methylation.
Recent studies have focused on mitochondrial DNA methylation [150, 151], identification of mechanistic pathways involved in outdoor air pollution-induced toxicity [152, 153], and in utero or childhood exposures [154–156] in connection to noncommunicable diseases, including cancer, in adults. For instance, Gruzieva et al. [157] provided evidence of a link between DNA methylation differences in several genes induced by particulate air pollution in newborns and airway disease, and Callahan et al. [158] reported that lifetime exposure women to ambient air pollution at first birth was associated with aberrant methylation of tumor suppressor genes in breast tumors. Additional studies have investigated gene-specific methylation, specifically “clock genes” associated with the circadian rhythm [159], and methylation of genes encoding inflammatory cytokines [160].
5.1.2.2. Histone Modifications.
A recent report showed air pollution exposure impairs brain chromatin silencing by decreasing histone posttranslational modifications, reducing DNA integrity overall [161]. Additionally, early-life ambient air pollution exposure was associated with cord plasma histone H3 modifications [162].
5.1.2.3. Non-Coding RNAs.
No human in vivo studies of epigenetic effects of outdoor air pollution investigated long non-coding RNAs; all non-coding RNA publications focus on miRNA. The latest miRNA research has centered on the identification of miRNA biomarkers for air pollution health risk assessment [163], prenatal exposure inducing inflammatory responses and carcinogenesis [164, 165], and miRNA-mediated mechanisms of air pollution biological insults [166].
Overall, the evidence of the role that epigenetic alterations may play in cancer hazard of outdoor air pollution is rapidly evolving. Because of the complexities of modeling outdoor air pollution in the laboratory environment, most studies published to date have been conducted in human cohorts. While such evidence in exposed humans is most relevant to environmental health decisions, challenges remain in comparing the results between studies or generalizing broadly because of the deficiencies in exposure assessment and spatial and temporal variability in pollutant concentrations.
5.2. Engine exhaust, diesel.
5.2.1. Routes of exposure, associated cancers, and genotoxicity.
Diesel and gasoline represent major energy sources used to power motor vehicles. Most human exposures occur in occupational (e.g. mining, construction, manufacturing) and traffic settings. Significant positive correlations exist between diesel engine emission exposure and the occurrence of lung and/or bladder cancer within humans. Diesel engine exhaust-associated carcinogenesis has been linked to genotoxic mechanisms including DNA damage, mutations and chromosomal-level events, gene expression modulation, and production of reactive oxygen species (e.g., increased inflammatory response). Lung carcinogenicity following diesel engine emission exposure is suspected to be amplified by the other known and suspected human carcinogens within diesel engine exhaust. Further exposure and other information are detailed elsewhere [167].
5.2.2. DNA methylation.
Workers chronically exposed to diesel engine exhaust exhibited significant hypomethylation of p16, RASSF1A, and MGMT (DNA damage response-related genes) and increased cytokinesis-block micronucleus cytome index, a biomarker of carcinogenesis [168]. A second human in vivo study, evaluating peripheral blood mononuclear cell DNA methylation levels, demonstrated significant global hypomethylation of CpG sites involved in inflammation responses, repetitive elements, and miRNA [169]. Human umbilical vein endothelial cells (HUVECs) exhibited increased reactive oxygen species generation and DNA damage following diesel exhaust particulate matter exposure [170]. Furthermore, aberrant gene methylation was reported.
Several human in vivo and in vitro studies have analyzed diesel exhaust and allergen co-exposures. For example, human bronchial epithelial cells exposed to diesel exhaust particles and house dust mite allergens resulted in increased TET1 and DNMT1 expression, leading to DNA methylation and hydroxymethylation changes in inflammation and epithelial repair response genes [171]. In contrast, human in vivo exposure to diesel exhaust particles, allergens, or co-exposure only demonstrated minor DNA methylation changes [172]. Only when allergens and diesel exhaust were experienced in sequential insults were significant CpG site methylation levels noted, with the order of exposure determining unique epigenetic changes. An additional study identified airway hyperresponsive patients with naturally high TET expression and global hypermethylation as a susceptible population to diesel exhaust-associated exposures [173].
Several rodent studies in vivo investigated the epigenetic effects of diesel exhaust exposure, specifically in utero exposures. For example, pregnant C57BL/6J mice were exposed to diesel exhaust; their offspring were subsequently necropsied for brain tissue at either day 1 or day 21 [174]. The genome-wide DNA methylation status was disrupted at both time points investigated, suggesting potential future health insults and disease induction due to altered developmental pathways. Additional studies demonstrated in utero exposure within murine models caused altered methylation within cardiomyocytes, modifying cardiac transcriptional responses [175] and metabolic capability of the cells [176]. Aberrant CpG island methylation of p16INK4A was noted within diesel exhaust-associated tumors, indicating tumorigenesis may involve p16 suppression [177]. Furthermore, the inactivation of p16 supports a possible role for oxidative stress and inflammation in the initiation of lung cancer. Lung inflammation following diesel exhaust exposure was further explored in Sprague-Dawley rats [177]. Diesel exhaust particle exposure suppressed Clara cell secretory protein (CC16) levels through aberrant methylation of the CCAAT/enhancer-binding protein alpha (C/EBPa) promoter.
5.2.3. Histone Modifications.
Human bronchial epithelial cells exposed to diesel exhaust particles showed enhanced expression of COX-2, a key mediator of stress-induced inflammatory responses; this effect was attributed to chromatin modifications [178]. Specifically, increased acetylation of H4 occurred through the degradation of HDAC1 (COX-2 transcription regulator).
5.2.4. Non-Coding RNAs.
Several studies utilizing human cells have demonstrated diesel exhaust exposure altered miRNA and gene expression [179–181]. For example, diesel exhaust exposure increased miRNA 21 expression and subsequently activated the PTEN/P13K/AKT pathway, a potential mechanism for diesel exhaust-associated carcinogenesis [182]. A second study demonstrated diesel exhaust exposure repressed miR382-5p expression, increasing CXCL12/MMP9 and triggering pulmonary inflammation [183].
Human in vivo studies investigating diesel exposure have shown dysregulated miRNA and associated genes induce bronchial immune responses [184] and oxidative stress [185]. Additionally, peripheral blood mononuclear cells collected from diesel exhaust-exposed finishing workers showed significant DNA adduct formation and miRNA dysregulation [186]; aberrantly expressed miRNAs were associated with carcinogenesis apoptosis and antioxidant effects.
Overall, there is moderate level of evidence for epigenetic alterations related to exposure to diesel engine exhaust, studies of these effects are available from different levels of biological organization, from in vitro to exposed humans. Most compelling evidence links transcriptional changes associated to these exposures with epigenetic mechanisms of controlling gene expression.
5.3. Lindane.
5.3.1. Routes of exposure, associated cancers, and genotoxicity.
The International Organization for Standardization (ISO) common name of “lindane” refers to any material containing > 99% γ-hexachlorocyclohexane (γ-HCH), a stable isomer of hexachlorocyclohexane. Lindane is primarily used as an insecticide within forestry and agricultural settings but is used in some niche pharmaceutical scenarios to treat head lice and scabies (mites). Adverse effects from lindane exposure have been noted within the liver, kidney, and hematopoietic systems along with testicular toxicity. Lindane is suggested to be immunosuppressive by increasing oxidative stress and genotoxic by inducing chromosomal and micronuclei frequency alterations. Specifically, the IARC Monographs working group concluded that the evidence is moderate that lindane is genotoxic. Information on exposure is detailed elsewhere [187].
5.3.2. Epigenetic effects.
One study reported epigenetic effects following lindane exposure, analyzing both DNA methylation and histone modifications. Pregnant female albino Wistar rats were exposed to lindane (0.25 mg/kg) and corn oil [188]. The resulting male offspring were further divided into three subgroups based on age (9, 18, or 27 weeks). These three subgroups were separated into subsequent treatment cohorts: lindane (5 mg/kg by weight, orally, 5 consecutive days) or corn oil (5 mg/kg by weight, orally, 5 consecutive days). Results from this study revealed upregulation in histone H3 acetylation and CpG methylation of promoter regions of cerebral cytochrome P450s (CYPs). The authors concluded the specific CpG alterations following lindane exposure may suggest the occurrence of imprinting of cerebral CYPs, which may account for enhanced responsiveness of these indicated CYPs. More robust rat model studies were recommended to validate these hypotheses.
Overall, the evidence of epigenetic alterations related to exposure to lindane is very limited, only one study in rats has been identified. Additional research is necessary to understand the potential of lindane exposure to alter the epigenome.
5.4. Pentachlorophenol.
5.4.1. Routes of exposure, associated cancers, and genotoxicity.
Pentachlorophenol is produced through (i.) stepwise chlorination of phenols in the presence of ferric chloride or anhydrous aluminum chloride (catalysts) or (ii.) alkaline hydrolysis of hexachlorobenzene. While the initial production of the chemical was for wood preservation, the compound is now used as an herbicide, fungicide, germicide, and algicide; in tannery; in production of paints and adhesives; and in general construction such as insulation and brickwork. Due to its primary uses, human exposure commonly occurs in occupational settings and has been linked to non-Hodgkin lymphoma, soft tissue sarcoma, and acute lymphocytic leukemia. Human in vitro data has indicated pentachlorophenol induces oxidative stress and genotoxicity; non-mammalian models report increased oxidative stress markers and DNA adduct levels. Information on exposure is detailed elsewhere [149].
5.4.2. Epigenetic effects.
One study reported epigenetic effects following pentachlorophenol, specifically modifications of non-coding RNAs. Pregnant ICR mice were exposed to pentachlorophenol (0.02, 0.2, or 2 mg/kg by weight) during gestation day (GD) 0.5 to GD 8.5 and then compared to controls [189]. Autophagy was increased within exposed mouse placentas as indicated by upregulated mRNA expression of P62, LC3-II/LC3-I, and Beclin1. To further investigate the increased autophagy found in pentachlorophenol-exposed placenta samples, the expression of miR-30a-5p (an expression inhibitor of Beclin1) was evaluated. Increased pentachlorophenol exposure was significantly correlated with the downregulation of miR-30a-5p expression and induction of autophagy through Beclin1 upregulation. These results characterize the developmental toxicity of pentachlorophenol, completed through autophagy.
Overall, the evidence of epigenetic alterations related to exposure to pentachlorophenol is very limited, only one study in mice has been identified. Additional research is necessary to understand the potential of pentachlorophenol exposure to alter the epigenome.
5.5. Polychlorinated biphenyls.
References for polychlorinated biphenyls (PCB) have been separated into the non-dioxin-like ortho-substituted PCB category, in which their two rings are not aligned in the same plane, and the more toxic dioxin-like category, with meta and para-substituents whose co-planar aromatic rings are aligned like those of the highly toxic polychlorinated dibenzodioxins. Dioxin-like PCB include PCBs 77, 81, 105, 114, 118, 123, 126, 156, 157, 167, 169, and 189, as listed within IARC Monograph 107 [190].
5.5.1. Routes of exposure, associated cancers, and genotoxicity.
PCB are ubiquitous environmental contaminants found in all elements (earth, water, air), wildlife, and in most humans. The carcinogens are produced both naturally and during industrial processes. Exposures may occur via ingestion, inhalation, and dermal absorption. Occupational sources include fires, recycling processes, and waste incineration. Exposure to PCB in human populations has been significantly associated with malignant melanoma, non-Hodgkin lymphoma, and cancer within the breast tissue. Multiple modes of actions have been identified for PCB and their metabolites. Specifically, the IARC Monographs working group concluded that PCBs occur and act in complex mixtures eliciting both genotoxic and nongenotoxic effects associated with carcinogenesis, tumor promotion, and progression. Less chlorinated congeners induce oxidative stress and genotoxicity following oxidative metabolism. Highly chlorinated congeners interact with receptors associated with xenobiotic and androgen metabolism. PCB also affect plasma membrane-associated proteins, causing dysregulation of cell processes, and promotion of tumorigenesis. Information on exposure is detailed elsewhere [190].
5.5.2. Non-Dioxin Like PCB.
5.5.2.1. DNA Methylation.
A large number of studies (39 publications) were identified for non-dioxin-like PCB effects on DNA methylation; therefore, only human in vivo data are summarized below. Studies investigating DNA methylation following PCB exposure in human cells and rodent/alternative species models can be reviewed within the HAWC assessment: https://hawcproject.org/assessment/1063/.
Several studies have reported an inverse relationship between global DNA methylation and PCB blood plasma levels collected from human subjects [191, 192]. A cross-sectional analysis identified global DNA hypermethylation within elderly individuals following PCB exposure, potentially identifying a susceptible population [193]. A study of genome-wide methylation following exposure to non-dioxin-like PCB reported a positive correlation between increased aberrant methylation and intake of fish that likely served as a source of non-dioxin-like PCB [194].
Other studies have implicated potential sex-specific effects of epigenomic responses induced by PCB exposure. For example, global DNA hypomethylation was denoted within an Alu assay for men and DNA hypermethylation within a LINE-1 assay for women [195]. Additionally, significant enrichment was noted within specific X-chromosome sites within prenatally PCB-exposed males [196]. Global hypermethylation was noted following PCB exposure within sperm DNA [197].
Several reports have investigated maternal PCB exposure and resulting prenatal toxicity [198–200]. Aberrant methylation of several genes was detected in blood of newborns perinatally exposed to high concentrations of PCB [201]; this study is the first to indicate PCB exposure persistence in an in utero model and may provide further insight into the underlying epigenetic mechanisms. One study, utilizing placental samples collected from PCB-exposed women, highlighted a positive association between total PCB levels and H19 expression [202].
Differentially methylated genes following PCB exposure in humans may provide further mechanistic evidence to better understand resulting carcinogenesis [203, 204]. Additionally, the methylation of specific genes may offer epigenetic biomarkers for early disease or adverse health effect detection [205, 206]. For example, hypermethylation of MGMT displayed an inverted U-shaped association following PCB exposure, indicating a potential non-monotonic dose-response [207].
5.5.2.2. Histone Modifications.
Prolonged exposure to Aroclor 1254 within human neuroblastoma cells (SH-Sy5Y) demonstrated dose-dependent cytotoxicity; cell death was proposed to occur via repressor element 1-silencing transcription factor (REST) decreasing synapsin1 and H3 and H4 acetylation [208]. Early-life PCB exposure within a rat model decreased H4K16ac and H3K4me3 levels via the induction of SirtT1 and Jarid1b (chromatin-modifying enzymes) and downregulation of androgen receptors [209].
5.5.2.3. Non-Coding RNAs.
PCB exposure was shown to affect the implantation function of human endometrial cells by impairing expression of miRNAs that are involved in the epithelial-mesenchymal transition processes [210]. The authors hypothesized that miRNAs may play a role in PCB-associated endometrial toxicity. As novel prenatal biomarkers, placental miRNA profiles were suggested as predictors of in utero PCB exposure [211], providing insight into the role of the placental epigenome in human reproduction and development.
Additional studies identified miRNAs and target genes with modified expression following PCB exposure were associated with various types of cancer [212] and provide key insights into mechanisms of PCB toxicity [213] within humans. Rodent in vitro and in vivo models implicated dysregulated miRNA levels following PCB exposure in the induction of several biological outcomes, including neurodevelopmental disorders [214, 215], atherosclerosis [216], and cardiac development [217].
Overall, most of the evidence demonstrating the ability of non-dioxin-like PCBs to elicit epigenetic effects comes from studies of global and gene-specific methylation patterns. These changes have been suggested as biomarkers of exposure and may be related to a number of secondary mechanisms of carcinogenesis. It is noteworthy that the majority of such alterations were measured and observed in human studies.
5.5.3. Dioxin-like PCBs.
5.5.3.1. DNA Methylation.
Global methylation levels were significantly decreased following dioxin-like PCB exposure within several cross-sectional human in vivo studies, demonstrating that exposure to carcinogenic environmental pollutants induces change in the methylome [192, 218]. Further investigation into the epigenetic modifications associated with exposures to dioxin-like PCBs in humans showed significant changes in CpG methylation [219]. In contrast to human in vivo results, human carcinoma cell lines (HepG2 and HeLa) treated with PCB 126 showed no apparent differences within methylation levels of specific CpG locations following exposure [220]. The study further investigated the expression of CYP1A1 following PCB 126 treatment. It was shown that cell-specific variation in treatment-induced chromatin accessibility of CYP1A1 promoter may be an important epigenetic regulatory pathway for cell-specific CYP1A1 expression. PCB 126 exposure within a zebrafish model showed significantly altered global methylation, with most modifications occurring within genes associated with development, metabolism, and regeneration pathways [221].
PCB118 (2,3′,4,4′,5-pentachlorobiphenyl) is the most individually studied dioxin-like polychlorinated biphenyl due to its high toxicity rate and rapid bioaccumulation capability. Three in vivo studies utilizing pregnant mice orally dosed with PCB118 identified altered methylation rates of imprinted genes and modified expression of Dnmt1, Dnmt3a, Dnmt3l, Uhrf1, and Tet3 following exposure [222]. Specifically, methylation patterns within genes H19 and Gtl2 of male offspring from the exposed dams were altered, identifying sex-variation in response to PCB118 exposure. Within the exposed dams, PCB118 dosage caused dysregulated oocyte maturation [222], defective uterine morphology, and decreased implantation rates [223].
5.5.3.2. Histone Modifications.
Human vascular endothelial cells treated with PCBs 77 or 126 showed increased expression of jumonji domain-contain protein 2b (JMJD2b). These affects were correlated with decreased H3K9me3 repression marks and significantly increased H3K4me3 activation marks in the promoter of this gene; these histone modifications were also associated with increased expression of inflammatory genes following dioxin-like PCB treatment [224]. Human endothelial cells exposed to relevant levels of PCB 126 and/or epigallocatechin-3-gallate (EGCG-an anti-inflammatory polyphenol) were evaluated for epigenetic modifications. Co-exposure of PCB 126 and EGCC decreased PCB-induced NF-kB activation through chromatin binding and histone H3 hypo-acetylation [225]. Mouse Hepatoma-1c1c7 cells treated with various dioxin-like PCBs (77, 169, and 153) demonstrated PCB-induced histone modifications depend upon the activation of AHR by ligand before chromatin modifications critical for active transcription can occur [226].
5.5.3.3. Non-Coding RNAs.
Human in vivo studies have shown PCB exposure induces significantly modulated miRNA and target gene expression, with the specified miRNA locations may serve as potential biomarkers for diagnosis of subsequent biological effects [212, 227]. Human liver cells (L-02) exposed to PCB 156, a compound associated with non-alcoholic fatty liver disease, was found to modulate the expression of mRNAs and lncRNAs associated with metabolic and inflammatory processes [228]. These identified effects may be used as potential biomarkers for early detection of the PCB-induced lipid metabolism disorder.
Overall, similar to the database on the non-dioxin-like PCBs, the dioxin-like PCBs affect global and gene-specific methylation patterns in exposed humans. Most of the reported effects relate to metabolism- and chromatin remodeling-related genes. Importantly, these changes in DNA methylation have been corroborated by findings of histone modifications and chromatin remodeling upon exposure to a number of dioxin-like PCBs.
5.6. Trichloroethylene.
5.6.1. Routes of exposure, associated cancers, and genotoxicity.
Trichloroethylene, a solvent used in medical, industrial, and consumer markets, is widely distributed in the environment (soil, air, water). The main source of exposure is during degreasing processes (an occupational exposure). Additional information on exposure is detailed elsewhere [229].
5.6.2. DNA Methylation.
The vast majority of publications concerning modifications to the methylome following trichloroethylene treatment utilized mouse in vivo models. Many of these studies focused on the trichloroethylene exposure-associated CD4+ T cell-mediated autoimmunity. For example, MRL mice were exposed to trichloroethylene via drinking water until adulthood; continuous exposure throughout development showed significant differentially methylated binding sites of PcG proteins, potentially representing a mechanism by which trichloroethylene alters the function of effector/memory CD4+ cells and induced immunotoxicity [230–232]. Further investigation into this mechanism demonstrated hypermethylation of CpG sites within the IFN-γ promoter of effector/memory CD4+ T cells, representing especially sensitive sites to chronic trichloroethylene exposure [233]. DNA methylation-induced activation, silencing, and reactivation of proinflammatory genes may represent a mechanism by which autoimmune pathology occurs following trichloroethylene exposure.
Additionally, reports in mouse in vivo studies focused on trichloroethylene-induced DNA methylation, and resulting gene expression changes, within the liver. For example, B6C3F1 mice orally dosed with trichloroethylene for five days demonstrated dose-dependent gene expression dysregulation, specifically of Utrf1, Tet2, DNMT1, DNMT3a, and DNMT3b, potentially leading to tumorigenesis [234]. Trichloroethylene was found to decrease global methylation levels and hypomethylated the promoter regions of protooncogenes, c-jun, and c-myc; these mechanisms are associated with increased cell proliferation and tumor promotion [235]. Several studies utilized trichloroethylene metabolites, dichloroacetic acid, and trichloroacetic acid. These studies concluded that trichloroethylene metabolite exposure induced DNA hypomethylation [236, 237] and increased expression of c-jun and c-myc genes [238] within liver tumors. Dichloroacetic acid-associated hypomethylation was prevented by methionine co-administration [239]. Additionally, methionine, an amino acid, may represent a potential treatment following accidental trichloroethylene exposure.
Human in vitro studies of the epigenetic modifications induced by trichloroethylene treatment have predominately focused on the identification of biomarkers of exposure and global DNA methylation changes. For example, human lymphoblastoid (TK6) cells demonstrated global DNA methylation as an early event in the genotoxic pathway of trichloroethylene [240]. Additionally, trichloroethylene exposure induced significant hypomethylation, subsequently inhibiting DNMT activity [241, 242] while increasing expression of SET [243] within L-02 cells. Utilizing single-molecular monitoring, it was uncovered trichloroethylene-associated DNA hypomethylation could be partially explained by disrupted DNMT3A-DNA associations [202]; in a dose-dependent manner, DNMT3A oligomers dissociated from heterochromatin, resulting in significant loss of 5-methylcytosine (5mC) and enhancement of TET enzymes. In total, these results present a mechanistic understanding of trichloroethylene toxicology and may provide the framework for comparable investigation of other environment toxicants.
5.6.3. Histone Modifications.
Multiple studies used L-02 cells to demonstrate histone modifications following trichloroethylene exposure. For example, trichloroethylene exposure increased the histone chaperone SET, enhancing H2AK9ac through HDAC1 inhibition, a mechanism associated with SET-mediated hepatic cytotoxicity [244]. Further investigation into trichloroethylene-induced hepatotoxicity suggested relative expression level changes of H3K79me2 and H3K79me3 were induced through DNA damage [245].
Naturally high levels of SET can induce significant modifications of histone deacetylases, suggesting SET is involved in trichloroethylene-induced cytotoxicity and epigenetic alterations [246]. An in-depth investigation into the mediation of trichloroethylene-induced hepatic cytotoxicity observed SET promoting acetylation of H2AK9 by suppressing HDAC1 levels [244]. This type of investigation could be utilized to further examine underlying mechanisms in which SET modulates histone acetylation following trichloroethylene exposure.
An additional study in human hepatocytes demonstrated a negative correlation between existing DNA hypomethylation and histone expression of tumor-related genes following trichloroethylene exposure; specifically, trichloroethylene increased histone H3 lysine 9 acetylation levels [247]. H3K4me2, a mark in sperm conserved across species and linked to transgenerational epigenetic inheritance, was significantly decreased in the regulatory protein kinase A signaling pathway genes following trichloroethylene exposure [248]. This epigenetic medication may account for TCE-induced spermatozoal toxicity with H3K4me2 being a potential target for trans-generational effects.
5.6.4. Non-Coding RNAs.
Trichloroethylene was found to modulate miRNA expression within multiple species; the identified locations may represent potential biomarkers for toxicity prediction [249–251]. Occupational exposure to trichloroethylene resulted in decreased serum levels of miRNAs involved in the B-cell receptor pathways and the innate immune response [252]; these results may increase understanding of the etiologic relevance of miRNAs in trichloroethylene-associated Non-Hodgkin’s Lymphoma. Human hepatocytes treated with trichloroethylene showed miR-199b suppression [253]. Expression of miR-182-5p was upregulated following trichloroethylene exposure and was associated with increased cell proliferation [254, 255]. Alterative animal models identified miRNA locations that mediate trichloroethylene-induced reactive oxygen species generation [251] and dysregulation of cell proliferation [250, 251, 256].
Overall, evidence of epigenetic alterations associated with exposure to TCE is extensive. Almost all studies have been performed in mice in vivo and some in human and other species cell lines. The reported findings of DNA methylation, histone modifications and non-coding RNA changes are concordant with dysregulation of the pathways known to be involved in carcinogenesis and other adverse effects of TCE. Therefore, the overall database of epigenetic effects of TCE is supportive of its cancer hazard classification.
5.7. Welding Fumes.
5.7.1. Routes of exposure, associated cancers, and genotoxicity.
Welding fumes are generated when metals reach temperatures above their melting points, vaporize, and then condense into fumes primarily consisting of fine, solid particles (diameter > 1 um). Welders represent the highest risk population, as they experience contemporaneous exposures of welding fumes, gases, radiation (ionizing and non-ionizing), and co-exposures of solvents and asbestos. Exposure has been positively associated with cancer of the lung and kidney, as well as correlated to other biological effects such as ocular melanoma, leukemia, and non-Hodgkin lymphoma. Evidence of welding fumes-induced genotoxicity included chromosomal aberrations, dysregulated sister-chromatid exchange rates, DNA damage, and anomalous micronuclei frequency. Overall, the IARC Monographs working group concluded that there is moderate evidence that welding fumes are genotoxic. Additional information on exposure is detailed elsewhere [257].
5.7.2. DNA Methylation.
Several studies reported findings from the same study population. Li et al. [258] reported DNA methylation in 101 welders and 127 control workers (all non-smoking males) from southern Sweden [258–260]. Blood sampling showed increased concentration of measurable respirable dust was positively associated with hypomethylation of F2RL3, a marker for cardiovascular disease progression and mortality [259]. A second study [260] reported decreased methylation within the mitochondrial regulatory region D-loop and MT-TF (a tRNA encoding gene) when compared to controls, indicating welding fume exposure may be associated with oxidative stress. Human THP-1 and small human airway epithelial (SAEC) cells and murine (RAW254.7) cells exposed to steel welding fumes showed aberrant methylation of LINE-1, SINE, and Alu elements (transposable elements) and modification of their transcription [261]. Furthermore, the expression of DNA methyltransferases and TET genes were differentially regulated in a cell-, chemical-, and dose-dependent manner. Male Fischer-344 exposed to welding fumes by inhalation showed DNA hypermethylation, increased telomere length, and expression of neurodegeneration markers within brain tissue [262]. By contrast, similarly exposed Sprague-Dawley rats demonstrated no significant differences in DNA methylation patterns between welding fume and control groups, but increased telomere length was detected [263]; these two studies highlight inter-strain variance in the epigenetic effects of welding fume inhalation.
5.7.3. Histone modifications.
No studies of histone modifications were identified.
5.7.4. Non-Coding RNAs.
A cross-sectional study of 30 male welders was conducted to understand the association between metal particles from welding fumes exposure and circulating miRNA concentrations [264]. The presence of certain metals, specifically chromium and nickel, within blood and urine samples were significantly associated with the downregulation of expression of miR-21, -146a, and -155 (biomarkers of renal function injury).
Overall, there is limited evidence for epigenetic effects of welding fumes. A small number of studies in exposed humans and rats, as well as in vitro models, showed changes in global and gene-specific DNA methylation. Only one study of non-coding RNAs in welding fume-exposed workers was published and no reports investigating potential effects on histone modifications were published to date.
6. Summary.
This systematic literature review updates the 2016 review of the published evidence on epigenetic effects related to Group 1 human carcinogens that are genotoxic and were included in IARC Monograph volume 100F [11]. Since 2015, the number of epigenetic studies on the same chemicals reviewed at that time nearly doubled, indicating a very brisk pace of research on epigenetics as a key mechanism in chemical exposure-associated cancer (Figure 4A). Benzo[a]pyrene, benzene, and aflatoxins remain the most studied of the genotoxic carcinogens included in IARC Monograph volume 100F, with 49, 40, and 21 studies, respectively, of epigenetic effects published in the last five years. In 2015, no studies of epigenetic effects of coal tar pitch were available, but several recent publications reported such effects. By contrast, no additional research has been published concerning epigenetic modifications following exposure to benzidine or 4,4’-methylenebis-(2-chloroaniline) (MOCA). Of the agents with strong or moderate evidence of genotoxicity classified as Group 1 carcinogens by IARC in Monograph volumes 105-118 (Figure 4B), the evidence of epigenetic effects is similarly uneven across agents. Outdoor air pollution is by far the most studied, with respect to epigenetics as a key characteristic of human carcinogens. Over 160 publications reported epigenetic effects of exposure to outdoor air pollution. Notably, most of these newly classified carcinogens had more than 10 published reports of epigenetic alterations.
Figure 4.
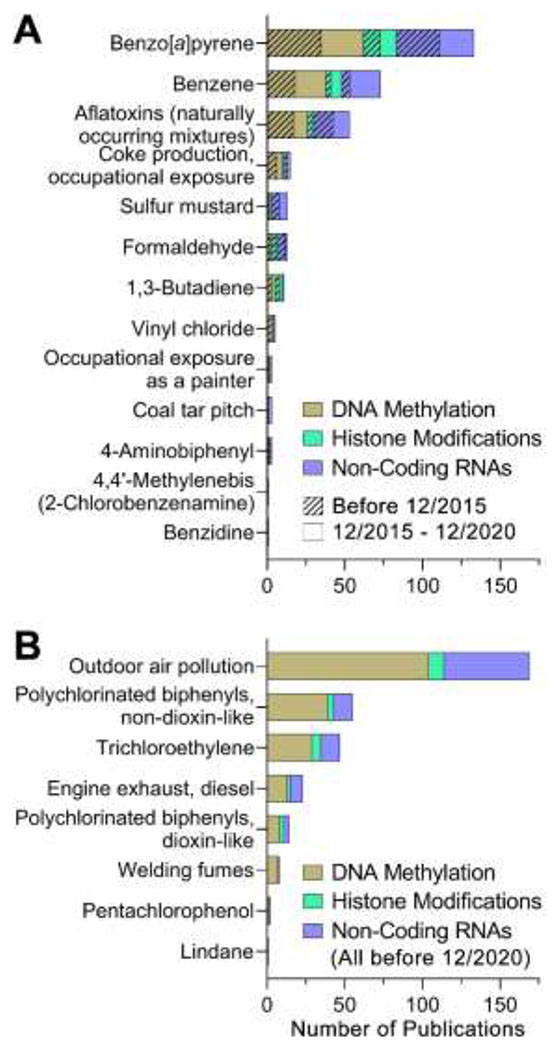
The number of publications reporting epigenetic effects of exposures to the agents, related occupations, or exposure scenarios included in IARC Monographs 100F (A) and 105-118 (B). Entries are sorted in a descending order. Colors refer to the type of the epigenetic endpoints: DNA methylation, histone modifications, and non-coding RNA are colored in gold, green, and purple, respectively (see color legend inset). In (A), studies published before December 2015 (and were included in Chappell et al [11] systematic literature review) are denoted by hashed segments. Studies published from December 2015 to December 2020 are denoted by solid segments. In (B), all studies published before December 2020 are included.
Of note is also the distribution of the types of epigenetic effects that were the subject of publications. This systematic literature review provides data to better understand the distribution of the endpoints, models and study types investigated within the included scientific reports (Figure 5). Interestingly, the relative proportion of the epigenetic endpoints remains relatively consistent for the comparison of the exposures classified in the Monograph volume 100F. Studies of DNA methylation remain of great interest to the scientific community, and non-coding RNA is an increased focus in the last 5 years. Investigation of histone modifications and chromatin state alterations in toxicology are far fewer in number, as compared to the other two epigenetic modifications. This disparity is likely because of the variety and technical demands for the assays of histones or chromatin [265], as compared to more straightforward sequencing-based assays for DNA methylation and non-coding RNAs. A similar disparity is evident for the agents identified as human carcinogens in Monograph volumes 105-118; almost two-thirds of studies focused on DNA methylation.
Figure 5.
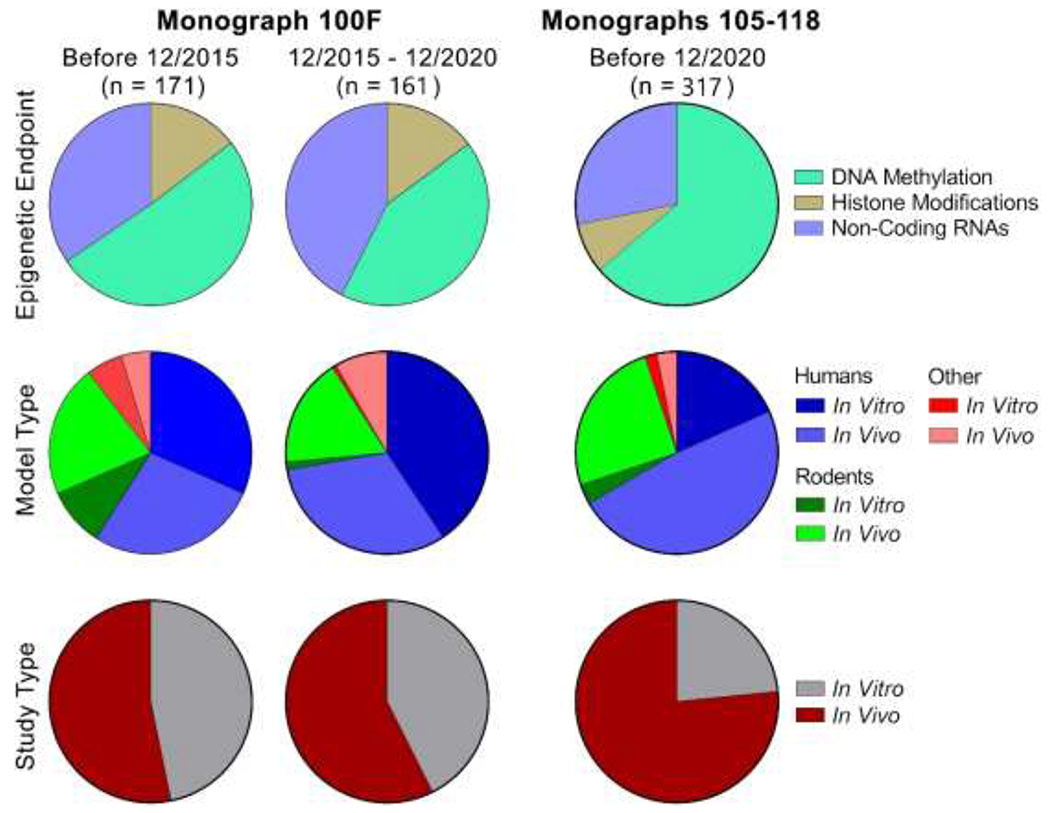
A relative comparison of epigenetic endpoints, model types (human, rodent, other), and study types (in vitro or in vivo) for the chemicals, related occupations, or types of exposure included in IARC Monographs 100F (left) and 105-118 (right). The pie charts for each type of comparison are color coded as shown in the legends.
It is also noteworthy that most studies of epigenetic effects of the agents reviewed herein were performed in human models, more than half of which were conducted in exposed humans. Studies in rodents represent the second most common type of investigation in this review, most of which were conducted in vivo. Evidence for epigenetic effects associated with environmental exposures in observational studies of humans is highly relevant and useful to human cancer hazard evaluations, as the data represent the target of interest [1]. However, it should be noted that while epidemiological data provide valuable evidence, the evidence is nonetheless associative in nature. Causal relationships between exposure and effect (or marker of effect) are more clearly delineated in mechanistic studies in controlled experimental scenarios, which do not necessarily represent “real life” exposure settings. Thus, an evidence base that includes and incorporates both types of data represents that which is the most informative for determining the likelihood that a chemical operates through a specific mechanism. As the number of studies of epigenetic effects in humans exposed to the agents reviewed herein increases, a trend that has been demonstrated through this review, the ability to replicate and/or validate the epigenetic effects in various cohorts will similarly increase and promote the confidence and applicability of such data in hazard assessment.
Even among in vitro studies, human cells are the most commonly used. The availability of the data in human cells provides an opportunity to fill gaps in knowledge when samples from human exposures are unavailable. Even though the evidence for associations between chemicals and epigenetic effects were not deemed to be “strong” in the recent IARC monographs that utilized the key characteristics approach in the evaluation of the mechanistic evidence [266], the information provided in this review demonstrates that epigenetic effects observed in studies in vitro are concordant with those from in vivo studies, an important point in support of the field of toxicology moving toward placing more weight on mechanistic data in hazard and risk assessments.
It is important to recognize that epigenetic alterations, as well as the methods to evaluate them, vary widely. The evidence presented herein is no exception to this trend. The epigenetic alterations that have a well-characterized regulatory action and are measure in relation to specific genes may inform the understanding of underlying mechanisms of a specific exposure or type of cancer to a greater degree than global changes to the epigenome. It is also true that global changes that result in genome instability are informative, albeit less specific and potential an indirect influence on carcinogenic outcomes. These nuances and limitations of epigenetics data should be considered in any assessment or integration of epigenetics data.
It is also important to note that a formal quality assessment to determine or rank the reliability of the data in studies discussed herein was not conducted as a part of this review. Nor was a weight-of-evidence framework determined or applied; thus, designations of strength of evidence (e.g., “weak” or “strong”) were not a component of this review. Rather, the review that was conducted and the results reported herein provides an overview of the landscape and state of the available science for epigenetic alterations for the Group 1 agents and exposure scenarios reviewed. Quality scoring and predetermined criteria would be necessary for a more in-depth weight-of-the-evidence assessment of any individual agent reviewed herein.
Overall, this review highlights the considerable progress in epigenetic research and its relevance to elucidating mechanisms of chemical carcinogenesis. Several important future research needs were identified. In particular, increased dose- and time-dependent testing in studies of epigenetic endpoints would improve interpretation. Further study on the persistence of effects following cessation of exposure is another important data gap, especially considering the dynamic nature of most epigenetic alterations. Finally, exploration of more diverse study populations, using both in vivo and in vitro models, will facilitate greater understanding of the variability in toxic response in exposed populations. In all, these results lay important groundwork for the application of epigenetic data in hazard and risk assessment exercises.
Supplementary Material
8. Acknowledgements.
This work was funded, in part, by grants from the National Institute of Environmental Health Sciences (R01 ES029911 and T32 ES026568). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The views expressed in this manuscript do not necessarily represent those of the U.S. Food and Drug Administration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Declaration of Interest.
The authors declare no relevant conflicts of interest with respect to the content of this review.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Samet JM, Chiu WA, Cogliano V, Jinot J, Kriebel D, Lunn RM, Beland FA, Bero L, Browne P, Fritschi L, Kanno J, Lachenmeier DW, Lan Q, Lasfargues G, Le Curieux F, Peters S, Shubat P, Sone H, White MC, Williamson J, Yakubovskaya M, Siemiatycki J, White PA, Guyton KZ, Schubauer-Berigan MK, Hall AL, Grosse Y, Bouvard V, Benbrahim-Tallaa L, El Ghissassi F, Lauby-Secretan B, Armstrong B, Saracci R, Zavadil J, Straif K, Wild CP, The IARC Monographs: Updated Procedures for Modern and Transparent Evidence Synthesis in Cancer Hazard Identification, J Natl Cancer Inst, 112 (2020) 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Smith MT, Guyton KZ, Gibbons CF, Fritz JM, Portier CJ, Rusyn I, DeMarini DM, Caldwell JC, Kavlock RJ, Lambert PF, Hecht SS, Bucher JR, Stewart BW, Baan RA, Cogliano VJ, Straif K, Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis, Environ Health Perspect, 124 (2016) 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chiu WA, Guyton KZ, Martin MT, Reif DM, Rusyn I, Use of high-throughput in vitro toxicity screening data in cancer hazard evaluations by IARC Monograph Working Groups, ALTEX, 35 (2018) 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Smith MT, Guyton KZ, Kleinstreuer N, Borrel A, Cardenas A, Chiu WA, Felsher DW, Gibbons CF, Goodson WH 3rd, Houck KA, Kane AB, La Merrill MA, Lebrec H, Lowe L, McHale CM, Minocherhomji S, Rieswijk L, Sandy MS, Sone H, Wang A, Zhang L, Zeise L, Fielden M, The Key Characteristics of Carcinogens: Relationship to the Hallmarks of Cancer, Relevant Biomarkers, and Assays to Measure Them, Cancer Epidemiol Biomarkers Prev, 29 (2020) 1887–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].La Merrill MA, Vandenberg LN, Smith MT, Goodson W, Browne P, Patisaul HB, Guyton KZ, Kortenkamp A, Cogliano VJ, Woodruff TJ, Rieswijk L, Sone H, Korach KS, Gore AC, Zeise L, Zoeller RT, Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification, Nat Rev Endocrinol, 16 (2020) 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Arzuaga X, Smith MT, Gibbons CF, Skakkebaek NE, Yost EE, Beverly BEJ, Hotchkiss AK, Hauser R, Pagani RL, Schrader SM, Zeise L, Prins GS, Proposed Key Characteristics of Male Reproductive Toxicants as an Approach for Organizing and Evaluating Mechanistic Evidence in Human Health Hazard Assessments, Environ Health Perspect, 127 (2019) 65001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Luderer U, Eskenazi B, Hauser R, Korach KS, McHale CM, Moran F, Rieswijk L, Solomon G, Udagawa O, Zhang L, Zlatnik M, Zeise L, Smith MT, Proposed Key Characteristics of Female Reproductive Toxicants as an Approach for Organizing and Evaluating Mechanistic Data in Hazard Assessment, Environ Health Perspect, 127 (2019) 75001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rusyn I, Arzuaga X, Cattley RC, Corton JC, Ferguson SS, Godoy P, Guyton KZ, Kaplowitz N, Khetani SR, Roberts RA, Roth RA, Smith MT, Key Characteristics of Human Hepatotoxicants as a Basis for Identification and Characterization of the Causes of Liver Toxicity, Hepatology, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lind L, Araujo JA, Barchowsky A, Belcher S, Berridge BR, Chiamvimonvat N, Chiu WA, Cogliano VJ, Elmore S, Farraj AK, Gomes AV, McHale CM, Meyer-Tamaki KB, Posnack NG, Vargas HM, Yang X, Zeise L, Zhou C, Smith MT, Key Characteristics of Cardiovascular Toxicants, Environ Health Perspect, 129 (2021) 95001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jones PA, Baylin SB, The epigenomics of cancer, Cell, 128 (2007) 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chappell G, Pogribny IP, Guyton KZ, Rusyn I, Epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens: A systematic literature review, Mutat Res Rev Mutat Res, 768 (2016) 27–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jones PA, Baylin SB, The fundamental role of epigenetic events in cancer, Nat.Rev.Genet, 3 (2002) 415–428. [DOI] [PubMed] [Google Scholar]
- [13].Feinberg AP, Ohlsson R, Henikoff S, The epigenetic progenitor origin of human cancer, Nature Reviews Genetics, 7 (2006) 21–33. [DOI] [PubMed] [Google Scholar]
- [14].Martin EM, Fry RC, Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations, Annu Rev Public Health, 39 (2018) 309–333. [DOI] [PubMed] [Google Scholar]
- [15].Inbar-Feigenberg M, Choufani S, Butcher DT, Roifman M, Weksberg R, Basic concepts of epigenetics, Fertil Steril, 99 (2013) 607–615. [DOI] [PubMed] [Google Scholar]
- [16].Pogribny IP, Rusyn I, Environmental toxicants, epigenetics, and cancer, Adv Exp Med Biol, 754 (2013) 215–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Herceg Z, Lambert MP, van Veldhoven K, Demetriou C, Vineis P, Smith MT, Straif K, Wild CP, Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation, Carcinogenesis, 34 (2013) 1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shapiro AJ, Antoni S, Guyton KZ, Lunn RM, Loomis D, Rusyn I, Jahnke GD, Schwingl PJ, Mehta SS, Addington J, Guha N, Software Tools to Facilitate Systematic Review Used for Cancer Hazard Identification, Environ Health Perspect, 126 (2018) 104501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen T, Li E, Establishment and maintenance of DNA methylation patterns in mammals, Curr Top Microbiol Immunol, 301 (2006) 179–201. [DOI] [PubMed] [Google Scholar]
- [20].Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond, Nature Reviews Genetics, 18 (2017) 517–534. [DOI] [PubMed] [Google Scholar]
- [21].Ito S, Kuraoka I, Epigenetic modifications in DNA could mimic oxidative DNA damage: A double-edged sword, DNA Repair (Amst), 32 (2015) 52–57. [DOI] [PubMed] [Google Scholar]
- [22].Bird AP, CpG-rich islands and the function of DNA methylation, Nature, 321 (1986) 209–213. [DOI] [PubMed] [Google Scholar]
- [23].Mohandas T, Sparkes RS, Shapiro LJ, Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation, Science, 211 (1981) 393–396. [DOI] [PubMed] [Google Scholar]
- [24].Gartler SM, Riggs AD, Mammalian X-chromosome inactivation, Annu Rev Genet, 17 (1983) 155–190. [DOI] [PubMed] [Google Scholar]
- [25].Reik W, Collick A, Norris ML, Barton SC, Surani MA, Genomic imprinting determines methylation of parental alleles in transgenic mice, Nature, 328 (1987) 248–251. [DOI] [PubMed] [Google Scholar]
- [26].Pan Y, Liu G, Zhou F, Su B, Li Y, DNA methylation profiles in cancer diagnosis and therapeutics, Clin Exp Med, 18 (2018) 1–14. [DOI] [PubMed] [Google Scholar]
- [27].Man CH, Fung TK, Wan H, Cher CY, Fan A, Ng N, Ho C, Wan TS, Tanaka T, So CW, Kwong YL, Leung AY, Suppression of SOX7 by DNA methylation and its tumor suppressor function in acute myeloid leukemia, Blood, 125 (2015) 3928–3936. [DOI] [PubMed] [Google Scholar]
- [28].Min HL, Kim J, Kim WH, Jang BG, Kim MA, Epigenetic Silencing of the Putative Tumor Suppressor Gene GLDC (Glycine Dehydrogenase) in Gastric Carcinoma, Anticancer Res, 36 (2016) 179–187. [PubMed] [Google Scholar]
- [29].Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R, Induction of tumors in mice by genomic hypomethylation, Science, 300 (2003) 489–492. [DOI] [PubMed] [Google Scholar]
- [30].Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP, Alterations in DNA methylation: a fundamental aspect of neoplasia, Adv Cancer Res, 72 (1998) 141–196. [PubMed] [Google Scholar]
- [31].Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L, Van Criekinge W, De Meyer T, van Engeland M, Analysis of DNA methylation in cancer: location revisited, Nat Rev Clin Oncol, 15 (2018) 459–466. [DOI] [PubMed] [Google Scholar]
- [32].Gonzalgo ML, Hayashida T, Bender CM, Pao MM, Tsai YC, Gonzales FA, Nguyen HD, Nguyen TT, Jones PA, The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines, Cancer Res, 58 (1998) 1245–1252. [PubMed] [Google Scholar]
- [33].Homma N, Tamura G, Honda T, Matsumoto Y, Nishizuka S, Kawata S, Motoyama T, Spreading of methylation within RUNX3 CpG island in gastric cancer, Cancer Sci, 97 (2006) 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deng G, Chen A, Hong J, Chae HS, Kim YS, Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression, Cancer Res, 59 (1999) 2029–2033. [PubMed] [Google Scholar]
- [35].Licchesi JD, Van Neste L, Tiwari VK, Cope L, Lin X, Baylin SB, Herman JG, Transcriptional regulation of Wnt inhibitory factor-1 by Miz-1/c-Myc, Oncogene, 29 (2010) 5923–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, Harris CC, Herman JG, SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity, Nat Genet, 28 (2001) 29–35. [DOI] [PubMed] [Google Scholar]
- [37].Michaud DS, Ruan M, Koestler DC, Alonso L, Molina-Montes E, Pei D, Marsit CJ, De Vivo I, Malats N, Kelsey KT, DNA Methylation-Derived Immune Cell Profiles, CpG Markers of Inflammation, and Pancreatic Cancer Risk, Cancer Epidemiol Biomarkers Prev, 29 (2020) 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gouil Q, Keniry A, Latest techniques to study DNA methylation, Essays Biochem, 63 (2019) 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kurdyukov S, Bullock M, DNA Methylation Analysis: Choosing the Right Method, Biology (Basel), 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Olkhov-Mitsel E, Bapat B, Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers, Cancer Med, 1 (2012) 237–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yong WS, Hsu FM, Chen PY, Profiling genome-wide DNA methylation, Epigenetics Chromatin, 9 (2016) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Audia JE, Campbell RM, Histone Modifications and Cancer, Cold Spring Harb Perspect Biol, 8 (2016) a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Saha S, Histone Modifications and Other Facets of Epigenetic Regulation in Trypanosomatids: Leaving Their Mark, MBio, 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fujisawa T, Filippakopoulos P, Functions of bromodomain-containing proteins and their roles in homeostasis and cancer, Nat Rev Mol Cell Biol, 18 (2017) 246–262. [DOI] [PubMed] [Google Scholar]
- [45].Josling GA, Selvarajah SA, Petter M, Duffy MF, The role of bromodomain proteins in regulating gene expression, Genes (Basel), 3 (2012) 320–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Eissenberg JC, Shilatifard A, Histone H3 lysine 4 (H3K4) methylation in development and differentiation, Dev Biol, 339 (2010) 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kouzarides T, Chromatin modifications and their function, Cell, 128 (2007) 693–705. [DOI] [PubMed] [Google Scholar]
- [48].Herz HM, Garruss A, Shilatifard A, SET for life: biochemical activities and biological functions of SET domain-containing proteins, Trends Biochem Sci, 38 (2013) 621–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK, Global histone modification patterns predict risk of prostate cancer recurrence, Nature, 435 (2005) 1262–1266. [DOI] [PubMed] [Google Scholar]
- [50].Bannister AJ, Kouzarides T, Regulation of chromatin by histone modifications, Cell Res, 21 (2011) 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Venkatesh S, Workman JL, Histone exchange, chromatin structure and the regulation of transcription, Nat Rev Mol Cell Biol, 16 (2015) 178–189. [DOI] [PubMed] [Google Scholar]
- [52].Waters R, van Eijk P, Reed S, Histone modification and chromatin remodeling during NER, DNA Repair (Amst), 36 (2015) 105–113. [DOI] [PubMed] [Google Scholar]
- [53].O’Hagan HM, Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations, Environ Mol Mutagen, 55 (2014) 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pierozan P, Cattani D, Karlsson O, Perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) induce epigenetic alterations and promote human breast cell carcinogenesis in vitro, Archives of Toxicology, 94 (2020) 3893–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Costa FF, Non-coding RNAs, epigenetics and complexity, Gene, 410 (2008) 9–17. [DOI] [PubMed] [Google Scholar]
- [56].Amaral PP, Dinger ME, Mercer TR, Mattick JS, The eukaryotic genome as an RNA machine, Science, 319 (2008) 1787–1789. [DOI] [PubMed] [Google Scholar]
- [57].Ghildiyal M, Zamore PD, Small silencing RNAs: an expanding universe, Nature Reviews Genetics, 10 (2009) 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ruvkun G, Molecular biology. Glimpses of a tiny RNA world, Science, 294 (2001) 797–799. [DOI] [PubMed] [Google Scholar]
- [59].Mishra S, Yadav T, Rani V, Exploring miRNA based approaches in cancer diagnostics and therapeutics, Crit Rev Oncol Hematol, 98 (2016) 12–23. [DOI] [PubMed] [Google Scholar]
- [60].Lee YS, Dutta A, MicroRNAs in cancer, Annu Rev Pathol, 4 (2009) 199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM, MicroRNA expression and function in cancer, Trends Mol Med, 12 (2006) 580–587. [DOI] [PubMed] [Google Scholar]
- [62].Jonas S, Izaurralde E, Towards a molecular understanding of microRNA-mediated gene silencing, Nature Reviews Genetics, 16 (2015) 421–433. [DOI] [PubMed] [Google Scholar]
- [63].Paraskevopoulou MD, Hatzigeorgiou AG, Analyzing MiRNA-LncRNA Interactions, Methods Mol Biol, 1402 (2016) 271–286. [DOI] [PubMed] [Google Scholar]
- [64].Huarte M, The emerging role of lncRNAs in cancer, Nat Med, 21 (2015) 1253–1261. [DOI] [PubMed] [Google Scholar]
- [65].Wang KC, Chang HY, Molecular mechanisms of long noncoding RNAs, Mol Cell, 43 (2011) 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang Y, Tang L, The Application of lncRNAs in Cancer Treatment and Diagnosis, Recent Pat Anticancer Drug Discov, 13 (2018) 292–301. [DOI] [PubMed] [Google Scholar]
- [67].Rathinasamy B, Velmurugan BK, Role of lncRNAs in the cancer development and progression and their regulation by various phytochemicals, Biomed Pharmacother, 102 (2018) 242–248. [DOI] [PubMed] [Google Scholar]
- [68].Zhao Y, Sun H, Wang H, Long noncoding RNAs in DNA methylation: new players stepping into the old game, Cell Biosci, 6 (2016) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jin Y, Xu P, Liu X, Zhang C, Tan C, Chen C, Sun X, Xu Y, Cigarette Smoking, BPDE-DNA Adducts, and Aberrant Promoter Methylations of Tumor Suppressor Genes (TSGs) in NSCLC from Chinese Population, Cancer Invest, 34 (2016) 173–180. [DOI] [PubMed] [Google Scholar]
- [70].Jin F, Thaiparambil J, Donepudi SR, Vantaku V, Piyarathna DWB, Maity S, Krishnapuram R, Putluri V, Gu F, Purwaha P, Bhowmik SK, Ambati CR, von Rundstedt FC, Roghmann F, Berg S, Noldus J, Rajapakshe K, Godde D, Roth S, Storkel S, Degener S, Michailidis G, Kaipparettu BA, Karanam B, Terris MK, Kavuri SM, Lerner SP, Kheradmand F, Coarfa C, Sreekumar A, Lotan Y, El-Zein R, Putluri N, Tobacco-Specific Carcinogens Induce Hypermethylation, DNA Adducts, and DNA Damage in Bladder Cancer, Cancer Prev Res (Phila), 10 (2017) 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jiang CL, He SW, Zhang YD, Duan HX, Huang T, Huang YC, Li GF, Wang P, Ma LJ, Zhou GB, Cao Y, Air pollution and DNA methylation alterations in lung cancer: A systematic and comparative study, Oncotarget, 8 (2017) 1369–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].He Z, Li D, Ma J, Chen L, Duan H, Zhang B, Gao C, Li J, Xing X, Zhao J, Wang S, Wang F, Zhang H, Li H, Chen S, Zeng X, Wang Q, Xiao Y, Zheng Y, Chen W, (TRIM36 hypermethylation is involved in polycyclic aromatic hydrocarbons-induced cell transformation), Environ Pollut, 225 (2017) 93–103. [DOI] [PubMed] [Google Scholar]
- [73].Xia B, Yang LQ, Huang HY, Pang L, Yang XF, Yi YJ, Ren XH, Li J, Zhuang ZX, Liu JJ, Repression of Biotin-Related Proteins by Benzo[a]Pyrene-Induced Epigenetic Modifications in Human Bronchial Epithelial Cells, Int J Toxicol, 35 (2016) 336–343. [DOI] [PubMed] [Google Scholar]
- [74].Bhargava A, Kumari R, Khare S, Shandilya R, Gupta PK, Tiwari R, Rahman A, Chaudhury K, Goryacheva IY, Mishra PK, Mapping the Mitochondrial Regulation of Epigenetic Modifications in Association With Carcinogenic and Noncarcinogenic Polycyclic Aromatic Hydrocarbon Exposure, Int J Toxicol, 39 (2020) 465–476. [DOI] [PubMed] [Google Scholar]
- [75].Bojang P Jr., Ramos KS, Epigenetic reactivation of LINE-1 retrotransposon disrupts NuRD corepressor functions and induces oncogenic transformation in human bronchial epithelial cells, Mol Oncol, 12 (2018) 1342–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zeng Z, Lu J, Wu D, Zuo R, Li Y, Huang H, Yuan J, Hu Z, Poly(ADP-ribose) glycohydrolase silencing-mediated H2B expression inhibits benzo(a)pyrene-induced carcinogenesis, Environ Toxicol, 36 (2021) 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zeng Z, Liu H, Yuan J, Ren X, Deng Y, Dai W, Wu Y, Huang Y, Huang R, Liu J, Huang H, Hu J, Poly (ADP-ribose) glycohydrolase silencing-mediated maintenance of H2A and downregulation of H2AK9me protect human bronchial epithelial cells from benzo(a)pyrene-induced carcinogenesis, Toxicol Lett, 295 (2018) 270–276. [DOI] [PubMed] [Google Scholar]
- [78].Li L, Chen J, Wang Y, Zhou C, Ma X, Fu J, Yao B, Zhao P, MicroRNA expression profiling and the role of ALCAM modulating tumor growth and metastasis in benzo[a]pyrene-transformed 16HBE cells, Toxicology, 442 (2020) 152539. [DOI] [PubMed] [Google Scholar]
- [79].Malik DE, David RM, Gooderham NJ, Mechanistic evidence that benzo[a]pyrene promotes an inflammatory microenvironment that drives the metastatic potential of human mammary cells, Archives of Toxicology, 92 (2018) 3223–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Malik DE, David RM, Gooderham NJ, Interleukin-6 selectively induces drug metabolism to potentiate the genotoxicity of dietary carcinogens in mammary cells, Archives of Toxicology, 93 (2019) 3005–3020. [DOI] [PubMed] [Google Scholar]
- [81].Chanyshev MD, Koval OA, Nushtaeva AA, Gulyaeva LF, Effect of benzo[a]pyrene on the expression of miR-126, miR-190a and their target genes EGFL7, TP53INP1 and PHLPP1 in primary endometrial cells, J Biochem Mol Toxicol, 33 (2019) e22314. [DOI] [PubMed] [Google Scholar]
- [82].Wu Y, Niu Y, Leng J, Xu J, Chen H, Li H, Wang L, Hu J, Xia D, Wu Y, Benzo(a)pyrene regulated A549 cell migration, invasion and epithelial-mesenchymal transition by up-regulating long non-coding RNA linc00673, Toxicol Lett, 320 (2020) 37–45. [DOI] [PubMed] [Google Scholar]
- [83].Fu Y, Wang W, Li X, Liu Y, Niu Y, Zhang B, Nie J, Pan B, Wang R, Yang J, LncRNA H19 interacts with S-adenosylhomocysteine hydrolase to regulate LINE-1 Methylation in human lung-derived cells exposed to Benzo[a]pyrene, Chemosphere, 207 (2018) 84–90. [DOI] [PubMed] [Google Scholar]
- [84].Liu D, Chen Y, Sun P, Bai W, Gao A, STAT3 methylation in white blood cells as a novel sensitive biomarker for the toxic effect of low-dose benzene exposure, Toxicol Res (Camb), 5 (2016) 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang DP, Cai DY, Yang XL, Lu X, Lin DF, Li PM, Zhang ZM, Zhang YF, Zhang W, [Study of methylation of mitochondrial MT-COI of benzene poisoning], Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi, 38 (2020) 664–668. [DOI] [PubMed] [Google Scholar]
- [86].Jamebozorgi I, Majidizadeh T, Pouryagoub G, Mahjoubi F, Aberrant DNA Methylation of Two Tumor Suppressor Genes, p14(ARF) and p15(INK4b), after Chronic Occupational Exposure to Low Level of Benzene, Int J Occup Environ Med, 9 (2018) 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Silvestre RT, Bravo M, Santiago F, Delmonico L, Scherrer L, Otero UB, Liehr T, Alves G, Chantre-Justino M, Ornellas MH, Hypermethylation in Gene Promoters Are Induced by Chronic Exposure to Benzene, Toluene, Ethylbenzene and Xylenes, Pak J Biol Sci, 23 (2020) 518–525. [DOI] [PubMed] [Google Scholar]
- [88].Ren JC, Wang T, Wu H, Zhang GH, Sun D, Guo K, Li H, Zhang F, Wu W, Xia ZL, Promoter hypermethylation in CSF3R induces peripheral neutrophil reduction in benzene-exposure poisoning, Environ Mol Mutagen, 61 (2020) 786–796. [DOI] [PubMed] [Google Scholar]
- [89].Zheng M, Lin F, Hou F, Li G, Zhu C, Xu P, Xing C, Wang Q, Association between Promoter Methylation of Gene ERCC3 and Benzene Hematotoxicity, Int J Environ Res Public Health, 14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ren J, Cui JP, Luo M, Liu H, Hao P, Wang X, Zhang GH, The prevalence and persistence of aberrant promoter DNA methylation in benzene-exposed Chinese workers, PLoS One, 14 (2019) e0220500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhang GH, Lu Y, Ji BQ, Ren JC, Sun P, Ding S, Liao X, Liao K, Liu J, Cao J, Lan Q, Rothman N, Xia ZL, Do mutations in DNMT3A/3B affect global DNA hypomethylation among benzene-exposed workers in Southeast China?: Effects of mutations in DNMT3A/3B on global DNA hypomethylation, Environ Mol Mutagen, 58 (2017) 678–687. [DOI] [PubMed] [Google Scholar]
- [92].Zhang H, Yuan Q, Pan Z, Ling X, Tan Q, Wu M, Zheng D, Xie P, Xie D, Liu L, Up-regulation of DNMT3b contributes to HOTAIRM1 silencing via DNA hypermethylation in cells transformed by long-term exposure to hydroquinone and workers exposed to benzene, Toxicol Lett, 322 (2020) 12–19. [DOI] [PubMed] [Google Scholar]
- [93].Yu CH, Li Y, Zhao X, Yang SQ, Li L, Cui NX, Rong L, Yi ZC, Benzene metabolite 1,2,4-benzenetriol changes DNA methylation and histone acetylation of erythroid-specific genes in K562 cells, Archives of Toxicology, 93 (2019) 137–147. [DOI] [PubMed] [Google Scholar]
- [94].Yu CH, Cui NX, Wang Y, Wang Y, Liu WJ, Gong M, Zhao X, Rong L, Yi ZC, Changes in DNA methylation of erythroid-specific genes in K562 cells exposed to catechol in long term, Toxicol In Vitro, 43 (2017) 21–28. [DOI] [PubMed] [Google Scholar]
- [95].Tang KY, Yu CH, Jiang L, Gong M, Liu WJ, Wang Y, Cui NX, Song W, Sun Y, Yi ZC, Long-term exposure of K562 cells to benzene metabolites inhibited erythroid differentiation and elevated methylation in erythroid specific genes, Toxicol Res (Camb), 5 (2016) 1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Chen J, Zheng Z, Chen Y, Li J, Qian S, Shi Y, Sun L, Han Y, Zhang S, Yu K, Histone Deacetylase Inhibitors Trichostatin A and MCP30 Relieve Benzene-Induced Hematotoxicity via Restoring Topoisomerase IIalpha, PLoS One, 11 (2016) e0153330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Li J, Xing X, Zhang X, Liang B, He Z, Gao C, Wang S, Wang F, Zhang H, Zeng S, Fan J, Chen L, Zhang Z, Zhang B, Liu C, Wang Q, Lin W, Dong G, Tang H, Chen W, Xiao Y, Li D, Enhanced H3K4me3 modifications are involved in the transactivation of DNA damage responsive genes in workers exposed to low-level benzene, Environ Pollut, 234 (2018) 127–135. [DOI] [PubMed] [Google Scholar]
- [98].Song MK, Ryu JC, Blood miRNAs as sensitive and specific biological indicators of environmental and occupational exposure to volatile organic compound (VOC), Int J Hyg Environ Health, 218 (2015) 590–602. [DOI] [PubMed] [Google Scholar]
- [99].Hu D, Peng X, Liu Y, Zhang W, Peng X, Tang H, Yuan J, Zhu Z, Yang J, Overexpression of miR-221 in peripheral blood lymphocytes in petrol station attendants: A population based cross-sectional study in southern China, Chemosphere, 149 (2016) 8–13. [DOI] [PubMed] [Google Scholar]
- [100].Liu Y, Chen X, Bian Q, Shi Y, Liu Q, Ding L, Zhang H, Zhu B, Analysis of plasma microRNA expression profiles in a Chinese population occupationally exposed to benzene and in a population with chronic benzene poisoning, J Thorac Dis, 8 (2016) 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Jiang R, Huang H, Lian Z, Hu Z, Lloyd RS, Fang D, Li Y, Xian H, Yuan J, Sha Y, Wang S, Hu D, Exosomal miR-221 derived from hydroquinone-transformed malignant human bronchial epithelial cells is involved in cell viability of recipient cells, J Appl Toxicol, 40 (2020) 224–233. [DOI] [PubMed] [Google Scholar]
- [102].Liang B, Chen Y, Yuan W, Qin F, Zhang Q, Deng N, Liu X, Ma X, Zhang X, Zhang B, Deng Q, Huang M, Tang H, Liu L, Chen W, Xiao Y, Down-regulation of miRNA-451a and miRNA-486-5p involved in benzene-induced inhibition on erythroid cell differentiation in vitro and in vivo, Archives of Toxicology, 92 (2018) 259–272. [DOI] [PubMed] [Google Scholar]
- [103].Ma X, Zhang X, Luo J, Liang B, Peng J, Chen C, Guo H, Wang Q, Xing X, Deng Q, Huang H, Liao Q, Chen W, Hu Q, Yu D, Xiao Y, MiR-486-5p-directed MAGI1/Rap1/RASSF5 signaling pathway contributes to hydroquinone-induced inhibition of erythroid differentiation in K562 cells, Toxicol In Vitro, 66 (2020) 104830. [DOI] [PubMed] [Google Scholar]
- [104].Gui Z, Zhang H, Tan Q, Ling X, Liu Z, Peng J, Shao J, Wu M, Yuan Q, Li J, Pan Z, Zhong B, Liu L, Poly(ADP-ribose) polymerase-1 promotes expression of miR-155 by the up-regulation of methyl-CpG binding domain protein 2 in TK6 cells exposed to hydroquinone, Toxicol In Vitro, 55 (2019) 51–57. [DOI] [PubMed] [Google Scholar]
- [105].Luo H, Liang H, Chen Y, Chen S, Xu Y, Xu L, Liu J, Zhou K, Peng J, Guo G, Lai B, Song L, Yang H, Liu L, Peng J, Liu Z, Tang L, Chen W, Tang H, miR-7-5p overexpression suppresses cell proliferation and promotes apoptosis through inhibiting the ability of DNA damage repair of PARP-1 and BRCA1 in TK6 cells exposed to hydroquinone, Chem Biol Interact, 283 (2018) 84–90. [DOI] [PubMed] [Google Scholar]
- [106].Wang TS, Tian W, Fang Y, Guo KR, Li AQ, Sun Y, Wu HT, Zheng GQ, Feng NN, Xing CH, Au WW, Sun DY, Xia ZL, Changes in miR-222 expression, DNA repair capacity, and MDM2-p53 axis in association with low-dose benzene genotoxicity and hematotoxicity, Sci Total Environ, (2020) 142740. [DOI] [PubMed] [Google Scholar]
- [107].Chen Y, Zhang W, Guo X, Ren J, Gao A, lncRNAVNN3 mediated benzene-induced hematotoxicity through promoting autophagy and apoptosis, Ecotoxicol Environ Saf, 185 (2019) 109672. [DOI] [PubMed] [Google Scholar]
- [108].Sun P, Wang J, Guo X, Chen Y, Xing C, Gao A, Benzene and its metabolite decreases cell proliferation via LncRNA-OBFC2A-mediated anti-proliferation effect involving NOTCH1 and KLF15, Oncotarget, 8 (2017) 40857–40871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Guo X, Zhang W, Ren J, Chen Y, Wang J, Zhu C, Zhang Y, Gao A, LncRNA-OBFC2A targeted to Smad3 regulated Cyclin D1 influences cell cycle arrest induced by 1,4-benzoquinone, Toxicol Lett, 332 (2020) 74–81. [DOI] [PubMed] [Google Scholar]
- [110].Yuan Q, Zhang H, Pan Z, Ling X, Wu M, Gui Z, Chen J, Peng J, Liu Z, Tan Q, Huang D, Xiu L, Chen W, Shi Z, Liu L, Regulatory loop between lncRNA FAS-AS1 and DNMT3b controls FAS expression in hydroquinone-treated TK6 cells and benzene-exposed workers, Environ Pollut, 261 (2020) 114147. [DOI] [PubMed] [Google Scholar]
- [111].Rieswijk L, Claessen SM, Bekers O, van Herwijnen M, Theunissen DH, Jennen DG, de Kok TM, Kleinjans JC, van Breda SG, Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma, Toxicology, 350-352 (2016) 31–39. [DOI] [PubMed] [Google Scholar]
- [112].Tryndyak V, Kindrat I, Dreval K, Churchwell MI, Beland FA, Pogribny IP, Effect of aflatoxin B1, benzo[a]pyrene, and methapyrilene on transcriptomic and epigenetic alterations in human liver HepaRG cells, Food and Chemical Toxicology, 121 (2018) 214–223. [DOI] [PubMed] [Google Scholar]
- [113].Tryndyak V, Borowa-Mazgaj B, Beland FA, Pogribny IP, Gene expression and cytosine DNA methylation alterations in induced pluripotent stem-cell-derived human hepatocytes treated with low doses of chemical carcinogens, Archives of Toxicology, 93 (2019) 3335–3344. [DOI] [PubMed] [Google Scholar]
- [114].Zhang B, Dai Y, Zhu L, He X, Huang K, Xu W, Single-cell sequencing reveals novel mechanisms of Aflatoxin B1-induced hepatotoxicity in S phase-arrested L02 cells, Cell Biol Toxicol, 36 (2020) 603–608. [DOI] [PubMed] [Google Scholar]
- [115].Soni P, Ghufran MS, Olakkaran S, Puttaswamygowda GH, Duddukuri GR, Kanade SR, Epigenetic alterations induced by aflatoxin B1: An in vitro and in vivo approach with emphasis on enhancer of zeste homologue-2/p21 axis, Sci Total Environ, 762 (2021) 143175. [DOI] [PubMed] [Google Scholar]
- [116].Soni P, Ghufran MS, Kanade SR, Aflatoxin B1 induced multiple epigenetic modulators in human epithelial cell lines, Toxicon, 151 (2018) 119–128. [DOI] [PubMed] [Google Scholar]
- [117].Wu XM, Xi ZF, Liao P, Huang HD, Huang XY, Wang C, Ma Y, Xia Q, Yao JG, Long XD, Diagnostic and prognostic potential of serum microRNA-4651 for patients with hepatocellular carcinoma related to aflatoxin B1, Oncotarget, 8 (2017) 81235–81249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Livingstone MC, Johnson NM, Roebuck BD, Kensler TW, Groopman JD, Serum miR-182 is a predictive biomarker for dichotomization of risk of hepatocellular carcinoma in rats, Mol Carcinog, 58 (2019) 2017–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Shirani K, Riahi Zanjani B, Mehri S, Razavi-Azarkhiavi K, Badiee A, Hayes AW, Giesy JP, Karimi G, miR-155 influences cell-mediated immunity in Balb/c mice treated with aflatoxin M1, Drug Chem Toxicol, 44 (2021) 39–46. [DOI] [PubMed] [Google Scholar]
- [120].Zhang Z, Tang D, Wang B, Wang Z, Liu M, Analysis of miRNA-mRNA regulatory network revealed key genes induced by aflatoxin B1 exposure in primary human hepatocytes, Mol Genet Genomic Med, 7 (2019) e971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Liu X, Kumar Mishra S, Wang T, Xu Z, Zhao X, Wang Y, Yin H, Fan X, Zeng B, Yang M, Yang D, Ni Q, Li Y, Zhang M, Zhu Q, Chen F, Li D, AFB1 Induced Transcriptional Regulation Related to Apoptosis and Lipid Metabolism in Liver of Chicken, Toxins (Basel), 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Liu Y, Li X, Zhang B, Fu Y, Yang A, Zhang H, Zhang H, Niu Y, Nie J, Yang J, CYP1A1 methylation mediates the effect of smoking and occupational polycyclic aromatic hydrocarbons co-exposure on oxidative DNA damage among Chinese coke-oven workers, Environ Health, 18 (2019) 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Yang J, Liu Y, Zhang H, Zhang H, Wang W, Fan Y, Urinary 1-hydroxypyrene and smoking are determinants of LINE-1 and AhRR promoter methylation in coke oven workers, Mutat Res Genet Toxicol Environ Mutagen, 826 (2018) 33–40. [DOI] [PubMed] [Google Scholar]
- [124].He Z, Zhang R, Chen S, Chen L, Li H, Ye L, Li Q, Wang Z, Wang Q, Duan H, Niu Y, Xiao Y, Dong G, Li D, Yu D, Zheng Y, Xing X, Chen W, FLT1 hypermethylation is involved in polycyclic aromatic hydrocarbons-induced cell transformation, Environ Pollut, 252 (2019) 607–615. [DOI] [PubMed] [Google Scholar]
- [125].Xing X, He Z, Wang Z, Mo Z, Chen L, Yang B, Zhang Z, Chen S, Ye L, Zhang R, Zheng Y, Chen W, Li D, Association between H3K36me3 modification and methylation of LINE-1 and MGMT in peripheral blood lymphocytes of PAH-exposed workers, Toxicol Res (Camb), 9 (2020) 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhang Z, Chen L, Xing X, Li D, Gao C, He Z, Li J, Zhu X, Xiao X, Wang S, Wang F, Ren Z, Xiao Y, Dharmage SC, Dong G, Zheng Y, Chen W, Specific histone modifications were associated with the PAH-induced DNA damage response in coke oven workers, Toxicol Res (Camb), 5 (2016) 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Zhang Z, Xing X, Jiang S, Qiu C, Mo Z, Chen S, Chen L, Wang Q, Xiao Y, Dong G, Zheng Y, Chen W, Li D, Global H3K79 di-methylation mediates DNA damage response to PAH exposure in Chinese coke oven workers, Environ Pollut, 268 (2021) 115956. [DOI] [PubMed] [Google Scholar]
- [128].Huang S, Deng Q, Feng J, Zhang X, Dai X, Li L, Yang B, Wu T, Cheng J, Polycyclic Aromatic Hydrocarbons-Associated MicroRNAs and Heart Rate Variability in Coke Oven Workers, J Occup Environ Med, 58 (2016) e24–31. [DOI] [PubMed] [Google Scholar]
- [129].Gao C, He Z, Li J, Li X, Bai Q, Zhang Z, Zhang X, Wang S, Xiao X, Wang F, Yan Y, Li D, Chen L, Zeng X, Xiao Y, Dong G, Zheng Y, Wang Q, Chen W, Specific long non-coding RNAs response to occupational PAHs exposure in coke oven workers, Toxicol Rep, 3 (2016) 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Simons T, Steinritz D, Bolck B, Schmidt A, Popp T, Thiermann H, Gudermann T, Bloch W, Kehe K, Sulfur mustard-induced epigenetic modifications over time - a pilot study, Toxicol Lett, 293 (2018) 45–50. [DOI] [PubMed] [Google Scholar]
- [131].Khafaei M, Samie S, Mowla SJ, Alvanegh AG, Mirzaei B, Chavoshei S, Dorraj GS, Esmailnejad M, Tavallaie M, Nourani M, Evaluation of miR-9 and miR-143 expression in urine specimens of sulfur mustard exposed patients, Interdiscip Toxicol, 8 (2015) 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Gharbi S, Khateri S, Soroush MR, Shamsara M, Naeli P, Najafi A, Korsching E, Mowla SJ, MicroRNA expression in serum samples of sulfur mustard veterans as a diagnostic gateway to improve care, PLoS One, 13 (2018) e0194530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Salimi S, Noorbakhsh F, Faghihzadeh S, Ghaffarpour S, Ghazanfari T, Expression of miR-15b-5p, miR-21-5p, and SMAD7 in Lung Tissue of Sulfur Mustard-exposed Individuals with Long-term Pulmonary Complications, Iran J Allergy Asthma Immunol, 18 (2019) 332–339. [DOI] [PubMed] [Google Scholar]
- [134].Rothmiller S, Wolf M, Worek F, Steinritz D, Thiermann H, Schmidt A, Alteration of miRNA expression in a sulfur mustard resistant cell line, Toxicol Lett, 293 (2018) 38–44. [DOI] [PubMed] [Google Scholar]
- [135].Rana T, Ahmad A, Zafar I, Mariappan N, Chandrashekar DS, Hamid T, Husain M, Varambally S, Ahmad S, Ahmad A, MicroRNA-mediated inflammation and coagulation effects in rats exposed to an inhaled analog of sulfur mustard, Ann N Y Acad Sci, 1479 (2020) 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Barbosa E, Dos Santos ALA, Peteffi GP, Schneider A, Muller D, Rovaris D, Bau CHD, Linden R, Antunes MV, Charao MF, Increase of global DNA methylation patterns in beauty salon workers exposed to low levels of formaldehyde, Environ Sci Pollut Res Int, 26 (2019) 1304–1314. [DOI] [PubMed] [Google Scholar]
- [137].Chen D, Fang L, Mei S, Li H, Xu X, Des Marais TL, Lu K, Liu XS, Jin C, Regulation of Chromatin Assembly and Cell Transformation by Formaldehyde Exposure in Human Cells, Environ Health Perspect, 125 (2017) 097019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lewis L, Chappell GA, Kobets T, O’Brian BE, Sangaraju D, Kosyk O, Bodnar W, Tretyakova NY, Pogribny IP, Rusyn I, Sex-specific differences in genotoxic and epigenetic effects of 1,3-butadiene among mouse tissues, Archives of Toxicology, 93 (2019) 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Lewis L, Borowa-Mazgaj B, de Conti A, Chappell GA, Luo YS, Bodnar W, Konganti K, Wright FA, Threadgill DW, Chiu WA, Pogribny IP, Rusyn I, Population-Based Analysis of DNA Damage and Epigenetic Effects of 1,3-Butadiene in the Mouse, Chem Res Toxicol, 32 (2019) 887–898. [DOI] [PubMed] [Google Scholar]
- [140].Israel JW, Chappell GA, Simon JM, Pott S, Safi A, Lewis L, Cotney P, Boulos HS, Bodnar W, Lieb JD, Crawford GE, Furey TS, Rusyn I, Tissue- and strain-specific effects of a genotoxic carcinogen 1,3-butadiene on chromatin and transcription, Mamm Genome, 29 (2018) 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Chappell GA, Israel JW, Simon JM, Pott S, Safi A, Eklund K, Sexton KG, Bodnar W, Lieb JD, Crawford GE, Rusyn I, Furey TS, Variation in DNA-Damage Responses to an Inhalational Carcinogen (1,3-Butadiene) in Relation to Strain-Specific Differences in Chromatin Accessibility and Gene Transcription Profiles in C57BL/6J and CAST/EiJ Mice, Environ Health Perspect, 125 (2017) 107006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Qiu YL, Xu ZB, Wang Q, Hu JY, Zhang L, Chen SQ, Lyu Y, Wei CL, Yan XY, Wang T, Association between methylation of DNA damage response-related genes and DNA damage in hepatocytes of rats following subchronic exposure to vinyl chloride, Chemosphere, 227 (2019) 323–328. [DOI] [PubMed] [Google Scholar]
- [143].Feng NN, Fang Y, Zhang YN, Xu XW, Li Y, Wang JW, Li YL, Brandt-Rauf P, Xia ZL, Analysis of microRNA expression and micronuclei frequency in workers exposed to vinyl chloride monomer in China, Epigenomics, 9 (2017) 1093–1104. [DOI] [PubMed] [Google Scholar]
- [144].Sisto R, Capone P, Cerini L, Sanjust F, Paci E, Pigini D, Gatto MP, Gherardi M, Gordiani A, L’Episcopo N, Tranfo G, Chiarella P, Circulating microRNAs as potential biomarkers of occupational exposure to low dose organic solvents, Toxicol Rep, 6 (2019) 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Duan S, Yuan H, Yu S, Wei X, Zhou X, Wang W, Feng F, Qu L, Wu Y, Epigenetic-Based Biomarkers in the Malignant Transformation of BEAS-2B Cells Induced by Coal Tar Pitch Extract, Medicina (Kaunas), 57 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Li Z, Wang W, Meng L, Zhang Y, Zhang J, Li C, Wu Y, Feng F, Zhang Q, Identification and analysis of key lncRNAs in malignant-transformed BEAS-2B cells induced with coal tar pitch by microarray analysis, Environ Toxicol Pharmacol, 79 (2020) 103376. [DOI] [PubMed] [Google Scholar]
- [147].Li Z, Zhang Y, Meng L, Yang S, Zhang P, Zhang J, Li C, Feng F, Zhang Q, LncRNA-ENST00000501520 promotes the proliferation of malignant-transformed BEAS-2B cells induced with coal tar pitch mediated by target genes, Environ Toxicol, 34 (2019) 869–877. [DOI] [PubMed] [Google Scholar]
- [148].Lin HD, Wang FZ, Lee CY, Nien CY, Tseng YK, Yao CL, Chen SC, 4-Aminobiphenyl inhibits the DNA homologous recombination repair in human liver cells: The role of miR-630 in downregulating RAD18 and MCM8, Toxicology, 440 (2020) 152441. [DOI] [PubMed] [Google Scholar]
- [149].IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Outdoor Air Pollution, IARC Monogr Eval Carcinog Risks Hum, 109 (2016) 9–444. [PMC free article] [PubMed] [Google Scholar]
- [150].Vos S, Nawrot TS, Martens DS, Byun HM, Janssen BG, Mitochondrial DNA methylation in placental tissue: a proof of concept study by means of prenatal environmental stressors, Epigenetics, 16 (2021) 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Wang M, Zhao J, Wang Y, Mao Y, Zhao X, Huang P, Liu Q, Ma Y, Yao Y, Yang Z, Yuan W, Cui W, Payne TJ, Li MD, Genome-wide DNA methylation analysis reveals significant impact of long-term ambient air pollution exposure on biological functions related to mitochondria and immune response, Environ Pollut, 264 (2020) 114707. [DOI] [PubMed] [Google Scholar]
- [152].Eze IC, Jeong A, Schaffner E, Rezwan FI, Ghantous A, Foraster M, Vienneau D, Kronenberg F, Herceg Z, Vineis P, Brink M, Wunderli JM, Schindler C, Cajochen C, Roosli M, Holloway JW, Imboden M, Probst-Hensch N, Genome-Wide DNA Methylation in Peripheral Blood and Long-Term Exposure to Source-Specific Transportation Noise and Air Pollution: The SAPALDIA Study, Environ Health Perspect, 128 (2020) 67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Wang C, O’Brien KM, Xu Z, Sandler DP, Taylor JA, Weinberg CR, Long-term ambient fine particulate matter and DNA methylation in inflammation pathways: results from the Sister Study, Epigenetics, 15 (2020) 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Wang C, Plusquin M, Ghantous A, Herceg Z, Alfano R, Cox B, Nawrot TS, DNA methylation of insulin-like growth factor 2 and H19 cluster in cord blood and prenatal air pollution exposure to fine particulate matter, Environ Health, 19 (2020) 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Merid SK, Bustamante M, Standl M, Sunyer J, Heinrich J, Lemonnier N, Aguilar D, Anto JM, Bousquet J, Santa-Marina L, Lertxundi A, Bergstrom A, Kull I, Wheelock AM, Koppelman GH, Melen E, Gruzieva O, Integration of gene expression and DNA methylation identifies epigenetically controlled modules related to PM2.5 exposure, Environ Int, 146 (2021) 106248. [DOI] [PubMed] [Google Scholar]
- [156].Feng F, Huang L, Zhou G, Wang J, Zhang R, Li Z, Zhang Y, Ba Y, GPR61 methylation in cord blood: a potential target of prenatal exposure to air pollutants, Int J Environ Health Res, (2020) 1–10. [DOI] [PubMed] [Google Scholar]
- [157].Gruzieva O, Xu CJ, Yousefi P, Relton C, Merid SK, Breton CV, Gao L, Volk HE, Feinberg JI, Ladd-Acosta C, Bakulski K, Auffray C, Lemonnier N, Plusquin M, Ghantous A, Herceg Z, Nawrot TS, Pizzi C, Richiardi L, Rusconi F, Vineis P, Kogevinas M, Felix JF, Duijts L, den Dekker HT, Jaddoe VWV, Ruiz JL, Bustamante M, Anto JM, Sunyer J, Vrijheid M, Gutzkow KB, Grazuleviciene R, Hernandez-Ferrer C, Annesi-Maesano I, Lepeule J, Bousquet J, Bergstrom A, Kull I, Soderhall C, Kere J, Gehring U, Brunekreef B, Just AC, Wright RJ, Peng C, Gold DR, Kloog I, DeMeo DL, Pershagen G, Koppelman GH, London SJ, Baccarelli AA, Melen E, Prenatal Particulate Air Pollution and DNA Methylation in Newborns: An Epigenome-Wide Meta-Analysis, Environ Health Perspect, 127 (2019) 57012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Callahan CL, Bonner MR, Nie J, Han D, Wang Y, Tao MH, Shields PG, Marian C, Eng KH, Trevisan M, Beyea J, Freudenheim JL, Lifetime exposure to ambient air pollution and methylation of tumor suppressor genes in breast tumors, Environ Res, 161 (2018) 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Cantone L, Tobaldini E, Favero C, Albetti B, Sacco RM, Torgano G, Ferrari L, Montano N, Bollati V, Particulate Air Pollution, Clock Gene Methylation, and Stroke: Effects on Stroke Severity and Disability, Int J Mol Sci, 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Sun Y, Huang J, Zhao Y, Xue L, Li H, Liu Q, Cao H, Peng W, Guo C, Xie Y, Liu X, Li B, Liu K, Wu S, Zhang L, Inflammatory cytokines and DNA methylation in healthy young adults exposure to fine particulate matter: A randomized, double-blind crossover trial of air filtration, J Hazard Mater, 398 (2020) 122817. [DOI] [PubMed] [Google Scholar]
- [161].Calderon-Garciduenas L, Herrera-Soto A, Jury N, Maher BA, Gonzalez-Maciel A, Reynoso-Robles R, Ruiz-Rudolph P, van Zundert B, Varela-Nallar L, Reduced repressive epigenetic marks, increased DNA damage and Alzheimer’s disease hallmarks in the brain of humans and mice exposed to particulate urban air pollution, Environ Res, 183 (2020) 109226. [DOI] [PubMed] [Google Scholar]
- [162].Vrijens K, Trippas AJ, Lefebvre W, Vanpoucke C, Penders J, Janssen BG, Nawrot TS, Association of Prenatal Exposure to Ambient Air Pollution With Circulating Histone Levels in Maternal Cord Blood, JAMA Netw Open, 3 (2020) e205156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Mancini FR, Laine JE, Tarallo S, Vlaanderen J, Vermeulen R, van Nunen E, Hoek G, Probst-Hensch N, Imboden M, Jeong A, Gulliver J, Chadeau-Hyam M, Nieuwenhuijsen M, de Kok TM, Piepers J, Krauskopf J, Kleinjans JCS, Vineis P, Naccarati A, microRNA expression profiles and personal monitoring of exposure to particulate matter, Environ Pollut, 263 (2020) 114392. [DOI] [PubMed] [Google Scholar]
- [164].Tsamou M, Nawrot TS, Carollo RM, Trippas AJ, Lefebvre W, Vanpoucke C, Vrijens K, Prenatal particulate air pollution exposure and expression of the miR-17/92 cluster in cord blood: Findings from the ENVIRONAGE birth cohort, Environ Int, 142 (2020) 105860. [DOI] [PubMed] [Google Scholar]
- [165].Li J, Wang T, Wang Y, Xu M, Zhang L, Li X, Liu Z, Gao S, Jia Q, Fan Y, Wang Z, Wu N, Zhang X, Dai Y, Kong F, Wang W, Duan H, Particulate matter air pollution and the expression of microRNAs and pro-inflammatory genes: Association and mediation among children in Jinan, China, J Hazard Mater, 389 (2020) 121843. [DOI] [PubMed] [Google Scholar]
- [166].Chen H, Xu Y, Rappold A, Diaz-Sanchez D, Tong H, Effects of ambient ozone exposure on circulating extracellular vehicle microRNA levels in coronary artery disease patients, J Toxicol Environ Health A, 83 (2020) 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Diesel and Gasoline Engine Exhausts and Some Nitroarenes. Iarc Monographs on the Evaluation of Carcinogenic Risks to Humans, IARC Monogr Eval Carcinog Risks Hum, 105 (2014) 9–699. [PMC free article] [PubMed] [Google Scholar]
- [168].Zhang X, Li J, He Z, Duan H, Gao W, Wang H, Yu S, Chen W, Zheng Y, Associations between DNA methylation in DNA damage response-related genes and cytokinesis-block micronucleus cytome index in diesel engine exhaust-exposed workers, Archives of Toxicology, 90 (2016) 1997–2008. [DOI] [PubMed] [Google Scholar]
- [169].Jiang R, Jones MJ, Sava F, Kobor MS, Carlsten C, Short-term diesel exhaust inhalation in a controlled human crossover study is associated with changes in DNA methylation of circulating mononuclear cells in asthmatics, Part Fibre Toxicol, 11 (2014) 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Lei X, Muscat JE, Zhang B, Sha X, Xiu G, Differentially DNA methylation changes induced in vitro by traffic-derived nanoparticulate matter, Toxicology, 395 (2018) 54–62. [DOI] [PubMed] [Google Scholar]
- [171].Zhang X, Chen X, Weirauch MT, Zhang X, Burleson JD, Brandt EB, Ji H, Diesel exhaust and house dust mite allergen lead to common changes in the airway methylome and hydroxymethylome, Environ Epigenet, 4 (2018) dvy020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Clifford RL, Jones MJ, MacIsaac JL, McEwen LM, Goodman SJ, Mostafavi S, Kobor MS, Carlsten C, Inhalation of diesel exhaust and allergen alters human bronchial epithelium DNA methylation, J Allergy Clin Immunol, 139 (2017) 112–121. [DOI] [PubMed] [Google Scholar]
- [173].Li H, Ryu MH, Rider CF, Tse W, Clifford RL, Aristizabal MJ, Wen W, Carlsten C, Predominant DNMT and TET mediate effects of allergen on the human bronchial epithelium in a controlled air pollution exposure study, J Allergy Clin Immunol, (2020). [DOI] [PubMed] [Google Scholar]
- [174].Tachibana K, Takayanagi K, Akimoto A, Ueda K, Shinkai Y, Umezawa M, Takeda K, Prenatal diesel exhaust exposure disrupts the DNA methylation profile in the brain of mouse offspring, J Toxicol Sci, 40 (2015) 1–11. [DOI] [PubMed] [Google Scholar]
- [175].Goodson JM, Weldy CS, MacDonald JW, Liu Y, Bammler TK, Chien WM, Chin MT, In utero exposure to diesel exhaust particulates is associated with an altered cardiac transcriptional response to transverse aortic constriction and altered DNA methylation, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 31 (2017) 4935–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Goodson JM, MacDonald JW, Bammler TK, Chien WM, Chin MT, In utero exposure to diesel exhaust is associated with alterations in neonatal cardiomyocyte transcription, DNA methylation and metabolic perturbation, Part Fibre Toxicol, 16 (2019) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Belinsky SA, Snow SS, Nikula KJ, Finch GL, Tellez CS, Palmisano WA, Aberrant CpG island methylation of the p16(INK4a) and estrogen receptor genes in rat lung tumors induced by particulate carcinogens, Carcinogenesis, 23 (2002) 335–339. [DOI] [PubMed] [Google Scholar]
- [178].Cao D, Bromberg PA, Samet JM, COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1, Am J Respir Cell Mol Biol, 37 (2007) 232–239. [DOI] [PubMed] [Google Scholar]
- [179].Jardim MJ, Fry RC, Jaspers I, Dailey L, Diaz-Sanchez D, Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways, Environ Health Perspect, 117 (2009) 1745–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Yadav S, Singh N, Shah PP, Rowbotham DA, Malik D, Srivastav A, Shankar J, Lam WL, Lockwood WW, Beverly LJ, MIR155 Regulation of Ubiquilin1 and Ubiquilin2: Implications in Cellular Protection and Tumorigenesis, Neoplasia, 19 (2017) 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Bleck B, Grunig G, Chiu A, Liu M, Gordon T, Kazeros A, Reibman J, MicroRNA-375 regulation of thymic stromal lymphopoietin by diesel exhaust particles and ambient particulate matter in human bronchial epithelial cells, J Immunol, 190 (2013) 3757–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Zhou F, Li S, Jia W, Lv G, Song C, Kang C, Zhang Q, Effects of diesel exhaust particles on microRNA-21 in human bronchial epithelial cells and potential carcinogenic mechanisms, Mol Med Rep, 12 (2015) 2329–2335. [DOI] [PubMed] [Google Scholar]
- [183].Zhang X, Zhang Y, Meng Q, Sun H, Wu S, Xu J, Yun J, Yang X, Li B, Zhu H, Xue L, Li X, Chen R, MicroRNA-382-5p is involved in pulmonary inflammation induced by fine particulate matter exposure, Environ Pollut, 262 (2020) 114278. [DOI] [PubMed] [Google Scholar]
- [184].Rider CF, Yamamoto M, Gunther OP, Hirota JA, Singh A, Tebbutt SJ, Carlsten C, Controlled diesel exhaust and allergen coexposure modulates microRNA and gene expression in humans: Effects on inflammatory lung markers, J Allergy Clin Immunol, 138 (2016) 1690–1700. [DOI] [PubMed] [Google Scholar]
- [185].Yamamoto M, Singh A, Sava F, Pui M, Tebbutt SJ, Carlsten C, MicroRNA expression in response to controlled exposure to diesel exhaust: attenuation by the antioxidant N-acetylcysteine in a randomized crossover study, Environ Health Perspect, 121 (2013) 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Rynning I, Arlt VM, Vrbova K, Neca J, Rossner P Jr., Klema J, Ulvestad B, Petersen E, Skare O, Haugen A, Phillips DH, Machala M, Topinka J, Mollerup S, Bulky DNA adducts, microRNA profiles, and lipid biomarkers in Norwegian tunnel finishing workers occupationally exposed to diesel exhaust, Occup Environ Med, 76 (2019) 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, in: DDT, Lindane, and 2,4-D, Lyon (FR), 2018. [Google Scholar]
- [188].Agrahari A, Singh A, Srivastava A, Jha RR, Patel DK, Yadav S, Srivastava V, Parmar D, Overexpression of cerebral cytochrome P450s in prenatally exposed offspring modify the toxicity of lindane in rechallenged offspring, Toxicology and applied pharmacology, 371 (2019) 20–37. [DOI] [PubMed] [Google Scholar]
- [189].Huang X, Han X, Huang Z, Yu M, Zhang Y, Fan Y, Xu B, Zhou K, Song L, Wang X, Lu C, Xia Y, Maternal pentachlorophenol exposure induces developmental toxicity mediated by autophagy on pregnancy mice, Ecotoxicol Environ Saf, 169 (2019) 829–836. [DOI] [PubMed] [Google Scholar]
- [190].IARC working Group on the Evaluation of Carcinogenic Risks to Humans, Polychlorinated Biphenyls and Polybrominated Biphenyls, IARC Monogr Eval Carcinog Risks Hum, 107 (2016) 9–500. [PMC free article] [PubMed] [Google Scholar]
- [191].Rusiecki JA, Baccarehi A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC, Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit, Environ Health Perspect, 116 (2008) 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Itoh H, Iwasaki M, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Yoshida T, Yokoyama K, Tsugane S, Association between serum organochlorines and global methylation level of leukocyte DNA among Japanese women: a cross-sectional study, Sci Total Environ, 490 (2014) 603–609. [DOI] [PubMed] [Google Scholar]
- [193].Lind L, Penell J, Luttropp K, Nordfors L, Syvanen AC, Axelsson T, Salihovic S, van Bavel B, Fall T, Ingelsson E, Lind PM, Global DNA hypermethylation is associated with high serum levels of persistent organic pollutants in an elderly population, Environ Int, 59 (2013) 456–461. [DOI] [PubMed] [Google Scholar]
- [194].van den Dungen MW, Murk AJ, Kampman E, Steegenga WT, Kok DE, Association between DNA methylation profiles in leukocytes and serum levels of persistent organic pollutants in Dutch men, Environ Epigenet, 3 (2017) dvx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Lee MH, Cho ER, Lim JE, Jee SH, Association between serum persistent organic pollutants and DNA methylation in Korean adults, Environ Res, 158 (2017) 333–341. [DOI] [PubMed] [Google Scholar]
- [196].Leung YK, Ouyang B, Niu L, Xie C, Ying J, Medvedovic M, Chen A, Weihe P, Valvi D, Grandjean P, Ho SM, Identification of sex-specific DNA methylation changes driven by specific chemicals in cord blood in a Faroese birth cohort, Epigenetics, 13 (2018) 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Consales C, Toft G, Leter G, Bonde JP, Uccelli R, Pacchierotti F, Eleuteri P, Jonsson BA, Giwercman A, Pedersen HS, Strucinski P, Goralczyk K, Zviezdai V, Spano M, Exposure to persistent organic pollutants and sperm DNA methylation changes in Arctic and European populations, Environ Mol Mutagen, 57 (2016) 200–209. [DOI] [PubMed] [Google Scholar]
- [198].Ouidir M, Mendola P, Buck Louis GM, Kannan K, Zhang C, Tekola-Ayele F, Concentrations of persistent organic pollutants in maternal plasma and epigenome-wide placental DNA methylation, Clin Epigenetics, 12 (2020) 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Kobayashi S, Sata F, Miyashita C, Miura R, Azumi K, Kobayashi S, Goudarzi H, Araki A, Ishizuka M, Todaka T, Kajiwara J, Hori T, Kishi R, Gender-specific association of exposure to non-dioxin-like polychlorinated biphenyls during pregnancy with methylation levels of H19 and long interspersed nuclear element-1 in cord blood in the Hokkaido study, Toxicology, 390 (2017) 135–145. [DOI] [PubMed] [Google Scholar]
- [200].Kim S, Cho YH, Lee I, Kim W, Won S, Ku JL, Moon HB, Park J, Kim S, Choi G, Choi K, Prenatal exposure to persistent organic pollutants and methylation of LINE-1 and imprinted genes in placenta: A CHECK cohort study, Environ Int, 119 (2018) 398–406. [DOI] [PubMed] [Google Scholar]
- [201].Su KY, Li MC, Lee NW, Ho BC, Cheng CL, Chuang YC, Yu SL, Guo YL, Perinatal polychlorinated biphenyls and polychlorinated dibenzofurans exposure are associated with DNA methylation changes lasting to early adulthood: Findings from Yucheng second generation, Environ Res, 170 (2019) 481–486. [DOI] [PubMed] [Google Scholar]
- [202].Cui Y, Choudhury SR, Irudayaraj J, Epigenetic Toxicity of Trichloroethylene: A Single-Molecule Perspective, Toxicol Res (Camb), 5 (2016) 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Georgiadis P, Gavriil M, Rantakokko P, Ladoukakis E, Botsivali M, Kelly RS, Bergdahl IA, Kiviranta H, Vermeulen RCH, Spaeth F, Hebbels D, Kleinjans JCS, de Kok T, Palli D, Vineis P, Kyrtopoulos SA, c. EnviroGenomarkers, DNA methylation profiling implicates exposure to PCBs in the pathogenesis of B-cell chronic lymphocytic leukemia, Environ Int, 126 (2019) 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Wang JH, Wang Y, Du LQ, Xu C, Liu Q, [Study on the exposure of polychlorinated biphenyl contamination and DNA methylation in male employees in an e-waste dismantling area in Tianjin], Zhonghua Yu Fang Yi Xue Za Zhi, 53 (2019) 376–381. [DOI] [PubMed] [Google Scholar]
- [205].Lee JY, Lee KM, Lee DH, Kim DS, Association of low-dose exposure to persistent organic pollutants with E-cadherin promoter methylation in healthy Koreans, Biomarkers, 23 (2018) 293–298. [DOI] [PubMed] [Google Scholar]
- [206].Eguchi A, Nishizawa-Jotaki S, Tanabe H, Rahmutulla B, Watanabe M, Miyaso H, Todaka E, Sakurai K, Kaneda A, Mori C, An Altered DNA Methylation Status in the Human Umbilical Cord Is Correlated with Maternal Exposure to Polychlorinated Biphenyls, Int J Environ Res Public Health, 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Park SY, Kim KS, Lee YM, Kim MJ, Jacobs DR Jr., Porta M, Kim DS, Lee DH, Persistent organic pollutants and promoter hypermethylation of the O(6)-methylguanine-DNA methyltransferase gene, Biomarkers, 20 (2015) 136–142. [DOI] [PubMed] [Google Scholar]
- [208].Formisano L, Guida N, Cocco S, Secondo A, Sirabella R, Ulianich L, Paturzo F, Di Renzo G, Canzoniero LM, The repressor element 1-silencing transcription factor is a novel molecular target for the neurotoxic effect of the polychlorinated biphenyl mixture aroclor 1254 in neuroblastoma SH-SY5Y cells, The Journal of pharmacology and experimental therapeutics, 338 (2011) 997–1003. [DOI] [PubMed] [Google Scholar]
- [209].Casati L, Sendra R, Colciago A, Negri-Cesi P, Berdasco M, Esteller M, Celotti F, Polychlorinated biphenyls affect histone modification pattern in early development of rats: a role for androgen receptor-dependent modulation?, Epigenomics, 4 (2012) 101–112. [DOI] [PubMed] [Google Scholar]
- [210].Cai JL, Liu LL, Hu Y, Jiang XM, Qiu HL, Sha AG, Wang CG, Zuo ZH, Ren JZ, Polychlorinated biphenyls impair endometrial receptivity in vitro via regulating mir-30d expression and epithelial mesenchymal transition, Toxicology, 365 (2016) 25–34. [DOI] [PubMed] [Google Scholar]
- [211].Li Q, Kappil MA, Li A, Dassanayake PS, Darrah TH, Friedman AE, Friedman M, Lambertini L, Landrigan P, Stodgell CJ, Xia Y, Nanes JA, Aagaard KM, Schadt EE, Murray JC, Clark EB, Dole N, Culhane J, Swanson J, Varner M, Moye J, Kasten C, Miller RK, Chen J, Exploring the associations between microRNA expression profiles and environmental pollutants in human placenta from the National Children’s Study (NCS), Epigenetics, 10 (2015) 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Krauskopf J, de Kok TM, Hebels DG, Bergdahl IA, Johansson A, Spaeth F, Kiviranta H, Rantakokko P, Kyrtopoulos SA, Kleinjans JC, MicroRNA profile for health risk assessment: Environmental exposure to persistent organic pollutants strongly affects the human blood microRNA machinery, Sci Rep, 7 (2017) 9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Wahlang B, Petriello MC, Perkins JT, Shen S, Hennig B, Polychlorinated biphenyl exposure alters the expression profile of microRNAs associated with vascular diseases, Toxicol In Vitro, 35 (2016) 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Lesiak A, Zhu M, Chen H, Appleyard SM, Impey S, Lein PJ, Wayman GA, The environmental neurotoxicant PCB 95 promotes synaptogenesis via ryanodine receptor-dependent miR132 upregulation, J Neurosci, 34 (2014) 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Topper VY, Walker DM, Gore AC, Sexually dimorphic effects of gestational endocrine-disrupting chemicals on microRNA expression in the developing rat hypothalamus, Mol Cell Endocrinol, 414 (2015) 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Shan Q, Qu F, Chen N, 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and Polychlorinated Biphenyl Coexposure Alters the Expression Profile of MicroRNAs in the Liver Associated with Atherosclerosis, Biomed Res Int, 2020 (2020) 2652756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Zhu C, Yu ZB, Zhu JG, Hu XS, Chen YL, Qiu YF, Xu ZF, Qian LM, Han SP, Differential expression profile of MicroRNAs during differentiation of cardiomyocytes exposed to polychlorinated biphenyls, Int J Mol Sci, 13 (2012) 15955–15966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Kim KY, Kim DS, Lee SK, Lee IK, Kang JH, Chang YS, Jacobs DR, Steffes M, Lee DH, Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans, Environ Health Perspect, 118 (2010) 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Pittman GS, Wang X, Campbell MR, Coulter SJ, Olson JR, Pavuk M, Birnbaum LS, Bell DA, Dioxin-like compound exposures and DNA methylation in the Anniston Community Health Survey Phase II, Sci Total Environ, 742 (2020) 140424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Vorrink SU, Hudachek DR, Domann FE, Epigenetic determinants of CYP1A1 induction by the aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126), Int J Mol Sci, 15 (2014) 13916–13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Aluru N, Karchner SI, Krick KS, Zhu W, Liu J, Role of DNA methylation in altered gene expression patterns in adult zebrafish (Danio rerio) exposed to 3, 3′, 4, 4′, 5-pentachlorobiphenyl (PCB 126), Environ Epigenet, 4 (2018) dvy005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].He QL, Wei XY, Han XY, Zhou Q, Wang HQ, Ding NZ, Meng XQ, Schatten H, Sun QY, Liu SZ, Effects of 2,3′,4,4′5-pentachlorobiphenyl exposure during pregnancy on epigenetic imprinting and maturation of offspring’s oocytes in mice, Archives of Toxicology, 93 (2019) 2575–2592. [DOI] [PubMed] [Google Scholar]
- [223].Qu XL, Ming Z, Yuan F, Wang H, Zhang YZ, Effect of 2,3′,4,4′,5-Pentachlorobiphenyl Exposure on Endometrial Receptivity and the Methylation of HOXA10, Reprod Sci, 25 (2018) 256–268. [DOI] [PubMed] [Google Scholar]
- [224].Liu D, Perkins JT, Petriello MC, Hennig B, Exposure to coplanar PCBs induces endothelial cell inflammation through epigenetic regulation of NF-kappaB subunit p65, Toxicology and applied pharmacology, 289 (2015) 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Liu D, Perkins JT, Hennig B, EGCG prevents PCB-126-induced endothelial cell inflammation via epigenetic modifications of NF-kappaB target genes in human endothelial cells, J Nutr Biochem, 28 (2016) 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Ovesen JL, Schnekenburger M, Puga A, Aryl hydrocarbon receptor ligands of widely different toxic equivalency factors induce similar histone marks in target gene chromatin, Toxicological sciences, 121 (2011) 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Guida M, Marra ML, Zullo F, Guida M, Trifuoggi M, Biffali E, Borra M, De Mieri G, D’Alessandro R, De Felice B, Association between exposure to dioxin-like polychlorinated biphenyls and miR-191 expression in human peripheral blood mononuclear cells, Mutat Res, 753 (2013) 36–41. [DOI] [PubMed] [Google Scholar]
- [228].Chen N, Shan Q, Qi Y, Liu W, Tan X, Gu J, Transcriptome analysis in normal human liver cells exposed to 2, 3, 3′, 4, 4′, 5 - Hexachlorobiphenyl (PCB 156), Chemosphere, 239 (2020) 124747. [DOI] [PubMed] [Google Scholar]
- [229].IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Trichloroethylene, Tetrachloroethylene, and Some Other Chlorinated Agents, IARC Monogr Eval Carcinog Risks Hum, 106 (2014) 1–512. [PMC free article] [PubMed] [Google Scholar]
- [230].Byrum SD, Washam CL, Patterson JD, Vyas KK, Gilbert KM, Blossom SJ, Continuous Developmental and Early Life Trichloroethylene Exposure Promoted DNA Methylation Alterations in Polycomb Protein Binding Sites in Effector/Memory CD4(+) T Cells, Front Immunol, 10 (2019) 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Gilbert KM, Blossom SJ, Reisfeld B, Erickson SW, Vyas K, Maher M, Broadfoot B, West K, Bai S, Cooney CA, Bhattacharyya S, Trichloroethylene-induced alterations in DNA methylation were enriched in polycomb protein binding sites in effector/memory CD4(+) T cells, Environ Epigenet, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Gilbert KM, Blossom SJ, Erickson SW, Reisfeld B, Zurlinden TJ, Broadfoot B, West K, Bai S, Cooney CA, Chronic exposure to water pollutant trichloroethylene increased epigenetic drift in CD4(+) T cells, Epigenomics, 8 (2016) 633–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Gilbert KM, Blossom SJ, Erickson SW, Broadfoot B, West K, Bai S, Li J, Cooney CA, Chronic exposure to trichloroethylene increases DNA methylation of the Ifng promoter in CD4(+) T cells, Toxicol Lett, 260 (2016) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Jiang Y, Chen J, Tong J, Chen T, Trichloroethylene-induced gene expression and DNA methylation changes in B6C3F1 mouse liver, PLoS One, 9 (2014) e116179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Tao L, Ge R, Xie M, Kramer PM, Pereira MA, Effect of trichloroethylene on DNA methylation and expression of early-intermediate protooncogenes in the liver of B6C3F1 mice, J Biochem Mol Toxicol, 13 (1999) 231–237. [DOI] [PubMed] [Google Scholar]
- [236].Tao L, Kramer PM, Ge R, Pereira MA, Effect of dichloroacetic acid and trichloroacetic acid on DNA methylation in liver and tumors of female B6C3F1 mice, Toxicological sciences, 43 (1998) 139–144. [DOI] [PubMed] [Google Scholar]
- [237].Tao L, Li Y, Kramer PM, Wang W, Pereira MA, Hypomethylation of DNA and the insulin-like growth factor-II gene in dichloroacetic and trichloroacetic acid-promoted mouse liver tumors, Toxicology, 196 (2004) 127–136. [DOI] [PubMed] [Google Scholar]
- [238].Tao L, Yang S, Xie M, Kramer PM, Pereira MA, Hypomethylation and overexpression of c-jun and c-myc protooncogenes and increased DNA methyltransferase activity in dichloroacetic and trichloroacetic acid-promoted mouse liver tumors, Cancer Lett, 158 (2000) 185–193. [DOI] [PubMed] [Google Scholar]
- [239].Pereira MA, Wang W, Kramer PM, Tao L, Prevention by methionine of dichloroacetic acid-induced liver cancer and DNA hypomethylation in mice, Toxicological sciences, 77 (2004) 243–248. [DOI] [PubMed] [Google Scholar]
- [240].Tabish AM, Poels K, Hoet P, Godderis L, Epigenetic factors in cancer risk: effect of chemical carcinogens on global DNA methylation pattern in human TK6 cells, PLoS One, 7 (2012) e34674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Zhang H, Hong WX, Ye J, Yang X, Ren X, Huang A, Yang L, Zhou L, Huang H, Wu D, Huang X, Zhuang Z, Liu J, Analysis of trichloroethylene-induced global DNA hypomethylation in hepatic L-02 cells by liquid chromatography-electrospray ionization tandem mass spectrometry, Biochem Biophys Res Commun, 446 (2014) 590–595. [DOI] [PubMed] [Google Scholar]
- [242].Lai C, Gao J, Zhu Z, Yuan J, Zhang W, Yang J, DNA methyltransferase expression and DNA hypomethylation status in human hepatocytes following trichloroacetic acid exposure, Biochem Biophys Res Commun, 511 (2019) 266–273. [DOI] [PubMed] [Google Scholar]
- [243].Ruan JW, Chen ZH, Lu WX, Zhang H, Ren XH, Huang XF, Yuan JH, Liu YG, Liu JJ, [Trichloroethylene-induced abnormal methylation on promoter region of SET in hepatic L-02 cells], Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi, 36 (2018) 165–168. [DOI] [PubMed] [Google Scholar]
- [244].Lu W, Chen Z, Ren X, Liu W, Deng R, Yuan J, Huang X, Zhu W, Liu J, SET promotes H2Ak9 acetylation by suppressing HDAC1 in trichloroethylene-induced hepatic cytotoxicity, Environ Toxicol Pharmacol, 59 (2018) 125–131. [DOI] [PubMed] [Google Scholar]
- [245].Deng RX, Ren XH, Ruan JW, Zheng J, Zhong JC, Lu WX, Zou XQ, Liu JJ, [An investigation of trichloroethylene-induced effects on histone methylation in L-02 hepatic cells], Zhonghua Yu Fang Yi Xue Za Zhi, 51 (2017) 347–352. [DOI] [PubMed] [Google Scholar]
- [246].Xie GS, Liu JJ, Hong WX, Zhang H, Sun Y, Zhu WG, [Effect of SET deficiency on the trichloroethylene-induced alteration of cell proliferation and cell apoptosis and DNA methylation in human hepatic L-02 cells], Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi, 34 (2016) 161–165. [DOI] [PubMed] [Google Scholar]
- [247].Lai C, Wu F, Wang Y, Wang W, Li Y, Zhang G, Gao J, Zhu Z, Yuan J, Yang J, Zhang W, Specific epigenetic microenvironment and the regulation of tumor-related gene expression by trichloroethylene in human hepatocytes, Ecotoxicol Environ Saf, 208 (2021) 111453. [DOI] [PubMed] [Google Scholar]
- [248].Stermer AR, Wilson SK, Klein D, Hall SJ, Boekelheide K, Trichloroethylene exposure alters dimethylated histone three lysine four in protein kinase A signaling pathway chromatin of rat spermdagger, Biol Reprod, 101 (2019) 875–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Aoki H, Tani H, Nakamura K, Sato H, Torimura M, Nakazato T, MicroRNA biomarkers for chemical hazard screening identified by RNA deep sequencing analysis in mouse embryonic stem cells, Toxicology and applied pharmacology, 392 (2020) 114929. [DOI] [PubMed] [Google Scholar]
- [250].Harting T, Stubbendorff M, Willenbrock S, Wagner S, Schadzek P, Ngezahayo A, Murua Escobar HM, Nolte I, The effect of dichloroacetate in canine prostate adenocarcinomas and transitional cell carcinomas in vitro, Int J Oncol, 49 (2016) 2341–2350. [DOI] [PubMed] [Google Scholar]
- [251].Huang Y, Jiang B, Xia Y, Wang J, Ji C, Tong J, Chen T, Jiang Y, Downregulation of miR-133a contributes to the cardiac developmental toxicity of trichloroethylene in zebrafish, Chemosphere, 251 (2020) 126610. [DOI] [PubMed] [Google Scholar]
- [252].Lee KM, Bassig BA, Zhang L, Vermeulen RC, Hu W, Wong JYY, Qiu C, Wen C, Huang Y, Purdue MP, Ji BT, Li L, Tang X, Rothman N, Smith MT, Lan Q, Association between occupational exposure to trichloroethylene and serum levels of microRNAs: a cross-sectional molecular epidemiology study in China, Int Arch Occup Environ Health, 92 (2019) 1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Ren X, Chen Z, Ruan J, Zhong J, Deng R, Liu W, Huang X, Yang X, Liu Y, Liu J, Trichloroethylene-induced downregulation of miR-199b-5p contributes to SET-mediated apoptosis in hepatocytes, Cell Biol Toxicol, 35 (2019) 565–572. [DOI] [PubMed] [Google Scholar]
- [254].Jiang Y, Chen J, Yue C, Zhang H, Tong J, Li J, Chen T, The Role of miR-182-5p in Hepatocarcinogenesis of Trichloroethylene in Mice, Toxicological sciences, 156 (2017) 208–216. [DOI] [PubMed] [Google Scholar]
- [255].Jiang Y, Zhou Z, Fei R, Zhou X, Wang J, Tao Y, Li J, Chen T, Role of miR-182-5p overexpression in trichloroethylene-induced abnormal cell cycle functions in human HepG2 cells, J Toxicol Environ Health A, 82 (2019) 920–927. [DOI] [PubMed] [Google Scholar]
- [256].Harting TP, Stubbendorff M, Hammer SC, Schadzek P, Ngezahayo A, Murua Escobar H, Nolte I, Dichloroacetate affects proliferation but not apoptosis in canine mammary cell lines, PLoS One, 12 (2017) e0178744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, in: Welding, molybdenum trioxide, and indium tin oxide, Lyon (FR), 2018. [PubMed] [Google Scholar]
- [258].Li H, Hedmer M, Wojdacz T, Hossain MB, Lindh CH, Tinnerberg H, Albin M, Broberg K, Oxidative stress, telomere shortening, and DNA methylation in relation to low-to-moderate occupational exposure to welding fumes, Environ Mol Mutagen, 56 (2015) 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Hossain MB, Li H, Hedmer M, Tinnerberg H, Albin M, Broberg K, Exposure to welding fumes is associated with hypomethylation of the F2RL3 gene: a cardiovascular disease marker, Occup Environ Med, 72 (2015) 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Xu Y, Li H, Hedmer M, Hossain MB, Tinnerberg H, Broberg K, Albin M, Occupational exposure to particles and mitochondrial DNA - relevance for blood pressure, Environ Health, 16 (2017) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Lu X, Miousse IR, Pirela SV, Melnyk S, Koturbash I, Demokritou P, Short-term exposure to engineered nanomaterials affects cellular epigenome, Nanotoxicology, 10 (2016) 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Shoeb M, Mustafa GM, Kodali VK, Smith K, Roach KA, Boyce G, Meighan T, Roberts JR, Erdely A, Antonini JM, A possible relationship between telomere length and markers of neurodegeneration in rat brain after welding fume inhalation exposure, Environ Res, 180 (2020) 108900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Shoeb M, Kodali VK, Farris BY, Bishop LM, Meighan TG, Salmen R, Eye T, Friend S, Schwegler-Berry D, Roberts JR, Zeidler-Erdely PC, Erdely A, Antonini JM, Oxidative Stress, DNA Methylation, and Telomere Length Changes in Peripheral Blood Mononuclear Cells after Pulmonary Exposure to Metal-Rich Welding Nanoparticles, NanoImpact, 5 (2017) 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Amrani I, Haddam N, Garat A, Allorge D, Zerimech F, Schraen S, Taleb A, Merzouk H, Edme JL, Lo-Guidice JM, Exposure to metal fumes and circulating miRNAs in Algerian welders, Int Arch Occup Environ Health, 93 (2020) 553–561. [DOI] [PubMed] [Google Scholar]
- [265].Zhao Z, Shilatifard A, Epigenetic modifications of histones in cancer, Genome Biol, 20 (2019) 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Guyton KZ, Rusyn I, Chiu WA, Corpet DE, van den Berg M, Ross MK, Christiani DC, Beland FA, Smith MT, Application of the key characteristics of carcinogens in cancer hazard identification, Carcinogenesis, 39 (2018) 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.