

Abstract
The influenza M2 protein forms a drug-targeted tetrameric proton channel to mediate virus uncoating and carries out membrane scission to mediate virus release. While the proton channel function of M2 has been extensively studied, the mechanism by which M2 conducts membrane scission is still not well understood. Previous fluorescence and electron microscopy studies indicated that M2 tetramers concentrate at the neck of the budding virus in the host plasma membrane. However, molecular evidence for this clustering is scarce. Here, we use 19F solid-state NMR to investigate M2 clustering in phospholipid bilayers. By mixing equimolar amounts of 4F-Phe47 labeled M2 and CF3-Phe47 labeled M2 and measuring F-CF3 cross peaks in 2D 19F-19F correlation spectra, we show that M2 tetramers form nanometer-scale cluster in lipid bilayers. This clustering is stronger in cholesterol-containing membranes and phosphatidylethanolamine (PE) membranes than in cholesterol-free phosphatidylcholine and phosphatidylglycerol membranes. The observed correlation peaks indicate that Phe47 sidechains from different tetramers are less than ~2 nm apart. 1H-19F correlation peaks between lipid chain protons and fluorinated Phe47 indicates that Phe47 is more deeply inserted into the lipid bilayer in the presence of cholesterol than in its absence, suggesting that Phe47 preferentially interacts with cholesterol. Static 31P NMR spectra indicate that M2 induces negative Gaussian curvature in the PE membrane. These results suggest that M2 tetramers cluster at cholesterol- and PE-rich regions of cell membranes to cause membrane curvature, which in turn can facilitate membrane scission in the last step of virus budding and release.
Graphical Abstract
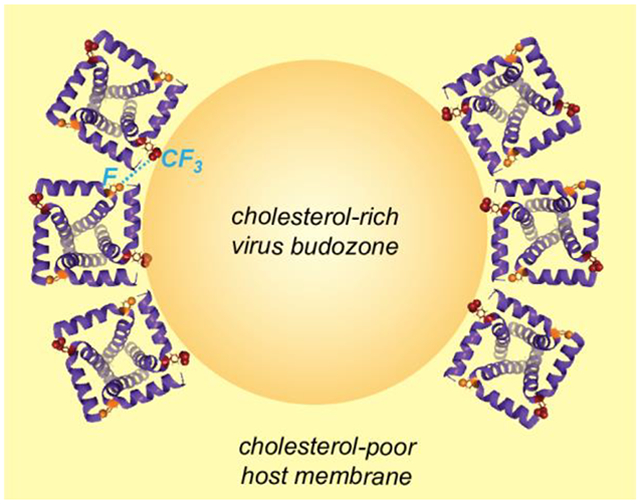
19F spin diffusion NMR reveals that tetrameric influenza M2 peptides cluster around cholesterol- and phosphoethanolamine-rich membrane domains.
1. Introduction
Influenza A and B infections are responsible for an average of 35,000 deaths per year in the U.S. in recent years [1]. The 97-residue M2 protein of the influenza A virus plays several important roles in the virus life cycle. After the influenza virus enters a cell by endocytosis, the acidic environment of the endosome activates the tetrameric proton channel, acidifying the virion, which triggers fusion of the virus lipid envelope and the endosomal membrane and subsequent release of the viral ribonucleoproteins into the host cell [2–4]. The mechanism of proton conduction by M2 has been extensively characterized [5–12], and the mechanism of channel inhibition by adamantane drugs has also been elucidated using many biophysical methods [13–18]. In addition to the proton channel function, M2 mediates influenza virus budding and release by carrying out membrane scission independent of the host ESCRT machinery [19–21]. M2-deletion viruses can form buds on the plasma membrane but cannot be released, demonstrating this scission function [19].
Several lines of evidence suggest that M2 and other membrane proteins may cluster to carry out their functions. Electron micrographs of immunogold labeled M2 on the ~100 nm scale show that the protein is clustered to the neck of the budding virions in influenza-infected MDCK cells [19]. Fluorescence microscopy images of rhodamine-labeled M2 on the 10 μm scale show that M2 preferentially localizes at the boundary of liquid-ordered (Lo) and liquid-disordered (Ld) phases in phase-separated giant unilamellar vesicles [19]. Functionally important cluster formation has also been reported for other membrane proteins. For example, fluorescence microscopy data revealed Gag-restricted clustering of the HIV Env protein upon virus maturation [22]. Co-clustering of the SNARE component syntaxin-1A and the pore-forming subunit of the Ca2+ channel Cav1.2 was associated with the interactions between these proteins in exocytotic neurotransmitter release [23].
Although biophysical data have shown that M2 co-localizes on the micron and ~100 nm scale, whether these tetramers associate on the molecular length scale of a few nanometers has not been established experimentally. Coarse-grained molecular dynamics simulations found that M2 tetramers spontaneously assemble into compact clusters or linear aggregates in response to entropic driving forces, and these clusters occur at the neck of the budding virus where membrane lateral stress is high [24]. Deuterium NMR spectra of M2(22–46) peptides containing deuterated Ala29 in DOPC : DOPE (4:1) bilayers showed that increasing the peptide/lipid molar ratio increased the quadrupolar couplings. Simulations of these 2H NMR lineshapes indicate that the uniaxial rotational diffusion rates of the tetramers slowed by 25-fold [25], suggesting protein clustering. However, since protein diffusion rates also decrease with increasing membrane viscosity as more protein is added to the membrane, this dynamics change does not provide unambiguous evidence of clustering of the tetramers.
Cholesterol binding to M2 might provide a molecular mechanism for M2 clustering [26–29]. The virus budding sites or “budozones” to which M2 is partitioned to contain higher concentrations of cholesterol than the rest of the plasma membrane [19, 24], and the Lo phase is also rich in cholesterol compared to the Ld phase [19]. Recent solid-state NMR experiments that measure 13C-19F distances between 13C-labeled M2(22–61) and fluorinated cholesterol showed that the methyl-rich β-face of cholesterol interacts with the methyl-rich Ile and Leu residues in the M2 transmembrane (TM) helix. Meanwhile, the polar hydroxyl group of cholesterol interacts with the polar and aromatic residues in the amphipathic helix (AH) [28]. These 13C-19F distance data further showed that on average two cholesterol molecules are bound to each M2 tetramer, forming an asymmetric protein-cholesterol complex. It was hypothesized that this sub-stoichiometric complex might facilitate the recruitment of M2 tetramers to the virus budozone because of the higher cholesterol concentration at the budozone compared to the plasma membrane. More recent 19F and 13C spin diffusion NMR data revealed that cholesterol molecules exist as dimers and tetramers in lipid bilayers, with the smooth α-face preferentially interacting with each other [30]. This means that the rough β-face could interact with proteins, suggesting that cholesterol dimers might act as the “glue” that clusters multiple M2 tetramers.
Here we employ 19F magic-angle-spinning (MAS) NMR spectroscopy to investigate whether M2 tetramers form nanoscale clusters in lipid bilayers, and we characterize the lipid composition and amino acid sequence requirements for clustering. We synthesize a 40-residue M2 peptide (residues 22–61) that contains both the transmembrane (TM) and amphipathic helix (AH) domains. We also synthesize a 28-residue M2 peptide (residues 22–49) that contains the TM domain and the 47FFK49 hinge. We incorporate either 4F-labeled Phe47 or 4-CF3 labeled Phe47 into these peptides. Using equimolar mixtures of the two peptides in lipid membranes, we conduct 2D 19F-19F spin diffusion correlation NMR experiments to measure the spatial proximity of the differently labeled peptides. 19F NMR provides a unique opportunity for probing nanometer-scale spatial proximity of membrane proteins. The lack of 19F background in proteins allows us to incorporate and detect sparsely fluorinated residues [31, 32]. 19F has large isotropic and anisotropic chemical shifts, thus it is sensitive to molecular conformation and dynamics. Most importantly, 19F has a large gyromagnetic ratio (γ), which increases its dipolar coupling strengths compared to low-γ nuclei. Thus, 19F NMR is well suited for measuring long interatomic distances [33–36]. Recently, 19F spin diffusion NMR experiments have been shown to be able to detect inter-fluorine distances up to ~2 nm [37–40]. Spin diffusion between a CF3 and a CF group is even more efficient than CF-CF spin diffusion, because of the simultaneous polarization transfer of three fluorines to a CF group. Using this 19F spin diffusion NMR approach, we demonstrate that M2 tetramers indeed form clusters in lipid membranes, and this clustering is facilitated by cholesterol and PE lipids. We also probe the depth of insertion of Phe47 at the junction of the TM and AH using 1H-19F 2D correlation experiments [41], and correlate these observations with M2 clustering.
2. Materials and Methods
2.1. Peptide synthesis and purification
All M2 peptides in this study were synthesized using Fmoc solid-phase chemistry on a custom designed flow peptide synthesizer [42]. Two peptides were synthesized. M2CD (residues 22–61) contain both the TM helix and the amphipathic helix and has the amino acid sequence H-22SSDPLVVAA SIIGILHLIL WILDRLFFKS IYRRLKYGLK R61-NH2. The segment 53RRLKY57 in the amphipathic helix corresponds to the Weybridge strain [28], while the commonly studied Udorn strain of M2 has 53RFFEH57 at the corresponding positions. Both sequences interact with cholesterol [28]. M2TM (residues 22–49) has the amino acid sequence H-22SSDPLVVAA SIIGILHLIL WILDRLFFK49-NH2. This peptide is three residues (47FFK49) longer than the commonly studied TM peptide (residues 22–46). Each peptide contained either a 4-CF3-Phe47 label or a 4F-Phe47 label. In addition, M2CD also contained 13C, 15N-labeled G34 and I51 and CD3-labeled A29, while M2TM also contained 13C, 15N-labeled V27, A30 and G34 (Table 1). These 13C, 15N labeled residues allow us to verify the secondary structure of these peptides in the lipid membranes.
Table 1:
Isotopic labeling and membrane compositions of the influenza AM2 samples used in this study. 4-CF3 or 4-F-labeled Phe47 residues are underlined and shown in red. 13C, 15N-labeled residues are bolded and shown in blue. CD3-labeled Ala is italicized.

|
Each M2 peptide was synthesized on the 0.05 mmol scale using H-Rink amide ChemMatrix resin (0.1 g at 0.05 mmol/g loading size). The resin was swelled in the reaction vessel for 5 min in ~5 mL of N,N-dimethylformamide (DMF) at 70°C. A 10-fold excess (0.5 mmol) of unlabeled amino acid was singly coupled with a coupling time of 50 s, using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) as the coupling agent. After the final coupling step, the peptide was deprotected and cleaved from the resin by addition of trifluoroacetic acid (TFA) / phenol / water / triisopropyl silane solution (88:5:5:2 by volume) in a fritted syringe under vortex agitation at room temperature for 3 hours. The resin was filtered off, and the crude peptide was precipitated and triturated three times with ~11 mL cold diethyl ether. The remaining solvent was allowed to evaporate from the crude peptide in the fume hood overnight.
To purify M2CD, we dissolved crude peptide in 30% acetonitrile and conducted reverse-phase HPLC using a Vydac C4 column (22 mm × 250 mm, 10 μm particle size). All HPLC procedures were run in the presence of 0.1% TFA. A linear gradient of 30–70% acetonitrile was applied over 44 min at a flow rate of 15 mL/min. The peptide eluted at 64% acetonitrile. The collected HPLC fractions were dried under nitrogen to remove most of the acetonitrile, then lyophilized overnight to obtain a homogeneous powder. MALDI mass spectrometry data verified the mass of CF3-Phe47 labeled M2CD to be 4766.8 Da, in good agreement with the calculated mass of 4764.8 Da. For 4F-Phe47 labeled M2CD, the measured mass was 4715.2 Da, in good agreement with the calculated mass of 4713.8 Da.
4F-Phe47 labeled M2TM peptide was purified by HPLC using a linear gradient of 30–100% acetonitrile in water over 65 min at a flow rate of 10 mL/min. The peptide eluted at 81% acetonitrile. MALDI-MS analysis verified the mass to be 3180.1 Da, in excellent agreement with the calculated mass of 3179.9 Da. HPLC fractions were dried under nitrogen gas and lyophilized overnight to obtain a powder. The total synthesis and purification yield was 5%. For CF3-Phe47 labeled M2TM, crude peptide was dissolved in 60% acetonitrile, and reverse-phase HPLC used a linear gradient of 80–99% acetonitrile in water over 54 min at a flow rate of 10 mL/min. The peptide eluted at 83% acetonitrile. MALDI-MS analysis verified the mass to be 3228.3 Da, in good agreement with the calculated mass of 3229.9 Da. The total yield was 10%. For all peptides, only HPLC fractions with purities higher than 90% were used.
2.2. Preparation of proteoliposomes
We used three phospholipids in this study: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE). All lipids and cholesterol were purchased from Avanti Polar Lipids and used without further purification. The POPX (X=C, G, E) lipids were chosen to mimic the acyl chain compositions of biological membranes. These lipids were mixed with cholesterol (chol) to produce three membranes: POPC : POPG (4:1), POPC : POPG : chol (4:1:1, i.e. 17 mol% cholesterol), and POPE. NMR samples contained 4.5–8.0 mg of peptide and 12–48 mg of lipids and cholesterol. The peptide/lipid molar ratios (P/L) refer to the ratio of M2 monomers to all phospholipids, excluding cholesterol. P/L values of 1:15 and 1:30 were used in this study. These 1:30 and 1:15 samples are more dilute compared to the lipid membranes in the recent 2H NMR study, which had a P/L ratio of 1:7.5 [25] .
For each membrane sample, we dissolved the appropriate mass of M2 peptide in 500 μL trifluoroethanol (TFE), and dissolved phospholipids and cholesterol in 200–500 μL chloroform. One Pasteur pipet drop of methanol was added to POPE to aid dissolution. The homogeneous lipid solution was added to the peptide TFE solution, incubated at room temperature for at least 10 min, then the organic solvent was removed using nitrogen gas. The dried peptide-lipid film was resuspended in 2 mL buffer (10 mM pH 7.4 HEPES/NaOH buffer, 1 mM EDTA, and 0.1 mM NaN3) and homogenized by one of two methods. The POPC : POPG membranes with or without cholesterol were subjected to 10–15 cycles of freeze-thawing between liquid nitrogen and a warm (37–50°C) water bath. The POPE vesicle suspension, which does not homogenize well by freeze-thawing, was sonicated at room temperature using a bath sonicator for at least three rounds of seven minutes each until the suspension appeared homogeneous. All proteoliposome suspensions were transferred to Thermo Scientific Snakeskin™ dialysis tubing with a 3.5 kD MWCO and dialyzed for 8–12 hours in sample buffer to remove residual TFE. TFE removal was important for preventing its 19F NMR signal from interfering with the measurement of the peptide 19F-19F cross peaks. The success of the TFE/TFA removal was confirmed by the absence of a 19F signal at −78 ppm in the 1D 19F direct polarization (DP) spectra. The proteoliposome suspensions were ultracentrifuged to obtain the membrane pellets. Some samples were spun down using a Beckman Coulter Optima LE-80K centrifuge equipped with a SW-60 swinging bucket rotor at 40,000–45,000 rpm (143,000–272,000 × g) at 4°C for 4–5 hours. Other samples were spun down at 55,000 rpm (max 186,000 × g) in a Beckman Coulter Optima Max preparative ultracentrifuge equipped with a TLA-55 rotor for two hours. The wet membrane pellets were allowed to dry in a desiccator or under nitrogen gas to reach 40–50 wt% water out of the total pellet mass. The hydration level was measured gravimetrically. When the volume of excess water was large, brief lyophilization was used to remove some of the bulk water. The samples were spun into a 4 mm MAS rotor using a 5 mL pipette tip in a Thermo Sorvall ST 16R centrifuge.
2.3. Solid-state NMR experiments
MAS NMR experiments were conducted on a Bruker 400 MHz (1H Larmor frequency) wide-bore AVANCE III-HD spectrometer using a 4 mm Bruker HFX MAS probe tuned to 1H, 19F and 13C Larmor frequencies. 31P chemical shifts were referenced to the hydroxyapatite signal at 2.73 ppm on the phosphoric acid scale. 19F chemical shifts were referenced to the Teflon signal at −122 ppm. 1H chemical shifts were referenced to the POPC headgroup Hγ signal at 3.26 ppm on the TMS scale, or the lipid chain-end methyl 1H signal at 0.9 ppm in POPE samples, since POPE has no headgroup γ protons. 13C chemical shifts were calibrated using the 13Cα signal of glycine at 43.65 ppm on the TMS scale. Typical radiofrequency (RF) field strengths were 50–63 kHz for 19F, 50–71 kHz for 1H, and 50 kHz for 13C. 1D 13C and 2D 13C-13C correlation experiments used a 1H excitation pulse RF field strength of 71 kHz. Static 31P NMR spectra were measured using a Bruker 4 mm 1H/31P probe at 298 K and 310 K. The 31P RF field strength was 62.5 kHz, and 1024 scans were measured per spectrum.
The 2D 19F-19F correlation spectra were measured using a CORD (Combined R2nν-driven) [43] spin diffusion mixing time of 500 ms. Cross peak intensities between 4-CF3-Phe47 and 4F-Phe47 report the distance between the two fluorines. The 2D 19F correlation experiment begins with 1H-19F cross polarization (CP), followed by 19F chemical shift evolution under 1H TPPM decoupling [44] and the CORD mixing period before detection. The unsynchronized 2D 19F-19F correlation spectra were measured under 8.5 kHz MAS. The indirect dimension spectral width was 211 ppm, and the maximum t1 evolution time was 0.567 ms. For each sample, four to eight blocks of 2D spectra were coadded to obtain sufficient signal-to-noise ratios (SNRs) and to correct for field drift between blocks. The total measuring time was 70 – 123 hours for each sample (Table 2). To increase the spectral sensitivity, we also measured rotor synchronized 2D 19F spectra that remove the spinning sidebands in the indirect dimension. For these experiments, the indirect dimension spectral width was set to 23 ppm and the MAS frequency was 8641 Hz, so that the t1 increment was one rotor period, or 115.73 μs. A total of 28 complex t1 time points were collected to give a maximum evolution time of 1.62 ms. With the small spectral width in the indirect dimension, the 4F-Phe47 peak was folded to −71 ppm. Four to eight blocks of 2D spectra were coadded for each sample to obtain sufficient SNRs, giving a total measuring time of 88 – 105 hours per sample (Table 2).
Table 2:
Parameters for the 2D 19F-19F and 1H-19F NMR experiments.
| Experiment | NMR Parameters | Experimental Time (hrs) | Membrane Sample |
|---|---|---|---|
| rotor synchronized 2D FF CORD | B0 = 9.4 T, Tset=243–245 K, νr = 8641 Hz, SWH1 = 8641 Hz (=23 ppm), SWH2 = 93.75 kHz, 19F carrier frequency = −68 ppm, NS per block =1024, τrd=1.7 s, t1,max= 1.62 ms, t1,inc= 115.8 μs, τdwell=5.3 μs, τacq = 4.3 ms, τHF=1 ms, τCORD = 500 ms, v1H,acq = 71.4 kHz (samples 3,4, and 6) and 62.5 kHz (samples 5 and 7). | 105 | 3 |
| 105 | 4 | ||
| 105 | 5 | ||
| 87.5 | 6 | ||
| 87.5 | 7 | ||
| 2D FF CORD without rotor synchronization | B0 = 9.4 T, Tset= 243 K, νr = 8500 Hz, SWH1 = 79.4 kHz, SWH2 = 93.75 kHz, 19F carrier frequency = −90.2 ppm, NS per block = 256, τrd = 2 s, t1,max = 0.567 ms, t1,inc= 12.60 μs, τdwell = 5.3 μs, τacq = 4.27 ms, τHF= 1 ms, τCORD = 500 ms, v1H,acq = 71.4 kHz. | 122.5 | 1 |
| 105 | 2 | ||
| 140 | 3 | ||
| 70 | 4 | ||
| 2D 1H-19F HETCOR with 100 ms 1H spin diffusion | B0 = 9.4 T, Tset = 262–265 K for samples 3, 4, 6, 7 and 283 K for sample 5, SWH1 = 5 kHz, SWH2 = 93.75 kHz, 19F carrier frequency = −97.6 ppm, NS per block = 1024, τrd = 1.5 s, t1,max= 4.8 ms, t1,inc= 200 μs, τdwell=5.3 μs, τacq = 4.3 ms, τHF= 1 ms, T2-filter duration = 117.6 μs, τmix = 100 ms, v1H,acq= 71.4 kHz for sample 3 and 50 kHz for samples 4–7. | 88 | 3 |
| 88 | 4 | ||
| 88 | 5 | ||
| 88 | 6 | ||
| 88 | 7 |
Symbols: B0 = magnetic field; Tset = thermocouple-reported bearing gas temperature; νr = MAS frequency; SWH1: spectral width of the ω1 dimension of the 2D spectra; SWH2: spectral width of the ω2 dimension of the 2D spectra; ns = number of scans per t1 slice of the 2D spectra; τrd = recycle delay; t1,max = maximum t1 evolution time; t1,inc = increment or dwell time for the t1 evolution period; τdwell = dwell time during direct acquisition of the FID; τacq = maximum acquisition time during direct detection; τHF = 1H-19F CP contact time; τCORD = CORD mixing time. v1H, acq = 1H decoupling field strength during detection.
To reduce peptide motion and ensure that the measured 19F-19F cross peaks reflect intermolecular proximities, these 2D 19F-19F correlation experiments were conducted at ~243 K (−30° C) using an FTS unit, which cooled house air to −85 to −78°C and outputted it to the MAS probe at a flow rate of 1700–1850 l/h. All reported temperatures are direct thermocouple readings, and are ~5°C lower than the actual sample temperature estimated based on the water 1H chemical shifts [45].
To investigate the depth of insertion of Phe47 in the lipid membrane, we measured 2D 1H-19F heteronuclear correlation (HETCOR) spectra with 1H spin diffusion [41]. The experiment begins with four 19F 90° pulses spaced by 2 ms each to saturate the 19F magnetization. Then a 1H 90° excitation pulse and a 1H T2 filter of 2*112.6 μs = 225.2 μs are used to select the water and lipid 1H magnetization. The ensuing 1H chemical shift evolution is followed by a 1H mixing time of 100 ms during which the water and lipid 1H magnetization is transferred to the peptide and is detected on 19F after 1H-19F CP. These 2D HETCOR experiments used 48 t1 time points to reach a maximum 1H t1 evolution time of 4.8 ms, and 1,024 scans per t1 slice. Typical spectra were coadded from four blocks of experiments, giving signal-averaging times of 88 hours per sample. For different lipid membranes, we chose sample temperatures of 262 to 283 K (−11 to +12° C; Fig. 6) to obtain similar lipid CH2 linewidth of about 350 Hz, as assessed from 1D 1H MAS spectra. This ensures that the lipid chain dynamics are similar between the different membranes, thus giving similar 1H spin diffusion coefficients. All NMR spectra were plotted from TopSpin 3.6.1. Peak intensities were analyzed in Microsoft Excel v16.52.
Figure 6.
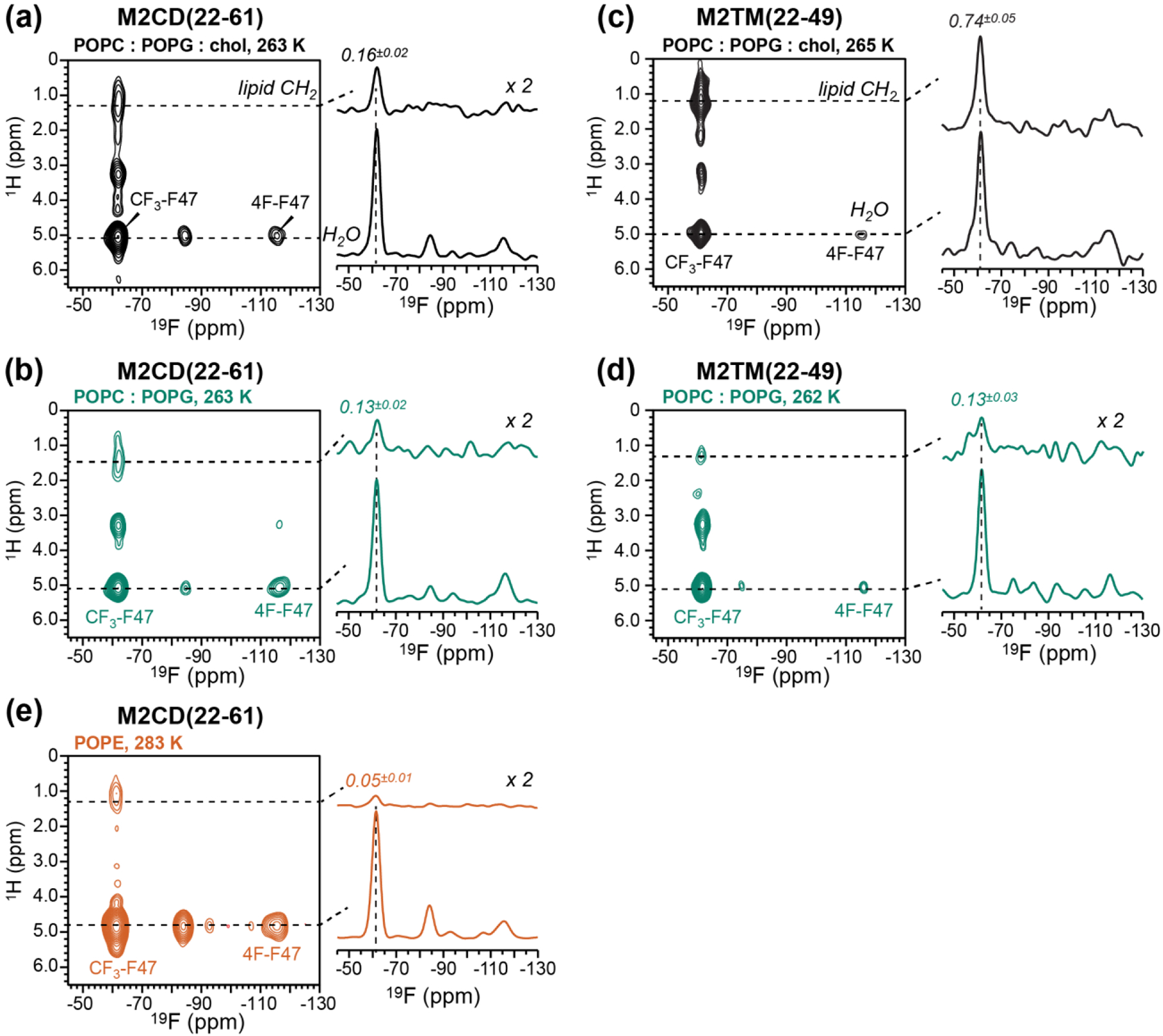
2D 1H-19F HETCOR spectra of M2 peptides in different lipid membranes. The spectra were measured at sample temperatures that give similar lipid chain 1H linewidths. All samples have a P/L of 1:30. (a, b, e) M2CD spectra. (c, d) M2TM spectra. (a) Spectrum of M2CD in POPC : POPG : chol membranes measured at 263 K. (b) Spectrum of M2CD in POPC : POPG membranes measured at 263 K. (c) Spectrum of M2TM in POPC : POPG : chol membranes measured at 265 K. (d) Spectrum of M2TM in POPC : POPG membranes measured at 262 K. (e) Spectrum of M2CD in POPE membrane measured at 283 K. Water and lipid CH2 cross sections are shown to the right of each 2D spectrum. The intensity ratio of the lipid–CF3 cross peak relative to the water-CF3 cross peak is the highest for M2TM in the POPC : POPG : chol membrane and the lowest for M2CD in the POPE membrane.
3. Results
In this study, we investigate M2 clustering in lipid membranes as a function of the peptide/lipid ratio, the presence or absence of cholesterol, and PC-rich membranes versus the PE membrane. We also compare M2 peptides that contain both the TM domain and the amphipathic helix (M2CD) versus only the TM domain (M2TM). The POPC : POPG (4:1) membrane composition and the peptide length were chosen to allow comparison of our data with previous biochemical studies of the membrane scission function of M2 [19, 46–48] and with NMR [27, 28] and EPR [49–51] studies of M2 interaction with cholesterol.
Our strategy for detecting M2 tetramer association is to mix 4-CF3-Phe47 labeled peptide with 4F-Phe47 labeled peptide at a 1:1 molar ratio and measure CF3-F cross peaks in 2D 19F-19F correlation spectra. Phe47 is chosen because it lies at the four corners of the tetramer in both M2CD and M2TM, with maximal separation within each tetramer and potentially the shortest distance between two tetramers (Fig. 1). Based on the eight lowest energy structures of M2CD (residues 22–62) in DOPC : DOPE bilayers (PDB: 2L0J) [52] solved at 30°C, the distances between the para hydrogens of the nearest-neighbor Phe47 sidechains within each tetramer range from 21 Å to 33 Å. The average distance, 28 Å, is too long to be measured by 19F spin diffusion NMR, whose detection upper limit is ~20 Å [38]. For M2TM, a solution NMR structure of M2(19–49) in DPC micelles [53] indicates a distance of ~23 Å between the para hydrogens of F47 on different chains (PDB 2MUV), again indicating that intra-tetramer Phe47-Phe47 distances are too long to be measurable. Thus, the hinge residue of Phe47 is a good candidate for probing the presence or absence of tetramer clustering for both M2CD and M2TM.
Figure 1.
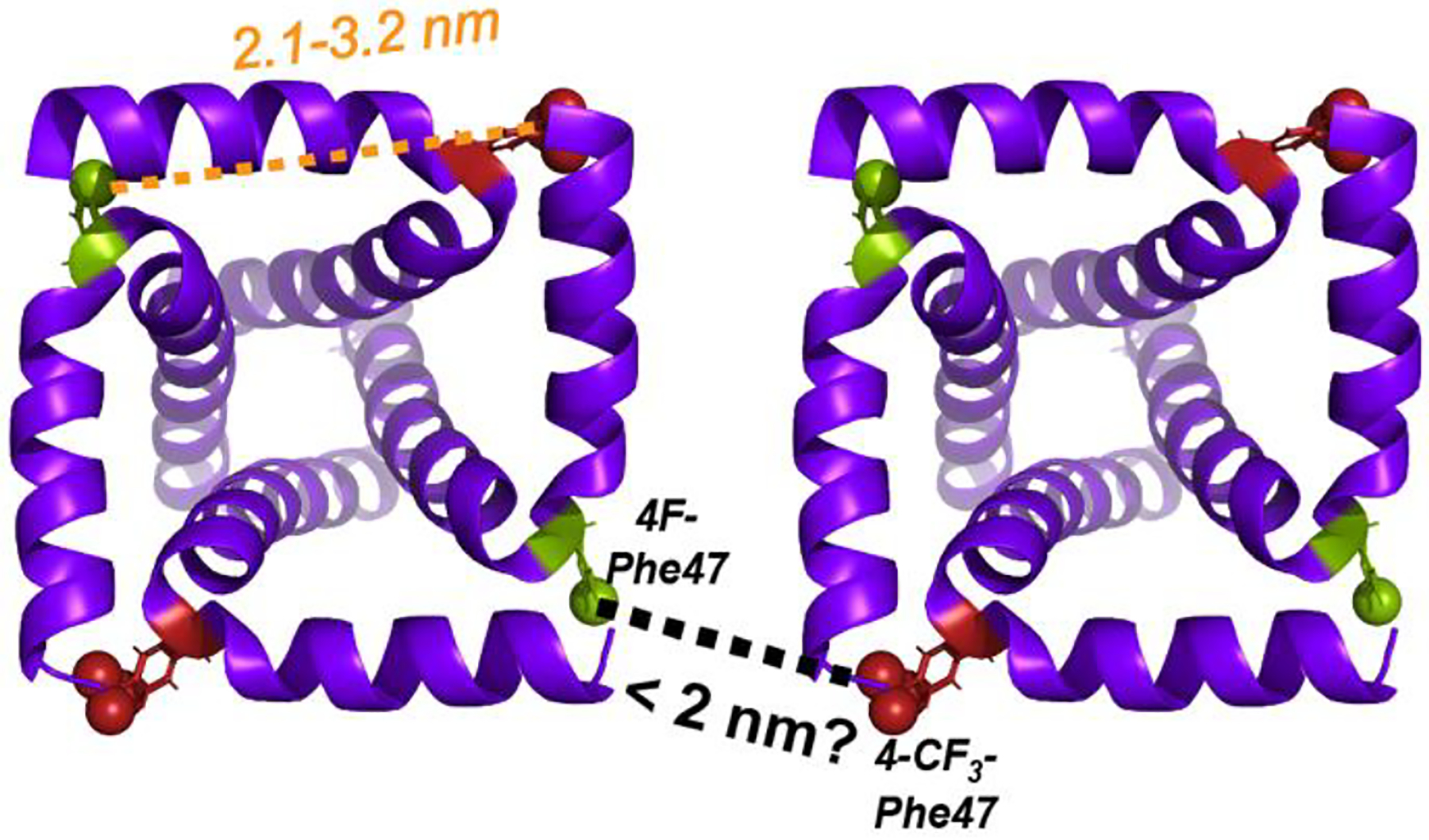
Schematic diagram of M2 clustering in lipid membranes. Two copies of the solid-state NMR orientational structure of M2(22–62) (PDB: 2L0J), manually positioned to illustrate how two 4F-Phe47 and CF3-Phe47 residues on two neighboring tetramers might approach each other within ~2 nm. This close approach is required to explain the observation of 19F-19F cross peaks in 2D 19F spin diffusion spectra. Intra-tetramer nearest-neighbor distances between Phe47 sidechain para-hydrogens are 2.1 – 3.2 nm.
We next estimate the closest approach between M2 tetramers if the tetramers are uniformly distributed in the membrane. Molecular dynamics simulations of gel-phase POPC membranes indicate a headgroup area per lipid of 59 Å2 [54]. For POPG the area/lipid is 60.6 Å2 based on scattering density profiles [55, 56]). If we assume that all M2 tetramers are parallel aligned, which would give rise to the shortest approach between Phe47 residues of two adjacent tetramers, then at a P/L ratio of 1:30, the center-to-center distance between two nearest-neighbor tetramers is about 66 Å. When P/L increases to 1:15, the center-to-center distance decreases to 51 Å. Since each M2CD tetramer has a side length of about 28 Å, these values correspond to closest-approach distances of ~39 Å (P/L 1:30) and ~23 Å (P/L 1:15) between the para hydrogens of Phe47 of two different tetramers. These inter-tetramer separations for homogeneously distributed M2 are beyond the detection limit of 19F spin diffusion NMR. If M2 tetramers are randomly aligned, with both antiparallel and parallel orientations possible, then the closest-approach distance between two tetramers will be even longer and hence undetectable for this homogeneous distribution model. However, if the tetramers cluster together in the membrane, then CF3-F cross peaks can become observable. We test this hypothesis by measuring 2D 19F-19F correlation spectra as a function of P/L and membrane compositions.
3.1. M2 tetramers cluster in a membrane-dependent manner
We first measured the 13C chemical shifts of site-specifically labeled M2CD and M2TM peptides to verify that Phe47 fluorination does not perturb the α-helical conformation of the peptide (Fig. 2). The 13C chemical shifts of G34 and I51 in M2CD and V27, A30 and G34 in M2TM all correspond to the α-helical conformation in both POPC : POPG membranes and the POPE membrane, confirming that the helical conformation of the TM and AH domains is preserved. This α-helicity is consistent with previously reported chemical shifts of M2 peptides in a variety of lipid membranes [16, 28, 57, 58] and with the periodicity of orientation-dependent 15N-1H dipolar couplings of M2CD in PC and PE membranes [52]. The 19F CP spectra of the peptides show the expected CF3-Phe and 4F-Phe peaks at −61 ppm and −116 ppm, respectively. CF3 spinning sidebands are also observed, but there is no detectable signal of TFE or TFA, indicating successful removal of the fluorinated solvents. The 4F-Phe peak is broader and lower in intensity than the CF3 peak: the linewidths are 4.0–5.5 ppm for 4F-Phe and 3.0–3.7 ppm for CF3 under these experimental conditions. These differences are expected because the para-fluorine group is more sensitive to sidechain conformational disorder than the fast-rotating methyl group. The CF3 spinning sideband intensity of M2CD is higher in the POPE membrane than in the POPC : POPG membranes, suggesting that the Phe47 sidechain is more immobilized in the POPE membrane.
Figure 2.
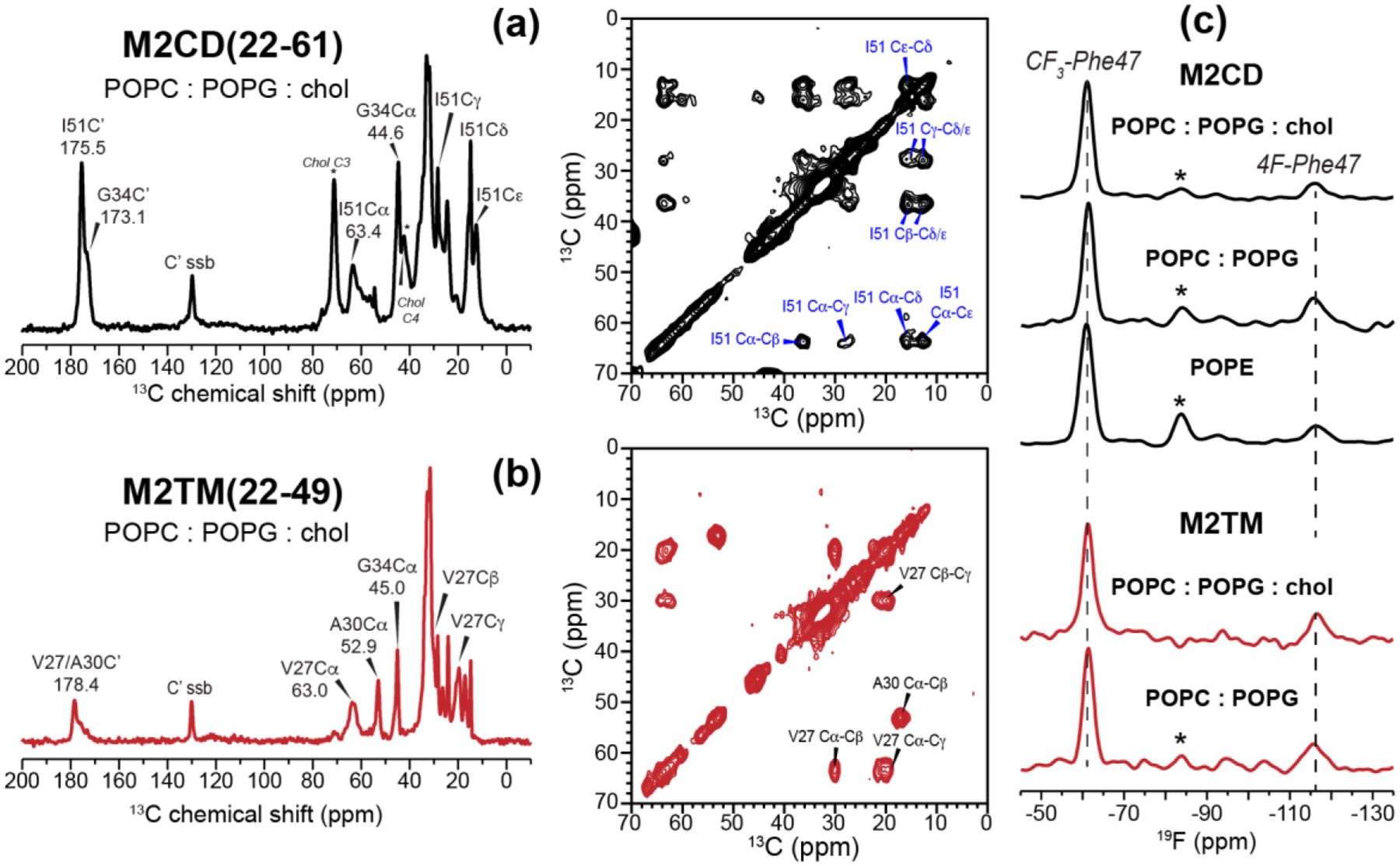
13C and 19F NMR spectra of membrane-bound M2 peptides. (a) 1D 13C CP and 2D 13C-13C correlation spectra of M2CD at P/L 1:15 in POPC : POPG : chol membranes. The spectra were measured at 243 K under 10 kHz MAS. Peaks that are not annotated in the 1D spectra are lipid 13C signals. The sample contains C3, C4-labeled cholesterol (peaks marked with * in the M2CD 1D spectrum). (b) 1D and 2D 13C spectra of M2TM at P/L 1:30 in POPC : POPG : chol membranes. The spectra were measured at 253 K under 9 kHz MAS. The peptide 13C chemical shifts confirm the α-helical conformation of M2CD and M2TM. (c) 19F CP spectra of membrane-bound M2CD and M2TM. Similar 19F linewidths and intensity distributions are observed for both M2CD and M2TM, indicating that the two peptides exhibit similar conformational dynamics under these experimental conditions. The spectra in (c) were measured between 262 K and 283 K under 8.5 kHz MAS. Asterisks indicate the spinning sideband of the CF3 peak.
Fig. 3 shows the 500 ms 2D 19F-19F CORD spectra of M2CD in POPC : POPG membranes with and without cholesterol. At a high P/L of 1:15, we observed a clear F-CF3 cross peak in the POPC : POPG : chol membrane but a weaker cross peak in the POPC : POPG membrane (Fig. 3a, c). When the peptide concentration was decreased two-fold to P/L 1:30 (Fig. 3b, d), the F-CF3 cross peak persisted in the POPC : POPG : chol membrane. Moreover, its intensity is similar between the low and high peptide concentrations (Fig. 3e, top), suggesting that M2 tetramers cluster rather than distributing homogeneously in the cholesterol-containing membrane. In contrast, dilution of the peptide in the POPC : POPG membrane completely suppressed the F-CF3 cross peak, indicating that the intra-tetramer Phe47-Phe47 distances are too long to be measurable. If the 2D experiment is mainly detecting intra-tetramer cross peaks, it would be difficult to explain the dependence of these cross peak intensities on the membrane composition and peptide concentration. Therefore, these results indicate that M2 tetramers cluster in the cholesterol-containing membrane, and this clustering is not an artifact of the high peptide concentration but reflects specific protein-protein and protein-cholesterol interactions.
Figure 3.
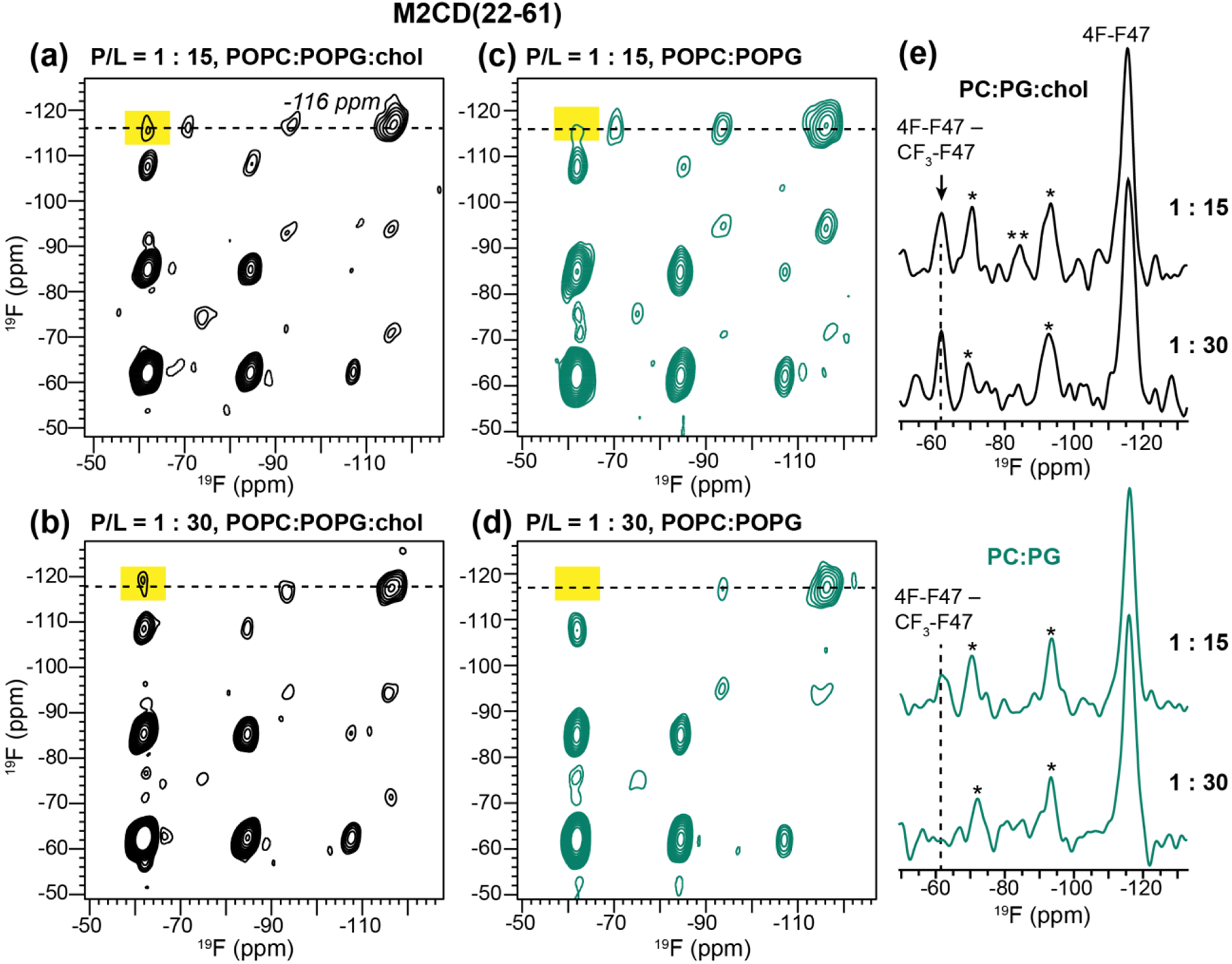
500 ms 2D 19F-19F CORD spectra of mixed Phe47-fluorinated M2CD in lipid membranes. The spectra were measured at 243 K under 8.5 kHz MAS. The −116 ppm cross section of 4F-Phe47 are shown on the right. Cross peak between 4F-Phe47 and 4-CF3-Phe47 (−62 ppm) indicates inter-tetramer contact. F-CF3 cross peaks are observed at high P/L in both membranes, but only in the cholesterol-containing membrane at the low P/L of 1:30. (a) 2D spectrum of M2CD in POPC : POPG : chol membranes, at P/L 1:15. (b) 2D spectrum of M2CD in POPC : POPG : chol membranes at P/L 1:30. (c) 2D spectrum of M2CD in POPC : POPG membranes, at P/L 1:15. (d) 2D spectrum of M2CD in POPC : POPG membranes at P/L 1:30. (e) Cross sections of the spectra in (a-d) at the 4F-Phe ω1 chemical shift of −116 ppm (marked with dashed lines in the 2D spectra). Dashed lines guide the eye to the 4F – CF3 cross peak. Single asterisks (*) denote spinning sidebands of the 4F-Phe47 peak while double asterisks (**) denote the CF3 spinning sideband.
To reproduce the F-CF3 cross peaks with higher sensitivity, we measured rotor-synchronized 2D 19F correlation spectra of the 1:30 M2CD samples, and quantified the intensity of the F-CF3 cross-peak relative to the total intensities in the −71 ppm cross section of the 4F-Phe47 peak (Fig. 4). The cross-peak intensities varied significantly between different membrane samples. In the POPC : POPG : chol sample, the F-CF3 cross peak intensity is 14±3% of the total 4F-Phe47 intensity (Fig. 4a), whereas in the POPC : POPG membrane, the cross peak relative intensity decreased to 6±3% of the total intensity (Fig. 4b), confirming that cholesterol facilitates clustering. To investigate whether M2 can also cluster in other membranes, we measured the 2D 19F-19F correlation spectrum of M2CD bound to the POPE membrane. PE lipids are enriched in influenza virus lipid envelopes relative to the host cell membranes from which they are derived [59, 60], and their negative curvature is known to play an important role in membrane fusion and fission in general [61, 62]. Fig. 4c shows the 2D 19F spin diffusion spectrum of POPE-bound M2CD. A strong CF3-F cross peak is observed, whose relative intensity, 26±3%, is even higher than that of the POPC : POPG : chol sample, indicating that M2CD clusters more tightly in the POPE membrane.
Figure 4.
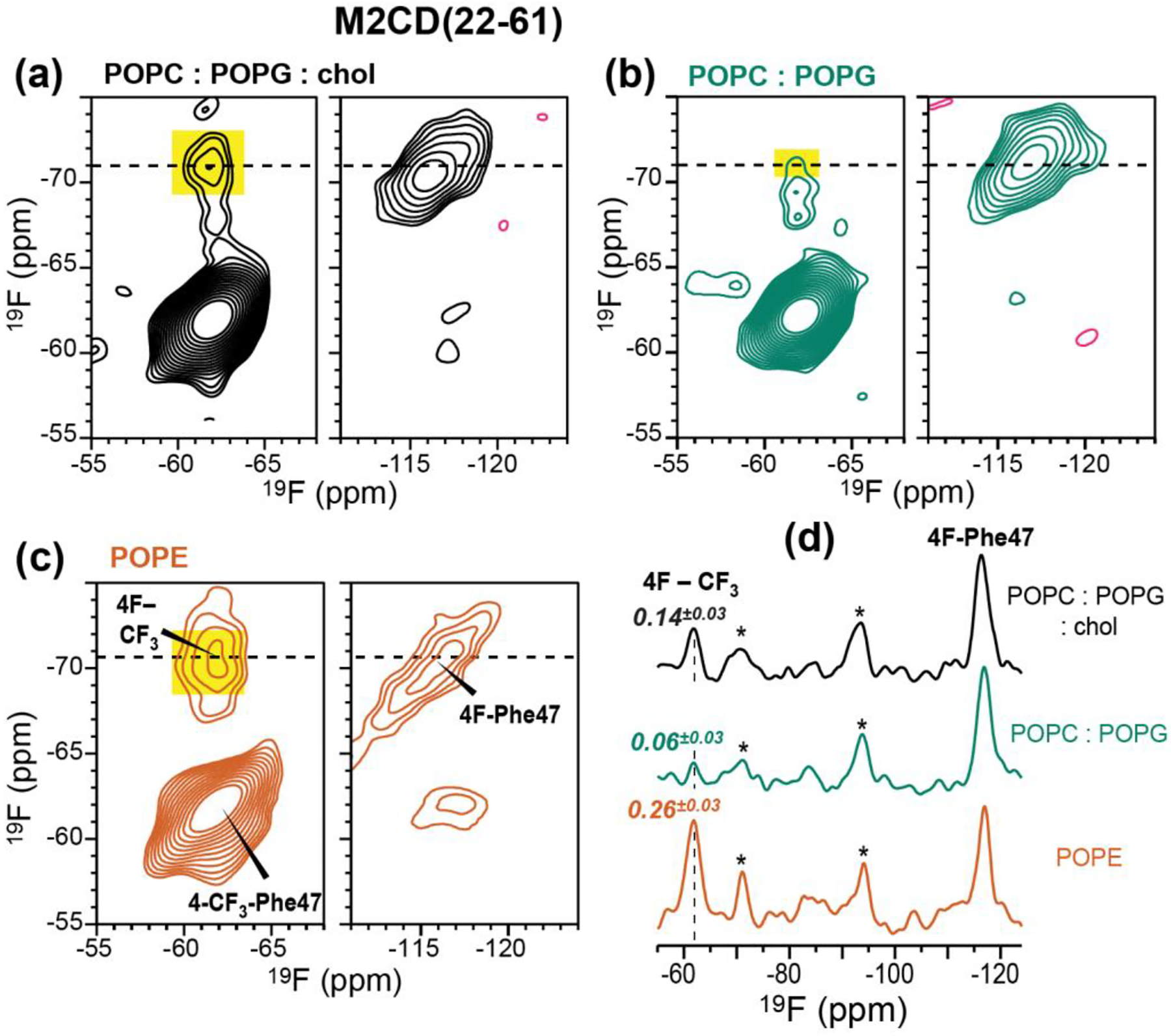
2D 19F-19F correlation spectra of mixed Phe47 fluorinated M2CD peptides in three membranes at P/L 1:30. The spectra were measured with 500 ms CORD mixing at 243 K under 8641 Hz MAS. (a) 2D spectrum of the peptide in the POPC : POPG : chol membrane. (b) 2D spectrum of the peptide in the POPC : POPG membrane. (c) 2D spectrum of the peptide in the POPE membrane. (d) 1D cross sections at the folded 4F-Phe47 chemical shift of −71 ppm. The CF3 cross peak (dashed line) intensity relative to the sum of all peak intensities in the cross section decreases in the order of POPE > POPC : POPG : chol > POPC : POPG. Asterisks (*) denote spinning sidebands of the 4F-Phe47 signal.
To determine whether the amphipathic helix is required for M2 clustering, we also measured the 2D 19F spin diffusion spectra of M2TM(22–49) in POPC : POPG membranes with and without cholesterol. At P/L 1:30, we observed a clear F-CF3 cross peak in the POPC : POPG : chol membrane but the cross peak is much weaker in the POPC : POPG membrane (Fig. 5). This trend is similar to M2CD, indicating that the TM peptide is able to cluster in the absence of the AH residues 50–62, moreover this clustering is also facilitated by cholesterol. The F-CF3 cross peak intensities of M2TM are stronger than that of M2CD (Fig. 5c), suggesting that M2TM peptides form tighter clusters than M2CD.
Figure 5.
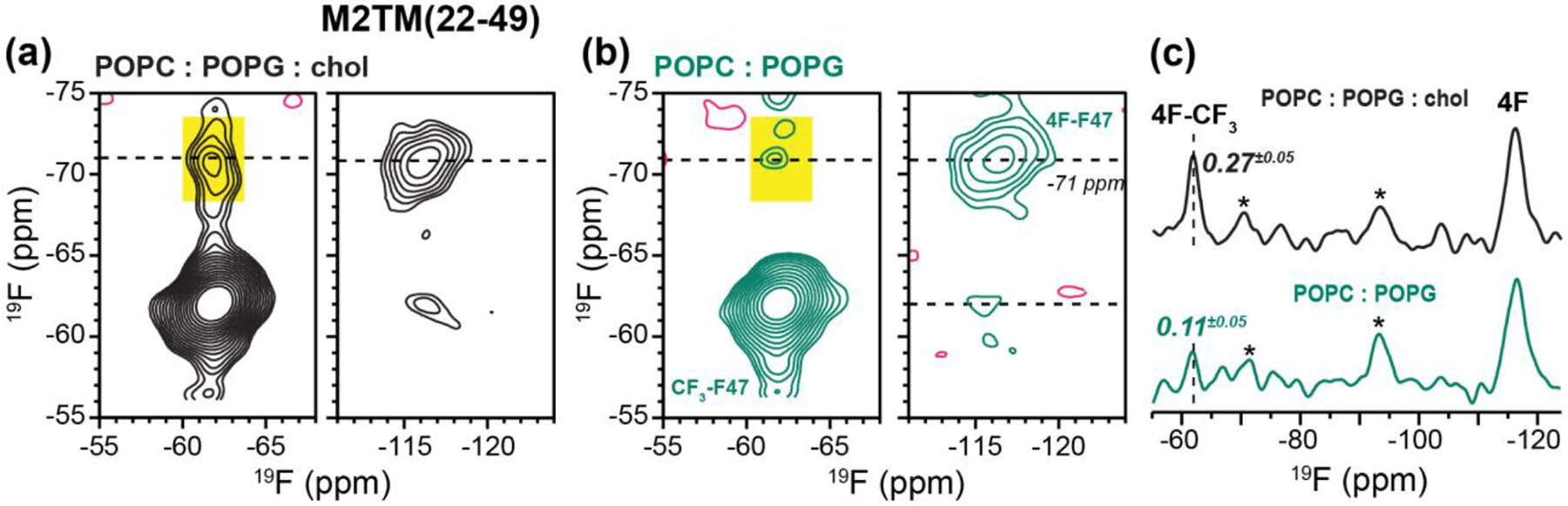
2D 19F-19F correlation spectra of mixed fluorinated M2TM peptides at P/L 1 : 30. The spectra were measured with a CORD mixing time of 500 ms at 243 K under 8641 Hz MAS. (a) 2D spectrum of the peptide in the POPC : POPG : chol membrane. (b) 2D spectrum of the peptide in the POPC : POPG : membrane. (c) 1D cross section of the two 2D spectra at the folded 4F-Phe47 chemical shift of −71 ppm. The F-CF3 cross peak (dashed line) intensity is much higher in the cholesterol-containing membrane than in the cholesterol-free membrane. Asterisks (*) denote spinning sidebands of the 4F-Phe47 signal.
3.2. Depth of insertion of Phe47 in lipid membranes
Using fluorinated Phe47, we next investigated the depth of insertion of the 47FFK49 hinge between the TM and AH domains in different membranes. We used the 2D 1H-19F correlation experiment with 1H spin diffusion for this purpose [41]. By detecting the cross peaks of CF3-Phe47 with the water 1H signal at 4.8–5.1 ppm and with the lipid 1H signal at 1.3 ppm, we obtain information about the depth of insertion of the TM-AH junction in different lipid membranes. The 4F-Phe47 signal at −116 ppm reports the same information, but due to its low intensity we do not analyze its signal. Fig. 6 compares the 100 ms 2D 1H-19F HETCOR spectra of the different membrane samples. All samples exhibit strong water-Phe47 cross peaks, as expected because of the surface-exposed position of this residue [50, 52]. The depth of insertion of Phe47 is manifested by the lipid-CF3 cross peak intensity relative to the water-CF3 cross peak, ICH2/IH2O. For M2CD, the intensity ratios are similar (0.16 and 0.13) between the POPC : POPG : chol membrane and POPC : POPG membranes (Fig. 6a, b). This indicates that the Phe47 sidechain is moderately inserted into both membranes. In dramatic contrast, the lipid-to-water intensity ratio is much higher, 0.74, for M2TM in the POPC : POPG : chol membrane (Fig. 6c), indicating that Phe47 in the TM peptide is in close contact with the lipid chains. Repeating this measurement for M2TM in the cholesterol-free POPC : POPG membrane decreased the lipid-CF3 cross peak intensity to 0.13 (Fig. 6d). These spectra were measured at temperatures at which the lipid chains have similar 1H linewidths. Thus, the high intensity of the lipid-Phe47 cross peak in the POPC : POPG : chol membrane cannot be attributed to different spin diffusion coefficients, but reflect deeper insertion of the Phe47 sidechain of the TM peptide in the cholesterol-containing membrane. Finally, we measured the 2D 1H-19F HETCOR spectrum of M2CD in the POPE membrane. In contrast to the POPC : POPG samples, the spectrum shows negligible lipid cross peak (Fig. 6e), indicating that Phe47 is much more shallowly inserted into the POPE membrane than the POPC : POPG membranes.
3.3. M2CD induces curvature to POPE membranes
Membrane scission requires negative Gaussian curvature (NGC), wherein every point on the surface of the membrane has principal curvatures of opposite signs. This NGC is manifested as a broad isotropic peak in the static 31P NMR spectra [63] and as diffraction peaks at characteristic Q positions in small-angle X-ray scattering (SAXS) data [64]. A previous SAXS study showed that M2CD and full-length M2 promote cubic phases, which possess NGC’s, in PE-rich membranes [62]. We previously measured 31P NMR spectra of oriented DMPC/DHPC (3:1) bicelles and found that M2CD, but not M2TM (residues 22–46), induced an isotropic 31P peak, indicating the formation of a high-curvature phase [65]. To investigate if clustering of M2 tetramers is correlated with membrane curvature formation, we measured the static 31P spectra of the three membranes containing M2CD at P/L 1:30 (Fig. 7). Neither of the two POPC : POPG membranes showed an isotropic peak, indicating that these two membranes maintain their lamellar morphologies. In comparison, the POPE membrane exhibits a small isotropic peak at 310 K, indicating the generation of NGC at this temperature. By comparison, pure POPE liposomes manifest a uniaxial powder pattern with no isotropic peak (Fig. 7d). Thus, the POPE membrane environment induces the most M2 tetramer clustering, the least Phe47 membrane insertion, and the strongest peptide-driven membrane curvature.
Figure 7.
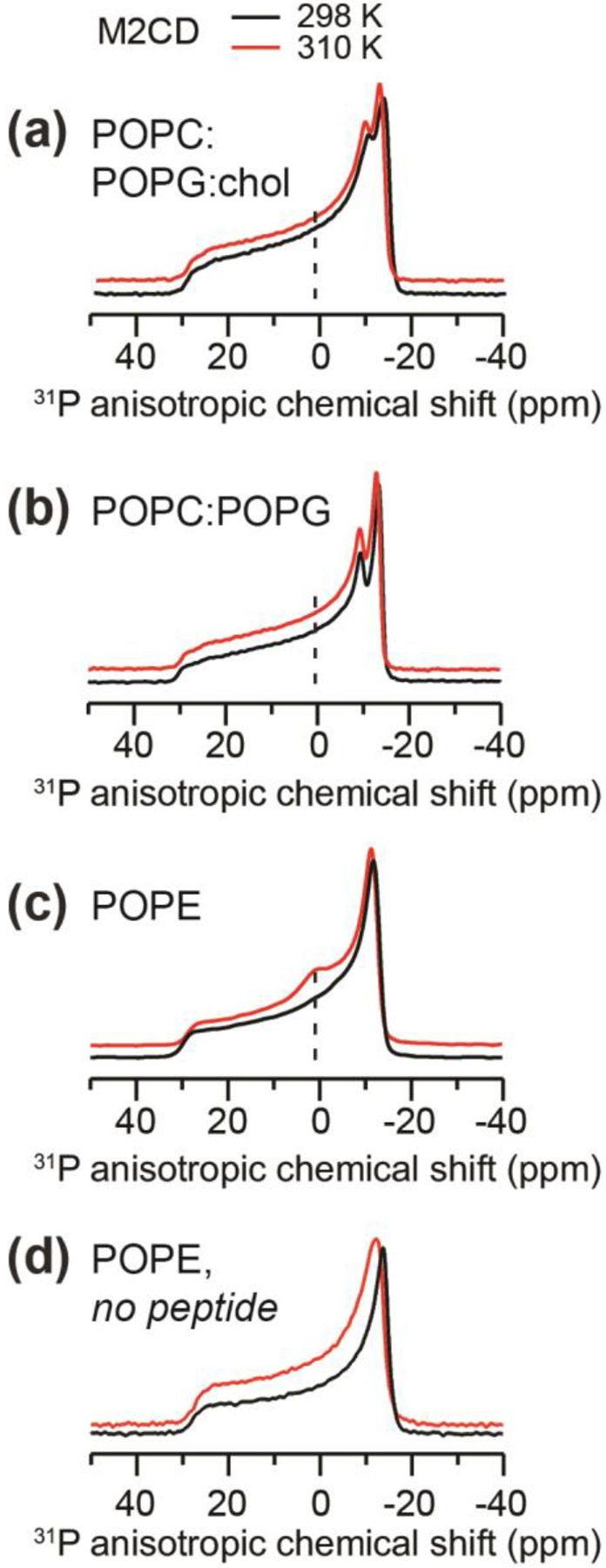
Static 31P NMR spectra of proteoliposomes containing M2CD at P/L 1:30. The spectra were measured at 298 K (black lines) and 310 K (red lines). (a) POPC : POPG : chol membranes. (b) POPC : POPG membranes. (c) POPE membranes. Only the M2CD-containing POPE membrane exhibits an isotropic peak, which is indicative of NGC. (d) Static 31P spectra of POPE membranes without any peptide. No isotropic peak is observed, indicating that the POPE membrane maintains a lamellar morphology in the absence of M2 peptides.
4. Discussion
M2 clustering depends on membrane environment
The 19F NMR data shown here, taken together, indicate that M2 tetramers cluster on the molecular length scale in lipid membranes, and this clustering is stronger in cholesterol-containing membrane and POPE membranes than in cholesterol-free POPC : POPG membranes. At the low temperature (−30°C) of the 19F spin diffusion experiments, the peptide motion is frozen and the lipid dynamics are also largely suppressed. Thus, the different F-CF3 cross peak intensities in the various membranes cannot be attributed to motional differences of the peptide. By varying the peptide/lipid molar ratios and lipid compositions of the membrane, we can attribute the observed F-CF3 cross peaks predominantly to tetramer-tetramer interactions, whereas Phe47-Phe47 contacts within a tetramer make a negligible contribution. This is because the F-CF3 cross peaks are not present in all samples: in the cholesterol-free POPC : POPG membrane at P/L ratios of 1:30 and 1:15, the F-CF3 cross peak of M2CD is either absent or weak, indicating that these samples exhibit neither intra-tetramer nor inter-tetramer contacts. Therefore, the Phe47-Phe47 distances within each tetramer are longer than measurable by 19F spin diffusion, which is consistent with the estimated average nearest-neighbor distance of ~2.8 nm within each tetramer [52]. In contrast, clear cross peaks are observed in the POPC : POPG : cholesterol membrane, indicating that cholesterol facilitates clustering of the tetramers.
To assess whether the full AH domain is necessary for clustering, we measured 2D 19F-19F correlation spectra of the M2TM(22–49) peptide. The Phe47-Phe47 distances within each tetramer in this shorter peptide remain sufficiently large to prevent intra-tetramer contacts from interfering with the clustering analysis. The presence of 47FFK49, which resides at the TM-AH junction, should retain the ability of the peptide to bind cholesterol [27, 28]. We observed clear F-CF3 cross peak in POPC : POPG : chol membrane-bound M2TM(22–49), indicating that the TM peptides, in the absence of the AH, are able to form clusters in cholesterol-containing membranes.
These 19F NMR data are consistent with, and more direct than, 2H NMR spectra of the Ala29 CD3 group for demonstrating clustering [25]. In that study, increasing the P/L ratio from 1:20 to 1:7.5 in the DOPC : DOPE membrane increased the uniaxial rotational correlation time of M2TM tetramers from 0.55 μs to 13.9 μs. This significant slowing of uniaxial rotation suggests clustering of the M2 tetramers. This hypothesis was further tested using coarse-grained molecular dynamics simulations, which found that M2TM tetramers cluster in an approximately linear fashion, and the tetramers can adopt both parallel and antiparallel orientations with respect to each other. Additional coarse-grained and atomistic simulations found that such M2 clusters are formed entropically at the boundary of liquid-ordered and liquid-disordered phases of the membrane, and clustering can induce membrane curvature [24].
EPR data of spin-labeled M2TM(residues 22–46) and M2C(residues 23–60) peptides showed an equilibrium between two conformations [51, 66, 67]. Both conformations are found in all membranes tested, but their relative populations change with the membrane composition. Cholesterol, thicker lipid bilayers, and PE lipids favor a “tight” conformer, which is characterized by (1) closer proximity between residues on neighboring monomers in both the TM and AH domains, (2) slower mobility of the tetramer, and (3) shallower insertion of the tetramer into the membrane and higher solvent accessibility. The first effect was demonstrated by differential spin-spin coupling between neighboring spin-labeled residues in X-band EPR spectra of M2TM [66, 67] and M2TMC [51]. The second effect was demonstrated by continuous-wave EPR spectra of M2TMC [51]. The reduced membrane insertion effect was manifested by power saturation EPR data that probe the membrane with O2 and the solvent with NiEDDA [51]. Interestingly, our previously used VMS membrane (SM : DPPC : DPPE : Chol) increased the population of the immobilized conformer [51]. To ensure that the two-state equilibrium reflects the property of full-length M2, these EPR studies were extended to wild-type M2 that is nitroxide-labeled at residue 59 in the AH. Saturation recovery EPR curves exhibit a clear biexponential form that is characteristic of conformational exchange. As with other constructs, cholesterol shifted the TM and AH equilibria toward the immobile and more tightly packed conformation [68].
These EPR spectra were measured at ambient temperature (~20°C) where cholesterol and PE can modulate the membrane viscosity and lateral pressure, which can in turn affect the tetramer conformation. However, the possibility of M2 clustering in these samples cannot be ruled out, even at the low peptide concentrations used in these studies. Even if this conformational distribution exclusively reflects intra-tetramer structural variation, it may already be reflected in the ensemble of orientational structures solved by solid-state NMR, in which the nearest-neighbor Phe47-Phe47 distances varied but never fell below 2.1 nm [52]. These considerations support the model that the measured 19F-19F cross peaks in the 2D 19F spin diffusion NMR spectra predominantly result from inter-tetramer clustering rather than intra-tetramer interactions.
M2 hinge region’s depth of insertion in lipid membranes
The fluorinated Phe47 allowed us to probe the depth of insertion of the junction between the TM and AH. While most spectra showed a modest lipid-Phe47 cross peak (Fig. 6), we observed an unexpectedly strong lipid-Phe47 cross peak for M2TM in the POPC : POPG : chol membrane. This result cannot be explained by differential dynamics of the lipids, since we measured these HETCOR spectra at temperatures that give similar lipid 1H linewidths and hence similar lipid chain dynamics. Instead, we attribute this strong lipid-Phe47 cross peak to preferential interaction of the Phe47 sidechain with cholesterol, which anchors this residue to the membrane. The propensity of the Phe47 sidechain to insert into the membrane is present in M2CD as well but may be partly restricted by the rest of the amphipathic helix. The phenylalanine residues of AH have been previously reported to interact with cholesterol. In DMPC : DMPG : chol (16 : 4 : 5) membranes, Phe 13C-labeled full-length M2 and 13C2,3,4-labeled cholesterol showed cross peaks in 2D 13C-13C correlation spectra [29]. Further 13C-19F REDOR experiments on 13C-labeled protein and fluorinated cholesterol showed that an M2TM(22–46) peptide that lacks the 47FFK49 motif does not bind cholesterol, whereas an M2CD(22–61) peptide that contains multiple Phe residues does [28]. 2D 13C-13C correlation spectra of 13C-labeled M2(21–97) in POPC : POPG : chol membranes measured with dynamic nuclear polarization (DNP) showed cross peaks between Phe aromatic carbons and cholesterol C3 and C9 carbons [27]. Finally, docking simulations constrained by the observed intermolecular distances and cholesterol orientations found that Phe47 was the only Phe that could bind cholesterol. These multiple lines of evidence strongly suggest that Phe47 directly interacts with cholesterol, which could explain the strong lipid-Phe47 cross peak.
We did not observe a significant difference in the depth of insertion of Phe47 in M2CD between the POPC : POPG : chol membrane and the POPC : POPG membrane (Fig. 6a, b). This result differs from power saturation EPR data of M2TMC in POPC : POPG membranes, which found that addition of 30% cholesterol caused AH residues 46, 48, 51 and 55 to be more surface exposed than they were in the non-cholesterol membrane [51]. Three experimental differences could cause this discrepancy. First, our 2D 1H-19F HETCOR spectra were measured in a membrane containing only 17% cholesterol. This low level of cholesterol might not cause a sufficiently large change in the AH insertion depth. Second, we conducted the HETCOR experiments at −10°C whereas the EPR experiments were conducted at ambient temperature. Since the M2 structure is highly sensitive to the membrane properties, the low temperature at which these HETCOR spectra were measured might reduce the difference in the insertion depth of the AH. Finally, at the temperature of these 2D HETCOR experiments, the lipid chain proton linewidth of the POPC : POPG : chol membrane was ~480 Hz, which was moderately broader than the POPC : POPG lipid linewidth of ~370 Hz. Thus, the lipids may be slightly more immobilized in the cholesterol-containing membrane. If this is the case, then it is possible that the true insertion depth of Phe47 is slightly shallower in the cholesterol-containing membrane.
Interestingly, the HETCOR spectra indicate that Phe47 is less inserted in the POPE membrane than in the POPC : POPG membranes (Fig. 6e). This result is in good agreement with atomic force microscopy (AFM) data that showed that adding POPE into POPC bilayers suppressed the ability of an AH-only peptide to insert into the membrane and to modulate bilayer structure [69]. Differential insertion depth of the AH domain may allow M2 to adapt to different membrane environments, and may help modulate the interfacial energy at the Lo/Ld boundary, which is important for inducing membrane curvature [70]. Fluorescence microscopy data of M2-infected cells have shown that M2 congregates at the edge of the budding virion in the host cell membrane [19]. POPE may be recruited to budozones, as suggested by the significant POPE enrichment in influenza virions relative to the cell membranes from which they bud [59]. This POPE can promote NGC, both due to its own negative curvature and due to its ability to promote M2 clustering and regulate the depth of insertion of M2. Thus, cholesterol, PE lipids, and M2 may jointly drive membrane scission by associating at the budozone to cause membrane curvature.
In conclusion, the current 19F and 31P NMR data indicate that both M2CD and M2TM tetramers form clusters in lipid membranes in the presence of cholesterol and PE lipids. Based on the CF3-F cross peaks between Phe47 residues on different peptide chains, we find the M2 tetramers to be more tightly clustered in the POPE membrane than in the 17% cholesterol POPC : POPG : chol membrane. In both membranes, the inter-tetramer distances must be shorter than 2 nm to account for the observed 19F-19F cross peaks. The membrane with the tightest M2 clusters, POPE, also exhibits the highest NGC and the shallowest insertion of the Phe47 sidechain. These results provide experimental evidence that M2 proteins cluster at the edge of the cholesterol- and PE-rich budding virion to induce membrane curvature, allowing an ever-narrower stalk to achieve membrane scission.
Highlights.
19F spin diffusion NMR shows that M2 tetramers cluster in lipid bilayers.
M2 clustering is facilitated by membrane cholesterol and by POPE lipids.
2D 1H-19F correlation experiments show that cholesterol promotes insertion of Phe47 sidechain into the membrane.
Acknowledgement
This work is supported by NIH grant GM088204 to M.H. This study made use of NMR spectrometers at the MIT-Harvard Center for Magnetic Resonance, which is supported by NIH grant P41 GM132079.
References
- [1].Center for Disease Control and Prevention, Past Seasons Estimated Influenza Disease Burden, 2020, https://www.cdc.gov/flu/about/burden/past-seasons.html?web=1&wdLOR=c42518B89-D610-A343-B6AB-ED30749B0F5A
- [2].Hong M, DeGrado WF, Structural basis for proton conduction and inhibition by the influenza M2 protein, Protein science : a publication of the Protein Society, 21 1620–1633 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pinto LH, Holsinger LJ, Lamb RA, Influenza virus M2 protein has ion channel activity, Cell, 69 517–528 (1992). [DOI] [PubMed] [Google Scholar]
- [4].Pinto LH, Lamb RA, The M2 proton channels of influenza A and B viruses, The Journal of biological chemistry, 281 8997–9000 (2006). [DOI] [PubMed] [Google Scholar]
- [5].Wang C, Lamb RA, Pinto LH, Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue, Biophys. J, 69 1363–1371 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tang Y, Zaitseva F, Lamb RA, Pinto LH, The Gate of the Influenza Virus M2 Proton Channel Is Formed by a Single Tryptophan Residue, The Journal of biological chemistry, 277 39880–39886 (2002). [DOI] [PubMed] [Google Scholar]
- [7].Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF, A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity, Proc. Natl. Acad. Sci. USA, 94 11301–11306 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hu F, Luo W, Hong M, Mechanisms of proton conduction and gating by influenza M2 proton channels from solid-state NMR, Science, 330 505–508 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hu F, Schmidt-Rohr K, Hong M, NMR detection of pH-dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel, J. Am. Chem. Soc,, 134 3703–3713 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA, Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity, Proc. Natl. Acad. Sci. U.S.A, 103 6865–6870 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li C, Yi M, Hu J, Zhou HX, Cross TA, Solid-state NMR and MD simulations of the antiviral drug amantadine solubilized in DMPC bilayers, Biophys. J, 94 1295–1302 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor’kov PL, Brey WW, Cross TA, M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes, Angew. Chem. Int. Ed. Engl, 51 8383–8386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M, Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers, Nature, 463 689–692 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ohigashi Y, Ma C, Jing X, Balannick V, Pinto LH, Lamb RA, An amantadine-sensitive chimeric BM2 ion channel of influenza B virus has implications for the mechanism of drug inhibition, Proc. Natl. Acad. Sci. U. S. A, 106 18775–18779 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF, Structural basis for the function and inhibition of an influenza virus proton channel, Nature, 451 596–599 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cady SD, Mishanina TV, Hong M, Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding, J. Mol. Biol, 385 1127–1141 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cady SD, Wang J, Wu Y, DeGrado WF, Hong M, Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy J. Am. Chem. Soc, 133 4274–4284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andreas LB, Barnes AB, Corzilius B, Chou JJ, Miller EA, Caporini M, Rosay M, Griffin RG, Dynamic nuclear polarization study of inhibitor binding to the M2(18–60) proton transporter from influenza A, Biochemistry, 52 2774–2782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rossman JS, Jing X, Leser GP, Lamb RA, Influenza virus M2 protein mediates ESCRT-independent membrane scission, Cell, 142 902–913 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bruce EA, Medcalf L, Crump CM, Noton SL, Stuart AD, Wise HM, Elton D, Bowers K, Digard P, Budding of filamentous and non-filamentous influenza A virus occurs via a VPS4 and VPS28-independent pathway, Virology, 390 268–278 (2009). [DOI] [PubMed] [Google Scholar]
- [21].Chen BJ, Lamb RA, Mechanisms for enveloped virus budding: can some viruses do without an ESCRT?, Virology, 372 221–232 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chojnacki J, Staudt T, Glass B, Bingen P, Engelhardt J, Anders M, Schneider J, Müller B, Hell SW, Kräusslich HG, Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy, Science, 338 524–528 (2012). [DOI] [PubMed] [Google Scholar]
- [23].Sajman J, Trus M, Atlas D, Sherman E, The L-type Voltage-Gated Calcium Channel co-localizes with Syntaxin 1A in nano-clusters at the plasma membrane, Sci Rep, 7 11350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Madsen JJ, Grime JMA, Rossman JS, Voth GA, Entropic forces drive clustering and spatial localization of influenza A M2 during viral budding, Proc Natl Acad Sci U S A, 115 E8595–e8603 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Paulino J, Pang X, Hung I, Zhou HX, Cross TA, Influenza A M2 Channel Clustering at High Protein/Lipid Ratios: Viral Budding Implications, Biophys J, 116 1075–1084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schroeder C, Heider H, Möncke-Buchner E, Lin TI, The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein, Eur Biophys J, 34 52–66 (2005). [DOI] [PubMed] [Google Scholar]
- [27].Elkins MR, Sergeyev IV, Hong M, Determining Cholesterol Binding to Membrane Proteins by Cholesterol (13)C Labeling in Yeast and Dynamic Nuclear Polarization NMR, J. Am. Chem. Soc, 140 15437–15449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Elkins MR, Williams JK, Gelenter MD, Dai P, Kwon B, Sergeyev IV, Pentelute BL, Hong M, Cholesterol-binding site of the influenza M2 protein in lipid bilayers from solid-state NMR, Proc. Natl. Acad. Sci. U. S. A, 114 12946–12951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ekanayake EV, Fu R, Cross TA, Structural Influences: Cholesterol, Drug, and Proton Binding to Full-Length Influenza A M2 Protein, Biophys J, 110 1391–1399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Elkins MR, Bandara A, Pantelopulos GA, Straub JE, Hong M, Direct Observation of Cholesterol Dimers and Tetramers in Lipid Bilayers, J Phys Chem B, 125 1825–1837 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kitevski-LeBlanc JL, Prosser RS, Current applications of 19F NMR to studies of protein structure and dynamics, Prog. Nucl. Magn. Reson. Spectrosc, 62 1–33 (2012). [DOI] [PubMed] [Google Scholar]
- [32].Sharaf NG, Gronenborn AM, 19F-Modified Proteins and 19F-Containing Ligands as Tools in Solution NMR Studies of Protein Interactions, in: Kelman Z (Ed.) Methods in Enzymology, vol. 565, Academic Press, 2015, pp. 67–95. [DOI] [PubMed] [Google Scholar]
- [33].Shcherbakov AA, Medeiros-Silva J, Tran N, Gelenter MD, Hong M, From Angstroms to Nanometers: Measuring Interatomic Distances in Solid-State NMR, Chem. Rev, in press (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shcherbakov AA, Hong M, Rapid Measurement of Long-Range Distances in Proteins by Multidimensional 13C-19F REDOR NMR under Fast Magic-Angle Spinning, J. Biomol. NMR, 71 31–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shcherbakov AA, Mandala VS, Hong M, High-Sensitivity Detection of Nanometer 1H-19F Distances for Protein Structure Determination by 1H-Detected Fast MAS NMR, J. Phys. Chem. B, 123 4387–4391 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shcherbakov AA, Roos M, Kwon B, Hong M, Two-dimensional 19F-13C correlation NMR for 19F resonance assignment of fluorinated proteins, J. Biomol. NMR, 74 193–204 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Roos M, Mandala VS, Hong M, Determination of Long-Range Distances by Fast Magic-Angle-Spinning Radiofrequency-Driven (19)F-(19)F Dipolar Recoupling NMR, J. Phys. Chem. B, 122 9302–9313 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roos M, Wang T, Shcherbakov AA, Hong M, Fast Magic-Angle-Spinning (19)F Spin Exchange NMR for Determining Nanometer (19)F-(19)F Distances in Proteins and Pharmaceutical Compounds, J. Phys. Chem. B, 122 2900–2911 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lu M, Wang M, Sergeyev IV, Quinn CM, Struppe J, Rosay M, Maas W, Gronenborn AM, Polenova T, (19)F Dynamic Nuclear Polarization at Fast Magic Angle Spinning for NMR of HIV-1 Capsid Protein Assemblies, J. Am. Chem. Soc, 141 5681–5691 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang M, Lu M, Fritz M, Quinn C, Byeon IJ, Byeon CH, Struppe J, Maas W, Gronenborn AM, Polenova T, Fast Magic Angle Spinning 1⁹F NMR of HIV-1 Capsid Protein Assemblies, Angew. Chem. Int. Ed. Engl, 57 16375–16379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huster D, Yao X, Hong M, Membrane protein topology probed by (1)H spin diffusion from lipids using solid-state NMR spectroscopy, J Am Chem Soc, 124 874–883 (2002). [DOI] [PubMed] [Google Scholar]
- [42].Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, Berger T, Policarpo RL, Zhang C, Zou Y, Liao X, Spokoyny AM, Jensen KF, Pentelute BL, Rapid flow-based peptide synthesis, Chembiochem, 15 713–720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hou G, Yan S, Trébosc J, Amoureux JP, Polenova T, Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids, J Magn Reson, 232 18–30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG, Heteronuclear decoupling in rotating solids, J. Chem. Phys, 103 6951–6958 (1995). [Google Scholar]
- [45].Böckmann A, Gardiennet C, Verel R, Hunkeler A, Loquet A, Pintacuda G, Emsley L, Meier BH, Lesage A, Characterization of different water pools in solid-state NMR protein samples, J. Biomol. NMR, 45 319–327 (2009). [DOI] [PubMed] [Google Scholar]
- [46].Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA, Influenza virus M2 ion channel protein is necessary for filamentous virion formation, J. Virol, 84 5078–5088 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Martyna A, Bahsoun B, Badham MD, Srinivasan S, Howard MJ, Rossman JS, Membrane remodeling by the M2 amphipathic helix drives influenza virus membrane scission, Sci. Rep, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Martyna A, Bahsoun B, Madsen JJ, Jackson F, Badham MD, Voth GA, Rossman JS, Cholesterol Alters the Orientation and Activity of the Influenza Virus M2 Amphipathic Helix in the Membrane, J. Phys. Chem. B, 124 6738–6747 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nguyen PA, Soto CS, Polishchuk A, Caputo GA, Tatko CD, Ma C, Ohigashi Y, Pinto LH, DeGrado WF, Howard KP, pH-induced conformational change of the influenza M2 protein C-terminal domain, Biochemistry, 47 9934–9936 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huang S, Green B, Thompson M, Chen R, Thomaston J, DeGrado WF, Howard KP, C-terminal juxtamembrane region of full-length M2 protein forms a membrane surface associated amphipathic helix, Protein Sci, 24 426–429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kim SS, Upshur MA, Saotome K, Sahu ID, McCarrick RM, Feix JB, Lorigan GA, Howard KP, Cholesterol-Dependent Conformational Exchange of the C-Terminal Domain of the Influenza A M2 Protein, Biochemistry, 54 7157–7167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA, Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer, Science, 330 509–512 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wu Y, Canturk B, Jo H, Ma C, Gianti E, Klein ML, Pinto LH, Lamb RA, Fiorin G, Wang J, DeGrado WF, Flipping in the pore: discovery of dual inhibitors that bind in different orientations to the wild-type versus the amantadine-resistant S31N mutant of the influenza A virus M2 proton channel, J Am Chem Soc, 136 17987–17995 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Leekumjorn S, Sum AK, Molecular characterization of gel and liquid-crystalline structures of fully hydrated POPC and POPE bilayers, J. Phys. Chem. B, 111 6026–6033 (2007). [DOI] [PubMed] [Google Scholar]
- [55].Pan J, Heberle FA, Tristram-Nagle S, Szymanski M, Koepfinger M, Katsaras J, Kucerka N, Molecular structures of fluid phase phosphatidylglycerol bilayers as determined by small angle neutron and X-ray scattering, Biochim Biophys Acta, 1818 2135–2148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kučerka N, Nieh MP, Katsaras J, Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature, Biochim Biophys Acta, 1808 2761–2771 (2011). [DOI] [PubMed] [Google Scholar]
- [57].Cady SD, Wang T, Hong M, Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein, J. Am. Chem. Soc, 133 11572–11579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Andreas LB, Eddy MT, Chou JJ, Griffin RG, Magic-angle-spinning NMR of the drug resistant S31N M2 proton transporter from influenza A, J. Am. Chem. Soc, 134 7215–7218 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ivanova PT, Myers DS, Milne SB, McClaren JL, Thomas PG, Brown HA, Lipid composition of viral envelope of three strains of influenza virus - not all viruses are created equal, ACS Infect Dis, 1 399–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang J, Pekosz A, Lamb RA, Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins, J Virol, 74 4634–4644 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chernomordik LV, Kozlov MM, Protein-lipid interplay in fusion and fission of biological membranes, Annu Rev Biochem, 72 175–207 (2003). [DOI] [PubMed] [Google Scholar]
- [62].Schmidt NW, Mishra A, Wang J, DeGrado WF, Wong GC, Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission, J Am Chem Soc, 135 13710–13719 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yang Y, Yao H, Hong M, Distinguishing bicontinuous lipid cubic phases from isotropic membrane morphologies using (31)P solid-state NMR spectroscopy, J Phys Chem B, 119 4993–5001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yao H, Lee MW, Waring AJ, Wong GC, Hong M, Viral fusion protein transmembrane domain adopts beta-strand structure to facilitate membrane topological changes for virus-cell fusion, Proc. Natl. Acad. Sci. USA, 112 10926–10931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang T, Hong M, Investigation of the curvature induction and membrane localization of the influenza virus M2 protein using static and off-magic-angle spinning solid-state nuclear magnetic resonance of oriented bicelles, Biochemistry, 54 2214–2226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Duong-Ly KC, Nanda V, Degrado WF, Howard KP, The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment, Protein Sci, 14 856–861 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Saotome K, Duong-Ly KC, Howard KP, Influenza A M2 protein conformation depends on choice of model membrane, Biopolymers, 104 405–411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Herneisen AL, Sahu ID, McCarrick RM, Feix JB, Lorigan GA, Howard KP, A Budding-Defective M2 Mutant Exhibits Reduced Membrane Interaction, Insensitivity to Cholesterol, and Perturbed Interdomain Coupling, Biochemistry, 56 5955–5963 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pan J, Dalzini A, Song L, Cholesterol and phosphatidylethanolamine lipids exert opposite effects on membrane modulations caused by the M2 amphipathic helix, Biochim. Biophys. Acta, 1861 201–209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].García-Sáez AJ, Chiantia S, Schwille P, Effect of line tension on the lateral organization of lipid membranes, The Journal of biological chemistry, 282 33537–33544 (2007). [DOI] [PubMed] [Google Scholar]