

Abstract
The pancreatic beta cell is the only cell type in the body responsible for insulin secretion, and thus plays a unique role in the control of glucose homeostasis. The loss of beta-cell mass and function plays an important role in both type 1 and type 2 diabetes. Thus, using chemical biology to identify small molecules targeting the beta cell could be an important component to developing future therapeutics for diabetes. This strategy provides an attractive path toward increasing beta-cell numbers in vivo. A regenerative strategy involves enhancing proliferation, differentiation, or neogenesis. On the other hand, protecting beta cells from cell death, or improving maturity and function, could preserve beta-cell mass. Here, we discuss the current state of chemical matter available to study beta-cell regeneration, and how they were discovered.
Induction of beta-cell proliferation
An attractive strategy to regenerating beta-cell mass has been to stimulate these cells to divide. Unlike, say, the liver, where hepatocytes can repopulate the organ, cells resident in pancreatic islets appear to be fixed, leading to the hypothesis that increasing beta-cell division with small molecules could help treat, for example, type 1 diabetes. In recent years, a substantial portion of the efforts to apply chemical biology to beta cells has been in the area of beta-cell proliferation. The question of how beta cells propagate in the pancreas—whether through self-duplication, differentiation from a stem cell population, or some other means—was under debate for many years. A seminal study in 2004 showed that beta cells in mice can themselves replicate [1]. A few years later, an evaluation of islets from juvenile humans demonstrated that replication is the primary means by which beta-cell mass increases from birth to adulthood [2]. The dramatic decrease in Ki67-positive beta cells observed after adolescence, however, suggested that proliferation was not accessible as a natural mechanism of regeneration in adult humans. For example, pregnancy causes a dramatic increase in beta-cell mass in mice [3,4], but in the only such study ever undertaken in humans, beta-cell area only increased 1.4-fold during pregnancy [5]. Further, data suggested that this increase was due to neogenesis from ductal cells in the pancreas, and not proliferation.
Thus, for many years, the prevailing wisdom remained that adult human beta cells could not be induced to divide. However, three studies in 2015 and 2016 pointed to inhibition of the kinase DYRK1A as a new mechanism capable of promoting human beta-cell proliferation (Figure 1) [6–8••]. Interestingly, different approaches to discovery were used in each case. Wang et al. performed a high-throughput screen for inducers of MYC expression in HepG2 cells; the natural product harmine emerged as a strong hit and was found to promote beta-cell proliferation through its inhibition of DYRK1A [8••]. Another effort took a more targeted approach, evaluating an aminopyrazine dual inhibitor of DYRK1A and GSK3β in beta cells [7••]. Finally, a third approach examined the effects of 5-iodotubercidin (5-IT), originally thought to result in phenotypic activity due to inhibiting adenosine kinase [9]. Instead, target identification efforts in human islets showed that, remarkably, 5-ITalso inhibits DYRK1A to promote beta-cell proliferation [6••]. Subsequent work to evaluate combinations of compounds showed that this effect of DYRK1A inhibitors (e.g., harmine, 5-IT, leucettine-41, INDY) in human islets can be enhanced by TGFβ inhibitors [10] or by GLP-1 agonists [11]. In fact, rather than just acting on DYRK1A alone, the activity of this class of compounds was shown to involve inhibition of both DYRK1A and DYRK1B, two closely related isoforms [12]. Importantly, these compounds have been demonstrated to restore glucose homeostasis in several mouse models of diabetes, including partial pancreatectomy and streptozotocin-induced diabetes. The fact that nearly all kinase inhibitors in this area inhibit both kinases allows for a more robust induction of beta-cell proliferation, an effect which could not be achieved by genetic means alone, emphasizing the power of leveraging polypharmacology to promote phenotypes of interest [13].
Figure 1.
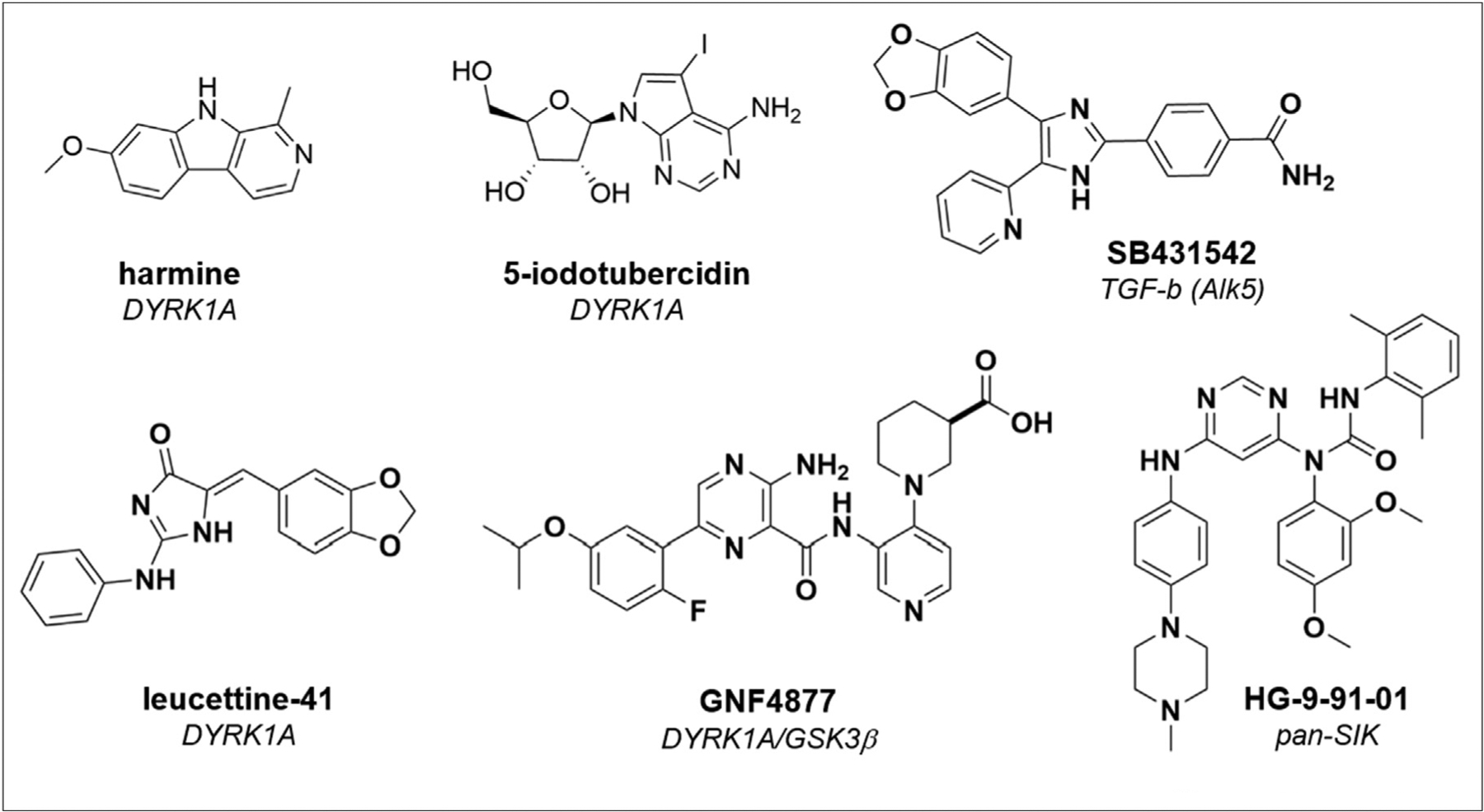
Representative small molecules reported to increase beta-cell proliferation. The mechanism of action for each compound is indicated below the name, and details about the biology of these compounds are included in the text.
In order to expand the chemical matter available in islet biology, several groups developed screening platforms for unbiased identification of compounds and new targets for beta-cell proliferation [14–20]. While some screening efforts focused on rat islets, which are more readily available [15], the challenge of this approach is the attrition of many compounds when translating findings to human islets, due to the additional brakes on the cell cycle applied in the latter tissue [21]. As a result, more attention has been paid to screening dissociated human islet cells, either using chemical libraries [14,19,20] or RNAi reagents [17•]. The use of EdU labeling can provide a larger signal-to-noise ratio over time in culture than staining for Ki67, which provides a snapshot of cell division. The use of zebrafish in whole-organism screens for beta-cell proliferation, similar to the cases of developmental and neurobiological screening [22,23], has also yielded good results. Using a FUCCI-expressing [24] zebrafish model for screening revealed compounds affecting the serotonin and retinoic acid pathways, as well as well-known glucocorticoids [18]. More recently, inhibition of salt-inducible kinase (SIK) by the pan-SIK inhibitor HG-9-91-01 resulted in a strong induction of proliferation in zebrafish beta cells, with a present (but milder) increase in human beta cells [16]. Together, these approaches will enhance our ability to expand beta cells as well as our understanding of the mechanisms required.
Open questions remain in this field. Key among them: first, can we find new targets to promote beta-cell division? The TGFβ pathway has been especially scrutinized, as small-molecule inhibition of Alk5 reduces p16INK4A expression and Smad3 transcriptional activity [25]. Modulation of the E-prostanoid receptors EP3 and EP4, which are regulated by the proliferation-associated FoxM1, with small molecules has also been shown to promote human beta-cell proliferation [26]. Further targets may emerge from screening, but most efforts have converged on these common threads of biology. A second major question is the specificity of these compounds to beta-cell proliferation; in addition to concerns about inducing oncogenesis, recent work questioning the effects on alpha cells and other cell types have raised specificity concerns [27]. Thus, selective delivery of compounds to beta cells, mostly focused on leveraging the very high levels of zinc in these cells, has been proposed as a safer therapeutic approach for the future. A third key question in the field regards whether beta-cell maturity is maintained in these proliferating cells. A mouse model in which insulin expression was reduced by w50% demonstrated a two-fold increase in beta-cell proliferation, accompanied by downregulation of master regulators of beta-cell identity and, somewhat surprisingly, a coordinate increase in glucagon expression [28]. Similarly, an evaluation of Myc overexpression in beta cells showed that the proliferating cells have lost some of their maturity. Further study of these effects, including whether this effect can be reversed after beta-cell expansion, are required to determine the ultimately translational impact of this approach.
Promoting beta-cell survival
Because beta cells are lost in both type 1 and type 2 diabetes, much energy has been put into finding small molecules that can protect beta-cell mass in the first place. Given the relative simplicity of a viability phenotype for high-throughput screening, small-molecule discovery in an effort to promote beta-cell survival has a slightly longer history. In a fashion similar to cancer biology, assay readouts detecting cell numbers (e.g., CellTiter-Glo, MTT) or induction of apoptosis (e.g., caspase activation) have been used. However, the opposite direction is desired here: rather than identify compounds that induce apoptosis in cancer cell lines, screening here aims to detect compounds that enable beta cells to remain impervious to the perturbation. Three primary models have been used for small-molecule suppressor screening. For compounds relevant to type 1 diabetes, inflammatory cytokines (usually a combination of IL-1β + IFN-γ or IL-β + IFN-γ + TNFα) are used. For compounds relevant to type 2 diabetes, free fatty acids, such as sodium palmitate, in the presence of high glucose (typically called “glucolipotoxicity”, or GLT) provide a model of overnutrition and hyperglycemia. Finally, several efforts have aimed to more specifically suppress ER stress in the beta cell, using perturbations such as tunicamycin or thapsigargin; theoretically, this modality could be applicable to either T1D or T2D, as ER stress appears to play an important role in cell death in both cases.
Suppression of inflammatory cytokines
In T1D, the immune system recognizes the beta cell as foreign, resulting in beta-cell death and thus the need for insulin therapy. IL-1β and TNF-α induce NFκB expression and nitric oxide (NO) signaling, increasing ER stress pathways [29]. IFN-γ-induced STAT1 signaling [29,30] works together with NFκB activation to abolish insulin secretion and to induce beta-cell apoptosis. Protein-based receptor antagonists had previously progressed to clinical trials, but have not yet led to approved therapies [31]. Cell replacement therapies, involving the implantation of stem cell-derived beta cells, are showing great promise, but routine clinical implementation is still a ways off, and many approaches will also require immunosuppression. On the other hand, discovery of small-molecule suppressors of cytokine-induced beta-cell apoptosis could lead to the identification of new cellular targets as well as candidate therapeutics (Figure 2a).
Figure 2.
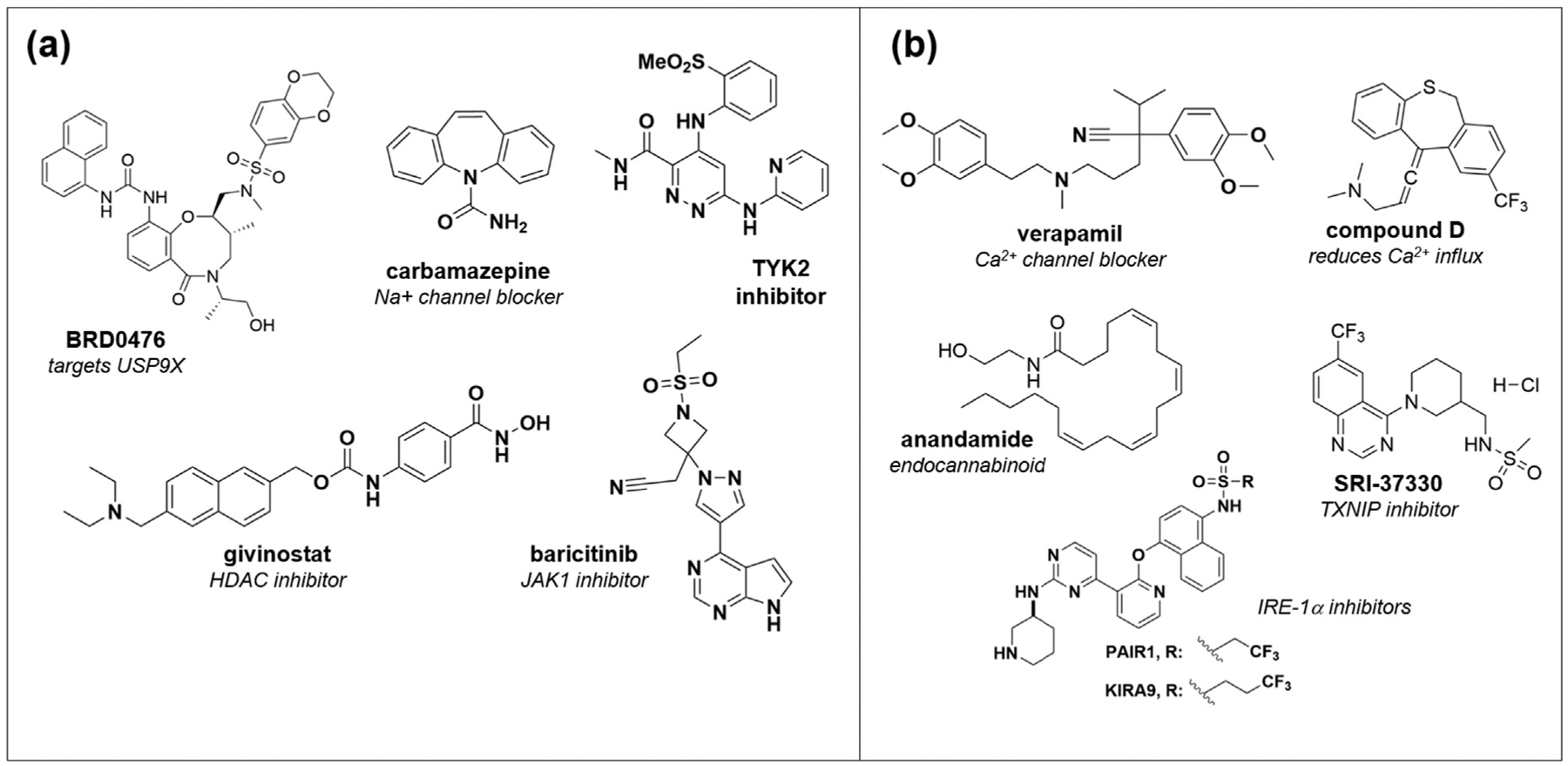
Representative small molecules that protect beta cells from death induced by (a) inflammatory cytokines or (b) glucolipotoxicity and ER stress.
Numerous natural products with anti-inflammatory properties have been shown to protect beta cells from cytokine-induced apoptosis, including resveratrol [32], silymarin [33], and wedelolactone in zebrafish [34]. To systematically identify novel synthetic suppressors of cytokine-based beta-cell death, we performed a high-throughput screen of nearly 400,000 compounds, using the rat INS-1E insulinoma cell line, leading to the discovery of BRD0476 [35–37•], a molecule derived from diversity-oriented synthesis (DOS) [38]. BRD0476 suppressed the activity of the triple cytokine cocktail in cell lines as well as human islets, and mechanism-of-action studies led to the finding that BRD0476 binds the deubiquitinase (DUB) USP9X, leading to a decrease in IFN-γ-induced STAT1 signaling through JAK2 [36]. Study of the interaction between USP9X and JAK2 is ongoing, and highlights the power of a phenotypic screening approach, as this DUB had never been associated with beta cells or even with diabetes in the past. In a more target-focused approach, we also found that inhibition of HDAC3 is able to suppress beta-cell death in the presence of both cytokine treatment and GLT conditions [39,40], likely due to more general anti-inflammatory properties. These inhibitors can also reduce the development of diabetes in nonobese diabetic (NOD) mice, a model of autoimmune disease [41]. The more general involvement of chromatin-modifying enzymes in ER stress has been explored in great depth by the Mandrup–Poulsen lab, who first identified the role of histone deacetylases [42••] and, more recently, lysine demethylases [43] in beta-cell apoptosis induced by cytokines.
Other groups have taken high-content screening approaches to discover small molecules that can protect beta cells [44,45]. For example, Yang et al. used a four-parameter live-cell imaging assay to measure several aspects of apoptosis in the mouse MIN6 insulinoma cell line [44]. Screening the Prestwick Chemical Library, they focused on the hit carbamazepine, a sodium channel inhibitor used to treat epilepsy, and found that the compound’s use-dependent inhibition was responsible for activity. Carbamazepine also inhibited ER signaling, so it is not clear whether this mechanism is specific to cytokine signaling. In other areas, most recently, kinase signaling downstream of IFN-α signaling has been explored, with inhibitors of JAK1 and the JAK-related kinase TYK2 undergoing preclinical evaluation [46,47•].
Suppression of glucolipotoxicity
Of the phenotypes typically explored in beta cells, it seems the largest number of screens have been performed to suppress beta-cell apoptosis induced by ER stress, including that induced by palmitate (Figure 2b). For example, 17,600 compounds were screened to find suppressors of tunicamycin in the mouse βTC6 cell line [48], leading to a series of 2,4-diaminoquinazolines [49]. Importantly, these compounds were also evaluated for their effects on proinsulin misfolding, a vulnerability in beta cells due to the massive insulin protein production needed to have a ready pool for secretion [49]. Other screening efforts identified GSK3β inhibitors and endocannabinoids such as anandamide [50]. And in a direct effort to counteract ER stress pathways in the beta cell, the Maly and Papa labs have developed reagents inhibiting IRE1α kinase/RNase activity (KIRAs) or partial antagonists of its RNase activity (PAIRs) [51–53•]. These approaches have shown promise in vitro and in vivo. Oxidative stress in the beta cell also plays an important role in cell death due to glucolipotoxicity [54,55]. For example, N-acetyl cysteine [56] and other antioxidants such as rosmarinic acid [57] can protect the beta cell from oxidative stress due to glucose and palmitate treatment.
An examination of the literature describing suppressors of ER stress, oxidative stress, and glucolipotoxicity reveals a common theme that inhibition of calcium influx can protect beta cells. Shalev et al. first described that the cardiovascular drug verapamil inhibits TXNIP expression (thioredoxin-interacting protein, important to cellular redox control) and protects beta cells both in culture and in ob/ob mice [58]. Indeed, a later analysis of patients taking verapamil revealed a mild reduction in type 2 diabetes [59], and a randomized double-blind placebo-controlled phase 2 trial of verapamil in new-onset type 1 diabetes found that patients had an improved C-peptide response to a mixed meal and reduced insulin requirements [60]. The same lab later screened ~300,000 compounds, focused on reducing TXNIP expression in islets, and found SRI-37330, which also prevented fatty liver and inhibited gluconeogenesis in mice, though perhaps through a different mechanism [61••]. Related to calcium signaling, NMDA receptors also conduct inward calcium currents, and dextromethorphan, a centrally acting cough suppressant and noncompetitive NMDAR antagonist, was shown to improve islet insulin content, insulin secretion, and viability in mouse and human islets [62,63]•. In another effort to protect beta cells in the presence of palmitate, a group composed of researchers across industry screened 312,000 compounds and identified a chemical series that reduced cytosolic calcium overload by reducing calcium influx [64•]. However, unlike nifedipine, a specific L-type calcium channel inhibitor, this series, including “compound D,” resulted in maintenance of optimal calcium levels in the beta cell. Although the target remains unknown, this work provides further evidence that calcium levels in the beta cell are so important for viability. This realization is leading to more focused efforts to screen directly for, for example, compounds that inhibit ER calcium depletion to stabilize ER-resident proteins [65]. Together, these results suggest that modulating calcium dynamics in the beta cell may result in greater viability and function.
Maintaining beta-cell maturity and function
Coupled with efforts to protect beta-cell viability are studies to find compounds that help keep beta cells mature and functional (i.e., showing appropriate glucose-stimulated insulin secretion). Previous studies have called into question whether beta cells undergo cell death or dedifferentiation during the development of diabetes [66•], and new chemical probes could help shed light on this area (Figure 3a). Thus far, relatively few unbiased efforts have been reported. We screened human PANC-1 ductal cancer cells for compounds that could induce PDX1 expression, and reported the discovery of BRD7552 [67], while the Johnson lab found that carbamazepine, the sodium channel inhibitor that protected beta cells from ER stress as described above, also positive modulates Ins1 and Ins2 expression in mouse islets [68]. Further, an evaluation of the reversal of dedifferentiation, focused on the urocortin 3 gene as a biomarker of beta-cell identity, found that TGFβ inhibitors can restore beta-cell differentiation over extended time in cell culture [69•]. As in the case of beta-cell proliferation, high-content screens in zebrafish for beta-cell function have also yielded tool compounds for follow-up [70,71].
Figure 3.
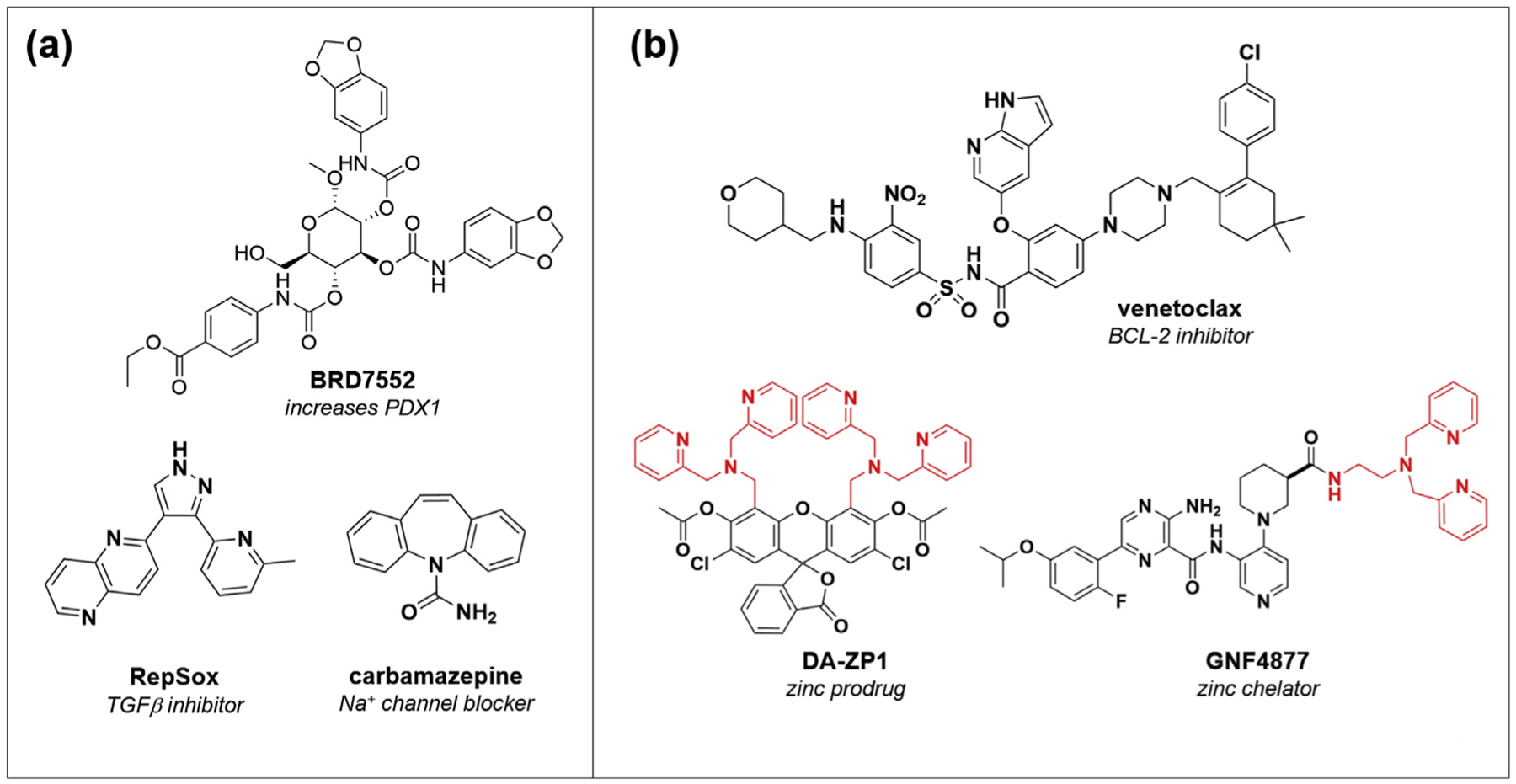
(a) Small molecules and mechanisms that enhance beta-cell maturity and function. (b) Emerging strategies to target senescent beta cells (Venetoclax) or to enable selective delivery to beta cells, due to their high levels of zinc, either through compound chelation or prodrug strategies.
Ultimately, the primary role of the beta cell is to secrete insulin in response to glucose, so direct screening for insulin secretagogues is of great interest to the beta-cell field. Historically, scarce methods available for high-throughput detection of insulin secretion limited discovery. Nonetheless, a number of mechanisms have been pursued to develop small molecules that can promote insulin secretion, including activation of glucokinase [72], activation of the GPCR free fatty acid receptor 1 (GPR40) [73], and inhibitors of the ATP-dependent potassium channel [74,75]. However, many of these mechanisms remain glucose-independent, so finding new glucose-dependent activities is an important next step in the field. Our group reported a luciferase-based assay, in which Gaussia luciferase was inserted into the C-peptide portion of proinsulin, that is highly correlated with traditional ELISA methods and enables HTS for this phenotype [76••]. Several screens have been performed using this modality; so far inhibitors but no inducers of insulin secretion have been reported [77–79]. The development of small molecule-based GLP-1R agonists, for example, may represent the best chance at success in this area.
Conclusions and outlook
An increasing appreciation for the power of small molecules to provide new insights into beta-cell biology has led to the exploration of additional phenotypes. For example, beta-cell senescence is an emerging area of great interest; recent reports indicate that insulin resistance induces a senescent state [80], and senolysis with Navitoclax or Venetoclax results in improved glucose metabolism, beta-cell function, and even prevention of autoimmune diabetes (Figure 3b) [80,81••]. New compounds in this exciting area will improve our understanding of this process. From a cell-based standpoint, development of sophisticated three-dimensional culture systems, for example from stem cell-derived beta cells [82], will allow the discovery of new compounds with strong physiological relevance. Finally, the beta-cell field will benefit tremendously from integrating new and emerging chemical tools; an outstanding recent review provides a summary of the chemical toolbox available for studying beta-cell function [83••]. Therapeutically, delivery mechanisms that provide beta-cell specificity will improve the safety of mechanisms like inducing proliferation (Figure 3b). Due to the high levels of zinc in the beta cell, for example, zinc-based approaches to either sequester compounds [84] or develop a prodrug system [85,86•] have shown early promise. In conclusion, a greater integration of the principles of beta-cell biology with the tools available from chemical biology (Figure 4) will accelerate the study of this important cell type in diabetes.
Figure 4.
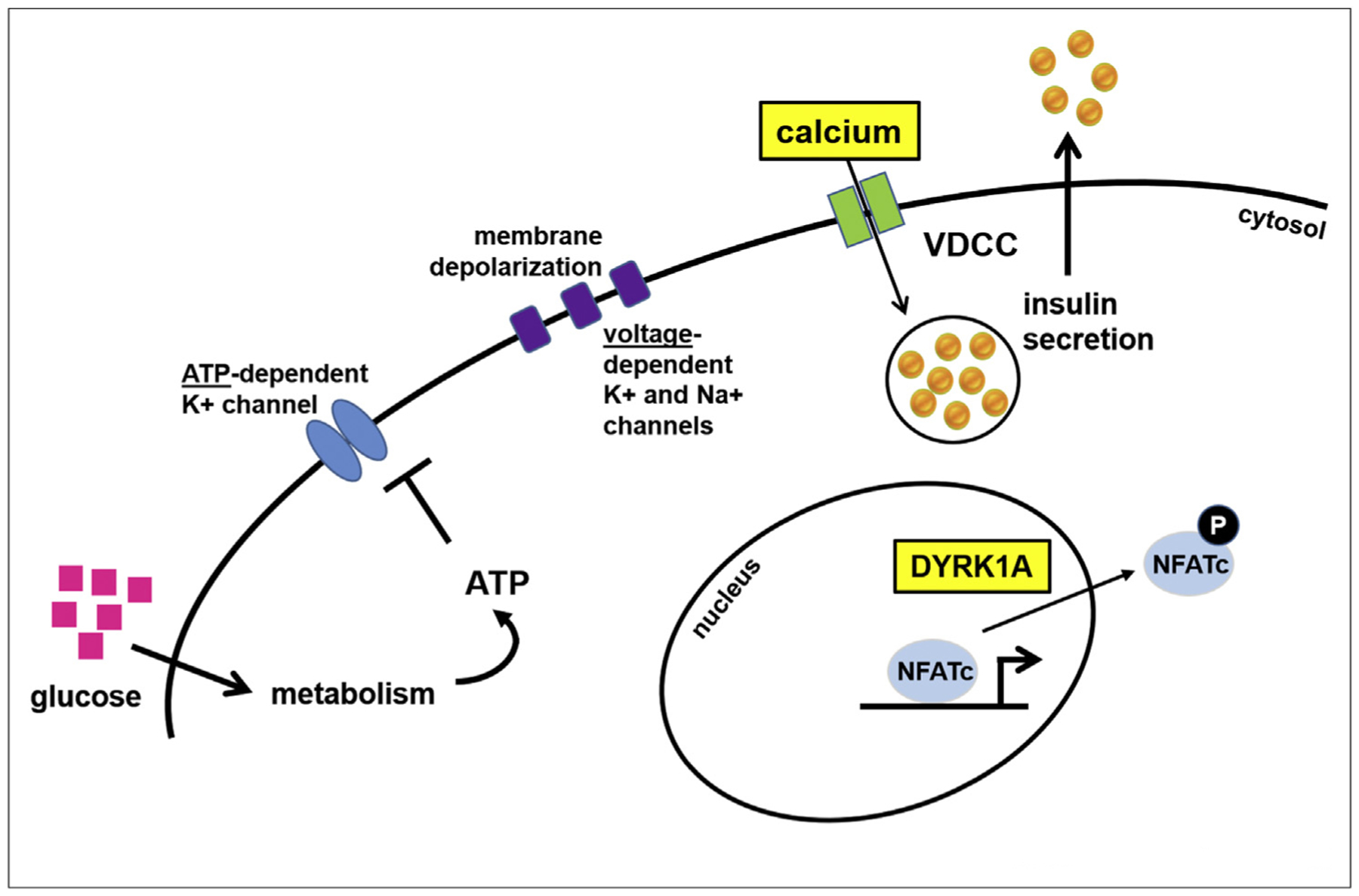
Summary of representative pathways affected in the beta cell. Two key pathways modulated by multiple compounds are highlighted: activation and inhibition of calcium influx into the beta cell, and inhibition of the kinase DYRK1A. VDCC, voltage-dependent calcium channel.
Funding source
B.K.W. is supported by U01-DK123717 and R01-DK129464 (NIDDK/NIH) and a Strategic Research Agreement with JDRF. B.K.W. is a Merkin Institute Fellow at the Broad Institute of MIT and Harvard.
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships that may be considered as potential competing interests: Bridget Wagner has patent inhibitors of histone deacetylase issued to Bridget Wagner. Bridget Wagner has patent compounds and methods for treating autoimmune diseases issued to Bridget Wagner. Bridget Wagner has patent targeted delivery to beta cells pending to Bridget Wagner.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* * of outstanding interest
- 1.Dor Y, Brown J, Martinez OI, Melton DA: Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429:41–46. [DOI] [PubMed] [Google Scholar]
- 2.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC: Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57: 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsons JA, Brelje TC, Sorenson RL: Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992, 130:1459–1466. [DOI] [PubMed] [Google Scholar]
- 4.Sorenson RL, Brelje TC: Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 1997, 29: 301–307. [DOI] [PubMed] [Google Scholar]
- 5.Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC: Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010, 53:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.••.Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dancik V, Burns SM, Gilbert TJ, Olson DE, et al. : Inhibition of DYRK1A stimulates human beta-cell proliferation. Diabetes 2016, 65:1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]; Along with Shen and Wang below, this paper provides evidence that DYRK1A inhibitors promote beta-cell proliferation in human islets, using 5-iodotubercidin in cell culture as well as in islets transplanted into humanized mouse models.
- 7.••.Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, Kamireddy A, Swafford A, Powers AF, Walker J, et al. : Inhibition of DYRK1A and GSK3β induces human beta-cell proliferation. Nat Commun 2015, 6:8372. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a series of aminopyrazines that increase beta-cell proliferation in human islets in culture as well as in vivo. Further evaluation showed that these compounds inhibit both DYRK1A and GSK-3β.
- 8.••.Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, et al. : A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med 2015, 21:383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]; Starting from a high-throughput screen for inducers of Myc, this paper demonstrates that the DYRK1A inhibitor harmine promotes beta-cell proliferation in human islets, also showing effects to stimulate NFAT signaling. Importantly, this group uses three different mouse models of beta-cell regeneration to show that harmine promotes glucose homeostasis.
- 9.Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA: Adenosine kinase inhibition selectively promotes rodent and porcine islet beta-cell replication. Proc Natl Acad Sci U S A 2012, 109:3915–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, Wilson J, Gonzalez BJ, Bender A, Takane KK, et al. : Combined inhibition of DYRK1A, SMAD, and trithorax pathways synergizes to induce robust replication in adult human beta cells. Cell Metabol 2019, 29:638–652. e635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ackeifi C,Wang P, Karakose E, Manning Fox JE, Gonzalez BJ, Liu H, Wilson J, Swartz E, Berrouet C, Li Y, et al. : GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human beta cell regeneration. Sci Transl Med 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackeifi C, Swartz E, Kumar K, Liu H, Chalada S, Karakose E, Scott DK, Garcia-Ocana A, Sanchez R, DeVita RJ, et al. : Pharmacologic and genetic approaches define human pancreatic beta cell mitogenic targets of DYRK1A inhibitors. JCI Insight 2020, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner BK, Schreiber SL: The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem Biol 2016, 23:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aamodt KI, Aramandla R, Brown JJ, Fiaschi-Taesch N, Wang P, Stewart AF, Brissova M, Powers AC: Development of a reliable automated screening system to identify small molecules and biologics that promote human beta-cell regeneration. Am J Physiol Endocrinol Metab 2016, 311:E859–E868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdolazimi Y, Zhao Z, Lee S, Xu H, Allegretti P, Horton TM, Yeh B, Moeller HP, Nichols RJ, McCutcheon D, et al. : CC-401 promotes beta-cell replication via pleiotropic consequences of DYRK1A/B inhibition. Endocrinology 2018, 159:3143–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charbord J, Ren L, Sharma RB, Johansson A, Agren R, Chu L, Tworus D, Schulz N, Charbord P, Stewart AF, et al. : In vivo screen identifies a SIK inhibitor that induces beta cell proliferation through a transient UPR. Nat Metab 2021, 3:682–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.•.Robitaille K, Rourke JL, McBane JE, Fu A, Baird S, Du Q, Kin T, Shapiro AM, Screaton RA: High-throughput functional genomics identifies regulators of primary human beta cell proliferation. J Biol Chem 2016, 291:4614–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a complementary approach to chemical screening, using RNAi screening. The authors observed that silencing of cell cycle-dependent kinase inhibitors p18 and p21 promotes human beta-cell proliferation, and provides a path forward to identifying new targets for modulation.
- 18.Tsuji N, Ninov N, Delawary M, Osman S, Roh AS, Gut P, Stainier DY: Whole organism high content screening identifies stimulators of pancreatic beta-cell proliferation. PLoS One 2014, 9, e104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walpita D, Hasaka T, Spoonamore J, Vetere A, Takane KK, Fomina-Yadlin D, Fiaschi-Taesch N, Shamji A, Clemons PA, Stewart AF, et al. : A human islet cell culture system for high-throughput screening. J Biomol Screen 2012, 17:509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walpita D, Wagner BK: Evaluation of compounds in primary human islet cell culture. Curr Protoc Chem Biol 2014, 6: 157–168. [DOI] [PubMed] [Google Scholar]
- 21.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, Troxell R, Wills R, Tanwir M, Casinelli G, Cox AE, Takane KK, Scott DK, et al. : Human pancreatic beta-cell G1/S molecule cell cycle atlas. Diabetes 2013, 62:2450–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruni G, Rennekamp AJ, Velenich A, McCarroll M, Gendelev L, Fertsch E, Taylor J, Lakhani P, Lensen D, Evron T, et al. : Zebrafish behavioral profiling identifies multitarget antipsychotic-like compounds. Nat Chem Biol 2016, 12: 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rihel J, Prober DA, Arvanites A, Lam K, Zimmerman S, Jang S, Haggarty SJ, Kokel D, Rubin LL, Peterson RT, et al. : Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 2010, 327:348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugiyama M, Sakaue-Sawano A, Iimura T, Fukami K, Kitaguchi T, Kawakami K, Okamoto H, Higashijima S, Miyawaki A: Illuminating cell-cycle progression in the developing zebrafish embryo. Proc Natl Acad Sci U S A 2009, 106:20812–20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhawan S, Dirice E, Kulkarni RN, Bhushan A: Inhibition of TGFbeta signaling promotes human pancreatic beta-cell replication. Diabetes 2016, 65:1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carboneau BA, Allan JA, Townsend SE, Kimple ME, Breyer RM, Gannon M: Opposing effects of prostaglandin E2 receptors EP3 and EP4 on mouse and human beta-cell survival and proliferation. Mol Metabol 2017, 6:548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maachi H, Ghislain J, Tremblay C, Poitout V: Pronounced proliferation of non-beta cells in response to beta-cell mitogens in isolated human islets of Langerhans. Sci Rep 2021, 11: 11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabat M, Page MM, Panzhinskiy E, Skovso S, Mojibian M, Fernandez-Tajes J, Bruin JE, Bround MJ, Lee JT, Xu EE, et al. : Reduced insulin production relieves endoplasmic reticulum stress and induces beta cell proliferation. Cell Metabol 2016, 23:179–193. [DOI] [PubMed] [Google Scholar]
- 29.Gysemans CA, Ladriere L, Callewaert H, Rasschaert J, Flamez D, Levy DE, Matthys P, Eizirik DL, Mathieu C: Disruption of the gamma-interferon signaling pathway at the level of signal transducer and activator of transcription-1 prevents immune destruction of beta-cells. Diabetes 2005, 54:2396–2403. [DOI] [PubMed] [Google Scholar]
- 30.Moore F, Naamane N, Colli ML, Bouckenooghe T, Ortis F, Gurzov EN, Igoillo-Esteve M, Mathieu C, Bontempi G, Thykjaer T, et al. : STAT1 is a master regulator of pancreatic {beta}-cell apoptosis and islet inflammation. J Biol Chem 2011, 286: 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY: Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007, 356:1517–1526. [DOI] [PubMed] [Google Scholar]
- 32.Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, Park JW, Kwon KB, Park BH: Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 2009, 58:344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda T, Ferreri K, Todorov I, Kuroda Y, Smith CV, Kandeel F, Mullen Y: Silymarin protects pancreatic beta-cells against cytokine-mediated toxicity: implication of c-Jun NH2-terminal kinase and janus kinase/signal transducer and activator of transcription pathways. Endocrinology 2005, 146:175–185. [DOI] [PubMed] [Google Scholar]
- 34.Delgadillo-Silva LF, Tsakmaki A, Akhtar N, Franklin ZJ, Konantz J, Bewick GA, Ninov N: Modelling pancreatic beta-cell inflammation in zebrafish identifies the natural product wedelolactone for human islet protection. Dis Model Mech 2019, 12, dmm036004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou DH, Bodycombe NE, Carrinski HA, Lewis TA, Clemons PA, Schreiber SL, Wagner BK: Small-molecule suppressors of cytokine-induced beta-cell apoptosis. ACS Chem Biol 2010, 5: 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.•.Chou DH, Vetere A, Choudhary A, Scully SS, Schenone M, Tang A, Gomez R, Burns SM, Lundh M, Vital T, et al. : Kinase-independent small-molecule inhibition of JAK-STAT signaling. J Am Chem Soc 2015, 137:7929–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a small-molecule suppressor of cytokinase-induced beta-cell apoptosis, and determines an unusual mechanism of inhibition of the JAK-STAT pathway without kinase inhibition. By modulating the deubiquitinase USP9X, this compound changes the balance between mutually exclusive phosphorylation and ubiquitination of JAK2, alleviating inflammatory signaling.
- 37.Chou DHC, Duvall JR, Gerard B, Liu H, Pandya BA, Suh BC, Forbeck EM, Faloon P, Wagner BK, Marcaurelle LA: Synthesis of a novel suppressor of β-cell apoptosis via diversity-oriented synthesis. ACS Med Chem Lett 2011, 2:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerry CJ, Schreiber SL: Chemical probes and drug leads from advances in synthetic planning and methodology. Nat Rev Drug Discov 2018, 17:333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou DH, Holson EB, Wagner FF, Tang AJ, Maglathlin RL, Lewis TA, Schreiber SL, Wagner BK: Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem Biol 2012, 19:669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagner FF, Lundh M, Kaya T, McCarren P, Zhang YL, Chattopadhyay S, Gale JP, Galbo T, Fisher SL, Meier BC, et al. : An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in beta-cell protection. ACS Chem Biol 2016, 11:363–374. [DOI] [PubMed] [Google Scholar]
- 41.Dirice E, Ng RWS, Martinez R, Hu J, Wagner FF, Holson EB, Wagner BK, Kulkarni RN: Isoform-selective inhibitor of histone deacetylase 3 (HDAC3) limits pancreatic islet infiltration and protects female nonobese diabetic mice from diabetes. J Biol Chem 2017, 292:17598–17608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.••.Larsen L, Tonnesen M, Ronn SG, Storling J, Jorgensen S, Mascagni P, Dinarello CA, Billestrup N, Mandrup-Poulsen T: Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia 2007, 50: 779–789. [DOI] [PubMed] [Google Scholar]; This paper is the first report that HDAC inhbition can protect beta cells from cytokine-induced apoptosis. This work opened up a field of study of epigenetic mechanisms for beta-cell protection, and subsequent efforts have shown that HDAC inhibitors can also protect beta cells from glucolipotoxicity.
- 43.Backe MB, Andersson JL, Bacos K, Christensen DP, Hansen JB, Dorosz JJ, Gajhede M, Dahlby T, Bysani M, Kristensen LH, et al. : Lysine demethylase inhibition protects pancreatic beta cells from apoptosis and improves beta-cell function. Mol Cell Endocrinol 2018, 460:47–56. [DOI] [PubMed] [Google Scholar]
- 44.Yang YH, Vilin YY, Roberge M, Kurata HT, Johnson JD: Multi-parameter screening reveals a role for Na+ channels in cytokine-induced beta-cell death. Mol Endocrinol 2014, 28: 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong L, Tran T, Baguley TD, Lee SJ, Henke A, To A, Li S, Yu S, Grieco FA, Roland J, et al. : A novel inhibitor of inducible NOS dimerization protects against cytokine-induced rat beta cell dysfunction. Br J Pharmacol 2018, 175: 3470–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colli ML, Ramos-Rodriguez M, Nakayasu ES, Alvelos MI, Lopes M, Hill JLE, Turatsinze JV, Coomans de Brachene A, Russell MA, Raurell-Vila H, et al. : An integrated multi-omics approach identifies the landscape of interferon-alpha-mediated responses of human pancreatic beta cells. Nat Commun 2020, 11:2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.•.Coomans de Brachene A, Castela A, Op de Beeck A, Mirmira RG, Marselli L, Marchetti P, Masse C, Miao W, Leit S, Evans-Molina C, et al. : Preclinical evaluation of tyrosine kinase 2 inhibitors for human beta-cell protection in type 1 diabetes. Diabetes Obes Metabol 2020, 22:1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper focuses on a new target for beta-cell protection, TYK2, and the effects of a selective kinase inhibitor. Related to JAK1, TYK2 inhibition has promise as a new target for type 1 diabetes.
- 48.Tran K, Li Y, Duan H, Arora D, Lim HY, Wang W: Identification of small molecules that protect pancreatic beta cells against endoplasmic reticulum stress-induced cell death. ACS Chem Biol 2014, 9:2796–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duan H, Lee JW, Moon SW, Arora D, Li Y, Lim HY, Wang W: Discovery, synthesis, and evaluation of 2,4-diaminoquinazolines as a novel class of pancreatic beta-cell-protective agents against endoplasmic reticulum (ER) stress. J Med Chem 2016, 59:7783–7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SH, Cunha D, Piermarocchi C, Paternostro G, Pinkerton A, Ladriere L, Marchetti P, Eizirik DL, Cnop M, Levine F: High-throughput screening and bioinformatic analysis to ascertain compounds that prevent saturated fatty acid-induced beta-cell apoptosis. Biochem Pharmacol 2017, 138:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.•.Feldman HC, Ghosh R, Auyeung VC, Mueller JL, Kim JH, Potter ZE, Vidadala VN, Perera BGK, Olivier A, Backes BJ, et al. : ATP-competitive partial antagonists of the IRE1alpha RNase segregate outputs of the UPR. Nat Chem Biol 2021, 17: 1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the development of PAIRs (partial antagonists of IRE1α RNase activity), which can serve as potent inhibitors of apoptotic ER stress, as opposed to adaptive ER stress. This group previously developed dual inhibitors of IRE1α kinase and RNase activity, and through careful medicinal chemistry, found compounds that can dissect these two activities.
- 52.Feldman HC, Vidadala VN, Potter ZE, Papa FR, Backes BJ, Maly DJ: Development of a chemical toolset for studying the paralog-specific function of IRE1. ACS Chem Biol 2019, 14: 2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, Mehdizadeh M, Ghosh R, Wang L, Colon-Negron K, et al. : Targeting ABL-IRE1alpha signaling spares ER-stressed pancreatic beta cells to reverse autoimmune diabetes. Cell Metabol 2017, 25:883–897. e888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stancill JS, Corbett JA: The role of thioredoxin/peroxiredoxin in the beta-cell defense against oxidative damage. Front Endocrinol 2021, 12:718235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreadi A, Bellia A, Di Daniele N, Meloni M, Lauro R, Della-Morte D, Lauro D: The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: a target for new therapies against cardiovascular diseases. Curr Opin Pharmacol 2022, 62:85–96. [DOI] [PubMed] [Google Scholar]
- 56.Alnahdi A, John A, Raza H: N-acetyl cysteine attenuates oxidative stress and glutathione-dependent redox imbalance caused by high glucose/high palmitic acid treatment in pancreatic Rin-5F cells. PLoS One 2019, 14, e0226696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Govindaraj J, Sorimuthu Pillai S: Rosmarinic acid modulates the antioxidant status and protects pancreatic tissues from glucolipotoxicity mediated oxidative stress in high-fat diet: streptozotocin-induced diabetic rats. Mol Cell Biochem 2015, 404:143–159. [DOI] [PubMed] [Google Scholar]
- 58.Xu G, Chen J, Jing G, Shalev A: Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 2012, 61: 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin T, Kuo SC, Chang YY, Chen YT, Wang KK: Verapamil use is associated with reduction of newly diagnosed diabetes mellitus. J Clin Endocrinol Metab 2017, 102:2604–2610. [DOI] [PubMed] [Google Scholar]
- 60.Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, Li P, Shalev A: Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med 2018, 24:1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.••.Thielen LA, Chen J, Jing G, Moukha-Chafiq O, Xu G, Jo S, Grayson TB, Lu B, Li P, Augelli-Szafran CE, et al. : Identification of an anti-diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metabol 2020, 32:353–365. e358. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a large-scale screen to identify compounds that decrease TXNIP expression for diabetes treatment. The compound they describe has surprising activity on expression, beta-cell survival, and glucagon activity from alpha cells, suggesting multiple effects emerging from this mechanism.
- 62.•.Marquard J, Otter S, Welters A, Stirban A, Fischer A, Eglinger J, Herebian D, Kletke O, Klemen MS, Stozer A, et al. : Character ization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat Med 2015, 21:363–372. [DOI] [PubMed] [Google Scholar]; This paper identifies NMDA receptors as new targets in the beta cell. Dextromethorphan, a commonly used cough suppressant, also showed impressive activity in improving islet insulin content, insulin secretion, and viability in mouse and human islets.
- 63.Scholz O, Otter S, Welters A, Wormeyer L, Dolensek J, Klemen MS, Pohorec V, Eberhard D, Mrugala J, Hamacher A, et al. : Peripherally active dextromethorphan derivatives lower blood glucose levels by targeting pancreatic islets. Cell Chem Biol 2021, 28:1474–1488. e1477. [DOI] [PubMed] [Google Scholar]
- 64.•.Vogel J, Yin J, Su L, Wang SX, Zessis R, Fowler S, Chiu CH, Wilson AC, Chen A, Zecri F, et al. : A phenotypic screen identifies calcium overload as a key mechanism of beta-cell glucolipotoxicity. Diabetes 2020, 69:1032–1041. [DOI] [PubMed] [Google Scholar]; This paper describes a large-scale screen to identify compounds that protect beta cells from glucolipotoxicity. The compound they focus on inhibits calcium influx, causing an overall reduction in calcium overload in the beta cell. Importantly, this compound does not appear to be a conventional channel blocker, and future mechanistic studies may yield a new target for modulation.
- 65.Henderson MJ, Trychta KA, Yang SM, Back S, Yasgar A, Wires ES, Danchik C, Yan X, Yano H, Shi L, et al. : A target-agnostic screen identifies approved drugs to stabilize the endoplasmic reticulum-resident proteome. Cell Rep 2021, 35:109040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.•.Talchai C, Xuan S, Lin HV, Sussel L, Accili D: Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150:1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper, important to the diabetes field, was one of the first to suggest that beta-cell death is not actually the primary driver of beta-cell failure in diabetes. Instead, the authors provide evidence that beta-cell dedifferentiation is what occurs and results in loss of insulin secretion and other functions.
- 67.Yuan Y, Hartland K, Boskovic Z, Wang Y, Walpita D, Lysy PA, Zhong C, Young DW, Kim YK, Tolliday NJ, et al. : A small-molecule inducer of PDX1 expression identified by high-throughput screening. Chem Biol 2013, 20:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Szabat M, Modi H, Ramracheya R, Girbinger V, Chan F, Lee JT, Piske M, Kamal S, Carol Yang YH, Welling A, et al. : High-content screening identifies a role for Na(+) channels in insulin production. R Soc Open Sci 2015, 2:150306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.•.Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL, Melton DA: Reversal of beta cell de-differentiation by a small molecule inhibitor of the TGFbeta pathway. Elife 2014, 3, e02809. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the loss of urocortin 3, a beta-cell marker, during early stages of type 2 diabetes, providing more evidence that beta-cell dedifferentiation is important to pathogenesis. Screening growth factors and other known pathways revealed that TGFβ inhibition can reverse this process, providing a path toward new candidate therapeutics.
- 70.Helker CSM, Mullapudi ST, Mueller LM, Preussner J, Tunaru S, Skog O, Kwon HB, Kreuder F, Lancman JJ, Bonnavion R, et al. : A whole organism small molecule screen identifies novel regulators of pancreatic endocrine development. Development 2019, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matsuda H, Mullapudi ST, Yang YHC, Masaki H, Hesselson D, Stainier DYR: Whole-organism chemical screening identifies modulators of pancreatic beta-cell function. Diabetes 2018, 67:2268–2279. [DOI] [PubMed] [Google Scholar]
- 72.Sarabu R, Bizzarro FT, Corbett WL, Dvorozniak MT, Geng W, Grippo JF, Haynes NE, Hutchings S, Garofalo L, Guertin KR, et al. : Discovery of piragliatin - first glucokinase activator studied in type 2 diabetic patients. J Med Chem 2012, 55: 7021–7036. [DOI] [PubMed] [Google Scholar]
- 73.Ito R, Tsujihata Y, Matsuda-Nagasumi K, Mori I, Negoro N, Takeuchi K: TAK-875, a GPR40/FFAR1 agonist, in combination with metformin prevents progression of diabetes and beta-cell dysfunction in Zucker diabetic fatty rats. Br J Pharmacol 2013, 170:568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson B, Satin LS: Beta-cell ion channels and their role in regulating insulin secretion. Compr Physiol 2021, 11:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin RJ, Yen YK, Lee CH, Hsieh SL, Chang YC, Juan YS, Long CY, Shen KP, Wu BN: Eugenosedin-A improves obesity-related hyperglycemia by regulating ATP-sensitive K(+) channels and insulin secretion in pancreatic beta cells. Biomed Pharmacother 2022, 145:112447. [DOI] [PubMed] [Google Scholar]
- 76.**.Burns SM, Vetere A, Walpita D, Dancik V, Khodier C, Perez J, Clemons PA, Wagner BK, Altshuler D: High-throughput luminescent reporter of insulin secretion for discovering regulators of pancreatic Beta-cell function. Cell Metabol 2015, 21:126–137. [DOI] [PubMed] [Google Scholar]; This paper describes a new luciferase-based assay system to measure insulin secretion for high-throughput screening. Historically, insulin secretion was measured by ELISA, and new miniaturized methods can speed the identification of new compounds for glucose-dependent enhancement of beta-cell function.
- 77.Hager R, Pitsch J, Kerbl-Knapp J, Neuhauser C, Ollinger N, Iken M, Ranner J, Mittermeier-Klessinger V, Dawid C, Lanzerstorfer P, et al. : A high-content screen for the identification of plant extracts with insulin secretion-modulating activity. Pharmaceuticals 2021, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kalwat MA, Hwang IH, Macho J, Grzemska MG, Yang JZ, McGlynn K, MacMillan JB, Cobb MH: Chromomycin A2 potently inhibits glucose-stimulated insulin secretion from pancreatic beta cells. J Gen Physiol 2018, 150:1747–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalwat MA, Wichaidit C, Nava Garcia AY, McCoy MK, McGlynn K, Hwang IH, MacMillan JB, Posner BA, Cobb MH: Insulin promoter-driven Gaussia luciferase-based insulin secretion biosensor assay for discovery of beta-cell glucose-sensing pathways. ACS Sens 2016, 1:1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, Bonner-Weir S: Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metabol 2019, 30:129–142. e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.••.Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A: Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metabol 2019, 29:1045–1060. e1010. [DOI] [PubMed] [Google Scholar]; This paper showed that beta-cell senescence is an important component of the loss of beta-cell mass in type 1 diabetes, in part due to the senescence-associated secretory phenotype (SASP). This process places the beta cell in a more active role in T1D, rather than merely a passive victim of immune attack. They used a BCL-2 inhibitor to eliminate these senescent beta cells and to protect the nonobese diabetic mouse model of autoimmune diabetes.
- 82.Ghazizadeh Z, Kao DI, Amin S, Cook B, Rao S, Zhou T, Zhang T, Xiang Z, Kenyon R, Kaymakcalan O, et al. : ROCKII inhibition promotes the maturation of human pancreatic beta-like cells. Nat Commun 2017, 8:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.••.Huey J, Keutler K, Schultz C: Chemical biology toolbox for studying pancreatic islet function - a perspective. Cell Chem Biol 2020, 27:1015–1031. [DOI] [PubMed] [Google Scholar]; This comprehensive review provides an overview of emerging chemical and genetic engineering technologies for evaluating islet function in vitro and in vivo, enabling the study of crosstalk between heterogeneous islet cell types.
- 84.Horton TM, Allegretti PA, Lee S, Moeller HP, Smith M, Annes JP: Zinc-chelating small molecules preferentially accumulate and function within pancreatic beta cells. Cell Chem Biol 2019, 26: 213–222. e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kahraman S, Manna D, Dirice E, Maji B, Small J, Wagner BK, Choudhary A, Kulkarni RN: Harnessing reaction-based probes to preferentially target pancreatic beta-cells and beta-like cells. Life Sci Alliance 2021, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.•.Lee M, Maji B, Manna D, Kahraman S, Elgamal RM, Small J, Kokkonda P, Vetere A, Goldberg JM, Lippard SJ, et al. : Native zinc catalyzes selective and traceless release of small molecules in beta-cells. J Am Chem Soc 2020, 142:6477–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the generation of a zinc-based prodrug system, by which the extraordinarily high levels of zinc in beta cells catalyzes hydrolysis and the scarless release of native small molecules only in beta cells. This work could have an impact especially on phenotypes where beta-cell selectivity is paramount (such as proliferation).